

Abstract
Immune therapies have had limited efficacy in high grade serous ovarian cancer (HGSC), as the cellular targets and mechanism(s) of action of these agents in HGSC are unknown. Here we performed immune functional and single cell RNA-seq transcriptional profiling on novel HGSC organoid/immune cell co-cultures treated with a unique bispecific anti-PD-1/PD-L1 antibody compared to monospecific anti-PD-1 or anti-PD-L1 controls. Comparing the functions of these agents across all immune cell types in real time identified key immune checkpoint blockade (ICB) targets that have eluded currently available monospecific therapies. The bispecific antibody induced superior cellular state changes in both T and NK cells. It uniquely induced NK cells to transition from inert to more active and cytotoxic phenotypes, implicating NK cells as a key missing component of the current ICB-induced immune response in HGSC. It also induced a subset of CD8 T cells to transition from naïve to more active and cytotoxic progenitor-exhausted phenotypes post-treatment, revealing the small, previously uncharacterized population of CD8 T cells responding to ICB in HGSC. These state changes were driven partially through bispecific antibody-induced downregulation of the bromodomain-containing protein BRD1. Small molecule inhibition of BRD1 induced similar state changes in vitro and demonstrated efficacy in vivo, validating the co-culture results. Our results demonstrate that state changes in both NK and a subset of T cells may be critical in inducing an effective anti-tumor immune response and suggest that immune therapies able to induce such cellular state changes, such as BRD1 inhibitors, may have increased efficacy in HGSC.
Keywords: Ovarian cancer, Immune therapy, T cell exhaustion, BRD1, NK cells
INTRODUCTION:
High grade serous ovarian cancer (HGSC) is the fifth leading cause of cancer death in women in the United States (1). Patients are diagnosed at late stage due to limited early detection methods, and are typically treated with combinations of surgery and chemotherapy (2). Though immune checkpoint blockade (ICB) agents demonstrate success in other solid tumor types, monospecific ICB antibodies exhibit minimal efficacy in HGSC (3,4). Many theories explore the contributions of different immune cell types to HGSC outcome and possibly ICB response. Poor HGSC outcomes associate with an increased proportion of tumor-associated macrophages (5). Positive outcomes associate with increased ratios of CD8 T cells to CD4 Tregs (6,7) and increased fractions of NK cells in ascites (8,9). Limited mechanistic data explain these findings. Based on work in ICB-responsive solid tumor types, it is likely that the quality of the T and NK cells, rather than their quantity, matters in eliciting ICB activity (10,11).
In most solid tumors, the tumor microenvironment induces dysfunction in T and possibly NK cells (12,13). In CD8 T cells, this dysfunction, called ‘exhaustion,’ is mediated by stable epigenetic reprogramming producing subsets of differentially functional cells including progenitor and terminally exhausted T cells (10,14,15). The contribution of naïve versus exhausted CD8 T cell subsets to ICB response, and the mechanism of NK cell dysfunction and ICB-triggered response, are still being defined in all solid tumor types, including HGSC (10,11,13,16).
A better understanding of cells critical for ICB response in HGSC and the effect of cell state on their response to ICB agents is needed. Organoid co-cultures containing patient-matched tumor and all intra-tumoral immune cells represent a human model that can be studied over time using ICB treatments to ask questions about ICB efficacy and mechanism of action (17,18).
Here we utilize short term patient derived HGSC organoid co-cultures containing tumor organoids and the full complement of intra-tumoral immune cells to functionally assess the mechanism of action of ICB agents in every type of immune cell in the culture. To detect key cellular and mechanistic targets evading current therapies, we compared the action of a novel bispecific anti-PD-1/PD-L1 ICB antibody (19) to its monospecific anti-PD-1 and anti-PD-L1 controls. We show that the bispecific antibody uniquely induces state changes in NK cells from inert to active states and most strongly induces a state change in CD8 T cells from naïve to cytotoxic progenitor exhausted states, with neither target previously demonstrated as critical for ICB response in HGSC. We demonstrate that both state changes are driven partially through down regulation of the bromodomain-containing protein BRD1. BRD1 inhibition by a small molecule inhibitor, BAY-299, recapitulates these state changes for T and NK cells in vitro and in vivo, and demonstrates efficacy in vivo, thereby validating the co-culture results and suggesting BRD1 as a possible immune therapy target. Taken together our data indicate that efficacy of immune therapies in HGSC can be driven by transition of NK and CD8 T cell subsets into active and cytotoxic states likely through epigenetic changes driven, in part, by BRD1 downregulation or inhibition removing some tumor microenvironment driven dysfunction.
MATERIALS AND METHODS:
For additional and detailed materials and methods, please see supplementary materials and methods.
Patient Samples:
HGSC tumor tissue was collected from twelve patients undergoing primary debulking surgery at Brigham and Women’s Hospital (BWH) and Dana-Farber Cancer Institute (DFCI) for organoid co-culture generation and functional testing between December 2019 and April 2020. Tumor tissue from patient 20–22 was obtained under BWH/Partners IRB approved discarded tissue protocol 2016P000559. Written informed consent was obtained for the remaining subjects on either BWH/Partners IRB approved protocol 2016P002819 or DFCI IRB approved protocol 02–051. All studies in this work were conducted in accordance with the U.S. Common Rule and Belmont Report and approved by the DFCI and BWH/Partners IRBs.
Co-culture generation:
Tumors were mechanically dissociated, diluted in DMEM (Life Tech # 11965–092), 10% FBS (Sigma # F2442–500), 1% Penicillin Streptomycin (Life Tech # 15140–122), and 2.5mg/mL Type II Collagenase (Life Technologies # 17101015), and shaken on a horizontal platform for 20 minutes at 37°C. The homogenate was filtered through a 100μm filter (Corning # 352360), pelleted, and washed in 1X Red Blood Cell Lysis buffer (Biolegend #420301). The cells were then resuspended in DMEM/10% FBS, counted, and diluted to a concentration of 6×106 cells (or organoids)/mL in DMEM, 10% FBS, 1% Pen/Strep, and 30ng/mL of IL-2 (Peprotech #200–02) mixed with 15% Matrigel (Corning #356231). This cell suspension was then plated into 48 well plates (USA Scientific # CC7672–7548) at 40μL per well and incubated at 37°C to allow for settling, then drug containing media was added to each well. All drug preparations were prepared in DMEM, 10% FBS, 1% Pen/Strep, and 30ng/mL of IL-2. Drugs included Anti-PD-1 (Selleck #A2005), IgGEn Isotype Control (provided by Eli Lilly), anti-PD-L1 (LY3300054), bispecific anti-PD-1/PD-L1 (LY3434172) (all antibodies used at 10μg/mL), and BAY-299 (MedChemExpress # HY-107424; used at 1μM). Co-cultures were harvested 96 hours after plating for all functional assays detailed in the supplementary methods.
Single Cell RNA sequencing:
Single cell RNA sequencing (scRNA-seq) experiments were performed by the BWH Single Cell Genomics Core. For scRNA-seq analysis of the organoid co-cultures, viable CD45+ cells from organoid co-cultures were isolated by FACS. Cells were stained with a distinct barcoded antibody (Cell-Hashing antibody, TotalSeq-A, Biolegend) as previously described (20). Next, 7,000 cells from each condition were resuspended in 0.4% BSA in PBS at a concentration of 1,000 cells per μL, pooled together, then loaded onto a single lane (Chromium chip, 10X Genomics) followed by encapsulation in a lipid droplet (Single Cell 3′kit V3, 10X Genomics) followed by cDNA and library generation according to the manufacturer’s protocol. For the scRNA-seq analysis of the parent tumor, 6,000 viable CD45+ cells and 4,000 CD45- (tumor and stromal cells) were pooled together and resuspended in 0.4% BSA in PBS at a concentration of 1,000 cells per μL. 10,000 cells were loaded onto a single lane (Chromium chip, 10X Genomics) followed by encapsulation in a lipid droplet (Single Cell 3′kit V3, 10X Genomics) followed by cDNA and library generation according to the manufacturer’s protocol.
mRNA libraries were sequenced to an average of 50,000 reads per cell and HTO (Cell Hashing antibodies) libraries sequenced to an average of 5,000 reads per cell, both using Illumina Novaseq. Please see supplementary materials and methods for single-cell RNA-seq data analysis methods.
In Vivo Study:
Massachusetts General Hospital Institutional Care and Use Committee (IACUC) protocol 2017N0000236 was used for this study. STOSE cells were cultured and expanded as described previously (21). Twenty 7–9 week-old FVB-N mice obtained from Jackson Labs (Bar Harbor, ME) were intraperitoneally injected with 1×107 STOSE cells in 250uL of PBS. Eighteen days post-injection, mice were weighed and randomized into two arms (10/arm). Randomization was done so that the average weights, weight gained compared to day 0, and standard deviations were close between the two arms upon starting treatment. The mice were treated with either vehicle (10% NMP (Fisher Cat # 390682500), 90%PEG400 (Sigma Cat # 202398–500G)) or 150mg/kg BAY-299 (MedChemExpress HY-107424) by oral gavage once per day for eighteen days. Animals were monitored by weight every three to four days and dosing was adjusted by weight. At the end of the study, mice were euthanized, and ascites and tumors were harvested. Preparation of ascites and tumor cell suspensions for analysis is described in supplementary methods.
Accession Number:
The sequencing data discussed in this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) database and are accessible through GEO Series accession number GSE160755.
RESULTS:
HGSC organoid-immune cell co-cultures resemble the immune microenvironment of their parent tumors:
Short term co-cultures containing tumor organoids and the full complement of intra-tumoral immune cells were generated from solid tumors from 12 HGSC patients, with two sites for one patient, making 13 total cultures (Figures 1 and S1A). Co-cultures were maintained in a limited growth matrix with minimal growth factor enrichment of the media; experiments occurred immediately after surgery and only lasted 96 hours. These strategies help prevent confounding alterations to the microenvironment which might occur in longer cultures such as clonal selection of T cells (22,23). Co-cultures were validated as matching parent tumors through visual, flow cytometry, and sequencing methods.
FIGURE 1: HGSC organoid co-cultures accurately mimic the parent tumors from which they were derived:
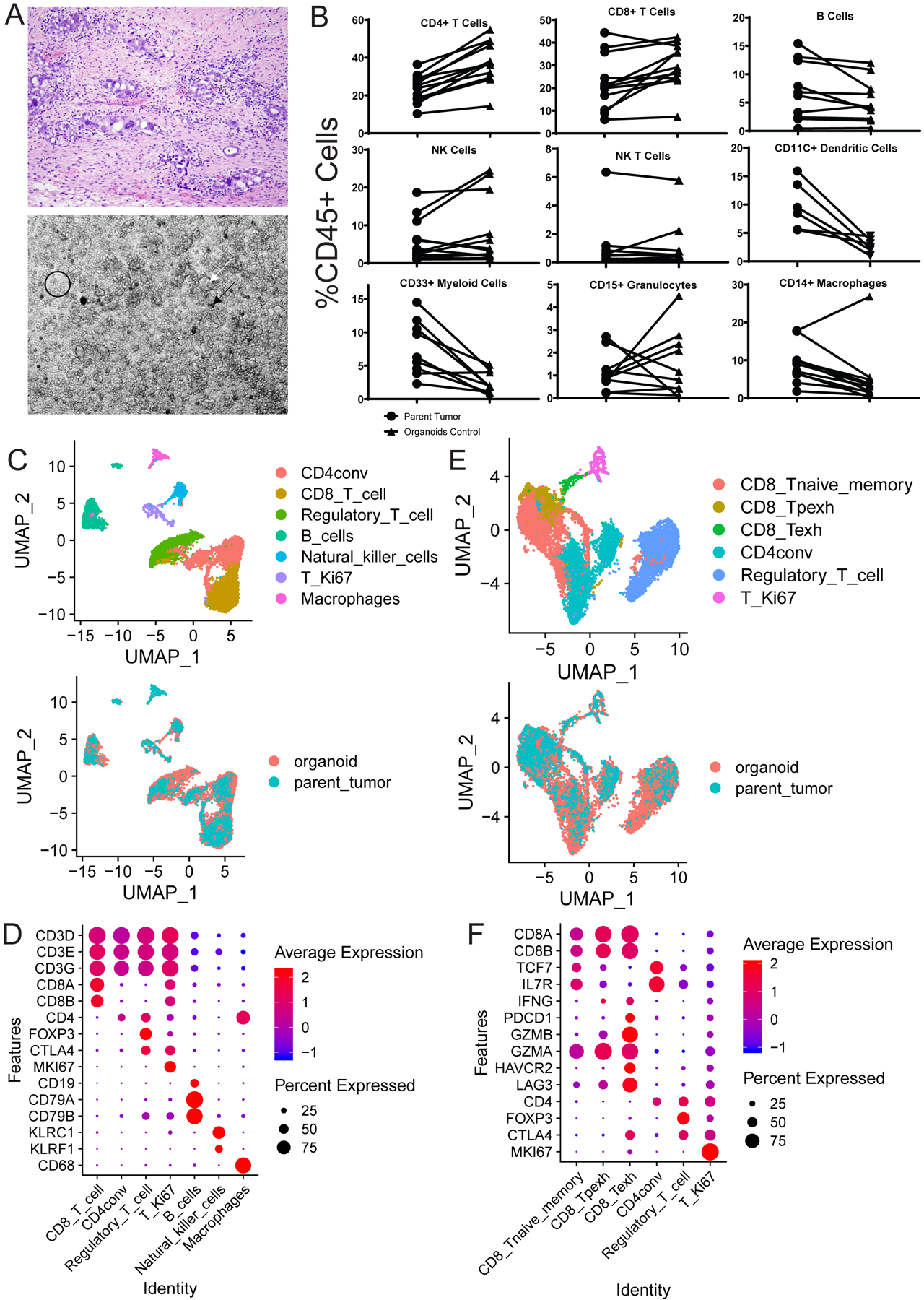
A) Organoid co-cultures (bottom) physically resemble the diverse cellular environment of the parent tumor (top) which includes cancer, stromal, and immune cells. The co-cultures contain tumor spheres (white arrow), clusters of single immune and stromal cells (black circle), and psammoma body calcifications (black arrow). B) Flow cytometry analysis for all immune cell types was performed on parent tumors and control treated organoids. Comparisons of each individual immune cell type between parent tumor (circles) and control treated organoid co-culture (triangles) as a percentage of viable CD45 positive cells are shown. C-F) scRNA-seq analysis results comparing the parent tumor and treated organoid co-cultures for patient 20–11. C) UMAPs are shown here to demonstrate concordance across all immune cell types between the parent tumor and organoid co-culture. All populations detected are shown on top with a color key on the right, and an overlay of these populations in the parent tumor (blue) and organoid co-cultures (orange) is shown below. D) Markers (Y axis) used to define each of the individual immune cell populations (X axis) in C are shown here along with the expression level in each defined cell type. The average expression level (colors) is shown in the percentage of cells (sphere) expressing each marker for each cell type. E) UMAPs are shown here to demonstrate concordance across all T cell types between the parent tumor and organoid co-culture. All populations detected are shown on top with a color key on the right, and an overlay of these populations in the parent tumor (blue) and organoid co-cultures (orange) is shown below. F) Markers (Y axis) used to define each of the different T cell populations (X axis) in E are shown here along with the expression level in each defined cell type. The average expression level (colors) is shown in the percentage of cells (sphere) expressing each marker for each cell type. CD8_Tnaive_memory= Naïve and memory CD8 T cells, CD4conv=Conventional non-regulatory CD4 T cells, Regulatory_T_cell=Regulatory CD4 T cells, T_Ki67=Proliferating T cells, CD8_Texh=Terminally exhausted CD8 T cells, CD8_Tpexh=Progenitor exhausted CD8 T cells
Brightfield microscopy revealed the co-cultures ‘look’ similar to their parent tumors and contain psammoma body calcifications and groups of tumor spheres permeated by sheets and clusters of immune cells (Figure 1A). Flow cytometry analysis of the parent tumors revealed a significant population of T cells with CD4 cells comprising the majority in most cases, similar to other HGSC studies (24), and revealed a significant population of NK and NK T cells (Figure S1B, S1C, and S2A). Flow cytometry comparison of the organoids and parent tumors revealed similar amounts of each immune cell type present in the co-cultures with overall proliferation of T and NK cells and an expected drop in myeloid cells over time (Figures 1B, S1C, S2A, and S2B). Significant PD-1, PD-L1, and TIM3 expression was found on relevant immune populations and tumor cells in the co-cultures (Figures S3 (A-D), S4 (A-C), and S5 (A-C)).
Single cell RNA sequencing (scRNA-seq) analysis comparing a single matched parent tumor and control treated organoid co-culture (20–11) revealed similar annotation of all immune cell types (Figure 1C) defined by standard immune markers (Figure 1D) with a decrease in macrophages mirroring flow cytometry (Figure 1B and S1C). Standard T cell subsets were detected (Figure 1E) using common subset markers (Figure 1F).
Overall, these flow cytometry and scRNA-seq results confirm that the organoid co-cultures authentically model the parent tumor immune microenvironment.
ICB antibodies reproducibly induce IFNγ production in co-cultures:
We next tested the immune activation capacity of ICB antibodies in our system. We sought to compare the efficacy and mechanism of action of a novel bispecific anti-PD-1/PD-L1 antibody (LY3434172) (19) to its monospecific controls, anti-PD-1 (Pembrolizumab) and anti-PD-L1 (LY3300054) (25) (Figure S6A). Monospecific ICB antibody combinations demonstrate some clinical success (26), but bispecific antibodies are hypothesized to be more effective given their ability to engage two different ICB receptors in close proximity (19,27). Indeed, compared to monospecific controls, the bispecific anti-PD-1/PD-L1 antibody (LY3434172) has been shown to increase T cell stimulation in a different in vitro system and anti-tumor efficacy in vivo in humanized ovarian and other tumor xenograft mouse models through unclear mechanisms (19). Based on this, we hypothesized that studying the function of the bispecific anti-PD-1/PD-L1 antibody, compared to controls, in our co-culture system could help dissect critical targets not being affected by available monospecific ICB antibodies.
To measure overall immune response, we performed IFNγ ELISA analysis on media from the 13 co-cultures post-treatment with a physiologic dose of each of the four antibodies individually. Pilot ELISA studies indicated 96 hours post-treatment as the best timepoint (Figure S6B). In six cases, media containing anti-PD-1 and anti-PD-L1 combined or an equivalent amount of IgG (IgG+IgG) was tested to determine whether increased bispecific antibody efficacy was due to an additive effect.
For every tumor analyzed, all three ICB antibodies induced significantly increased IFNγ production compared to the isotype control (Figures 2A, 2B, and S6C). The bispecific antibody induced the strongest IFNγ production, not attributable to an additive effect as evidenced by the lower IFNγ production for the anti-PD-1+anti-PD-L1 combination (Figure 2A and S6D).
FIGURE 2: ICB antibodies induce detectable IFNγ production in proportion to the parent tumor aggregate immune state, increased CD4, CD8, and NK cell activity, and a killing phenotype in CD8 T and NK cells in HGSC co-cultures:
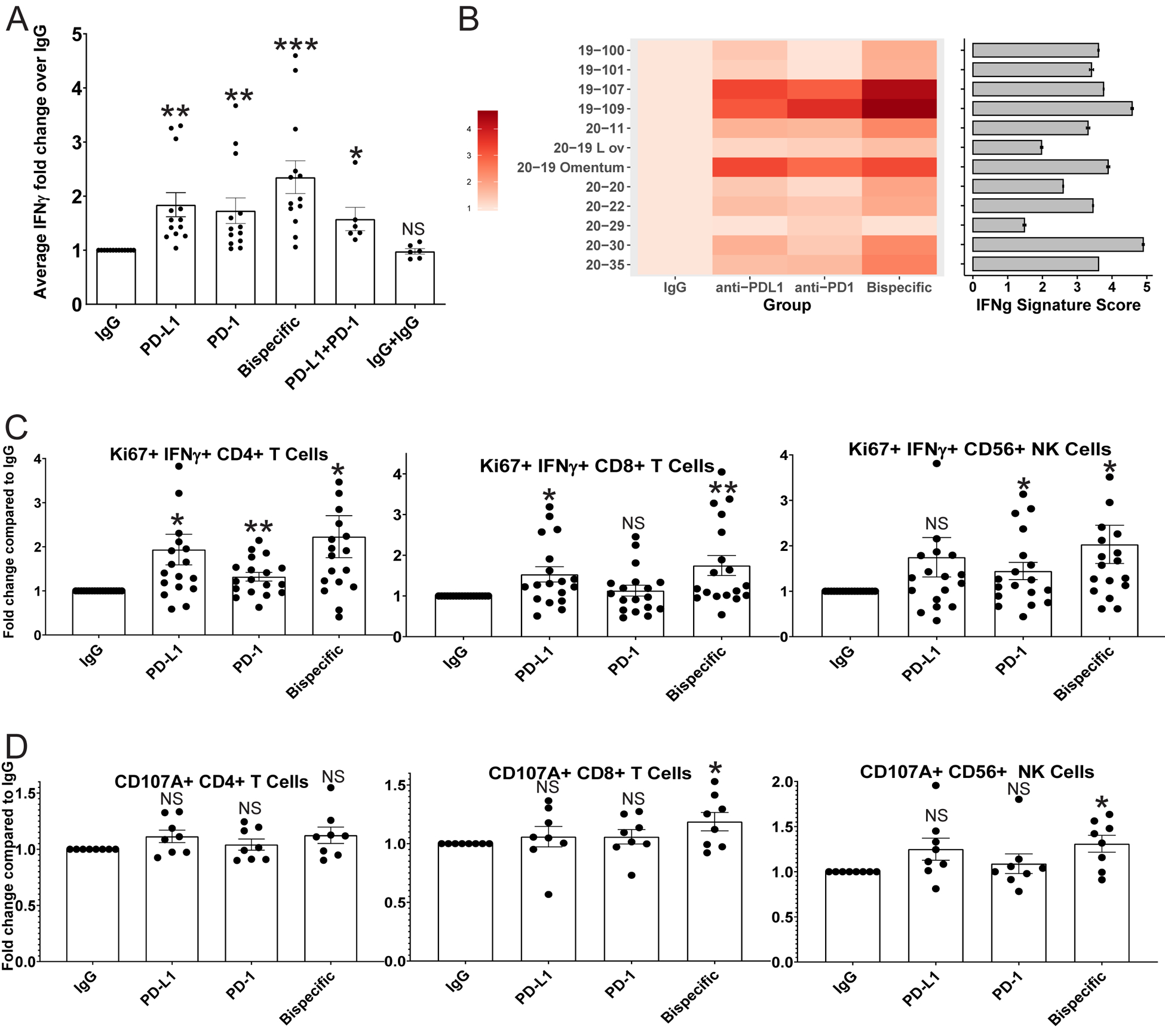
A) IFNγ ELISA analysis was performed on media from organoid co-cultures treated with IgG control, anti-PD-L1, anti-PD-1, anti-PD-1/PD-L1 (bispecific), anti-PD-1+anti-PD-L1, and IgG+IgG. Average IFNγ amounts normalized to the IgG control are shown here across all experiments with error bars representing standard error of the mean. p-values were calculated for all comparisons using a paired t-test. Comparisons of key antibodies to the IgG control are shown. *<0.05, **<0.005, ***<0.0005. p-values for the significance of other treatment comparisons are shown in Figure S6. B) A heatmap is shown for the normalized ELISA results of each individual tumor for IgG, anti-PD-L1, anti-PD-1, and the bispecific antibody. The color code is shown on the left. In addition, bulk RNA sequencing was performed on the parent tumors used to generate the co-cultures, and an IFNG signature score was generated. Each tumor was sequenced twice, and the average IFNG score from each parent tumor is shown to the right of the heatmap as a horizontal bar graph with the number score key below. Error bars represent standard deviation. C) Flow cytometry analysis for IFNγ/Ki67 double positive CD4, CD8, and CD56 positive NK cells across treatments normalized to the IgG control. D) Flow cytometry analysis for CD107A expression on CD4, CD8, and CD56 positive NK cells across treatments normalized to the IgG control. *p<0.05, **p<0.005, NS=Not significant. Error bars represent standard error of the mean.
To compare the aggregate immune activation state of the parent tumors and co-cultures, we generated IFNG expression signature scores for each parent tumor as a measure of the bulk tumor immune state, and compared the scores to the co-culture ELISA results as a measure of bulk organoid immune state (Figure 2B) (28). The co-cultures with the highest IFNγ ELISA signals derived from parent tumors with the highest IFNG scores (19–107, 19–109, 20–19 omentum), those with medium IFNγ ELISA signals were from parent tumors with medium scores (19–100, 19–101, 20–11, 20–20, 20–22, and 20–35), and the cultures with low IFNγ ELISA signal derived from the lowest scoring parent tumors (20–29 and 20–19 Left ovary) (Figure 2B). Patient 20–30 is an outlier with a parent tumor with a high IFNG score but co-cultures with a medium IFNγ signal by ELISA (Figure 2B), potentially due to sampling discrepancies between tissue used for culture and sequencing. Overall, these results indicate that the parent tumor immune activation state is largely matched by the co-cultures and supports the hypothesis that the quality of the immune cells in a tumor rather than the quantity dictate ICB response.
Given our ability to detect immune responses in the co-cultures, we next asked which cells each antibody targets to induce this response.
Different ICB antibodies induce varying degrees of activation in unique immune cell types:
Treated organoid co-cultures were analyzed by flow cytometry to determine which cell population(s) each antibody affected by searching for changes in cell numbers and activation markers.
Cell numbers for each lineage were unaffected by the antibodies. There were no changes in T or NK cell numbers, CD69 positive active T cell numbers, or the ratio of CD4:CD8 T cells across all treatments (Figure S7 (A-C)). There were no changes in any myeloid cell type except for a small but statistically significant decrease in CD14+ macrophages induced by the bispecific antibody compared to the isotype control (Figure S7A).
We assessed all cell types for markers of proliferation (Ki67), antigen stimulation (IFNγ), and cytotoxic phenotype (granzyme B (GZMB) and CD107A) (Figures 2C and 2D, Figure S8 (A-D)). All three ICB antibodies induced a statistically significant increase in antigen stimulated proliferating Ki67/IFNγ double positive CD4 and CD8 T cells and NK cells, with the bispecific antibody induction the strongest for all three cell types (Figure 2C). Only the bispecific antibody induced an increased percentage of CD107A positive CD8 T cells and NK cells over the IgG control (Figure 2D). Finally, the bispecific antibody induced a small but discernible increase in GZMB positive CD4 and CD8 T cells and NK cells (Figure S8D). Taken together with the ELISA results, these data indicate that the bispecific antibody induces both an increase in T and NK cell activation and cytotoxicity more efficiently than the controls. To better understand the mechanism of ICB antibody-driven immune activation in these cell types we performed scRNA-seq analysis.
scRNA-seq of treated organoid co-cultures reveals differential expression in all immune lineages after multiple treatment modalities:
To study the effects of each ICB antibody on every kind of immune cell in a tumor, we performed scRNA-seq analysis on organoid co-cultures from an untreated HGSC omental metastasis (20–11) 96 hours post-treatment, with isotype control, anti-PD-1, anti-PD-L1, or the bispecific antibody (Figure 3A). Viable CD45+ cells from each treatment were sorted, hashed (20), harvested for library preparation, and sequenced (Figure 3A).
FIGURE 3: scRNA-seq analysis of treated organoid co-cultures offers a comprehensive assessment of all immune cell types post-ICB treatment:
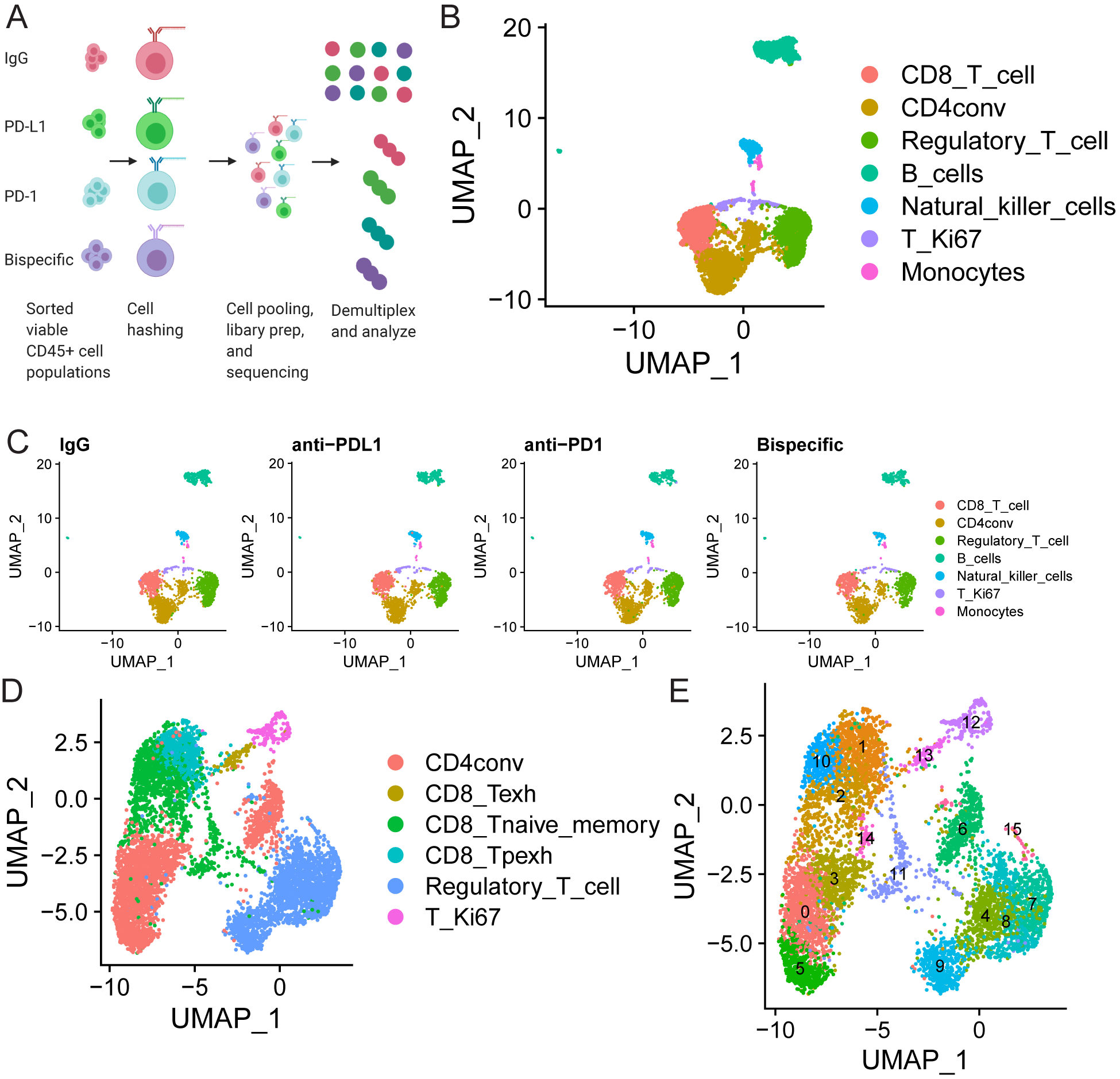
A) Schematic of scRNA-seq experiment. A single organoid co-culture was treated with isotype control, anti-PD-L1, anti-PD-1, or anti-PD-1/PD-L1. Viable CD45+ cells were sorted 96 hours later, hashed with different barcodes for each treatment, mixed in equal proportion, and submitted for 10X Genomics library preparation and subsequent sequencing analysis. B-E) scRNA-seq analysis comparing results in the organoid co-cultures across treatments. B) UMAP demonstrating all immune cells detected in the pool of mixed cells from all treatments from the organoid co-cultures. The color code for each cell type is shown on the right. C) UMAPs demonstrating the cells detected in organoid co-cultures from each treatment in the populations in B shown separately to demonstrate equal distribution of all lineages across treatment. Treatment is indicated above the graph and cell type is indicated by a color code on the right. D and E) UMAPs are shown to demonstrate (D) all T cell subsets detected across the mixture of cells analyzed across all four treatments and (E) that within each of these subsets there are 15 separate clusters with unique transcriptional states. Cell types are indicated by color codes on the right, and clusters are numbered in E. CD4conv=conventional non-regulatory CD4 T cells, Regulatory_T_cell=regulatory CD4 T cells, T_Ki67=proliferating T cells, CD8_Tpexh=Progenitor exhausted CD8 T cells, CD8_Tnaive_memory=Naïve and memory CD8 T cells, Regulatory_T_cell=Regulatory CD4 T cells, CD8_Texh=Terminally exhausted CD8 T cells
Analysis and cell type annotation of the pooled samples revealed every immune cell type was detectable (Figure 3B) with equal numbers of each immune cell type present across all four treatment groups (Figure 3C). All T cell subtypes were present (Figure 3D), and unsupervised clustering by Seurat (29) identified 15 unique T cell subsets/clusters each with distinct transcriptional states (Figure 3E). All immune cell types showed differential expression across treatments (Figures S9 (A-N), S10 (A-D), and S11 (A-K); Tables S1–S4).
Gene expression analysis reveals increased cytotoxicity in T and NK cells and decreased exhaustion in T cells induced most strongly by the bispecific antibody:
We analyzed CD8 T and NK cells for activity or cytotoxicity marker expression. For NK cells, we analyzed expression across a panel of 22 NK cell activation genes to generate activation scores for each treatment (Figure 4A; Table S5) (30). The highest and only statistically significant score was for the bispecific antibody compared to the isotype control. Furthermore, we analyzed expression of genes associated with NK cell cytotoxicity, activation, metabolism, and Myc signaling (30–33). Upregulation of c-Myc or its targets and downregulation of Myc degraders (FBXW7) causes NK cell expansion and metabolic activation (32,34). The bispecific antibody induced the greatest increase in expression of all cytotoxicity, activation, metabolism and Myc targets and the strongest decrease of FBXW7 (Figure 4B). Bispecific antibody upregulation of GZMB was independently verified in sorted NK cells from a different patient’s treated organoid co-cultures (Figures S10D). These results suggest a state change from inert to highly active. Neither monospecific antibody induced NK cell activation suggesting that NK cell activation is a unique bispecific antibody target.
FIGURE 4: scRNA-seq analysis reveals that the bispecific antibody induces an increased cytotoxic phenotype in CD8 T cells and NK cells and a decreased exhaustion and naivety phenotype in CD8 T cells:
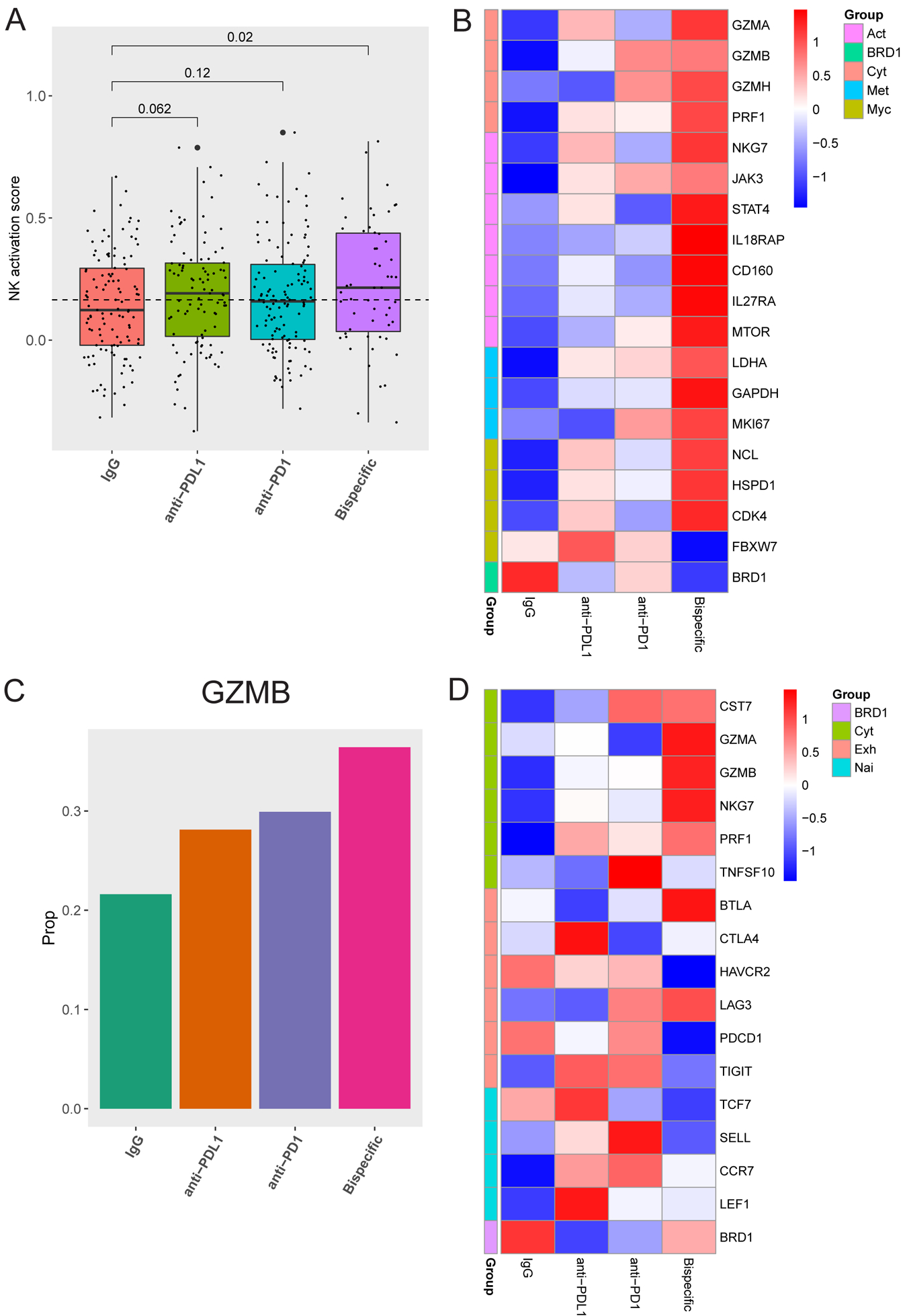
A) Activation scores were generated by assessing a panel of 22 genes associated with NK cell activation. Box plots of the average scores for each treatment are shown with p-values on top generated using a one-tailed t-test compared to the isotype control. B) A heatmap demonstrating Z scores generated from mean expression in NK cells for genes related to cytotoxicity (cyt, top), activation, (act, second from top), metabolism (met, second from bottom), Myc signaling (Myc, third from bottom), and BRD1 (bottom) is shown comparing IgG, anti-PD-L1, anti-PD-1, and the bispecific antibody. C) The positive rate of Granzyme B (GZMB) expression across all treatments in all CD8 T cell populations combined is shown. D) A heatmap demonstrating Z scores generated from mean expression in all CD8 T cell populations combined for genes related to cytotoxicity (cyt, top), exhaustion (exh, second from top), naivety (nai, second from bottom), and BRD1 (bottom) is shown comparing IgG, anti-PD-L1, anti-PD-1 and the bispecific antibody.
Gene expression analysis of bulk CD8 T cells revealed that all ICB agents induced GZMB expression, most strongly the bispecific antibody (Figure 4C), validating flow cytometry findings (Figure S8D). The bispecific antibody induced a larger increase in expression of multiple cytotoxicity markers, including GZMA, GZMB, and PRF1 (Figure 4D). GZMB upregulation was validated in CD8 T cells sorted from organoid co-cultures from a different patient (Figure S11G). Additionally, the bispecific antibody induced an overall decrease in expression of a subset of exhaustion markers (PDCD1=PD-1 and HAVCR2=TIM3) and naïve T cell markers (TCF7 and SELL), suggesting a shift of the CD8 T cells from naïve and exhausted states to active (Figure 4D). However, shifts in the bulk CD8 T cell analysis were small and sometimes inconsistent across functional groups (Figure 4D). In other tumor types, small subsets of CD8 T cells respond to ICB therapies (10,11), prompting us to re-examine activation and exhaustion marker expression in each CD8 subset to determine the reason for the smaller shifts we were observing in the bulk CD8 T cell analysis and if this also occurs in HGSC (Figures 4C and 4D).
CD8 T cell trajectory analysis suggests a state transition induced by ICB antibodies:
To identify which CD8 T cells respond to ICB agents, we focused on three subsets including naïve, progenitor exhausted, and terminally exhausted, all defined by varying expression of activation, naivety, and exhaustion markers (Figure 5A) (10,11). Examination of these subsets across the four treatment groups reveals 1) an increase in progenitor exhausted and a decrease in naïve groups both most prominent after bispecific antibody treatment, and 2) small decreases within the terminally exhausted group after anti-PD-1 and bispecific antibody treatment (Figure 5A). This suggested a state transition within these groups in response to ICB (10,11).
FIGURE 5: scRNA-seq analysis reveals state changes in CD8 T cells between distinct subsets:
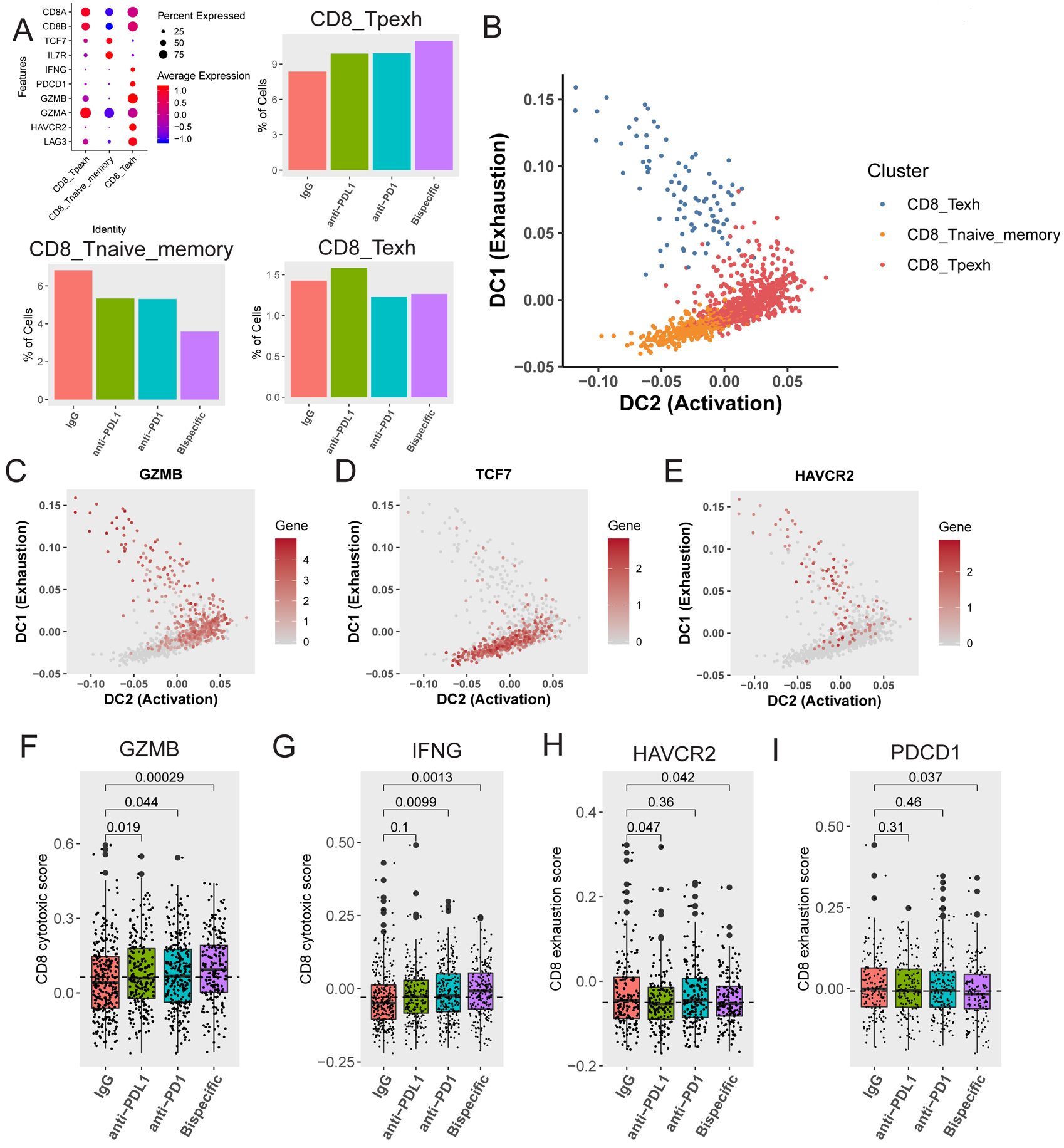
A) Definition of and percentage of CD8 T cells in naïve (CD8_Tnaive_memory), progenitor exhausted (CD8_Tpexh), and terminally exhausted (CD8_Texh) CD8 T cell groups (mapped in Figure 3D–E). On the top left is a bubble map demonstrating markers used to define these three groups. Markers (Y axis) used to define each of the different groups (X axis) are shown here along with the expression level in each defined cell type. The average expression level (colors) is shown in the percentage of cells (sphere) expressing each marker for each cell type. Bar graphs demonstrating the proportion of CD8 T cells in progenitor exhausted (top right), naïve (bottom left), and terminally exhausted (bottom right) CD8 T cell groups across antibody treatments are shown. B) Diffusion map demonstrating transition between 1) naïve (orange) and progenitor exhausted (red) cells, and 2) terminally exhausted (blue) and progenitor exhausted (red) cells over pseudotime. The X axis represents increasing activation, and the Y axis represents increasing exhaustion. The color code for the different clusters/subgroups is shown on the top right. C, D, E) Diffusion maps demonstrating transition between naïve and progenitor exhausted cells and terminally exhausted and progenitor exhausted cells (mapped in B) over pseudotime for C) the activation marker GZMB (Granzyme B), D) the naivety marker TCF7, and E) the exhaustion marker HAVCR2 (TIM3). The X axis represents increasing activation, and the Y axis represents increasing exhaustion. The color code for gene expression level is shown on the right. F and G) Activation scores were generated by assessing a panel of 50 genes associated with (F) GZMB (Granzyme B) or (G) IFNG expression in CD8 T cells in naïve and progenitor exhausted CD8 T cells. Box plots of the average scores for each treatment are shown with p-values compared to the IgG control for (F) GZMB and (G) IFNG. H and I) Exhaustion scores were generated by assessing a panel of 50 genes associated with (H) HAVCR2 (TIM3) expression or (I) PDCD1 (PD-1) expression in CD8 T cells in both naïve and progenitor exhausted cells. Box plots of the average scores for each treatment are shown with p-values compared to the control for (H) HAVCR2 and (I) PDCD1. All p-values were generated using a one-tailed t-test.
To define the direction of the dynamic state shifts between these three CD8 T cell groups in response to treatment, we utilized diffusion maps of activation (GZMB, PRF1, IFNG), naivety (TCF7, SELL), and exhaustion (HAVCR2 (TIM3)) markers ordered in pseudotime (Figure 5B–E, S11H–J) (11,35). The activation markers increase between naive and progenitor exhausted, and terminally exhausted and progenitor exhausted cells (Figures 5B, S11H, S11J), the naivety markers decrease from naive into progenitor exhausted cells (Figures 5D, S11I), and the exhaustion markers decrease from terminally exhausted into progenitor exhausted cells and from progenitor exhausted into naïve cells (Figure 5E). These transitions are most significantly induced by the bispecific antibody (Figure 5A). Overall, this suggests that naïve CD8 T cells might give rise to the progenitor exhausted active subset, and that some terminally exhausted cells shift to the more active progenitor exhausted state (Figure 5B), both changes observed in other tumor types in response to ICB (10,11).
Having defined the naïve and progenitor exhausted groups as the ICB responders, we re-examined the exhaustion and activation marker gene panels which revealed only small shifts in the bulk CD8 T cells (Figure 4D). By assessing expression of the top 50 genes associated with either GZMB (Figure 5F) or IFNG (Figure 5G) in the combined progenitor exhausted and naïve CD8 T cell groups, we generated activation scores for these cells across treatments and found the largest statistically significant increases were for the bispecific antibody over the isotype control for both GZMB and IFNG (Figures 5F and 5G). Similarly, we assessed expression across the top 50 genes associated with either HAVCR2 (TIM3) (Figure 5H) or PDCD1 (PD-1) (Figure 5I) in the same combined CD8 groups to generate exhaustion scores across treatments. We found a statistically significant decrease for anti-PD-L1 and the bispecific antibody over the isotype control for HAVCR2-associated genes (Figure 5H) and for the bispecific antibody for PDCD1-associated genes (Figure 5I). These results validated our findings regarding the decreased exhaustion and overall active state changes the bispecific antibody in particular induced within these CD8 groups (Figure 5B).
We next sought to determine the mechanism of how the bispecific and other ICB antibodies induce these changes.
The bispecific antibody induces decreased T and NK cell exhaustion by downregulating BRD1 expression in immune cells:
We examined the differentially expressed genes for the bispecific antibody compared to the controls in the CD8 T and NK cell populations, searching for cell state control genes with small molecule therapies. We focused on the bromodomain-containing protein BRD1 which is known to regulate CD8 T and other cell development (36,37) and has a small molecule inhibitor, BAY-299 (38). BRD1 expression was downregulated by the bispecific antibody in NK cells in the scRNA-seq data (Figures 4B, 6A, S10A and Table S4) and in bulk RNA sequencing of sorted NK cells from treated organoid co-cultures from a different patient (Figure 6B). All ICB antibodies induced a decrease in BRD1 expression in bulk (Figure 4D) and combined ICB-responding progenitor exhausted and naïve CD8 T cells (Figure 6C), and by anti-PD-L1 in terminally exhausted CD8 T cells (Figure S11K). In diffusion analysis over pseudotime, BRD1 expression decreased from naïve into progenitor exhausted and progenitor into terminally exhausted cells (Figure 6D) suggesting the depletion was leading to an increase in activation and decrease in exhaustion. Based on these data, we hypothesized that BRD1 may negatively regulate T and NK cells and that BRD1 downregulation or inhibition may lead to enhanced anti-tumor immune function.
FIGURE 6: The bispecific antibody acts, in part, through strongly depleting BRD1 expression leading to increased immune activity through activation and state changes in T and NK cells:
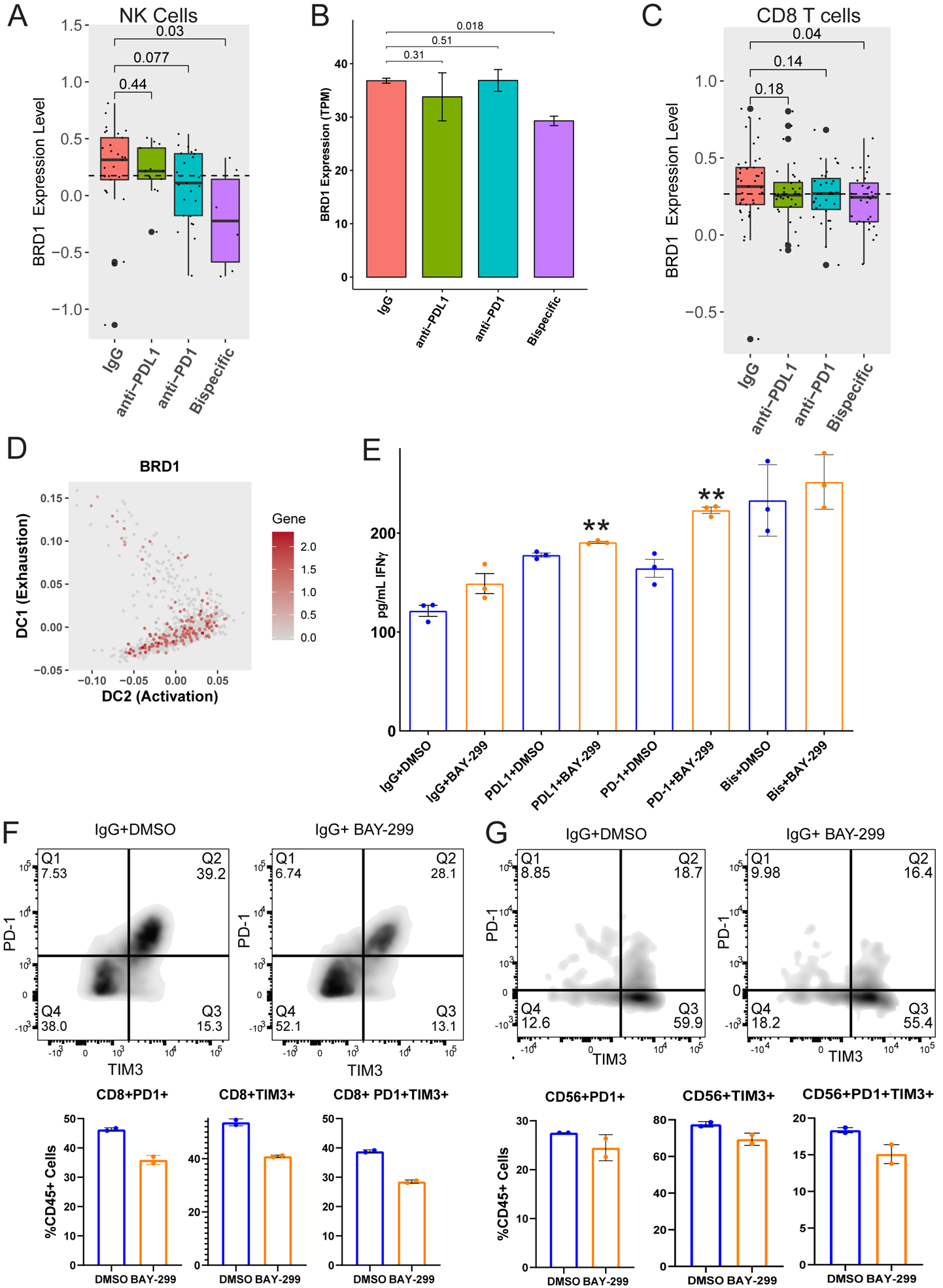
A) BRD1 expression was analyzed across different treatment groups from the scRNA-seq experiment (Figure 3) for NK cells. Bar graph demonstrating the expression level of BRD1 in NK cells for each ICB antibody compared to the IgG control. p-values were generated using a one-tailed t-test. B) To verify the BRD1 depletion induced by the bispecific antibody in NK cells, another organoid co-culture from an untreated HGSC patient (20–35) was treated with isotype control, anti-PD-1, anti-PD-L1, or anti-PD-1/PD-L1. NK cells were sorted from the treated cultures at the 96 hour timepoint and sent for bulk RNA sequencing. The BRD1 expression across treatments is shown here as transcripts per million (TPM). p-values were generated using a one-tailed t-test. C) BRD1 expression was analyzed across different treatment groups from the scRNA-seq experiment (Figure 3) for the naïve and progenitor exhausted CD8 T cell subgroups combined. Shown here is a bar graph demonstrating the expression level of BRD1 in the combined ICB responsive naïve and progenitor exhausted CD8 T cells compared to the IgG control. p-values were generated using a one-tailed t-test. D) Diffusion map demonstrating BRD1 expression in CD8 T cell subgroups transitioning between naïve and progenitor exhausted CD8s and terminally exhausted and progenitor exhausted CD8s (mapped in Figure 5B). The X axis represents increasing activation, and the Y axis represents increasing exhaustion. The color code for gene expression level is shown on the right. E) The same organoid co-culture from figure 6B (20–35) was treated with isotype control, anti-PD-1, anti-PD-L1, or anti-PD-1/PD-L1 combined with either DMSO (blue) or the BRD1 inhibitor BAY-299 (orange). The co-culture media supernatants underwent IFNγ ELISA analysis shown here as the average pg/mL of IFNγ for the treatment with error bars representing standard error. **p<0.005 F and G) The treated organoid co-cultures from Figure 6B and 6E underwent flow cytometry analysis for PD-1 and TIM3 single positive and double positive cells. Only the IgG and PD-L1 treated cultures could be analyzed here because the anti-PD-1 and bispecific antibody treated co-cultures show decreased PD-1 due to the treatment antibody blocking the flow antibody. F) The flow quadrant plots for IgG+DMSO and IgG+BAY-299 are shown for CD8 T cells on the top. PD-1 is on the Y axis and TIM3 is on the X axis. The percentage of PD-1 (Q1) and TIM3 (Q3) single positive and double positive (Q2) CD8 T cells is shown on the bottom as a percent of CD45 positive cells. Error bars represent standard error across two replicates. G) The flow quadrant plots for IgG+DMSO and IgG+BAY-299 are shown for NK cells on the top. PD-1 is on the Y axis and TIM3 is on the X axis. The percentage of PD-1 (Q1) and TIM3 (Q3) single positive and double positive (Q2) NK cells is shown on the bottom as a percent of CD45 positive cells. Error bars represent standard error across two replicates.
We examined BRD1 expression in TIMER (Tumor Immune Estimation Resource), a compilation of expression profiling of immune cells across multiple tumor types to determine if BRD1 expression in HGSCs in vivo correlates with immune cell inhibition (39). We found that BRD1 expression is low in tumor cells and high in immune cells in HGSC (Figure S12). High BRD1 expression correlated with significant downregulation of T and NK cell activity markers (GZMA, GZMB, IFNG, and NKG7) and upregulation of the naïve T cell marker TCF7 supporting our hypothesis that BRD1 is a negative regulator of T and NK cell activity (Figure S12).
To confirm that BRD1 is a negative immune regulatory gene, we tested the BRD1 inhibitor BAY-299 (38) in HGSC organoid co-cultures. We observed that BAY-299 combined with isotype control, anti-PD-1, or anti-PD-L1 leads to a statistically significant increase in IFNγ levels over any antibody alone indicating increased immune activation (Figure 6E). BAY-299 addition generated only a small increase in IFNγ levels over bispecific antibody alone (Figure 6E), as expected, given the bispecific antibody-induced depletion of BRD1 in key cell types (Figures 6A–6C).
Given the state transitions induced by the bispecific antibody (Figures 4 and 5), we wondered if BRD1 inhibition caused increased immune efficacy through a cell state change. We examined exhaustion markers TIM3 and PD-1 (40) on T and NK cell populations from HGSC organoid co-cultures treated with our ICB antibody panel +/− BAY-299 by flow cytometry. BAY-299 treatment decreased the number of TIM3 or PD-1 single positive and TIM3/PD-1 double positive CD3, CD4, and CD8 T cells and NK cells (Figures 6F, 6G, and S13 (A-B)), indicating that BRD1 inhibition may indeed be leading to a cell state change from an exhausted to an active phenotype as observed for all ICB antibodies, most significantly the bispecific antibody (Figures 4, 5, and 6). Thus, an underlying mechanism of action for the increased efficacy of the bispecific antibody is potentially an induction of BRD1 depletion in NK and T cells, promoting active states (Figures 5 and 6). The next question was how BRD1 depletion induces this state change and if it increases tumor cell killing.
BRD1 inhibition leads to increased NK cell activation and tumor cell killing partially through altering chromatin access for key immune transcription factors:
To study the mechanism of BRD1 inhibition-induced immune cell state changes, we applied BAY-299 to an NK cell line, KHYG1 (41). BAY-299 caused a small decrease in BRD1 protein levels in the KHYG1 cells after a 96-hour exposure (Figure S14A). In a growth rate corrected sensitivity analysis, BAY-299 was not overtly toxic to HGSC tumor, NK, or T cell or HGSC organoid lines (Figure S14B) (42).
ATAC-seq analysis of BAY-299 treated KHYG1 cells revealed significant alterations to chromatin accessibility across the genome (Figure 7A and S14C). The major chromatin alterations showed significant overlap with binding sites for key NK cell development regulatory transcription factors such as GATA3, TBX21, and TBXT which were all associated with down peaks (Figure 7B) (43). Several genes linked to these transcription factors with important biological functions were in altered peaks. For example, BAY-299 caused alterations in the chromatin accessibility of the promoter region of CDK9 (Figure 7C bottom). CDK9 is, in part, recruited to immune genomic loci by TBX21 as part of the PTef-b complex (44), and CDK9 inhibition allows global reactivation of epigenetically silenced genes leading to increased IFNγ activity and sensitivity to ICB agents in tumor cells (45). In addition, BAY-299 reduces chromatin accessibility of EMB (Figure 7C top), another TBX21 regulated gene recently identified as a marker of immature NK cells and as part of an immature NK cell signature (46). Taken together, these findings suggest that a possible BRD1 inhibitor mechanism of action is altering chromatin accessibility for major NK cell regulatory transcription factors at key immune modulatory genes to allow for a mature (EMB) active and cytotoxic (CDK9) state.
Figure 7: A BRD1 inhibitor causes increased anti-tumor immunity in HGSC by altering immune cell chromatin state:
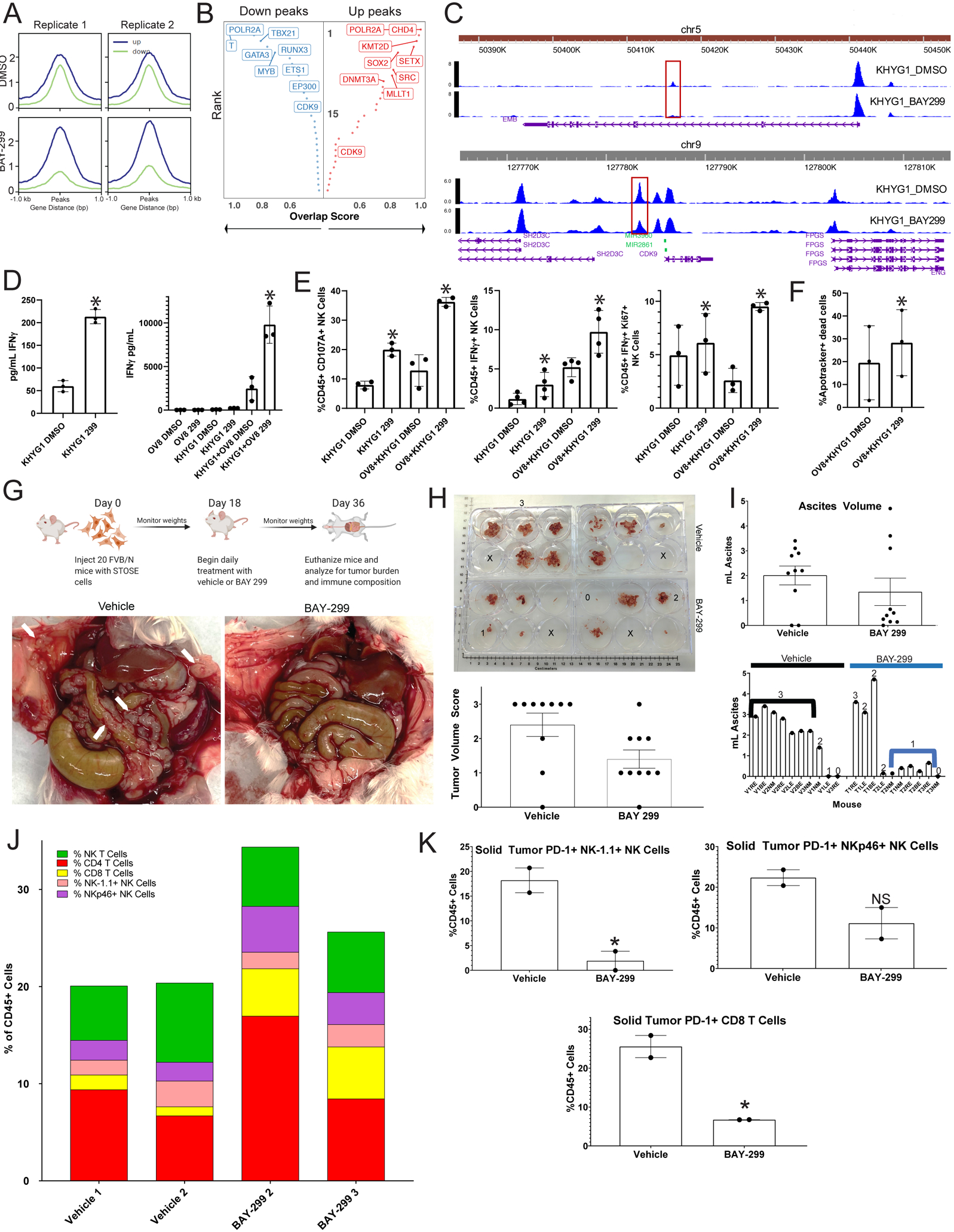
A) ATAC-seq was performed in duplicate on KHYG1 cells treated with either vehicle or BAY-299. Aggregated reads within 1kb on either side of center for up (blue) and down (green) differentially accessible chromatin sites for the two replicates for DMSO (top) and BAY-299 (bottom) treated cells are shown here. B) The transcription factors (TF) associated with the most strongly altered up (right red) and down (left blue) peaks are shown here. The Y axis represents rank of the transcription factor from 1 (highest rank at top) to 30 (lowest rank at bottom) for number of overlapping sites, and the X axis represents the overlap score increasing from left to right for up peaks and right to left for down peaks. Highest ranking TFs are on the top left for down peaks and top right for up peaks. C) Chromatin peaks surrounding and within the EMB (top) and CDK9 (bottom) genomic locus for DMSO treated KHYG1 cells (top in each panel) and BAY-299 treated KHYG1 cells (bottom in each panel). The taller the peak the more open the chromatin. The scale for peak size is on the y-axis and the x axis represents location in the genome. D) KHYG1 cells treated with DMSO vehicle or BAY-299 (299) were plated either alone or in co-culture with OVCAR8 (OV8) tumor cells, and the media was subsequently tested for IFNγ presence by ELISA. Bar graphs for the ELISA for KHYG1 cells alone is shown on the left and for the co-cultures on the right. The error bars represent the standard deviation between three replicates of the experiment *=p<0.05 using a paired t-test. E) KHYG1 cells treated with vehicle DMSO or BAY-299 (299) were plated either alone or in co-culture with OVCAR8 (OV8) tumor cells, and the cultures were analyzed by flow cytometry for NK cell CD107A expression (left), IFNγ expression (middle), and IFNγ/Ki67co-expression (right). Error bars represent standard deviation between 3–4 replicates. *=p<0.05 using a paired t-test. F) KHYG1 cells treated with DMSO vehicle or BAY-299 (299) were plated in co-culture with OVCAR8 (OV8) tumor cells, and six hours later the OVCAR8 cells were analyzed for apoptotic death. The percentage of non-viable apoptotic cells (Apotracker+ Dead Cells) from three separate experiments is shown here for each group with error bars representing standard deviation. *=p<0.05 using a paired t-test. G) A schematic of the in vivo experiment is shown in the top panel. In the bottom panel, gross images of the tumor burden in vehicle and BAY-299 treated mice are shown with white arrows pointing to solid tumor deposits on the peritoneum and bowel. The animals shown are representative of the most common tumor burden levels in each group. H) Grossly visible solid tumors were dissected from each mouse in each group, and a photo of tumor volume is shown in the top panel. Solid tumors from each animal were placed in a well of a six well plate for visual volume scoring. A three represents high tumor burden, a two medium tumor burden, a one limited tumor burden, and a zero no tumor burden. Numbers are placed in representative 3, 2, 1, and 0 wells, and an X is placed in empty wells. In the bottom panel, the tumor volume scores are shown for all ten animals per group in a bar graph with error bars representing standard error of the mean (SEM). I) Ascites was aspirated from each animal and the volume measured. The top panel represents a bar graph of the ascites volumes for all animals in each group with the error bars representing SEM. The bottom panel shows the individual animal ascites volumes with the numeric tumor volume score for each animal over the volume bar. Black and blue lines mark the vehicle (V) and treatment (T) groups most common volume and tumor burdens. J) Solid tumors were harvested from each animal in both the vehicle and treatment groups. For each treatment group, the tumors for 3–4 animals were combined. There were only enough cells from two combined groups each for vehicle and BAY-299 to perform flow analysis. The single cell suspensions of the solid tumors were analyzed for T and NK composition which is shown here. Each color represents a cell type and each bar represents a group. The color code is at the top left. K) PD-1 expression was analyzed in both solid tumor treatment groups on NK-1.1+ NK cells (top left), NKp46+ NK cells (top right), and CD8 T cells (bottom), and the percent of PD-1+ T or NK cells for each treatment group is shown here as a percentage of CD45+ cells. Error bars represent standard deviation. *=p<0.05 generated using an unpaired t-test, NS=not significant with an unpaired t-test.
To test for this possibility, we functionally assessed BAY-299 treated NK-tumor cell co-cultures. BAY-299 led to increased IFNγ production by the NK cells alone and at even higher levels when in co-culture with the HGSC cell line OVCAR8 or organoid line 17–116 (47) indicating increased immune activity (Figure 7D and S14D). Accordingly, paired flow cytometry analysis of the NK cells after BAY-299 treatment either alone or in co-culture demonstrated increased IFNγ+, Ki67+/IFNγ+ double positive cells, and CD107A+ NK cells, more pronounced when these cells were cultured with OVCAR8 or organoid tumor cells (Figure 7E and S14E). Finally, BAY-299 increased tumor cell killing over vehicle in KHYG1 and OVCAR8 cell co-cultures (Figure 7F). Altogether, these results indicate that BAY-299 induced chromatin remodeling causes a more cytotoxic and active state in NK cells in vitro and begs the question of whether BAY-299 can lead to enhanced anti-tumor killing by NK and possibly other cytotoxic immune cells in vivo.
BRD1 inhibition by BAY-299 shows efficacy in vivo:
To assess BRD1 inhibitor efficacy in vivo, BAY-299 was tested in a syngeneic PAX8 positive ovarian cancer mouse model generated with STOSE cells (21). A maximum tolerated dose study was performed in female FVB/N mice. The drug was well tolerated with no side effects at the maximum dose.
For the in vivo experiment (Figure 7G top), twenty FVB/N female mice were injected with STOSE cells on day 1, and eighteen days later daily vehicle or BAY-299 treatments were initiated. Eighteen days after treatment initiation the mice were weighed, euthanized, and the tumor burden and immune composition analyzed (Figure 7G).
Final animal weights were similar in both treatment groups (Figure S15A). Grossly visible solid tumor was dissected from all animals and appeared histologically similar in both groups forming sheets and clusters of neoplastic epithelioid cells with cytomorphologic features consistent with a poorly differentiated Müllerian carcinoma (Figure S15B). Tumor volumes were given a score of 0–3 (three being highest), and most of the vehicle treated mice scored highest while the majority of the BAY-299 scored lowest (Figure 7H). Most of the vehicle treated animals had high volume ascites correlating with high solid tumor burden, while most of the BAY-299 treated animals had little to no ascites correlating with low tumor burden (Figure 7I).
We characterized solid tumors, ascites, and spleens for immune composition (T and NK cell quantity), exhaustion marker expression (TIM3, PD-1, and PD-L1), and activation marker expression (IFNγ, GZMB, Ki67) (Figure 7J, 7K and Figure S15 (C-I)). Given the low tumor burden and ascites volume in BAY-299 treated animals, samples were combined from multiple animals for analysis for each individual tissue/treatment type. Flow cytometry analysis of the spleens did not demonstrate shifts in T or NK cell populations (Figure S15C and S15E).
Flow cytometry analysis of the solid tumors demonstrated an increase in NK cells and CD8 T cells after BAY-299 treatment (Figure 7J). NK cell antibodies for markers NK-1.1 and NKp46 were utilized (48) with limited overlap in the CD3-/ NK-1.1+ and CD3-/NKp46+ groups. The NKp46+ NK cells expanded after BAY-299 treatment in the solid tumors (Figures 7J and S15F). Assessment of exhaustion markers post-treatment demonstrated a decrease in PD-1 expression on CD8 T and both NK-1.1 and NKp46+ NK cells (Figure 7K) similar to bispecific antibody (Figure 5) and BAY-299 treatments (Figure 6) in organoid co-cultures. TIM3 and PD-1/TIM3 co-expression and activation and killing marker expression was similar between treatments.
Conversely, in the ascites, only two BAY-299 treated samples showed CD8 T cell expansion and NK-1.1+ NK cells expanded post-treatment (Figure S15D and S15G). TIM3 expression decreased post-treatment on both NK-1.1 and NKp46+ NK cells (Figure S15H) similar to the bispecific antibody (Figure 5) and BAY-299 treatments (Figure 6) in organoid co-cultures. TIM3 and PD-1/TIM3 co-expression was similar between treatments. Activation and killing markers were assessed, and CD107A mildly increased in both the NK-1.1 and NKp46+ NK cells post-treatment (Figure S15I).
Altogether these results suggest that BRD1 depletion is likely an underlying mechanism for the superiority of the bispecific antibody and that, like the bispecific antibody, BRD1 inhibition leads to decreased immune exhaustion and increased immune activation, particularly in NK cells, which may make it an effective immune target in HGSC.
DISCUSSION:
The major cellular and mechanistic targets of ICB therapies in HGSC have not been defined making designing more effective therapies and identifying patients who might benefit from immune therapy difficult. Using real time functional analysis of novel HGSC organoid-immune cell co-cultures treated with a novel bispecific antibody and its monospecific controls, we identified three critical cellular and mechanistic immune therapy targets in HGSC. This led to the identification of two novel immune therapies that may have increased activity in HGSC patients, a bispecific anti-PD-1/PD-L1 antibody and BRD1 inhibitors, both of which show in vivo anti-tumor efficacy suggesting further therapeutic exploration of these agents may be merited (Figure 7 and (19)).
Key to understanding the molecular functions of ICB agents in HGSC was our ability to gain a comprehensive appreciation of the effects of these agents on every cell in the tumor in novel HGSC organoid co-cultures (Figures 2–6). Our most striking findings were in NK and T cells. Mechanisms of tumor microenvironment-induced NK cell dysfunction and NK response to ICB agents are undefined (8,16). We found that the bispecific antibody and the BRD1 inhibitor BAY-299 induced strong NK cell activation through induction of a state change from an inert and exhausted to a more active and cytotoxic phenotype which correlated with in vivo efficacy of these agents (Figures 2, 4, 5, 6, 7)(19). In so doing, we define NK cell activation as a key component of therapeutic efficacy for immune therapy agents in HGSC that is lacking with currently used ICB agents.
In addition, our analysis demonstrated which CD8 T cells are most critical for ICB response in HGSC. We find that all three ICB agents, particularly the bispecific antibody, induced a transition from 1) naïve to cytotoxic progenitor exhausted groups, and 2) terminally exhausted to progenitor exhausted cytotoxic groups (Figure 5). This suggests that the ICB-driven naïve to cytotoxic progenitor transition may drive response in the long term and suggest that identifying therapies that can induce this state change may be important.
We determined that these state changes were being driven, in part, by bispecific antibody downregulation of BRD1 expression (Figure 6). Bromodomain proteins such as BRD1 and BRD4 play roles in immune and hematologic cell development and modulation of tumor inflammation (36,37,49) but are often targeted with the goal of affecting changes within the tumor cells as a means of therapeutic efficacy (38,49,50). Our findings led us to explore the novel role of BRD1 and BRD1 inhibitors in the immune cells instead.
Here we show BRD1 inhibition induces NK and some CD8 T cells into more active states by reducing exhaustion, and that this can enhance the activity of ICB agents like pembrolizumab in vitro (Figure 6). The mechanism of action of BRD1 inhibition in these changes likely involves key epigenetic alterations (Figures 7A–C); however, further work will be needed to fully understand this complex process. Additionally, BAY-299 demonstrated increased efficacy in decreasing tumor burden in vivo through decreasing exhaustion of NK and CD8 T cells (Figure 7). Overall, these findings suggest that BRD1 inhibition may be effective at enhancing the anti-tumor immune response and merit further exploration as a therapeutic option either alone or in relevant combinations in HGSC. In addition, this discovery in organoid co-cultures highlights the importance of studying the effects of any agent, even those thought to target intrinsic tumor cell properties, on every cell in a tumor. This may lead to unexpected discoveries that open up a broader array of small molecule therapies in the immune space due to unanticipated effects of therapeutic agents on the immune compartment.
Overall, this work suggests the potential for HGSCs to be effectively targeted with immune therapies if the therapies engage the correct mechanistic pathways in the right immune cell types. These findings highlight a common immune therapy problem across HGSCs. It is not a tumor cell genomic or molecular defect making only some HGSCs responsive to these agents, rather it is a state of dysfunction in different subsets of CD8 T cells and NK cells driven by the solid tumor microenvironment blocking current ICB response across HGSCs. By gaining a better understanding of the mechanisms driving these dysfunctional states and how to overcome them, as we have begun to do here using a novel model system and novel therapeutic tool, we have identified available therapies to offer HGSC patients, like BRD1 inhibitors, which effectively target these pathways. Mechanism driven therapies have the potential to someday make immune therapy effective in HGSC, a deadly disease where it has not been effective before.
Supplementary Material
SIGNIFICANCE:
This study indicates that increased efficacy of immune therapies in ovarian cancer is driven by state changes of NK and small subsets of CD8 T cells into active and cytotoxic states.
ACKNOWLEDGEMENTS:
We would like to express our gratitude to all the patients who donated tissue for this work. LY3300054, LY3434172, and the IgG control used in this work were provided by Eli Lilly and Company. Financial Support: S.J.H was supported by the 2019 AACR-AstraZeneca Ovarian Cancer Research Fellowship (Grant Number 19-40-12-HILL), a DOD OCRP Pilot Award (W81XWH-19-1-0123), a Tina’s Wish Rising Star Grant, NIH K08 CA241093, NIH DP5 OD029637, and a sponsored research agreement from Eli Lilly and Company. B.R. was supported by the Advanced Medical Research Foundation and the Vincent Memorial Hospital Foundation. X.S.L was supported by NIH R01 CA234018. Rui Xu was supported by the China Scholarship Council (Grant Number 201908610120). Myles Brown was supported by a DOD OCRP Pilot Award (W81XWH-19-1-0147).
Footnotes
Competing interests: M.B. receives sponsored research support from Novartis. M.B. serves as a consultant and scientific advisor to Kronos Bio, H3 Biomedicine and GV20 Therapeutics. In the past M.B. served as a scientific advisor to GTx, Inc. and as consultant to Aleta BioTherapeutics. U.A.M. is a consultant for Novartis and Merck, is on the scientific advisory boards for 2X Oncology and the Clearity Foundation, and is an uncompensated advisor for AstraZeneca. X.S.L. is a cofounder and board member of GV20 Oncotherapy, on the scientific advisory board of 3DMed Care, a consultant for Genentech, and a stock holder of BMY, TMO, WBA, ABT, ABBV, and JNJ. S.J.H. receives sponsored research support from Eli Lilly and Company and AstraZeneca. B.R. is an uncompensated scientific advisory board member for VincenTech. B.R. receives sponsored research support from Palleon Pharmaceuticals and Mercy Bioanalytics. Neither is directly related to the proposed research.
REFERENCES:
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34 [DOI] [PubMed] [Google Scholar]
- 2.Narod S Can advanced-stage ovarian cancer be cured? Nat Rev Clin Oncol 2016;13:255–61 [DOI] [PubMed] [Google Scholar]
- 3.Ghisoni E, Imbimbo M, Zimmermann S, Valabrega G. Ovarian Cancer Immunotherapy: Turning up the Heat. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Odunsi K Immunotherapy in ovarian cancer. Ann Oncol 2017;28:viii1–viii7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity 2018;48:812–30 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005;102:18538–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 2003;348:203–13 [DOI] [PubMed] [Google Scholar]
- 8.Hoogstad-van Evert JS, Bekkers R, Ottevanger N, Jansen JH, Massuger L, Dolstra H. Harnessing natural killer cells for the treatment of ovarian cancer. Gynecol Oncol 2020 [DOI] [PubMed] [Google Scholar]
- 9.Hoogstad-van Evert JS, Maas RJ, van der Meer J, Cany J, van der Steen S, Jansen JH, et al. Peritoneal NK cells are responsive to IL-15 and percentages are correlated with outcome in advanced ovarian cancer patients. Oncotarget 2018;9:34810–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 2019;20:326–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 2019;25:1251–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015;15:486–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res 2014;2:410–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thommen DS, Schumacher TN. T Cell Dysfunction in Cancer. Cancer Cell 2018;33:547–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017;545:452–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun H, Sun C. The Rise of NK Cell Checkpoints as Promising Therapeutic Targets in Cancer Immunotherapy. Front Immunol 2019;10:2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov 2018;8:196–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018;175:1972–88 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kotanides H, Li Y, Malabunga M, Carpenito C, Eastman SW, Shen Y, et al. Bispecific Targeting of PD-1 and PD-L1 Enhances T-cell Activation and Antitumor Immunity. Cancer Immunol Res 2020 [DOI] [PubMed] [Google Scholar]
- 20.Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 2018;19:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCloskey CW, Goldberg RL, Carter LE, Gamwell LF, Al-Hujaily EM, Collins O, et al. A new spontaneously transformed syngeneic model of high-grade serous ovarian cancer with a tumor-initiating cell population. Front Oncol 2014;4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gitto SB, Kim H, Rafail S, Omran DK, Medvedev S, Kinose Y, et al. An autologous humanized patient-derived-xenograft platform to evaluate immunotherapy in ovarian cancer. Gynecol Oncol 2020;156:222–32 [DOI] [PubMed] [Google Scholar]
- 23.Friese C, Harbst K, Borch TH, Westergaard MCW, Pedersen M, Kverneland A, et al. CTLA-4 blockade boosts the expansion of tumor-reactive CD8(+) tumor-infiltrating lymphocytes in ovarian cancer. Sci Rep 2020;10:3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol 2012;22:33–40 [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Carpenito C, Wang G, Surguladze D, Forest A, Malabunga M, et al. Discovery and preclinical characterization of the antagonist anti-PD-L1 monoclonal antibody LY3300054. J Immunother Cancer 2018;6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zamarin D, Burger RA, Sill MW, Powell DJ Jr., Lankes HA, Feldman MD, et al. Randomized Phase II Trial of Nivolumab Versus Nivolumab and Ipilimumab for Recurrent or Persistent Ovarian Cancer: An NRG Oncology Study. J Clin Oncol 2020:JCO1902059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov 2019;18:585–608 [DOI] [PubMed] [Google Scholar]
- 28.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive Integration of Single-Cell Data. Cell 2019;177:1888–902 e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med 2016;8:357ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Del Zotto G, Marcenaro E, Vacca P, Sivori S, Pende D, Della Chiesa M, et al. Markers and function of human NK cells in normal and pathological conditions. Cytometry B Clin Cytom 2017;92:100–14 [DOI] [PubMed] [Google Scholar]
- 32.Dong H, Adams NM, Xu Y, Cao J, Allan DSJ, Carlyle JR, et al. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat Immunol 2019;20:865–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer 2016;16:7–19 [DOI] [PubMed] [Google Scholar]
- 34.Loftus RM, Assmann N, Kedia-Mehta N, O’Brien KL, Garcia A, Gillespie C, et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun 2018;9:2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haghverdi L, Buettner F, Theis FJ. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 2015;31:2989–98 [DOI] [PubMed] [Google Scholar]
- 36.Mishima Y, Miyagi S, Saraya A, Negishi M, Endoh M, Endo TA, et al. The Hbo1-Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood 2011;118:2443–53 [DOI] [PubMed] [Google Scholar]
- 37.Mishima Y, Wang C, Miyagi S, Saraya A, Hosokawa H, Mochizuki-Kashio M, et al. Histone acetylation mediated by Brd1 is crucial for Cd8 gene activation during early thymocyte development. Nat Commun 2014;5:5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bouche L, Christ CD, Siegel S, Fernandez-Montalvan AE, Holton SJ, Fedorov O, et al. Benzoisoquinolinediones as Potent and Selective Inhibitors of BRPF2 and TAF1/TAF1L Bromodomains. J Med Chem 2017;60:4002–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol 2016;17:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A 2010;107:14733–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yagita M, Huang CL, Umehara H, Matsuo Y, Tabata R, Miyake M, et al. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia 2000;14:922–30 [DOI] [PubMed] [Google Scholar]
- 42.Hafner M, Niepel M, Chung M, Sorger PK. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat Methods 2016;13:521–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang D, Malarkannan S. Transcriptional Regulation of Natural Killer Cell Development and Functions. Cancers (Basel) 2020;12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hertweck A, Evans CM, Eskandarpour M, Lau JC, Oleinika K, Jackson I, et al. T-bet Activates Th1 Genes through Mediator and the Super Elongation Complex. Cell Rep 2016;15:2756–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Pandey S, Travers M, Sun H, Morton G, Madzo J, et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018;175:1244–58 e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang C, Siebert JR, Burns R, Zheng Y, Mei A, Bonacci B, et al. Single-cell transcriptome reveals the novel role of T-bet in suppressing the immature NK gene signature. Elife 2020;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ Jr., et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov 2018;8:1404–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walzer T, Blery M, Chaix J, Fuseri N, Chasson L, Robbins SH, et al. Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc Natl Acad Sci U S A 2007;104:3384–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White ME, Fenger JM, Carson WE 3rd. Emerging roles of and therapeutic strategies targeting BRD4 in cancer. Cell Immunol 2019;337:48–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018;33:401–16 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.