

Abstract
The Neuroepithelial transforming gene 1 (Net1) is a RhoA subfamily guanine nucleotide exchange factor that is overexpressed in a number of cancers and contributes to cancer cell motility and proliferation. Net1 also plays a Rho GTPase independent role in mitotic progression, where it promotes centrosomal activation of Aurora A and Pak2, and aids in chromosome alignment during prometaphase. To understand regulatory mechanisms controlling the mitotic function of Net1, we examined whether it was phosphorylated by the mitotic kinase Cdk1. We observed that Cdk1 phosphorylated Net1 on multiple sites in its N-terminal regulatory domain and C-terminus in vitro. By raising phospho-specific antibodies to two of these sites, we also demonstrated that both endogenous and transfected Net1 were phosphorylated by Cdk1 in cells. Substitution of the major Cdk1 phosphorylation sites with aliphatic or acidic residues inhibited the interaction of Net1 with RhoA, and treatment of metaphase cells with a Cdk1 inhibitor increased Net1 activity. Cdk1 inhibition also increased Net1 localization to the plasma membrane and stimulated cortical F-actin accumulation. Moreover, Net1 overexpression caused spindle polarity defects that were reduced in frequency by acidic substitution of the major Cdk1 phosphorylation sites. These data indicate that Cdk1 phosphorylates Net1 during mitosis and suggest that this negatively regulates its ability to signal to RhoA and alter actin cytoskeletal organization.
Keywords: Net1, RhoA, Cdk1, mitosis, F-actin, spindle polarity
Introduction
The neuroepithelial transforming gene 1 (Net1) is a RhoA/RhoB-specific guanine nucleotide exchange factor (GEF) that is overexpressed in a number of cancers and is required for cell proliferation [1-6]. We have shown previously that Net1 contributes to mitotic progression, where it controls centrosomal activation of Aurora A and Pak2, and chromosome alignment during prometaphase [7]. Importantly, the ability of Net1 to regulate mitosis is independent of its ability to stimulate RhoA subfamily activation, as knockdown of RhoA and RhoB did not phenocopy Net1 knockdown, and expression of catalytically-inactive Net1 was sufficient to rescue mitosis in Net1 knockdown cells [7].
As cells enter mitosis, they often detach from the extracellular matrix and assume a roughly spherical shape. This occurs through the disassembly of focal adhesions, an increase in cortical actin, and an increase in osmotic pressure [8-12]. RhoA activation contributes to this process by promoting the formation of a cortical actin network that provides rigidity to the plasma membrane [13]. Plasma membrane activation of RhoA is stimulated by the release of the nuclear RhoGEF Ect2 following nuclear envelope breakdown in prophase [14]. This allows for mammalian Diaphanous 1 (mDia1)-dependent formation of F-actin filaments, and Sterile 20-like kinase (SLIK)-dependent phosphorylation of ERM proteins, which connects the newly formed actin filaments to the plasma membrane [15, 16]. The formation of a rigid plasma membrane is necessary for positioning of the mitotic spindle and proper partitioning of chromosomes to the daughter cells [15, 16]. Thus, it follows that cells must precisely regulate the temporal and spatial activation of RhoA during mitosis.
Net1 is one of two RhoGEFs that predominantly localize to the nucleus in interphase cells, the other being Ect2 [17-19]. Because Net1 is expressed during mitosis, it would be released from the nucleus following nuclear envelope breakdown and could contribute to RhoA activation. However, it is clear that Ect2 is sufficient to control RhoA activation during prophase [14], so it follows that cells must have one or more mechanisms to negatively regulate the ability of Net1 to activate RhoA. Previously we have shown that the serine/threonine kinase Pak1 phosphorylates Net1 in interphase cells to inhibit its catalytic activity toward RhoA [20]. Thus, we reasoned that one or more mitotic kinases might phosphorylate Net1 and play an analogous role during mitosis. In the present work, we show that the mitotic master kinase Cdk1 robustly phosphorylates Net1 and that this inhibits its its ability to activate RhoA. Moreover, inhibition of Cdk1 activity towards Net1, or mutation of the Cdk1 phosphorylation sites in Net1, results in aberrant cortical F-actin polymerization and mitotic spindle polarity defects. Thus, phosphorylation of Net1 by Cdk1 during mitosis constitutes an important mechanism to to precisely regulate mitotic RhoA signaling and cortical actin formation.
Materials and Methods
Cell culture and transfections
HeLa cells were maintained in Dulbecco's modified Eagle medium (DMEM) (Hyclone), supplemented with 10% fetal bovine serum (FBS) plus 100 U/ml penicillin and 100 μg/ml streptomycin (Hyclone, Chicago IL). All cells were grown in a humidified 10% CO2 incubator at 37°C. For synchronization in G0, the cells were washed 3x with PBS and then cultured in DMEM plus penicillin/streptomycin without FBS for 24 hours. For synchronization in S-phase, the cells were incubated overnight with complete media plus 2 mM thymidine (Thermo Fisher, St. Louis, MO). The next morning the thymidine-containing media was removed and the cells were washed 3x with PBS. Normal growth media was added for 4 hours, and then the cells were incubated a second time with complete media plus 2 mM thymidine. The cells were collected by the next morning. For prometaphase synchronization, cells were incubated with complete media plus 200 ng/ml nocodazole (Thermo Fisher) overnight. Prometaphase cells were dislodged by shaking and the cells in the media were collected by centrifugation (500 x g, 5 min.). Roscovitine was purchased from MilliporeSigma (557360). To inhibit Cdk1 phosphorylation, it was used at 10 μM for 2 hours.
For plasmid transfections, cells were seeded in 35 mm or 60 mm dishes 1 day before transfection. Plasmids were transfected using deacetylated polyethylenimine (PEI) 2200 reagent (provided by Dr. Guangwei Du, UT-HSC) or Lipofectamine with Plus reagent (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. Short interfering RNAs (siRNAs) were transfected using INTERFERin Reagent (PolyPlus), according to the manufacturer’s instructions.
Plasmids, siRNAs, and antibodies
pEF-HA/Net1 was as described previously [20]. pCMV5M/NLS-β-Galactosidase was as described previously [21]. 6xHis-Ubiquitin was a kind gift from Catherine Denicourt, UT-HSC. HA- or GST-epitope tagged wild type and mutant Net1 constructs were generated by PCR and confirmed by DNA sequencing. For bacterial expression, Net1 mutants were subcloned into pGEX-KG. Control and human Net1-specific siRNAs were purchased from Sigma-Aldrich. Sequences were as described [22].
The following commercial antibodies were used. Mouse anti-Net1 (sc-271941), mouse anti-HA ( sc-7392), mouse anti-GAPDH (sc-47724), rabbit anti-Myc (sc-789), mouse anti-GST (sc-138), rabbit anti-Lamin A/C (sc-20681) were from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-HA (600-401-384) was from Rockland Immunochemicals (Pottstown, PA). Rabbit anti-phospho-Lamin A/C (13448), rabbit anti-phospho-Cdc2 T161 (9114), mouse anti-Cdc2 (9116), mouse anti-Cyclin A (4656), mouse anti-phospho-S10 H3 (9706), and rabbit anti-H3 (4499) were from Cell Signaling Technology (Beverly, MA). Rabbit anti-Net1 (HPA020068) and mouse anti-α-Tubulin clone DMA1 (T6199) were from Sigma-Aldrich (St. Louis, MO). Mouse anti-Myc epitope clone 9E10 was from the National Cell Culture Center (Minneapolis, MN).
To generate antibodies specific for phosphorylated S131 and T146, peptides corresponding to mouse pS131 (RGDHR-pS-PASAQC) and mouse pT146 (RSTVP-pT-PTKRRC) were synthesized with C-terminal cysteines, coupled to keyhole limpet hemocyanin (KLH), and injected into New Zealand white rabbits along with Freund’s incomplete adjuvant (Sigma-Genosys, St. Louis, MO).
Recombinant protein expression, in vitro kinase reactions, and mass spectrometry
Plasmids for expression of glutathione S-transferase (GST)-Net1 fusion proteins were transformed into Rosetta 2/DE3 E. coli (EMD Millipore Chemicals, Burlington MA). 1 L cultures were grown at 37°C to OD600 = 0.5, 17g of NaCl was added, and the bacteria were cultured at 37°C to OD600 = 0.8. Protein expression was induced by addition of isopropyl 1-thio-β-D-galactopyranoside (IPTG) to 100 μM, and cultures were allowed to grow overnight at 25°C. For expression of GST or GST-A17RhoA proteins, cultures of BL21(DE3) E. coli bearing the relevant plasmids were cultured to O.D.600 = 0.8 and protein expression was induced for 12 to 16 h at room temperature following the addition of 50 μM isopropyl-β-D-thiogalactopyranoside (IPTG). All GST fusion proteins were purified using glutathione-agarose affinity resin (Sigma Aldrich), as described previously [18].
For kinase assays, purified GST fusion proteins were incubated with CDK1-cyclinB (P6020, New England BioLabs, Ipswich, MA) in kinase buffer (20 mM Tris-HCl [pH 8.0], 10 mM MgCl2, 1 mM dithiothreitol, 100 μM ATP, 2 μCi of [γ32P]-ATP (PerkinElmer Life Sciences, Waltham, MA)) for 30 minutes at 30°C. Proteins were resolved by 10% sodium dodecyl sulfate-polyacrylamide electrophoresis (SDS-PAGE) and visualized with Coomassie staining. After drying the gel, phosphorylated proteins were visualized by autoradiography. Radioactive phosphate incorporated into GST-Net1 was determined by scintillation counting of the excised protein bands.
For analysis of Cdk1 phosphorylation sites in Net1 by mass spectrometry, GST-Net1 was incubated with CDK1-CyclinB as described above, without radioactive ATP. The GST-Net1 was resolved by SDS-PAGE, silver stained, and excised. The excised band was then sent to the Taplin Mass Spectrometry Facility (https://taplin.med.harvard.edu/home), where it was eluted, digested with trypsin, and analyzed by LC/MS/MS, as per core facility protocols.
Immunoprecipitation and Western blotting
For immunoprecipitation of HA-Net1, HeLa cells were transfected, arrested in pro-metaphase with nocodazole (200 ng/ml) overnight, and collected by mitotic shake-off. Cells were lysed in RIPA (0.1% SDS, 50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1.0% Triton X-100, 80 mM β-glycerophosphate, 0.5% Deoxycholate, 1 mM Na3VO4, 50 mM NaF, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 10 μg/ml aprotinin, 1 mM PMSF), incubated on ice for 10 min, and pelleted by centrifugation (16,000 x g, 10 min, 4°C). Soluble lysates were incubated with 2 μg of mouse anti-HA for 1h at 4°C with rotation and incubated further with Protein A-Sepharose for 1h at 4°C. Immunoprecipitates were washed with three times with wash buffer (20 mM Tris-HCl [pH 8.0], 500 mM NaCl, 1.0% Triton X-100), resuspended in 2 X Laemmi sample buffer, and boiled for 5 min. Insoluble cell pellets were lysed in SDS lysis buffer (2% SDS, 20 mM Tris-HCl [pH 8.0], 10 mM NaCl, 1mM EDTA, 80 mM β-glycerophosphate, 0.5% Deoxycholate, 1 mM Na3VO4, 50 mM NaF, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 10 μg/ml aprotinin, 1 mM PMSF), sonicated, and boiled in 5x Laemmi sample buffer. Equal amounts of total protein were resolved by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membrane (GE Healthcare, Chicago, IL), and analyzed by western blotting.
For western blotting of whole cell lysates, cells were lysed in SDS buffer (2% SDS, 20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 80 mM β-glycerophosphate, 50 mM NaF, 1 mM sodium orthovanadate, 10 μg/ml pepstatin A, 10 μg/ml leupeptin, 10 μg/ml aprotinin), sonicated, and resolved by SDS-PAGE. After transfer to PVDF membrane, blots were blocked by incubation in Tris-buffered saline (TBST) + 0.05% Tween 20 + 5% nonfat milk at room temperature for 1 hr. Blots were then incubated with primary antibody diluted in TBST plus 0.25% non-fat milk for 1 hr at 37°C, or overnight at 4°C. When blotting for phosphorylation of Net1 on S131 or T146, blots were blocked in TBST + 1% bovine serum albumin (BSA). Anti-S 131 and anti-T146 antibodies were diluted in TBST + 1% BSA and incubated overnight at 4°C. After primary antibody incubation, blots were washed with TBST and then incubated with horseradish-peroxidase-conjugated anti-mouse or anti-rabbit antibodies (KPL, Milford MA, or ThermoFisher) diluted in TBST + 0.025% non-fat milk for 30 min. at room temperature. After washing with TBST, blots were developed with enhanced chemiluminescence and detected with X-ray film.
GST-A17RhoA pulldown assay
HeLa cells were transfected with the indicated plasmids and cells were arrested with nocodazole (200 ng/ml) overnight. Cells were collected by mitotic shake-off and GST-A17RhoA pulldown assays were performed as previously described [23]. Briefly, cells were washed with PBS and lysed in Trition lysis buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1% Triton X-100, 1 mM DTT, 1 mM PMSF, and 10 μg/ml each of aprotinin, leupeptin, and pepstatin A), sonicated for 30s, and clarified by centrifugation (16,100 x g, 10 min., 4°C). Lysate concentrations were determined by bicinchoninic acid assay (Thermo Fisher) and equal amounts of lysate were mixed for 1 h at 4°C with 20 μg of GST or GST-A17RhoA beads. Beads were pelleted by centrifugation and washed 3 times in lysis buffer, resuspended in 25 μl Laemmli sample buffer, boiled for 5 minutes, separated by SDS-PAGE, and transferred to PVDF membrane for western blot analysis.
Protein half-life measurements and Ni-NTA-Ubiquitin pulldowns
Untransfected HeLa cells, or HeLa cells transfected with the relevant HA-Net1 expression plasmids were synchronized in pro-metaphase by treatment with 200 ng/ml nocodazole (Thermo Fisher) overnight. Protein synthesis was then halted by treatment with cycloheximide (10 μg/ml) for different times, after which the mitotic cells were collected by shakeoff and lysed in a 2% SDS buffer (2% SDS, 20 mM Tris-HCl pH 8.0, 50 mM NaF, 80 mM β-glycerophosphate, 1 mM sodium orthovanadate, 10 μg/ml pepstatin A, 10 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mM PMSF). After boiling the samples, proteins were resolved by SDS-PAGE and endogenous or transfected Net1 proteins were detected by western blotting. Amounts of Net1 proteins remaining were quantified by densitometry (NIH ImageJ).
To measure Net1 ubiquitylation, HeLa cells were transfected with a 6xHis-Ubiquitin and HA-Net1 expression plasmids, and then arrested with nocodazole (200 ng/ml) overnight. Cells were treated 10 μM MG132 (Thermo Fisher) for 1h at 37°C. Cells were collected by mitotic shake-off and resuspended in PBS containing 5 mM N-ethyl maleimide and incubated with Ni-NTA agarose beads (Qiagen, Hilden, Germany) in denaturing buffer (6 M guanidium chloride, 0.2 M Na2HPO4, 0.2 M NaH2PO4, 10 mM Tris-HCl [pH 8.0], 10 mM β-mercaptoethanol, 5 mM N-ethyl maleimide, 5 mM imidazole) for 16h at 4°C. 6xHis-Ub-conjugated proteins were sequentially washed for 5 min each with buffer A (6 M guanidium chloride, 0.2 M Na2HPO4, 0.2 M NaH2PO4, 10 mM Tris-HCl [pH 8.0], 10 mM β-mercaptoethanol), buffer B (8 M urea, 0.2 M Na2HPO4, 0.2 M NaH2PO4, 10 mM Tris-HCl [pH 8.0], 10 mM β-mercaptoethanol), buffer C (8 M urea, 0.2 M Na2HPO4, 0.2 M NaH2PO4, 10 mM Tris-HCl [pH 8.0], 10 mM β-mercaptoethanol, 0.2% Triton X-100) and buffer D (8 M urea, 0.2 M Na2HPO4, 0.2 M NaH2PO4, 10 mM Tris-HCl [pH 8.0], 10 mM β-mercaptoethanol, 0.1% Triton X-100). Ni-NTA-bound proteins were eluted, boiled at 95°C for 5 min, separated by 8% SDS–PAGE, and transferred to PVDF membrane. Ubiquitylated proteins were detected by western blotting.
Immunofluorescence staining and confocal microscopy
HeLa cells were plated onto fibronectin coated coverslips and transfected with plasmids encoding Myc-tagged β-Galactosidase, HA-tagged wild-type Net1, or mutant Net1. Two days after transfection, the cells were fixed in 4% paraformaldehyde (Thermo Fisher, Waltham, MA, 286906) in PBS and permeabilized with 0.2% Triton X-100 in PBST (PBS plus 0.05% Tween 20). Immunofluorescence staining was performed using antibodies for the Myc-epitope (1:100 dilution), HA-epitope (1:1000 dilution), and alpha-tubulin (1:1000), and cells were incubated with primary antibodies for 1 h at 37°C. Cells were washed 3x with PBST for 5 min each, and then incubated with secondary anti-rabbit or anti-mouse Alexa Fluor-488, and anti-mouse or anti-rabbit Alexa Fluor-594 (Thermo Fisher), diluted 1:1000 in PBST, plus 1 μg/ml DAPI (4′,6-diamidino-2-phenylindole) (Sigma Aldrich). F-actin staining was performed using acti-stain 670 Phalloidin (Cytoskeleton, Denver, CO). Cells were washed in PBST as before, rinsed with distilled water, and mounted onto glass slides with Fluormount reagent (Thermo Fisher).
Cells were imaged using a Nikon A1R confocal microscope at 60X magnification and z-stacks were acquired with 0.5 μm steps. To obtain intensity profiles of Net1 and F-actin at the plasma membrane, z-stacks were analyzed using CellProfiler image analysis software (version 3.0). Measurement of mitotic spindle angles was performed using NIS Elements software, where a maximum intensity projections of the z-stacks were created to identify the spindle poles. Then, a horizontal line was generated between the two spindle poles and the spindle angle was measured relative to the substrate, where the reference horizontal angle was α=0.
Graphing and statistical analysis
All experiments we performed a minimum of three separate times, as denoted in figure legends. Data was graphed and analyzed for significance by unpaired Student’s T test using Prism software (GraphPad, San Diego, CA).
Results
1.1. Identification of Cdk1 phosphorylation sites in Net1 in vitro.
We have shown previously that Net1 regulates mitotic progression in a RhoA-independent manner [7]. These data indicated that Net1 functions as a scaffold to control mitosis, and suggested that the cell may employ mechanisms to suppress the exchange factor activity of Net1 towards RhoA. Since many mitotic proteins are regulated by Cdk1, we reasoned that Cdk1 may phosphorylate Net1 to control its activity. To test whether this was the case, we produced mouse Net1 as a recombinant GST fusion protein in E. coli and incubated the purified GST-Net1 with recombinant, purified Cdk1/Cyclin B in an ATP-containing buffer. The GST-Net1 was then resolved by SDS-PAGE and the excised band was analyzed by mass spectrometry (LC/MS/MS) to identify phosphorylated peptides. Through this analysis, we observed that six residues in mouse Net1 were phosphorylated by Cdk1 in vitro (Fig. 1A, B). Of these, only T35 was not conserved in human Net1. The most highly phosphorylated sites were S131, S106, and T35, respectively, based on the number of phosphorylated peptides identified and the A-Score. An A-score predicts the likelihood of correctly identifying the correct phosphorylated residue using mass spectrometry [24].
Figure 1. Identification of Cdk1 phosphorylation sites in Net1 in vitro.
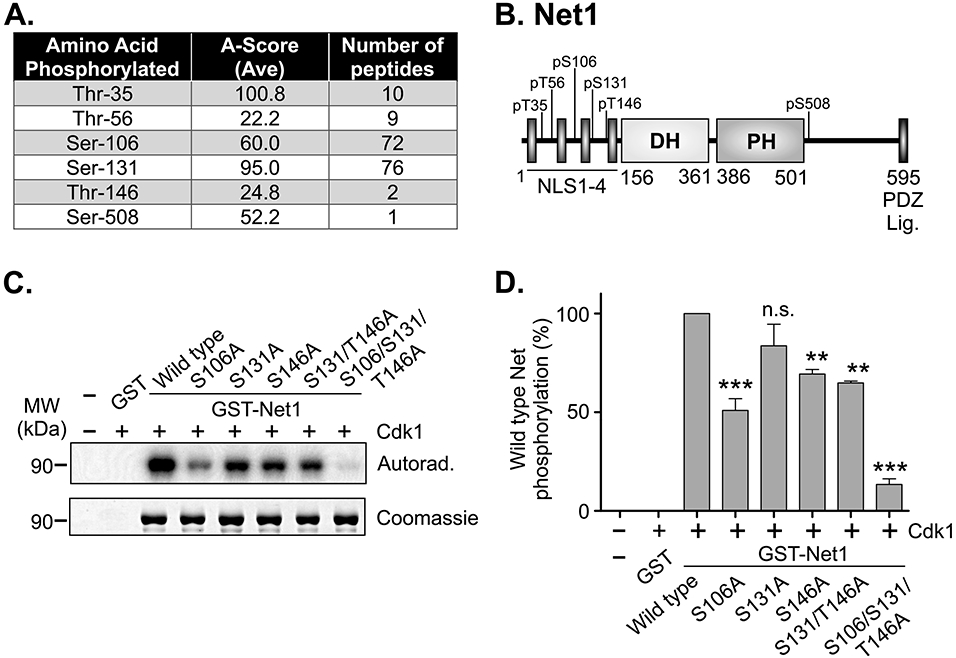
(A) Table of phosphopeptides from LC/MS/MS analysis of recombinant mouse GST-Net1 phosphorylated by purified Cdk1-Cyclin B in vitro. Note that T35 is not conserved in human Net1. (B) Schematic of the domain structure of Net1, and Cdk1 phosphorylation sites. (C) In vitro Cdk1-dependent phosphorylation of recombinant GST-Net1 proteins with the mutations shown. Phosphorylation was detected by incorporation of 32P-labeled phosphate and autoradiography. Equal loading of proteins was monitored by Coomassie staining. Shown is a representative experiment. (D) Quantification of phosphorylation of GST-Net1 proteins by purified Cdk1 in vitro. Shown is the average of 3 independent experiments. Errors are standard error of the mean (SEM). ** = p<0.01; *** = p<0.001; n.s. = not significant.
To determine whether we had identified the majority of Cdk1 phosphorylation sites, we mutated one or more of the conserved sites to alanines and produced the mutants as GST fusion proteins in E. coli. For this analysis, we focused on the most highly phosphorylated sites, S106 and S131, as well as T146, which was predicted to be the highest affinity Cdk1 site in two different prediction programs (ELM and Scansite 4.0). The purified GST-Net1 fusion proteins were then for Cdk1 phosphorylation using [γ32P]-labelled ATP, and the degree of phosphorylation was measured using autoradiography and scintillation counting. We observed that mutation of S106 to alanine had the greatest effect among the individual mutations (Fig. 1C, D). Importantly, mutation of S106, S131, and T146 together eliminated nearly 90% of Net1 phosphorylation, indicating that these are the major sites for Cdk1 phosphorylation in vitro.
1.2. Cdk1 phosphorylates S131 and T146 in mitotic cells
To determine whether these sites were phosphorylated in cells, we attempted to raise phosphorylation-specific antibodies to each of the sites. We were successful in generating antibodies specific for pS131 and pT146, but not pS106. To determine the specificity of these antibodies, we expressed HA-epitope tagged wild type or mutant Net1 proteins in HeLa cells, synchronized the cells in pro-metaphase using nocodazole, immunoprecipitated the HA-tagged proteins, and tested them for recognition of each phosphorylation site by western blotting. We observed that the pS131 antibody recognized wild type Net1, and to a lesser extent Net1 T146A, but was unable to recognize Net1 S131A or Net1 S131A/T146A (Fig. 2A). The decreased phosphorylation of T146 in the S131A mutant may reflect a dependence of C-terminal phosphorylation sites on prior, N-terminal phosphorylation, which has been observe for other Cdk1 phosphorylated proteins [25, 26]. Alternatively, the pT146 antibody efficiently recognized wild type Net1 and Net1 S131A, but not Net1 T146A or Net1 S131A/T146A (Fig. 2A). Thus, the phospho-specific antibodies were reasonably specific for each of their intended phosphorylation sites.
Figure 2. Characterization of Net1 phosphorylation on S131 and T146 in cells.
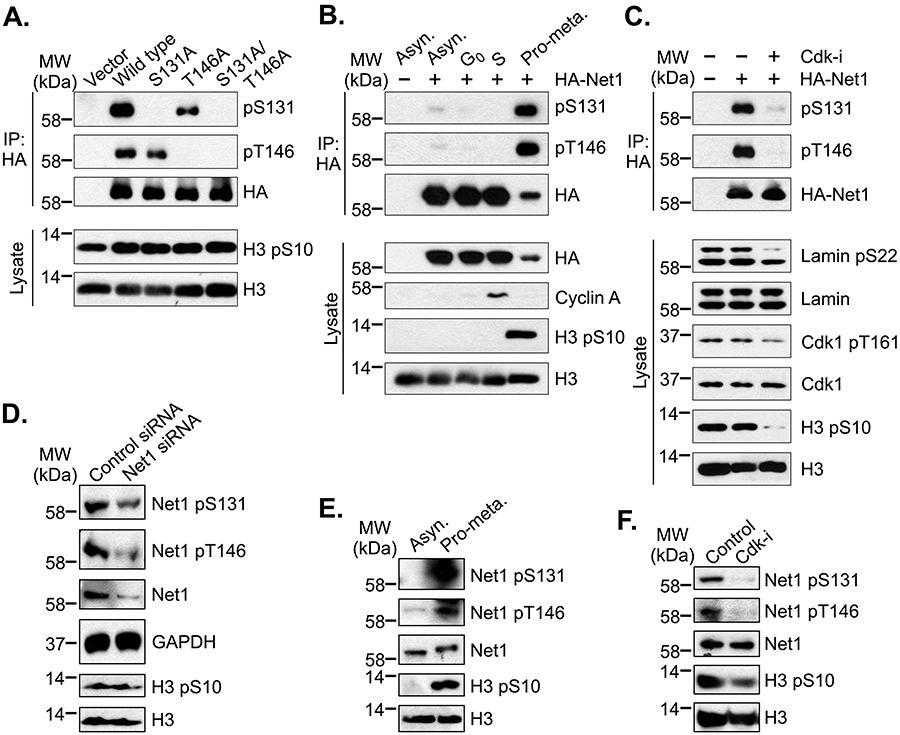
(A) Characterization of pS131- and pT146-specific antibodies. HeLa cells were transfected with HA-epitope tagged wild type or mutant Net1 expression vectors and synchronized in prometaphase by overnight treatment with nocodazole. Mitotic cells were isolated by shakeoff, lysed, and HA-tagged proteins were immunoprecipitated. Immunoprecipitated proteins were resolved by SDS-PAGE and examined by western blotting. Shown is a representative experiment from 6 independent experiments. (B) Phosphorylation of S131 and T146 in synchronized cells. HeLa cells were transfected with HA-tagged Net1 and synchronized in G0 by serum starvation, S phase by double thymidine treatment, of pro-metaphase by nocodazole treatment. HA-Net1 was then immunoprecipitated and analyzed by western blotting. Shown is a representative experiment from 4 independent experiments. (C) pS131 and pT146 is sensitive to Cdk inhibition. HeLa cells were transfected with HA-Net1, synchronized in pro-metaphase, and treated with vehicle or the Cdk inhibitor roscovitin (10 μM) for 2 hours. HA-Net1 was then immunoprecipitated and analyzed by Western blotting. Phosphorylation of Lamin A/C on the Cdk1 site S22 and Cdk1 on the activation loop site T161 was assessed in the lysate to confirm inhibitor function. Shown is a representative experiment from 3 independent experiments. (D) Assessment of specificity of pS131 and pT146 antibodies for endogenous Net1. HeLa cells transfected with control or Net1-specific siRNAs and then synchronized in prometaphase. Cells were lysed and tested for pS131 and pT146 by western blotting. Shown is a representative experiment from 3 independent experiments. (E) Phosphorylation of endogenous Net1 on S131 and T146 is enriched in mitotic cells. HeLa cells were grown as an asynchronous population or synchronized in pro-metaphase. Cells were then lysed in an SDS-containing buffer and tested for phosphorylation of endogenous Net1 on S131 and T146 by western blotting. Shown is a representative experiment from 3 independent experiments. (F) Endogenous Net1 phosphorylation on S131 and T146 is sensitive to Cdk inhibition. HeLa cells were synchronized in pro-metaphase and treated with vehicle or roscovitin (10 μM) for 2 hours. Phosphorylation of Net1 was assessed by western blotting. Shown is a representative experiment from 4 independent experiments.
We then examined whether these sites were preferentially phosphorylated in mitotic cells, and if this was dependent on Cdk1 activity. Thus, cells were transfected with HA-epitope tagged, wild type Net1 and left as an asynchronous population, or enriched for cells in G0, S-phase, or pro-metaphase (G2/M). HA-Net1 was then immunoprecipitated and tested for phosphorylation on S131 and T146 by western blotting. Synchronization in each population was determined by western blotting for cyclin A (S-phase marker) and pS10-histone H3 (mitosis marker). G0 cells should not be positive for either of these markers. We observed that Net1 was weakly phosphorylated on each site in an asynchronous population, and that this phosphorylation was not detectable in the G0 or S-phase cell populations. However, Net1 was strongly phosphorylated on S131 and T146 in the prometaphase synchronized cells, indicating that these sites were primarily phosphorylated during mitosis (Fig. 2B). To determine whether Cdk1 was responsible for phosphorylating these sites, cells were transfected with wild type Net1, synchronized in pro-metaphase, and then treated with vehicle or the Cdk1/2/5 inhibitor roscovitine for 2 hours. HA-Net1 was then immunoprecipitated and tested for phosphorylation on S131 and T146 by western blotting. We observed that treatment with roscovitine dramatically reduced Net1 phosphorylation on both sites (Fig. 2C). The roscovotine was effective at inhibiting Cdk1 activity, as the phosphorylation of Lamin A/C on the Cdk1 phosphorylation site S22 was reduced by inhibitor treatment (Fig. 2C, lysate lane 3, upper band). These data indicate that Net1 phosphorylation on S131 and T146 is subject to rapid turnover. Moreover, because Cdk1 is the major cyclin dependent kinase that is active during prometaphase [27, 28], these data indicate that S131 and T146 are primarily phosphorylated by Cdk1 in mitotic cells.
We then tested whether the pS131 and pT146 antibodies were capable of detecting endogenous Net1 phosphorylation during mitosis. We first tested the specificity of each antibody in western blots following Net1 knockdown. Thus, cells were transfected with control or Net1-specific siRNAs, synchronized in prometaphase with nocodozole, and tested for Net1 phosphorylation on S131 and T146 by western blotting. We observed that Net1 knockdown reduced the pS131 and pT146 signals commensurate with the degree of Net1 knockdown (Fig. 2D). Thus, the antibodies are specific for endogenous Net1. We then tested whether endogenous Net1 was preferentially phosphorylated on the Cdk1 sites during mitosis. Cells were grown as an asynchronous population or synchronized in prometaphase, lysed, and tested for phosphorylation by western blotting. We found that endogenous Net1 was highly phosphorylated on S131 and T146 in prometaphase cells, but not in the asynchronous cell population (Fig. 2E). To test whether this phosphorylation was dependent on Cdk1 activity, prometaphase cells were treated with vehicle or roscovitine for 2 hours, and Net1 phosphorylation was monitored by western blotting. Similar to transfected HA-Net1, endogenous Net1 phosphorylation in S131 and T146 was sensitive to Cdk1 inhibition (Fig. 2F).
1.3. Cdk1 phosphorylation does not affect Net1 stability
The stability of many mitotic proteins is regulated by Cdk1-dependent phosphorylation [29, 30]. To test whether Net1 stability was controlled by Cdk1, cells were transfected with wild type Net1 or Net1 with alanine or acidic substitutions at S106, S131, and T146 (SST/AAA or SST/EDE, respectively). The cells were then synchronized in pro-metaphase and treated with the protein synthesis inhibitor cycloheximide for different times. Loss of Net1 expression over time was monitored by western blotting. Wild type HA-Net1 displayed a predicted half-life of approximately 14 hours in prometaphase cells and this was not appreciably changed by acidic or alanine substitution of the Cdk1 phosphorylation sites (Fig. 3A, B). The stability of Net1 during pro-metaphase was very similar to that of endogenous Net1, which also displayed a predicted half-life of 14 hours (Fig. 3C, D). These values are in good agreement with the half-life of Net1 in interphase cells [31]. We also examined whether Net1 was ubiquitylated in mitotic cells, and if this was regulated by Cdk1 phosphorylation. Thus, cells were transfected with wild type HA-Net1 or the triple alanine or acidic Net1 mutants, plus hexa-histidine tagged ubiquitin. After synchronization in prometaphase, the cells were lysed in a urea-based buffer and ubiquitylated proteins were isolated using Ni-NTA-agarose. Net1 was detected by Western blotting for the HA epitope. We observed that Net1 was ubiquitylated in pro-metaphase cells, but that this was not significantly altered by substitution of the Cdk1 phosphorylation sites (Fig. 3E). Taken together, these data indicate that Cdk1 phosphorylation of Net1 does not alter the ubiquitylation or stability of Net1 during mitosis.
Figure 3. Substitution of the Cdk1 phosphorylation sites does not affect Net1 stability.
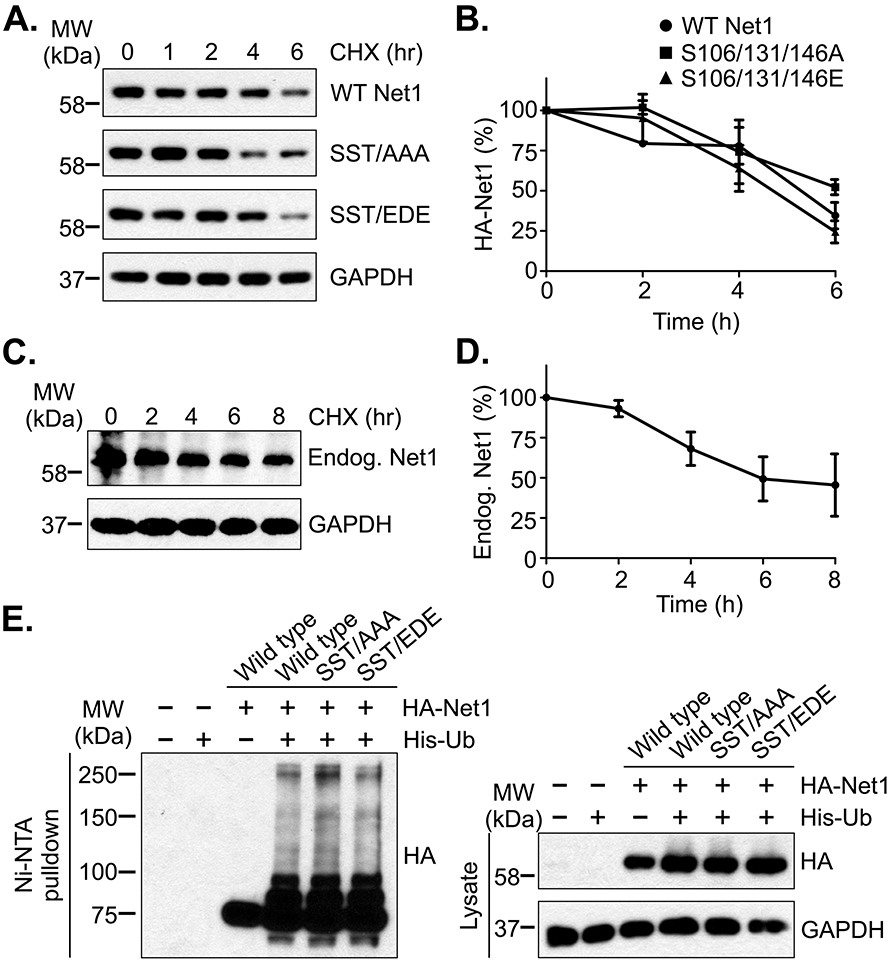
(A) Stability of wild type and mutant HA-Net1 proteins in prometaphase. HeLa cells were transfected with the Net1 expression vectors shown, synchronized in prometaphase, and protein synthesis was halted by addition of cycloheximide (CHX) for the times shown. Cells were lysed in SDS-containing buffer and HA-Net1 expression was examined by western blotting. Shown is a representative experiment from 3 independent experiments. (B) Quantification of transfected Net1 stability in pro-metaphase. Errors are SEM. (C) Stability of endogenous Net1 in prometaphase. HeLa cells were synchronized in prometaphase and cycloheximide was added for the times shown. Endogenous Net1 expression was detected by western blotting. Shown is a representative experiment from 4 independent experiments. (D) Quantification of endogenous Net1 stability in pro-metaphase. Errors are SEM. (E) Substitution of the Cdk1 phosphorylation sites in Net1 does not affect its ubiquitylation. HeLa cells were transfected with the HA-tagged Net1 expression plasmids shown, plus a vector encoding 6xHis-Ubiquitin, synchronized in prometaphase, and treated with MG132 for 1 hour. The cells were then lysed in a urea-containing buffer, ubiquitylated proteins were isolated using Ni-NTA-agarose, and ubiquitylation was assessed by western blotting for the HA-epitope. Shown is a representative experiment from 3 independent experiments.
1.4. Cdk1 phosphorylation reduces Net1 catalytic activity and prevents Net1 from promoting cortical actin polymerization.
We have shown previously that Pak1-dependent phosphorylation of Net1 on S152 and S153 in interphase cells inhibits its guanine nucleotide exchange activity towards RhoA [20]. These sites are adjacent to an N-terminal extension of the catalytic Dbl domain, which in the related RhoGEF LARG make contact with key residues in RhoA [32], suggesting why phosphorylation of sites outside the catalytic domain affects Net1 enzymatic activity. Because the Cdk1 sites are close to the Pak1 phosphorylation sites, we tested whether alanine or acidic substitution of these sites also affected Net1 activity. A convenient way to measure the activation state of a RhoA GEF is to test for interaction with A17RhoA, which is a nucleotide-free version of RhoA that simulates the transition state immediately after release of GDP, and which only binds tightly to active RhoA GEFs [33]. Thus, HeLa cells were transfected with wild type Net1, Net1 SST/AAA or Net1 SST/EDE, synchronized in pro-metaphase, lysed, and incubated with GST-A17RhoA. Active Net1 proteins interacting with GST-A17RhoA were isolated by glutathione-agarose pulldown and detected by western blotting. We observed that mutation of the Cdk1 sites to alanines, and even more so to acidic residues, significantly reduced the interaction with GST-A17RhoA, indicating that these substitutions reduced the fraction of active Net1 in cells (Fig. 4A, B). This indicates that Net1 activation is sensitive to the amino acids present at the Cdk1 phosphorylation sites, and that substitution of these sites with acidic, phospho-mimetic amino acids is more deleterious that alanine substitution. To determine whether Cdk1 inhibition increased endogenous Net1 catalytic activity, cells were synchronized in pro-metaphase using nocodzaole, and then treated with vehicle or roscovitine for 2 hours. Under these conditions, Net1 phosphorylation on S131 and T146 was largely abolished (Fig. 2F). We observed that roscovitine treatment significantly increased endogenous Net1 activity (Fig. 4C, D), consistent with the idea that Cdk1 phosphorylation of Net1 inhibits its ability to activate RhoA.
Figure 4. Effects of Cdk1 on Net1 activity and localization.

(A) Substitution of the Cdk1 phosphorylation sites downregulates Net1 activity. HeLa cells were transfected with the HA-Net1 expression plasmids shown, synchronized in prometaphase, and active Net1 proteins were isolated by GST-A17RhoA pulldown (top blots). Lysates were also probed with GST alone as a control (middle blots). Proteins present in the lysates are shown in the bottom blots. Shown is a representative experiment. (B) Quantification of the activation state of Net1 proteins in prometaphase cells. Bars are median values. * = p<0.05; *** = p<0.001. (C) Treatment of prometaphase cells with rosocovitin (10 μM, 2 hrs) increases endogenous Net1 activation, as measured in GST-A17RhoA pulldowns. Shown is a representative experiment from 5 independent experiments. (D) Quantification of endogenous Net1 activation state in prometaphase cells treated with roscovitin. Bars are median values. ** = p<0.01. (E) Treatment of cells with the Cdk inhibitor roscovitine (10 μM, 2 hrs) causes increased plasma membrane localization of endogenous Net1 and increased cortical F-actin formation. Cells were fixed and stained for endogenous Net1 (green), DNA (blue), and F-actin (magenta), and imaged using confocal microscopy. Shown are representative z-plane images. Scale bar = 10 μm. (D) Quantification of endogenous Net1 intensity at the plasma membrane in vehicle and roscovitin (Cdk-i) treated, prometaphase cells. Each point represents one cell. Data are aggregated from 3 independent experiments. Bars are median values. ** = p<0.01. (E) Quantification of plasma membrane F-actin staining in vehicle and roscovitin treated, prometaphase cells. Each point represents one cell. Data are aggregated from 3 independent experiments. Bars are median values. * = p<0.05.
To determine whether Cdk1 regulates Net1 localization or signaling in cells, we treated asynchronous HeLa cells with roscovitine for 2 hours, and then fixed and stained the cells for endogenous Net1, F-actin, and DNA. We tested for effects on F-actin content, as active RhoA promotes F-actin accumulation by stimulating mDia-dependent F-actin polymerization, by promoting actomyosin contraction, and by inhibiting cofilin-dependent F-actin severing [34, 35]. We observed that endogenous Net1 was localized to the cytoplasm and the mitotic spindle in metaphase cells (Fig. 4E). Interestingly, treatment with the Cdk1 inhibitor did not displace Net1 from the spindle. However, it did lead to a small but statistically significant increase in Net1 localization to the plasma membrane (Fig. 4E-G, left panels). Cdk1 inhibition also caused a robust increase in cortical F-actin staining (Fig. 4E-G, middle panels). These data suggest that Cdk1 displaces Net1 from the plasma membrane where it would otherwise activate RhoA and promote excess cortical actin polymerization.
To determine whether overexpressed Net1 behaved similarly to the endogenous protein, HeLa cells were transfected with wild type HA-Net1 and then treated with vehicle or roscovitine for two hours. The localization of HA-Net1 and cortical F-actin content in metaphase cells were then quantified. Surprisingly, we observed that overexpressed HA-Net1 did not localize significantly to the mitotic spindle. However, the plasma membrane localization of HA-Net1 was increased by Cdk1 inhibition, and this was accompanied by an increase in cortical F-actin staining (Fig. 5A-C). Thus, overexpressed Net1 did faithfully recapitulate the effects of Cdk1 regulation on endogenous Net1 signaling to the actin cytoskeleton, which in interphase cells is a RhoA-dependent event [17, 18, 20] (Fig. 4).
Figure 5. Expression of Net1 with alanine substitutions of the Cdk1 phosphorylation sites causes increased cortical F-actin accumulation in prometaphase cells.

(A) Transfected HA-Net1 responds similarly to Cdk inhibition as endogenous Net1. HeLa cells were transfected with wild type HA-Net1 and treated with vehicle or roscovitin (10 μM, 2 hrs). The cells were then fixed and stained for HA-Net1 (green), DNA (blue), and F-actin (magenta), and imaged by confocal microscopy. Shown are representative z-plane images. Scale bar = 10 μm. (B) Quantification of HA-Net1 plasma membrane localization. Each point represents one cell. Data are aggregated from 3 independent experiments. Bars are median values. ** = p<0.01. (C) Quantification of cortical F-actin staining. Each point represents one cell. Data are aggregated from 3 independent experiments. Bars are median values. * = p<0.05. (D) Net1 SST/AAA expression increases cortical F-actin staining. HeLa cells were transfected with expression vectors for Myc epitope tagged β-Galactosidase, HA-wild type Net1, or HA-Net1 SST/AAA. The cells were then fixed and stained for HA-Net1 or Myc-β-Galactosidase (green), DNA (blue), and F-actin (magenta), and imaged by confocal microscopy. Shown are representative z-plane images. Scale bar = 10 μm. (E) Quantification of plasma membrane association of expressed proteins. Each point represents one cell. Data are aggregated from 3 independent experiments. Bars are median values. ** = p<0.01. (C) Quantification of F-actin at the plasma membrane. Each point represents one cell. Data are aggregated from 3 independent experiments. Bars are median values. *** = p<0.001; n.s. = not significant. (F) Quantification of cortical F-actin staining. Each point represents one cell. Data are aggregated from 3 independent experiments. Bars are median values. * = p<0.05; n.s. = not significant.
To determine whether phosphorylation of Net1 on the Cdk1 phosphorylation sites was necessary for these effects, HeLa cells were transfected with expression vectors for a control protein (β-galactosidase), wild type Net1, or Net1 SST/AAA. We observed that overexpressed, wild type Net1 localized to the plasma membrane to a greater extent than β-galactosidase. However, Net1 SST/AAA exhibited reduced plasma membrane localization relative to wild type Net1 (Fig. 5D, E). This suggests that the Cdk1 phosphorylation sites were somehow important for Net1 plasma membrane localization, and that Cdk1 inhibition was not increasing Net1 plasma membrane localization by modulating phosphorylation on S106, S131, or T146. However, the Net1 SST/AAA mutant did elevate cortical F-actin intensity relative to wild type Net1 (Fig. 5D, F), consistent with Cdk1 phosphorylation negatively regulating the RhoA GEF activity of Net1 (Fig. 4A-D).
1.5. Overexpression of Net1 causes spindle polarity defects that are corrected by acidic substitution of the Cdk1 phosphorylation sites.
Cells precisely regulate their plasma membrane stiffness during mitosis by controlling cortical actin structure. This is critically important, as disruption of cortical actin causes aberrant positioning of the mitotic spindle relative to the plane of the cell [15, 16]. The nuclear Rho GEF Ect2, which is released from the nucleus following nuclear envelope breakdown in prophase, has been shown to be necessary and sufficient to regulate RhoA-dependent cortical actin polymerization as mitosis begins [14]. Because Net1 is also a nuclear Rho GEF, and it is possible that Net1 overexpression would overwhelm negative regulation by Cdk1 and result in spindle polarity defects. To test this possibility, we transfected HeLa cells with vectors expressing the control protein β-galactosidase, wild type Net1, Net1 SST/AAA, or Net1 SST/EDE. Spindle polarity was then measured in metaphase cells (Fig. 6B). In these experiments, we observed that expression of β-galactosidase did not cause spindle polarity defects (Fig. 6A, top panels). However, overexpression of wild type Net1 caused aberrant spindle polarization in approximately 10% of the transfected cells, as did Net1 SST/AAA overexpression (Fig. 6A, C). Importantly, this defect occurred less frequently in cells transfected with Net1 SST/EDE (Fig. 6A, C), consistent with the reduced catalytic activity of this mutant (Fig. 4A, B). These data indicate that Net1 overexpression can cause spindle polarity defects, but that phosphomimetic substitution of the Cdk1-dependent phosphorylation sites in Net1 can alleviate this effect.
Figure 6. Acidic substitution of the Cdk1 phosphorylation sites reduces spindle polarity defects caused by Net1 overexpression.

(A) Imaging of spindle polarity defects. HeLa cells were transfected with expression plasmids for the proteins shown, and fixed and stained for HA-Net1 or Myc-β-Galactosidase (green), DNA (blue), and α-Tubulin (red), and imaged by confocal microscopy. Shown are representative z-plane images in the X-Y or X-Z axis. Scale bar = 10 μm. (B) Schematic depicting the method for quantification of spindle angle. Lines in green are the mitotic spindle. Cells are imaged in the Z-X plane and an imaginary line is drawn through the spindle poles. The angle with which this line intersects the horizontal-axis is defined as the spindle angle. (C) Quantification of spindle angle. Each point represents one cell. Data are aggregated from 3 independent experiments. Bars are median values. ** = p<0.01; *** = p<0.001.
Discussion
We have previously shown that Net1 regulates chromosome alignment during pro-metaphase in a RhoGEF-independent manner [7]. This suggested that mechanisms may exist to negatively regulate Net1 activity towards RhoA during mitosis. In the current work, we demonstrate that Net1 is phosphorylated by Cdk1 on multiple sites in vitro. We were able to raise phospho-specific antibodies to two of these sites, S131 and T146, and demonstrated that both transfected as well as endogenous Net1 is phosphorylated on these sites in mitotic cells. We also demonstrated that substitution of these sites with aliphatic or phosphomimetic residues reduced the fraction of active Net1 in cells, and that treatment of mitotic cells with a Cdk1 inhibitor increased endogenous Net1 activity towards RhoA, enhanced its localization to the plasma membrane, and resulted in an increase in cortical F-actin accumulation. Moreover, overexpression of Net1 increased the frequency of spindle misorientation, which was reduced by acidic substitution of the Cdk1 phosphorylation sites within Net1. These data indicate that Cdk1 phosphorylates Net1 during mitosis to negatively regulate its ability to stimulate cortical F-actin accumulation at the plasma membrane, and suggest a cogent explanation for how cells may prevent aberrant Net1 function during mitosis.
We were surprised to find that overexpressed HA-Net1 did not completely mimic the localization of endogenous Net1 in mitotic cells, as it did not substantially localize to the mitotic spindle (compare Fig. 4E and 5A). However, Cdk1 regulation of HA-Net1 phosphorylation on S131 and T146 mirrored that of endogenous Net1 (Fig. 2). Moreover, the cellular activity of overexpressed Net1 was still sensitive to Cdk1 inhibition, as roscovitin treatment increased the plasma membrane localization of HA-Net1 in metaphase cells and promoted cortical F-actin accumulation (Fig. 5A). These data suggest that overexpressed HA-Net1 was still a valuable tool for measuring Cdk1 regulation of Net1 activity towards RhoA.
Thus, it was curious that the Net1 SST/AAA mutant did not localize to the plasma membrane, similar to what occurred in Cdk-inhibitor treated cells expressing wild type Net1. This may suggest that Cdk1 inhibition promotes plasma membrane localization of Net1 through an indirect mechanism, such as affecting the localization of one or more Net1 binding proteins. The identity of such a protein is unclear, as mitotic Net1 interacting proteins have not been identified. Net1 has been shown to bind to a number of scaffold proteins in interphase cells, including Dlg1 [31, 36]. Intriguingly, Dlg1 has been shown to localize to the cell cortex in Drosophila S2 cells and Drosophila follicular epithelium, where it links spindle orientation to apical-basal polarity [37, 38]. Although this has not yet been demonstrated to occur in mammalian cells, and HeLa cells do not exhibit apical-basal polarity, it is possible that one or more Dlg1 family proteins link Net1 to the plasma membrane in Cdk1 inhibitor treated cells.
The consequences of over activation of RhoA during mitosis can be severe. Cells must precisely regulate their plasma membrane stiffness during mitosis to achieve proper spindle positioning. This occurs through regulation of both cortical actin structure and osmotic pressure [10-12]. Moreover, it appears that mitotic cells achieve a maximal cortical tension during mitosis that is impaired by either decreasing or increasing cortical actin polymerization [39]. Thus, the increase in cortical F-actin accumulation and concurrent defects in mitotic spindle positioning occurring in Net1 overexpressing cells are likely connected. Cells regulate spindle positioning through parallel pathways. The predominant mode of regulation in HeLa cells is through the LGN complex, which allows for attachment of astral microtubules to the cell cortex without the requirement for cortical actin. However, additional actin-dependent pathways exist [40, 41]. For example, Myosin-10 can link the astral microtubules to the cortical actin network [42]. Similarly, the actin binding protein Afadin has been shown to link the actin cytoskeleton to LGN to control spindle positioning [43]. In addition, the formation of motile, subcortical actin clouds has been shown to affect spindle positioning in an Arp3-dependent manner [44]. We were unable to detect changes in localization of the LGN complex members NUMA and p150-glued in Net1 overexpressing cells (not shown), suggesting that Net1 does not affect spindle positioning through the canonical LGN complex. Rather, it is likely that Net1 affects spindle positioning through one or more of the actin-dependent pathways describe above.
Although altered spindle positioning is generally not detrimental in cultured cells, it can have extreme consequences in development and disease [41, 45, 46]. A normal epithelium divides in a spatially oriented manner to maintain tissue structure. In cancer, this organization is lost and aberrant spindle positioning can contribute to random proliferation of tumor cells in all orientations. It can also contribute to aneuploidy by promoting improper chromosome division between daughter cells. Net1 is overexpressed in a number of human cancers, including breast cancer [1-6]. It is intriguing to speculate that Net1 overexpression may contribute to aneuploidy through its role in controlling chromosome alignment during metaphase [7], as well as by improper spindle positioning. Future studies will be required to ascertain whether this is, in fact, the case.
Highlights.
Cdk1 phosphorylates Net1 on multiple sites during mitosis
Phosphorylation of Net1 by Cdk1 inhibits its GEF activity towards RhoA
Inhibition of Net1 phosphorylation by Cdk1 results in excess cortical F-actin accumulation and mitotic spindle polarity defects.
Acknowledgments
We would like to thank members of the Cheng, Cunha, Denicourt, Dessauer, and Frost Labs for helpful discussions. We would also like to thank Catherine Denicourt for the kind gift of the 6xHis-Ubiquitin expression plasmid. This work was funded by grants CA116356 and CA172129 to J.A.F.
Abbreviations
- Arp3
Actin related protein 3
- Cdk1
Cyclin dependent kinase 1
- Dlg1
Discs large gene 1
- Dbl
Diffuse B cell lymphoma
- Ect2
Epithelial cell transforming 2
- ERM
Ezrin Radixin Moesin
- GEF
Guanine nucleotide exchange factor
- GST
Glutathione-S-transferase
- LGN
Leu-gly-asn repeat protein
- mDia1
mammalian Diaphanous 1
- Net1
Neuroepithelial transforming gene 1
- NUMA
Nuclear mitotic apparatus protein
- Pak1
p21 activated kinase 1
- Pak2
p21 activated kinase 2
- RhoA
Ras-related homolog A
- RhoB
Ras-related homolog B
- SLIK
Sterile 20-like kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Leyden J, Murray D, Moss A, Arumuguma M, Doyle E, McEntee G, O'Keane C, Doran P, MacMathuna P, Net1 and Myeov: computationally identified mediators of gastric cancer, British journal of cancer 94(8) (2006) 1204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shen SQ, Li K, Zhu N, Nakao A, Expression and clinical significance of NET-1 and PCNA in hepatocellular carcinoma, Medical oncology (Northwood, London, England) 25(3) (2008) 341–5. [DOI] [PubMed] [Google Scholar]
- [3].Gilcrease MZ, Kilpatrick SK, Woodward WA, Zhou X, Nicolas MM, Corley LJ, Fuller GN, Tucker SL, Diaz LK, Buchholz TA, Frost JA, Coexpression of alpha6beta4 integrin and guanine nucleotide exchange factor Net1 identifies node-positive breast cancer patients at high risk for distant metastasis, Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 18(1) (2009) 80–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tu Y, Lu J, Fu J, Cao Y, Fu G, Kang R, Tian X, Wang B, Over-expression of neuroepithelial-transforming protein 1 confers poor prognosis of patients with gliomas, Japanese journal of clinical oncology 40(5) (2010) 388–94. [DOI] [PubMed] [Google Scholar]
- [5].Dutertre M, Gratadou L, Dardenne E, Germann S, Samaan S, Lidereau R, Driouch K, de la Grange P, Auboeuf D, Estrogen regulation and physiopathologic significance of alternative promoters in breast cancer, Cancer research 70(9) (2010) 3760–70. [DOI] [PubMed] [Google Scholar]
- [6].Zhang Z, Lee JH, Ruan H, Ye Y, Krakowiak J, Hu Q, Xiang Y, Gong J, Zhou B, Wang L, Lin C, Diao L, Mills GB, Li W, Han L, Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer, Nature communications 10(1) (2019) 4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Menon S, Oh W, Carr HS, Frost JA, Rho GTPase-independent regulation of mitotic progression by the RhoGEF Net1, Molecular biology of the cell 24(17) (2013) 2655–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yamakita Y, Totsukawa G, Yamashiro S, Fry D, Zhang X, Hanks SK, Matsumura F, Dissociation of FAK/p130(CAS)/c-Src complex during mitosis: role of mitosis-specific serine phosphorylation of FAK, The Journal of cell biology 144(2) (1999) 315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Suzuki K, Takahashi K, Reduced cell adhesion during mitosis by threonine phosphorylation of beta1 integrin, Journal of cellular physiology 197(2) (2003) 297–305. [DOI] [PubMed] [Google Scholar]
- [10].Dao VT, Dupuy AG, Gavet O, Caron E, de Gunzburg J, Dynamic changes in Rap1 activity are required for cell retraction and spreading during mitosis, Journal of cell science 122(Pt 16) (2009) 2996–3004. [DOI] [PubMed] [Google Scholar]
- [11].Stewart MP, Helenius J, Toyoda Y, Ramanathan SP, Muller DJ, Hyman AA, Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding, Nature 469(7329) (2011) 226–30. [DOI] [PubMed] [Google Scholar]
- [12].Zlotek-Zlotkiewicz E, Monnier S, Cappello G, Le Berre M, Piel M, Optical volume and mass measurements show that mammalian cells swell during mitosis, The Journal of cell biology 211(4) (2015) 765–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Maddox AS, Burridge K, RhoA is required for cortical retraction and rigidity during mitotic cell rounding, The Journal of cell biology 160(2) (2003) 255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Matthews HK, Delabre U, Rohn JL, Guck J, Kunda P, Baum B, Changes in Ect2 localization couple actomyosin-dependent cell shape changes to mitotic progression, Developmental cell 23(2) (2012) 371–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Carreno S, Kouranti I, Glusman ES, Fuller MT, Echard A, Payre F, Moesin and its activating kinase Slik are required for cortical stability and microtubule organization in mitotic cells, The Journal of cell biology 180(4) (2008) 739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kunda P, Pelling AE, Liu T, Baum B, Moesin controls cortical rigidity, cell rounding, and spindle morphogenesis during mitosis, Current biology : CB 18(2) (2008) 91–101. [DOI] [PubMed] [Google Scholar]
- [17].Schmidt A, Hall A, The Rho exchange factor Net1 is regulated by nuclear sequestration, The Journal of biological chemistry 277(17) (2002) 14581–8. [DOI] [PubMed] [Google Scholar]
- [18].Qin H, Carr HS, Wu X, Muallem D, Tran NH, Frost JA, Characterization of the biochemical and transforming properties of the neuroepithelial transforming protein 1, The Journal of biological chemistry 280(9) (2005) 7603–13. [DOI] [PubMed] [Google Scholar]
- [19].Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T, Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis, The Journal of cell biology 147(5) (1999) 921–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Alberts AS, Qin H, Carr HS, Frost JA, PAK1 negatively regulates the activity of the Rho exchange factor NET1, The Journal of biological chemistry 280(13) (2005) 12152–61. [DOI] [PubMed] [Google Scholar]
- [21].Oh W, Frost JA, Rho GTPase independent regulation of ATM activation and cell survival by the RhoGEF Net1A, Cell cycle (Georgetown, Tex.) 13(17) (2014) 2765–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Carr HS, Zuo Y, Oh W, Frost JA, Regulation of focal adhesion kinase activation, breast cancer cell motility, and amoeboid invasion by the RhoA guanine nucleotide exchange factor Net1, Molecular and cellular biology 33(14) (2013) 2773–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Carr HS, Morris CA, Menon S, Song EH, Frost JA, Rac1 controls the subcellular localization of the Rho guanine nucleotide exchange factor Net1A to regulate focal adhesion formation and cell spreading, Molecular and cellular biology 33(3) (2013) 622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP, A probability-based approach for high-throughput protein phosphorylation analysis and site localization, Nature biotechnology 24(10) (2006) 1285–92. [DOI] [PubMed] [Google Scholar]
- [25].Örd M, Möll K, Agerova A, Kivi R, Faustova I, Venta R, Valk E, Loog M, Multisite phosphorylation code of CDK, Nature structural & molecular biology 26(7) (2019) 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Valk E, Venta R, Ord M, Faustova I, Kõivomägi M, Loog M, Multistep phosphorylation systems: tunable components of biological signaling circuits, Molecular biology of the cell 25(22) (2014) 3456–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lindqvist A, Rodriguez-Bravo V, Medema RH, The decision to enter mitosis: feedback and redundancy in the mitotic entry network, The Journal of cell biology 185(2) (2009) 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Enserink JM, Kolodner RD, An overview of Cdk1-controlled targets and processes, Cell division 5 (2010) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Reed SI, The ubiquitin-proteasome pathway in cell cycle control, Results and problems in cell differentiation 42 (2006) 147–81. [DOI] [PubMed] [Google Scholar]
- [30].Sivakumar S, Gorbsky GJ, Spatiotemporal regulation of the anaphase-promoting complex in mitosis, Nature reviews. Molecular cell biology 16(2) (2015) 82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Carr HS, Cai C, Keinanen K, Frost JA, Interaction of the RhoA exchange factor Net1 with discs large homolog 1 protects it from proteasome-mediated degradation and potentiates Net1 activity, The Journal of biological chemistry 284(36) (2009) 24269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kristelly R, Gao G, Tesmer JJ, Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor, The Journal of biological chemistry 279(45) (2004) 47352–62. [DOI] [PubMed] [Google Scholar]
- [33].Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K, Analysis of activated GAPs and GEFs in cell lysates, Methods in enzymology 406 (2006) 425–37. [DOI] [PubMed] [Google Scholar]
- [34].Paterson HF, Self AJ, Garrett MD, Just I, Aktories K, Hall A, Microinjection of recombinant p21rho induces rapid changes in cell morphology, The Journal of cell biology 111(3) (1990) 1001–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jaffe AB, Hall A, Rho GTPases: biochemistry and biology, Annual review of cell and developmental biology 21 (2005) 247–69. [DOI] [PubMed] [Google Scholar]
- [36].Garcia-Mata R, Dubash AD, Sharek L, Carr HS, Frost JA, Burridge K, The nuclear RhoA exchange factor Net1 interacts with proteins of the Dlg family, affects their localization, and influences their tumor suppressor activity, Molecular and cellular biology 27(24) (2007) 8683–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Johnston CA, Hirono K, Prehoda KE, Doe CQ, Identification of an Aurora-A/PinsLINKER/Dlg spindle orientation pathway using induced cell polarity in S2 cells, Cell 138(6) (2009) 1150–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bergstralh DT, Lovegrove HE, St Johnston D, Discs large links spindle orientation to apical-basal polarity in Drosophila epithelia, Current biology : CB 23(17) (2013) 1707–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chugh P, Clark AG, Smith MB, Cassani DAD, Dierkes K, Ragab A, Roux PP, Charras G, Salbreux G, Paluch EK, Actin cortex architecture regulates cell surface tension, Nature cell biology 19(6) (2017) 689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lu MS, Johnston CA, Molecular pathways regulating mitotic spindle orientation in animal cells, Development (Cambridge, England) 140(9) (2013) 1843–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].di Pietro F, Echard A, Morin X, Regulation of mitotic spindle orientation: an integrated view, EMBO reports 17(8) (2016) 1106–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kwon M, Bagonis M, Danuser G, Pellman D, Direct Microtubule-Binding by Myosin-10 Orients Centrosomes toward Retraction Fibers and Subcortical Actin Clouds, Developmental cell 34(3) (2015) 323–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Carminati M, Gallini S, Pirovano L, Alfieri A, Bisi S, Mapelli M, Concomitant binding of Afadin to LGN and F-actin directs planar spindle orientation, Nature structural & molecular biology 23(2) (2016) 155–63. [DOI] [PubMed] [Google Scholar]
- [44].Mitsushima M, Aoki K, Ebisuya M, Matsumura S, Yamamoto T, Matsuda M, Toyoshima F, Nishida E, Revolving movement of a dynamic cluster of actin filaments during mitosis, The Journal of cell biology 191(3) (2010) 453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pease JC, Tirnauer JS, Mitotic spindle misorientation in cancer--out of alignment and into the fire, Journal of cell science 124(Pt 7) (2011) 1007–16. [DOI] [PubMed] [Google Scholar]
- [46].Noatynska A, Gotta M, Meraldi P, Mitotic spindle (DIS)orientation and DISease: cause or consequence?, The Journal of cell biology 199(7) (2012) 1025–35. [DOI] [PMC free article] [PubMed] [Google Scholar]