

Summary
Long-range movement of organelles within the cytoplasm relies on coupling to microtubule motors, a process that is often mediated by adaptor proteins. In many cases, this coupling involves organelle- or adaptor-induced activation of the microtubule motors by conformational reversal of an autoinhibited state. Herein we show that a similar regulatory mechanism operates for an adaptor protein named SKIP (also known as PLEKHM2). SKIP binds to the small GTPase ARL8 on the lysosomal membrane to couple lysosomes to the anterograde microtubule motor kinesin-1. Structure-function analyses of SKIP reveal that the C-terminal region comprising three PH domains interacts with the N-terminal region comprising ARL8- and kinesin-1-binding sites. This interaction inhibits coupling of lysosomes to kinesin-1 and, consequently, lysosome movement toward the cell periphery. We also find that ARL8 does not just recruit SKIP to the lysosomal membrane, but also relieves SKIP autoinhibition, promoting kinesin-1-driven, anterograde lysosome transport. Finally, our analyses show that the largely disordered middle region of SKIP mediates self-association, and that this self-association enhances the interaction of SKIP with kinesin-1. These findings indicate that SKIP is not just a passive connector of lysosome-bound ARL8 to kinesin-1, but is itself subject to intra- and inter-molecular interactions that regulate its function. We anticipate that similar organelle- or GTPase-induced conformational changes could regulate the activity of other kinesin adaptors.
Graphical Abstract
eTOC blurb
Anterograde transport of lysosomes involves coupling to the microtubule motor kinesin-1 via the small GTPase ARL8 and the adaptor protein SKIP. Keren-Kaplan and Bonifacino show that ARL8 does not just recruit SKIP to lysosomes but also promotes SKIP activation through relief of an autoinhibited state.
Introduction
Many cellular processes depend on the ability of organelles to move within the cytoplasm [1]. Long-range organelle movement occurs along microtubules through coupling to dynein-dynactin and kinesin microtubule motors. Whereas dynein-dynactin drives transport from the plus end to the minus end of microtubules (i.e., retrograde transport), most kinesins drive transport in the opposite direction (i.e., anterograde transport). In general, coupling of organelles to microtubule motors is not direct but depends on a variety of adaptor proteins. The crucial importance of this machinery in cell and organismal physiology is underscored by the existence of many human diseases that result from mutations in genes encoding microtubule motors (e.g., dynein-dynactin [2, 3]; kinesins [4–6]) or adaptors ([e.g., RAB7 [7]; FYCO1 [8]; SKIP [9]; BORCS5 [10]; BICD2 [11]).
Lysosomes are well-known examples of organelles that undergo bidirectional transport along microtubules. Coupling of lysosomes to dynein-dynactin is mediated by several adaptor systems, including the small GTPase RAB7 and its effector RILP [12], the transmembrane protein TMEM55B and the adaptor protein JIP4 [13], the related adaptor protein JIP3 [14], and the Ca2+ channel TRPML1 and penta-EF-hand protein ALG2 [15]. Anterograde lysosome transport also involves several adaptor systems that engage different kinesins. For instance, the accessory protein KAP3 is required for coupling of lysosomes to kinesin-2 motors [16]. RAB7 and another effector, FYCO1, couple lysosomes as well as autophagosomes to kinesin-1 motors [17]. An ensemble of the hetero-octameric complex BORC, the small GTPase ARL8 and the adaptor protein SKIP (also known as PLEKHM2) constitute another adaptor system that couples lysosomes to kinesin-1 motors [18–21]. Finally, BORC and ARL8 (without the participation of SKIP) couple lysosomes to kinesin-3 motors [22]. BORC/ARL8-dependent coupling of lysosomes to kinesin-1 and kinesin-3 motors allows lysosomes to move along microtubule tracks having distinct tubulin post-translational modifications and distributions in the cytoplasm of both non-neuronal cells [22] and neurons [23, 24].
The coupling of lysosomes and other organelles to microtubule motors is highly regulated. One mode of regulation involves autoinhibition of the motors by intramolecular interactions [25]. A well-studied example is kinesin-1, a tetrameric complex of two kinesin heavy chains (KHCs) (KIF5A, KIF5B or KIF5C paralogs) and two kinesin light chains (KLCs) (KLC1, KLC2, KLC3 or KLC4 paralogs) [25]. The KHCs comprise, in N- to C-terminal direction, motor, neck, stalk and tail domains [25]. The motor domain binds microtubules whereas the tail domain interacts with cargo. The KLCs comprise an N-terminal heptad repeat domain that binds to KHCs, and a C-terminal TPR domain that binds cargo [25]. Both KHCs and KLCs have been shown to be autoinhibited: KHCs by interaction between the motor and tail domains [26–30] and KLCs by an interaction between a Leu-Phe-Pro-acidic (LFP-acidic) motif in the unstructured region that connects the heptad repeat and TPR domains, and the cargo-binding site in the TPR domain [31]. The autoinhibition of both KHCs and KLCs is reversed by interaction with cargos or adaptors, activating kinesin-1 for movement along microtubule tracks [26, 32–35].
Whereas autoinhibitory regulation is well-established for kinesin-1 [26–35] and other kinesins [36–39], to date it has not been shown to occur for the adaptor proteins that mediate coupling to the corresponding organelles. Herein we present evidence for such a mechanism for the adaptor protein SKIP (Figure 1A). Our studies show that the SKIP C-terminal region interacts with the N-terminal region to decrease the interaction of SKIP with kinesin-1 and the ability of SKIP to mediate anterograde lysosome movement. Furthermore, we find that ARL8 not only recruits SKIP to lysosomes but also relieves SKIP autoinhibition. This mechanism ensures that SKIP remains in an autoinhibited state until it couples to lysosomes via ARL8. The existence of this mechanism for SKIP raises the possibility that other cargo-kinesin adaptors may be similarly regulated.
Figure 1. Structural Organization of SKIP.
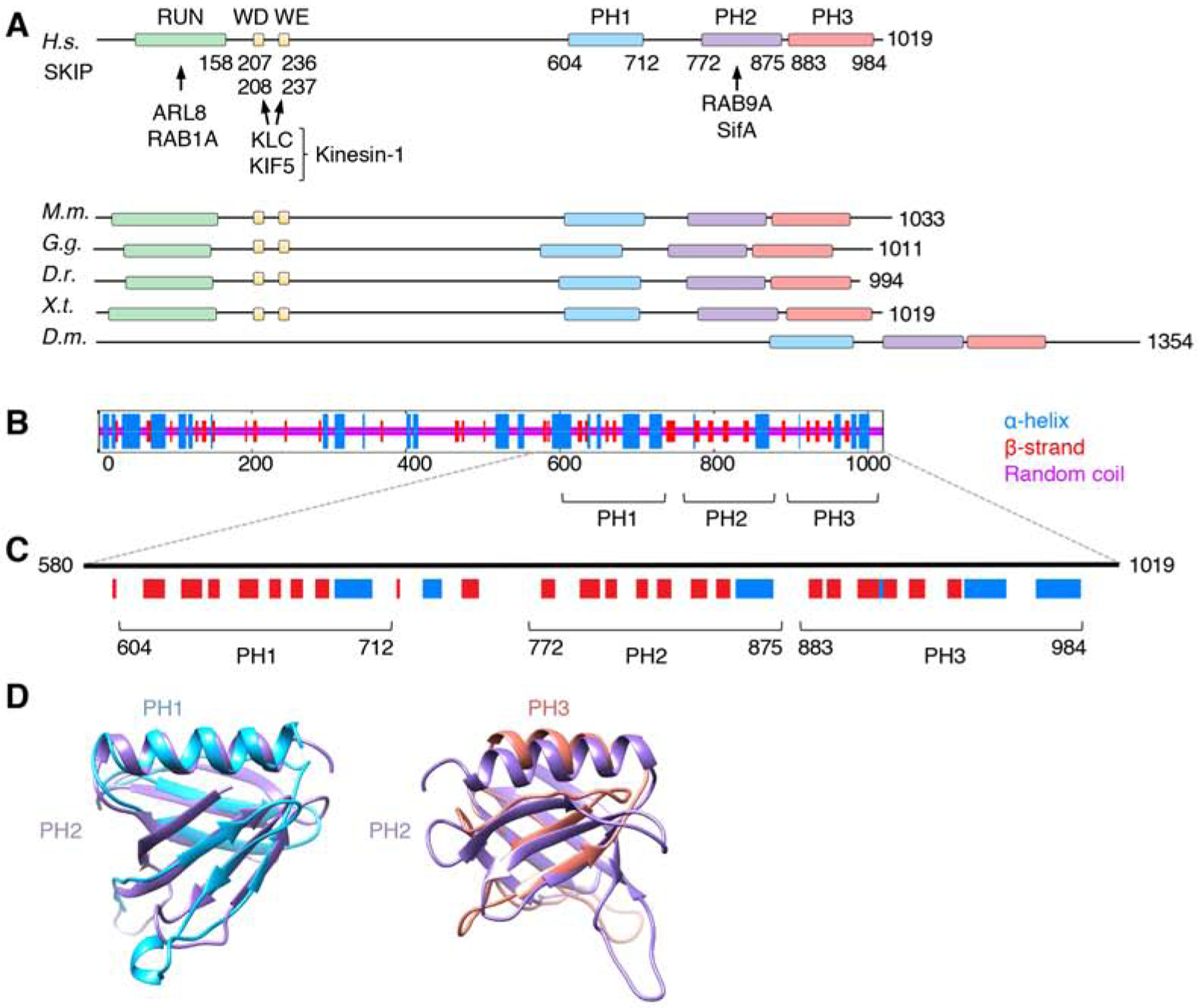
(A) Domain organization of SKIP from different animal species. Boxes indicate domains and motifs. RUN: RPIP8, UNC-14 and NESCA domain; WD: Trp-Asp motif; WE: Trp-Glu motif; PH: pleckstrin homology domain. Amino-acid numbers are indicated. Arrows indicate interactions with binding partners. H.s.: Homo sapiens, M.m.: Mus musculus, G.g.: Gallus gallus, D.r.: Danio rerio, X.t.: Xenopus tropicalis, D.m.: Drosophila melanogaster. Notice that Drosophila SKIP does not contain a RUN domain or WD/WE motifs, but its C-terminal domain is well conserved. The RUN domain mediates ARL8-dependent coupling to lysosomes [20] and RAB1A-dependent coupling to melanosomes [40]. Because melanosomes are not present in the cells used in this study, the role of RAB1A was not analyzed.
(B) Consensus secondary structure prediction of SKIP generated at the NPS-IBCP server (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_seccons.html).
(C) PH domain predictions by the PredictProtein server (https://www.predictprotein.org/). Notice the similarity in the secondary structure of the predicted PH1 and PH3 to the known PH2. HHpred (https://toolkit.tuebingen.mpg.de/tools/hhpred) also predicts PH1 and PH3 to be structurally similar to PH domains with high probabilities (95.2% for PH1 and 91.8% for PH3).
(D) Structural models of PH1 and PH3 according to the PHYRE2 Protein Fold Recognition Server superimposed on the known structure of PH2 (PDB ID: 3CXB). Confidence levels for PH1 and PH3 were 99.3% and 30.56%, respectively. Models were represented with UCSF Chimera.
Results
The Middle Region, but not the PH Domains, of SKIP Contributes to Anterograde Lysosome Movement
The initial goal of this study was to examine the requirement of the different domains of SKIP for the ability of SKIP to associate with lysosomes and to move them toward the cell periphery. SKIP comprises an N-terminal RUN domain that interacts with the GTP-bound forms of ARL8 [20] and RAB1A [40], followed by a largely disordered region harboring Trp-Asp (WD) and Trp-Glu (WE) motifs that interact with KLCs and KHCs [19, 41, 42], and a C-terminal pleckstrin homology domain (PH) (henceforth denoted PH2) that interacts with the GTP-bound form of RAB9A [43] and the Salmonella virulence factor SifA [18] (Figure 1A–C). Our bioinformatic analyses revealed that the C-terminal domain of SKIP contains not just one but three predicted PH domains designated PH1–3 (Figure 1A–D). To assess the functional importance of these structural elements, we conducted a structure-function analysis involving overexpression of various Myc-tagged SKIP deletion mutants in HeLa cells (Figure 2A). As previously reported [18, 20–22], we observed that overexpression of full-length SKIP resulted in a dramatic redistribution of lysosomes, labeled for LAMTOR4 and decorated with SKIP, to peripheral cell protrusions (herein referred to as “vertices”) (Figure 2B–D), where the plus ends of microtubule bundles are most concentrated. Deletion of residues 1–300, comprising the RUN domain and WD-WE motifs (Figure 2A, SKIP301–1019 construct), resulted in cytosolic localization of the truncated SKIP and normal distribution of lysosomes (Figure 2B–D), consistent with the requirement of these structural elements for interactions of SKIP with ARL8 and kinesin-1, respectively [20, 41]. In contrast, sequential deletion of the PH domains (Figure 2A, SKIP1–883, SKIP1–772 and SKIP1–603 constructs) had no significant effect on the association of SKIP with lysosomes and on the SKIP-induced redistribution of lysosomes to cell vertices (Figure 2B–D). These observations indicated that the three C-terminal PH domains are dispensable for the effects of overexpressed SKIP on lysosome positioning. A longer C-terminal deletion spanning amino acids 301–1019 (Figure 2A, SKIP1–300 construct), however, resulted in loss of both SKIP association with lysosomes and SKIP-induced lysosome redistribution to cell vertices (Figure 2B–D). This latter observation confirmed previous findings by Rosa-Ferreira and Munro (2011) using the same SKIP1–300 construct. Dumont et al. (2010) showed that a slightly longer construct, SKIP1–310, caused peripheral redistribution of lysosomes, but in only 25% of the cells, in contrast to full-length SKIP, which did so in 84% of the cells. The results with the SKIP1–300 and SKIP1–310 were intriguing, since they comprise the RUN domain that interacts with ARL8 and WD-WE motifs that interact with kinesin-1 (Figure 1A), and should therefore have all the elements needed to couple lysosomes to kinesin-1. These findings pointed to the existence of an additional element in the middle region spanning amino acids 301–603 that contributes to the function of SKIP in the regulation of lysosome positioning.
Figure 2. Effect of SKIP Deletions on Lysosome Positioning.
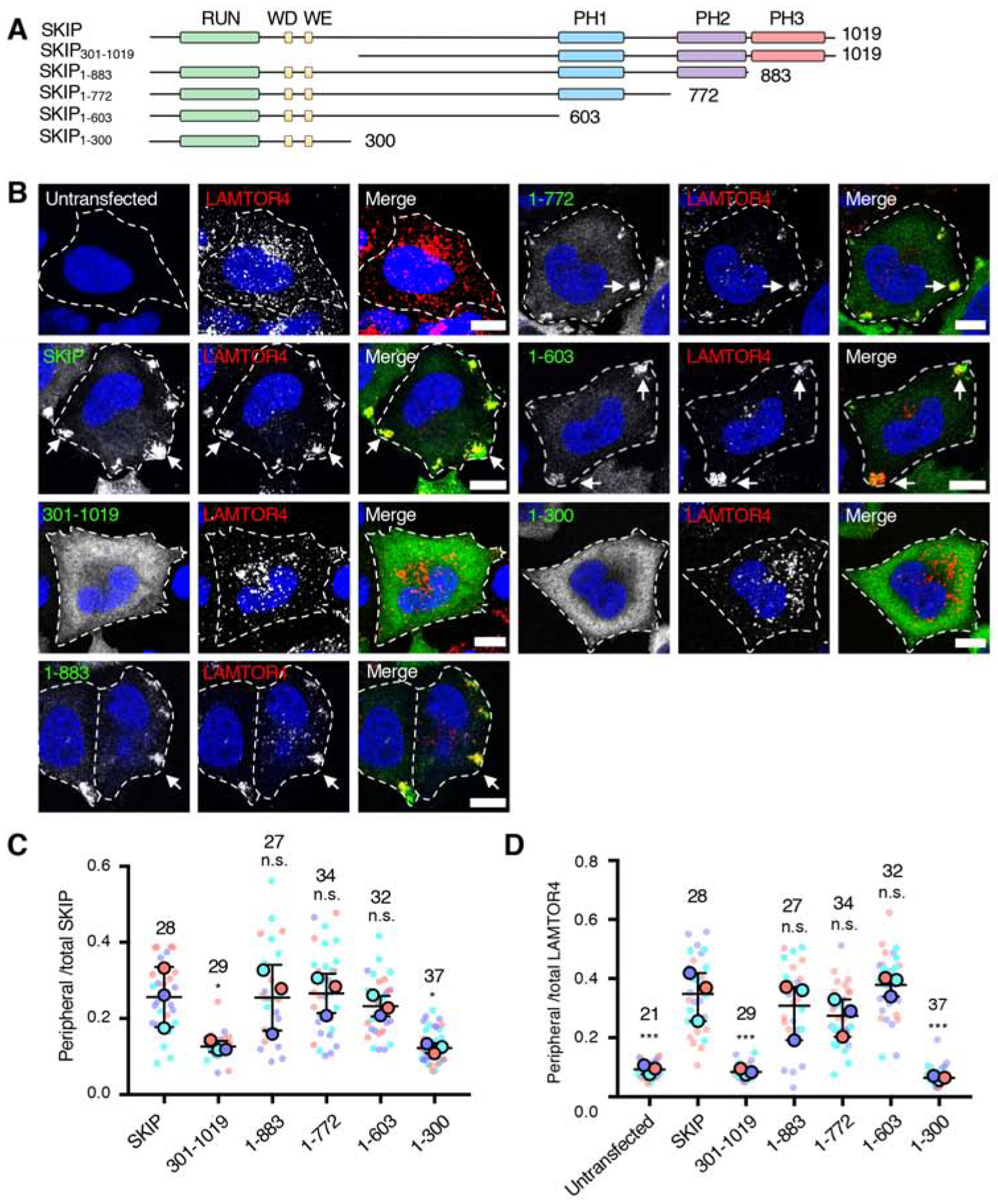
(A) Schematic representation of the SKIP deletion constructs used in these experiments. Domain organization of SKIP is as described in the legend to Figure 1. The constructs were tagged with a Myc epitope at the N-terminus.
(B) Confocal immunofluorescence microscopy of WT HeLa cells transfected with plasmids encoding the Myc-SKIP constructs represented in panel A, and co-immunostained with antibodies to the Myc epitope (green channel) and to the lysosomal marker LAMTOR4 (red channel). Nuclei were stained with DAPI (blue). Single channels are shown in grayscale with nuclei in blue. Cell edges are outlined. Arrows point to accumulation of Myc-SKIP and LAMTOR4 at cell vertices. Scale bars: 10 μm.
(C,D) Quantification of the ratio of peripheral to total Myc-SKIP constructs (C) and endogenous LAMTOR4 (D) from experiments such as those shown in panel B, represented as SuperPlots [60]. Peripheral fluorescence refers to the presence of the proteins at cell vertices. Horizonal lines indicate the mean ± SD of the means from 3 independent experiments. Experiments are color coded. Big circles represent the mean and small dots the individual data points from each experiment. The total number of cells analyzed is indicated on top of each value. Statistical significance was calculated by one-way ANOVA followed by multiple comparisons to Myc-SKIP-expressing cells using Dunnett’s test. Not significant (n.s.) P>0.05, *P<0.05, ***P<0.001.
ARL8, but not RAB9A, is Necessary for SKIP-Induced Lysosome Redistribution
To complement the deletion analysis of SKIP, we compared the requirement of ARL8 (which interacts with the RUN domain [20]) and RAB9A (which interacts with the PH2 domain [43]) (Figure 1A) for the localization and function of SKIP. To this end, we used CRISPR-Cas9 to perform double knock out (KO) of the ARL8A and ARL8B paralogs of ARL8 (henceforth referred to as ARL8-KO) (Figure 3A) or single KO of RAB9A in HeLa cells (Figure 3B). We observed that while ARL8-KO abrogated the ability of overexpressed SKIP to associate with lysosomes and to redistribute them to the cell periphery, RAB9A KO had no significant effect (Figure 3C–E). Moreover, we found that, whereas overexpression of ARL8B-GFP rescued SKIP function in ARL8-KO cells, overexpression of GFP-RAB9A did not, even though it associated with perinuclear lysosomes (Figure 3F–H). The effect of ARL8B-GFP was nucleotide dependent, as SKIP function was rescued by GTP-bound mutant (Q75L) but not GDP-bound mutant (T34N) ARL8B-GFP (Figure S1). We concluded that ARL8, but not RAB9A, is necessary for SKIP effects on lysosome positioning, and that overexpression of RAB9A cannot compensate for the absence of ARL8. This conclusion is in line with the requirement of the ARL8-binding RUN domain but not the RAB9A-binding PH2 domain for peripheral redistribution of lysosomes by SKIP (Figure 2B–D).
Figure 3. Analysis of the Requirement of ARL8 and RAB9A for SKIP function.
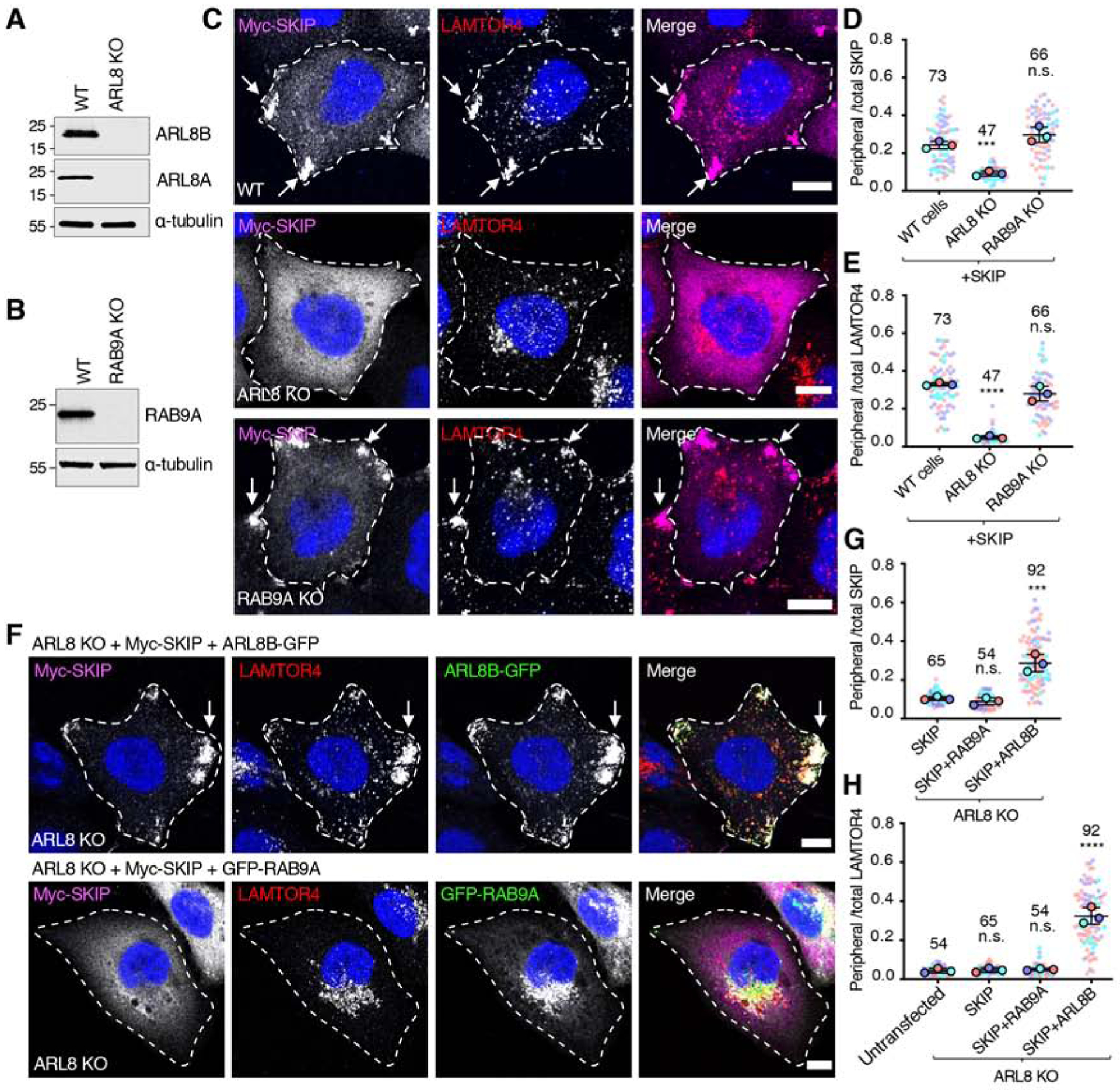
(A) Immunoblot analysis of WT and ARL8A-ARL8B-KO (referred to as ARL8-KO) HeLa cells with antibodies to ARL8A, ARL8B and α-tubulin (loading control).
(B) Immunoblot analysis of WT and RAB9A-KO HeLa cells with antibodies to RAB9A and α-tubulin (loading control). In A and B, numbers on the left indicate the positions of molecular mass markers (in kDa).
(C) Confocal immunofluorescence microscopy of WT, ARL8-KO and RAB9A-KO HeLa cells transfected with a plasmid encoding Myc-SKIP and co-immunostained with antibodies to the Myc epitope (magenta) and the lysosomal marker LAMTOR4 (red). Nuclei were stained with DAPI (blue). Single channels are shown in grayscale with nuclei in blue. Cell edges are outlined. Arrows indicate accumulation of Myc-SKIP and LAMTOR4 at cell vertices. Scale bars: 10 μm.
(D,E) Quantification of the ratio of peripheral to total Myc-SKIP (D) and endogenous LAMTOR4
(E) from experiments such as those shown in panel C, calculated and represented as described in the legend to Figure 2C. Not significant (n.s.) P>0.05, ***P<0.001, ****P<0.0001.
(F) Confocal immunofluorescence microscopy of ARL8-KO cells co-transfected with plasmids encoding Myc-SKIP together with ARL8B-GFP or GFP-RAB9A. Cells were co-immunostained with antibodies to the Myc epitope (magenta) and to the lysosomal marker LAMTOR4 (red). GFP fluorescence is shown in the green channel. Nuclei were stained with DAPI (blue). Single channels are shown in grayscale with nuclei in blue. Cell edges are outlined. Arrows indicate accumulation of Myc-SKIP and LAMTOR4 at cell vertices. Scale bars: 10 μm. The related Figure S1 shows the effect of expressing constitutively active ARL8B-GFP-Q75L and inactive ARL8BGFP-T34N on Myc-SKIP function in ARL8-KO cells.
(G,H) Quantification of the ratio of peripheral to total Myc-SKIP (G) and endogenous LAMTOR4 (H) from experiments such as those shown in panel F, calculated and represented as described in the legend to Figure 2C. Not significant (n.s.) P>0.05, ***P<0.001, ****P<0.0001.
The Middle Region of SKIP Contributes to Interaction with Kinesin-1 via Self-Association
We next addressed how the middle region (i.e., amino acids 301–603) contributes to the function of SKIP. To determine whether this region promotes the interaction of SKIP with kinesin-1, HEK293T cells were transfected with plasmids encoding Myc-tagged, full-length SKIP, SKIP1–300 and SKIP1–603. Cell lysates were immunoprecipitated with antibody to Myc, and immunoprecipitates were analyzed by SDS-PAGE and immunoblotting with antibodies to the endogenous KIF5B and KLC2 subunits of kinesin-1 (i.e., the ubiquitous form of kinesin-1 expressed in non-neuronal cells) (Figure 4A–C). As expected from previous work [18, 42], we observed that both KIF5B and KLC2 co-precipitated with full-length SKIP. Importantly, co-precipitation of KIF5B and KLC2 was reduced for Myc-SKIP1–300 but increased for SKIP1–603 relative to full-length SKIP (Figure 4A–C). These experiments thus indicated that the 301–603 region is needed for optimal binding of SKIP to kinesin-1, consistent with its requirement for the function of SKIP in moving lysosomes to the cell periphery (Figure 2). These findings were again surprising since the WD-WE motifs involved in kinesin-1 binding are located N-terminally to this region (i.e., amino acids 207–208 and 236–237 in human SKIP) (Figure 1A), pointing to the existence of additional elements that contribute to the interaction of SKIP with kinesin-1. Moreover, the increased co-precipitation of kinesin-1 with SKIP1–603 relative to full-length SKIP suggested that the segment comprising the PH domains (amino acids 604–1019) partially inhibited the interaction of SKIP with kinesin-1.
Figure 4. SKIP Self-Association Mediated by the Middle Region Enhances Interaction with Kinesin-1.
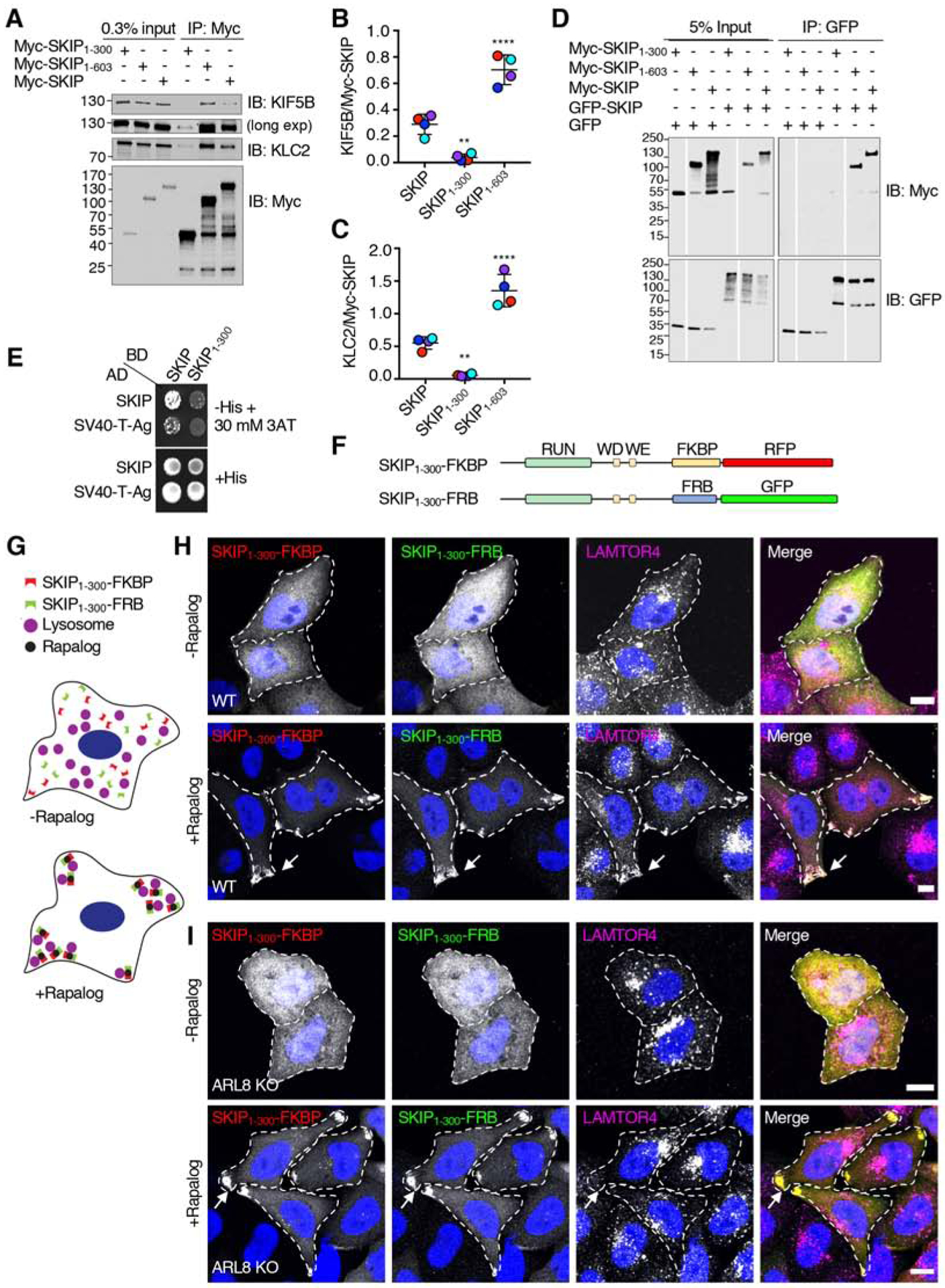
(A) Co-immunoprecipitation of Myc-SKIP constructs with endogenous kinesin-1. HEK293T cells were transfected with plasmids encoding the indicated Myc-SKIP constructs, subjected to immunoprecipitation with antibody to the Myc epitope and analyzed by SDS-PAGE and immunoblotting for the Myc epitope and the endogenous KIF5B and KLC2 subunits of kinesin-1. Immunoblotting for KIF5B is shown in short and long exposures.
(B,C) Quantification of experiments shown in panel A. Signal intensity was quantified using Image J. The ratio of KIF5B (B) and KLC2 (C) to immunoprecipitated Myc-SKIP was calculated. Values are the mean ± SD from 4 independent experiments. Statistical significance was calculated by one-way ANOVA followed by multiple comparisons to full-length SKIP using Dunnett’s test., **P<0.01, ****P<0.0001. Independent experiments are color coded.
(D) Co-immunoprecipitation of SKIP constructs. HEK293T cells were co-transfected with plasmids encoding the indicated Myc-SKIP constructs and full-length GFP-SKIP or GFP (control), subjected to immunoprecipitation with antibody to GFP, and analyzed by SDS-PAGE and immunoblotting with antibodies to the Myc epitope and GFP. Data are representative of 2 independent experiments with similar results. Vertical white lines indicate places where blots were cut to remove irrelevant lanes. All lanes are from the same blots in the same experiment. In A and D, numbers on the left indicate the positions of molecular mass markers (in kDa).
(E) Yeast two-hybrid analysis of the interaction of full-length SKIP or the SV40 T-antigen (control) fused to the β-Gal activation domain (AD), and full-length SKIP or SKIP1–300 fused to the β-Gal DNA-binding domain (BD). Growth in the absence of histidine (-His) indicates binding between the proteins. Thirty mM 3AT was added to reduce non-specific interactions. Data are representative of 3 experiments with similar results.
(F) Schematic representation of the constructs used to perform rapalog-induced dimerization of SKIP1–300 as previously described for microtubule motors [61]. Domains of SKIP are as described in the legend to Figure 1. SKIP constructs were C-terminally appended with FKBP-RFP or FRB-GFP.
(G) Cartoon representing the hypothetical effect of rapalog addition to WT HeLa cells co-expressing the constructs shown in panel F if dimerization of SKIP1–300 enables coupling of lysosomes to kinesin-1. SKIP1–300 plasmids tagged with FKBP-RFP or FRB-GFP are cytosolic in the absence of rapalog. Addition of rapalog induces the dimerization of FKBP with FRB, promoting the accumulation of SKIP1–300 with lysosomes at cell vertices.
(H) Confocal immunofluorescence microscopy of WT HeLa cells co-transfected with plasmids encoding the constructs shown in panel F, without (top panel) and with rapalog (bottom panel), and immunostained with antibody to the lysosomal marker LAMTOR4 (magenta channel). SKIP1–300-FKBP-RFP and SKIP1–300-FRB-GFP fluorescence is shown in the red and green channel, respectively. Nuclei were stained with DAPI (blue). Single channels are shown in grayscale with nuclei in blue. Cell edges are outlined. Arrows point to accumulation of SKIP1–300 constructs and LAMTOR4 at cell vertices. Scale bars: 10 μm.
(I) Same as panel H but using ARL8-KO HeLa cells.
The related Figure S2 shows the amino-acid sequence conservation of the SKIP middle region in different animal species.
The middle region exhibits a high level of amino-acid sequence conservation (~67% similarity) among SKIP proteins from different vertebrate species (Figure S2), despite being predicted to be largely unstructured and not having any known functional motifs. We hypothesized that this region could mediate SKIP self-association, increasing the valency of the interactions with other partners. Indeed, co-immunoprecipitation experiments showed that full-length Myc-SKIP and Myc-SKIP1–603, but not Myc-SKIP1–300, co-precipitated with full-length GFP-SKIP (Figure 4D). Yeast two-hybrid (Y2H) analysis confirmed the requirement of the middle region for self-association by showing that full length SKIP interacted with itself, but not with SKIP1–300 (Figure 4E). These experiments thus demonstrated that the 301–603 region of SKIP is required for self-association.
Self-Association Promotes the Function of SKIP in Lysosome Dispersal
To determine if SKIP self-association contributes to its function, we examined the effect of forcing rapalog-induced dimerization of SKIP1–300-RFP and SKIP1–300-GFP fused to the FKBP (FK506- and rapamycin-binding protein) and FRB (FKBP-rapamycin binding) modules, respectively [44] (Figure 4F,G). We observed that, in the absence of rapalog, SKIP1–300-FKBP-RFP and SKIP1–300-FRB-GFP were cytosolic and did not affect the distribution of lysosomes (Figure 4H) in (wild-type) WT HeLa cells. Addition of rapalog, however, resulted in the redistribution of both SKIP1–300-FKBP-RFP and SKIP1–300-FRB-GFP, as well as lysosomes, to cell vertices (Figure 4H). Notably, rapalog-induced dimerization of SKIP1–300-FKBP-RFP with SKIP1–300-FRB-GFP in ARL8-KO HeLa cells also resulted in accumulation of these constructs at cell vertices, but lysosomes remained unperturbed (Figure 4I). From these experiments, we concluded that inducing dimerization of SKIP1–300 in WT cells is sufficient to rescue movement of this construct toward the cell periphery together with lysosomes. In ARL8-KO cells, on the other hand, SKIP1–300 dimerization enabled movement of this construct to cell vertices but did not alter the distribution of lysosomes because of the inability of these organelles to bind SKIP in the absence of ARL8. Therefore, the requirement of the middle region for function can be explained by its ability to mediate SKIP self-association.
The C-Terminus of SKIP Negatively Regulates its Association with Kinesin-1
Although the C-terminal region comprising the three PH domains (amino acids 604–1019) was dispensable for the ability of SKIP to move lysosomes toward the cell periphery (Figure 2A–D), two observations suggested that it could have an inhibitory role. First, deletion of this region increased the co-precipitation of SKIP with kinesin-1 (Figure 4A–C). Second, whereas a dimer of SKIP1–300-FKBP-RFP with SKIP1–300-FRB-GFP (lacking the C-terminal region) associated with cell vertices in ARL8-KO cells (Figure 4I), full-length Myc-SKIP was cytosolic in those cells (Figure 3C). To further investigate the function of the SKIP C-terminal region, we examined the behavior of full-length SKIP and SKIP1–603 in WT cells (Figure 5A) and various KO cell lines (Figure 5B–E) (quantification is shown in Figure 5F). We observed that in WT cells both full-length SKIP and SKIP1–603 associated with lysosomes and moved them to the cell periphery (Figure 5A,F), as shown before (Figure 2B–D). In KIF5B-KO cells, full-length SKIP and SKIP1–603 also associated with lysosomes, but were unable to move lysosomes to the cell periphery (Figure 5B,F), consistent with the requirement of kinesin-1 for the effects of SKIP on lysosome positioning [20, 22]. In contrast, KO of the kinesin-3 KIF1B, which moves lysosomes in an ARL8-dependent but SKIP-independent manner [22], did not prevent the ability of both SKIP constructs to redistribute lysosomes to the cell periphery (Figure 5C,F). KO of ARL8B had an intermediate phenotype, with partial localization of both SKIP constructs and lysosomes to cell vertices (Figure 5D,F); the residual effect was probably due to the function of ARL8A. Indeed, double KO of ARL8A and ARL8B (i.e., ARL8 KO) prevented the peripheral redistribution of lysosomes by overexpression of either full-length SKIP or SKIP1–603 (Figure 5E,F). However, whereas full-length SKIP was predominantly cytosolic and failed to accumulate at cell vertices, SKIP1–603 localized to cell vertices in the majority of cells (Figure 5E,F). The SKIP1–603 that moved to cell vertices in ARL8-KO cells was probably not associated with any organelles, since SKIP is only known to bind to lysosomes via ARL8 (Rosa-Ferreira and Munro, 2011). These experiments thus demonstrated that that the C-terminus of SKIP prevents the movement of full-length SKIP in ARL8-KO cells, suggesting that ARL8 may relieve an inhibitory effect of the C-terminal domain (amino acids 604–1019). Additionally, these experiments demonstrated that the association of full-length SKIP with lysosomes is independent of kinesin-1 binding (Figure 5B), but kinesin-1 binding and the consequent accumulation of SKIP at cell vertices are dependent on ARL8 (Figure 5E).
Figure 5. Contribution of the C-Terminal Domain to the Function of SKIP in Various KO Cell Lines.
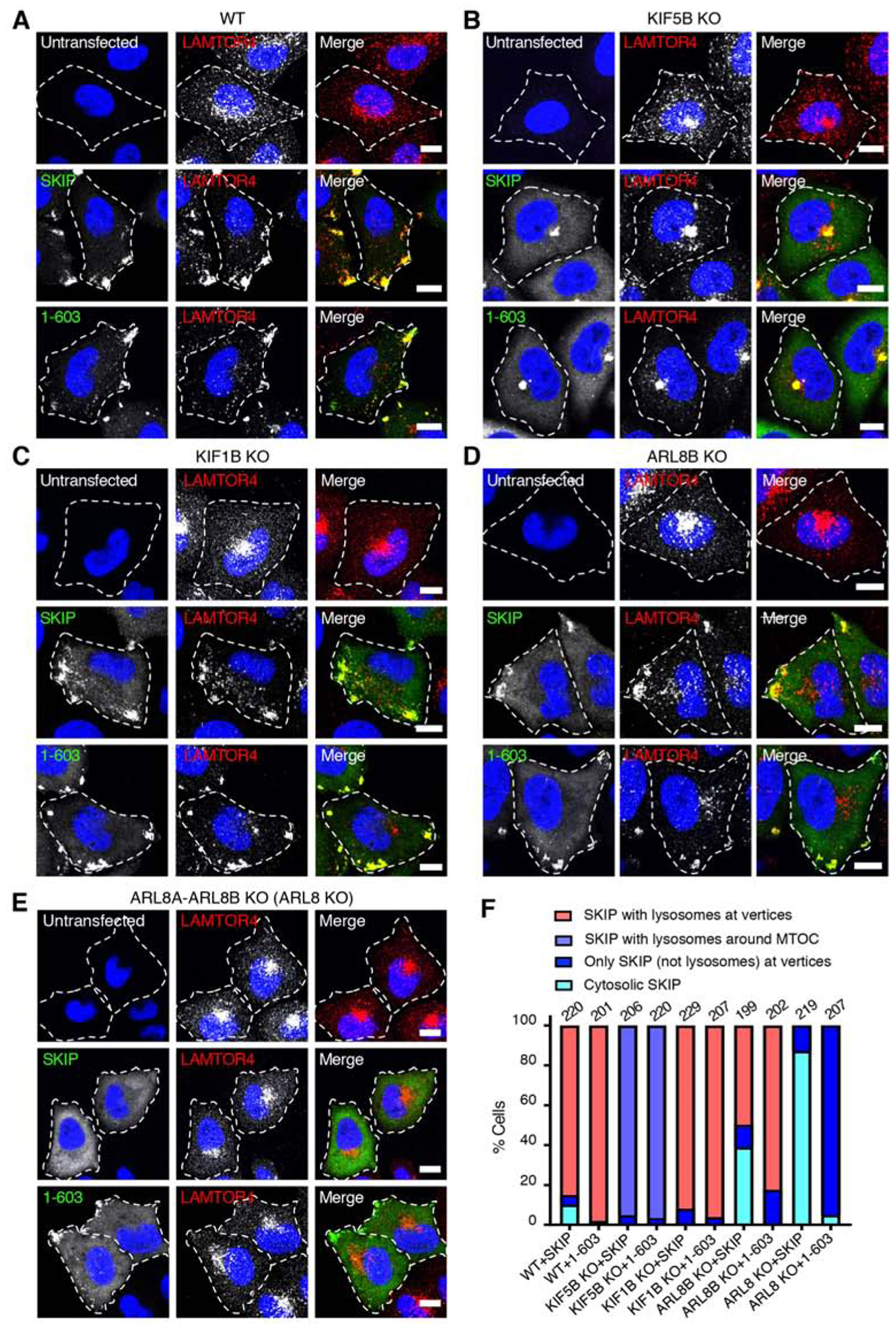
(A-E) Confocal immunofluorescence microscopy of Myc-SKIP and Myc-SKIP1–603 expressed by transfection in WT HeLa cells (A), and in HeLa cells with KO of KIF5B (B), KIF1B (C), ARL8B
(D) and ARL8A-ARL8B (ARL8 KO) (E). Untransfected cells were used as controls. Cells were immunostained with antibodies to the Myc epitope (green) and the lysosomal marker LAMTOR4 (red), and counterstained with DAPI (blue). Single channels are shown in grayscale with nuclei in blue. Cell edges are outlined. Scale bars: 10 μm.
(F) Quantification of the percentage of cells displaying the indicated phenotypes in two independent experiments such as those shown in panels A-E. The total number of cells examined in both experiments is indicated on top of each bar.
The related Figure S3 shows the localization of different Myc-SKIP constructs and their effect on lysosome positioning in ARL8-KO cells.
ARL8 Enhances Binding of SKIP to Kinesin-1 by Relieving the Inhibitory Effect of the C-Terminal Domain
To further investigate the function of the C-terminal domain of SKIP, we examined the localization of several SKIP truncation mutants in ARL8-KO cells. We observed that Myc-tagged SKIP1–603 (lacking PH1–3), SKIP1–772 (lacking PH2-PH3) and SKIP1–883 (lacking PH3) were all partially localized to cell vertices, whereas full-length SKIP was cytosolic (Figure S3). As expected from the absence of ARL8, none of these constructs altered the centrally clustered distribution of lysosomes (Figure S3). To determine if ARL8 influences the inhibitory effect of the SKIP C-terminal domain on the interaction of SKIP with kinesin-1 (Figure 4A–C), we compared the co-precipitation of different Myc-tagged SKIP constructs with endogenous KIF5B and KLC2 in WT and ARL8-KO cells (Figure 6A–C). These experiments showed that ARL8-KO did not affect the co-precipitation of SKIP1–603 but decreased the co-precipitation of full-length SKIP with kinesin-1 (Figure 6A–C). Conversely, overexpression of ARL8B-mCherry in WT cells increased the co-precipitation of kinesin-1 with full-length SKIP (Figure 6D–F). This effect was dependent on the nucleotide-bound state of ARL8B-mCherry, as the co-precipitation of kinesin-1 was increased by GTP-bound (Q75L) but not GDP-bound (T34N) ARL8B-mCherry in WT cells (Figure 6G–I) Therefore, ARL8-GTP activates SKIP for binding to kinesin-1 in the presence of the SKIP C-terminal domain, but not in the absence of this domain, when SKIP is already activated.
Figure 6. SKIP is Negatively Regulated by its C-Terminal Domain.
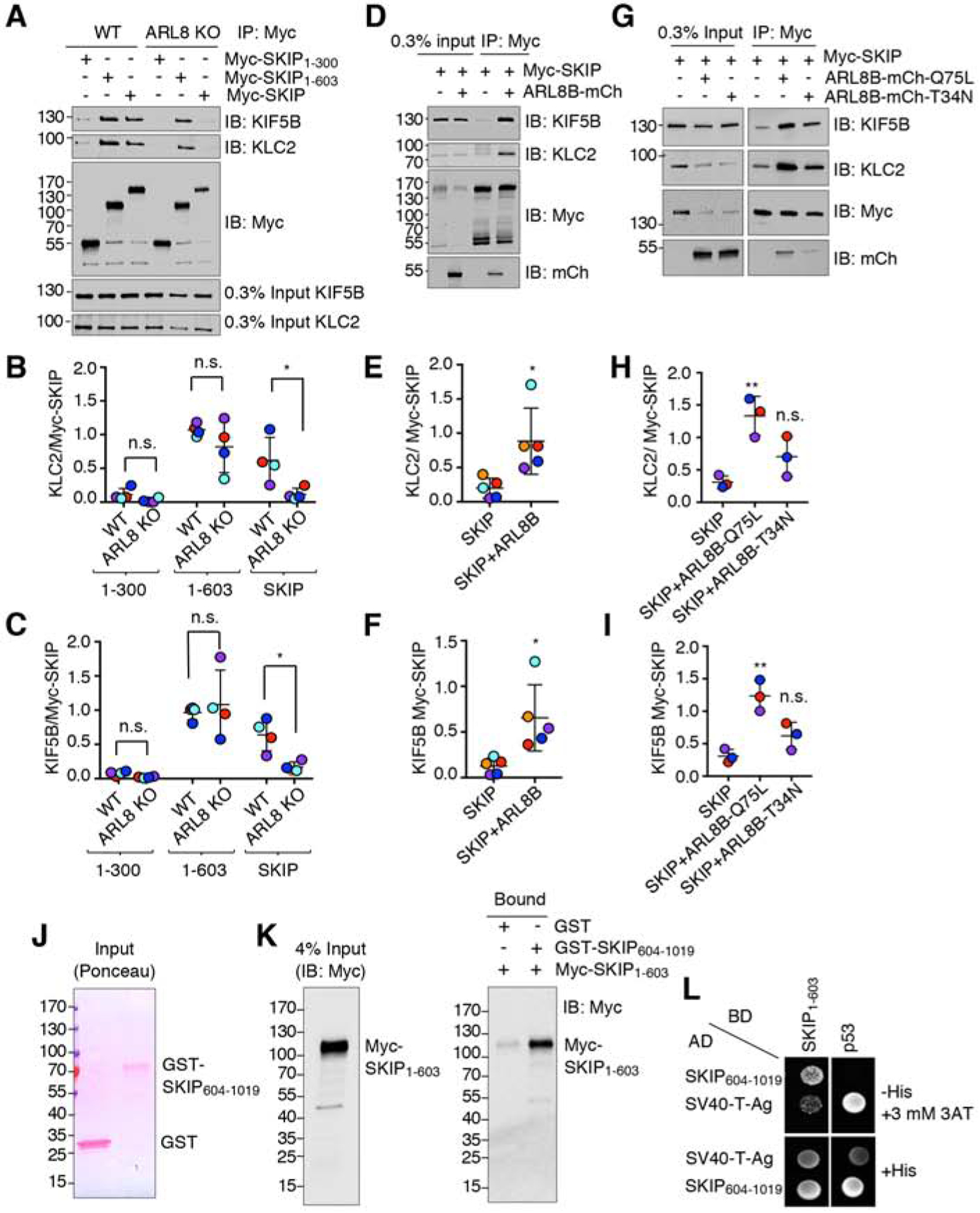
(A) Co-immunoprecipitation of full-length and truncated Myc-SKIP constructs with kinesin-1 in WT and ARL8-KO HeLa cells. Cells were transfected with plasmids encoding the indicated Myc-SKIP constructs, subjected to immunoprecipitation using an antibody to the Myc tag, followed by SDS-PAGE and immunoblotting with antibodies to the endogenous KIF5B and KLC2 subunits of kinesin-1 and the Myc tag.
(B,C) Quantification of experiments such as that shown in panel A. Signal intensity was quantified by Image J. The ratio of KLC2 (B) or KIF5B (C) to Myc-SKIP in the immunoprecipitates was calculated, and relative Myc-SKIP binding in WT HeLa was normalized to 1. Horizontal lines represent the mean ± SD from 4 independent experiments. Independent experiments are color-coded. Statistical significance was calculated using two-way ANOVA followed by multiple comparisons using Sidak’s test. Not significant (n.s.) P>0.05, *P<0.05.
(D) Co-immunoprecipitation of endogenous KIF5B and KLC2 with Myc-SKIP in the absence or presence of ARL8B-mCherry overexpressed by transfection in HEK293T cells. Cell extracts were subjected to immunoprecipitation using an antibody to the Myc tag followed by SDS-PAGE and immunoblotting with antibodies to the endogenous KIF5B and KLC2 subunits of kinesin-1 and the Myc tag.
(E,F) Quantification of experiments such as that shown in panel D. Signal intensity was quantified by Image J. The ratio of bound KLC2 (E) or KIF5B (F) to bound Myc-SKIP signals was calculated. Horizontal lines represent the mean ± SD of 5 independent experiments consisting of 6 and 9 replicates per sample for SKIP and SKIP+ARL8, respectively. Independent experiments are color-coded. Statistical significance was calculated using Student’s t-test. Not significant (n.s.) P>0.05, *P<0.05.
(G) Co-immunoprecipitation of endogenous KIF5B and KLC2 with Myc-SKIP in the absence or presence of ARL8B-mCherry T34N (GDP-bound) and Q75L (GTP-bound) overexpressed by transfection in HEK293T cells. Cell extracts were subjected to immunoprecipitation using an antibody to the Myc tag followed by SDS-PAGE and immunoblotting with antibodies to the endogenous KIF5B and KLC2 subunits of kinesin-1 and the Myc tag.
(H,I) Quantification of experiments such as that shown in panel G. Signal intensity was quantified by Image J. The ratio of bound KLC2 (H) or KIF5B (I) to bound Myc-SKIP signals was calculated. Horizontal lines represent the mean ± SD from 3 independent experiments consisting of 7 replicates per sample. Independent experiments are color-coded. Statistical significance was calculated by one-way ANOVA followed by multiple comparisons using Tukey’s test. Not significant (n.s.) P>0.05, *P<0.05, **P<0.01.
(J,K) Pulldown by purified, recombinant GST and GST-SKIP604–1019 of Myc-SKIP1–603 expressed by transfection in HEK293T cells. GST and GST-SKIP604–1019 were immobilized on glutathione-Sepharose and incubated with lysates of Myc-SKIP1–603 -expressing HEK293T cells. Samples were analyzed by SDS-PAGE; GST proteins were visualized by Ponceau staining (J) and Myc-SKIP1–603 by immunoblotting with antibody to the Myc epitope (K). In A, D, G, J and K, numbers on the left indicate the positions of molecular mass markers (in kDa).
(L) Yeast two-hybrid analysis of the interaction of SKIP604–1019 fused to the β-Gal activation domain (AD) with SKIP1–603 fused to the β-Gal DNA-binding domain (BD). Growth in the absence of histidine (−His) indicates binding between the proteins. Three mM 3AT was added to reduce non-specific growth.
Interaction Between the N- and C-Terminal Domains of SKIP
To explain the ARL8-dependence of the inhibitory role of the SKIP C-terminal domain, we hypothesized that the N- and C-terminal domains of SKIP could interact with each other, thus precluding binding of the WD-WE motifs to kinesin-1 and necessitating the intervention of ARL8 to break up the intramolecular interaction. Indeed, we observed that purified recombinant GST-SKIP604–1019 (i.e., C-terminal region construct), but not GST, was able to pull down Myc-SKIP1–603 (i.e., N-terminal region construct) expressed by transfection in HEK293T (Figure 6J,K). Moreover, Y2H assays confirmed the specific interaction of SKIP604–1019 with SKIP1–603 (Figure 6L). These experiments thus demonstrated an intramolecular interaction between the N- and C-terminal domains of SKIP.
ARL8 Promotes Activation of Kinesin-1 by SKIP
Binding of SKIP via Trp-acidic motifs to KLC reverses KHC autoinhibition, resulting in kinesin-1 activation for movement of lysosomes to the cell periphery [42]. To determine whether kinesin-1 activation is dependent on the activation state of SKIP, we co-transfected ARL8-KO cells with plasmids encoding GFP-KIF5B and HA-KLC2 in the absence or presence of ARL8B-mCherry and different Myc-SKIP constructs, and examined kinesin-1 activation by its ability to move toward cell vertices (Figure 7A,B). We observed that, in the absence of ARL8B-mCherry, GFP-KIF5B and HA-KLC2 were largely cytosolic, irrespective of the absence or presence of full-length Myc-SKIP (Figure 7A,B), consistent with kinesin-1 being autoinhibited under these conditions. In the presence of ARL8B-mCherry, on the other hand,, full-length Myc-SKIP, GFP-KIF5B and HA-KLC2 redistributed to cell vertices along with ARL8B-mCherry (Figure 7A,B). Finally, we observed that SKIP1–603 promoted GFP-KIF5B and HA-KLC2 movement to cell vertices in the absence of ARL8B-mCherry (Figure 7A,B). These findings indicated that kinesin-1 can be activated by full-length Myc-SKIP in the presence but not the absence of ARL8B-mCherry, or by SKIP1–603 even in the absence of ARL8B-mCherry, consistent with kinesin-1 activation depending on activated SKIP.
Figure 7. Kinesin-1 Activation by SKIP.
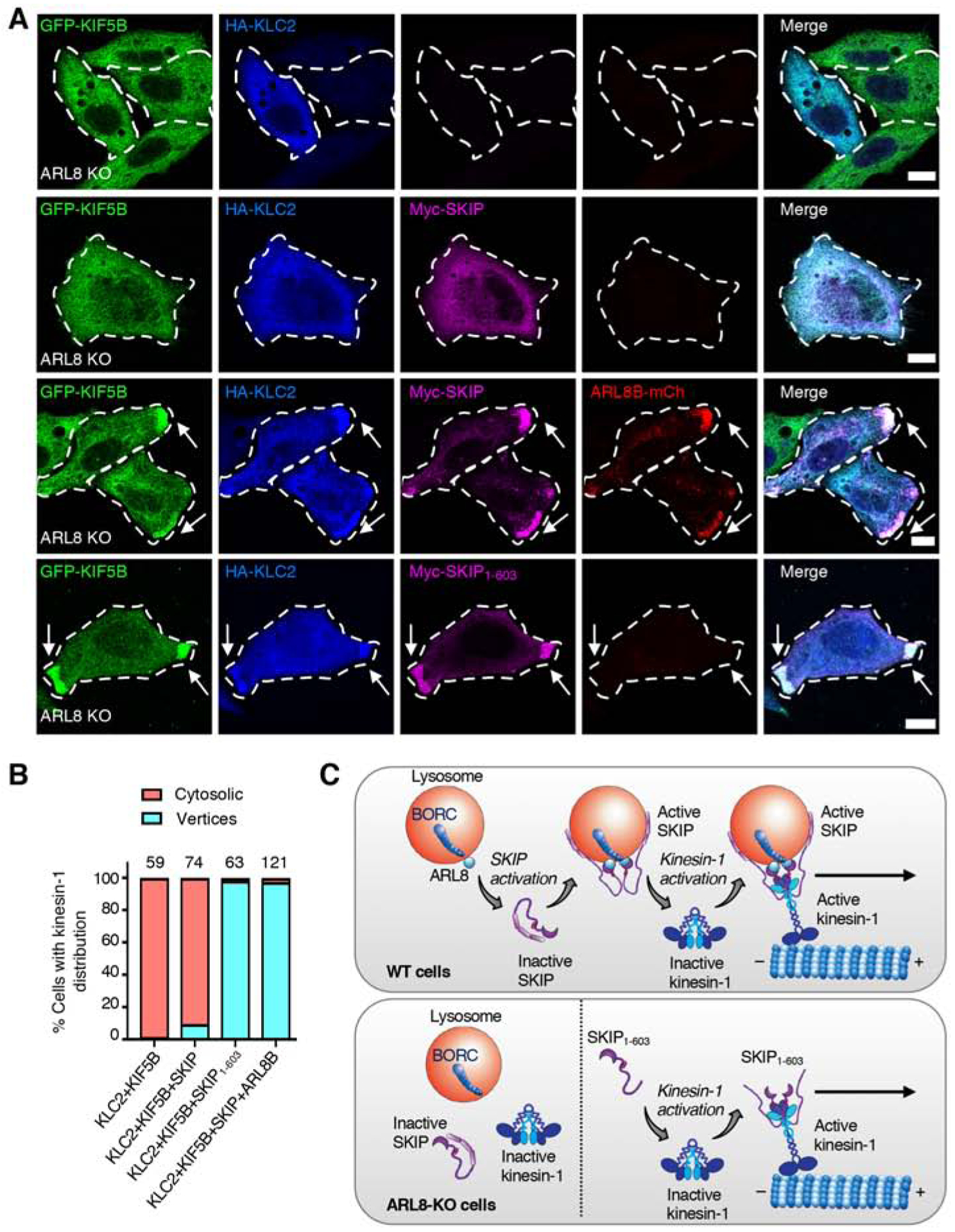
(A) Confocal immunofluorescence microscopy of ARL8-KO HeLa cells transfected with plasmids encoding the indicated proteins. Cells were immunostained with antibodies to KIF5B (green), HA epitope (blue) and Myc epitope (magenta). ARL8B was visualized by the mCherry tag (red). Single channels are shown in grayscale. Cell edges are outlined. Arrows indicate protein accumulation at cell vertices. Scale bars: 10 μm.
(B) Quantification of the percentage of cells showing localization of GFP-KIF5B and HA-KLC2 to cell vertices or the cytosol in two independent experiments such as that shown in panel A. The total number of cells examined in both experiments is indicated on top of each bar.
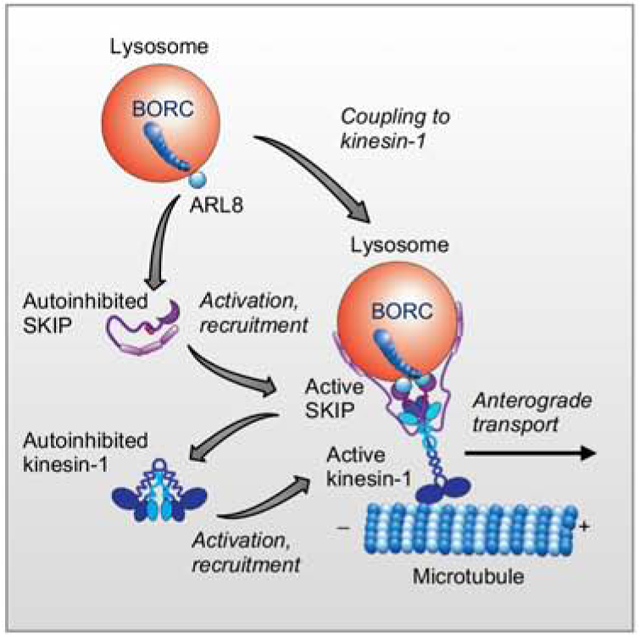
(C) Cartoon representing the regulation of SKIP by autoinhibition. Top. In WT cells, the lysosome-associated complex BORC recruits ARL8-GTP to lysosomes. Cytosolic SKIP exists in an autoinhibited state involving an interaction between the N- and C-terminal parts of the molecule. ARL8-GTP relieves this autoinhibition and recruits the activated SKIP to lysosomes. Active SKIP in turn promotes a conformational activation of kinesin-1 and recruits it to lysosomes. This triggers anterograde transport of lysosomes toward microtubule plus ends enriched in the cell periphery. Lysosome-bound SKIP is represented as a dimer assembled via the middle region because of evidence of self-association and for symmetry with kinesin-1; the exact stoichiometry of SKIP oligomers, however, has not been determined. Bottom. In ARL8-KO cells, SKIP remains autoinhibited and cannot bind to lysosomes and kinesin-1. SKIP1–603 cannot bind to lysosomes, but is constitutively activated, can activate kinesin-1 and move with kinesin-1 along microtubules toward the cell periphery.
Discussion
Our structure-function analysis of SKIP has revealed a novel regulatory mechanism for a kinesin adaptor, involving an autoinhibitory interaction between the N- and C-terminal regions of SKIP that is relieved by ARL8 (Figure 7C). This mechanism is analogous to that of several kinesins, whose autoinhibitory, intramolecular interactions are reversed by binding to adaptors or cargos [26–39]. For both SKIP and kinesins, autoinhibition ensures that the proteins exist as inactive forms in the cytosol, preventing futile interactions until they are needed for engagement in cargo movement along microtubules.
The C-Terminal Region of SKIP Mediates Autoinhibition
The function of the C-terminal region of SKIP was not known prior to this work. Previous studies had shown that this region encompassed a PH domain (herein named PH2) that interacted with RAB9A [43] and with the Salmonella effector SifA [45]. Our experiments showed that both PH2 and RAB9A are dispensable for association of SKIP with lysosomes and for the ability of SKIP to move lysosomes to the periphery (Figures 2 and 3), so the functional significance of the PH2-RAB9A interaction remains to be established. The interaction of PH2 with SifA, on the other hand, was shown to promote the formation and movement of kinesin-1-enriched vesicles derived from Salmonella-containing vacuoles, demonstrating that it plays a role in the context of intracellular bacterial infection [18]. Our bioinformatic analyses predicted that the C-terminal region of SKIP comprises two additional PH domains (named PH1 and PH3) flanking the previously known PH2 domain (Figure 1). In general, PH domains bind proteins or membrane lipids [46]. We recently found that SKIP binds phosphoinositides including PtdIns(4)P, which is enriched in phagolysosomes and regulates the emergence of membrane tubules in the process of phagolysosome resolution [47]. However, it remains to be determined if this binding is mediated by the PH domains. In any event, deletion of the entire C-terminal region did not prevent the ability of SKIP to associate with lysosomes and to move them toward the cell periphery (Figure 2). Rather, this deletion enhanced binding of SKIP to kinesin-1 (Figure 4A–C), consistent with the C-terminal region having an inhibitory role. Further analyses showed that the C-terminal region interacted with the N-terminal region of SKIP (Figure 6J–L), supporting a model in which an intramolecular interaction between the N- and C-terminal regions hinders binding of SKIP to kinesin-1 and thus negatively regulates the function of SKIP in lysosome movement. Although the molecular details of this autoinhibitory mechanism remain to be determined, we speculate that the intramolecular interaction likely blocks access of the WD-WE motifs of SKIP to the KLC-TPR and KHC-tail domains of kinesin-1.
ARL8 Relieves SKIP Autoinhibition
Our studies also revealed a mechanism by which autoinhibition of SKIP is relieved through interaction with ARL8. This GTPase was already known to recruit SKIP to lysosomes by virtue of its interaction with the RUN domain [20]. We found that ARL8 KO prevented the movement of full-length SKIP, but not SKIP1–603 (lacking the C-terminal region), to cell vertices (Figure 5). Moreover, ARL8 KO reduced the interaction of full-length SKIP, but not SKIP1–603, with kinesin-1, and ARL8 overexpression increased this interaction (Figure 6). Therefore, binding of ARL8 to the RUN domain of SKIP likely promotes a conformational change that frees the WD-WE motifs for interaction with kinesin-1. ARL8 thus serves both as an anchor for SKIP at the lysosomal membrane and as an activator of SKIP for binding to kinesin-1. This mechanism is reminiscent of the RAB6A-induced relief of BicD2 autoinhibition for the function of the latter as an adaptor/activator of dynein-dynactin [48]. We speculate that a similar mechanism may operate for other kinesin and dynein-dynactin adaptors that interact with small GTPases, as is the case for the FHF complex and RAB5 [49], FYCO1-RAB7 [17], protrudin-RAB7 [50], RILP-RAB7 [12, 51] and RAB11FIP3-RAB11 [52]. A GTPase-triggered chain of conformational activation events may be a common mechanism for the regulated coupling of various cargos to microtubule motors.
Connections to the Pathogenesis of DCM/LVNC
Two mutations in SKIP were recently identified as causes for the hereditary cardiac disorders autosomal-recessive dilated cardiomyopathy (DCM) and left ventricular noncompaction (LVNC) in humans [9]. These mutations remove either part of the PH1 domain or most of the C-terminal region [9], thus affecting the part of SKIP that is responsible for autoinhibition. It is therefore possible that dysregulation of SKIP inhibition/activation contributes to the pathogenesis of these disorders. However, fibroblasts from DCM/LVNC patients also exhibit reduced levels of the mutant SKIP proteins [9], probably due to destabilization and degradation. Future studies are needed to distinguish if the disease results from the dysregulation or reduced levels of SKIP. It also remains to be determined why these mutations cause specific defects in the heart, since SKIP is ubiquitously expressed [53]. Because ARL8 can recruit the kinesin-3 KIF1Bβ to lysosomes [22], it is possible that this alternative mechanism mitigates the reduction in ARL8-SKIP-kinesin-1 lysosome coupling in cells other than cardiomyocytes.
Functional Requirement for Self-Association Mediated by the Middle Region of SKIP
Our molecular dissection of SKIP also revealed a requirement for the middle region (amino acids 300–603) in SKIP function. Although this region is predicted to be largely disordered (Figure 1B) and does not have any known motifs, its high degree of conservation in vertebrates (Figure S2) was suggestive of a functional requirement. Indeed, we found that, despite having the reported binding sites for ARL8 (i.e., RUN domain) and kinesin-1 (i.e., WD-WE motifs), a SKIP1–300 construct was inactive, whereas a SKIP1–603 construct including the middle region was active in moving lysosomes toward the cell periphery (Figure 2). Moreover, SKIP1–603 bound kinesin-1 with greater avidity than SKIP1–300 (Figure 4A–C). Since other microtubule motor adaptors such as FYCO1 [17], protrudin [54], FEZ1 [55], RILP [56], JIP3 [57, 58] and RAB11FIP3 [59] function as dimers or oligomers, we hypothesized that the middle region of SKIP could similarly mediate self-association. Our experiments showed that this was indeed the case, as we observed co-immunoprecipitation and Y2H interaction of SKIP constructs having the middle region but not of those lacking this region (Figure 4D,E). Moreover, we demonstrated that self-association is critical for SKIP function by showing that forced dimerization of SKIP1–300 using a FKBP-FRB-rapalog system conferred on this otherwise inactive construct the ability to move lysosomes to the cell periphery (Figure 4F–I). Therefore, self-association via the middle region enhances the function of SKIP.
Concluding Remarks
Our studies have thus identified two novel structural features of SKIP that contribute to its function as an adaptor of lysosomes to kinesin-1: an interaction between the N- and C-terminal regions that inhibits SKIP function until bound by ARL8, and self-association mediated by the middle region that promotes interaction with kinesin-1. These findings indicate that SKIP is not just a passive link between lysosome-bound ARL8 and kinesin-1 but is subject to intra- and inter-molecular interactions that modulate its function as a kinesin adaptor. It will now be of interest to investigate if other kinesin adaptors are similarly regulated.
STAR Methods
Resource Availabilty
Lead Contact
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Juan S. Bonifacino (juan.bonifacino@nih.gov).
Materials Availability
ARL8- and RAB9A-knockout cells generated in this study are available upon request. Plasmids not covered by any restrictions like MTAs generated in this study are available upon request.
Data and Code Availability
This study did not generate any unique datasets or code.
Experimental Model and Subject Details
Cells
HeLa and HEK293T cells (ATCC, Manassas, VA) were cultured in complete DMEM (CDMEM) that consisted of Dulbecco’s Modified Eagle’s Medium (DMEM) (112-319-101, Quality Biological, Gaithersburg, MD) supplemented with 10% fetal bovine serum (35–011-CV, Corning, NY), 50 U/mL penicillin, 50 μg/mL streptomycin (30002-CL, Corning), and MycoZap Plus-CL (VZA-2011, Lonza, Basel, Switzerland) at 37°C and 5% CO2. For immunofluorescence microscopy, transfections were performed using 24-well plates (Corning) with 0.1–0.5 μg plasmid and 1μL Lipofectamine 2000 (11668019, Thermo Fisher scientific, Waltham, MA) according to the manufacturer’s protocol. Transfection medium was replaced with CDMEM after 1 h. Cells were analyzed ~24 h after transfection. For FKBP-FRB dimerization experiments (Figure 3H–I), 1 h after transfection, the medium was replaced with CDMEM supplemented with 0.5 μM A/C heterodimerizer (Takara Bio Inc., Shiga, Japan). For co-immunoprecipitation experiments, cells were grown in 10-cm dishes (Corning) and transfections were performed using 1–8 μg plasmid. Cells were kept in the transfection medium until they were harvested.
Method Details
Recombinant DNA Constructs
Constructs encoding SKIP truncations were generated from a pCMV-Myc-SKIP plasmid containing full-length SKIP (gift from Stéphane Méresse, Centre d’Immunologie de Marseille-Luminy, France) by insertion of a stop codon using PCR site-directed mutagenesis with back-to-back primer design. Sequences encoding His6-GFP fusion proteins were subcloned into the pET28a-His6-sf-GFP vector between KpnI and NotI endonuclease restriction sites. To create pGEX-6P-GST-SKIP, pGBT9-SKIP and pGADT7-SKIP, the coding sequence for SKIP was amplified by PCR from Myc-SKIP plasmid and subcloned into pGEX-6P, pGBT9 or pGADT7 vectors by Gibson assembly [62]. To create SKIP1–300-EGFP-FRB and SKIP1–300-RFP-FKBP, a SKIP1–300-encoding fragment was subcloned into the pEGFP-N1-FKBP-RFP and pEGFP-N1-FRB-EGFP plasmids between SacI and SalI endonuclease restriction sites. ARL8B-mCherry Q75L and T34N mutations were introduced by Quikchange site directed mutagenesis method (200521, Agilent Technologies, Santa Clara, CA). DNA sequences were verified by Sanger sequencing. Additional plasmids that were used in the study are listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| LAMTOR4 (C7orf59) (D4P6O), rabbit, used for IF at 1:200 | Cell Signaling Technology | Cat# 13140 |
| α-tubulin (DM1A), mouse, used for IB at 1:2,000 | Sigma-Aldrich | Cat# T-9026 |
| GFP-HRP, mouse, used for IB at 1:2,000 | Miltenyi Biotec | Cat# 130-091-833 |
| KIF5B [EPR10276(B)], rabbit, used for IB at 1:500 and for IF at 1:200 | Abcam | Cat# ab167429 |
| KLC2, rabbit, used for IB at 1:500 | Abcam | Cat# ab95881 |
| KLC2, rabbit, used for IB at 1:500 | Thermo Fisher Scientific | Cat# PA5–59168 |
| ARL8B, rabbit, used for IB at 1:1,000 | Proteintech | Cat# 13049–1-AP |
| ARL8A, rabbit, used for IB at 1:1,000 | Proteintech | Cat# 17060–1-AP |
| RAB9A (D52G8) XP, rabbit, used for IB at 1:500–1:1,000 | Cell Signaling Technology | Cat# 5118 |
| C-Myc epitope (9E10), HRP-conjugated, mouse, used for IB at 1:1000–1:1,0000 | Santa Cruz Biotechnology | Cat# Sc-40 HRP |
| C-Myc epitope (9E10), mouse, used for IB at 1:200 | Santa Cruz Biotechnology | Cat# Sc-40 |
| HA epitope, chicken, used for IF at 1:200 | Sigma-Aldrich | Cat# AB3254 |
| mCherry (16D7), rat, used for IB at 1:2,000 | Thermo Fisher Scientific | Cat# m11217 |
| Alexa Fluor 647-conjugated donkey anti-rabbit IgG, used for IF at 1:1,000 | Thermo Fisher Scientific | Cat# A31573 |
| Alexa Fluor 546-conjugated donkey anti-rabbit IgG, used for IF at 1:1000 | Thermo Fisher Scientific | Cat# A10040 |
| Alexa Fluor 488-conjugated donkey anti-mouse IgG, used for IF at 1:1,000 | Thermo Fisher Scientific | Cat# A21202 |
| Alexa Fluor 488-conjugated donkey anti-rabbit IgG, used for IF at 1:1,000 | Thermo Fisher Scientific | Cat# A21206 |
| Alexa Fluor 647-conjugated donkey anti-mouse IgG, used for IF at 1:1,000 | Thermo Fisher Scientific | Cat# A31571 |
| Alexa Fluor 405-conjugated goat anti-chicken IgY, used for IF at 1:1,000 | Abcam | Cat# ab175675 |
| HRP-conjugated goat anti-rabbit IgG (H+L), used for IB at 1:5,000–1:10,000 | Jackson ImmunoResearch | Cat# 111-035-003 |
| HRP-conjugated goat anti-rat IgG (H+L), used for IB at 1:5,000–1:10,000 | Jackson ImmunoResearch | Cat# 112-035-143 |
| HRP-conjugated donkey anti-mouse IgG (H+L), used for IB at 1:5,000–1:10,000 | Jackson ImmunoResearch | Cat# 715-035-150 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM culture medium | Quality Biological | Cat# 112-319-101 |
| Fetal bovine serum | Corning | Cat# 35–011-CV |
| MycoZap Plus-CL | Lonza | Cat# VZA-2011 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| Fluoromount G with DAPI | Electron Microscopy Sciences | Cat# 17984–24 |
| Fluoromount G | Electron Microscopy Sciences | Cat# 17984–25 |
| A/C heterodimerizer (Rapalog) | Takara | Cat# 635057 |
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 200521 |
| EDTA-free protease inhibitor capsule | Roche | Cat# 1836170 |
| Pierce™ anti-c-Myc magnetic beads | Thermo Fisher Scientific | Cat# 88842 |
| GFP-Trap magnetic agarose kit | ChromoTek | Cat# gtmak-20 |
| Glutathione-Sepharose 4B | GE Healthcare | Cat# 17-0756-05 |
| EZ-YEAST™ transformation kit | MP Biomedicals | Cat# 112100200 |
| Experimental Models: Cell Lines | ||
| HeLa | ATCC | N/A |
| HEK293T | ATCC | N/A |
| HeLa CRISPR-Cas9 RAB9A KO | This work | N/A |
| HeLa CRISPR-Cas9 KIF1B KO | [64] | N/A |
| HeLa CRISPR-Cas9 KIF5B KO | [64] | N/A |
| HeLa CRISPR-Cas9 ARL8B KO | [21] | N/A |
| HeLa CRISPR-Cas9 ARL8A-ARL8B KO | This work | N/A |
| Bacterial strains | ||
| BL21-CodonPlus(DE3) RP | Agilent Technologies | Cat# 230255 |
| Recombinant DNA | ||
| pmCherry-N1-ARL8B-mCherry, human | [24] | N/A |
| pmCherry-N1-ARL8B-mCherry-Q75L | This work | N/A |
| pmCherry-N1-ARL8B-mCherry-T34N | This work | N/A |
| pCMV-Myc-SKIP, human | This work | N/A |
| pCMV-Myc-SKIP1–300 | This work | N/A |
| pCMV-Myc-SKIP301–1019 | This work | N/A |
| pCMV-Myc-SKIP1–603 | This work | N/A |
| pCMV-Myc-SKIP1–772 | This work | N/A |
| pCMV-Myc-SKIP1–883 | This work | N/A |
| pEGFP-C1-GFP-SKIP | This work | N/A |
| SKIP1–300-FKBP-RFP in pEGFP-N1 backbone | This work | N/A |
| SKIP1–300-FRB-EGFP in pEGFP-N1 backbone | This work | N/A |
| pGEX-6P1-GST-SKIP | This work | N/A |
| pEGFP-C1-GFP-RAB9A, canine | This work | N/A |
| pcDNA3.1/CT-GFP-ARL8B, human | John Brumell, Hospital for Sick Children, Toronto, Canada | N/A |
| pcDNA3.1/CT-GFP-ARL8B-Q75L | This work | N/A |
| pcDNA3.1/CT-GFP-ARL8B-T34N | This work | N/A |
| pGADT7-SKIP | This work | N/A |
| pGBT9-SKIP | This work | N/A |
| pGBT9-SKIP1–300 | This work | N/A |
| pGADT7-SKIP604–1019 | This work | N/A |
| pGBT9-SKIP1–603 | This work | N/A |
| pGBT9-SKIP1–300 | This work | N/A |
| pTD1–1-SV40-T-Ag | Clontech | N/A |
| pVA3–1-p53 | Clontech | N/A |
| pGEX-6P1-GST-SKIP604–1019 | This work | N/A |
| pET28a-HIS6-GFP-SKIP1–603 | This work | N/A |
| pEGFP-C1 (without GFP) HA-KLC2, human | [22] | N/A |
| pGFP-C1 (A206K)-KIF5B, mouse | [22] | N/A |
| pSpCas9(BB)-2A-GFP (pX-458) | Addgene | Cat# 48138 |
| Software and Algorithms | ||
| Fiji/ImageJ | [65, 66] | https://imagej.nih.gov/ij/ |
| NPS@ consensus secondary structure prediction | [67] | https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_seccons.html |
| PredictProtein server | [68, 69] | https://www.predictprotein.org |
| HHPRED | [70] | https://toolkit.tuebingen.mpg.de/tools/hhpred |
| PHYRE2 | [71] | http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index |
| UCSF Chimera | [72] | https://www.cgl.ucsf.edu/chimera/ |
| Illustrator | Adobe | N/A |
| PRISM 7 | GraphPad | N/A |
Generation of CRISPR-Cas9 KO Cell Lines
To generate ARL8 double-KO cells, the ARL8A gene was knocked out in ARL8B-KO cells [21]. The following pairs of gRNAs targeting the ARL8A gene with a BbsI restriction site and PAM sequence were used: 5’-CACCGCGCGATCACGTTGACGAAGG-3’, 5’-AAACCCTTCGTCAACGTGATCGCGC-3’ and 5’-CACCGCTGGTCGGGCTTCAGTACT-3’, 5’-AAACGAGTACTGAAGCCCGACCAGC-3’ (Eurofins Genomics, Louisville, KY). Complementary gRNAs were annealed by PCR and subcloned into the pSpCas9(BB)-2A-GFP (pX-458) vector (Addgene plasmid #48138) between BbsI endonuclease restriction sites [63]. ARL8B-KO HeLa cells were co-transfected with plasmids encoding the two different gRNAs. GFP-positive cells were isolated by fluorescence-activated cell sorting (FACS), and 5% of the highest GFP-positive cells were collected. The collected cells were diluted to 3,000 cells per 10-cm dish. After 14 days, colonies were expanded, and cells analyzed by immunobloting with antibody to ARL8A. Positive colonies were verified by genomic DNA extraction followed by PCR amplification and sequencing. The ARL8-KO cells harbor, in addition to the ARL8B gene KO, an 8 base-pair deletion in the ARL8A gene encoding a 30-amino-acid protein instead of 186-amino-acid protein. RAB9A-KO was carried out as described for ARL8-KO using the following targeting gRNAs: 5’-CACCGCACCTATTGTATGGAAGAGC-3’, 5’-AAACGCTCTTCCATACAATAGGTG-3’ and 5’-CACCGAAAATTCCACACCTATTGTA-3’, 5’-AAACTACAATAGGTGTGGAATTTTC3’. Positive colonies were verified by PCR with the following primers: Forward: 5’-GAAGTGGATGGACATTTTGTTACC-3’ and Reverse: 5’-CAGGCAGCAGTCAGAACCTATGTAAAATGG-3’, and by immunoblotting with antibody to endogenous RAB9A. KIF5B-KO and KIF1B-KO HeLa cells [64] were previously described.
Immunofluorescence Microscopy
Cells were washed three times in PBS supplemented with 1 mM MgCl2 (351-033-721, Quality Biological) and 0.1 mM CaCl2 (21115, Sigma-Aldrich, St. Louis, MO) (PBS-CM), fixed with 4% paraformaldehyde (PFA) (15714-S, Electron Microscopy Sciences, Hatfield, PA), permeabilized with 0.2% Triton X-100, blocked with PBS-CM supplemented with 0.2% bovine serum albumin (BSA) (A-421, GoldBio, St. Louis, MO), stained and mounted with fluoromount-G with DAPI (17984–24, Electron Microscopy Sciences). Images were acquired on a Zeiss LSM780 confocal microscope fitted with a Plan-Apochromat 63X, 1.4 numerical aperture (NA) objective (Carl Zeiss, Oberkochen, Germany). Images were processed in ImageJ (Schneider et al., 2012).
Antibodies
Antibodies to the following antigens were used for immunostaining and immunoblotting: LAMTOR4 (D4P6O, Cell Signaling Technology, Danvers, MA), c-Myc (9E10, Santa Cruz Biotechnology, Santa Cruz, CA), c-Myc-HRP (9E10, Santa Cruz Biotechnology), HA (AB3254, Sigma-Aldrich), GFP-HRP (130-091-833, Miltenyi Biotec, Bergisch Gladbach, Germany), KIF5B (ab167429, Abcam, Cambridge, UK), KLC2 (a mixture of two antibodies: PA5–59168, Thermo Fisher Scientific, and ab95881, Abcam), RAB9A (D52G8) XP, Cell Signaling Technology), ARL8B (13049–1-AP, Proteintech, Rosemont, IL), ARL8A (17060–1-AP, Proteintech, Rosemont, IL), α-tubulin (DM1A, T-9026, Sigma-Aldrich) and mCherry (16D7, M11217, Thermo Fisher Scientific). The following fluorescently-labeled secondary antibodies were used for immunofluorescence microscopy: Alexa Fluor 546-conjugated donkey anti-rabbit IgG, Alexa Fluor 488-conjugated donkey anti-mouse IgG and Alexa Fluor 488-conjugated donkey anti-rabbit IgG, Alexa Fluor 647-conjugated donkey anti-mouse IgG and Alexa Fluor 647-conjugated donkey anti-rabbit IgG (Thermo Fisher Scientific) and Alexa Fluor 405-conjugated goat anti-chicken IgY (H+L) (Abcam). The following HRP-conjugated secondary antibodies were used for immunoblotting: HRP-conjugated goat anti-rabbit IgG (H+L) (111-035-003), HRP-conjugated goat anti-rat IgG (H+L) (112-035-143) and HRP-conjugated donkey anti-mouse IgG (H+L) (715-035-150) (Jackson ImmunoResearch, Westgrove, PA). More detailed antibody information can be found in the Key Resources Table.
Co-Immunoprecipitation
For co-immunoprecipitation of SKIP or SKIP mutants with KIF5B and KLC2 (Figures 4A, 6A, 6D, 6G), ~2–2.5×106 cells were plated on a 10-cm dish. The next day, cells were transfected with the indicated plasmids, and harvested ~24–36 h later. Cells were washed three times in ice-cold PBS, each time followed by centrifugation for 5 min at 500 × g and 4° C. Cells pellets were resuspended in 1 mL ice-cold buffer [25 mM Tris-HCl pH 7.4, 0.15 M NaCl, 1 mM EDTA, 1% NP40 (011332473001, Roche), 5% glycerol] supplemented with complete EDTA-free protease inhibitor capsule (1836170, Roche, Basel, Switzerland) at 4°C for 30 min with occasional mixing. The soluble fraction was separated by centrifugation for 10 min at 20,000 × g and 4°C. Samples were loaded onto 15 μL Pierce™ anti-c-Myc magnetic beads (88842, Thermo Fisher Scientific) and incubated on a rotating wheel for 2 h at 4°C. Following incubation, samples were washed three times in wash buffer (25 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.05 % Tween-20) and separated by centrifugation for 3 min at 500 × g and 4°C. Samples were eluted by heating for 10 min at 99°C with 1X Laemmli Sample Buffer (1610747, Bio-Rad, Hercules, CA). Co-immunoprecipitation experiments to analyze self-association (Figure 4D) were carried out with GFP-Trap magnetic agarose kit (gtmak-20, ChromoTek GmbH, Planegg-Martinsried, Germany). Samples were incubated on 15 μL GFP-trap resin and processed according to the manufacturer’s protocol.
Purification of Recombinant Proteins
Plasmids encoding GST and GST-SKIP604–1019 were transformed into BL21-CodonPlus (DE3) RP E. coli cells (230255, Agilent Technologies) and grown in 1 L Terrific Broth medium supplemented with antibiotics [100 μg/mL ampicillin (A1066, Sigma-Aldrich), 34 μg/mL chloramphenicol (C-6378, Sigma-Aldrich)] at 37°C, with constant shaking. Cultures were induced with 1 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) (I2481, GoldBio) and incubated for 16 h at 16°C. Bacteria were harvested by centrifugation for 20 min at 4,000 rpm and 4°C. Pellets were resuspended in buffer containing 50 mM Tris-HCl pH 8.0, 300 mM NaCl, 5% glycerol and 4 mM 1,4-dithiothreitol (DTT) (10708984001, Sigma-Aldrich) supplemented with lysozyme (VWRV0663, VWR, Radnor, PA), DNase I (LS002139, Worthington Biochemical Corporation, Lakewood, NJ) and complete EDTA-free protease inhibitor capsules (1836170, Roche). Final lysis was achieved by sonication. The soluble fraction was separated by centrifugation for 45 min at 16,000 rpm and 4°C and incubated with glutathione-Sepharose 4B (17-0756-05, GE Healthcare, Chicago, IL) for 2 h at 4 °C. Following washes with buffer (50–100 column volumes), sample was eluted with buffer composed of 50 mM Tris-HCl pH 8.0 and 10 mM glutathione. Samples were further purified by size-exclusion chromatography on a Superdex 200 10/300 column. Purified proteins were further concentrated, flash-frozen in liquid nitrogen and stored at −80 °C.
GST Pulldowns
HEK293T cells transiently transfected with plasmid encoding Myc-SKIP1–603 were lysed in 400 μL buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40, 0.09% sodium azide). Following incubation and centrifugation, the soluble fraction was diluted in 600 μL buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.018% sodium azide). Lysates (240 μL) were incubated with 10 μl glutathione-Sepharose 4B preloaded with equal amounts of bound GST and GST-SKIP604–1019. One-hundred μL of buffer (25 mM HEPES pH 7.4, 300 mM NaCl, 5mM MgCl2) was added to samples to a total reaction volume of 350 μL. Samples were incubated while rotating for 1 h at 4°C. Following incubation, the bound fraction was separated by centrifugation for 5 min at 500 × g and 4°C, and washed three times with 1 mL buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.018% sodium azide). Samples were boiled in Laemmli buffer and analyzed by SDS-PAGE and immunoblotting.
Yeast Two-Hybrid Assays
Transformation into the AH109 yeast strain was performed with EZ-YEAST™ transformation kit (112100200, MP Biomedicals, Santa Ana, CA) according to the manufacturer’s protocol, with the following modifications: after incubation at 42°C, yeast were centrifuged for 3 min at 10,000 × g, then residual PEG was removed, and the pellet was resuspended with 25 μL double-distilled water. The resuspended pellet was spread on drop-out media plates lacking leucine and tryptophan. Plates were incubated at 30°C for 4–6 days. For binding experiments, yeast colonies were resuspended in 200 μL double-distilled water. Samples were normalized to optical density at 600 nm of 0.05–0.1 with double-distilled water in a final volume of 100 μL. Samples of 4 μL were spotted onto plates lacking histidine, leucine and tryptophan supplemented with 0–30 mM 3AT (3-amino-1,2,4-triazole, Sigma-Aldrich). Plates were incubated at 30°C for 4–8 days.
Quantification and Statistical Analyses
KIF5B/Myc-SKIP and KLC2/Myc-SKIP ratios in co-immunoprecipitation experiments (Figures 4B, 4C, 6B, 6C, 6E, 6F, 6H and 6I) were calculated using ImageJ [65] of bound Myc-SKIP, KIF5B and KLC2 bands in immunoblots. Accumulation of SKIP constructs and lysosomes at cell vertices (Figures 2C, 2D, 3D, 3E, 3G and 3H) was quantified by collecting images from independent experiments at subsaturation settings and analyzing the images with ImageJ. For each channel, the intensity at the vertices was calculated relative to the total cell intensity. For untransfected cells, an equivalent region of interest at the vertices was used (~5–10% of total cell area). The region of vertices was defined as the peripheral area with accumulation of SKIP and lysosomes. Cells of similar size were selected for analysis when different cell lines were compared (Figure 3D–E). Data are presented as SuperPlots [60]. Statistical significance was calculated by one-way ANOVA followed by multiple comparisons using Dunnett’s test. Quantification of cells showing a phenotype (Figures 5F and 7B) was done by counting cells from each sample in two independent experiments. Percentages of cells showing a particular phenotype were calculated for each experiment, followed by calculation of the average percentage from the two experiments. Total numbers of cells counted are indicated on the figures.
Supplementary Material
Highlights.
SKIP is an adaptor that mediates regulated coupling of lysosomes to kinesin-1
SKIP is autoinhibited by intramolecular interaction of its N- and C-terminal parts
The GTPase ARL8 relieves SKIP autoinhibition besides recruiting SKIP to lysosomes
SKIP exemplifies the regulation of a kinesin adaptor by reversible autoinhibition
Acknowledgments
We thank Prabuddha Sengupta for gift of FKBP-RFP and FRB-GFP plasmids, Stéphane Méresse for gift of Myc-SKIP plasmid, John Brumell for gift of ARL8B-GFP plasmid, Chad Williamson for advice with microscopy, Rafael Mattera and Carlos Guardia for critical review of the manuscript, and other members of the Bonifacino lab for reagents, advice and support. We also thank the reviewers of the manuscript for their helpful suggestions. We apologize to colleagues whose work we could not cite because of space limitations. This work was funded by the Intramural Program of NICHD, NIH (ZIA HD001607).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.van Bergeijk P, Hoogenraad CC, and Kapitein LC (2016). Right Time, Right Place: Probing the Functions of Organelle Positioning. Trends Cell Biol 26, 121–134. [DOI] [PubMed] [Google Scholar]
- 2.Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, et al. (2003). Mutant dynactin in motor neuron disease. Nat Genet 33, 455–456. [DOI] [PubMed] [Google Scholar]
- 3.Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, Parrini E, Valence S, Pierre BS, Oger M, et al. (2013). Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet 45, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, Graham FL, Gaskell PC, Dearlove A, Pericak-Vance MA, et al. (2002). A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet 71, 1189–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riviere JB, Ramalingam S, Lavastre V, Shekarabi M, Holbert S, Lafontaine J, Srour M, Merner N, Rochefort D, Hince P, et al. (2011). KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet 89, 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayrakli F, Poyrazoglu HG, Yuksel S, Yakicier C, Erguner B, Sagiroglu MS, Yuceturk B, Ozer B, Doganay S, Tanrikulu B, et al. (2015). Hereditary spastic paraplegia with recessive trait caused by mutation in KLC4 gene. J Hum Genet 60, 763–768. [DOI] [PubMed] [Google Scholar]
- 7.Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer-Grumbach M, Kwon JM, FitzPatrick D, Schmedding E, De Vriendt E, Jacobs A, et al. (2003). Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet 72, 722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Ma Z, Jiao X, Fariss R, Kantorow WL, Kantorow M, Pras E, Frydman M, Pras E, Riazuddin S, et al. (2011). Mutations in FYCO1 cause autosomal-recessive congenital cataracts. Am J Hum Genet 88, 827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muhammad E, Levitas A, Singh SR, Braiman A, Ofir R, Etzion S, Sheffield VC, Etzion Y, Carrier L, and Parvari R (2015). PLEKHM2 mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction. Hum Mol Genet 24, 7227–7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charng WL, Karaca E, Coban Akdemir Z, Gambin T, Atik MM, Gu S, Posey JE, Jhangiani SN, Muzny DM, Doddapaneni H, et al. (2016). Exome sequencing in mostly consanguineous Arab families with neurologic disease provides a high potential molecular diagnosis rate. BMC Med Genomics 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neveling K, Martinez-Carrera LA, Holker I, Heister A, Verrips A, Hosseini-Barkooie SM, Gilissen C, Vermeer S, Pennings M, Meijer R, et al. (2013). Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am J Hum Genet 92, 946–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jordens I, Fernandez-Borja M, Marsman M, Dusseljee S, Janssen L, Calafat J, Janssen H, Wubbolts R, and Neefjes J (2001). The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr Biol 11, 1680–1685. [DOI] [PubMed] [Google Scholar]
- 13.Willett R, Martina JA, Zewe JP, Wills R, Hammond GRV, and Puertollano R (2017). TFEB regulates lysosomal positioning by modulating TMEM55B expression and JIP4 recruitment to lysosomes. Nat Commun 8, 1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drerup CM, and Nechiporuk AV (2013). JNK-interacting protein 3 mediates the retrograde transport of activated c-Jun N-terminal kinase and lysosomes. PLoS Genet 9, e1003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Rydzewski N, Hider A, Zhang X, Yang J, Wang W, Gao Q, Cheng X, and Xu H (2016). A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol 18, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown CL, Maier KC, Stauber T, Ginkel LM, Wordeman L, Vernos I, and Schroer TA (2005). Kinesin-2 is a motor for late endosomes and lysosomes. Traffic 6, 1114–1124. [DOI] [PubMed] [Google Scholar]
- 17.Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjorkoy G, and Johansen T (2010). FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol 188, 253–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dumont A, Boucrot E, Drevensek S, Daire V, Gorvel JP, Pous C, Holden DW, and Meresse S (2010). SKIP, the host target of the Salmonella virulence factor SifA, promotes kinesin-1-dependent vacuolar membrane exchanges. Traffic 11, 899–911. [DOI] [PubMed] [Google Scholar]
- 19.Dodding MP, Mitter R, Humphries AC, and Way M (2011). A kinesin-1 binding motif in vaccinia virus that is widespread throughout the human genome. EMBO J 30, 4523–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosa-Ferreira C, and Munro S (2011). Arl8 and SKIP act together to link lysosomes to kinesin-1. Dev Cell 21, 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pu J, Schindler C, Jia R, Jarnik M, Backlund P, and Bonifacino JS (2015). BORC, a multisubunit complex that regulates lysosome positioning. Dev Cell 33, 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guardia CM, Farias GG, Jia R, Pu J, and Bonifacino JS (2016). BORC Functions Upstream of Kinesins 1 and 3 to Coordinate Regional Movement of Lysosomes along Different Microtubule Tracks. Cell Rep 17, 1950–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niwa S, Tao L, Lu SY, Liew GM, Feng W, Nachury MV, and Shen K (2017). BORC Regulates the Axonal Transport of Synaptic Vesicle Precursors by Activating ARL-8. Curr Biol 27, 2569–2578 e2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farias GG, Guardia CM, De Pace R, Britt DJ, and Bonifacino JS (2017). BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc Natl Acad Sci U S A 114, E2955–E2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verhey KJ, Kaul N, and Soppina V (2011). Kinesin assembly and movement in cells. Annu Rev Biophys 40, 267–288. [DOI] [PubMed] [Google Scholar]
- 26.Coy DL, Hancock WO, Wagenbach M, and Howard J (1999). Kinesin’s tail domain is an inhibitory regulator of the motor domain. Nat Cell Biol 1, 288–292. [DOI] [PubMed] [Google Scholar]
- 27.Friedman DS, and Vale RD (1999). Single-molecule analysis of kinesin motility reveals regulation by the cargo-binding tail domain. Nat Cell Biol 1, 293–297. [DOI] [PubMed] [Google Scholar]
- 28.Hackney DD, and Stock MF (2000). Kinesin’s IAK tail domain inhibits initial microtubule-stimulated ADP release. Nat Cell Biol 2, 257–260. [DOI] [PubMed] [Google Scholar]
- 29.Cai D, Hoppe AD, Swanson JA, and Verhey KJ (2007). Kinesin-1 structural organization and conformational changes revealed by FRET stoichiometry in live cells. J Cell Biol 176, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaan HY, Hackney DD, and Kozielski F (2011). The structure of the kinesin-1 motor-tail complex reveals the mechanism of autoinhibition. Science 333, 883–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yip YY, Pernigo S, Sanger A, Xu M, Parsons M, Steiner RA, and Dodding MP (2016). The light chains of kinesin-1 are autoinhibited. Proc Natl Acad Sci U S A 113, 2418–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blasius TL, Cai D, Jih GT, Toret CP, and Verhey KJ (2007). Two binding partners cooperate to activate the molecular motor Kinesin-1. J Cell Biol 176, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun F, Zhu C, Dixit R, and Cavalli V (2011). Sunday Driver/JIP3 binds kinesin heavy chain directly and enhances its motility. EMBO J 30, 3416–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu MM, and Holzbaur EL (2013). JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J Cell Biol 202, 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Twelvetrees AE, Lesept F, Holzbaur ELF, and Kittler JT (2019). The adaptor proteins HAP1a and GRIP1 collaborate to activate the kinesin-1 isoform KIF5C. J Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imanishi M, Endres NF, Gennerich A, and Vale RD (2006). Autoinhibition regulates the motility of the C. elegans intraflagellar transport motor OSM-3. J Cell Biol 174, 931–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hammond JW, Blasius TL, Soppina V, Cai D, and Verhey KJ (2010). Autoinhibition of the kinesin-2 motor KIF17 via dual intramolecular mechanisms. J Cell Biol 189, 1013–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niwa S, Lipton DM, Morikawa M, Zhao C, Hirokawa N, Lu H, and Shen K (2016). Autoinhibition of a Neuronal Kinesin UNC-104/KIF1A Regulates the Size and Density of Synapses. Cell Rep 16, 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siddiqui N, Zwetsloot AJ, Bachmann A, Roth D, Hussain H, Brandt J, Kaverina I, and Straube A (2019). PTPN21 and Hook3 relieve KIF1C autoinhibition and activate intracellular transport. Nat Commun 10, 2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishida M, Ohbayashi N, and Fukuda M (2015). Rab1A regulates anterograde melanosome transport by recruiting kinesin-1 to melanosomes through interaction with SKIP. Sci Rep 5, 8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pernigo S, Lamprecht A, Steiner RA, and Dodding MP (2013). Structural basis for kinesin-1:cargo recognition. Science 340, 356–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanger A, Yip YY, Randall TS, Pernigo S, Steiner RA, and Dodding MP (2017). SKIP controls lysosome positioning using a composite kinesin-1 heavy and light chain-binding domain. J Cell Sci 130, 1637–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jackson LK, Nawabi P, Hentea C, Roark EA, and Haldar K (2008). The Salmonella virulence protein SifA is a G protein antagonist. Proc Natl Acad Sci U S A 105, 14141–14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi J, Chen J, Schreiber SL, and Clardy J (1996). Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 273, 239–242. [DOI] [PubMed] [Google Scholar]
- 45.Diacovich L, Dumont A, Lafitte D, Soprano E, Guilhon AA, Bignon C, Gorvel JP, Bourne Y, and Meresse S (2009). Interaction between the SifA virulence factor and its host target SKIP is essential for Salmonella pathogenesis. J Biol Chem 284, 33151–33160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lemmon MA (2007). Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levin-Konigsberg R, Montano-Rendon F, Keren-Kaplan T, Li R, Ego B, Mylvaganam S, DiCiccio JE, Trimble WS, Bassik MC, Bonifacino JS, et al. (2019). Phagolysosome resolution requires contacts with the endoplasmic reticulum and phosphatidylinositol-4-phosphate signalling. Nat Cell Biol 21, 1234–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huynh W, and Vale RD (2017). Disease-associated mutations in human BICD2 hyperactivate motility of dynein-dynactin. J Cell Biol 216, 3051–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo X, Farias GG, Mattera R, and Bonifacino JS (2016). Rab5 and its effector FHF contribute to neuronal polarity through dynein-dependent retrieval of somatodendritic proteins from the axon. Proc Natl Acad Sci U S A 113, E5318–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raiborg C, Wenzel EM, Pedersen NM, Olsvik H, Schink KO, Schultz SW, Vietri M, Nisi V, Bucci C, Brech A, et al. (2015). Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature 520, 234–238. [DOI] [PubMed] [Google Scholar]
- 51.Cantalupo G, Alifano P, Roberti V, Bruni CB, and Bucci C (2001). Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J 20, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Horgan CP, Hanscom SR, Jolly RS, Futter CE, and McCaffrey MW (2010). Rab11-FIP3 links the Rab11 GTPase and cytoplasmic dynein to mediate transport to the endosomal-recycling compartment. J Cell Sci 123, 181–191. [DOI] [PubMed] [Google Scholar]
- 53.Boucrot E, Henry T, Borg JP, Gorvel JP, and Meresse S (2005). The intracellular fate of Salmonella depends on the recruitment of kinesin. Science 308, 1174–1178. [DOI] [PubMed] [Google Scholar]
- 54.Pantakani DV, Czyzewska MM, Sikorska A, Bodda C, and Mannan AU (2011). Oligomerization of ZFYVE27 (Protrudin) is necessary to promote neurite extension. PLoS One 6, e29584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alborghetti MR, Furlan AS, Silva JC, Paes Leme AF, Torriani IC, and Kobarg J (2010). Human FEZ1 protein forms a disulfide bond mediated dimer: implications for cargo transport. J Proteome Res 9, 4595–4603. [DOI] [PubMed] [Google Scholar]
- 56.Wu M, Wang T, Loh E, Hong W, and Song H (2005). Structural basis for recruitment of RILP by small GTPase Rab7. EMBO J 24, 1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kelkar N, Gupta S, Dickens M, and Davis RJ (2000). Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol Cell Biol 20, 1030–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watt D, Dixit R, and Cavalli V (2015). JIP3 Activates Kinesin-1 Motility to Promote Axon Elongation. J Biol Chem 290, 15512–15525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horgan CP, Oleksy A, Zhdanov AV, Lall PY, White IJ, Khan AR, Futter CE, McCaffrey JG, and McCaffrey MW (2007). Rab11-FIP3 is critical for the structural integrity of the endosomal recycling compartment. Traffic 8, 414–430. [DOI] [PubMed] [Google Scholar]
- 60.Lord SJ, Velle KB, Mullins RD, and Fritz-Laylin LK (2020). SuperPlots: Communicating reproducibility and variability in cell biology. J Cell Biol. 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kapitein LC, Schlager MA, van der Zwan WA, Wulf PS, Keijzer N, and Hoogenraad CC (2010). Probing intracellular motor protein activity using an inducible cargo trafficking assay. Biophys J 99, 2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- 63.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jia R, Guardia CM, Pu J, Chen Y, and Bonifacino JS (2017). BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy 13, 1648–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Combet C, Blanchet C, Geourjon C, and Deleage G (2000). NPS@: network protein sequence analysis. Trends Biochem Sci 25, 147–150. [DOI] [PubMed] [Google Scholar]
- 68.Yachdav G, Kloppmann E, Kajan L, Hecht M, Goldberg T, Hamp T, Honigschmid P, Schafferhans A, Roos M, Bernhofer M, et al. (2014). PredictProtein--an open resource for online prediction of protein structural and functional features. Nucleic Acids Res 42, W337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rost B, Yachdav G, and Liu J (2004). The PredictProtein server. Nucleic Acids Res 32, W321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zimmermann L, Stephens A, Nam SZ, Rau D, Kubler J, Lozajic M, Gabler F, Soding J, Lupas AN, and Alva V (2018). A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J Mol Biol 430, 2237–2243. [DOI] [PubMed] [Google Scholar]
- 71.Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.