

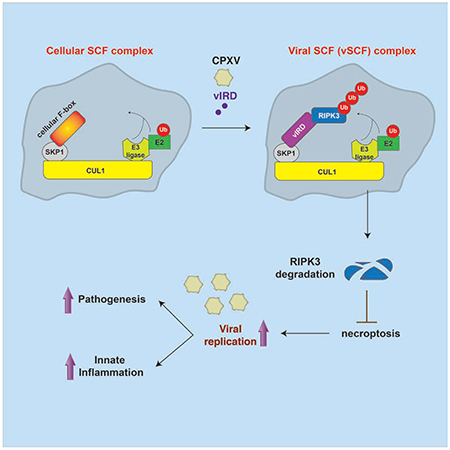
Summary
The vaccine strain against smallpox, vaccinia virus (VACV), is highly immunogenic yet causes relatively benign disease. These attributes are believed to be caused by gene loss in VACV. Using a targeted siRNA screen, we identified a viral inhibitor found in cowpox virus (CPXV) and other orthopoxviruses that bound to the host SKP1-Cullin1-F-box (SCF) machinery and the essential necroptosis kinase Receptor Interacting Protein Kinase 3 (RIPK3). This “viral inducer of RIPK3 degradation (vIRD)” triggered ubiquitination and proteasome-mediated degradation of RIPK3 and inhibited necroptosis. In contrast to orthopoxviruses, the distantly related leporipoxvirus Myxoma virus (MYXV), which infects RIPK3-deficient hosts, lacks a functional vIRD. Introduction of vIRD into VACV, which encodes a truncated and defective vIRD, enhanced viral replication in mice. Deletion of vIRD reduced CPXV-induced inflammation, viral replication and mortality, which were reversed in RIPK3- and MLKL-deficient mice. Hence, vIRD-RIPK3 drives pathogen-host evolution and regulates virus-induced inflammation and pathogenesis.
eTOC Blurb
The orthopoxvirus vaccinia virus sensitizes cells to TNF-induced necroptosis, but it is not known whether other orthopoxviruses also sensitizes cells to necroptosis. Liu et al. demonstrate that a family of orthopoxvirus viral inhibitors that targets RIPK3 for proteasomal degradation. This strategy critically controls viral replication and anti-viral innate immunity.
Graphical Abstract
Introduction
Interference of the host immune response is critical in determining the fitness and pathogenicity of viruses (Nailwal and Chan, 2018). Evasion of host cell death is a common strategy used by viruses to facilitate their replication within the host. While apoptosis is tolerogenic, certain forms of lytic cell death could promote anti-viral inflammation. Large DNA viruses in the herpesvirus and poxvirus families are adept at subverting host cell apoptosis. Inhibition of apoptosis can prime the infected cells to lytic cell death such as necroptosis, an inflammatory form of cell death mediated by the serine/threonine kinase Receptor Interacting Protein Kinase 3 (RIPK3) and its downstream effector mixed lineage kinase domain-like (MLKL) (Chan et al., 2015). Caspase 8 cleaves and inactivates the necroptosis adaptors RIPK1, RIPK3 and the de-ubiquitinase cylindromatosis (CYLD). Thus, optimal induction of necroptosis requires caspase 8 inhibition. The orthopoxvirus vaccinia virus encodes the CrmA-like orthologue B13R (aka Spi2) to inhibit caspase 8 and sensitize the infected cells to necroptosis (Chan et al., 2003; Cho et al., 2009; Li and Beg, 2000). Mice that are deficient in RIPK1 or RIPK3 fail to control VACV replication and often succumb to the infection (Cho et al., 2009; Polykratis et al., 2014).
In contrast to VACV, herpesviruses such as murine cytomegalovirus (MCMV), herpes simplex virus 1 (HSV1) and HSV2 encode inhibitors that block both caspase-dependent apoptosis and RIP kinase-mediated necroptosis (Guo et al., 2015; Huang et al., 2015; Upton et al., 2010, 2012; Yu et al., 2016). In these herpesviruses, the viral inhibitors encode “RIP homotypic interaction motif” (RHIM) that mediates binding to and sequestration of RIPK3 and another necroptosis adaptor ZBP1. Hence, unlike VACV, apoptosis and necroptosis are both inhibited in these herpesviruses. As such, despite the wide acceptance of necroptosis as an anti-viral host response (Mocarski et al., 2015; Nailwal and Chan, 2018; Upton and Chan, 2014), evidence that necroptosis plays a role in host response and viral pathogenesis remains limited.
VACV is a laboratory adapted orthopoxvirus of unknown origin. In contrast to other orthopoxviruses found in nature, VACV causes a relatively benign viral disease in human and in mice (Sanchez-Sampedro et al., 2015). The reduced pathogenicity renders VACV a superior choice as a vaccine vehicle. When compared to other orthopoxviruses, the genome of VACV contains many gene deletions (Bratke et al., 2013). It is widely believed that these missing genes regulate host immune responses and that their loss contribute to the relatively mild disease of VACV. However, the functions of the majority of these missing viral genes and their roles in pathogenesis have remained elusive.
Here, we report the identification of a viral inhibitor that we have termed “viral Inducer of RIPK3 Degradation” (vIRD). vIRD was identified from a targeted siRNA screen in CPXV-infected cells. During CPXV infection, vIRD bound to RIPK3 via its N-terminal ankyrin repeats and the cellular SKP1-CUL1 complex via its C-terminal F-box. vIRD promoted K48-linked RIPK3 ubiquitination and proteasome-mediated degradation, leading to necroptosis resistance of the infected cells. Deletion of vIRD, pharmacologic inhibition CUL1, or siRNA silencing of CUL1 reversed virus-induced RIPK3 degradation and restored sensitivity to necroptosis of infected cells. Reconstitution of wild type vIRD in a vIRD deletion mutant restored RIPK3 degradation and necroptosis resistance. In mouse infection, deletion of vIRD reduced chemokine expression, immune cell recruitment, tissue injury, viral replication and mortality of CPXV infection. These effects were reversed in mice expressing a loss-of-function RHIM-deleted RIPK3 mutant and Mlkl−/− mice, indicating that vIRD targets RIPK3- and MLKL-dependent necroptosis to promote pathogenesis. Reconstitution of an intact vIRD in VACV, which lacks an intact vIRD orthologue, increased viral replication in vivo. We further identified intact vIRD orthologues in other orthopoxviruses such as variola virus, monkeypox virus and ectromelia virus. The distantly related leporipoxvirus Myxoma virus (MYXV) lacks a functional vIRD but nonetheless causes deadly disease due to the host’s deficiency of RIPK3. These results indicate that vIRD critically regulates anti-viral inflammation, viral replication and pathogenesis by inhibiting RIPK3- and MLKL-dependent necroptosis. The loss of vIRD in poxviruses that infect RIPK3-deficient hosts suggests that the vIRD-RIPK3 interaction plays an important role in pathogen-host co-evolution.
Results
Cowpox Virus Actively Inhibits Necroptosis
VACV is a laboratory adapted orthopoxvirus that sensitizes infected cells to necroptosis (Cho et al., 2009). Whether other orthopoxviruses similarly sensitize cells to necroptosis has not been explored. To interrogate the effect of other orthopoxviruses on necroptosis, we infected the murine fibrosarcoma L929 with recombinant VACV, CPXV and MYXV expressing GFP. Infection of L929 stimulated autocrine Tnf expression (Fig. S1A). As expected, VACV caused cell death in infected L929 cells (Figure 1A). MYXV-infected cells similar underwent virus-induced cell death (Figure 1A). By contrast, cell death was not detected in CPXV-infected L929 cells (Figure 1A). We induced classical necroptosis with TNF, the Smac mimetic BV6 and the pan-caspase inhibitor zVAD-fmk (TBZ) and found that CPXV-infected cells were resistant to TBZ-induced necroptosis (Figure 1B). CPXV encodes the caspase 1 and caspase 8 inhibitor CrmA, which is widely thought to be critical for the switch from apoptosis to necroptosis. Caspase 8 cleavage was indeed inhibited in CPXV-infected L929 despite autocrine TNF production (Figure S1B). TNF and BV6 (TB) led to reduced but detectable caspase 8 cleavage (Figure S1B–S1C), indicating that the inhibitory activity of CrmA could be overcome by exogenous TNF stimulus. Consequently, CPXV-infected L929 underwent TB-induced apoptosis (Figure 1C). In contrast to L929, MEFs did not induce autocrine Tnf expression upon CPXV infection (Figure S1D). In MEFs, CPXV infection also conferred resistance to TBZ-induced necroptosis but had minimal effects on TB-induced apoptosis (Figure S1E and S1F). Furthermore, CPXV did not interfere with macrophage pyroptosis or cancer cell ferroptosis (Figure 1D and 1E). Thus, unlike VACV, CPXV specifically targets necroptosis.
Figure 1. CPXV confers resistance to RIPK3-dependent necroptosis.
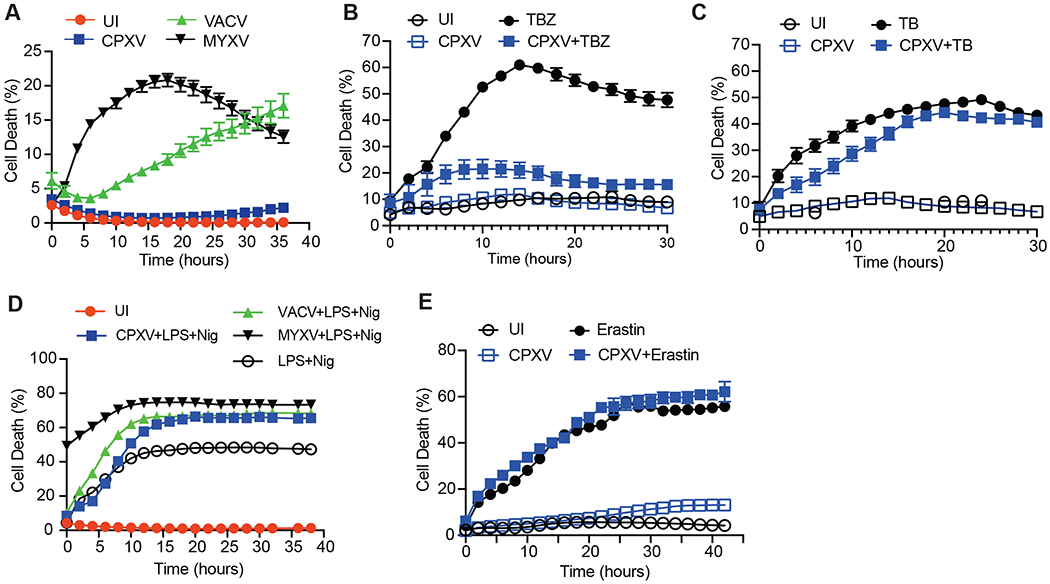
(A) L929 cells were infected with the indicated GFP-expressing recombinant virus. Cell death was determined by Incucyte live cell imaging. UI: uninfected cells. (B) CPXV-infected or uninfected (UI) L929 cells were stimulated with TNF, BV6 and zVAD-fmk. Necroptosis was measured by Incucyte. (C) CPXV-infected or uninfected (UI) L929 cells were treated with TNF and BV6. Cell death was measured by Incucyte. (D) Uninfected J2 virus-transformed macrophages or J2 macrophages infected with the indicated poxviruses were stimulated with LPS (20 μg/ml) and Nigericin (Nig, 10 μM). Cell death was detected using Incucyte. (E) Uninfected or CPXV-infected human colorectal carcinoma Colo205 were treated with Erastin (10 μM) and cell death was measured using Incucyte. See also Figure S1.
CPXV Induces Proteasomal Degradation of RIPK3
RIPK1, RIPK3 and MLKL are key adaptors for TBZ-induced necroptosis. We found that RIPK3 protein expression was completely abolished in CPXV-infected L929 cells (Figure 2A–C). By contrast, RIPK3 expression was not affected by MYXV or VACV (Figure 2A). CPXV-induced RIPK3 protein disappearance was also observed in murine J2 virus-transformed macrophages (Figure 2D–E), MEFs (Figure 2F–G), and human carcinoma HT29 and Colo205 (Figure S2A–S2D). In contrast, RIPK1 expression was not reduced by CPXV (Figure 2A–G). Modest reduction in MLKL, Caspase 8, cIAP2 and the TLR3 and TLR4 adaptor TRIF was also detected (Figure 2B–G, S2A–S2D). Consistent with these results, TBZ-induced MLKL phosphorylation, but not RIPK1 auto-phosphorylation at S166, was abrogated in CPXV-infected cells (Figure 2H).
Figure 2. CPXV causes proteasome-mediated degradation of RIPK3.
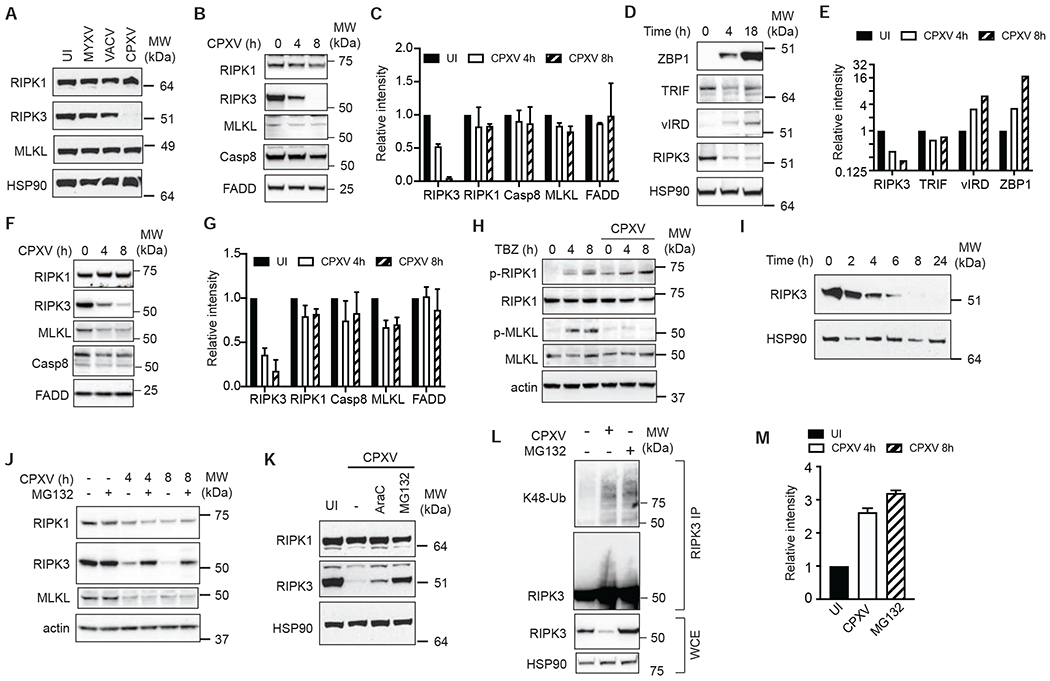
(A-G) Effect of CPXV on necroptosis signal adaptor expression. (A) L929 cells were infected with the indicated viruses for 18 hours and protein expression was determined by Western blot. (B-C) L929 (D-E) J2 virus-transformed macrophages and (F-G) MEFs were infected with CPXV for the indicated amount of time and protein expression was determined by Western blot. (C, E, G) Quantification of CPXV-induced changes of protein expression. Protein expression in uninfected cells at T=0 was normalized to “1”. (H) CPXV inhibited MLKL phosphorylation. Uninfected or CPXV-infected MEFs were treated with TNF, BV6 and zVAD for the indicated times. RIPK1 phosphorylation at S166 and MLKL phosphorylation at S345 were examined by Western blot. (I) Kinetics of RIPK3 protein degradation in CPXV-infected L929 cells. (J) The proteasome inhibitor MG132 inhibited CPXV-induced RIPK3 degradation. L929 cells were infected with CPXV for 2h, followed by MG132 (10 μM). Protein expression was determined by Western blot. (K) CPXV-infected L929 cells were treated with AraC or MG132 and their effect on RIPK3 degradation was determined by Western blot. (L, M) CPXV induces K48-linked RIPK3 ubiquitination. Cell lysates from CPXV-infected or MG132-treated L929 cells were denatured in 6M urea prior to immunoprecipitation with anti-RIPK3 antibody. Western blot was performed to determine the extent of RIPK3 K48-linked ubiquitination. The extent of RIPK3 ubiquitination was quantified in (M). See also Figure S2.
Poxviruses are known to inhibit transcription of certain host anti-viral genes (Harte et al., 2003; Myskiw et al., 2009; Sumner et al., 2014). However, Ripk3 mRNA was not reduced, but rather increased modestly in CPXV-infected cells (Figure S2E). The loss of RIPK3 protein expression was first detected 4 hours post-infection (Figure 2I). The rapid kinetics of protein loss without reduction in gene transcription suggests that proteolysis was involved. We tested different protease inhibitors and found that RIPK3 expression in CPXV-infected cells was rescued by the proteasome inhibitors MG132, lactacystin and epoxomicin, but not by other protease inhibitors (Figure 2J, S2F and S2G). In contrast, MG132 did not restore MLKL expression (Figure 2J), indicating that distinct mechanisms control RIPK3 and MLKL expression in response to CPXV.
Proteasome activity is required for optimal late poxviral gene expression (Satheshkumar et al., 2009; Teale et al., 2009). Thus, proteasome inhibitors could rescue RIPK3 expression via blockade of late poxviral gene expression. To determine if this was the case, we inhibited late poxvirus gene expression with the DNA synthesis inhibitor AraC (Weir and Moss, 1984). Both AraC and MG132 significantly reduced transcription of CPXV011 and CPXV016 but had no effect on CPXV017 transcription (Figure S2H). However, when compared to MG132, AraC only modestly rescued RIPK3 protein expression (Figure 2K). RIPK3 purified from CPXV-infected cells under 6M urea denaturing condition revealed that RIPK3 underwent K48-linked ubiquitination (Figure 2L–M). We therefore conclude that late gene transcription did not contribute to the disappearance of RIPK3 protein. Rather, CPXV induces K48-linked ubiquitination and subsequent proteasomal degradation of RIPK3.
siRNA Screen Identifies Viral Inducer of RIPK3 Degradation (vIRD)
When compared to other naturally occurring orthopoxviruses, the genome of VACV contains large segments of deletions and many fragmented genes. These missing genes are mostly located in the inverted terminal repeats, a region that encodes many immune modulatory genes (Sanchez-Sampedro et al., 2015). We postulated that CPXV might encode a “viral inducer of RIPK3 degradation” (vIRD) to promote RIPK3 degradation and necroptosis resistance. To this end, we surveyed the CPXV genome and identified genes that are either missing, deleted or fragmented in VACV. Our query returned 53 “unique” CPXV genes that matched our criteria (less than 60% identity with over 80% coverage, E value < 0.0001) (Table S1). We performed a focused siRNA screen against these genes and used Western blot to determine if any of the siRNAs would reverse CPXV-induced RIPK3 degradation. One of the most prominent hits from our screen was CPXV006, which encodes a protein with six ankyrin repeats at the N-terminus and a C-terminal F-box (Figure 3A). Deletion of CPXV006 (hereafter referred to as CPXV-ΔvIRD), but not the unrelated CPXV219, fully prevented CPXV-induced RIPK3 degradation (Figure 3B). L929 cells infected with CPXV-ΔvIRD underwent autocrine TNF-induced necroptosis (Figure 3C). In contrast, re-introduction of wild type vIRD into CPXV-ΔvIRD (CPXV-WT) restored resistance to autocrine TNF-induced necroptosis (Figure 3C). Time course analysis revealed that vIRD protein expression was detected as early as 4 hours post-infection (Figure 3D), which correlated with the time at which RIPK3 degradation became apparent (Figure 3D). CPXV006 orthologues in the Western Reserve and Copenhagen strains of VACV are truncated and missing the C-terminal F-box (Figure 3A), while other VACV strains lack vIRD orthologues that exhibit high sequence homology (https://4virology.net/). We therefore conclude that CPXV006 is the bona fide vIRD that promotes RIPK3 degradation.
Figure 3. vIRD interacts with cellular SCF complex components to promote RIPK3 degradation and necroptosis resistance.
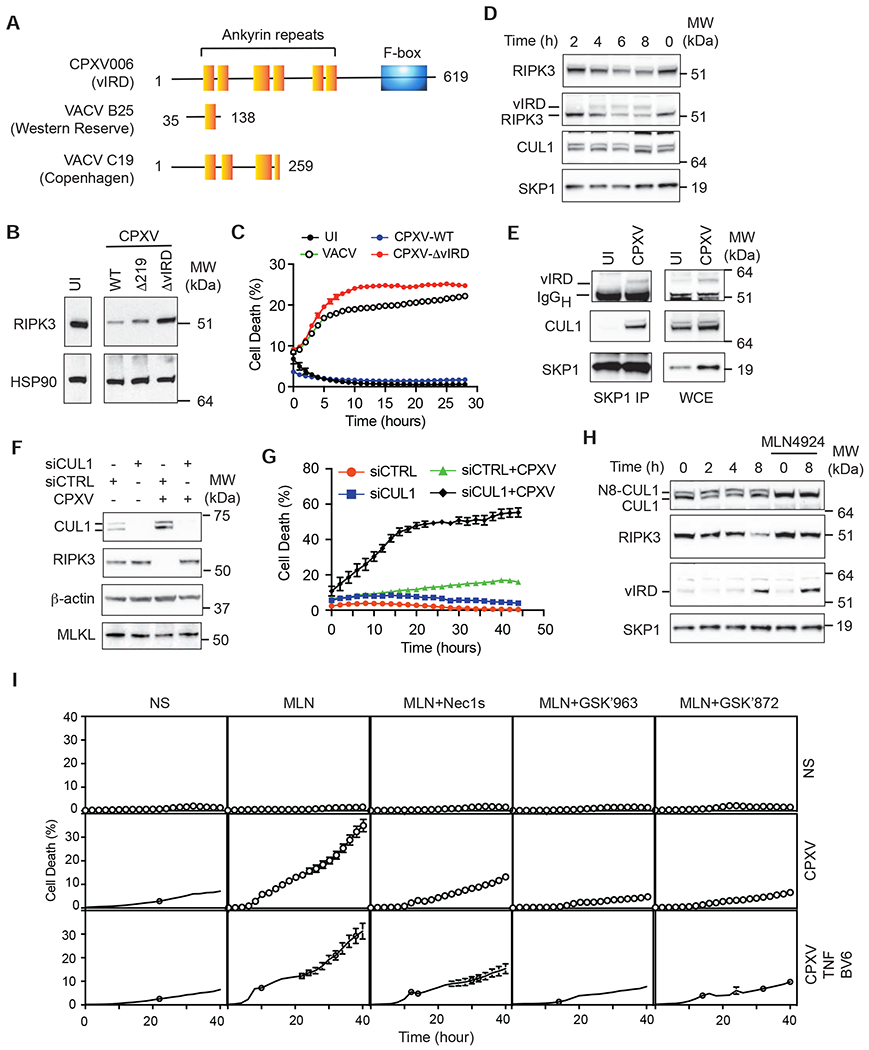
(A) Schematic diagram of the domain organization of vIRD from CPXV and the truncated vIRD orthologues from VACV Western Reserve and Copenhagen strains. (B) L929 cells infected with wild type or the indicated mutant CPXV were analyzed for RIPK3 expression by Western blot. (C) vIRD is essential for necroptosis resistance of CPXV-infected cells. L929 cells infected with VACV, vIRD-deleted CPXV (CPXV-ΔvIRD) or vIRD revertant (CPXV-WT) were monitored for cell death using Incucyte. (D) Kinetics of vIRD expression and RIPK3 degradation were determined by Western blot at the indicated times after CPXV infection. (E) Formation of the viral SCF complex. SKP1 was immunoprecipitated from CPXV-infected and uninfected L929 cells. vIRD and CUL1 recruitment upon CPXV infection was determined by Western blot. (F) CUL1 is essential for CPXV-induced RIPK3 degradation. L929 cells transfected with CUL1-specific or control scrambled siRNAs were monitored for RIPK3 degradation by Western blot. (G) CUL1 is essential for CPXV-mediated resistance to necroptosis. L929 cells transfected with the indicated siRNA were monitored for cell death by Incucyte live cell imaging. (H) Effect of CUL1 neddylation on CPXV-induced RIPK3 degradation. Uninfected or CPXV-infected L929 cells were treated with MLN4924 (1 μM) as indicated. The expression of vIRD and RIPK3 was determined by Western blot. (I) The effect of CUL1 neddylation on CPXV-induced necroptosis. Uninfected and CPXV-infected L929 cells were treated with the indicated inhibitors and/or stimulated with TNF and BV6. Cell death was monitored by Incucyte. See also Figure S3 and Tabls S1.
vIRD Co-opts the Cellular SKP-Cullin-F Box (SCF) Machinery to Promote RIPK3 Degradation
Mammalian F-box proteins function as bridging factors to recruit protein substrates to the SKP1-Cullin1-F box (SCF) complex, a multi-subunit complex that promotes protein ubiquitination and proteasome degradation (Lee and Diehl, 2014). We hypothesized that vIRD interacts with cellular SCF complex components to mediate RIPK3 degradation. Indeed, CPXV infection induced specific interaction between vIRD, SKP1 and CUL1 (Figure 3E). The binding of vIRD to CUL1 was specific, since vIRD did not interact with other Cullins (Figure S3B–C).
Silencing of CUL1 by siRNA reversed CPXV-induced RIPK3 degradation and restored sensitivity to autocrine TNF-induced necroptosis (Figure 3F–G). SCF activity is activated by CUL1 neddylation (Enchev et al., 2015). The neddylation inhibitor MLN4924 effectively converted all cellular CUL1 into the fast-migrating inactive de-neddylated form, reversed CPXV-induced RIPK3 degradation and restored sensitivity of CPXV-induced L929 cells to necroptosis (Figure 3H, 3I and S3A). CPXV-mediated cell death in the presence of MLN4924 was partially inhibited by anti-TNF neutralizing antibody (Figure S3A). Although we could not rule out contribution from other cytokines, these results indicate that autocrine TNF was at least partially responsible for CPXV-induced necroptosis. Regardless of the upstream signals, virus-induced cell death in the presence of MLN4924 was fully inhibited by the RIPK1 kinase inhibitors GSK’963 and Nec1s, and the RIPK3 kinase inhibitor GSK’872 (Figure 3I), indicating that vIRD targets RIPK3 for degradation to inhibit TNF-, RIPK1-, and RIPK3-induced necroptosis.
The ankyrin repeats mediate vIRD binding to RIPK3
F-box binds to SKP1 to recruit substrates to the SCF complex. We therefore postulated that vIRD recruits RIPK3 to the vSCF complex via the ankyrin repeats. Co-expression of vIRD with RIPK3 reduced RIPK3 expression (Figure 4A–B), suggesting that vIRD is sufficient to promote RIPK3 degradation in the absence of virus infection. Deletion of the F-box inhibited vIRD-mediated RIPK3 degradation, which facilitated detection of vIRD-RIPK3 interaction (Figure 4B, compare the first two lanes). Thus, the N-terminal ankyrin repeats were sufficient to mediate RIPK3 binding. Further mapping using various ankyrin repeats deletion mutants revealed that the first five ankyrin repeats are critical for RIPK3 binding (Figure 4B). Deletion of ankyrin repeat 6 did not affect RIPK3 binding, although this mutant also exhibited reduced RIPK3 degradative activity (Figure 4B), which we reasoned might be due to the proximity of the last ankyrin repeat to the essential F-box at the C-terminus.
Figure 4. The ankyrin repeats of vIRD mediate binding to RIPK3.
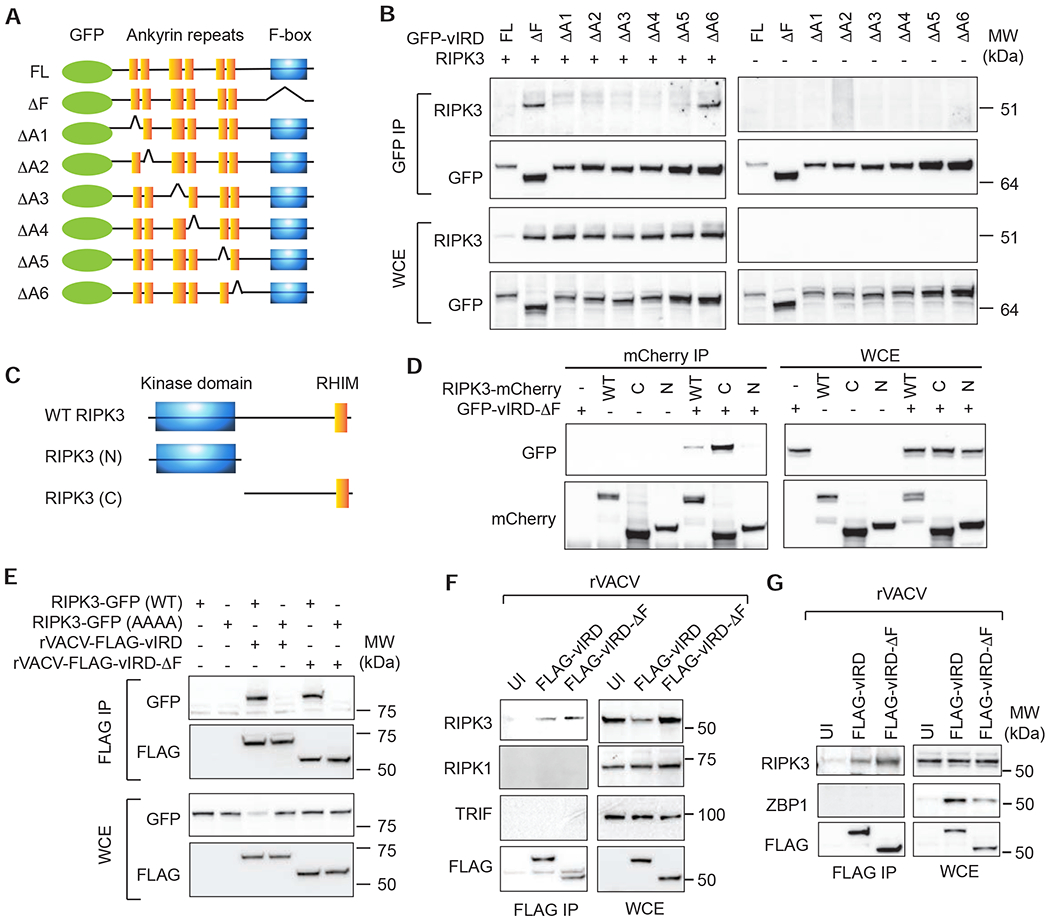
(A) Schematic diagram of the vIRD mutants used. (B) HEK293T cells were transfected with the indicated GFP-tagged vIRD and RIPK3. Immunoprecipitation and Western blot were performed using the indicated antibodies. (C) Schematic diagram of the RIPK3 domain structure and truncation mutants used. (D) An intact RHIM is required for RIPK3 binding to vIRD. HEK293T cells were transfected with the indicated plasmids. Interaction between RIPK3 mutants and vIRD was determined by immunoprecipitation and Western blot as indicated. (E) The RHIM is essential for RIPK3 binding to vIRD. HEK293T cells transfected with the indicated plasmids were infected with rVACV-vIRD or rVACV-vIRD-ΔF. Binding with vIRD was determined by immunoprecipitation and Western blot as indicated. (F) Specific interaction between vIRD and RIPK3. L929 cells were infected with rVACV-FLAG-vIRD or rVACV-FLAG-vIRD-ΔF. vIRD was pulled down with anti-FLAG antibody followed by Western blot detection of the indicated RHIM adaptors. (G) vIRD does not interact with ZBP1. J2 virus transformed macrophages were infected with the indicated rVACV. Immunoprecipitation and Western blot were performed as indicated.
RIPK3 contains an N-terminal kinase domain and a C-terminal RHIM, both of which are essential for necroptosis (Figure 4C). We found that the C-terminus containing the RHIM, but not the N-terminal kinase domain, interacted with vIRD-ΔF (Figure 4D). The RHIM is defined by a highly conserved tetra-peptide core flanked by hydrophobic residues. Tetra-alanine substitution of the core RHIM sequence in RIPK3 abolished its binding to FLAG-tagged vIRD expressed by recombinant VACV (rVACV) (Figure 4E). The binding to RIPK3-RHIM prompted us to ask whether other mammalian RHIM adaptors also interact with vIRD. However, no interaction was detected between vIRD and RIPK1, TRIF or ZBP1 (Figure 4F–G). Hence, the RHIM is necessary but not sufficient to mediate RIPK3 binding to vIRD.
vIRD is conserved in other orthopoxviruses
VACV lacks an intact vIRD and does not confer resistance to necroptosis. Consistent with the hypothesis that vIRD is sufficient to confer resistance to necroptosis, rVACV-vIRD greatly reduced virus-induced necroptosis compared with rVACV-infected L929 cells (Figure 5A). The reduced necroptosis was accompanied by the loss of RIPK3 expression and formation of the vIRD-SKP1-CUL1 complex (Figure 5B–C). In contrast, rVACV-vIRD-ΔF failed to inhibit necroptosis or induce RIPK3 degradation (Figure 5A–B), indicating that vIRD expression is sufficient to reverse the necroptosis sensitivity in VACV-infected cells.
Figure 5. vIRD is conserved in other orthopoxviruses.
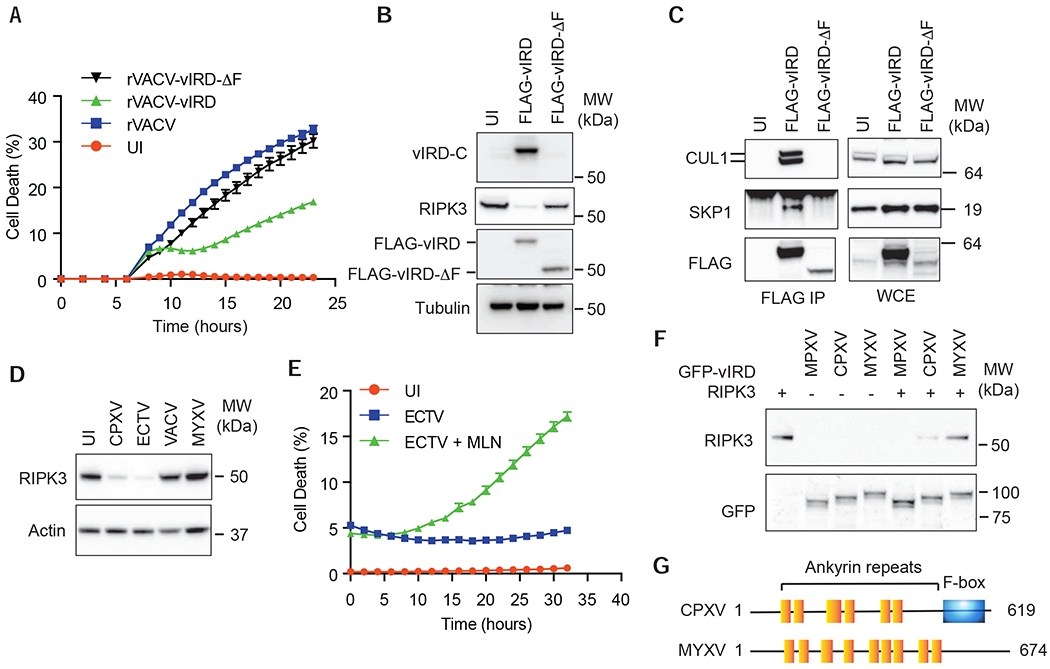
(A) Expression of vIRD inhibits rVACV-induced necroptosis. L929 cells infected with the indicated rVACV were monitored for cell death by Incucyte. (B) Expression of vIRD by VACV triggers RIPK3 degradation. Western blot analysis of RIPK3 expression in L929 cells infected with the indicated rVACV. vIRD-C is a rabbit polyclonal antibody specific for the C-terminus of vIRD. (C) Assembly of vIRD with SKP1 and CUL1 in L929 cells infected with the indicated rVACV expressing wild type or F-box deleted vIRD. (D) RIPK3 degradation by ECTV. L929 cells infected with the indicated viruses were examined for RIPK3 degradation by Western blot. (E) Resistance of ECTV-infected L929 cells to necroptosis is reversed by the neddylation inhibitor MLN4924. (F) The vIRD orthologues from MPXV and ECTV, but not MYXV, triggered RIPK3 degradation. HEK293T cells transfected with the indicated plasmids were determined for RIPK3 expression by Western blot. (G) Schematic diagram of the domain structure of MYXV-m148R compared to that of vIRD from CPXV. See also Figure S4 and S5.
The divergent effect of CPXV and VACV on RIPK3 degradation and necroptosis prompted us to ask whether other orthopoxviruses similarly employ vIRD as strategy to evade host cell necroptosis. We queried the genomes of orthopoxviruses and found that the majority of them, including the etiological agent for smallpox, the variola virus, encode highly conserved vIRD orthologues (Figure S4). Infection of cells with the mousepox ectromelia virus (ECTV), which encodes an intact vIRD orthologue, induced RIPK3 degradation (Figure 5D). Similar to CPXV, ECTV also conferred resistance to autocrine TNF-induced necroptosis in L929 cells, which was reversed by the CUL1 neddylation inhibitor MLN4924 (Figure 5E and S5A). As in the case of CPXV-vIRD, ECTV-vIRD was sufficient to induce RIPK3 degradation independent of virus infection, and deletion of the F-box abolished ECTV-vIRD-mediated RIPK3 degradation (Figure S5B). Moreover, the vIRD orthologue from Monkeypox virus (MPXV) also induced RIPK3 degradation (Figure 5F). These results indicate that vIRD is a highly conserved immune inhibitory mechanism within the orthopoxvirus genus.
In contrast to CPXV and ECTV but similar to VACV, L929 cells infected by the related leporipoxvirus MYXV were sensitive to virus-induced necroptosis (Figure 1A). Survey of the MYXV genome did not reveal any highly conserved vIRD orthologue. The closest ankyrin repeats-containing adaptor to vIRD in MYXV is m148R, which contains eight ankyrin repeats but no F-box (Figure 5G). The ankyrin repeats of m148R shares only ~30% sequence identity with the vIRD orthologues from CPXV (Figure S5C). Accordingly, MYXV-m148R did not induce RIPK3 degradation (Figure 5F). Avipoxviruses, which infect hosts that lack RIPK3 expression (Dondelinger et al., 2016; Petrie et al., 2019), also encode ankyrin repeats-containing adaptors with poor sequence conservation to the orthopoxvirus vIRDs (~30-40%). Collectively, these results strongly implicate vIRD-mediated RIPK3 degradation as an immune evasion strategy employed by orthopoxviruses.
vIRD Targets RIPK3 to Promote Viral Replication
VACV was chosen as the vaccine strain for smallpox because of its reduced pathogenicity when compared to other naturally found orthopoxviruses. The loss of vIRD in VACV prompted us to ask whether it might contribute to viral pathogenesis. RIPK3 deficiency greatly enhanced VACV replication in vivo (Cho et al., 2009). Introduction of an intact vIRD, but not the F-box-deleted vIRD, enhanced VACV replication in vivo (Figure 6A). In contrast, we found that viral load in wild type CPXV-infected mice was more than 100-fold higher than that of CPXV-ΔvIRD infected mice (Figure 6B). To distinguish whether the pathogenic effects of vIRD was due to RIPK3 targeting, we infected Ripk3ΔR/ΔR mice, which express a Ripk3 allele with deletion of the C-terminal RHM (ΔR) and are functionally equivalent to Ripk3−/− mice (Moriwaki et al., 2017). Replication of CPXV-ΔvIRD was restored in Ripk3ΔR/ΔR mice in the spleen and fat pad (Figure 6B). The rescue of CPXV-ΔvIRD replication in the liver of Ripk3ΔR/ΔR mice was statistically significant yet incomplete (Figure 6B). Wild type mice failed to clear CPXV and ultimately succumbed to the infection by day 5 (Figure 6C and S6A). In contrast, CPXV-ΔvIRD was efficiently cleared in wild type mice (Figure 6C and S6A). As expected, CPXV-ΔvIRD infection in Ripk3ΔR/ΔR mice resulted in uncontrolled viral replication and lethal disease (Figure 6C and S6A). Half of the WT CPXV-infected Ripk3ΔR/ΔR mice cleared the virus and survived the infection (Figure 6C and S6A), suggesting that RIPK3 in uninfected cells may also contribute to anti-viral response. These results indicate that RIPK3 is a major cellular target through which vIRD exerts its control on in vivo viral replication.
Figure 6. vIRD promotes viral replication through RIPK3 and necroptosis inhibition.
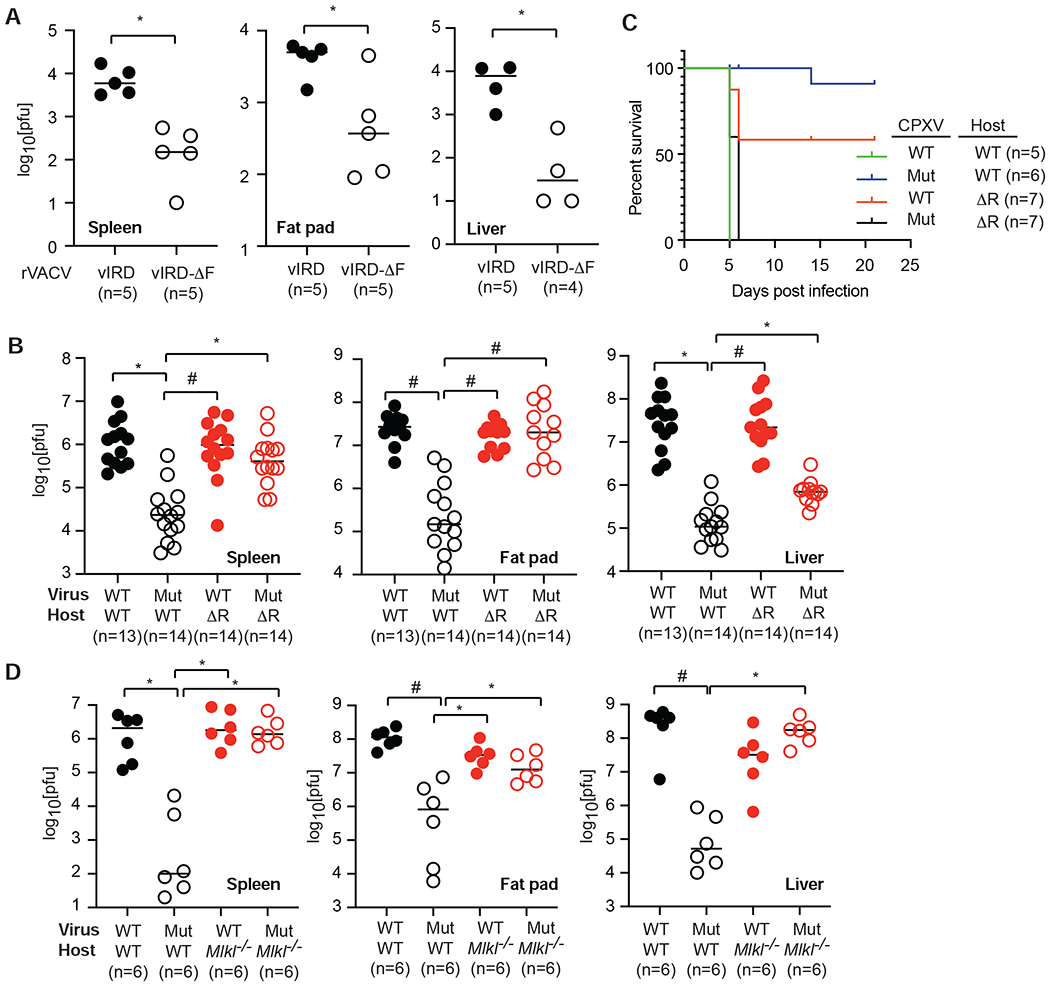
(A) Expression of vIRD enhanced VACV replication in mice. Mice were infected with rVACV-vIRD or rVACV-vIRD-ΔF. Three and a half days later, viral titers in the indicated tissues were determined by Vero cell plaque assay. (B) Ripk3gfpfl/fl (WT) or Ripk3ΔR/ΔR (ΔR) mice were infected with wild type (WT) or CPXV-ΔvIRD (Mut) via the intraperitoneal route as indicated. Viral load in the indicated tissues was determined by Vero cell plaque assay 3.5 days post-infection. (C) Survival of Ripk3gfpfl/fl (WT) or Ripk3ΔR/ΔR (ΔR) mice infected with the indicated wild type or ΔvIRD CPXV. (D) MLKL deficiency rescues defective viral replication of CPXV-ΔvIRD. Mlkl−/− and wild type littermates were infected by the indicated virus. Viral load in the indicated tissues was determined by Vero cell plaque assay 3.5 days post-infection. * p < 0.05, # p < 0.01. See also Figure S6.
Besides necroptosis, RIPK3 has also been implicated in necroptosis-independent signaling (Moriwaki and Chan, 2017). To determine whether necroptosis-dependent or necroptosis-independent function of RIPK3 was the target of vIRD interference, we infected Mlkl−/− mice with CPXV. Viral replication in Mlkl−/− mice was similar to that in Ripk3ΔR/ΔR mice (Figure 6D), indicating that the protective effect of RIPK3 is mainly mediated by necroptosis. However, replication of CPXV-ΔvIRD was fully restored in the liver of Mlkl−/− mice (Figure 6D). The similar pattern in Ripk3ΔR/ΔR and Mlkl−/− mice indicates that vIRD mainly targets the necroptosis-inducing function of RIPK3 to enhance viral pathogenesis.
vIRD Determines Virus-Induced Inflammation
Necroptosis is widely viewed as an inflammatory form of cell death. By targeting RIPK3 for degradation, vIRD would be predicted to suppress necroptosis-induced inflammation. In addition to RIPK3, vIRD might disrupt endogenous F-box adaptors and proteasomal degradation of other cellular substrates. Phosphorylated IκBα is a substrate of the SKP1-CUL1 complex. Indeed, TNF-induced IκBα degradation was inhibited in CPXV-infected MEFs (Figure S6B). Moreover, deletion of vIRD increased CPXV-induced IκBα phosphorylation and degradation (Figure S6C) (Mohamed et al., 2009). These results suggest that vIRD might inhibit virus-induced inflammation via blockade of necroptosis or NF-κB. Nanostring analysis of the visceral fat pad revealed that vIRD rather enhanced expression of the majority of inflammatory genes (Figure 7A, S7A and S7B). For instance, chemokines such as Ccl2 and Cxcl2 were differentially upregulated in wild type CPXV-infected mice but showed impaired induction in CPXV-ΔvIRD infected mice (Figure 7B, Table S2). The reduced Cxcl2 expression correlated with a decrease in neutrophil infiltration in the visceral fat pad of CPXV-ΔvIRD infected mice (Figure 7C). Reduced necrotic cell death and tissue damage was also observed in the visceral fat pad of CPXV-ΔvIRD infected mice (Figure 7D). Importantly, neutrophil infiltration and virus-induced tissue necrosis caused by CPXV-ΔvIRD were restored in Ripk3ΔR/ΔR and Mlkl−/− mice (Figure 7C–D and S7C). Similar pattern of Cxcl2 and Ccl2 expression was observed in Mlkl−/− mice (Figure 7E). Inflammatory gene expression was subdued in the liver when compared with that in the fat pad (Figure 7F). Moreover, expression of the top differentially expressed (DE) genes was not dependent on vIRD expression (Figure 7F, S7D and S7E). Taken together, these results indicate that vIRD-mediated inhibition of RIPK3 and necroptosis increased anti-viral inflammation in a tissue-specific manner.
Figure 7. vIRD regulates virus-induced tissue inflammation.
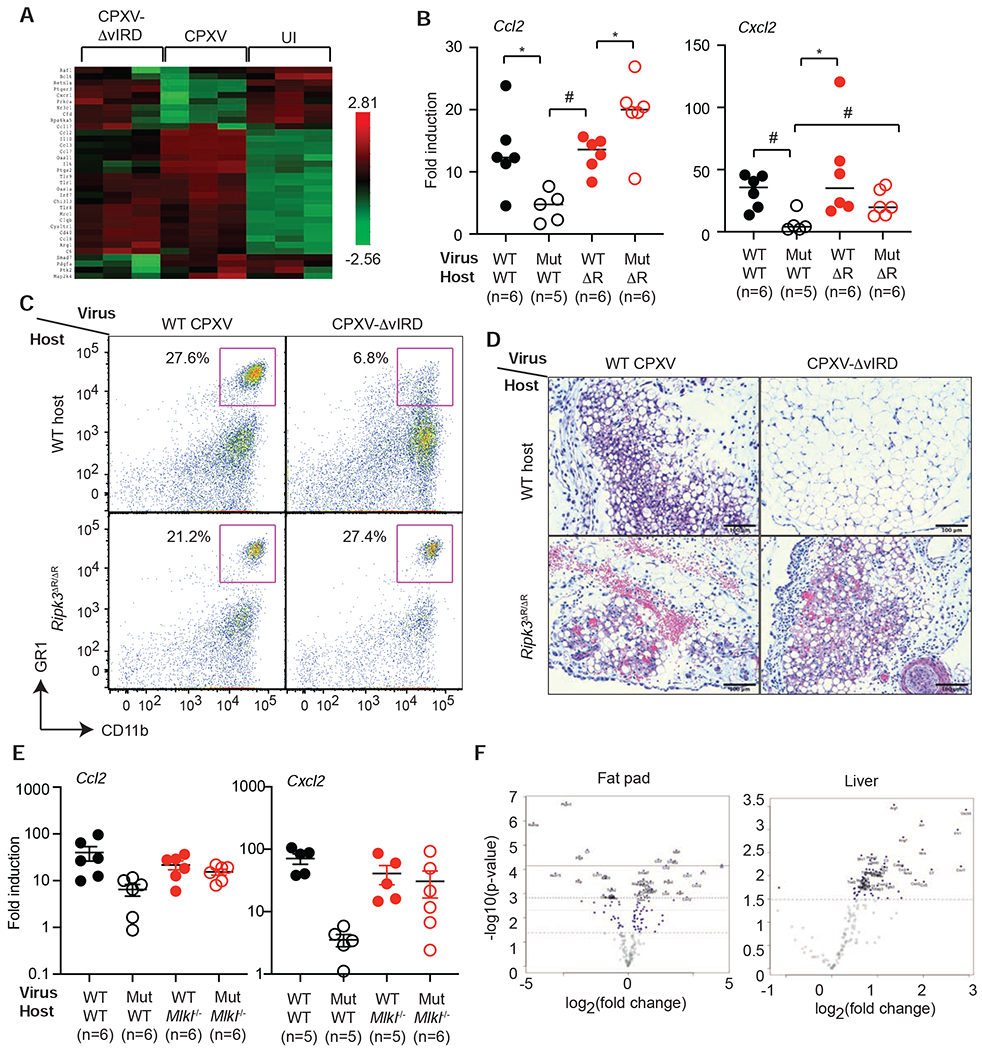
(A) Heat map of the expression of the top 20 DE genes in WT CPXV vs UI and CPXV-ΔvIRD vs UI groups were shown. (B) Q-PCR analysis of Ccl2 and Cxcl2 expression in infected mouse visceral fat pad of the indicated groups. (C) FACS analysis of CD11b+GR1+ neutrophils isolated from the visceral fat pad of the infected mice. (D) H&E staining of infected visceral fat pad from mice in the different groups. Scale bars represent 100 μm. (E) Chemokine expression in Mlkl−/− mice. Wild type or Mlkl−/− littermates were infected with wild type CPXV or CPXV-ΔvIRD and Ccl2 and Cxcl2 expression was determined 3.5 days post-infection. (F) Tissue-specific effects of vIRD on inflammatory gene expression. Wild type mice infected with wild type CPXV or CPXV-ΔvIRD were analyzed for inflammatory gene expression by Nanostring. The top DE genes in visceral fat pad and liver were shown as Volcano plots. * p < 0.05, # p < 0.01. See also Figure S7 and Table S2.
Discussion
In this study, we identified a class of viral necroptosis inhibitors that target RIPK3 for proteasomal degradation. Genetic complementation using vIRD-expressing or deficient viruses and infection in Ripk3ΔR/ΔR and Mlkl−/− mice showed that vIRD targeting of RIPK3 and necroptosis is a key mechanism by which orthopoxviruses enhance viral replication, innate inflammation and pathogenesis. This is contrary to the widely accepted view that necroptosis promotes inflammation (Nailwal and Chan, 2018). How might vIRD promote inflammation when necroptosis is inhibited? One possibility is that vIRD also targets other cellular substrates. Alternatively, vIRD might interfere with activity of the cellular SCF complex to indirectly alter gene expression. In this regard, turnover of certain components of the protein translation machinery that are SCF substrates could account for the effects of vIRD on innate immune gene expression (Yamasaki and Pagano, 2004).
The effect of vIRD on RIPK3 and necroptosis is intrinsic to the infected cells. However, results from mouse infection suggests that RIPK3 function in uninfected cells also contribute to innate immune protection. For example, expression of certain cytokines and chemokines such as Ccl8 and Il6 were modestly reduced in Ripk3ΔR/ΔR mice compared with wild type mice. A role for RIPK3 in cytokine expression independent of necroptosis is consistent with recent observations (Daniels et al., 2017; Najjar et al., 2016). Although RIPK3 deficiency did not affect viral replication on day 3.5, it ultimately compromised viral control by day 5-6 post-infection, leading to highly elevated viral load and lethal disease in some Ripk3ΔR/ΔR mice. Thus, the necroptosis-independent activity of RIPK3 was manifested late during the acute phase of the infection.
Koehler and colleagues recently reported that the antiviral E3 protein from VACV binds to another necroptosis adaptor ZBP1 via its Zα domain to inhibit ZBP1 and RIPK3-dependent necroptosis (Koehler et al., 2017). Cells infected with mutant VACV lacking the Zα domain of E3 were highly sensitive to interferon (IFN)-induced cell death. This ZBP1-RIPK3 axis is kept in check in CPXV and VACV infection and therefore will not be affected by vIRD. In contrast, infection stimulates rapid autocrine TNF production in certain cell types, which leads to RIPK1-RIPK3-MLKL-dependent necroptosis (Carpenter et al., 1994; Cho et al., 2009; Polykratis et al., 2014). Hence, vIRD and E3 act in synergy to protect cells from TNF and IFN, two distinct stress signals that promote necroptosis.
In contrast to most orthopoxviruses, MYXV does not encode a functional vIRD. Yet, MYXV causes deadly disease in its natural host rabbits. Since rabbits lack an intact Ripk3 gene, it is tempting to speculate that the loss of selective pressure from RIPK3 could have led to the loss of a functional vIRD in MYXV. Recently, the Murphy group identified several viral MLKL xenologs from avipoxviruses that bind to RIPK3 and inhibit necroptosis in transfection assays (Petrie et al., 2019). vMLKLs are found in avipoxviruses, but not orthopoxviruses, whose hosts also lack RIPK3 expression. Despite having a vMLKL (Petrie et al., 2019), MYXV-infected cells remained sensitive to autocrine TNF-induced necroptosis. This suggests that vMLKLs may have functions other than necroptosis inhibition. Unlike the majority of orthopoxviruses, VACV lacks a functional vIRD. Given that vIRD potently stimulates innate inflammation, it is tempting to speculate that vIRD may enhance immunogenicity of vaccines.
The poxvirus genomes encode several proteins with N-terminal ankyrin repeats and a C-terminal F-box. This architecture is not found in mammalian F-box or ankyrin repeats-containing adaptors, suggesting that this assembly might have arisen from gene recombination or gene shuffling during host-pathogen evolution. Identification of the cellular targets for these viral ankyrin repeats and F-box adaptors has been elusive. Our results identified RIPK3 as a bona fide substrate for one of these viral ankyrin repeats and F-box adaptors. The ankyrin repeat-RHIM binding between vIRD and RIPK3 represents a distinct mode of interaction, since previous studies showed that the RHIM exclusively mediates homotypic RHIM-RHIM interaction (Li et al., 2012). However, as other RHIM-containing adaptors do not interact with vIRD, additional elements are required for RIPK3 binding to vIRD. Necroptosis adaptors have critical functions in cell death and inflammation. Since manipulating the activity of vIRD could tune the magnitude and quality of host inflammation, the principles revealed from this study may inform the design of more efficacious and safer vaccines in the future.
Limitations of Study
Our study implicates that vIRD is a conserved mechanism that determines virulence of infection by orthopoxviruses. Although expression of several vIRD orthologues from other orthopoxviruses was sufficient to cause RIPK3 degradation, we did not experimentally test whether expression of vIRD in other orthopoxviruses indeed contribute to viral pathogenesis. Our results also revealed subtle differences in the roles of RIPK3 and MLKL in the control of viral replication, innate inflammation and survival in CPXV infection. These differences suggest distinct necroptosis-independent functions for these two necroptosis adaptors. The precise mechanism underlying their distinct roles were not investigated in this study.
STAR Methods
Detailed methods are provided in the online version of this paper and include the following:
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Francis Chan (fc98@duke.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Nanostring datasets are available at Mendeley (https://data.mendeley.com/datasets/tzdrrc59bk/draft?a=c617e139-67e9-4371-9493-e40dadeeb48c).
Experimental Models and Subjects Details
Mice
Ripk3gfpfl/fl mice (B6.129-Ripk3tm1.1Fkmc, Jackson Laboratory), Ripk3DR/DR and Mlkl−/− mice have been described before (Moriwaki et al., 2017; Murphy et al., 2013). All mice have been backcrossed to C57BL6/J background for at least 10 generations. Both male and female mice that were 8-10 weeks old were used. Genotyping primers for Ripk3gfpfl/fl are: 5’-agagatcatctaacccgatctgcct, 5’-tcatagcctgaagaacgagatcagc and 5‘-tcttcacctgctagctactgcgtc. Genotyping primers for Ripk3DR/DR are: 5’-atgagttactctggaggtgggaagg and 5’-cagtttgggtgtaggtccaactgtc. Genotyping primers for Mlkl−/− mice are: 5’-tatgaccatggcaactcacg, 5’-accatctccccaaactgtga and 5’-tccttccagcacctcgtaat. All mice were maintained in standard SPF facility at the Duke University. Infection experiments were performed in dedicated biocontainment facility. All animal experiments were reviewed and approved by the Duke Institutional Animal Care and Use Committee.
Viruses
Wild type CPXV, CPXV-ΔvIRD-GFP, CPXV-ΔvIRD vIRD GFP revertant (Brighton Red strain), VACV-GFP (Western Reserve strain) and MYXV-GFP (Lausanne strain) have been described (Mohamed et al., 2009). Ectromelia virus (ECTV) was obtained from ATCC (VR-1374). Viruses were passaged in BS-C-1 cells by infection at moi = 0.1. After 72 hours, cells were harvested by washing in 1X PBS twice. The resulting pellets were resuspended in PBS supplemented with 0.1% bovine serum albumin. The suspensions were subjected to 3 cycles of freeze and thaw. Recombinant VACV expressing FLAG-tagged wild type vIRD or vIRD-ΔF were generated in the vRB12 strain using the method of Blasco and Moss (Blasco and Moss, 1995). Briefly, FLAG-vIRD or FLAG-vIRD-ΔF were cloned into pRB21 using HiFi DNA assembly (New England Biolabs). BS-C-1 cells (250,000 cells per well) were plated on 12-well plate. Next day, cells were infected with vRB12 (moi = 0.2) in 1 ml of DMEM medium containing 2.5% fetal calf serum for 2 hours. Cells were then transfected with pRB21 plasmid encoding vIRD using TransIT®-LT1 Transfection Reagent (Mirus, MIR2300). After 48 hours, viral supernatants were harvested by 3 cycles of freeze and thaw. The resulting viral supernatants were used to infect BS-C-1 cells and overlayed with 2.5% methylcellulose. Forty-eight hours later, large plaques containing recombinant viruses were collected and subjected to 3 rounds of freeze and thaw. The resulting viral supernatants were subjected to two more rounds of selection in BS-C-1 cells to obtain monoclonal clones of recombinant virus. Viral titers were determined by plaque assay using Vero cells.
Method Details
Virus infection in tissue culture and cell death
Unless otherwise stated, cells were infected with the indicated viruses at moi = 2. Cells were incubated with virus in DMEM (L929, MEFs and J2 virus-transformed macrophages), RPMI 1640 (Colo205) or McCoy’s 5A Modified Medium (HT29) supplemented with 2.5% fetal calf serum for 2 hours before changing to medium with 10% fetal bovine serum. TBZ-induced necroptosis was induced in L929 cells 12 hours after infection by pretreatment with BV6 (1 μM) and zVAD-fmk (10 μM) for 30 minutes, followed by mTNF (20 ng/ml). Pyroptosis was induced in J2 virus-transformed macrophages. Twelve hours after infection, J2 virus-transformed macrophages were treated with LPS (20 ng/ml) for 3 hours followed by nigericin (10 μM) treatment. For ferroptois, human colorectal carcinoma Colo205 infected with CPXV-GFP were treated with erastin (10 μM) 12 hours after infection. Where indicated, after 2 hours infection, L929 cells were treated with MG132 (Sigma, 10 μM), MLN4924 (APExBIO, 1 μM), GSK’963 (kind gift of GSK, 1 μM), GSK’872 (kind gift of GSK, 1 μM), Nec1s (APExBIO, 1 μM) or TNF neutralizing antibody (R&D Systems, MAB4101 at 10 μg/ml). Cell death was tracked by the Incucyte live cell imaging using 100 nM Cytotox Red (Essen Bioscience #4632) or 100 nM Cytotox Green (Essen Bioscience #4633). Percentage cell death was calculated by dividing the Cytotox Red+ or Cytotox Green+ area (nuclear stain of dead cells) over the phase area (area of the whole cell). In some experiments, cell death was independently confirmed by flow cytometric staining with propidium iodide.
Immunoprecipitation and Western blot
HEK293T cells and L929 cells were plated at 2.5×106 cells per 10 cm or 15 cm dish, respectively. Cell lysates were prepared using RIPA lysis buffer (25 mM Tris-HCl, 150 mM NaCl, 0.5 mM EDTA, 0.1% SDS, 0.5% Sodium deoxycholate) supplemented with 1X Complete protease inhibitor cocktail (Roche) and 1X phosphatase inhibitor cocktail (Sigma). Insoluble material was removed via centrifugation at 15,000 rpm for 15 minutes at 4°C. Soluble lysates were transferred to new Eppendorf tubes. Protein concentrations were determined with the BCA Protein Assay (ThermoFisher). Cell lysates were mixed with 1 μg of the immunoprecipitation (IP) antibody. The antibody used are listed in the figure legends of each experiment. Depending on the species from which the IP antibody was derived from, lysates were also incubated with 30 μl of anti-rabbit IgG or anti-mouse IgG conjugated magnetic beads (New England Biolabs).
After rotation at 4°C overnight, the immune complex was washed 2X with RIPA lysis buffer, 2X with RIPA lysis buffer containing 500 mM NaCl, and 1X with RIPA lysis buffer. The resulting immune complex on magnetic beads were boiled in 2X SDS Laemmli lysis buffer and resolved on 4-20% SurePAGE, Bis-Tris precast gels (Genscript). The gel was transferred to nitrocellulose membrane using the Bio-Rad Trans-Blot Turbo system. Clarity or Clarity Max ECL Western blotting substrates were used for chemiluminescence detection.
Chemiluminescence signals were detected using the ChemiDoc MP System from Bio-Rad. For denaturing immunoprecipitation, cell lysates were prepared from L929 cells in 100 μl lysis buffer containing 6 M urea, 25 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10% glycerol, 1.5 mM MgCl2 and 2 mM N-ethylmaleimide for 30 minutes with rotation at room temperature. The denatured cell lysates were diluted with 1.5 ml of lysis buffer without urea, which brought the urea concentration to 0.4 M. After centrifugation at 15,000 rpm for 15 minutes, soluble lysates were transferred to new Eppendorf tubes and incubated with 1 μg rabbit polyclonal anti-mRIPK3 antibody (ProSci) and 30 μl anti-rabbit IgG magnetic beads (New Enlgand Biolabs). After overnight incubation at 4°C and four washes with lysis buffer, the immune complex was resolved on SDS-PAGE. Ubiquitinated RIPK3 was detected on Western blot using K48-linked ubiquitin-specific antibody (Cell Signaling).
Antibodies for Western blot used in the study include: mRIPK3 (ProSci #2283 and Genentech PUR135347), RIPK1 (BD #610459), ZBP1 (Adipogen AG-20B-0010), CUL1 (Life Technologies #718700), MLKL (EMD Millipore, MABC604), HSP90 (BD Bioscience #610418), K48-linked ubiquitin (CST #8081), Caspase 8 (Enzo #ALX-804-447-C100), FADD (Abcam, ab124812), c-Myc (Invitrogen, MA1-980-HRP), SKP1 (BD Bioscience #610530), TRIF (Genentech PUR136257), GFP (Santa Cruz sc-7868), FLAG (Sigma F1804), β-actin (Prosci #3779), HA (Invitrogen #26183), mCherry (Invitrogen M11217). The rabbit anti-CPXV006 antibody (PAS21389) was generated by ProSci using the peptide (CDYHLKSMLYGKNHYKHYPY) as immunogen. Chemiluminescence signals were detected using the ChemiDoc MP System from Bio-Rad.
DNA and siRNA transfection
For DNA plasmid transfections, HEK293T cells cultured in High glucose Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 2 mM glutamine, 10 mM HEPES (pH 7.2), 1X non-essential amino acids solution, 100 U/ml penicillin and 100 μg/ml streptomycin) were transfected with the indicated plasmids using the TransIT®-LT1 Transfection Reagent (Mirus, MIR2300). After 24 hours, cells were lysed with RIPA lysis buffer supplemented with Complete Protease inhibitors (Roche) and phosphatase inhibitor cocktail (Sigma). In Figure 4F and 4G, the transfected 293T cells were infected with rVACV-Flag-vIRD or rVACV-Flag-vIRD-ΔF at moi = 5 for 6h. Cell lysates were prepared using RIPA lysis buffer supplemented with protease and phosphatase inhibitors prior to immunoprecipitation. For siRNA screen, L929 cells were seeded at 2.5 x 105 cells per well on 12-well plate and transfected with 50 nM of siRNA with 6 μl lipofectamine RNAiMax reagent as per manufacturer’s protocol (Invitrogen). Twenty-four hours later, cells were infected with CPXV at moi = 2. Six hours after transfection, whole cell lysates were harvested for Western blot analysis. The siRNA sequences used are found in Table S1. CUL1 siRNA used are: 5’-AUCAGUGUGCAUCAGUCCAACCAAG and 5’-CAUGAUCACACGUAGUCAGGUUGGUUC.
Mouse infection and viral titer determination
All experimental procedures were approved by the Duke Institutional Animal Care and Use Committee. Ripk3-gfpfl/fl were used as wild type controls for Ripk3ΔR/ΔR mice. Wild type controls for Mlkl−/− mice were generated by crossing Mlkl−/− mice with wild type C57BL/6 mice. Male and female mice 8-10 weeks of age were infected intraperitoneally with 1 x 106 pfu of wild type CPXV-GFP or CPXV-ΔvIRD-GFP. Spleen, visceral fat pad, liver and peritoneal exudate cells (PECs) were harvested 3.5 days post-infection using a Omni tissue disruptor DMEM containing 2.5% fetal calf serum. Serial 10-fold dilutions were made for the tissue extracts and viral titers were determined by plaque assays on Vero cells. After 2 days of infection, Vero cells were fixed in 5% formaldehyde for 30 minutes, followed by staining with 0.1% crystal violet in 1% formalin (Chan et al., 2003). After rinsing with water, plaques were counted by eye.
Real time-PCR and Nanostring
Total RNA was isolated from visceral fat pads and liver with RNeasy kit from Qiagen per manufacturer’s instructions. cDNA was generated using iScript Reverse Transcription Supermix (Bio-Rad). Real time-PCR was performed using iQ™ SYBR Green Supermix (Bio-Rad) on the Bio-Rad CFX Connect Real-Time PCR Detection System. Cytokine and chemokine expression were normalized to the internal control Tbp1. The primers for the qPCR reactions are listed in the Key Resource Table. For Nanostring analysis, total RNA was extracted from the fat pad and liver with Qiagen RNeasy Kit. The nCounter Mouse Immunology v1 Gene Expression Panel was used to analyze inflammatory gene expression. Each well in the cartridge was loaded with 300 ng of RNA. Samples were analyzed on the Nanostring nCounter Max System. The nSolver 3.0 software was used for data analysis including background subtraction, normalization, generation of heat map and volcano plots. Nanostring datasets are available at (https://data.mendeley.com/datasets/tzdrrc59bk/draft?a=c617e139-67e9-4371-9493-e40dadeeb48c).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-human/mouse RIPK3 | ProSci | Cat# 2283; RRID: AB_203256 |
| Rat monoclonal anti-mouse RIPK3 | Genentech | Cat# PUR135347 |
| Mouse monoclonal anti-human/mouse RIPK1 | BD Biosciences | Cat# 610459; RRID: AB_397832 |
| Rabbit monoclonal phospho-RIPK1 (Ser166) (D1L3S) | Cell Signaling Technology | Cat#65746; RRID: AB_2799693 |
| Mouse monoclonal anti-human/mouse ZBP1 (Zippy-1) | AdipoGen | Cat# AG-20B-0010, RRID: AB_2490191 |
| Rabbit monoclonal anti-mouse MLKL (D6W1K) | Cell Signaling Technology | Cat# 37705, RRID: AB_2799118 |
| Rabbit monoclonal anti-human MLKL | Abcam | Cat# ab184718, RRID: AB_2755030 |
| Rabbit monoclonal anti-mouse MLKL (phospho S345) | Abcam | Cat# ab196436; RRID: AB_2687465 |
| Mouse monoclonal anti-human/mouse TICAM-1 (TRIF) | BioLegend | Cat# 657102, RRID: AB_2562543 |
| Rabbit monoclonal anti-mouse FADD | Abcam | Cat# ab124812, RRID: AB_10976310 |
| Mouse monoclonal anti-human/mouse cIAP-2/HIAP-1 | R&D Systems | Cat# MAB817, RRID: AB_2063932 |
| Goat polyclonal anti-human cIAP-1/HIAP-2 | R&D Systems | Cat# AF8181, RRID: AB_2259001 |
| Rat monoclonal anti-mouse Caspase-8 (clone 1G12) | Enzo | Cat# ALX-804-447-C100, RRID: AB_2050952 |
| Rabbit monoclonal anti-mouse cleaved Caspase-8 | Cell Signaling Technology | Cat# 8592; RRID: AB_10891784 |
| Rabbit polyclonal anti-CPXV006 | This paper | N/A |
| Mouse monoclonal anti-FLAG (Clone M2) | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Mouse monoclonal anti-HA Tag | Thermo Fisher Scientific | Cat# 26183, RRID: AB_10978021 |
| Rat monoclonal anti-mCherry | Thermo Fisher Scientific | Cat# M11217, RRID: AB_2536611 |
| Rabbit polyclonal anti-human/mouse beta-actin HRP conjugate | ProSci | Cat# 3779, RRID: AB_735551 |
| Rabbit monoclonal anti-K48-linkage Specific Polyubiquitin (D9D5) | Cell Signaling Technology | Cat# 8081; RRID: AB_10859893 |
| Rat monoclonal anti-mouse TNF-alpha (Clone MP6-XT22) | R&D Systems | Cat# MAB4101, RRID: AB_2240643 |
| Mouse monoclonal anti-human/mouse IκBα (Phospho Ser32/36) (5A5) | Cell Signaling Technology | Cat# 9246, RRID: AB_2267145 |
| Mouse monoclonal anti-human/mouse IκBα (Clone L35A5) | Cell Signaling Technology | Cat# 4814, RRID: AB_390781 |
| Mouse monoclonal anti-human c-Myc | Thermo Fisher Scientific | Cat# MA1-980-HRP, RRID: AB_2537628 |
| Mouse monoclonal anti-mouse SKP1 | BD Biosciences | Cat# 610530, RRID: AB_397887 |
| Mouse monoclonal anti-GFP (clone B-2) | Santa Cruz Biotechnology | Cat# sc-9996, RRID: AB_627695 |
| Mouse anti-human TNF RI/TNFRSF1A | R&D Systems | Cat# MAB225, RRID:AB_2204150 |
| Rabbit polyclonal anti-human/mouse Cullin 1 | Thermo Fisher Scientific | Cat# 71-8700, RRID: AB_2534002 |
| Mouse monoclonal anti-human/mouse Hsp90 | BD Biosciences | Cat# 610418, RRID: AB_397798 |
| Rabbit polyclonal anti-human/mouse alpha-tubulin | ProSci | Cat# 7599, RRID: AB_2316270 |
| Peroxidase-IgG Fraction Monoclonal Mouse Anti-Rabbit IgG, Light Chain Specific antibody | Jackson ImmunoResearch Labs | Cat# 211-032-171, RRID: AB_2339149 |
| Peroxidase-AffiniPure Goat Anti-Mouse IgG, Light Chain* Specific antibody | Jackson ImmunoResearch Labs | Cat# 115-035-174, RRID: AB_2338512 |
| Goat Anti-Mouse IgG Magnetic Beads | New England Biolabs | Cat# S1431S |
| Goat Anti-Rabbit IgG Magnetic Beads | New England Biolabs | Cat# S1432S |
| Rat anti-CD16/32 (2.4G2) | Acris Antibodies GmbH | Cat# AM08040FC-N, RRID: AB_1926133 |
| PE anti-mouse Ly-6C (HK1.4) | BioLegend | Cat# 128008, RRID: AB_1186132 |
| APC/Cyanine7 anti-mouse Ly-6G/Ly-6C (Gr-1) (RB6-8C5) | BioLegend | Cat# 108424, RRID: AB_2137485 |
| Pacific Blue anti-mouse CD3epsilon (145-2C11) | BioLegend | Cat# 100333, RRID: AB_2028473 |
| Pacific Blue anti-mouse F4/80 (BM8) | Thermo Fisher Scientific | Cat# MF48028, RRID: AB_10373419 |
| PerCP/Cyanine5.5 anti-mouse/human CD11b (M1/70) | BioLegend | Cat# 101228, RRID: AB_893232 |
| APC anti-mouse CD11c (N418) | BioLegend | Cat# 117309, RRID: AB_313778 |
| Bacterial and Virus Strains | ||
| CPXV (Brighton Red) | Grant McFadden | N/A |
| rCPXV-GFP (Brighton Red) | Grant McFadden | N/A |
| CPXV-ΔvIRD-GFP | Grant McFadden | N/A |
| CPXV-ΔvIRD-GFP vIRD revertant | Grant McFadden | N/A |
| MYXV-GFP (Lausanne) | Grant McFadden | N/A |
| CPXVΔ219 | Grant McFadden | N/A |
| ECTV | ATCC | Cat# VR-1374 |
| VACV (vRB12 strain) | Bernard Moss | N/A |
| rVACV-FLAG-vIRD | This paper | N/A |
| rVACV-FLAG-vIRD-ΔF | This paper | N/A |
| rVACV-GFP (Western Reserve) | Grant McFadden | N/A |
| NEB 5-alpha Competent E. coli | NEB | Cat# C2987 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MG-132 | Sigma-Aldrich | Cat# 474787 |
| AraC (Cytarabine) | APExBIO | Cat# A8405 |
| Nec1s | APExBIO | Cat# A4213 |
| zVAD-fmk | APExBIO | Cat# A1902 |
| CA074 | APExBIO | Cat# A1926 |
| BB-94 | APExBIO | Cat# A2577 |
| AEBSF | APExBIO | Cat# A2573 |
| zFA-fmk | APExBIO | Cat# A8170 |
| Bestatin | APExBIO | Cat# A2575 |
| E-64 | APExBIO | Cat# A2576 |
| PSI60 | APExBIO | Cat# A1900 |
| MLN4924 | APExBIO | Cat# A3629 |
| Erastin | APExBIO | Cat# B1524 |
| Epoxomicin | APExBIO | Cat# A2606 |
| Lactacystin | APExBIO | Cat# A2578 |
| Incucyte® Cytotox Green Reagent | Essen Bioscience | Cat# 4633 |
| Incucyte® Cytotox Red Reagent | Essen Bioscience | Cat# 4632 |
| propidium iodide | Sigma | Cat# P4864 |
| 10% Neutral Buffered Formalin | Leica Biosystems | Cat# 3800510 |
| Smac mimetic (TS) BV6 | Genentech | N/A |
| GSK ‘963 | GlaxoSmithKline | GSK3002963A |
| GSK ‘872 | GlaxoSmithKline | GSK2399872B |
| Nigericin | APExBIO | Cat# B7644 |
| TransIT®-LT1 Transfection Reagent | MIRUS Bio | Cat# MIR2300 |
| Lipofectamine™ RNAiMAX Transfection Reagent | Thermo Fisher Scientific | Cat# 13778075 |
| Recombinant mouse TNF-α | Biolegend | Cat# 575204 |
| Recombinant human TNF-α | Biolegend | Cat# 570106 |
| LPS-EB (LPS from E. coli O111:B4) | Invivogen | Cat# tlrl-eblps |
| Opti-MEM Reduced Serum Medium | GIBCO | Cat# 31985-070 |
| Fetal Bovine Serum (Characterized) | HyClone | Cat# SH30071.03 |
| Sodium pyruvate | Corning | Cat#: 25-000-CI |
| L-Glutamine | GIBCO | Cat#: 25030-164 |
| Pen Strep (Penicillin Streptomycin) | GIBCO | Cat# 15140-122 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M3148 |
| DNase I | Sigma-Aldrich | Cat# D7291 |
| Collagenase Type II | Sigma-Aldrich | Cat# C2-28 |
| N-Ethylmaleimide (NEM) | Calbiochem | Cat# 34115 |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma-Aldrich | Cat# E9884 |
| Complete Protease Inhibitor cocktail | Sigma-Aldrich | Ca #11697498001 |
| Phosphatase inhibitor cocktail | Sigma-Aldrich | Cat# P5726 |
| Critical Commercial Assays | ||
| nCounter Mouse Immunology v1 Gene Expression Panel |
Nanostring Technology | Cat# 115000082 |
| Master Kit--12 reactions | Nanostring Technology | Cat# 100052 |
| RNeasy kit | QIAGEN | Cat# 74104 |
| iScript™ cDNA Synthesis Kit | BIO-RAD | Cat# 4106228 |
| iQ™ SYBR® Green Supermix | BIO-RAD | Cat# 1708880 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Deposited Data | ||
| Nanostring datasets | This paper | https://data.mendeley.com/datasets/tzdrrc59bk/draft?a=c617e139-67e9-4371-9493-e40dadeeb48c |
| Experimental Models: Cell Lines | ||
| MEF | Generated in-house | N/A |
| L929 | ATCC | Cat# CCL-1 |
| HEK293T cell line | Invitrogen TM | Cat#: R70007 |
| Vero | ATCC | Cat# CRL-1586 |
| BS-C-1 | ATCC | Cat# CCL-26 |
| Colo205 | ATCC | Cat# CCL-222 |
| J2 virus-transformed macrophages | Generated in-house | N/A |
| THP-1 | ATCC | Cat# HIB-202 |
| HT29 | ATCC | Cat# HTB-38 |
| Experimental Models: Organisms/Strains | ||
| Ripk3gfpfl/fl mice | Jackson Laboratory | Cat# 030284 |
| Ripk3ΔR/ΔR mice | PMID: 28273458 | N/A |
| Mlkl−/− mice | PMID: 24012422 | N/A |
| Recombinant DNA | ||
| pEGFP-C1-CPXV006 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔANK1 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔANK2 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔANK3 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔANK4 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔANK5 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔANK6 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔF | This paper | N/A |
| pEGFP-C1-CPXV006-ΔF-Box-ΔANK1 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔF-Box-ΔANK2 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔF-Box-ΔANK3 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔF-Box-ΔANK4 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔF-Box-ΔANK5 | This paper | N/A |
| pEGFP-C1-CPXV006-ΔF-Box-ΔANK6 | This paper | N/A |
| pcDNA3-myc3-Cul1 | Addgene | Cat# 19896 |
| pcDNA3-myc3-Cul2 | Addgene | Cat# 19892 |
| pcDNA3-myc3-Cul3 | Addgene | Cat# 19893 |
| pcDNA3-myc3-Cul4a | Addgene | Cat# 19951 |
| pcDNA3-myc3-Cul4b | Addgene | Cat# 19922 |
| pcDNA3-myc3-Cul5 | Addgene | Cat# 19895 |
| pcDNA3-HA-Cul7 | Addgene | Cat# 20696 |
| pEGFP-N1-mZbp1 | This paper | N/A |
| pEGFP-C1-m148R | This paper | N/A |
| pEGFP-C1-MPXV-vIRD | This paper | N/A |
| pEGFP-C1-ECTV-vIRD | This paper | N/A |
| pEGFP-C1-ECTV-vIRD-ΔF | This paper | N/A |
| pmCherry-hRIPK3-C | This paper | N/A |
| pmCherry-hRIPK3-N | This paper | N/A |
| HA-mRIP3 WT-FLAG/pTRIPZ C-HA | Addgene | Cat# 73701 |
| HA-mRIP3 AAAA-FLAG/pTRIPZ C-HA | Addgene | Cat#73702 |
| pRB21 | Bernard Moss | N/A |
| pRB21-FLAG-vIRD | This paper | N/A |
| pRB21-FLAG-vIRD-ΔF | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism 8.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo v10 | FlowJo | https://www.flowjo.com/solutions/flowjo |
| Adobe Illustrator CC 2020 | Adobe | https://www.adobe.com/ |
| Fiji/ImageJ | Open source | fiji.sc |
| nSolver 3.0 | Nanostring Technology | https://www.nanostring.com/products/analysis-software/nsolver |
| Incucyte S3 Imaging Software version 2020B | Essen Bio | https://www.essenbioscience.com/en/products/incucyte/incucyte-s3/ |
| Other | ||
| siRNA sequences for CPXV screen | Table S1 | N/A |
| Q-PCR primers | Table S2 | N/A |
Flow cytometry analysis
Splenocytes were released from the tissues by physical disruption using glass slides. Peritoneal exudate cells (PECs) were collected by flushing the peritoneal cavity with 5 ml DPBS. Visceral fat pad was digested with DNase I (10 μg/ml), collagenase II (4 mg/ml) and CaCl2 (10 mM) for 30 minutes at 37°C to release the leukocytes. Red blood cells were lysed with ACK lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA). Tissue debris was removed by filtering on nylon mesh. Prior to staining with primary antibodies, cells were incubated with anti-Fc receptor 2.4G2 antibody for 10 minutes, followed by staining with the indicated antibodies for flow cytometric analysis on a BD FACSCanto™ II flow cytometer. The antibodies used are listed in the Key Resource Table. Data analysis was performed with Flowjo 9.9.6 (Treestar). Quantification of results were performed using Prism 8.0 (Graphpad).
Histology
Visceral fat pad was removed from the peritoneal cavity and fixed in 10% Neutral Buffered Formalin (Leica Biosystems), embedded in paraffin and cut in 5 μm sections. Tissue sections were stained with Hematoxylin and Eosin. Stained sections were imaged with Leica DMD108 microscope using 20X objective (for Ripk3gfpfl/fl and Ripk3ΔR/ΔR mice) or 10X objective (for Mlkl−/− mice). Representative images are shown in Figure 7D and S7C.
Statistical analysis
Statistical analyses were performed using Prism 8.0 (GraphPad). Line and bar graphs depict mean ± SEM for each data point. In Figure 6 and 7, data for each individual mouse were shown and the bars represent the mean of the group. Non-paired two-tailed Student’s t tests (95% confidence level) were used for comparison of 2 groups. n represents the number of animals used in each group. Statistical details can be found in the figure legends. P values of less than 0.05 were considered significant (* p < 0.05, # p < 0.01). Kaplan-Meier survival curve was plotted using Prism 8.0.
Supplementary Material
Highlights.
Cowpox virus renders infected cells resistant to necroptosis
RIPK3 is rapidly degraded upon infection with cowpox virus
Targeted siRNA screen identifies viral Inducer of RIPK3 degradation (vIRD)
vIRD targets necroptosis, regulates viral replication and anti-viral inflammation
Acknowledgement
We thank members of the Chan Lab for discussion. This work was supported by NIH grants AI 128197 (F.C.) and R01 AI080607 (G.M.). We thank the Duke University School of Medicine for the use of the Microbiome Core Facility for help with Nanostring data acquisition.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
A U.S. Provisional Patent Application No. 62/848,255 has been filed for this study.
References
- Blasco R, and Moss B (1995). Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene 158, 157–162. [DOI] [PubMed] [Google Scholar]
- Bratke KA, McLysaght A, and Rothenburg S (2013). A survey of host range genes in poxvirus genomes. Infection, genetics and evolution : journal of molecular epidemiology and evolutionary genetics in infectious diseases 14, 406–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter EA, Ruby J, and Ramshaw IA (1994). IFN-gamma, TNF, and IL-6 production by vaccinia virus immune spleen cells. An in vitro study. J Immunol 152, 2652–2659. [PubMed] [Google Scholar]
- Chan FK, Luz NF, and Moriwaki K (2015). Programmed necrosis in the cross talk of cell death and inflammation. Annual review of immunology 33, 79–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, and Lenardo MJ (2003). A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem 278, 51613–51621. [DOI] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, and Chan FK (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels BP, Snyder AG, Olsen TM, Orozco S, Oguin TH 3rd, Tait SWG, Martinez J, Gale M Jr., Loo YM, and Oberst A (2017). RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell 169, 301–313.e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJM, and Vandenabeele P (2016). An evolutionary perspective on the necroptotic pathway. Trends in cell biology 26, 721–732. [DOI] [PubMed] [Google Scholar]
- Enchev RI, Schulman BA, and Peter M (2015). Protein neddylation: beyond cullin-RING ligases. Nature reviews. Molecular cell biology 16, 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, and Mocarski ES (2015). Herpes simplex virus suppresses necroptosis in human cells. Cell host & microbe 17, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harte MT, Haga IR, Maloney G, Gray P, Reading PC, Bartlett NW, Smith GL, Bowie A, and O’Neill LA (2003). The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. The Journal of experimental medicine 197, 343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, Wu J, Zhuang Q, Chen C, Li J, et al. (2015). RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell host & microbe 17, 229–242. [DOI] [PubMed] [Google Scholar]
- Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, Upton JW, Mocarski ES, and Jacobs BL (2017). Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc Natl Acad Sci U S A 114, 11506–11511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EK, and Diehl JA (2014). SCFs in the new millennium. Oncogene 33, 2011–2018. [DOI] [PubMed] [Google Scholar]
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et al. (2012). The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, and Beg AA (2000). Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J Virol 74, 7470–7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarski ES, Guo H, and Kaiser WJ (2015). Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology 479-480, 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed MR, Rahman MM, Rice A, Moyer RW, Werden SJ, and McFadden G (2009). Cowpox virus expresses a novel ankyrin repeat NF-kappaB inhibitor that controls inflammatory cell influx into virus-infected tissues and is critical for virus pathogenesis. J Virol 83, 9223–9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki K, Balaji S, Bertin J, Gough PJ, and Chan FK (2017). Distinct Kinase-Independent Role of RIPK3 in CD11c(+) Mononuclear Phagocytes in Cytokine-Induced Tissue Repair. Cell Rep 18, 2441–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki K, and Chan FK (2017). The Inflammatory Signal Adaptor RIPK3: Functions Beyond Necroptosis. International review of cell and molecular biology 328, 253–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. [DOI] [PubMed] [Google Scholar]
- Myskiw C, Arsenio J, van Bruggen R, Deschambault Y, and Cao J (2009). Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-kappaB, and IRF3 pathways. J Virol 83, 6757–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nailwal H, and Chan FK (2018). Necroptosis in anti-viral inflammation. Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjar M, Saleh D, Zelic M, Nogusa S, Shah S, Tai A, Finger JN, Polykratis A, Gough PJ, Bertin J, et al. (2016). RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity 45, 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie EJ, Sandow JJ, Lehmann WIL, Liang LY, Coursier D, Young SN, Kersten WJA, Fitzgibbon C, Samson AL, Jacobsen AV, et al. (2019). Viral MLKL Homologs Subvert Necroptotic Cell Death by Sequestering Cellular RIPK3. Cell Rep 28, 3309–3319.e3305. [DOI] [PubMed] [Google Scholar]
- Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, Lee TH, Chan FKM, Pasparakis M, and Kelliher MA (2014). Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol 193, 1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Sampedro L, Perdiguero B, Mejias-Perez E, Garcia-Arriaza J, Di Pilato M, and Esteban M (2015). The evolution of poxvirus vaccines. Viruses 7, 1726–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satheshkumar PS, Anton LC, Sanz P, and Moss B (2009). Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J Virol 83, 2469–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner RP, Maluquer de Motes C, Veyer DL, and Smith GL (2014). Vaccinia virus inhibits NF-kappaB-dependent gene expression downstream of p65 translocation. J Virol 88, 3092–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teale A, Campbell S, Van Buuren N, Magee WC, Watmough K, Couturier B, Shipclark R, and Barry M (2009). Orthopoxviruses require a functional ubiquitin-proteasome system for productive replication. J Virol 83, 2099–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, and Chan FK (2014). Staying alive: cell death in antiviral immunity. Mol Cell 54, 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, and Mocarski ES (2010). Virus inhibition of RIP3-dependent necrosis. Cell host & microbe 7, 302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, and Mocarski ES (2012). DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell host & microbe 11, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir JP, and Moss B (1984). Regulation of expression and nucleotide sequence of a late vaccinia virus gene. J Virol 51, 662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki L, and Pagano M (2004). Cell cycle, proteolysis and cancer. Current opinion in cell biology 16, 623–628. [DOI] [PubMed] [Google Scholar]
- Yu X, Li Y, Chen Q, Su C, Zhang Z, Yang C, Hu Z, Hou J, Zhou J, Gong et al. (2016). Herpes Simplex Virus 1 (HSV-1) and HSV-2 Mediate Species-Specific Modulations of Programmed Necrosis through the Viral Ribonucleotide Reductase Large Subunit R1. J Virol 90, 1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Nanostring datasets are available at Mendeley (https://data.mendeley.com/datasets/tzdrrc59bk/draft?a=c617e139-67e9-4371-9493-e40dadeeb48c).