

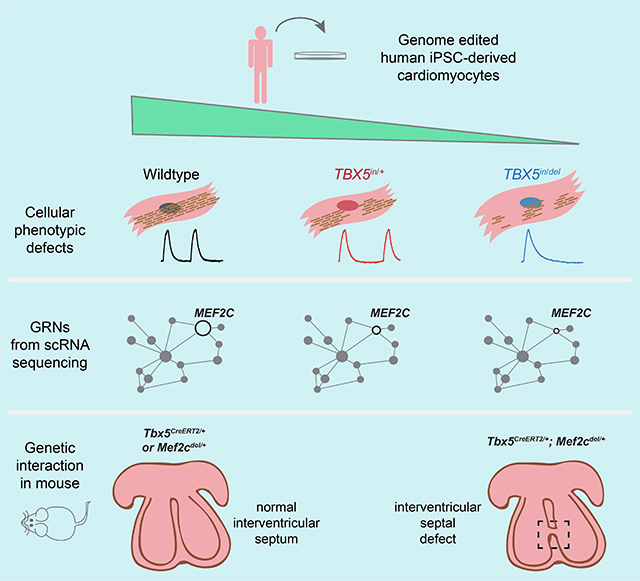
Summary
Haploinsufficiency of transcriptional regulators causes human congenital heart disease (CHD). However, underlying CHD gene regulatory network (GRN) imbalances are unknown. Here, we define transcriptional consequences of reduced dosage of the CHD transcription factor, TBX5, in individual cells during cardiomyocyte differentiation from human induced pluripotent stem cells (iPSCs). We discovered highly sensitive dysregulation of TBX5-dependent pathways—including lineage decisions and genes associated with heart development, cardiomyocyte function, and CHD genetics—in discrete subpopulations of cardiomyocytes. Spatial transcriptomic mapping revealed chamber-restricted expression for many TBX5-sensitive transcripts. GRN analysis indicated that cardiac network stability, including vulnerable CHD-linked nodes, is sensitive to TBX5 dosage. A GRN-predicted genetic interaction between Tbx5 and Mef2c was validated in mouse, manifesting as ventricular septation defects. These results demonstrate exquisite and diverse sensitivity to TBX5 dosage in heterogeneous subsets of iPSC-derived cardiomyocytes, and predicts candidate GRNs for human CHDs, with implications for quantitative transcriptional regulation in disease.
Keywords: Gene regulation, transcription factor, gene dosage, haploinsufficiency, gene regulatory networks, cardiomyocyte differentiation, congenital heart disease
eTOC
Reduced cardiac transcription factor dosage causes congenital heart disease. Kathiriya et al. interrogate genome-edited human iPSC-derived cardiomyocytes to model TBX5 haploinsufficiency. Single cell RNA-seq reveals discrete cell types with altered expression of disease-related genes. Gene regulatory networks disrupted by reduced TBX5 dosage identify nodes for congenital heart disease.
Graphical Abstract
Introduction
CHDs are a leading cause of childhood morbidity and mortality, and incidence of CHDs is estimated to be ten-fold higher in human fetuses of spontaneous termination (Hoffman, 1995; Hoffman and Kaplan, 2002). Many human mutations linked to CHDs are predicted to result in reduced dosage of transcriptional regulators, including transcription factors (TFs) and chromatin-modifying genes (Zaidi and Brueckner, 2017). Despite advances to elucidate the roles of individual factors in heart development and CHDs, how dosage of transcriptional regulators translates to altered GRNs is not known.
Heterozygous mutations in the T-box TF gene TBX5 cause Holt-Oram syndrome (HOS) (Basson et al., 1997; Li et al., 1997), which uniformly presents with upper limb defects and often with CHDs that include ventricular or atrial septal defects (VSD or ASDs), diastolic dysfunction, and arrhythmias. The majority of TBX5 mutations in patients with HOS, whether they are heterozygous null or missense mutations, are predicted to cause loss of function (LOF) (Mori and Bruneau, 2004) and do not predict phenotypes (Brassington et al., 2003). Thus, heterozygous TBX5 mutations lead to haploinsufficiency in the developing heart and limbs. Experiments in mice have revealed a stepwise sensitivity to reductions in TBX5 dosage (Bruneau et al., 2001; Mori et al., 2006). In humans, homozygous LOF mutations of TBX5 are not observed in the genome aggregation database of exomes and genomes from large-scale sequencing projects (Karczewski et al., 2020) and is presumed to cause fetal demise. Tbx5 null mice die of embryonic lethality from severely deformed hearts (Bruneau et al., 2001). These findings demonstrate that a reduction in TBX5 dosage perturbs downstream gene expression. However, the disrupted regulatory networks and mechanisms are not understood.
To build upon findings from mouse models, a tractable human model system is required to study human TBX5 haploinsufficiency. Human heart tissue from normal, living individuals is largely inaccessible for molecular analysis. As the estimated prevalence of Holt-Oram syndrome is 1:100,000, pathological or surgical specimens from affected patients are very limited. Genome editing in human induced pluripotent stem (iPS) cells enables targeted genetic manipulations in an isogenic background. These targeted mutant iPSCs can be differentiated into varied cardiac cell types, including cardiomyocytes, and then subjected to single cell RNA sequencing (RNA-seq). This in vitro system provides a human cellular platform for gene-centered cardiac disease modeling at single cell resolution. Although iPSC-derived cardiomyocyte differentiation lacks a three-dimensional context for patterning and organization of myriad cell types of the heart, it recapitulates key developmental steps, including mesoderm and cardiac precursors. Despite some caveats, human iPSC-derived cardiomyocytes can be used to discover molecular and genetic insights that may inform structural cardiac defects (Theodoris et al., 2015; Ang et al., 2016; Miao et al., 2020).
In considering how TBX5 haploinsufficiency might cause CHDs, at least two scenarios are possible. 1. Reduced dosage may only affect genes in vulnerable cell types in specific anatomical locations, such as the ventricular septum. 2. Reduced dosage may affect cardiac gene expression broadly, but altered programs manifest as morphologic defects only in anatomic structures most sensitive to the disturbance. The first scenario would be challenging to investigate in two-dimensional cultures, particularly if susceptible region-specific cell types are absent. The second predicts that changes in gene expression might be detected by bulk RNA-seq studies of heterozygous human iPS cell models of CHDs (Theodoris et al., 2015; Ang et al., 2016; Gifford et al., 2019), but relevant, discrete alterations in a complex cell mixture could be missed. Discerning between these scenarios requires a single cell analysis approach.
Here, we used an allelic series of TBX5 in engineered human iPS cells, comprising wildtype, and heterozygous or homozygous loss of function mutations, to investigate GRNs that are altered in response to reduced TBX5 dosage. We observed TBX5 dose-dependent cellular phenotypes reminiscent of anomalies in patients with TBX5 mutations. We deployed single cell RNA-seq (scRNA-seq) across a time course of differentiation and discovered discrete gene expression responses to reduced TBX5 dosage in cardiomyocyte subpopulations. From these data, we identified putative cardiac GRNs that help explain several cellular and developmental phenotypes related to human CHD. We validated one of these GRN-predicted genetic interactions in mice: Tbx5 and Mef2c interact to cause muscular VSDs, a common type of human CHD. We conclude that TBX5 dosage sensitivity, modeled in human iPS cells, reveals discrete gene regulation programs in an unanticipated variety of cardiomyocyte subtypes and informs the biology of human CHD.
Results
Impaired human cardiomyocyte differentiation and function by reduced TBX5 dosage
To determine a role for TBX5 dosage in human cardiac biology, we created an isogenic allelic series of human iPS cells mutant for TBX5, using CRISPR/Cas9-mediated genome editing to target exon 3 of TBX5 at the start of the essential T-box domain (Figure S1A, B). We isolated targeted iPS cell lines, including heterozygous (TBX5 in/+) and homozygous (TBX5in/del and TBX5PuR/PuR) mutants (Figure 1A, S1C–F). We also isolated a control (TBX5+/+) iPS cell line, which was exposed to CRISPR/Cas9 nuclease but not mutated at exon 3 of TBX5, to control for off-target effects and the sub-cloning procedure. Subsequently, we refer to wildtype and control collectively as “WT” when significant differences between them were not observed. TBX5 protein levels in cardiomyocytes differentiated from these lines were diminished in TBX5in/+ cells and absent in TBX5in/del and TBX5PuR/PuR cells (Figure 1B), consistent with a dosage-step allelic series of mutant TBX5 loss-of-function cell lines.
Figure 1. A human allelic series of TBX5 mutants model features of congenital heart disease.

(A) Experimental cell lines and strategy. (B) TBX5 and cTNT protein expression at day 15. (C) Differentiation efficiency by flow cytometry for cTNT+ cells (* p<0.05 by unpaired t test). (D) Onset of beating (** p<0.01, *** p<0.001 by unpaired t test). (E-I) Myofibrillar arrangement of cardiomyocytes (* p<0.05, **** p<0.0001 by Fisher’s exact test, compared to control). (J) Cell size (*** p<0.001 by unpaired t-test, compared to control). (K) Traces of calcium transients. (L) Time at 90% decay (t90 down) (* FDR<0.05, ** FDR<0.01). Error bars represent standard deviation (J, K) or standard error (L, M) of the mean.
Given an inherent variability of the directed differentiation system, we observed reduced cardiomyocyte differentiation efficiency and a delay in onset of beating by loss of TBX5, when compared to WT (Figure 1C, D). Worsening sarcomere disarray was seen by stepwise depletion of TBX5 (Figure 1E–I, S1G). We measured cell size of CMs, and TBX5in/del was larger than WTC11 and Control (Figure 1J). Notably, TBX5in/+ cell size was normal.
Patch clamp analysis of cardiomyocytes, which were predominantly ventricular in this differentiation method, revealed lengthened action potential duration (APD) in TBX5in/del cells (Figure S1H, I), consistent with previous findings (Karakikes et al., 2017; Churko et al., 2018). Although TBX5PuR/PuR cells showed high variability of APDs and were not statistically significantly different from WT, some recordings were distinctly abnormal, displaying striking APDs eight times greater than an average wildtype or control cell (Figure S1H, I). Calcium imaging of spontaneously beating cardiomyocytes revealed protracted calcium transient durations in TBX5in/del and TBX5PuR/PuR cells, with an intermediate defect in TBX5in/+ cells (Figure 1L, M), implying a potential impairment of cardiomyocyte relaxation. Together, these cellular findings recapitulated several pathological and physiological characteristics, which may contribute to diastolic dysfunction in HOS in mice and humans (Zhou et al., 2005; Zhu et al., 2008), and reflect generally impaired cellular differentiation.
Resolving susceptible cardiac cell types from reduced TBX5 dosage
To determine how TBX5 dosage alters gene expression in a heterogeneous cell population, we used a droplet-based single cell RNA-seq method with cells collected from parental WTC11, control TBX5+/+, and mutant TBX5 (TBX5in/+, TBX5in/del) genotypes. From three time points during cardiomyocyte differentiation, we interrogated 55,782 cells with an average normalized read depth of 88,234 reads per cell (Figure 2A–C). These datasets can viewed at the UCSC cell browser (https://cardiac-differentiation.cells.ucsc.edu). At day 6, we identified 11 cell clusters, representing at least four cell types, including POU5F1+ mesodermal cells, MESP1+ pre-cardiac mesoderm, ISL1+ cardiac precursors, and nascent TNNT2+ cardiomyocytes (Figure 2D). At day 11 and day 23, differentiated cell types were assigned and present in all genotypes (Figure 2E–H, S2A, B), based on cell type-specific marker genes (DeLaughter et al., 2016; Li et al., 2016). This included a diversity of TBX5+ cell types, comprising very few PLVAP+ endothelial cells or TTR+ endodermal cells, some COL1A1+ fibroblasts and, most abundantly, TNNT2+/IRX4+ ventricular cardiomyocytes (Figure 2E–G).
Figure 2. Human cardiomyocyte differentiation is sensitive to reduced TBX5 dosage.

(A-C) UMAPs display cells of TBX5 genotypes at day 6, day 11 or day 23. (D-F) Feature plots of selected marker genes, which represent major cell types at each timepoint. (G) Cell type assignments of iPSC-derived cells at day 23 by manual annotation. (H) Manhattan plot displays differentially expressed genes by cell type cluster at day 23. Example genes by cluster are shown. Manual annotation was based on expression of bolded genes. (I) Classifier prediction of cell types at day 23. Inset: Prediction probabilities. (J) Prediction probabilities of iPSC-derived cells classified as ventricular cardiomyocytes (vCM), atrial cardiomyocytes (aCM) or mixed (<0.95 probability difference of vCM and aCM). ** p<0.01, *** p<0.001, **** p<0.0001 by Fisher’s exact test.
We employed a machine learning algorithm (Pedregosa et al., 2011), to quantitatively evaluate the degree of similarity, if any, between iPSC-derived cells and cells from the developing human heart. A cell type classifier was trained on single cell gene expression from human fetal heart at 6.5–7 weeks gestation (Figure S2C) (Asp et al., 2019). This was used to predict cell type assignments for cells harvested at day 23 (Figure 2I, S2D, Table S1). Ventricular-like cardiomyocytes were the most commonly predicted cell type, constituting 52% of cells from all genotypes, with a high prediction probability average of 0.93, consistent with manual assignments by cell type-specific markers genes, such as TNNT2 and IRX4 (Figure 2G, I, S2B, D, E). Twenty-three percent of cells were assigned as fibroblast-like cells with 0.89 probability, 6% as epicardial-like cells with 0.89 probability, and 7% as cardiac cells of neural crest origin with 0.72 probability (Figure 2I, S2D, E). AFP+ or TTR+ cells, considered to be derived from endoderm or mesendoderm, were dispersed across several predicted cell assignments (Figure 2I, S2D). As expected, differentiation did not yield many other iPSC-derived cell types, including erythrocytes (1%) and immune cells (0.2%), which were sparsely represented with less than 0.5 prediction probability (Figure 2I, S2D). Taken together, the classifier’s predictions appeared to provide sufficient fidelity for assignments of iPSC-derived cells as in vivo-like cardiac cell types.
Although the cell type classifier was largely consistent with cell type assignments from manual annotations (Figure 2G, I, S2D), it predicted 9% of iPSC-derived cells from all genotypes as atrial-like cardiomyocytes with a 0.83 probability. Whereas the cell type classifier predicted a similar total number of high-probability (>0.7) cardiomyocytes for each TBX5 genotype (Figure S2E), more “atrial-like” cardiomyocytes were predicted for TBX5in/+ and TBX5in/del cells (p<0.0001 by Fisher’s exact test), than for WT (Figure S2E). Genes enriched in “atrial-like” cells included markers of the atrioventricular canal, such as TBX2, RSPO3 and BMP2. The classifier also uncovered a population of iPSC-derived cardiomyocytes with ‘mixed’ identity, of both ventricular- and “atrial-like” predictions. These ‘mixed’ cardiomyocyte predictions were more prevalent among TBX5in/+ (24.3%) and TBX5in/del cells (48%), than wildtype (20.9%) or control (17.0%) (Figure 2J). Predicted doublets by DoubletFinder (McGinnis et al., 2019) were distributed throughout the dataset and were not enriched in those cells predicted to be of ‘mixed’ identity (Figure S2F). This supports a notion that reduced TBX5 dosage may broadly perturb ventricular cardiomyocyte gene expression programs.
TBX5 protects human ventricular cardiomyocyte fate
To assess if reduced TBX5 dosage disturbs paths of directed differentiation, we evaluated supervised URD trajectories from all TBX5 genotypes and time points. URD predicts cellular chronology based on user-determined origins and ends (Farrell et al., 2018). We defined POU5F1+ cells, which were predominantly from a single cluster at day 6 (Figure 2D), as the root and day 23 clusters as the tips in the pseudotime tree (Figure 3A–E). Cells at day 6 were found near the top of the tree, while cells at day 11 were distributed mid-way, followed by day 23 cells at the user-defined tips (Figure S3A–C). This demonstrated a logical ordering of cells along pseudotime by URD. Since TBX5 transcripts were detected in all genotypes, including TBX5in/del cells, inferred lineage decisions for TBX5+ cell types in the absence of TBX5 could be examined. We focused on inferred trajectories of TBX5+ cells to ventricular cardiomyocytes. TBX5in/+ cells followed a path similar to WT (Figure 3B, C, dashed lines), but to a transcriptionally distinct endpoint. In contrast, TBX5in/del cells deviated from the WT differentiation path to ventricular cardiomyocytes (Figure 3D).
Figure 3. TBX5 loss disturbs cell trajectories to ventricular cardiomyocyte fate.

(A) URD developmental trajectories for day 6, 11 and 23 of cardiomyocyte differentiation. Pseudotime is displayed from root (top) to tips (bottom). TBX5 genotype is color-coded from light to dark, to indicate the time point. (B-D) Cells are highlighted by TBX5 genotype on the aggregate pseudotime dendrogram. Arrowheads: Enrichment or depletion of cells from one genotype at certain branch points to tips. Dashed lines show path to cardiomyocytes. (E) Expression of genes that define the major cell types. (F) Deduced paths to cardiomyocytes of WT (black dashed line) or TBX5in/del (blue dashed line), from intermediates (‘interm’) to tips. (G) Expression for each gene that displays no correlation with pseudotime in the WT path (above), but a positive or negative correlation (|rho|≥0.4, p<0.05 by two-sided t test, and Z-score≥15 by difference in rho) in the TBX5in/del path (below). (H) Feature plots for ventricular cardiomyocyte genes HAND1 and HEY2, and atrioventricular canal genes TBX2 and RSPO3. (I, J) Fluorescent in situ hybridization and quantification of cell numbers (** p<0.01 by Fisher’s exact test) for HAND1 or HEY2, or gene expression intensity for RSPO3 or TBX2 (**** p<0.0001 by unpaired t-test) of TNNT2+ cells. Scale bar: 50 microns. (K) Differential gene expression of inferred precursors for the cardiomyocyte branches (dashed ovals) (adj p<0.05 by Wilcoxon Rank Sum test). (L) Feature plots for IRX4 (absent in TBX5in/del path), SLN (enriched in TBX5in/del path), and WNT2 and NKX2–5 (delayed in TBX5in/del).
In order to explore gene expression changes that may have led to this deviation, we identified genes that change as a function of pseudotime in the WT or TBX5in/del paths (Figure 3F). We deduced 22 genes (e.g. electrophysiology-related NAV1 and TECRL, cardiomyopathy-associated LAMA4, and small peptide hormone NPPA), which were positively correlated with pseudotime in the WT/TBX5in/+ branch, but aberrant in the TBX5in/del branch, suggesting that these genes were not activated properly in TBX5in/del cells (Figure S3D). Conversely, five genes were negatively correlated in the WT/TBX5in/+ branch, but not in the TBX5in/del branch. Likewise, we identified 18 genes that positively (e.g. sarcomere DES, vascular adhesion VCAM1, and TF LBH) or negatively (e.g. TF HES1 and actomyosin binding CALD1) correlated with pseudotime in TBX5in/del cells, but were altered in wildtype cells (Figure 3G), signifying that these genes were inappropriately deployed in TBX5in/del cells.
A few ventricular markers (e.g. cardiac TFs IRX4 and HEY2) were reduced in TBX5in/del cells (Figure 3H, L), reminiscent of features from mouse (Bruneau et al., 2001). However, TBX5in/del cells still expressed other ventricular-enriched genes (e.g. cardiac TFs HAND1 and HAND2) (Figure 3H), consistent with their electrophysiologic characteristics as ventricular cardiomyocytes (Figure S1H). TBX5in/del cells also expressed markers of the atrioventricular canal (e.g. cardiac TF TBX2 and Wnt agonist RSPO3) (Figure 3H). Fluorescence in situ hybridization demonstrated that the quantity of TNNT2+/HAND1+ cells were unchanged in TBX5in/del, while TNNT2+/HEY2+ cells were reduced in number (Figure 3I, J). Intensity of expression of RSPO3 or TBX2 was also increased in TNNT2+ cells in TBX5in/del (Figure 3I, J). This suggests that TBX5 loss results in a disordered ventricular cardiomyocyte-like identity with ectopic gene expression. It is also possible that the absence of TBX5 expression led to differentiation of precursors that become cells of AV canal or outflow tract identity.
We tested differential gene expression between intermediate branches, to identify genes that determine TBX5-dependent ventricular cardiomyocyte differentiation. We considered these branches as potential precursors proximal to TBX5 genotype-specific tips. We compared these intermediate branches of cells that distinguish the cell trajectory route of WT and TBX5in/+ to TBX5in/del (Figure 3K). These included secreted factors or cell surface receptors (WNT2, FGFR1) and cardiac TFs (IRX4, HAND2). Of note, expression of the CHD cardiac transcription factor NKX2–5, a transcriptional partner of TBX5 (Bruneau et al., 2001; Hiroi et al., 2001; Luna-Zurita et al., 2016), was differentially expressed between genotype-enriched intermediate branches of the URD tree (Figure 3L). Onset of NKX2–5 expression was delayed in TBX5in/del cells. In conjunction, a module of genes (chromatin regulator PARP1, ribosome RPL37, junctional protein encoding KIAA1462 and Na+/K+ transport ATP1A1), were delayed concomitantly with NKX2–5 (Figure S3E, F). Delayed NKX2–5 expression could provide a potential molecular explanation, possibly via HEY2 (Anderson et al., 2018a), for the observed delay in the onset of beating by TBX5 loss (Figure 1D), and for the emergence of a mixed identity (Li et al., 2016). It is important to note that the instability of cell types is only highly pronounced with the complete loss of TBX5. Heterozygous mutant TBX5 cardiomyocytes largely preserve their identity.
Discrete transcriptional responses to reduced TBX5 dosage in cardiomyocytes
TBX5 genotype-specific clusters emerged among cardiomyocytes at day 11 (Figure 2B), and TBX5 genotype-specific segregation was more striking at day 23, particularly in TNNT2+ cells but not in other cell types (Figure 2C). This was observed in analyses of two independent experiments or as an integrated dataset corrected for batch effects between biological replicates (Figure 2C). Therefore, we focused on TNNT2+ clusters at day 23 (Figure 4A, B). First, we used a low resolution Louvain clustering to assess genes that are highly differential between TBX5 genotype-driven TNNT2+ clusters (Figure 4C). We detected 121 genes that were differentially expressed between WT and TBX5in/+-enriched clusters (Figure 4D, Table S2). Five hundred twenty genes showed differential expression between WT and TBX5in/del-enriched clusters (Figure 4E, Table S2). To identify stepwise TBX5 dose-dependent genes, we evaluated genes that were differentially expressed between WT vs. TBX5in/+-enriched clusters and WT vs. TBX5in/del-enriched clusters. We found 85 genes common to both lists with a multitude of expression patterns (Figure 4F, Table S2). Many genes displayed changes in both expression level and percentage of expressing cells (e.g. small peptide hormone NPPA, Wnt agonist RSPO3, arrhythmia-linked TECRL, sarcomere-associated DES) (Figure 4F). A few genes showed similar levels of gene expression, with changes to percentage of expressing cells (e.g. serine hydrolase MGLL or CHD TF ANKRD1 in TBX5in/+-enriched clusters). Some genes, such as NPPA, were highly sensitive to TBX5 dosage, with reduced expression in TBX5in/+ nearly comparable to that in TBX5in/del. In contrast, TECRL was partly reduced in TBX5in/+ cells and was further decreased in TBX5in/del. Notably, some genes were altered in TBX5in/+ cells but had elevated levels in TBX5in/del cells (e.g. TBX5, myosin light chain MYL9, cardiac TF HOPX, sarcomere DES). Specifically, TBX5 expression likely reflected apparent upregulation of non-mutated exons in TBX5in/del cells. Importantly, TBX5 protein expression was not detected (Figure 1B). In mouse, targeting exon 3 of Tbx5 leads to an upregulation of surrounding exons in TBX5 mutants (Mori et al, 2006). We presume that a similar phenomenon for the analogous targeting strategy in humans may be occurring. We speculate that expression of other genes with potentially counterintuitive behavior, such as DES, may reflect a type of regulatory network compensation or overcompensation, or perhaps indicate a disordered cell type.
Figure 4. Subsets of cardiomyocytes respond discretely by quantitative transcriptional perturbations to reduced TBX5 dosage.

(A-B) TNNT2+ clusters from day 23. UMAP shows cells colored by biological replicate (A) or TBX5 genotype (B). (C) TBX5 genotype-dominant clusters segregated at low resolution of Louvain clustering. (D) Clusters enriched for WT or TBX5in/+ were compared by differential gene expression. Top five upregulated or downregulated genes are displayed. EP (orange) or CHD (purple) genes are shown. *: Transcriptional regulators; bold: predicted direct targets of TBX5 (Table S5). Dot size corresponds to the percentage of cells expressing the gene in a cluster, while the color intensity represents scaled expression values in a cluster. Significance was determined by Wilcoxon Rank Sum test (adj p<0.05). (E) Clusters enriched for WT or TBX5in/del were compared by differential gene expression. (F) Common genes differentially expressed between WT- vs. TBX5in/+ clusters and WT vs. TBX5in/del clusters are shown. (G) Fluorescence in situ hybridization for TNNT2 (green) or NPPA (red) in day 23 cardiomyocytes. Scale bar: 100 microns. (H) Mean intensity of NPPA signal of individual double-positive TNNT2+/NPPA+ cells by TBX5 genotype (* p<0.05, **** p<0.0001 by unpaired t test). (I-K) Flow cytometry for cTNT+ or DES+ cells (**** p<0.0001 by Chi-Square test). (L) UMAP of higher resolution of Louvain clustering. (M) Relatedness of clusters using PC space. The proportion of cells in each cluster are colored by TBX5 genotype. (N) Hierarchically-sorted enriched genes in clusters 6 or 10. (O, Q, S, U) UMAPs highlight clusters used for pairwise comparisons. (P,R,T,V) Top five differentially expressed upgregulated or downregulated genes, with EP or CHD genes, from cluster pairs above in UMAPs.
We used orthogonal assays at single cell resolution to validate examples of TBX5-dependent genes. TBX5 dosage-dependent downregulation of NPPA was evident in cardiomyocytes by fluorescent in situ hybridization (Figure 4G, H), consistent with the TBX5-dependent rheostatic regulation of Nppa in mouse (Bruneau et al., 2001; Mori et al., 2006). By flow cytometry, DES protein was reduced in TBX5in/+ and upregulated in TBX5in/del cardiomyocytes, compared to wildtype (Figure 4I–K), corroborating this pattern of TBX5 dose-dependent expression.
To assess the heterogeneity among cardiomyocytes, we used a higher resolution Louvain clustering and constructed a phylogenetic cluster tree relating 16 different TNNT2+ cell clusters (Figure 4L, M). We considered these clusters as putative functional subpopulations of cardiomyocytes, since they could not be classified based on a conventional anatomy-based categorization. We found two clusters (clusters 6 and 10) that included a similar proportion of cells from each TBX5 genotype, implying that these putative cardiomyocyte subpopulations may be insensitive to reduced TBX5 dosage (Figure 4M, N). We then searched for differentially expressed genes by pairwise comparisons of related subpopulations between TBX5 genotypes (Figure 4O–V). For example, cluster 5 contains WT and TBX5in/+ cells (Figure 4Q), suggesting that these TBX5 heterozygous cells are indistinguishable from a subpopulation of WT. In contrast, cluster 7 is largely composed of TBX5in/+, suggesting that these TBX5 heterozygous cells are distinct. In addition to stepwise TBX5 dose-dependent genes (Figure 4F) that were often altered in many cluster-to-cluster comparisons, we detected additional common changes in gene expression amongst pairwise cluster comparisons of WT vs. TBX5in/+ clusters. These included the cardiac TF FHL2, the cardiomyopathy-linked sarcomere gene TTN, and the ventricular-enriched sarcomere gene MYL2 (Figure 4Q–V). We also discerned many differences in gene expression based on cluster-specific comparisons, implying varied transcriptional responses among subpopulations of cardiomyocytes to TBX5 haploinsufficiency (Figure 4Q–V) or TBX5 loss (Figure 4O, P).
HOS patients commonly have arrhythmias, and TBX5 is strongly associated with arrhythmias in GWAS (Pfeufer et al., 2010; Smith et al., 2011; Ellinor et al., 2012; Cordell et al., 2013a; 2013b; Hoed et al., 2013; Hu et al., 2013). Differentially expressed gene sets at day 23 were enriched for electrophysiology (EP) genes (Figure 4, Table S2–3), which are implicated in membrane depolarization (SCN5A), calcium handling (RYR2, ATP2A2, and PLN) and arrhythmias (TECRL) (Figure 4). These genes provide a molecular explanation for the EP defects observed upon TBX5 mutation. In addition, some TBX5-dependent genes that were previously associated with arrhythmias by genome-wide association studies (GWAS) were identified (Pfeufer et al., 2010; Smith et al., 2011; Ellinor et al., 2012; Cordell et al., 2013a; 2013b; Hoed et al., 2013; Hu et al., 2013). We uncovered 45 reported genes for arrhythmias (for example, PLN, HCN4, SCN5A, GJA1, PITX2 and TECRL; FDR<0.05) among TBX5-sensitive genes (Table S2–4).
In addition to genes related to sarcomere function and EP, several genes involved in myriad aspects of cardiac differentiation and morphogenesis had altered expression by reduced TBX5, and many encode genes implicated in CHD (Figure 4, Table S2–4) (McCulley and Black, 2012; Zaidi et al., 2013; Lalani and Belmont, 2014; Prendiville et al., 2014; Homsy et al., 2015; Priest et al., 2016; Sifrim et al., 2016; Jin et al., 2017). These included reduced expression of the cardiac TFs IRX4, HEY2, HAND2, and MYOCD. Also, of note, there was increased expression of TBX2, BMP2, BAMBI and HOPX. Several other regulators of heart development had altered expression (Table S2, S3). Furthermore, IGFBP7, MYH7B and SMCHD1 from CHD-GWAS were altered among TBX5-dependent genes. This dysregulation of important mediators of cardiac differentiation and morphogenesis indicate links to morphologically-relevant regulatory pathways.
Next, we examined the expression of TBX5-vulnerable genes from iPSC-derived cardiomyocytes during human fetal heart development using the dataset from (Asp et al 2019). The low resolution of the spatial transcriptomics data does not afford cell type specificity, but robustly provides relative locations of gene expression within a tissue sample. TBX5-sensitive genes were categorized based on spatiotemporally-conserved anatomic classifications from published spatial transcriptomics of human fetal hearts at 4.5–5, 6.5, and 9 weeks post-conception (Figure 5A–D, Table S4) (Asp et al., 2019). While some TBX5-dependent genes were uniformly expressed in the fetal human heart, many had expression restricted or enriched to specific anatomical regions. For example, COL2A1, GJA5 and PRKAG2 were enriched in the atria, IRX4 and HEY2 in the ventricle; MYOCD, NPPA, and SMYD1 in the myocardium of atria and the compact and trabecular layers of the ventricles; TBX2, SMAD6, and GATA4 in the AVC; and JAG1 and FN1 in the outflow tract (Figure 5C–J). Many of these are consistent with expression data from mouse, but some expression patterns were not previously known. This analysis provides an anatomic context of TBX5-sensitive gene expression during human cardiac morphogenesis, which may be relevant in the setting of HOS, or more broadly for CHDs.
Figure. 5. Anatomic context of TBX5-sensitive genes during human cardiac development.

(A) Histology from a human fetal heart at 6.5 PCW (Asp et al 2019). (B) An anatomic map of clusters from spatial gene expression data. (C, D) Heatmaps of downregulated or upregulated TBX5-dependent genes display expression (Z-scores) within clusters corresponding to anatomic regions from Asp et al., 2019. Selected TBX5-dependent genes for major anatomic clusters are shown. (E-I) Expression profiles by spatial position for selected TBX5-dependent genes. (J, K) Curated terms from gene set enrichment analyses of downregulated or upregulated TBX5-dependent genes. PCW: post-conceptual weeks, RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle. *: Transcriptional regulators.
We assessed if TBX5 dose-sensitive genes were largely direct or indirect targets of TBX5, by examining TBX5 occupancy in human iPSC-derived CMs from a published dataset (Ang et al., 2016). We found correlations of TBX5 occupancy near TBX5 dosage-vulnerable gene sets at day 23 (Figure 4, bolded genes; Figure S4A–F, Table S5). For example, 61 of 85 genes that showed stepwise dose-dependence were near TBX5 binding sites (Figure 4F, S4A), suggesting that these genes were predicted targets of TBX5. TBX5 cooperates with GATA4 for cardiac gene regulation (Garg et al., 2003; Luna-Zurita et al., 2016; Ang et al., 2016). We also observed a high association of GATA4 occupancy with TBX5 (Ang et al., 2016) near TBX5-dependent genes (Figure S4A–E, Table S5), indicating that GATA4 may have a role in modulating TBX5 dosage-sensitive genes.
Since modifiers in different genetic backgrounds can modulate phenotypic effects, we assessed alternatively targeted iPSC lines of TBX5 mutants in an independent genetic background (PGP1, from a Caucasian male (Lee et al., 2009), compared to WTC11 from a Japanese male (Miyaoka et al., 2014), Figure S5A, B). We independently evaluated comparisons between genotype-enriched subtype clusters in day 23 TNNT2+ cells from PGP1-derived cell lines (Figure S5C–G). Comparisons of lists of TBX5-dependent genes in day 23 TNNT2+ cells showed overlap between WTC11 vs. TBX5in/+ and PGP1 vs. TBX5+/in (p<5.219e-81 by hypergeometric test), or WTC11 vs. TBX5in/del and PGP1 vs. TBX5del/del (p<1.438e-172 by hypergeometric test).
We also integrated day 23 TNNT2+ cells from each genetic background into one combined dataset for analysis. Cells were largely indistinguishable in UMAP space regardless of experimental replicate or genetic background (Figure S5H). Importantly, we again observed segregation by TBX5 genotypes (Figure S5I). By comparing genotype-enriched subtype clusters (Figure S5J, K), we detected 148 genes between WTC11/Control/PGP1 and TBX5 heterozygous cells, and 457 genes between WTC11/Control/PGP1 and TBX5 homozygous cells (Figure S5L, M, Table S2). These results demonstrated robust TBX5 dosage-dependent gene expression alterations in cardiomyocytes from independent experiments, genetic backgrounds, and gene targeting strategies. Any differences in gene expression between biological replicates and genetic backgrounds likely reflected a combination of technical variability, biological stochasticity or genetic modifiers that, as in patients with TBX5 mutations (Basson et al., 1994), may explain variable expressivity of disease for a given mutation.
TBX5 dosage maintains cardiac gene network stability
CHD-associated and arrhythmia-related genes were enriched among TBX5-dependent genes in complex patterns of expression. We sought to independently, and without bias, assess the importance of TBX5 in a global cardiac gene regulatory network (GRN) beyond changes to gene expression. To evaluate the role of TBX5 dosage for regulating GRNs, we used bigSCale2 (Iacono et al., 2019) to independently infer putative GRNs without a priori knowledge (e.g. protein-protein interactions, known genetic associations, cardiac-enriched genes) from single cell expression data of TNNT2+ cells. GRNs are constructed by correlating transformed variables representing gene expression within a network for each time point and genotype. By applying the concept of “pagerank”, first devised to rank the importance by popularity of websites via numerical weighting (Brin and Page, 1998), we predicted quantitatively the biological importance (“centrality”) of genes in a GRN, even if a node’s gene expression was unchanged by TBX5 dosage.
Centrality is a node’s connectedness within a network. By comparing inferred networks of WTC11 and Control to TBX5in/+ or TBX5in/del within a given time point, we uncovered several candidate nodes that displayed loss of pagerank centrality in at least one TBX5 mutant genotype at any stage (Figure 6A–D, S6, Table S6). These included the calcium-handling gene RYR2, and twenty CHD genes (for example, TFs GATA6, HAND2, and SMAD2, p<2.2e-5 by hypergeometric test) (Figure 6E). This indicates an enrichment of CHD genes in TBX5 dosage-sensitive networks and is largely consistent with our analysis from differential gene expression. For example, at day 11, pagerank centrality of the CHD TF SMAD2 was absent in TBX5in/+ cells (Figure 6A–C, E), indicating a possible impairment of SMAD2 function from TBX5 haploinsufficiency. Pagerank centrality of the cardiac development-related TF MEF2C, which is necessary for mouse heart development (Lin et al., 1997), was substantially reduced by both heterozygosity and loss of TBX5 at day 11 (Figure 6A–E). Quantitative alterations to GRNs showed that TBX5 dosage may be critical for maintaining cardiac network stability, and potentially unveiled putative genetic interactions disrupted in TBX5-dependent CHDs.
Figure 6. TBX5 dosage preserves cardiomyocyte network stability.

(A-D) Gene regulatory networks (GRNs) of TNNT2+ cells at day 11 for TBX5 genotypes are displayed. Nodes of CHD (purple), heart development (red) or electrophysiology (orange) genes are shown. Node size represents quantitative importance of the gene, based on pagerank centrality. (E) Pagerank centrality for significantly altered (top 5% cutoff) nodes. (F) TBX5-dependent genes with a reduction of pagerank are correlated (correlation >0.5), anti-correlated (correlation <−0.5), or indeterminate (0.5<correlation<−0.5) with TBX5 expression in TNNT2+ cells. (G) Degree centrality for MEF2C in TBX5in/+ and TBX5in/del GRNs. (H) Correlations with MEF2C and TBX5-dependent genes with a reduction of degree centrality in TNNT2+ cells.
We used a complementary approach to identify gene-gene correlations with TBX5 expression in individual TNNT2+ cells across timepoints and TBX5 genotypes (Figure 6F). Genes highly co-expressed with TBX5 regardless of TBX5 genotype suggested potential positive regulation or possible cell autonomous effects by TBX5 dosage (for example, calcium-handling PLN and RYR2, and sarcomere TTN), while those with high anti-correlation suggested potential negative regulation or possibly non-cell autonomous effects (for example, HES1, TLE1, CBX1, ETV4, ID4, FGFR1). When considering the top 10 genes with the highest correlation with TBX5, MEF2C expression was among the highest correlated with TBX5 expression and demonstrated the greatest TBX5-dependent decrease of pagerank at day 11 (Figure 6E, F, Table S6), further suggesting MEF2C as a putative candidate for mediating TBX5 dose-sensitive regulatory effects in the GRN.
MEF2C gene expression itself was unchanged by reduced TBX5 dosage. Yet, MEF2C also displayed the greatest TBX5-dependent decrease in degree centrality, which indicates a node’s connections in a network (Figure 6G, Table S6). We found that multiple TBX5-sensitive genes, which correlated with MEF2C, displayed diminished levels of degree by reduced TBX5 dosage (e.g. transcriptional regulators SMYD1 and MYOCD, sarcomere TTN, calcium-handling RYR2, and kinase PDK1) (Figure 6H, Table S6). This likely reflects their altered gene expression by reduced TBX5 dosage and impacts connectivity in a TBX5-dependent network. Some genes (SMYD1 and MYOCD) are direct MEF2C targets in mice in vivo (Phan et al., 2005; Creemers et al., 2006). This suggested that these candidate genes with reduced degree may mediate putative MEF2C functional connectivity for TBX5 dosage-sensitive GRNs. It is also possible that a key node, such as MEF2C, could be functionally changed by changes to co-factors or access to chromatin.
Tbx5 and Mef2c cooperate for ventricular septation in vivo
Several potential genetic interactions were predicted by reduced pagerank from TBX5 dose-dependent human GRNs. A predicted genetic interaction between Tbx5 and Gata6 is known from mouse studies (Maitra et al., 2009). However, heterozygous loss of Tbx5 can lead to highly penetrant perinatal lethality based on mouse genetic background strains (Bruneau et al., 2001; Mori et al., 2006), making it difficult to evaluate genetic interactions based on postnatal lethality. Therefore, we further characterized a multifunctional allele of Tbx5 (Tbx5CreERT2IRES2xFLAG, abbreviated Tbx5CreERT2) (Devine et al., 2014), which appeared to be a hypomorphic Tbx5 allele, as a potential genetic tool for probing highly-sensitive in vivo genetic interactions with Tbx5 (Figure 7). Mice heterozygous for Tbx5CreERT2IRES2xFLAG (Tbx5CreERT2/+) survived to adulthood, and Mendelian ratios were recovered at weaning, as well as during embryonic development (Figure 7A). However, embryos homozygous for Tbx5CreERT2IRES2xFLAG (Tbx5CreERT2/CreERT2) could only be recovered until embryonic day 16.5 (E16.5), indicating that the Tbx5CreERT2 allele is hypomorphic (Figure 7B). Histological analysis of embryonic Tbx5CreERT2/CreERT2 hearts at E16.5 showed atrioventricular canal (AVC) defects, which include ASDs, VSDs and an atrioventricular valve (AVV), which were not present in wildtype or Tbx5CreERT2/+ mice, implicating CHDs as the cause of late embryonic lethality (Figure 7C–F).
Figure 7. Tbx5 and Mef2c cooperate in heart development.

(A) Numbers of P21 pups from matings of wildtype X Tbx5CreERT2/+ (n=94). (B) Numbers of embryos from Tbx5CreERT2/+ X Tbx5CreERT2/+ crosses (n=538). (C-F) Transverse sections of hearts at E16.5. Atrioventricular valves (AVV), ventricular septal defects (arrowhead), and atrial septal defects (arrow). LV, left ventricle; RV, right ventricle; IVS, interventricular septum; LA, left atrium; RA, right atrium; IAS, interatrial septum. (G) Numbers of P21 pups from matings of Tbx5CreERT2/+ X Mef2cdel/+ mice (n=64). (H, J, L, N, P) Transverse sections of hearts at embryonic day 14.5 (E14.5). (I, K, M, O, Q) Magnified views of the interventricular septum are shown. Muscular VSDs (arrowheads in M, O), a subaortic membranous VSD (Q, arrowhead) and dilated blood-filled atria (L, N, P) in compound heterozygotes (n=4). (R, S) Overlap of TBX5, MEF2a or MEF2c occupancy near mouse orthologs of human TBX5-dependent (R) or -independent genes (S) (Odds ratios, FDR<0.05, Table S7). (T, U) Browser tracks for ChIP-seq data from E12.5 hearts for TBX5, MEF2c, MEF2a and H3K27ac near TBX5-dependent genes, Hand2 (T) or Tecrl (U). Yellow shading: co-occupancy. (V) Odds ratios (FDR<0.05) of combinations of TBX5, MEF2a and MEF2c occupancy near mouse orthologs of TBX5-dependent human genes (Table S7).
We evaluated the GRN’s predicted genetic interaction between TBX5 and MEF2C in an in vivo mammalian context. MEF2C is a transcription factor that is required for cardiac development in mice and zebrafish (Lin et al., 1997; Ghosh et al., 2009). Additionally, combined knockdowns of tbx5 and mef2c lead to broad defects of early cardiac morphogenesis in zebrafish (Ghosh et al., 2009). Also, MEF2C is part of a cadre of cardiac TFs, including TBX5, that can induce reprogramming of cardiac fibroblasts to cardiomyocytes (Ieda et al., 2010; Qian et al., 2012; Song et al., 2012).
Using the hypomorphic allele of Tbx5 and a null allele of Mef2c (Mef2cdel) (Lin et al., 1997), we noted that Tbx5CreERT2/+;Mef2cdel/+ mice were underrepresented at weaning (Figure 7G). By histology, we detected a highly penetrant morphologic phenotype of VSDs (n=4 of 4), consisting of muscular (n=3 of 4) or membranous (n=1 of 4) VSDs, in compound heterozygous embryos at E14.5. VSDs were not observed in Tbx5CreERT2/+ or Mef2cdel/+ littermate embryos (Figure 7H–Q). Muscular VSDs are rarely observed in mouse models of CHD, making this observation particularly compelling, especially as they feature prominently in HOS (Basson et al., 1994; 1997; Sletten and Pierpont, 1996; Brassington et al., 2003). These findings demonstrate a highly-sensitive genetic interaction between Tbx5 and Mef2c in mouse in vivo, consistent with predictions from a human TBX5 dose-sensitive GRN.
We speculated that MEF2C may play a direct role to co-regulate TBX5-dependent gene expression during heart development. Using mice targeted with a FLAG-biotin (fl-bio) tag at specific TF loci, chromatin occupancy (Akerberg et al., 2019) of TBX5, MEF2C, and MEF2A (also predicted to be part of the TBX5-dependent GRN, Table S6) was highly correlated near mouse orthologs of TBX5-sensitive human genes (for example, HAND2, FHL2, TECRL, NPPA/NPPB; Figure 7R–V; Table S7, FDR<0.05 for multiple comparisons). This indicated that TBX5, MEF2C, and MEF2A were more likely to be found together on chromatin at TBX5-dependent genes than at TBX5-independent genes (Figure 7V, Table S7). Thus, direct co-regulation of target genes by TBX5, MEF2C, and MEF2A, in addition to previously known co-occupancy with NKX2–5 and GATA4 (Luna-Zurita et al., 2016; Ang et al., 2016), support the concept of a TF collective at TBX5-dependent genes, and may be a potential TBX5 dosage-dependent mechanism for TBX5 haploinsufficiency.
Discussion
Our studies with a human cellular model of TBX5 haploinsufficiency have defined consequences of reduced TBX5 dosage during cardiomyocyte differentiation at single cell resolution, indicating a dose-sensitive requirement of TBX5 for human ventricular cardiomyocyte differentiation and function. Our human cellular models of TBX5 haploinsufficiency utilize insertions that lead to premature termination and loss of function; these are similar to the nonsense mutations often found in HOS, but not exact reproductions of disease-associated point mutations. Of potential relevance to a range of anatomical and functional manifestations of TBX5 haploinsufficiency, we uncovered discrete responses to reduced TBX5 dosage in susceptible cardiomyocyte subsets. The quantitative specificity of TBX5-dependent cell types underscores cellular complexity in response to reduced transcription factor dosage. Many of the cellular phenotypes of this human disease model are cardiomyocyte-specific, intrinsic, and likely cell autonomous. Dysregulated gene expression of EP, CHD, and cardiac development genes provide potential molecular explanations for these cellular phenotypes, which are relevant to HOS, and more broadly to CHDs, in humans. While the iPS cell system is two-dimensional, the regulatory networks affected by reduced TBX5 dosage may be related to three-dimensional CHDs. Indeed, we observed a high degree of CHD-associated genes, many of which are seemingly unrelated by biological processes or cellular pathways, as vulnerable to reduced TBX5 dosage. Many are direct TBX5 targets. Overall, this evidence places TBX5 at a nexus of human CHD genes.
Susceptibility to TBX5 dosage-dependent gene expression in specific regions of the developing heart was apparent from studies modeling TBX5 haploinsufficiency in the mouse (Bruneau et al., 2001; Mori et al., 2006). The implication would be that discrete populations in the developing human heart would respond specifically to reduced TBX5 dosage. In support of this notion, with single cell resolution of gene expression in human iPSC-derived cardiomyocytes, we detected discrete changes to reduced TBX5 dosage in subpopulations of human cardiomyocytes. As well, we find that TBX5-sensitive genes are expressed in various anatomic regions of the developing human heart, consistent with a role for TBX5 in many aspects of cardiac differentiation, morphogenesis and function. While we confirm some altered genes that are known from mouse, such as NPPA (Bruneau et al., 2001; Mori et al., 2006), the richness of detail achieved here eclipses current knowledge of TBX5 haploinsufficiency from mouse models. For example, upregulation of the human atrioventricular canal gene TBX2 may have implications for abnormal cardiac morphogenesis in vivo. In mice, forced ectopic expression of Tbx2 suppressed the expression of cardiac chamber markers, such as Nppa, and induced ectopic endocardial mesenchyme markers, including Bmp2, in atrial and ventricular chamber myocardium (Singh et al., 2012). Therefore, altered gene expression of TBX2, NPPA, and BMP2 in the human cellular disease model may relate to atrioventricular canal defects, including ASDs and perimembranous VSDs. Reduced expression of ventricular factors, such as HAND1, IRX4 and HEY2, may be related to abnormal trabeculation and muscular VSDs observed in Holt-Oram syndrome patients.
Many TBX5-sensitive genes are related to heart function. CHDs are largely viewed as three-dimensional structural defects, but they are often accompanied by cardiac dysfunction, even after surgical correction. Arrhythmias and diastolic dysfunction are observed in patients with HOS (Basson et al., 1994; Mori and Bruneau, 2004; McDermott et al., 2008; Zhu et al., 2008), and in many other types of CHDs not related to TBX5 (Panesar and Burch, 2017). Furthermore, TBX5 is strongly associated with EP defects based on genome-wide association studies (Pfeufer et al., 2010; Smith et al., 2011; Ellinor et al., 2012). In addition, an enrichment of chromatin occupancy of TBX5 and cardiac transcription factors is observed near genes associated with EP-GWAS (Table S5) (Pfeufer et al., 2010; Smith et al., 2011; Ellinor et al., 2012; Cordell et al., 2013a; 2013b; Hoed et al., 2013; Hu et al., 2013). Several TBX5-sensitive electrophysiology genes implicated in arrythmias (e.g. GJA1, GJA3, GJA5, TECRL, SCN5A, DSP), might contribute to conduction defects in Holt-Oram syndrome patients. Only one of these, GJA5, is known from mouse.
TBX5 dosage has been shown to be necessary for preserving diastolic function in mice, by modulating SERCA2a-dependent calcium transients (Zhu et al., 2008), and regulating calcium cycling in atrial myocytes in the context of atrial fibrillation (Nadadur et al., 2016; Dai et al., 2019; Laforest et al., 2019). In our iPS cell model, calcium transients are prolonged significantly in TBX5in/+ cells (Figure 1L, M), which contributes to diastolic dysfunction in HOS. Cardiomyocytes showed TBX5 dose-sensitive slowing of decay of calcium transients and disarray of sarcomeres, likely reflecting impaired ventricular cardiomyocyte relaxation from reducing TBX5 dosage. In these human cells, concordant dysregulation of genes responsible for calcium cycling provide a potential molecular explanation for ventricular cardiomyocyte impairment and diastolic dysfunction in HOS (Eisner et al., 2020). For example, PLN inhibits calcium reuptake into the sarcoplasmic reticulum (SR) by SERCA2. PLN is upregulated by reduced TBX5 dosage, while the SERCA2-encoding gene ATP2A2 is downregulated by heterozygous loss of TBX5 (Figure 4V). Furthermore, RYR2 mediates release calcium from the SR, and the pagerank centrality of RYR2 is reduced in a TBX5-dependent GRN. Similarly, the arrhythmia-associated gene TECRL displayed stepwise dosage-dependent sensitivity to reduced TBX5 and is a predicted TBX5 target, and loss of TECRL in human iPSCs also leads to prolonged calcium transients (Devalla et al., 2016). An understanding of TBX5 function in calcium homeostasis may uncover new mechanisms for human arrhythmogenesis and potentially for ventricular cardiomyocyte relaxation.
Quantitative analysis of human TBX5 dose-sensitive GRNs predicted vulnerable nodes enriched for CHD, heart development and cardiac function-related genes, suggesting a vital role for TBX5 dosage to maintain cardiac network stability. The sensitivity of a GRN to transcription factor dosage has been observed in Drosophila embryo patterning (Stathopoulos and Levine, 2002), but has not been linked to human disease. From TBX5-sensitive GRNs, we discovered several altered nodes linking many CHD genes, often without altered expression levels. For example, reduced centrality of MEF2C in the TBX5-dependent GRN predicted an important and sensitive genetic link between these cardiac transcription factors. Consistent with this notion, double-knockdown of tbx5 and mef2c in zebrafish lead to severe defects in the looping heart tube (Ghosh et al., 2009). We observed a strikingly sensitive genetic interaction of Tbx5 and Mef2c in mice, unveiling a finely tuned role in mammalian heart development, beyond heart looping and chamber formation, for the process of ventricular septation. Of note, the Tbx5 and Mef2c genetic interaction in mouse yielded muscular VSDs, a very specific type of CHD rarely observed in mouse models but common in humans. The embryologic origin of muscular VSDs is different than membranous VSDs. The muscular ventricular septum is derived from remodeling at the interventricular groove, which has multiple origins (Moorman and Christoffels, 2003). The membranous ventricular septum is considered to be from the fusion of endocardial cushions (Anderson et al., 2014; 2018b). We anticipate that these and other genetic interactions will allow the discovery of molecular pathways and cellular processes that underlie specific CHDs.
Since genetic modifiers can influence the expressivity of disease, we use more than one genetic background and targeting strategy to model TBX5 haploinsufficiency and observe similar outcomes. While many TBX5-dependent genes were congruous across two ethnically diverse genetic backgrounds, there were some apparent differences. This is consistent with a notion that modifiers in genetic backgrounds can contribute to varying degrees of phenotypic expressivity for CHDs. Furthermore, variability in CHDs with monogenic inherited or de novo mutations could be explained by additional mutations or copy number variations of genes that form part of these functional regulatory networks, as illuminated by our findings, and as evidenced by oligogenic inheritance of CHD-causing variants (Gifford et al., 2019).
Our results point to a genomic framework that will guide genetic insights into the underpinnings of CHD. The biophysical rules relating to transcription factor binding and dosage sensitivity are only now becoming understood. Our results in a human cellular model of TBX5 haploinsufficiency may potentially bring immediate pertinence of human disease to this biological context.
STAR Methods
RESOURCE AVAILABILITY
Lead contact.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Benoit G. Bruneau (benoit.bruneau@gladstone.ucsf.edu).
Materials Availability.
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability.
scRNA-seq datasets have been deposited at NCBI GEO, under accession GSE137876. R and python scripts will be available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines.
Protocols for use of iPS cells were approved by the Human Gamete, Embryo and Stem Cell Research Committee, as well as the Institutional Review Board at UCSF. iPS cells harboring genome-edited mutations for TBX5 (TBX5in/+, TBX5+/in, TBX5in/del, TBX5del/del, TBX5PuR/PuR) were generated for this study.
Mice.
All mouse protocols were approved by the Institutional Animal Care and Use Committee at UCSF. Mice were housed in a barrier animal facility with standard husbandry conditions at the Gladstone Institutes. Tbx5CreERT2IRES2xFLAG (abbreviated here as Tbx5CreERT2) (Devine et al., 2014) mice were described previously. Mef2cdel/+ mice (Lin et al., 1997) were obtained from Brian Black. Tbx5CreERT2/+ and Mef2cdel/+ were maintained in the C57BL6/J background (Jackson Laboratory #664). Tbx5fl-bio/fl-bio (Waldron et al., 2016) mice were obtained from Frank Conlon. Mef2afl-bio and Mef2cfl-bio (Jackson Laboratory #025983) were described in (Akerberg et al., 2019). Rosa26BirA mice were obtained from the Jackson Laboratory (#010920) (Driegen et al., 2005). Both male and female embryos were collected and used at random for experiments.
METHOD DETAILS
Gene targeting and genotyping of human iPS cells mutant for TBX5.
sgRNAs for TBX5 exon 3 (sgRNA1, TCCTTCTTGCAGGGCATGGA) or exon 7 (sgRNA2, CCTTTGCCAAAGGATTTCG), which encode the T-box domain, were selected using crispr.genome-engineering.org, and cloned by annealing pairs of oligos into a plasmid containing humanized S. pyogenes Cas9 (Cong et al., 2013) (px330-U6-Chimeric_BB-CBh-hSpCas9 was a gift from Feng Zhang, Addgene #42230).
For WTC11-derivatives TBX5+/+ (control), TBX5in/+ or TBX5in/del, the induced pluripotent stem (iPS) cell line WTC11 (gift from Bruce Conklin, available at NIGMS Human Genetic Cell Repository/Coriell #GM25236) (Miyaoka et al., 2014) was electroporated (Lonza #VPH-5012) with a cloned nuclease construct containing a guide RNA (sgRNA1) targeting exon 3 of TBX5 (Mandegar et al., 2016; Miyaoka et al., 2014). Cells were plated on human ESC-grade Matrigel (Corning #354277) and cultured in mTeSR-1 (StemCell Technologies Cat #05850) with 10μM ROCK inhibitor (StemCell Technologies, Y-27632). For screening of TBX5 exon 3 non-homologous end-joining (NHEJ) mutations, genomic DNA flanking the targeted sequence was amplified by PCR (For1: ATGGCATCAGGCGTGTCCTATAA and Rev1: CCCACTTCGTGGAATTTTAGCCA), amplicons underwent digestion by NlaIII, and then were evaluated for loss of NlaIII by gel electrophoresis (wildtype band 800bp, mutant band 880bp). Clones with no change, a heterozygous or homozygous loss of NlaIII were sequenced (For1: ATGGCATCAGGCGTGTCCTATAA, Rev1: TTCCGGGCTTGAACTTCTGG, Seq1: ATAGCCTTGTGCTGATGGCA).
For generation of TBX5PuR/PuR, a puromycin resistance gene cassette (Frt-PGK-EM7-PuroR-bpA-Frt) containing homology arms of 469bp (5’ homology arm) and 466bp (3’ homology arm) around the sgRNA1 target site at +9bp from the start of TBX5 exon 3 was cloned by Cold Fusion (System Biosciences #MC010B) using amplicons from genomic DNA of WTC11 into a construct that was a modification of plasmid pEN114 (Nora et al., 2017). WTC11 cells were electroporated with a cloned nuclease construct containing a guide RNA targeting exon 3, along with the TBX5 exon3 homology arm-Frt-PGK-EM7-PuroR-bpA-Frt cassette and plated as a serial dilution in mTeSR-1 with Rock inhibitor (Mandegar et al., 2016). On day 2 and subsequent days, cells were grown in media containing mTeSR-1, Rock inhibitor and puromycin (0.5ug/mL), to select for puromycin-resistant cells. For screening of TBX5 exon 3 homology-directed repair (HDR) mutations, genomic DNA flanking the targeted sequence was amplified by PCR (For1: ATGGCATCAGGCGTGTCCTATAA, and Rev2: CCCACTTCGTGGAATTTTAGCCA for wildtype, 797 bp, For1: ATGGCATCAGGCGTGTCCTATAA, Rev3: GTTCTTGCAGCTCGGTGAC (Nora et al., 2017) for PuroR, 1631 bp). Positive 5’ arm clones were genotyped by PCR for the 3’ arm (For2: ATTGCATCGCATTGTCTGAG (Nora et al., 2017), Rev4: TTTGACAATCGGGTGGGACC, 829 bp).
For PGP1-derivatives TBX5+/in or TBX5del/del, the iPS cell line PGP1 (gift from George Church, available at NIGMS Human Genetic Cell Repository/Coriell #GM23338) (Lee et al., 2009) was electroporated with a cloned nuclease construct containing a guide RNA (sgRNA2) targeting exon 7 of TBX5 (Byrne and Church, 2015). For screening of TBX5 exon 7 NHEJ mutations, the targeted sequence was amplified using PCR primers (For3: GCTTCTTTTGGTTGCCAGAG, Rev5: CATTCTCCCCATTTCCATGT, Seq2: AGAGGCTGCATTTCCATGAT), Illumina compatible-libraries from clones were generated and multiplex-sequenced on a MiSeq for purity of homogeneity of clones for heterozygous or homozygous mutations (Byrne and Church, 2015).
Isolation of homogenous iPS cell clones.
Isolation of homogenous colonies for WTC11-derivatives TBX5+/+ (control), TBX5in/+ or TBX5in/del was performed by modification of methods described previously (Mandegar et al., 2016; Peters et al., 2008). Briefly, single cell suspension of electroporated iPS cells was plated on Matrigel-coated 6 well plates (WP) (BD Bioscience #351146). Once cultures were adherent and recovered to ~80% confluency, cells were detached by Accutase Cell Detachment Solution (Stemcell Technologies #07920), diluted with 1X DPBS without Ca2+/Mg2+ and singularized using a P1000 filtered tip, and centrifuged. The cell pellet was resuspended in mTeSR-1, Rock inhibitor and Gentamicin (Life Technologies #15750–060) media, incubated with DAPI (1:1000 from a 1mg/mL stock) for 5 min, centrifuged and resuspended at a concentration of at least 1.0E6 cells/mL in mTeSR-1, Rock inhibitor and Gentamicin media without DAPI. After filtering cells with a 40-micron mesh into FACS tubes, remaining cells (about 120,000 cells per well) were plated onto 6WP for maintenance. Single cells were then sorted for DAPI negativity using a BD FACS AriaII or AriaIII, with a 100-micron nozzle at the lowest flow rate available, into individual wells of a 96WP coated with Matrigel containing media of mTeSR-1, Rock inhibitor and Gentamicin. Upon recovery at 37°C, each well was evaluated one day later for no cells, one cell or more than one cell. All cells were maintained with mTeSR-1, Rock inhibitor and Gentamicin media for at least 5 days, then with mTeSR-1 alone for an additional 5–7 days. Each well at 25% confluency was harvested and replated upon singularization with P200 tips in 96WP for more efficient cell growth. When the cell confluency of each well from “single” cells was nearly 100%, then 90% of cells were harvested for genotyping using QuickExtract DNA lysis solution (Epicentre #QE0905T), while 10% of cells were re-plated for the next round of cell selection for wells of interest by FACS sorting again or by serial dilution of cells for manual picking of colonies (Mandegar et al., 2016; Miyaoka et al., 2014) from apparent “single” cells. Rounds were repeated until every daughter well showed the same genotype, consistent with homogeneity. Genomic DNA from individual wells of interest were amplified using high fidelity Taq polymerase, TA-cloned and sequenced to confirm genotype and homogeneity.
Isolation of homogenous colonies for PGP1-derivatives TBX5+/in or TBX5del/del was performed, as in (Byrne and Church, 2015). Isolation of homogenous colonies for WTC11-derivative TBX5PuR/PuR was performed, as in (Mandegar et al., 2016). After sequencing confirmation of respective genotypes, karyotypically-normal cells from each iPS cell line were expanded for subsequent studies.
Maintenance of iPS cells and differentiation to cardiomyocytes.
All iPS cell lines were transitioned to and maintained on growth factor-reduced basement membrane matrix Matrigel (Corning #356231) in mTeSR-1 medium. For directed cardiomyocyte differentiations, iPS cells were dissociated using Accutase and seeded onto 6WP or 12WP. The culture was allowed to reach 80–90% confluency and induced with the Stemdiff Cardiomyocyte Differentiation Kit (Stemcell Technologies #05010), according to the manufacturer’s instructions. Starting on day 7, differentiations were monitored daily for beating cardiomyocytes and onset of beating was recorded as the day when beating was first observed.
Flow Cytometry.
iPS-derived cardiomyocytes from WTC11, Control, TBX5in/+ and TBX5in/del lines were dissociated using Trypsin-EDTA 0.25% on day 15 or day 23 after induction of the differentiation protocol and fixed with 4% methanol-free formaldehyde. Cells were washed with PBS and permeabilized using FACS buffer (0.5% w/v saponin, 4% Fetal Bovine Serum in PBS). For evaluation of differentiation efficiency, cells were stained with a mouse monoclonal antibody for cardiac isoform Ab-1 Troponin at 1:100 dilution (ThermoFisher Scientific #MS-295-P) or the isotype control antibody (ThermoFisher Scientific #14–4714-82). For analyzing levels of Desmin protein, cells were co-stained with the mouse monoclonal antibody for cardiac isoform Ab-1 Troponin at 1:100 dilution and recombinant rabbit anti-Desmin antibody at 1:70 dilution (Abcam #ab32362), or normal rabbit IgG antibody (Millipore Sigma #NI01) for 1 hour at room temperature. After washing with FACS buffer, cells were stained with the following secondary antibodies - goat anti-mouse IgG Alexa 594 at 1:200 dilution (ThermoFisher Scientific #A-11005) and donkey anti-rabbit IgG Alexa 488 at 1:200 dilution (ThermoFisher Scientific #A21206) for 1 hour at room temperature. Cells were then washed with FACS buffer, stained with DAPI for 5 minutes, rinsed, and filtered with a 40-micron mesh. At least 10,000 cells were analyzed using the BD FACSAriaII or AriaIII (BD Bioscience), and results were processed using FlowJo (FlowJo, LLC).
Western blotting.
iPS-derived cardiomyocytes were harvested on day 15, pelleted and flash frozen. Protein was isolated from supernatant in RIPA buffer with EDTA-free protease and phosphatase inhibitor (ThermoFisher Scientific) after sonication (15 second pulse on, 15 second pulse off, for four pulses). After quantification by BCA assay (ThermoFisher Scientific), 150μg of total protein was loaded per well for each genotype. After running on SDS-PAGE and wet transfer with NuPage Transfer buffer (ThermoFisher Scientific) to a PVDF membrane, the blot was washed in PBST and incubated in primary antibodies of rabbit polyclonal anti-TBX5 at a 1:400 dilution (Sigma #HPA008786) and mouse monoclonal anti-cTNT at 1:1000 dilution (ThermoFisher Scientific #MS-295-P), followed by secondary antibody incubation with donkey anti-rabbit IgG IRDye680 at 1:2000 dilution (Licor #926–68073) and donkey anti-mouse IgG IRDye800 at 1:2000 dilution (Licor #926–32212). The blot was imaged on an Odyssey FC Dual-Mode Imaging system (Licor).
Fluorescent in situ hybridization.
iPS cell-derived cardiomyocytes from WTC11, Control, TBX5in/+, TBX5in/del and TBX5PuR/PuR were dissociated using Trypsin-EDTA 0.25% on day 23 after induction of the differentiation protocol, and 25,000–40,000 cells were plated on to 8-well chambered slides (Ibidi #80826), to obtain a relatively sparse monolayer of cardiomyocytes. Cells were fixed the following day with 10% Neutral Buffered Formalin for 15 minutes at room temperature. Cells were then serially dehydrated in 50%, 70% and 100% ethanol and stored at −20°C until ready to be hybridized. In situ hybridization was performed using the RNAscope Multiplex Fluorescent v2 Assay kit (Advanced Cell Diagnostics #323100) with probes for Hs-TNNT2 (#518991), Hs-NPPA (#531281), Hs-HAND1 (#429701), Hs-HEY2 (#441761), Hs-RSPO3 (#413711) and Hs-TBX2 (#509331). Slides were imaged at 10X and 40X magnification on the Keyence BZ-X710 All-in-One Fluorescence Microscope or at 20X magnification on the Olympus FV3000RS.
Replating cardiomyocytes for single cell electrophysiology.
iPS cell-derived cardiomyocytes (day 15 or older) from WTC11, Control, TBX5in/+, TBX5in/del and TBX5PuR/PuR were gently dissociated in Trypsin-EDTA 0.25% and quenched using StemDiff Maintenance Medium with 10% FBS. Cell suspension was centrifuged at 800 rpm for 5 minutes. The pellet was resuspended in StemDiff Maintenance Medium with Rock inhibitor at a 1:1000 dilution. Cardiomyocytes were counted, and 25,000–35,000 cells were plated on to growth factor-reduced Matrigel-coated 15mm round glass coverslips (Warner Instruments #64–0703) to obtain a sparse distribution. Cardiomyocytes were then maintained on coverslips in StemDiff Maintenance Medium.
Patch Clamp Electrophysiology.
Patch clamp recordings were made on single iPSC-derived cardiomyocytes using the perforated-patch configuration. Experiments were performed at 30°C under continuous perfusion of warmed Tyrode’s solution containing (in mM): 140 NaCl, 5.4 KCl, 1 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, with the pH adjusted to 7.4 with NaOH. Recordings were conducted using borosilicate glass pipettes (Sutter Instruments) with typical resistances of 2 to 4MW. The pipette solution consisted of (in mM): 150 KCl, 5 NaCl, 5 MgATP, 10 HEPES, 5 EGTA, 2 CaCl2, and 240 mg/mL amphotericin B, with the pH adjusted to 7.2 with KOH. Spontaneous action potentials were acquired in a zero-current current clamp configuration using an Axopatch 200B amplifier and pClamp 10 software (Axon Instruments). Data was digitized at 20 kHz and filtered at 1kHz. Action potential parameters from each cell were derived using Clampfit 10 software (Axon Instruments).
Calcium imaging.
iPSC-derived cardiomyocytes on glass coverslips were loaded with Ca2+ indicator dye Fluo-4 AM (Thermo Fisher Scientific #F14201) to record Ca2+ flux, as previously described (Spencer et al., 2014). Measurements were made on spontaneously firing single or small clusters of iPSC-derived cardiomyocytes using a 10X objective on a Zeiss Axio Observer Z1 inverted microscope. For experiments, cells were placed in Tyrode’s solution containing 1.8 mM Ca2+ within a 37°C heated stage-top imaging chamber (Okolab). Images were acquired at 100 fps using an ORCA-Flash 4.0 camera (Hamamatsu, Bridgewater, NJ). Data was processed using ZEN (Zeiss) or Image J software (http://rsbweb.nih.gov/ij/) and analyzed using custom in-house software (Hookway et al., 2019).
Immunostaining of cardiomyocytes.
iPSC-derived cardiomyocytes from WTC11, Control, TBX5in/+ and TBX5in/del were replated on coverslips placed in 12-well plates on day 23 above for replating for electrophysiology. Cells were fixed in 4% formaldehyde for 20 minutes at room temperature, followed by washes in PBS. Cells were then treated with a blocking buffer containing 5% goat serum and 0.1% Triton X-100 in PBS for 1 hour at room temperature. A mouse monoclonal antibody for cardiac isoform Ab-1 Troponin (ThermoFisher Scientific #MS-295-P) was added to the coverslip-containing wells at a 1:100 dilution in blocking buffer and incubated on a rocker for 2 hours at room temperature. Following washes with 0.1% Triton X-100 in PBS, coverslips were treated with a donkey anti-rabbit IgG Alexa 488 antibody (ThermoFisher Scientific #A21206) at a 1:200 dilution for 2 hours at room temperature. Coverslips were then washed with 0.1% Triton X-100 in PBS and stained with DAPI at a 1:1000 dilution for 2 minutes. Coverslips were washed and stored in PBS at 4C. Images were acquired on a Zeiss LSM 880 with Airyscan and processed by ImageJ (Abràmoff et al., 2004).
Cell harvesting for single cell RNA sequencing.
Briefly, each of the TBX5 genotypes was differentiated, harvested and prepared at the same time for each biological replicate. Therefore, each biological replicate represents an experimental batch. We made every effort to compare samples of different TBX5 genotypes from differentiations that were beating robustly and of similar efficiency by flow cytometry or RNA expression. Cells from day 6, day 11 or day 23 of the differentiation protocol were collected from 3 independent differentiations. Wells for dissociation were chosen based on typical differentiated morphology on day 6 or robust beating on day 11 and day 23. Cells were singularized with Trypsin-EDTA 0.25%. After quenching, the single cell suspension was centrifuged at 800 rpm for 5 minutes. The pellet was resuspended in 1X PBS with 0.04% w/v Ultrapure BSA (MCLAB #UBSA-500) and counted. A 30μL cell suspension containing 10,000 cells was used to generate single cell droplet libraries with the Chromium Single Cell 3′ GEM, Library & Gel Bead Kit v2 according to manufacturer’s instructions (10X Genomics). After KAPA qPCR quantification, a shallow sequencing run was performed on a NextSeq 500 (Illumina) prior to deep sequencing on a NextSeq 500, HiSeq 4000, or NovaSeq (Illumina) for a read depth of >100 million reads per cell.
Data processing using Cellranger.
All datasets were processed using Cellranger 2.0.2. FASTQ files were generated using the mkfastq function. Reads were aligned to hg19 reference (version 1.2.0). Cellranger aggr was used to aggregate multiple GEM libraries.
Seurat analysis.
Outputs from the Cellranger pipeline were analyzed using the Seurat package (version 2.3.4 or 3.1.4) (Butler et al., 2018; Satija et al., 2015; Stuart et al., 2019) in R (version 3.5.1) [R Core Team (2018). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/]. Datasets from day 6, day 11 or day 23 experiments were analyzed as separate Seurat objects. Seurat objects for day 6 or day 11 were generated using Seurat v2. Seurat objects for day 23 datasets with multiple biological replicates were generated using Seurat v3, unless otherwise noted.
Quality control steps were performed to remove dead cells or doublets, and cells with a UMI count between 10,000 to 80,000 were retained. After normalizing the data, sources of unwanted variation, such as differences in the number of UMI, number of genes, percentage of mitochondrial reads and differences between G2M and S phase scores were regressed using the ScaleData function. Next, principal component analysis (PCA) was performed using the most highly variable genes. Cells were then clustered based on the top 25–30 principal components and visualized using a dimensionality reduction method called Uniform Manifold Approximation and Projection (UMAP) (Becht et al., 2018). The resolution parameter was set, so that cluster boundaries largely separated the likely major cell types.
Two technical replicates at day 6 and day 11 for WTC11-derived cells (WTC11, control, TBX5in/+, TBX5in/del) were evaluated. For control at day 23, two technical replicates were evaluated. For WTC11, TBX5in/+ or TBX5in/del at day 23, two technical replicates from biological replicate 1 and one sample from biological replicate 2 were evaluated. For PGP1-derived cells (PGP1, TBX5+/in and TBX5del/del), one sample for each genotype was evaluated.
Major cell type categories were defined by their expression of select enriched genes in a given cluster--pluripotent cells (POU5F1) cardiomyocytes (TNNT2), dividing cardiomyocytes (CENPF+/TNNT2+), ventricular cardiomyocytes (TNNT2+/IRX4+), fibroblasts (COL1A1), epicardial cells (WT1+/TBX18+), neural crest-derived cells (MAFB+, MSX1+), endoderm (TTR alone or TTR+/AFP+) and endothelial cells (PLVAP). Clusters of cells not defined by any of these markers were labeled as “Undetermined”. The numbers of cells in each major cell type category in each genotype were then calculated. Sunburst plot was generated in Excel using the percentage of cells in each cell type category per genotype. We used FindAllMarkers to generate a list of top marker genes for each cluster and highlighted selected genes in a Manhattan plot to display potential diversity of subtypes among these major cell types.
Integration and Visualization of Datasets from Multiple Samples.
For the day 23 WTC11-derived cell line (biological replicate 1 and 2) analysis, we ran CellRanger to normalize sequencing depth variation between individual libraries. We then ran Seurat v3.1.4’s ‘Integration and Label Transfer-SCTransform’ workflow to resolve effects from experimental instances that are driven by cell-cell technical variations, including sequencing depth (Hafemeister and Satija, 2019; Stuart et al., 2019). Cells with lower than 10,000 UMIs and concurrently higher percentage of mitochondrial reads were removed. Potential doublets with higher than 75,000 UMIs were also removed. The dataset was then split into two Seurat objects using the biological replicate status. We ran CellCycleScoring(default) and SCTransform(vars.to.regress=c(“S.Score”, “G2M.Score”)) to regress out cell cycle variations. The remaining steps followed the ‘Integration and Label Transfer-SCTransform’ workflow. Briefly, these steps include finding 2,000 highly variable genes to create anchors that represent biologically common cells connected from opposing batches. After integration, Seurat set the active assay to ‘integrated’ for downstream data visualization analysis. UMAPs were created by running RunPCA(default) and RunUMAP(default).
We also evaluated genetic backgrounds from two iPSC lines. The WTC11-derived cell lines were considered genetic background 1, which included biological replicate 1 and 2. PGP1-derived cell lines were considered genetic background 2. We followed the same CellRanger aggregate and qc filtering. However, we used the genetic background status to make three Seurat objects and no variables were regressed when running the ‘Integration and Label Transfer-SCTransform’ workflow. UMAPs were created by running RunICA(default) and RunUMAP(reduction=”ica”, dims=1:40, min.dist=0.4, spread=0.9, repulsion.strength=6).
For day 23 cardiomyocyte datasets, TNNT2+ clusters were defined as containing a majority of cells expressing TNNT2 on a feature plot and extracted using the subset function and re-clustered. Subsequently, the resolution parameter was set to partition clusters enriched for a particular genotype. A phylogenetic tree was generated by relating the “average” cell from each cluster in PC space, using the BuildClusterTree function. Differential gene expression tests were run between closely related clusters, using the FindMarkers function with min.pct set to 0.1 and logfc.threshold set to 0.25. Selected differentially expressed genes with an adjusted p-value less than 0.05 from the Wilcoxon Rank Sum test were then displayed using the Dotplot function. As Seurat log normalizes gene expression counts and scales values for each gene (mean is 0, std dev of +/−1), dot plots and heatmaps are based on scaled expression values.
Cell Type Classifier by Machine Learning.
We applied machine learning to predict corresponding in vivo cell types in our WTC11-derived samples. A sklearn multiclass logistic regression model, using a one-vs-rest scheme and the cross-entropy loss cost function (Pedregosa et al., 2011), was trained on the in vivo scRNA-seq dataset published by (Asp et al., 2019). The training data contained eleven cardiac cell type classes (i.e. Fibroblast-like, atrial cardiomyocyte (aCM)-like, ventricular cardiomyocyte (vCM)-like, Cardiac neural crest-like, Sub-epicardial-like, Capillary endothelium/pericytes/adventitia-like, Smooth muscle/fibroblast-like, and Erythrocyte-like). The test data was the day 23 integrated WTC11-biological replicates.
We ran SCTransform(default) independently on the training and test data to remove sequencing depth bias, while preserving biological heterogeneity. To train our classifier on cell-type specific signals from both datasets, we used SCTransform Pearson residuals as the feature space for 1,538 genes. The genes were selected by taking the intersection of the top 3,000 highly variable genes (HVGs) from the training and test datasets (Table S1). We evaluated the cell type classifier using sklearn’s stratified 10-fold cross validation method; StratifiedKFold(n_splits=10, random_state=42). Each fold preserves the percentage of in vivo cell types. Thus, recapitulating true in vivo cardiac cell type composition in our training evaluation. For each fold of the cross validation, we used a sklearn logistic regression model to fit and predict on the fold’s training and test set; LogisticRegression(penalty=‘l2’, solver=‘lbfgs’, random_state=42). The cross validation model’s average performance measurements were: accuracy (96.9%), precision (97.8%), recall (97.4%), and f1 score (97.1%) (Figure S2C). Due to the strong cross validation performance, we trained our deployment model on the full in vivo dataset to increase cell type generalizability. The trained multinomial classifier was then deployed on our WTC11-derived samples.
We ran DoubletFinder to check if ‘mixed’ cell type predictions were doublets (McGinnis et al., 2019). The WTC11-derived biological replicate Seurat object was split by GEM lane to make eleven Seurat objects. We selected DoubletFinder parameters for each using the pK Identification version 3 (no ground-truth) method. The Poisson doublet formation rate was set assuming a 7.5% percent doublet formation rate per 10,000 cells, for each GEM lane, as suggested by 10X Genomics. Doublets were then predicted, using pK and nExp simulation parameters optimized to each GEM lane.
Congenital Heart Disease-Associated or Electrophysiology-Related Gene Lists and Cell Type Expression.
A list of 375 CHD candidate genes, including inherited, de novo, syndromic or non-syndromic CHD genes of interest, was manually curated from literature (Homsy et al., 2015; Jin et al., 2017; Lalani and Belmont, 2014; McCulley and Black, 2012; Prendiville et al., 2014; Priest et al., 2016; Sifrim et al., 2016; Zaidi et al., 2013). A list of 76 EP genes were manually curated. A list of cardiac development-related factors is from (Duan et al., 2019). Lists can be found in Table S3.
Cell trajectories and pseudotime analysis.
Pseudotime analysis was performed using the URD package (version 1.0.2) (Farrell et al., 2018). A single Seurat object (from Seurat v2), consisting of combined data from two technical replicates of three timepoints and four genotypes, was processed as described in the previous section, and then converted to an URD object using the seuratToURD function. Cell-to-cell transition probabilities were constructed by setting the number of nearest neighbors (knn) to 211 and sigma to 8. Pseudotime was then calculated by running 80 flood simulations with POU5F1+ clusters as the ‘root’ cells. Next, all day 23 clusters were set as ‘tip’ cells and biased random walks were simulated from each tip to build an URD tree.
We identified URD monotonic genes, which are genes that neither deviate from an increase or decrease in expression with pseudotime. Spearman rank correlation (Python v3.7.3, and libraries Pandas 0.25.0, Numpy 1.17.1, and SciPy 1.3.1) was used to find significant monotonic genes (p < 0.05). To determine if these monotonic relationships differ between WT and TBX5in/del paths to cardiomyocytes, we used a Fisher z-transformation to test the null hypothesis that there is no significant difference in correlation (Fisher, 1921). To illustrate these results, we use heatmaps for genes with a |rho|≥0.4 to pseudotime and Z-score≥15 as a difference between WT and TBX5in/del paths.
To identify differential expressed genes in inferred cardiac precursors (intermediate branches in the URD tree) that are affected by TBX5 loss, cell barcodes from each precursor segment (wildtype/control/TBX5in/+ path vs. TBX5in/del path) were extracted from the URD object and assigned new identities in the corresponding Seurat object. Differential gene test was then performed between the two segments using Wilcoxon Rank Sum test with min.pct set to 0.1 and logfc.threshold set to 0.25. Selected genes with an adjusted p-value less than 0.05 were plotted on the URD tree to visualize their expression.
To compare the trident (TNNT2+ distal branch for WTC11, control and TBX5in/+) and fork (TNNT2+ distal branch for TBX5in/del) during pseudotime, we subdivided the pseudotime from the common branchpoint to the tips of the trident and fork into twenty uniform windows. Within each window, we then calculated the t test, difference of means, and fold change between the trident and fork for all genes. We filtered the statistics by gene-window combinations with adjusted p-value<0.05 after Bonferroni-Holm multiple testing correction. Then, we hierarchically clustered the genes on t test p-values and plotted statistics using the R pheatmap library.
Cell browser implementation.
We created a cell browser session at cells.ucsc.edu that allows the user to interrogate the spatial distribution of metadata and expression across data, in multiple reduced dimensionality spaces including the URD trajectory. Using a Scanpy python pipeline, we generated PCA, tSNE, UMAP, PAGA, and drl transforms. We also imported the URD trajectory mapping and WGCNA transform from their respective packages. We ran the scoreCT algorithm to assign cell types to cell clusters using a marker gene set.
Spatial transcriptomics analysis.
We assessed spatial transcriptomics data of human fetal hearts at 4.5–5, 6.5, 9 post-conception weeks (Asp, et al., 2019; https://hdca-sweden.scilifelab.se/a-study-on-human-heart-development/) to evaluate the anatomic context of TBX5-dependent genes from iPSC-derived cardiomyocytes of TBX5in/+ or TBX5in/del at day 23. Downregulated or upregulated TBX5-dependent gene lists were assembled from differential tests between WTC11 and TBX5in/+ or WTC11 and TBX5in/del (Figure 4D–F, P, R, T, V). Spatial transcriptomics data was normalized using Seurat v3.1.4’s SCTransform method.
Gene expression was then aggregated and standard scaled across spatiotemporally-conserved anatomic region labels from (Asp et al., 2019). For each region, the top fifty genes were selected based on highest z-score. Overlapping genes were binned in the region with the higher z-score. The ‘myocardial’ gene set was defined as overlapping genes found in the top 175 of Z-scores for the ‘Atrial myocardium’, ‘Compact ventricular myocardium’, and ‘Trabecular ventricular myocardium’ regions. The ‘ventricular’ gene set was defined as overlapping genes found in the top 175 of Z-scores for the ‘Compact ventricular myocardium’ and ‘Trabecular ventricular myocardium’ regions. Genes from the ‘ventricular’ set that overlapped with ‘myocardial’, were assigned to ‘myocardial’. The resulting gene by region matrix was used to generate a clustermap using seaborn (v0.11.0).
Spatial transcriptomics spot data was normalized using Seurat v3.1.4’s SCTransform method and then scaled across all 19 tissue sections. The Matplotlib (v3.3.2) library was used to paint expression spots on corresponding histological structures. Hollow spots denote regions of no signal.
Gene ontology analysis.
Gene Ontology (GO) analysis for downregulated or upregulated TBX5-dpendent genes was performed with gseapy Enrichr (v0.10.1) (Chen et al., 2013; Kuleshov et al., 2016) using GO 2018 biological process terms. Downregulated or upregulated TBX5-dependent gene lists from iPSC-derived cardiomyocytes at day 23 were assembled from differential tests between WTC11 and TBX5in/+ or WTC11 and TBX5in/del (Figure 4D–F, P, R, T, V). Gene sets were filtered with a significance threshold set at an adjusted p-value < 0.05. Significant biologically-relevant pathways within the first thirty hits were visualized with bar plots produced using gseapy.plot (v0.10.1).
Gene regulatory network analysis.
bigSCale2 (https://github.com/iaconogi/bigSCale2) (Iacono et al., 2019; 2018) was used with default parameters to infer gene regulatory networks and “correlomes” from single cell RNA-seq expression data for TNNT2+ cells. Expression counts and gene names were used as input from two technical replicates at each time point and TBX5 genotype. Details of each dataset can be found in Table S6. To evaluate significant changes in pagerank or degree centrality, we computed all pairwise differential differences in pagerank or degree between baseline (wildtype and control) vs. TBX5 mutants (TBX5in/+ or TBX5in/del) (12 total differences, from 2 TBX5 mutants * 2 baselines * 3 stages) and used these values to determine the top 5% upper change cutoff from 8,704 genes of all networks. Classification of Pearson correlations were empirically chosen at >0.5 for correlation and <−0.05 for anti-correlation.
ChIP-seq.
Combined peaks of human TBX5 or GATA4 ChIP-seq from hiPSC-derived cardiomyocytes were used (Ang et al., 2016). bioChIP-seq of mouse TBX5, MEF2c and MEF2a from E12.5 hearts were from (Akerberg et al., 2019). Single replicates of TF bioChIP peaks, which were IDR normalized (IDR_THRESHOLD=0.05 between each set of replicates), were defined as the summit of the peak with the strongest ChIP signal ± 100bp of the individual replicate with the greatest peak intensity. Mouse H3K27ac ChIP-seq at E12.5 of embryonic cardiac ventricles was from (He et al., 2014).
QUANTIFICATION AND STATISTICAL ANALYSIS
Scoring of sarcomeric disarray.
Myofibrillar arrangement in cardiomyocytes was manually scored on a scale of 1–5, similar to (Judge et al., 2017). A score of 1 represents cells with intact myofibrils in a parallel arrangement. A score of 2 represents cells that have intact myofibrils, but many are not parallel. Scores of 3 and 4 include cells with increasing degrees of myofibrillar fragmentation or aggregation. A score of 5 represents cells without visible myofibrils. No cells were apparent among our samples with a score of 5. Violin plots were generated in Prism (GraphPad) to show distribution of scored cells from each group. Fisher’s exact test was used to determine statistical significance.
Measurement of cell size.
Cell size measurements were performed in ImageJ by drawing an outline around each cell and measuring the area using the ‘Measure’ tool. Mean area measurements between genotypes were then compared using an unpaired t-test.
Quantifying cell abundance or gene expression intensity by fluorescence in situ hybridization.
HEY2+ or HAND1+ cells were defined as cells with at least two HEY2 or HAND1 punctae, respectively. Number of TNNT2+/HEY2+ and TNNT2+/HAND1+ double positive cells at day 23 from multiple wells were counted manually in ImageJ (Abràmoff et al., 2004). Fisher’s exact test was used to calculate statistical significance between groups. Non-saturated mean intensity of NPPA+, TBX2+ or RSPO3+ signal was measured in ImageJ within each TNNT2+ cell from each genotype at day 23. Unpaired t-tests were used to calculate statistical significance. Brightness and contrast of images in Figures 3I and 4G have been adjusted to facilitate viewing of cells.
Graphing and statistics for electrophysiology.
For electrophysiology and calcium imaging experiments, graphs were generated using Prism 8.2.0 (GraphPad Software). Significance between parental and experimental groups was determined with a custom R-script using unpaired two-sided Welch’s t tests with Holm-Sidak correction for multiple comparisons (Holm, 1979). Adjusted p-value<0.05 was considered statistically significant.
Statistical analyses for correlations.
We evaluated the pairwise association among 38 variables, including all human genes, TBX5-dysregulated genes in human cardiomyocytes from day 23, CHD genes, EP genes, TBX5 or GATA4 binding (Ang et al., 2016), and genome-wide association (GWAS) genes for CHDs or arrhythmias (Cordell et al., 2013a; 2013b; Ellinor et al., 2012; Hoed et al., 2013; Hu et al., 2013; Pfeufer et al., 2010; Smith et al., 2011). Reported GWAS genes from https://www.ebi.ac.uk/gwas/ for the terms congenital heart disease, congenital heart malformation, congenital left sided heart lesions, conotruncal heart defect and aortic coarctation were used to define congenital heart disease-related (CHD) GWAS genes. Reported genes from terms such as cardiac arrhythmia, supraventricular ectopy, ventricular ectopy, premature cardiac contractions, atrial fibrillation, sudden cardiac arrest and ventricular fibrillation were considered as arrhythmia-related (EP-GWAS) genes. Two nearest genes within 100kb, by using GREAT (great.stanford.edu) (McLean et al., 2010), of TBX5 or GATA4 binding sites or of reported genes from each group of GWAS, were considered for the analysis. The natural logarithm odds of genes associating with each one of these variables versus the odds of genes associating with every other variable were estimated using generalized linear models with family=“binomial” setting in R. The resulting significance of these natural log odds ratios were adjusted for multiple testing by the Benjamini-Hochberg method (Benjamini and Hochberg, 1995). Significance was determined using an FDR threshold of 0.05 or less.
Additional correlations were evaluated between 26 variables, including human genes, human TBX5-dysregulated genes from day 23 cardiomyocytes, and TBX5, MEF2c or MEF2a binding sites from E12.5 mouse heart tissue (Akerberg et al., 2019). Human gene symbols were converted to mouse gene symbols, using the getLDS() function from the biomaRt package (https://www.r-bloggers.com/converting-mouse-to-human-gene-names-with-biomart-package/). Two nearest genes within 100kb of TBX5, MEF2c or MEF2a binding sites were considered for the analysis.
For assessment of associations between binding locations of TBX5, MEF2c and MEF2a transcription factors with genes dysregulated by TBX5, analyses were performed corresponding to binding regions of each of the three TFs. First, binding regions of each TF was evaluated for association with genes, defined by the nearest two genes within 100kb. Using the list of human TBX5-dysregulated genes, binding regions of each TF associated with a TBX5-dysregulated gene was determined. Identified binding regions of each TF that overlapped with at least 50% of the binding regions of each of the other two TFs was determined, using bedops --element-of - 50%. This approach defined three variables, including every binding region of the TF, if associated with a TBX5-dysregulated gene, or if it overlaps by at least 50% with the binding region of the other TFs, that were used for logistic regression in R. The resulting changes in odds are represented as natural logarithm odds ratios. Multiple testing correction was performed using the multtest package in R. All estimates are based on analyses for human TBX5-dysregulated genes.
Supplementary Material
Table S1. Gene features and weights for classifying cell type predictions. Related to Figure 2.
Table S2. TBX5-dependent genes in human iPSC-derived cardiomyocytes. Related to Figure 4, S5. Lists of differential genes from comparisons between TNNT2+ clusters, at day 23 by biological replicate or genetic background at day 23 are tabulated.
Table S3. Cardiac development or disease-related genes. Related to Figure 4, S5, S6, 7. Gene lists, which are used in this study, include electrophysiology (EP) genes, human congenital heart disease (CHD) genes, mouse CHD genes, cardiac development genes, CHD-associated GWAS genes and EP-related GWAS genes, are curated.
Table S4. Expression of TBX5-dependent genes in human fetal hearts. Related to Figure 5. Z-scores for expression of TBX5-dependent genes by anatomic region from spatial transcriptomics of human fetal hearts from 4.5-5, 6.5 and 9 weeks post-conception (Asp, et al., 2019) are compiled.
Table S5. Correlations near human TBX5-dependent genes. Related to Figures 4, S4, S5. Genes near TBX5 or GATA4 occupancy, congenital heart disease (CHD)-associated GWAS, electrophysiology (EP)-related GWAS, CHD genes, or EP genes are tallied. Odds ratios are displayed as natural logarithms. Statistical significance was determined by Benjamini-Hochberg multiple testing. Het: TBX5in/+; Hom: TBX5in/del; PGP1-Het: TBX5+/in; PGP1-Hom: TBX5del/del.
Table S6. TBX5-sensitive gene regulatory network analyses. Related to Figures 6, S6. Network parameters, including pagerank or degree, or correlations with TBX5 or MEF2C expression, are assessed by time point. D: day; C: Control; WT.CO: WTC11 and Control; WT: WTC11; DN: down; TBX5het: TBX5in/+; TBX5hom: TBX5in/del.
Table S7. Correlation of mouse orthologs near human TBX5-dependent genes. Related to Figure 7. Genes near TBX5, MEF2c or MEF2a occupancy, congenital heart disease (CHD)-associated GWAS, electrophysiology (EP)-related GWAS, CHD genes, or EP genes are tabulated. Odds ratios as natural logarithms are displayed. Statistical significance was determined by Benjamini-Hochberg multiple testing. het: TBX5in/+; hom: TBX5in/del.
Highlights.
Modeling human TBX5 haploinsufficiency manifests disease-related cellular defects.
Subpopulations of cardiomyocytes show distinct responses to reduced TBX5 dosage.
TBX5-sensitive genes are linked to congenital heart disease and cardiac function.
TBX5-dose dependent gene networks in humans predicts genetic interactions in mouse.
Acknowledgements.
We thank Dario Miguel-Perez and Sarah Winchester for mouse genotyping and colony maintenance, Jeff Farrell for input on URD, David Joy and Todd McDevitt for imaging software, Brian Black for mouse lines, Kathryn Claiborn for editorial assistance, and members of the Bruneau lab for discussions and comments. We also thank the Gladstone Bioinformatics, Genomics, Histology and Microscopy, and Stem Cell Cores, the UCSF Laboratory for Cell Analysis, the UCSF Center for Advanced Technology, the Salk Institutes Center of Excellence for Stem Cell Genomics, and the UCSC Stem Cell Data Center Hub. This work was supported by grants from the National Institutes of Health (NHLBI Bench to Bassinet Program UM1HL098179 to B.G.B. and UM1HL098166 to J.G.S., C.E.S. and W.T.P.; R01HL114948 to B.G.B., USCF CVRI 2T32HL007731-27 to S.K.H), the California Institute for Regenerative Medicine (RB4-05901 to B.G.B), the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Review Medical Research Program under Award No. W81XWH-17-1-0191 (B.G.B), the Foundation for Anesthesia Education and Research (Mentored Research Training Grant to I.S.K.), Society for Pediatric Anesthesia (Young Investigator Award to I.S.K.), Hellman Family Fund (I.S.K.), UCSF REAC Grant (I.S.K.) and UCSF Department of Anesthesia and Perioperative Care (New Investigator Award to I.S.K.). H.H. is a Miguel Servet (CP14/00229) researcher supported by the Spanish Institute of Health Carlos III (ISCIII) and Ministerio de Ciencia, Innovación y Universidades (SAF2017-89109-P; AEI/FEDER, UE). This work was also supported by an NIH/NCRR grant (C06 RR018928) to the J. David Gladstone Institutes, and the Younger Family Fund (B.G.B.).
Footnotes
Declaration of Interests: B.G.B. is a co-founder and shareholder of Tenaya Therapeutics. None of the work presented here is related to the interests of Tenaya Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES.
- Abràmoff MD, Magalhães PJ, and Ram SJ (2004). Image processing with ImageJ. Biophotonics International 11, 36–42. [Google Scholar]
- Akerberg BN, Gu F, VanDusen NJ, Zhang X, Dong R, Li K, Zhang B, Zhou B, Sethi I, Ma Q, et al. (2019). A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat Commun 10, 4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RH, Spicer DE, Brown NA, and Mohun TJ (2014). The Development of Septation in the Four-Chambered Heart. Anat Rec 297, 1414–1429. [DOI] [PubMed] [Google Scholar]
- Anderson DJ, Kaplan DI, Bell KM, Koutsis K, Haynes JM, Mills RJ, Phelan DG, Qian EL, Leitoguinho AR, Arasaratnam D, et al. (2018a). NKX2–5 regulates human cardiomyogenesis via a HEY2 dependent transcriptional network. Nat Commun 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RH, Spicer DE, Mohun TJ, Hikspoors JPJM, and Lamers WH (2018b). Remodeling of the Embryonic Interventricular Communication in Regard to the Description and Classification of Ventricular Septal Defects. Anat Rec 302, 19–31. [DOI] [PubMed] [Google Scholar]
- Ang Y-S, Rivas RN, Ribeiro AJS, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TMA, Fu J-D, Spencer CI, et al. (2016). Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell 167, 1734–1749.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asp M, Giacomello S, Larsson L, Wu C, Fürth D, Qian X, Wärdell E, Custodio J, Reimegård J, Salmén F, et al. (2019). A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 179, 1647–1660.e19. [DOI] [PubMed] [Google Scholar]
- Basson CT, Cowley GS, Solomon SD, Weissman B, Poznanski AK, Traill TA, Seidman JG, and Seidman CE (1994). The clinical and genetic spectrum of the Holt-Oram syndrome (heart-hand syndrome). N Engl J Med 330, 885–891. [DOI] [PubMed] [Google Scholar]
- Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, et al. (1997). Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 15, 30–35. [DOI] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Statistical Methodology 57, 289–300. [Google Scholar]
- Brassington A-ME, Sung SS, Toydemir RM, Le T, Roeder AD, Rutherford AE, Whitby FG, Jorde LB, and Bamshad MJ (2003). Expressivity of Holt-Oram syndrome is not predicted by TBX5 genotype. Am J Hum Genet 73, 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brin S, and Page L (1998). The anatomy of a large-scale hypertextual Web search engine. Computer Networks and Isdn Systems 30, 107–117. [Google Scholar]
- Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, et al. (2001). A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106, 709–721. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 48, 1070–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne SM, and Church GM (2015). Crispr-mediated Gene Targeting of Human Induced Pluripotent Stem Cells. Current Protocols in Stem Cell Biology 35, 5A.8.1–A.8.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churko JM, Garg P, Treutlein B, Venkatasubramanian M, Wu H, Lee J, Wessells QN, Chen S-Y, Chen W-Y, Chetal K, et al. (2018). Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat Commun 9, 326–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell HJ, Topf A, Mamasoula C, Postma AV, Bentham J, Zelenika D, Heath S, Blue G, Cosgrove C, Granados Riveron J, et al. (2013a). Genome-wide association study identifies loci on 12q24 and 13q32 associated with Tetralogy of Fallot. Hum Mol Genet 22, 1473–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell HJ, Bentham J, Töpf A, Zelenika D, Heath S, Mamasoula C, Cosgrove C, Blue G, Granados-Riveron J, Setchfield K, et al. (2013b). Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat Genet 45, 822–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creemers EE, Sutherland LB, McAnally J, Richardson JA, and Olson EN (2006). Myocardin is a direct transcriptional target of Mef2, Tead and Foxo proteins during cardiovascular development. Development 133, 4245–4256. [DOI] [PubMed] [Google Scholar]
- Dai W, Laforest B, Tyan L, Shen KM, Nadadur RD, Alvarado FJ, Mazurek SR, Lazarevic S, Gadek M, Wang Y, et al. (2019). A calcium transport mechanism for atrial fibrillation in Tbx5-mutant mice. eLife 8, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, Hinson JT, Homsy J, Gray J, Pu W, et al. (2016). Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev Cell 39, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devalla HD, Gélinas R, Aburawi EH, Beqqali A, Goyette P, Freund C, Chaix MA, Tadros R, Jiang H, Le Béchec A, et al. (2016). TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol Med 8, 1390–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine WP, Wythe JD, George M, Koshiba-Takeuchi K, and Bruneau BG (2014). Early patterning and specification of cardiac progenitors in gastrulating mesoderm. eLife 3, 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driegen S, Ferreira R, van Zon A, Strouboulis J, Jaegle M, Grosveld F, Philipsen S, and Meijer D (2005). A generic tool for biotinylation of tagged proteins in transgenic mice. Transgenic Res 14, 477–482. [DOI] [PubMed] [Google Scholar]
- Duan J, Li B, Bhakta M, Xie S, Zhou P, Munshi NV, and Hon GC (2019). Rational Reprogramming of Cellular States by Combinatorial Perturbation. CellReports 27, 3486–3499.e3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner DA, Caldwell JL, Trafford AW, and Hutchings DC (2020). The Control of Diastolic Calcium in the Heart: Basic Mechanisms and Functional Implications. Circ Res 126, 395–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, Arking DE, Müller-Nurasyid M, Krijthe BP, Lubitz SA, et al. (2012). Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 44, 670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell JA, Wang Y, Riesenfeld SJ, Shekhar K, Regev A, and Schier AF (2018). Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 360, eaar3131–eaar3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA (1921). On the“ Probable Error” of a Coefficient of Correlation Deduced from a Small Sample. Metron 1, 3–32. [Google Scholar]
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, et al. (2003). GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. 424, 443–447. [DOI] [PubMed] [Google Scholar]
- Ghosh TK, Song FF, Packham EA, Buxton S, Robinson TE, Ronksley J, Self T, Bonser AJ, and Brook JD (2009a). Physical interaction between TBX5 and MEF2C is required for early heart development. Mol Cell Biol 29, 2205–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford CA, Ranade SS, Samarakoon R, Salunga HT, de Soysa TY, Huang Y, Zhou P, Elfenbein A, Wyman SK, Bui YK, et al. (2019). Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 364, 865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He A, Gu F, Hu Y, Ma Q, Yi Ye L, Akiyama JA, Visel A, Pennacchio LA, and Pu WT (2014). Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat Commun 5, 4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroi Y, Kudoh S, Monzen K, Ikeda Y, Yazaki Y, Nagai R, and Komuro I (2001). Tbx5 associates with Nkx2–5 and synergistically promotes cardiomyocyte differentiation. Nat Genet 28, 276–280. [DOI] [PubMed] [Google Scholar]
- Hoed, den M, Eijgelsheim M, Esko T, Brundel BJJM, Peal DS, Evans DM, Nolte IM, Segrè AV, Holm H, Handsaker RE, et al. (2013). Identification of heart rate–associated loci and their effects on cardiac conduction and rhythm disorders. Nat Genet 45, 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JI (1995). Incidence of congenital heart disease: II. Prenatal incidence. Pediatr Cardiol 16, 155–165. [DOI] [PubMed] [Google Scholar]
- Hoffman JIE, and Kaplan S (2002). The incidence of congenital heart disease. J Am Coll Cardiol 39, 1890–1900. [DOI] [PubMed] [Google Scholar]
- Holm S (1979). A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics 6, 65–70. [Google Scholar]
- Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. (2015). De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hookway TA, Matthys OB, Mendoza-Camacho FN, Rains S, Sepulveda JE, Joy DA, and McDevitt TC (2019). Phenotypic Variation Between Stromal Cells Differentially Impacts Engineered Cardiac Tissue Function. Tissue Engineering Part A 25, 773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Shi Y, Mo X, Xu J, Zhao B, Lin Y, Yang S, Xu Z, Dai J, Pan S, et al. (2013). A genome-wide association study identifies two risk loci for congenital heart malformations in Han Chinese populations. Nat Genet 45, 818–821. [DOI] [PubMed] [Google Scholar]
- Iacono G, Mereu E, Guillaumet-Adkins A, Corominas R, Cuscó I, Rodríguez-Esteban G, Gut M, Pérez-Jurado LA, Gut I, and Heyn H (2018). bigSCale: an analytical framework for big-scale single-cell data. Genome Research 28, 878–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacono G, Massoni-Badosa R, and Heyn H (2019). Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol 20, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, and Srivastava D (2010). Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. 142, 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, et al. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet 49, 1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge LM, Perez-Bermejo JA, Truong A, Ribeiro AJS, Yoo JC, Jensen CL, Mandegar MA, Huebsch N, Kaake RM, So P-L, et al. (2017). A BAG3 chaperone complex maintains cardiomyocyte function during proteotoxic stress. JCI Insight 2, 83–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakikes I, Termglinchan V, Cepeda DA, Lee J, Diecke S, Hendel A, Itzhaki I, Ameen M, Shrestha R, Wu H, et al. (2017). A Comprehensive TALEN-Based Knockout Library for Generating Human-Induced Pluripotent Stem Cell-Based Models for Cardiovascular Diseases. Circ Res 120, 1561–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforest B, Dai W, Tyan L, Lazarevic S, Shen KM, Gadek M, Broman MT, Weber CR, and Moskowitz IP (2019). Atrial fibrillation risk loci interact to modulate Ca2+-dependent atrial rhythm homeostasis. J Clin Invest 412, 1825–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalani SR, and Belmont JW (2014). Genetic basis of congenital cardiovascular malformations. Eur J Med Genet 57, 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-H, Park I-H, Gao Y, Li JB, Li Z, Daley GQ, Zhang K, and Church GM (2009). A Robust Approach to Identifying Tissue-Specific Gene Expression Regulatory Variants Using Personalized Human Induced Pluripotent Stem Cells. PLoS Genet 5, e1000718–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T, et al. (1997). Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet 15, 21–29. [DOI] [PubMed] [Google Scholar]
- Li G, Xu A, Sim S, Priest JR, Tian X, Khan T, Quertermous T, Zhou B, Tsao PS, Quake SR, et al. (2016). Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Dev Cell 39, 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Schwarz J, Bucana C, and Olson EN (1997). Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 276, 1404–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MAH, He D, Small EM, Mileikovsky M, et al. (2016). Complex Interdependence Regulates Heterotypic Transcription Factor Distribution and Coordinates Cardiogenesis. Cell 164, 999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra M, Schluterman MK, Nichols HA, Richardson JA, Lo CW, Srivastava D, and Garg V (2009). Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. 326, 368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandegar MA, Huebsch N, Frolov EB, Shin E, Truong A, Olvera MP, Chan AH, Miyaoka Y, Holmes K, Spencer CI, et al. (2016). CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 18, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulley DJ, and Black BL (2012). Transcription factor pathways and congenital heart disease. Curr. Top. Dev. Biol. 100, 253–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott DA, Hatcher CJ, and Basson CT (2008). Atrial Fibrillation and Other Clinical Manifestations of Altered TBX5 Dosage in Typical Holt-Oram Syndrome. Circ Res 103, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Systems 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Tian L, Martin M, Paige SL, Galdos FX, Li J, Klein A, Zhang H, Ma N, Wei Y, et al. (2020). Intrinsic Endocardial Defects Contribute to Hypoplastic Left Heart Syndrome. Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaoka Y, Chan AH, Judge LM, Yoo J, Huang M, Nguyen TD, Lizarraga PP, So P-L, and Conklin BR (2014). Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Meth 11, 291–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman AFM, and Christoffels VM (2003). Cardiac chamber formation: development, genes, and evolution. Physiological Reviews 83, 1223–1267. [DOI] [PubMed] [Google Scholar]
- Mori AD, and Bruneau BG (2004). TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealed. Curr Opin Cardiol 19, 211–215. [DOI] [PubMed] [Google Scholar]
- Mori AD, Zhu Y, Vahora I, Nieman B, Koshiba-Takeuchi K, Davidson L, Pizard A, Seidman JG, Seidman CE, Chen XJ, et al. (2006). Tbx5-dependent rheostatic control of cardiac gene expression and morphogenesis. Dev. Biol. 297, 566–586. [DOI] [PubMed] [Google Scholar]
- Nadadur RD, Broman MT, Boukens B, Mazurek SR, Yang X, van den Boogaard M, Bekeny J, Gadek M, Ward T, Zhang M, et al. (2016). Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Science Translational Medicine 8, 354ra115–354ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton A-L, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–933.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panesar DK, and Burch M (2017). Assessment of Diastolic Function in Congenital Heart Disease. Front. Cardiovasc. Med. 4, 2254–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 12, 2825–2830. [Google Scholar]
- Peters DT, Cowan CA, and Musunuru K (2008). Genome editing in human pluripotent stem cells (Cambridge (MA): Harvard Stem Cell Institute; ). [PubMed] [Google Scholar]
- Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, Tarasov KV, Müller M, Sotoodehnia N, Sinner MF, et al. (2010). Genome-wide association study of PR interval. Nat Genet 42, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan D, Rasmussen TL, Nakagawa O, McAnally J, Gottlieb PD, Tucker PW, Richardson JA, Bassel-Duby R, and Olson EN (2005). BOP, a regulator of right ventricular heart development, is a direct transcriptional target of MEF2C in the developing heart. Development 132, 2669–2678. [DOI] [PubMed] [Google Scholar]
- Prendiville T, Jay PY, and Pu WT (2014). Insights into the genetic structure of congenital heart disease from human and murine studies on monogenic disorders. Cold Spring Harb Perspect Med 4, a013946–a013946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest JR, Osoegawa K, Mohammed N, Nanda V, Kundu R, Schultz K, Lammer EJ, Girirajan S, Scheetz T, Waggott D, et al. (2016). De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects. PLoS Genet 12, e1005963–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu J-D, and Srivastava D (2012). In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485, 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifrim A, Hitz M-P, Wilsdon A, Breckpot J, Turki Al, S.H., Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ, et al. (2016). Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet 48, 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Hoogaars WM, Barnett P, Grieskamp T, Rana MS, Buermans H, Farin HF, Petry M, Heallen T, Martin JF, et al. (2012). Tbx2 and Tbx3 induce atrioventricular myocardial development and endocardial cushion formation. Cell Mol Life Sci 69, 1377–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten LJ, and Pierpont ME (1996). Variation in severity of cardiac disease in Holt-Oram syndrome. Am J Med Genet 65, 128–132. [DOI] [PubMed] [Google Scholar]
- Smith JG, Magnani JW, Palmer C, Meng YA, Soliman EZ, Musani SK, Kerr KF, Schnabel RB, Lubitz SA, Sotoodehnia N, et al. (2011). Genome-Wide Association Studies of the PR Interval in African Americans. PLoS Genet 7, e1001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Nam Y-J, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, et al. (2012). Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CI, Baba S, Nakamura K, Hua EA, Sears MAF, Fu C-C, Zhang J, Balijepalli S, Tomoda K, Hayashi Y, et al. (2014). Calcium Transients Closely Reflect Prolonged Action Potentials in iPSC Models of Inherited Cardiac Arrhythmia. Stem Cell Reports 3, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopoulos A, and Levine M (2002). Dorsal Gradient Networks in the Drosophila Embryo. 246, 57–67. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM III, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoris CV, Li M, White MP, Liu L, He D, Pollard KS, Bruneau BG, and Srivastava D (2015). Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell 160, 1072–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron L, Steimle JD, Greco TM, Gomez NC, Dorr KM, Kweon J, Temple B, Yang XH, Wilczewski CM, Davis IJ, et al. (2016). The Cardiac TBX5 Interactome Reveals a Chromatin Remodeling Network Essential for Cardiac Septation. Dev Cell 36, 262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. (2013). De novo mutations in histone-modifying genes in congenital heart disease. 498, 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S, and Brueckner M (2017). Genetics and Genomics of Congenital Heart Disease. Circ Res 120, 923–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y-Q, Zhu Y, Bishop J, Davidson L, Henkelman RM, Bruneau BG, and Foster FS (2005). Abnormal cardiac inflow patterns during postnatal development in a mouse model of Holt-Oram syndrome. Am J Physiol Heart Circ Physiol 289, H992–H1001. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Gramolini AO, Walsh MA, Zhou Y-Q, Slorach C, Friedberg MK, Takeuchi JK, Sun H, Henkelman RM, Backx PH, et al. (2008). Tbx5-dependent pathway regulating diastolic function in congenital heart disease. Proceedings of the National Academy of Sciences 105, 5519–5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Gene features and weights for classifying cell type predictions. Related to Figure 2.
Table S2. TBX5-dependent genes in human iPSC-derived cardiomyocytes. Related to Figure 4, S5. Lists of differential genes from comparisons between TNNT2+ clusters, at day 23 by biological replicate or genetic background at day 23 are tabulated.
Table S3. Cardiac development or disease-related genes. Related to Figure 4, S5, S6, 7. Gene lists, which are used in this study, include electrophysiology (EP) genes, human congenital heart disease (CHD) genes, mouse CHD genes, cardiac development genes, CHD-associated GWAS genes and EP-related GWAS genes, are curated.
Table S4. Expression of TBX5-dependent genes in human fetal hearts. Related to Figure 5. Z-scores for expression of TBX5-dependent genes by anatomic region from spatial transcriptomics of human fetal hearts from 4.5-5, 6.5 and 9 weeks post-conception (Asp, et al., 2019) are compiled.
Table S5. Correlations near human TBX5-dependent genes. Related to Figures 4, S4, S5. Genes near TBX5 or GATA4 occupancy, congenital heart disease (CHD)-associated GWAS, electrophysiology (EP)-related GWAS, CHD genes, or EP genes are tallied. Odds ratios are displayed as natural logarithms. Statistical significance was determined by Benjamini-Hochberg multiple testing. Het: TBX5in/+; Hom: TBX5in/del; PGP1-Het: TBX5+/in; PGP1-Hom: TBX5del/del.
Table S6. TBX5-sensitive gene regulatory network analyses. Related to Figures 6, S6. Network parameters, including pagerank or degree, or correlations with TBX5 or MEF2C expression, are assessed by time point. D: day; C: Control; WT.CO: WTC11 and Control; WT: WTC11; DN: down; TBX5het: TBX5in/+; TBX5hom: TBX5in/del.
Table S7. Correlation of mouse orthologs near human TBX5-dependent genes. Related to Figure 7. Genes near TBX5, MEF2c or MEF2a occupancy, congenital heart disease (CHD)-associated GWAS, electrophysiology (EP)-related GWAS, CHD genes, or EP genes are tabulated. Odds ratios as natural logarithms are displayed. Statistical significance was determined by Benjamini-Hochberg multiple testing. het: TBX5in/+; hom: TBX5in/del.
Data Availability Statement
scRNA-seq datasets have been deposited at NCBI GEO, under accession GSE137876. R and python scripts will be available upon request.