

Abstract
The thalamus, a significant part of the diencephalon, is a symmetrical and bilateral central brain structure. The thalamus is subdivided into three major groups of nuclei based on their function: sensorimotor nuclei (or principal/relay nuclei), limbic nuclei and nuclei bridging these two domains. Anatomically, nuclei within the thalamus are described by their location, such as anterior, medial, lateral, ventral, and posterior. In this review, we summarize the role of medial and midline thalamus in cognition, ranging from learning and memory to flexible adaptation. We focus on the discoveries in animal models of alcohol-related brain damage, which identify the loss of neurons in the medial and midline thalamus as drivers of cognitive dysfunction associated with alcohol use disorders. Models of developmental ethanol exposure and models of adult alcohol-related brain damage and are compared and contrasted, and it was revealed that there are similar (anterior thalamus) and different (intralaminar [adult exposure] versus ventral midline [developmental exposure]) thalamic pathology, as well as disruptions of thalamo-hippocampal and thalamo-cortical circuits. The final part of the review summarizes approaches to recover alcohol-related brain damage and cognitive and behavioral outcomes. These approaches include pharmacological, nutritional and behavioral interventions that demonstrated the potential to mitigate alcohol-related damage. In summary, the medial/midline thalamus is a significant contributor to cognition function, which is also sensitive to alcohol-related brain damage across the life span, and plays a role in alcohol-related cognitive dysfunction.
Key terms: Fetal alcohol, alcoholism, thalamus, neuropathology, intervention
1. Introduction:
The thalamus, a significant part of the diencephalon, is a symmetrical and bilateral central brain structure. Nuclei within the thalamus are described by their anatomical location, using terms such as anterior, medial, lateral, ventral, and posterior. The medial and midline thalamus are key sites influencing memory and cognition. Even these restricted regions of the thalamus are heterogeneous with several key nuclei (anterior, medial dorsal, intralaminar and midline ventral [nucleus reuniens - Re, rhomboid - Rh]) having critical diversity in connectivity and function. The medial and midline thalamus comprise what has been coined the “cognitive thalamus” (see Cassel and Pereira de Vasconcelos 2015; Wolff and Vann 2019), which has positioned the thalamus’ status beyond its early description as a relay station. Sherman and Guillery (1996) suggested that the thalamus contains at least two types of nuclei: first-order nuclei which transfer sensory and subcortical information, and higher-order nuclei that significantly alter the nature of the information relayed to the cortex (see also Sherman 2016).
The study of alcohol-related brain damage revealed that the thalamus is critical for memory and cognition. This review will focus on the vulnerability of the thalamus to alcohol exposure, highlighting findings from animal models of developmental and adult alcohol-related brain damage. Interestingly, models of adult alcohol-related brain damage and models of developmental ethanol exposure display similar (anterior thalamus) and different (intralaminar [adult exposure] versus ventral midline [developmental exposure]) thalamic pathology, as well as disruptions of thalamo-hippocampal and thalamo-cortical circuits. In humans, heavy adult alcohol consumption with associated thiamine deficiency can progress into the amnestic disorder of Korsakoff Syndrome (KS), a type of diencephalic amnesia. Extensive research, including patients with, and animal models of, KS have been conducted to determine the structural underpinnings of amnesia and failing executive functioning. Studies using rodent models of KS have revealed that the anterior and midline intralaminar nuclei are consistently damaged, with minimal damage to the medial dorsal nucleus (Mair et al. 2015, Savage et al. 2012). Models of third-trimester alcohol exposure display increased cell death and shrinkage in the anterior thalamus (Farber et al. 2010; Wozniak et al. 2004). In addition, there is selective cell loss in the Re, with sparing of surrounding midline thalamic regions (Gursky et al. 2019). Prior to summarizing the critical literature on alcohol-related thalamic pathology, we will first review the neuroanatomy and functional consequences of neuronal loss to these critical medial and midline thalamic regions, based primarily on studies in rodents.
There is strong correspondence in alcohol-related thalamic pathology across humans and animal studies. While the thalamus has been the target of human neuroimaging studies of human patients diagnosed with FASD or adult alcohol use disorders (AUDs), past technical limitations have prevented analysis of individual thalamic nuclei. Despite this, most studies do recognize significant reductions in thalamic volume throughout childhood and adolescence, greater than can be accounted for gross microcephaly associated with FASD (Lebel et al. 2008; Mattson et al. 1992; Meintjes et al. 2014; Nardelli et al. 2011; Treit et al. 2013). While some studies do not observe alcohol-induced changes in whole-thalamus volume in humans (Archibald et al. 2001) or rodents (Akers et al. 2011), rodent studies suggest that this may be due to nucleus-specific alterations to thalamus (Akers et al. 2011; Gursky et al. 2019). It is possible that both alcohol-induced cell death (Gursky et al. 2020) and altered development of the 3rd ventricle (Petrelli et al. 2018) may contribute to thalamic malformation in FASD in both humans and in rodent models of the disorder, but these hypotheses have yet to be explored in depth. The specificity and details of these alterations will be discussed further in Section 4 (“Animal models of developmental alcohol exposure”).
Imaging studies in adult patients with severe AUD have supported post-mortem histopathological analysis (Harding et al, 2000; Victor et al, 1971) that the medial thalamus is affected in both uncomplicated patients with severe AUD and those with AUD and thiamine deficiency; However, patients with KS displaying the greatest thalamic pathology (Pitel et al., 2014; Sullivan and Pfefferbaum, 2008). A recent study (Segobin et al, 2019), with a more detailed structural and connectivity analyses, revealed that chronic and excessive consumption of alcohol is associated with structural and connectivity alterations in the mediodorsal and anterior thalamic nuclei. However, atrophy of the anterior thalamic was relatively specific to individuals with severe AUD that progressed into KS. However, neurohistological analyses of thalamic pathology in cases of KS display cell loss in both the anterior and medial dorsal thalamic nuclei (Harding et al, 2000; Victor et al, 1971). Animal studies modeling adult alcohol-related pathology have demonstrated extensive thalamic pathology following thiamine deficiency, but not after chronic ethanol exposure. These studies confirm that thiamine-deficiency-induced lesions are limited to the ventral portion of medial dorsal, but more extensive cell loss is observed in several anterior and midline intralaminar nuclei (see Savage 2012 et al, for a review). Thus, our focus will be on alcohol-related pathology in the anterior and midline thalamic regions.
2. Evidence that the medial and midline thalamus are key elements of the cognitive thalamus.
This section summarizes the thalamic nucleus-specific contributions to behaviors by highlighting three functional midline thalamic groups (Groenewegen 1988): the anterior nuclei, the intralaminar nuclei, and the ventral midline thalamus, which includes the Re and rhomboid (Rh) nuclei. One major challenge to discussing the consequences of thalamic damage in any animal models, including models of alcohol exposure, is the technical difficulty in identifying unique versus overlapping contributions of each nucleus to behavior. Understanding the unique versus redundant contributions of each nucleus to behavior provides a foundation for functional implications of nucleus-specific neuroanatomical alterations, such as those observed in models of fetal alcohol spectrum disorders (FASD; discussed in Section 4) and adult alcohol use disorders (discussed in Section 5).
The traditional subdivisions of midline thalamic nuclei at times appears discrepant with recent findings of functional subdivisions within each nucleus (Gao et al. 2020), obtained from correlational observations of neuroanatomy and neuronal activity markers (e.g., Vann et al. 2000) or in vivo electrophysiological function (Dollemann-van der Weel et al. 1997; Ito et al. 2015), which provide the most precise delineation of individual nuclei, their subdivisions, and their function during behavior. Distinct functional properties can be challenging to infer at the level of an individual thalamic nucleus for the following three reasons: small size or uneven shape of the nucleus, overlap in afferent/efferent targets between nuclei, and heterogeneity of neuronal function within each structure. Although there are technical limitations to uncovering these specific contributions, the concurrent use of multiple techniques to provide convergent evidence for the role of thalamic nuclei in cognition. Understanding the specificity of thalamic influence on cortico-thalamic circuitry and behavior provides a strong foundation to understand the functional consequences of alcohol-related brain damage.
2.1. Anterior thalamus:
The anterior thalamus is comprised of the anterodorsal (AD), anteroventral (AV), and anteromedial (AM) nuclei, and is part of the rostral dorsomedial thalamus (Jankowski et al. 2013). These subregions have unique connectivity to the hippocampus, parahippocampus, retrosplenial cortex, medial prefrontal cortex (mPFC), and therefore contribute to spatial mapping and are associated with spatial learning and memory. The AD nucleus (60% neurons) and the AV nucleus (39% neurons) were first discovered to house head direction (HD) cells (Taube 2007; Tsanov et al. 2011; Clark and Taube 2012). Later, HD cells were also found near the Re (Jankowski et al. 2014). HD cells fire when the head moves in specific directions (not location), and encode primary information to organize spatial orientation within the environment (for review see Taube 2007; Clark and Taube 2012). Although HD cells rely on vestibular and proprioceptive information, these neurons have been called “the compass of the brain”, as they respond when an animal points its head in a particular direction (Jankowski et al. 2014). These neurons allow organisms to keep track of their movements and position, even in a cue-free environment (Knierim et al. 1995). Anterior thalamic projections to the postsubiculum are critical for the establishment of HD cell activity throughout the parahippocampal circuit (Preston-Ferrer et al. 2016). Hippocampal place cells and thalamic HD cells are strongly coupled and are involved in spatial representation (Knierim et al. 1995). Place cells use the signal from the HD system to establish and maintain place-field activity (Dudchenko et al. 2019). Lesions of brain areas that convey the HD signal, such as the anterior thalamus, can degrade place fields, leading to place-field instability (Carlton et al. 2003). Place cells fire selectively at one or few locations in the environment and express current as well as past and future spatial experiences (Moser et al. 2015). HD signals from the anterior thalamus are necessary for the generation and function of grid cell activity (Winter et al. 2015). There are a limited number of grid-like cells in the AM, with the majority residing in the entorhinal cortex (EC). Grid cells have multiple firing fields with clear regions of silence between the fields, creating a triangular or hexagonal grid-like pattern. HD cells, place cells and grid cells form a network from which spatial representations are formed and remembered (Moser et al. 2015).
Within the anterior thalamus, theta-on cells were found within the AV (94%), AD (77%) and AM (71%), and approximately 75% of cells of the AV fire in synchrony with hippocampal theta rhythm (Vertes et al. 2001). It should be noted that AD, AV and AM have varying amounts of HD cells (AD>AV>AM) and theta activity (AM≈AV>AD) (Perry and Mitchell 2019). It has been suggested that theta serves as an important signal involved in the differential processing of HD activity to selectively guide spatial behaviors (Vertes et al. 2001). The crossover between theta and HD signals support the conclusion that anterior thalamus integrates information related to heading and movement, and is critical for spatial information processing (Tsanov et al. 2011).
It is therefore not surprising that rodents with anterior thalamic nuclei (ATN) lesions show substantial spatial memory deficits (Aggleton et al. 1996; Byatt and Dalrymple-Alford 1996; Savage et al. 2011; Sziklas and Petrides 1999). We found that complete lesions of the ATN impaired performance on spontaneous alternation and delayed alternation, measures of hippocampal-dependent learning and memory, and such lesions severely disrupted behaviorally-evoked acetylcholine (ACh) efflux within the hippocampus (Savage et al. 2011). These data supports the notion that damage to the anterior thalamus directly down-regulate function within the hippocampus. There is also evidence that the anterior thalamus may be required for other hippocampal and cortical processing, such as contextual learning (Dumont et al. 2014; Law & Smith. 2012; Marchand et al. 2014), as well as attentional control (Nelson et al. 2018), as these behaviors are disrupted following ATN lesions. There is some supporting evidence that different ATN support different cognitive factors: The AD nucleus is critical for direction information, whereas the AV nucleus is more involved in place/location learning and memory, and the AM nucleus appears to be necessary for attentional and temporal information (Aggleton and Nelson 2015).
2.2. Intralaminar thalamus:
The intralaminar thalamic nuclei are located lateral to the mediodorsal nucleus, surrounding it in a U shape and are embedded within the myelin sheath of the internal medullary lamina. It is the least explored of the medial/midline thalamic groups. Often the intralaminar nuclei are classified into two groups based on their anterior to posterior location (see Van der Werf et al. 2002; Vertes et al. 2015a): The rostral group consists of the central medial (CM), paracentral (PC), and central lateral (CL) nuclei; whereas the caudal group is composed of the parafascicular (PF) and subparafascicular nuclei (SPF). This collection of nuclei project to different cortical regions, but all have the dorsal striatum as a common target. Key projection targets of the intralaminar nuclei are the dorsal striatum and cortex, with other critical projections including the ventral striatum and amygdala. The lateral (PC, CL) and caudal (PF, SPF) nuclei mostly innervate the sensorimotor cortex and the dorsal striatum, whereas CM has more diverse innervation including the cingulate, perirhinal and orbital frontal cortices, amygdala, ventral striatum, in addition to innervation of the dorsal striatum and premotor cortex (Benendese and Groenewegen 1991; Van der Werf et al. 2002; Yasukawa et al. 2004; Vertes et al. 2015a). The PF and CM nuclei differ from other intralaminar nuclei as they have denser projections to the basal ganglia, mostly the caudate and putamen, but also innervate the sensory and motor cortices.
There is substantial brainstem input to the intralaminar nuclei. There is dense innervation from the reticular formation, the cholinergic pedunculopontine and the laterodorsal tegmental nuclei, the serotonergic axons from the dorsal raphe, dopaminergic input from the substantia nigra, and noradrenergic input from the locus coeruleus nucleus (Krout et al. 2002). Frontal cortical regions, including the dorsal prelimbic, anterior cingulate, orbital frontal and premotor areas, project to the intralaminar nuclei (Berendse and Groenewegen 1991). Furthermore, the basal ganglia return direct projections to the caudal intralaminar complex (Parent et al. 1983; Sidibe et al. 1997). Through critical reciprocal thalamo-cortico-striatal connectivity patterns, the intralaminar nuclei play a significant role in coordinating the activity across cortical, limbic, and basal ganglia circuits, which influences a variety of cognitive functions. Furthermore, intralaminar neurons burst discharge to a variety of salient stimuli and likely play an essential role in shifting attention and responses to changing environmental conditions (Matsumoto et al. 2001).
Lesion studies have shed light on the role of the functional outputs of the intralaminar nuclei. The main findings are that lesions to the intralaminar nuclei produce impairment on frontal cortical, hippocampal and dorsal striatal tasks and difference across studies likely reflect differences in lesion size that can include different nuclei across the rostral-caudal axis. Frontal cortical-like dysfunction following intralaminar lesions has been reported on operant delayed match to position task (Bailey and Mair 2005; Burk and Mair 1998). Intralaminar lesions impair re-acquisition and short-term delayed recall of spatial location on the operant non-matching-to-position (NMTP) task (Savage et al. 1997), with little recovery, even 8 months post-lesion. In addition, persistent impairments of go/no go responding in continuous olfactory delayed nonmatching task occur following intralaminar lesions (Koger and Mair 1994, Mair et al. 1998, Zhang et al. 1998). These studies demonstrate that both spatial and nonspatial working memory, which are dependent on the frontal cortex, are impaired by lesions of the intralaminar nuclei. Furthermore, attentional set-shifting, known to rely on the mPFC, in which the strategy for successful discrimination is changed from visual cues to a place-based strategy was impaired following immunotoxin lesion of CL (Kato et al. 2018).
When rats with lesions of the intralaminar nuclei were tested on hippocampal-dependent tasks, such as the radial arm maze delayed nonmatching to position task where choices were drawn at random (to prevent egocentric solutions) or learning the location of a hidden platform in the Morris water maze, significant impairment was observed (Mair et al. 1998, Mair et al. 2003; Savage et al. 1998). Rats with lesions that encompassed all intralaminar nuclei (CL, CM, PC, PF) and some midline (AM, interoanteromedial [IAM]) regions were impaired at learning the location of a hidden platform in the water maze task. In contrast, rats that had limited lesions, restricted to the lateral nuclei (CL, PC, and PF) were not impaired on the Morris water maze (Savage et al. 1998). Rostral intralaminar lesions did not alter acquisition or short-term recall (5 days post-training) in the water maze task, but such lesions did impair remote memory (25 days post-training) on this task (Lopez et al. 2009). However, in that study, it was unclear whether the intralaminar nuclei participate in consolidation or long-term recall.
Damage to the caudal intralaminar nuclei can also lead to impairments on striatal-dependent tasks. Such intralaminar lesions disrupted performance on a visuospatial reaction time task, which is sensitive to striatal damage (Mair et al. 2002). Lesions or inactivation of the PF and other lateral intralaminar nuclei impaired reversal of rewarded place learning and prevented the rise in ACh in the dorsal striatum during the rule shift (Brown et al. 2010).
A working hypothesis is that medial/intralaminar thalamus is a diverse region that coordinates prefrontal cortical neural activity with other cortical, limbic, and basal ganglia circuits that modulate complex cognitive functions such as attentional processes, behavioral flexibility, and working memory (Saalmann 2014). Although there appears to be a functional dissociation across the rostral-caudal axis of the intralaminar nuclei, such distinction can be difficult to adequately distinguish given the size and close proximity of nuclei in this grouping.
2.3. Nucleus Reuniens:
The Re is often called the ventral midline thalamus as it resides below the IAM, CM and Rh nuclei and directly above the third ventricle. It is the largest of the midline/intralaminar nuclei. It is commonly subdivided into a central/medial portion and lateral “wings” referred to as the peri-reuniens nucleus (Dolleman-van der Weel et al. 2019). In many studies, the Re is associated with the Rh nucleus, as their proximity can make it difficult to assess the distinct function of each in experimental preparations. To highlight the difficulty imposed by the shape and size of these nuclei, Figure 1 demonstrates the outlines of Re and Rh nucleus, taken from two sections approximately 0.5 mm apart. It is obvious that Rh is less than 0.5 mm tall (dorsal-ventral) at all points in Figure 1A (shorter dotted outline) but is very wide (medial-lateral), while it is more evenly shaped in Figure 1B. In contrast, Re is more evenly shaped in Figure 1A (longer dashed outline), while it is impinged upon by the xiphoid nucleus (resulting in a substantial concave curvature of its ventral surface) in Figure 1B. The irregular shapes and small sizes of these nuclei make it infeasible to comprehensively target Rh by injection (e.g., using tracers, drugs) without diffusion into Re across the ventral border of Rh, or the IAM nucleus (or CM, depending on the rostral-caudal location) immediately dorsal to Rh. In the rodent, many midline “limbic” thalamic nuclei have a wide and flat shape (Vertes et al. 2015b). While this makes it difficult to establish whether a single nucleus is necessary for a given behavior, manipulations often determine the role of a group of nuclei in conjunction, such as the common grouping of Rh and Re into the singular “ventral midline thalamus” (e.g., Hallock et al. 2016; Klein et al. 2019; Linley et al. 2016).
Figure 1.
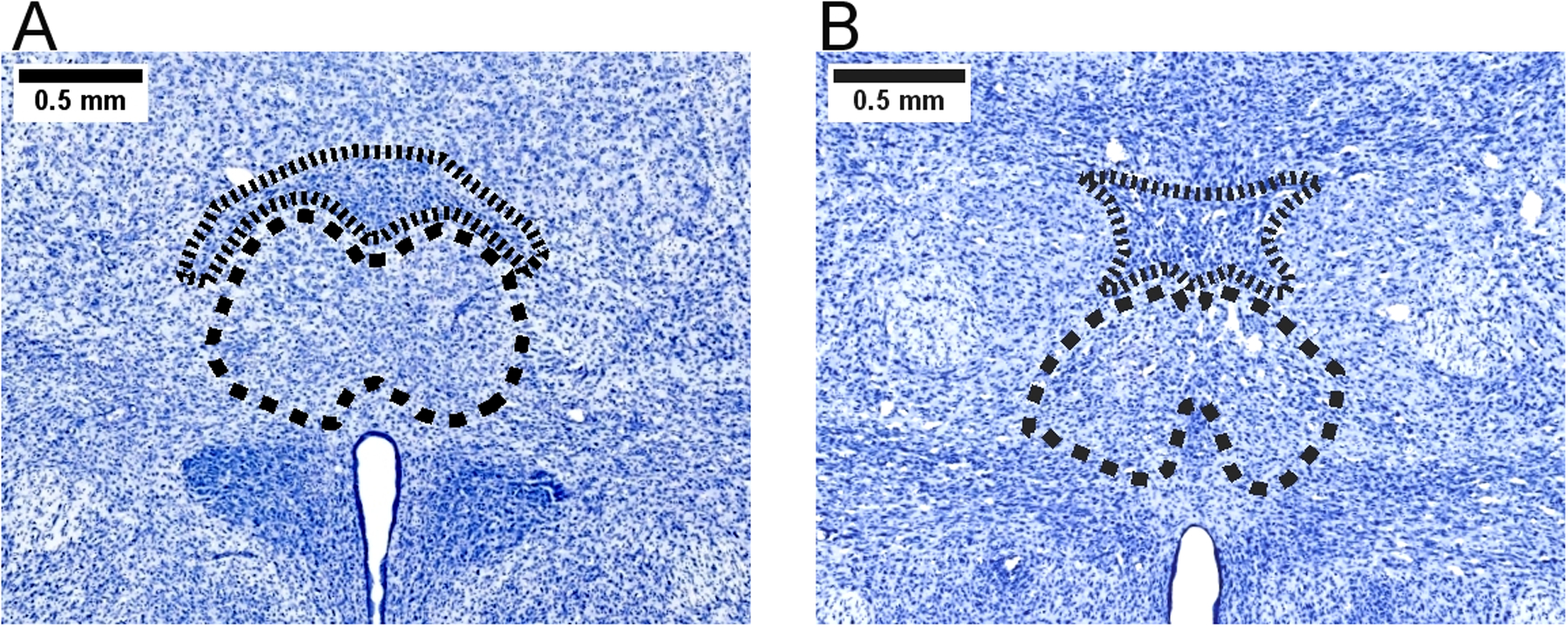
Outlines of the nucleus reuniens (Re) and rhomboid nucleus (Rh) in Nissl-stained sections of rat thalamus. The photomicrographs demonstrate anatomically delineated contours of thalamic Re (large dashed outlines) and Rh (small dotted outline). The section in panel A is approximately 2.0 mm caudal to bregma, while the section in panel B is approximately 2.5 mm caudal to bregma. Tissue is stained with Cresyl Fast Violet. Scale bar = 0.5mm.
Afferents into the Re are diverse: A large range of sites that include the brainstem (periaqueductal gray, and commissure nuclei), hypothalamus (median and lateral preoptic nucleus, lateral hypothalamic area, supramammillary nucleus), amygdala (medial, basomedial, and posterior nuclei), basal forebrain (lateral septum and diagonal band), claustrum, and entire limbic cortex (McKenna and Vertes 2004) send inputs into the Re. Vertes (2006) found that all four subregions of the mPFC (prelimbic [PL], infralimbic [IL], medial orbital [MO], anterior cingulate [AC] cortices) project densely to the Re. Furthermore, the Re also receives inputs from presubiculum, postsubiculum, and retrosplenial cortex, and these sites contain HD cells (McKenna and Vertes 2004). This pattern of connectivity suggests various potential sources for the heading information to merge - into the Re. Projections into the rostral Re and caudal Re are relatively comparable, with the exception that the rostral region receives heavier inputs, than the caudal region, from subcortical limbic regions, such as the basal forebrain, diencephalon, and brainstem, as well as from the CA1/subiculum hippocampal sectors (McKenna and Vertes 2004).
In contrast, the efferents from Re are more restricted to limbic sites such as the orbital and mPFC, the retrosplenial cortex, as well as the parahippocampal and hippocampal regions (Vertes 2006; Vertes et al. 2006). Within the prefrontal cortex, the Re, specifically the caudal and peri-Re regions, have dense projections to layers 1, 5/6 of the IL, PL, and AC cortices (Hoover and Vertes 2012). The Re also strongly projects to the EC (Vertes et al. 2006).
The Re, particularly the rostral pole, selectively sends projections to the CA1 region and subiculum of the hippocampus, with the ventral hippocampus receiving significantly greater innervation than the dorsal hippocampus (Hoover and Vertes 2012). Fibers from the Re form excitatory contacts with pyramidal cells dendrites in stratum lacunosum-moleculare of CA1; thus, the Re exerts significant excitatory actions within this region (Dollemann-Van der Weel et al. 1997; Bertram and Zhang 1999; Wouterlood et al. 1990). Some projections from Re and EC converge onto the same pyramidal cells in CA1 (Dolleman-van der Weel et al. 2017), forming points of synergism. Re stimulation evokes long-term potentiation in CA1, demonstrating that the Re modulates hippocampal plasticity (Dolleman-Van Der Weel et al. 1997). It should be noted that the Re does not send projections to the dentate gyrus nor the CA2/CA3 hippocampal sectors.
Of functional interest, 5%–10% of Re neurons have axon collaterals that project to both the hippocampus and prefrontal cortex (Hoover and Vertes 2012; Varela et al. 2014). It has been suggested that this feature is critical to the synchronization and coordination of the activity between the prefrontal cortex and hippocampus. In support of this hypothesis, Re, through theta and delta oscillations, has been associated with the effective coupling between the mPFC and hippocampus (Roy et al., 2017).
Rh efferents, however, distribute more widely over the cortex to limbic and non-limbic regions, less spread to the hippocampus and more spread subcortically than Re projections (Vertes et al. 2015b). While there have been studies characterizing the anatomical connectivity of Rh (e.g., Varela et al. 2014), we have been unable to find a study examining nucleus’ unique functional or behavioral correlates that cannot be attributed to Re. This scarcity of literature represents a gap in our knowledge of nucleus-specific contributions of the ventral midline thalamic nuclei to behavior.
Although the hippocampus projects to mPFC, there are no direct return projections from the mPFC to the hippocampus (Hoover and Vertes 2012; Jay and Witter 1991). Rather, Re is the main link returning projections from the mPFC to the hippocampus, completing the circuit: hippocampus → mPFC → Re → hippocampus. Connections mediated between mPFC and the hippocampus via the Re appear to be necessary for the synchronization of slow oscillations between mPFC and hippocampus. When Re cells are inactivated, the coherence between corticohippocampal slow oscillations is impaired (Hauer et al. 2019). In contrast, the coordination between mPFC and Re neurons is enhanced by CA1 theta rhythm when rats approached the choice point in a T-maze (Ito et al. 2018). However, optogenetic manipulation of the temporal coordination in mPFC-Re-CA1 suggests that these interdependencies are dependent on their connections with the supramammillary nucleus (Ito et al. 2018).
Re also contributes to head directionality during light-dark transitions, and transitions into novel or new environments (Cholvin et al. 2018). It appears that one function of the Re is to monitor the specificity of distinct environmental representations. Lesions to Re/Rh region decrease of place field stability and increase their firing variability (Cholvin et al. 2018). However, the role of the Re in spatial navigation is complicated, because lesions or inactivation of Re impairs the animal’s performance in some navigation tasks (Cholvin et al. 2013; Mei et al. 2018), but not on other tasks (Dolleman-van der Weel et al. 2009; Ito et al. 2015). It has been hypothesized that Re is not responsible for spatial reference memory, but rather response or strategy selection in a spatial context (Cassel 2013; Davoodi et al. 2009; Xu and Sudhof 2013).
The Re is uniquely positioned at the apex of a higher-order cortico-thalamo-cortical circuit and appears to bridge memory and executive function (Dolleman-van der Weel 2019).
Theta rhythm suppression within the Re has been shown to induce working memory deficits (Duan et al. 2015). Lesions to the Re/Rh region impair delayed performance on the radial arm maze, and operant delayed nonmatching to position, similar to what was observed after either mPFC or hippocampal lesions (Hembrook and Mair 2011; 2012). Muscimol inactivation of the Re/Rh region impaired delayed alternation performance, but spared nondelayed visual discrimination (Hallock et al. 2013; Layfield et al. 2015). It should be noted that on the delayed alternation) task, there was significant hippocampal–prefrontal theta-gamma coupling and mPFC theta-gamma coupling when rats were at the maze choice point, but not during the discrimination task. However, when Re was selectively inhibited at the sample, delay or choice phase of delayed non-match-to-position, only inhibition at the sample phase impaired short-term spatial memory (Maisson et al. 2018). Thus, the mPFC-Re-hippocampus circuit may be critical for encoding information for later successful short-term memory performance. However, suppression of circuit activity with Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) within the mPFC → Re projection impaired odor sequence memory in rats (Jayachandran et al. 2019), and a sequential lag analysis (item order) showed that the mPFC→Re pathway contributes to a working memory retrieval strategy. Thus, the exact contributions of the Re to working memory may be dependent on task demands.
There is also evidence the Re may be involved in behavioral flexibility because reversal learning is impaired. Re inactivation, impaired the ability of rats to switch strategies from response to place responding in a double H water maze (Cholvin et al. 2013). In an attentional set-shifting odor/texture discrimination task, it was found that rats with Re lesions were significantly impaired in reversal learning (Linley et al. 2016). This behavioral deficit in behavioral flexibility may be a factor of the Re connections with the orbital frontal cortex, rather than the mPFC-hippocampal circuit.
3. Normal development of cortico-thalamic circuitry (including critical periods)
The most recent classification of thalamic nuclei includes the midline nuclei into “limbic group” (Lara-Vásquez et al. 2016; Vertes et al. 2015b). These midline nuclei either span the whole rostrocaudal length of midline complex (paraventricular) or are located more anteriorly (paratenial, Re) or posteriorly (intermediodorsal [IMD]), or lie just dorsal to Re and overlap with approximately the caudal two-thirds of Re (Berendse and Groenewegen 1991; Vertes et al. 2015a). Developmentally, the midline nuclei are considered later-forming structures within the rodent thalamus. The birth of neurons there occurs between embryonic day (E) E16 - E17.5 (Altman and Bayer 1979), whereas the generation of neurons in mediodorsal nucleus peaks by E15 and even earlier (E14) in the reticular nucleus. With such chronological gradient in generation, differentiation and maturation of thalamic neurons, it is natural that these cells have different levels of vulnerability to teratogenic (abnormal development caused by environment) insults depending on the timing of such insults. Time of neuronal birth in a particular thalamic nucleus is only one variable that defines the resilience or vulnerability, the others include (but not limited) the number of neurons in interconnected structures and formation of appropriate connections (Mooney and Miller 2010).
Thalamocortical projections of midline thalamic neurons undergo significant remodeling during the first postnatal weeks by a gradual massive reduction in the contralateral cortical projections, such that ipsilateral innervation dominates in the adult brain (Minciacchi and Granato 1989). Thalamic projections reach cortical layers by postnatal day (PD) 1, whereas the cortical projections to midline thalamus develop later, starting as early as PD 4 and displaying adult pattern by PD 8 (van Eden et al. 1990). Thus, teratogenic insult during the first ten postnatal days (3rd trimester-equivalent brain development period in rodents also known as “brain growth spurt” (see Dobbing and Sands 1979) would occur when the generation of neurons within mPFC-Re-hippocampus circuitry has ended (Altman and Bayer 1979; Bayer 1980), and would target cortico-thalamic neurons and their reciprocal connections. The afferent midline thalamic projection neurons within hippocampus and mPFC are undergoing extensive differentiation and synaptogenesis (Ikonomidou et al. 2000) as well as naturally occurring apoptosis (White and Barone 2001) during this postnatal period. Given the vulnerability of hippocampus and mPFC to alcohol exposure due to these synaptogenic periods (Semple et al. 2013; Rice and Barone 2000), and high interconnectivity of these structures with Re, it is likely that the Re is also damaged following early postnatal alcohol exposure. Indeed, the recent publication from our group indicates that Re is selectively vulnerable to alcohol exposure during early postnatal rat brain development (Gursky et al. 2019).
Overall, our understanding of the long-term consequences of developmental alcohol exposure for frontal cortical structure, connectivity and function is far from complete. To our knowledge, only three papers address alcohol’s effects on the development of cortico-thalamic circuitry: A rat model of 2nd trimester alcohol exposure evaluated somatosensory cortical projections’ damage (Granato et al. 1995; Santarelli et al. 1995) and a rat model of 3rd trimester alcohol exposure was employed to estimate the vulnerability of two midline thalamic nuclei, Re and Rh (Gursky et al. 2019). Two earlier publications from the Klintsova lab have demonstrated that postnatal alcohol exposure produces long-term effects on the dendritic organization of pyramidal neurons within the mPFC by decreasing dendritic complexity and the total length of dendrites (Hamilton et al. 2010; Whitcher and Klintsova 2008).
Animal studies have shown that, following alcohol-exposure either during gestation or within the postnatal “brain growth spurt,” there are dysmorphogenesis, abnormal migration of neuroblasts and reduced cortical thickness (Dunty et al. 2002; Aronne et al. 2011; Kotkoskie and Norton 1989a, 1989b), reduced neuronal recruitment during behavior (Hamilton et al. 2010), inhibitory deficits (Riley et al. 1986; Mihalick et al. 2001) and impairments on tests of spatial ability (Christie et al. 2005; Gabriel et al. 2002), working memory (Thomas et al. 1996; Nagahara and Handa 1997), and reversal learning (Lee and Rabe 1999; Mihalick et al. 2001).
4. Animal models of developmental alcohol exposure
Rodent models of developmental alcohol exposure are imperative for uncovering and predicting the deficits associated with prenatal alcohol exposure in humans and for the development of targeted therapeutic interventions. Shortly following the clinical categorization of Fetal Alcohol Syndrome (FAS) in 1973 (Jones and Smith 1973), rodent models were developed to conduct translational research for better understand better the physical and cognitive deficits observed in children with prenatal alcohol exposure and to study the mechanisms of alcohol-induced brain damage during early development. Evidence provided by animal models of developmental alcohol exposure highlighted the important correlation between the timing of ethanol exposure and the peak of blood alcohol concentration (BAC). Neuronal degeneration indicates increased teratogenic vulnerability of the cerebellum, hippocampus, thalamus, and cortex to developmental ethanol exposure (Bonthius and West 1990, 1991; Ikonomidou et al. 2000). Indeed, it is evident that there are dose-dependent patterns of damage to neurons and neuroglia that may persist into adolescence and adulthood and contribute to the complex etiology of developmental ethanol exposure (Bielawski and Abel 2002; Terasaki and Schwarz 2016; Madden et al. 2020). Further, clinical and animal model data suggest that early gestational alcohol exposure results in severe cognitive impairments and craniofacial dysmorphologies, whereas alcohol exposure during late-stage human pregnancy, modeled by the early postnatal period in rodents, results in a range of learning and memory deficits without the identifiable dysmorphologies (Stanton and Goodlett 1998; Godin et al. 2010; Lipinski et al. 2012; Klintsova et al. 2013). It should be noted that while the outcomes of moderate ethanol exposure during gestation are more subtle, there are long-lasting neurobehavioral deficits, including alterations in learning, memory, social behavior and motor coordination. Importantly, these behavioral changes are accompanied by alterations to neurotransmitter systems and synaptic plasticity (Valenzuela et al. 2012). These studies prompted the update of the clinical diagnostic criteria and terminology to the umbrella term Fetal Alcohol Spectrum Disorder (FASD; Hoyme et al. 2016).
4.1. Rodent Models of FASD and their Contributions to Cortico-Thalamic Pathology
Comparative neuroanatomical studies demonstrate that the development and the rate of growth of different brain structures are species-dependent (Dobbing and Sands 1979). Indeed, specific neurodevelopmental stages spanning rodent gestation and early postnatal life are well-described in relation to their human analogues. In accordance with this perspective, multiple animal models of FASD have been developed to assess the teratogenicity of ethanol exposure across stages of neurodevelopment. The advantages of using rat and mouse models of FASD include their highly translational nature, feasibility for controlled experimentation critical for the identification of neuropathological mechanisms, short lifespan, and well-defined patterns of reproduction and development (Cudd 2005).
Few studies have investigated the impact of developmental ethanol exposure on cortico-thalamic maturation or morphology. The proper function of these circuits is critical for spatial memory, executive function, and learning. The following sub-sections characterize key findings from rodent model studies examining the teratogenic impact of ethanol on the morphology of thalamic nuclei and cortical structures.
4.2. Gestational Models
Given the significance of the timing and amount of neonatal alcohol exposure on symptomology, the major differences between rodent models of FASD include gestational versus postnatal ethanol exposure and the use of varying methods of ethanol administration. Ethanol exposure via liquid diet to pregnant dams constitutes one type of gestational rodent model of developmental alcohol exposure (comparable to the first two trimesters of human pregnancy). It is argued that this method more closely mimics the human paradigm of prenatal alcohol exposure and produces the least amount of maternal stress. However, it is impossible to quantify the individual degree of ethanol exposure of fetal pups in utero. It was demonstrated that maternal exposure to an ethanol-containing liquid diet resulted in an altered ratio of apoptotic markers in the cortices, but not the thalami of exposed rat pups from PD 0–30; however, there was a latent effect on neurogenesis in the ventrobasal thalamus (Mooney and Miller 2001, 2010). Additionally, adult rats that were exposed to ethanol via liquid diet throughout gestation showed reduced plasticity in the barrel cortex associated with a striking depression of sensory responses: spontaneous activity and evoked responses were reduced by 70 and 80%, respectively (Rema and Ebner 1999). Offspring of pregnant mice with ad libitum access to a 25% ethanol solution has been shown to exhibit damage to intraneocortical connections mirrored by altered gene expression of RZR, Id2, and Cadherin8, genes critical for cortical patterning (El Shawa et al. 2013). Functional connectivity of the rodent cortex is altered by prenatal ethanol exposure, a brief daily exposure to 5% ethanol throughout gestation was sufficient to induce changes in “functional network connectivity” of the cortex through adulthood (Rodriguez et al. 2016).
Experiments controlling for the timing and amount of alcohol administration to pregnant dams employ a forced gavage technique through intragastric intubation. This method produces an increased, albeit transient, BAC peak compared to the lower, yet consistent, BAC produced by an ethanol-containing liquid diet or vapor inhalation and may be interpreted as a binge-like exposure. Maternal gavage with 6 g/kg of ethanol to pregnant rat dams resulted in the reduction of total and individual barrel field sizes in the primary somatosensory cortices of ethanol -exposed juvenile and adult offspring (Chappell et al. 2007). It was shown that moderate levels of prenatal ethanol exposure via a 2 g/kg gavage from E 10 to birth significantly increased cytokine production and the expression of Cideb1, a cell death activator, in the fetal (E 17) hippocampus and cortex and resulted in exaggerated interleukin 1-β (IL-1β) expression in the prefrontal cortex of ethanol-exposed offspring in response to an immune challenge in adulthood, ultimately impacting object recognition memory (Terasaki and Schwarz 2016).
4.3. Postnatal Models
Artificial rearing, colloquially referred to as the “pup-in-a-cup” method, was the first method developed to assess the impact of postnatal ethanol exposure on development and includes gastrostomization of pups for permanent implantation of an adjustable gastric tube for ethanol administration (West et al. 1984). This technique increased the experimenter control over the timing and rate of ethanol administration, significantly contributing to peak BAC that could be correlated with brain growth and neurodegeneration. However, it became apparent that the limitations of this approach outweighed its advantages. The highly invasive technique was criticized for inducing stress associated with maternal separation and social isolation; both of which have been shown to negatively impact neurodevelopment (Francis et al. 1999).
A similar intragastric intubation method, as described above as maternal gavage, can be applied to pups during the first few postnatal weeks (human fetal third-trimester equivalent). This method allows for individual quantification of peak BAC without inducing nutritional insufficiency and enables researchers to target distinct neurodevelopmental events such as the rapid brain growth spurt period characterized in humans and rodents (Goodlett et al. 1987; Stanton and Goodlett 1998; Tomlinson et al. 1998; Kelly and Lawrence 2008; Klintsova et al. 2013; Idrus et al. 2017). It is less invasive than the surgical technique required in the artificial rearing paradigm. The number of neurons and the total volume of the Re but not the Rh is significantly reduced in adult rodents with postnatal ethanol exposure from PD 4–9 (Gursky et al. 2019). Further, this exposure paradigm has been shown to alter connectivity in the mPFC by reducing spine density of pyramidal neurons in layers III (Whitcher and Klintsova 2008) and dendritic complexity in proximal basilar dendrites of Layer II/III neurons (Hamilton et al. 2010). Single-day ethanol exposure during this period can lead to severe apoptotic neurodegeneration in the AD, followed by learning and memory deficits in late adolescence (Wozniak et al. 2004). Such neurodegeneration in AD is also prevalent in non-human primate models of FASD (Farber et al. 2010). While the exact timing of exposure used across studies occasionally differs, there are consistent findings that adolescent males display memory deficits following just a single day of early postnatal ethanol exposure (Thomas et al. 2004; Wozniak et al. 2004).
Another widely used technique is known as ethanol vaporization during which pregnant dams or pups receive ethanol exposure through atmospheric inhalation. While this method produces similar behavioral and cognitive deficits, the neuropathological mechanism of damage is distinguishable from the methods previously described (Heaton et al. 2000; Morton et al. 2014). Using this model, Newville et al. (2017) demonstrated that ethanol exposure on PD 3–15 resulted in a persistent disruption of the white matter integrity of the corpus callosum that was correlated with a decrease in the expression of myelin basic protein. This study proved highly translational given the identified structural abnormalities of the corpus callosum in children diagnosed with FASD (Wozniak and Muetzel 2011).
Further research involving animal models of developmental alcohol exposure will be necessary for continued exploration of the teratogenicity of alcohol on the development of midline thalamic nuclei and associated cortical circuits. The use of multimodal techniques to assess concurrent alterations to neuroanatomy, physiology, and behavior will be paramount for the discovery of biomolecular mechanisms of damage impacting cortico-thalamic circuitry.
5. Animal models of alcohol exposure during adulthood and thalamic pathology
Adult ethanol exposure with and without and thiamine deficiency has been used to model alcohol-related brain damage in rodents. Ethanol-induced and thiamine deficiency-induced alterations in brain may differ in intensity and region, which can be a direct function of the extent of ethanol exposure and the duration of thiamine deficiency treatment. Although both conditions produce alterations on the hippocampus, basal forebrain, frontal cortical grey and white matter, the thalamus and mammillary bodies are considered the primary sites and target of thiamine deficiency (see review Vetreno et al. 2011). Studies have shown that thiamine deficiency, relative to chronic alcohol, has a greater impact on the thalamus, including gross lesions, compared to the minor cell loss reported following chronic ethanol exposure (see Nunes et al. 2019). In this section, the literature regarding the animal models of alcohol exposure and thiamine deficiency during adulthood will be reviewed in relation to both ethanol-induced and thiamine deficiency-induced thalamic pathology.
Studies using animal models of ethanol exposure during adulthood have demonstrated that the degree of molecular and behavior consequences of chronic ethanol exposure depends on the duration of ethanol consumption and the number of withdrawal episodes. Chronic ethanol treatment (CET) is a paradigm used to model long-term drinking behavior, with forced daily ethanol administration (10% – 20% v/v) that can last from weeks to months (Henriques et al. 2018; Nunes et al. 2019; Vetreno et al. 2011). Although a pre-clinical study using prolonged ethanol exposure in mice found neuronal loss and volume reduction on thalamic nuclei (medial dorsal and anterior nuclei), after 7 months of forced ethanol consumption (12% (v/v) ethanol in drinking water; Beracochea et al. 1987), there is little evidence for further ethanol-induced thalamic pathology using experimental animals. However, we found that following 6 months of CET alone, the active form of thiamine was reduced (Toledo Nunes et al. 2019). Furthermore, the alcohol-related brain damage within the thalamus has been documented mostly in studies using thiamine deficiency models as a critical feature for understanding neuropathological and neurochemical mechanisms involved in KS, Wernicke’s Encephalopathy (WE) and Wernicke-Korsakoff Syndrome (WKS) (Hazell and Butterworth, 2009; Hazell et al. 2010; Savage et al. 2012).
5.1. Animal models of adult ethanol-related brain damage: Thiamine deficiency emerges as necessary for thalamic pathology in ethanol-related used disorders.
Experimental rodent models of thiamine deficiency have been used to mimic the progression of neurological/behavioral symptoms involved in the pathology described in WE and WKS patients. The well-established rodent pyrithiamine-induced thiamine deficiency (PTD) model of WKS is a model that induces thiamine deficiency through the combination of thiamine-free food and i.p. injections of pyrithiamine, a thiamine pyrophosphokinase inhibitor. This treatment produces a characteristic thalamic damage centered on the anterior thalamus, internal medullary lamina, with consistent lesions on rostral intralaminar and moderate damage to ventral portions of the mediodorsal nucleus, similar to what is observed in WKS patients (See Figure 2; Langlais et al. 1988, 1996; Mair et al. 1985, 1988). Furthermore, the timing of the PTD treatment is crucial in the extension of the thalamic pathology (Hazell and Butterworth 2009; Savage et al. 2012; Zhang et al. 1995). In rodents submitted to PTD treatment, the clinical symptoms of thiamine deficiency begin 10 days into treatment, with loss of weight, followed by a decrease in food and water intake (Nunes et al. 2019; Zhang et al. 1995). Between the days 12 and 13, the loss of righting reflexes is observed in the PTD animals, and the first neuronal loss is reported in thalamic nuclei, such as the gelatinosus, AV and ventral posterolateral nuclei. Rapid neuronal loss is observed between day 13 and 16, when seizures are observed. The progression of the lesions in multiple thalamic nuclei depends on the duration of PTD treatment. In brief, PTD-induced thalamic damage can be divided into 3 stages. During the early acute stage (EAS), the gelatinosus nuclei and AV nuclei show severe neuronal degeneration within 1 to 2-hours after seizure onset. Within 3–5 hours following seizure onset, the middle acute stage (MAS) is characterized by lesions on ventral, intralaminar and central nuclei of thalamus. At the late acute stage (LAS), 6 to 14 hours after seizure, multiple thalamic nuclei (anterior, medial dorsal, intralaminar, posterior) have a complete neuronal loss (Savage et al. 2012; Zhang et al. 1995).
Figure 2.
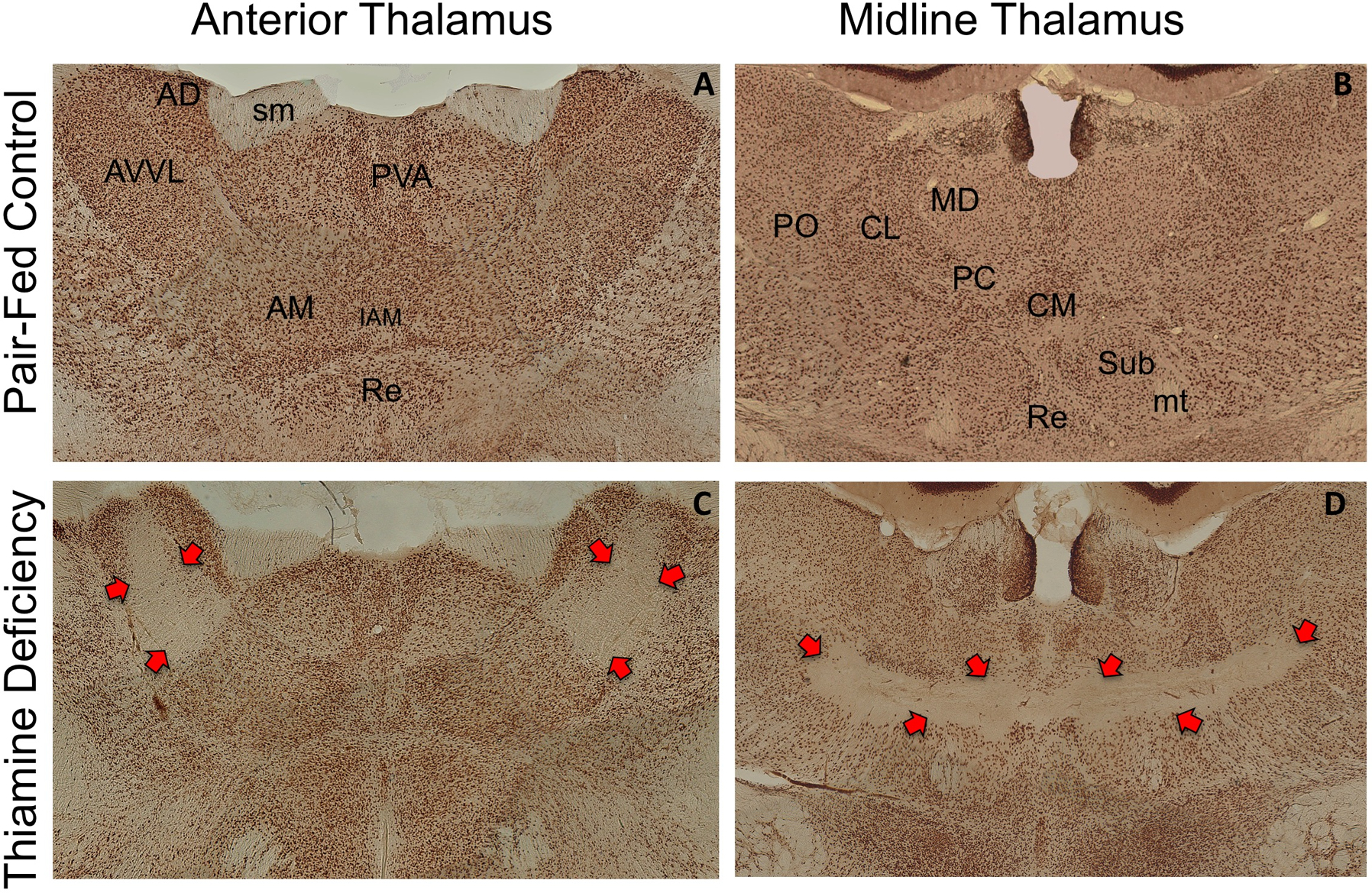
Medial and midline thalamus in the Rat. Sections are stained with nuclear specific stain (NeuN) that stains neuronal cell bodies. The top panels (A, B) present the thalamus of a control rat. The bottom panels (C, D) demonstrates the neuronal cell loss (red arrows) induced by thiamine deficiency. It should be noted that there is cells loss in anteroventral ventrolateral nucleus (AVVL), with sparing of anterior dorsal (AD) nucleus. Furthermore, there is extensive cell loss in the intralaminar region, with sparing of medial dorsal and ventral midline nuclei. Abbreviations AM (anteromedial thalamic nucleus), CL (central lateral thalamic nucleus), CM (central medial thalamic nucleus), IAM (interoanteromedial thalamic nucleus), MD (medial dorsal nucleus), mt (mammillothalamic tract), PC (paracentral thalamic nucleus), PO (posterior thalamic nucleus), PVA, Re (nucleus reuniens), Sub (nucleus submediius), sm (stria medullaris)
Studies using histological and immunohistochemical assessment have confirmed the thalamic lesions with neuronal cell loss in brains of PTD rats. Vemuganti and colleagues (2006) observed neuronal loss (44 to 83%) in many thalamic nuclei (gelatinosum, mediodorsal, AD, AV, ventroposteriolateral, ventroposteriomedial and dorsolateral geniculate) of rats with loss of righting reflex on day 14. At this time, the PTD rats did not exhibit apparent seizures (Vemuganti et al. 2006). A significant change in thalamus has also consistently documented in studies from our laboratory. Using NeuN immunohistochemistry, Anzalone and colleagues (2010) observed in PTD-treated rats after 1 month of recovery, complete neuronal loss in the AD thalamic nucleus, however, the neuronal loss within dorsal AV and AD nuclei was less. Evident cell loss was also observed in several midline intralaminar nuclei extending into the posterior thalamic nucleus, and some loss of neurons in the most ventral portion of the medial dorsal complex was revealed. Also, approximately 20–30% of the thalamic mass was lost in PTD rats, when compared with pair-fed animals (Anzalone et al. 2010; Hall and Savage 2016). Bobal and Savage (2015) also observed neuronal loss in the anteroventral ventrolateral (AVVL), gelatinosus, internal medullary laminar nuclei, with sparing of neurons within the ventral midline nuclei, specifically the Rh and Re. Interestingly, that study demonstrated that the ventral midline thalamus is critical for the recovery of spatial behavior following thiamine deficiency when ACh levels are increased with the hippocampus and frontal cortex. Several studies suggest that thalamic damage after PTD treatment is associated with spatial behavior dysfunction and the degree of thalamic tissue loss predicts the extent of learning and memory dysfunction (Langlais and Savage 1995; Robinson and Mair 1992).
Previous work using two stages of PTD treatment (EAS and MAS), CET (rats consumed a 20% v/v solution of ethanol over 6 months), moderate PTD followed by CET (PTD-CET) and moderate PTD during CET (CET-PTD), showed that PTD-MAS, PTD-EAS and PTD-CET treatments all produce thalamic lesions (Vedder et al. 2015). As expected, PTD-MAS rats had largest areas of thalamic lesion, and PTD-EAS rats had the same thalamic lesion size as PTD-CET, showing that the synergism between treatments was not able to exacerbate thalamic lesion observed in PTD animals. Also, 6 months of CET alone and the treatment with thiamine deficiency during CET did not result in a thalamic lesion. These findings suggest that there is an influence of order in which rats were treated with CET and thiamine deficiency to produce lesions on thalamus. We hypothesized that the exposure to ethanol before PTD altered the number of glutamate receptors resulting in neurons less susceptible to excitotoxicity caused by thiamine deficiency (Vedder et al. 2015).
The glutamate-mediated excitotoxicity, oxidative stress, compromised glucose metabolism and neuroinflammation are mechanisms involved in PTD-induced neuronal degeneration within the thalamus (Hazell and Butterworth 2009; Hazell et al. 2013). Magnetic resonance imaging (MRI) studies of PTD-treated rats confirmed lesions in the thalamus through T2 maps which are indicative of inflammation and neurodegeneration processes (Dror et al. 2010; Sarkar et al. 2016). Both studies showed that these animals have abnormal T2 relaxation values. Dror and colleagues (2010) showed massive lesions in the thalamus during the symptomatic stage (between days 12–14), which recovered over time. Although Sarkar and colleagues (2016) also verified abnormal T2 relaxation in PTD-treated rats after 3 weeks of recovery, they observed that severe neurodegeneration in the medial thalamic area, medial geniculate nuclei, and inferior olive were concomitant with vascular damage and microglial activation (Sarkar et al. 2016). In addition, a recent study (Bowyer et al. 2018) revealed a clear progression of microglial activation and vascular responses associated with the early phase of thalamic neurodegeneration in PTD-treated mice. It verified that activated microglia start before the astrocyte activation, neurodegeneration, and signs of vascular dysfunction in the thalamus. This investigation suggests that glutamate excitotoxicity was not the principal mechanism involved in the early neurodegenerative response to thiamine deficiency.
In contrast, considerable evidence for the role of excitotoxicity on neurodegeneration from thiamine deficiency treatment has been demonstrated in PTD-treated rats during the later symptomatic stages (day 14 after the onset of seizure) of PTD-treatment (Hazel et al. 1993; Langlais and Zhang 1993; Todd and Butterworth 1998). High levels of extracellular glutamate were observed in thalamic regions that were consistent with PTD-induced lesion within the thalamus (Armstrong-James et al. 1988; Hazell et al. 1993; Langlais and Zhang 1993 Zhang et al. 1995). Zhang and colleagues (1995) revealed that neurocytopathological changes cited above during stages of PTD treatment are identical to those observed in glutamate-induced excitotoxic lesions. These findings have shown that mechanisms involved in thalamic neurodegeneration are dependent on the time and stage of PTD treatment.
Investigation of the molecular mechanisms in cell death observed in the thalamus of PTD rats has revealed changes to the expression in a range of genes and proteins involved in neuropathology, such as markers of oxidative stress (eNOS, ferritin), neuroinflammation (interleukin-6 [IL-6], IL-1β, tumor necrosis factor-ɑ [TNF-ɑ]) and regulation of neuronal death (apolipoprotein E [Apoe], synuclein alpha [Snca], clusterin [Clu], leucine-rich repeat kinase 2 [Lrrk2], Vimentin) (Karuppagounder et al. 2007; Nunes et al. 2018; Toledo Nunes et al. 2019; Vemuganti et al. 2006). Differential changes in neuroinflammation genes and protein were observed in thalamus of PTD-treated rats in different stages: At the presymptomatic (day 10) stage (Karuppagounder et al. 2007), day 14 when loss of righting reflex is observed (Vemuganti et al. 2006), during acute and severe PTD treatment (1 or 4 hours after seizure), and following recovery (24-hour or 3-weeks post-PTD treatment; Toledo Nunes et al. 2019). Our recent study showed that the largest neuroimmune response was observed within the thalamus during PTD treatments (PTD-EAS and PTD-MAS), with profound increase in neuroimmune genes and proteins (IL-6, IL-1β, TNF-ɑ, and nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha [IκBɑ], which recovered within 24 hrs. This study also investigated the effect of CET and the synergism between both conditions and verified that ethanol showed small fluctuations in inflammatory genes (Toledo Nunes et al. 2019). Vemuganti and colleagues (2006) also observed the upregulation of genes in that thalamus involved in neuroinflammation or immune response, stress, metabolic perturbation, cell death and repair. They confirmed thalamic pathology during the first symptomatic stage (loss of righting reflex on day 14) of PTD treatment. In addition, proteins involved in oxidative stress, neurotransmitter synthesis and synaptic vesicle cycle were also altered in the thalamus of PTD-treated rats after 3-weeks post-treatment (Nunes et al. 2018). Thus, the identification of genes and proteins, such as those involved in neuroinflammation (IL6, IL-1β, TNF-ɑ) and synaptic vesicle recycling (complexin 2 [gene:Cplx2 – accession: CPLX2], dynamin 3 [gene: Dnm3 –accession: DYN3], and vacuolar adenosinetriphosphatase [V-ATP]) changed by thiamine deficiency in vulnerable brain regions may be important markers of the neuronal death process, which can contribute to the severity of thiamine deficiency-induced thalamic lesions. Therefore, the investigations of neuroimmune makers after CET have focused on other vulnerable brain regions to ethanol, such as hippocampus and cortex.
There is limited evidence in animal models that prolonged ethanol exposure results in thalamic lesions. Furthermore, clinicopathological studies have shown that the thalamus is more severely damaged in alcoholics with a diagnosis of thiamine deficiency (i.e., KS), than in alcoholics patients without severe neurological impairments, or uncomplicated alcoholics (Pitel et al. 2012, 2015; Sullivan and Pfefferbaum 2009). A recent study revealed that two brain networks, implicated in uncomplicated alcohol use disorder and KS, are dissociated by pathology to different thalamic nuclei (Segobin et al. 2019). Atrophy of mediodorsal nuclei, nuclei involved in the frontocerebellar circuit, was observed in both uncomplicated alcoholics and patients with KS. In addition, uniquely in KS patients, there was atrophy of anterior thalamus, nuclei involved on Papez circuit, and there was also a decrease in the connectivity between anterior nuclei and hippocampus. These findings suggest that anterior nuclei are a crucial marker to the pathophysiology in alcohol use disorders (Segobin et al. 2019). Thus, data across several studies, involving both human and animal models, point towards thiamine deficiency as being the critical mediator of pathology to the thalamus in adult alcohol-related brain damage and severe memory dysfunction.
6. Approaches to recover alcohol-related brain damage and cognitive and behavioral outcomes
Given the prevalence of developmental and adult alcohol-related disorders, extensive efforts have been put forward to determine whether brain and behavioral dysfunction can be improved or restored. Despite the negative consequences of heavy drinking, with abstinence, there is some degree of recovery. Furthermore, behavioral interventions, modeled in preclinical studies, have revealed the unique adaptations and plasticity that can occur within and across neural circuits. Furthermore, changing brain structure are associated with improvements in cognition.
6.1. Recovery of developmental alcohol exposure brain damage and behavioral dysfunction.
Various types of adolescent and adult interventions have been successful in mitigating some of the neuroanatomical and behavioral deficits resulting from developmental ethanol exposure in rodent models of FASD. While these therapeutic strategies remain promising, there exists a barrier to their effectiveness in humans. Additional research in humans and animal models will be necessary for the refinement of these interventions.
6.1.1. Pharmaceutical and nutritional interventions for developmental ethanol exposure
Few pharmaceutical interventions have proven efficacious in the mitigation of the cognitive and behavioral deficits associated with developmental alcohol exposure. To avoid pharmaceutical exposure in the pediatric population and reduced prevalence of harmful adverse side effects, researchers have implemented nutrient-rich dietary supplementation as a natural and affordable alternative (Fuglestad et al. 2013).
Resveratrol, a phenol found in berries and nuts, acts as a potent antioxidant and anti-inflammatory agent. Co-administration of resveratrol with ethanol in milk substitute to rat pups in a postnatal model of FASD has been shown to reduce ethanol-induced oxidative stress and inflammation in the cerebral cortex, hippocampus and cerebellum, and rescue aberrant behavior on the Morris water maze and elevated plus maze tasks (Tiwari and Chopra 2011). Pretreatment with resveratrol attenuated impairment in hippocampal neurogenesis and depletion of neural precursor cells resulting from one-day ethanol exposure on PD 7 (Xu et al. 2015). Furthermore, resveratrol prevented ethanol-induced apoptosis in the granule layer of the cerebellum and increased the survival of cerebellar granule cells (Kumar et al. 2011). Human studies indicate that resveratrol may induce hippocampal plasticity in adulthood (Dias et al. 2016), but its use as a therapeutic agent has not been tested in infants diagnosed with FASD.
Dietary supplementation with choline, an essential nutrient for cell membrane integrity and a robust methyl group donor, is found in leafy greens and eggs and is essential for cortical plasticity following brain injury (Conner et al. 2005). Specifically, choline supplementation attenuates some of the neurobiological and behavioral deficits associated with FASD as demonstrated in both human and rodent model studies. It is hypothesized that fetal ethanol exposure perturbs natural choline metabolism, resulting in an altered epigenetic profile and cellular apoptosis in the developing brain (Light et al. 1989; Fagerlund et al. 2006; Zeisel 2011). It has been demonstrated using a gestational rodent model of FASD that low maternal choline dietary intake during pregnancy exacerbates the negative impact of prenatal ethanol exposure on offspring behavior (Idrus et al. 2017). Further, it was shown that choline supplementation concurrent with postnatal alcohol exposure or in adolescence mitigates ethanol-induced learning and memory deficits and reverses damage to the global epigenome of the hippocampus and prefrontal cortex in ethanol -exposed rats (Otero et al. 2012; Perkins et al. 2015; Schneider and Thomas 2016). Human studies of maternal and infant choline supplementation during or directly following pregnancy demonstrate an increase in infant cognition speed (Caudill et al. 2018). However, this transient benefit may not be maintained through late childhood and adolescence (Wozniak et al. 2015).
Taken together, these studies indicate a possible protective mechanism linked to nutritional supplementation, indirectly suggesting that neonatal ethanol exposure causes metabolic imbalance inside the developing offspring. As such, nutrition-based therapies are functional –preventing extensive neurodegeneration and rescuing neurotransmission –and difficult to maintain given the varying physiological demands of the developing offspring. Additionally, due to the complex impact of ethanol exposure on the developing brain, the lasting efficacy of these treatments remains promising, yet unclear.
6.1.2. Behavioral Interventions to mitigate developmental alcohol-related damage
Several environmental and behavioral interventions have produced promising results, indicated by attenuation of ethanol-related learning and memory deficits in adolescence and adulthood. Highly feasible and translational, these therapies stimulate neuroplasticity by upregulating neurotrophic concentration and inducing neurogenesis, synaptogenesis, and angiogenesis. These interventions include voluntary aerobic exercise, motor skill training, and environmental enrichment. Moreover, the combination of these interventions to produce a “super-intervention” has yielded some synergist effects benefitting behavioral outcome.
Both human and animal studies indicate that physical activity enhances experience-dependent brain plasticity and improves learning and memory (reviewed in Voss et al. 2013). Indeed, exercise reduces the impact of aging and vascular disease on cognitive capacity and restores neural function in the diseased brain (Cotman et al. 2007; Voss et al. 2013; Vivar and van Praag 2017). Forced and voluntary aerobic exercise enhances adult hippocampal neurogenesis in the dentate gyri of adult rodents with neonatal ethanol exposure (Thomas et al. 2008; Hamilton et al. 2011, 2015) and increases activity-dependent synaptic plasticity in the normal brain. However, exercise alone was not sufficient to promote cerebellar synaptic plasticity in the rat model of FASD (Hamilton et al. 2012).
Long-lasting deficits in behaviors that are known to depend on cerebellar function have been documented in animal models, including tests of gait, balance and coordinated motor performance (Goodlett and Lundahl 1996; Thomas et al. 1998) and eyeblink classical conditioning (Stanton and Goodlett 1998). These deficits model effects linked to cerebellar damage in FAS/FASD children (Luo 2015) including deficits in motor performance, eyeblink conditioning, gait and balance (Jirikowic et al. 2013; Cheng et al. 2015; Taggart et al. 2017). Complex motor skill training (learning to traverse an obstacle course) was used as a successful rehabilitation treatment for brain damage induced in a rat model of binge ethanol exposure during the early postnatal brain growth spurt (Klintsova et al. 1998, 2002). In normal adult rats, complex motor skill training increased the number of synapses in specific regions in the cerebellum and motor cortex. Furthermore, it was found that 20 days of complex motor skill training (a typical duration used in the normal adult studies) significantly increased the number of parallel fiber synapses per Purkinje cell in the paramedian lobule of the cerebellum in both normal control rats and ethanol-exposed rats and that acquisition of complex motor tasks, but not mere exercise, could rehabilitate the motor deficits occurring as a result of developmental exposure to ethanol (Klintsova et al. 2002).
Rearing in a complex or enriched (EE) environment during adolescence and adulthood supports synaptic and cellular re-modeling in the brain (Uylings et al. 1978; Mohammed et al. 2002; Olson et al. 2006). EE are characterized by their inclusion of novel sensory stimuli and ability to house 6–12 same-sex rodents simultaneously (reviewed in Gursky and Klintsova 2017; Hannan 2014). When applied to rodent models of developmental alcohol exposure, EE induces neuroplasticity. In one experiment using a prenatal model of FASD, exposure to EE for about 5 weeks following weaning on PD 20 was shown to rescue alcohol-induced microglial activation and protect against alterations to dopaminergic cell soma volume in the ventral tegmental area (Aghaie et al. 2019). Moderate prenatal ethanol exposure in mice has also been shown to prevent the survival and differentiation of proliferating progenitor cells in the dentate gyrus of the hippocampus following EE exposure for 8–12 weeks post-weaning (Choi et al. 2005). Finally, both exercise intervention and EE contribute to enhanced dendritic complexity of immature neurons in the hippocampi of rats with postnatal ethanol exposure (Boschen et al. 2017).
Few studies have investigated the synergistic effects of increased exercise with exposure to EE on neuroanatomy and behavior in rodent models of developmental ethanol exposure. By exposing rodents to exercise before incorporation into EE, adult hippocampal neurogenesis is upregulated, and these cells are integrated into existing circuits, enhancing maturation and survival of the cells. This “Super-intervention” promotes the survival of newly born hippocampal neurons compared to exercise alone in a postnatal model of FASD (Hamilton et al. 2012). However, ethanol-induced alterations to context-dependent memory were not rescued (Schreiber et al. 2013). The impact of either EE alone or the “super-intervention” on thalamic and cortical plasticity is understudied. Future research will be necessary to evaluate the therapeutic effects of non-invasive interventions on damage and function of these structures.
In sum, it is evident that while these interventions play a pivotal role in treating the symptoms of developmental ethanol exposure, they lack a targeted approach in which they might prevent either behavioral deficits or neuroanatomical damage. Further, it is clear that developmental ethanol exposure hinders neuroplasticity. A comprehensive understanding of the teratogenic damage of ethanol to developing circuits will be the first step in enhancing therapeutic strategy by supporting the creation or repair of neural connections.
6.2. Recovery of adult ethanol-related brain damage and behavioral dysfunction.
A number of approaches (exercise, EE, pharmacology) have been successful for recovering cognitive performance in animal models of adult alcohol-related brain damage and diencephalic amnesia. Exercise and EE have a diversity of effects; changes are observed across molecular, cellular and behavioral scales, and are widespread across the brain (Kempermann 2019; van Praag 2009). The effects of EE that can include exercise are complex with nascent properties that are modulated by both the disease state and individual experience (Kempermann 2019). Thus, it is inherently challenging to pin point the exact mechanisms that lead to recovery when an organism engaged in exercise or was exposed to an EE. When the data from studies that evoke exercise and/or EE to recover brain pathology and behavior in adult models of alcohol-related brain damage or diencephalic amnesia are reviewed, it is clear that there is no recovery of thalamic pathology (Hall and Savage 2016: Dalrymple-Alford et al. 2015). However, instead there are other plasticity changes in the extended hippocampal and frontocortical circuits contribute to system adaptation and behavioral recovery.
6.2.1. Recovery of neurotrophin deficits can lead to circuit adaptation and behavioral recovery.
It has been well established that exercise, commonly induced in rodents by attaching a running wheel to their home cage, increases neurotrophin levels that can persist for weeks (Berchtold et al. 2010). Several growth factors (brain-derived neurotrophic factor [BDNF], vascular endothelial growth factor [VEGF], nerve growth factor [NGF]) are up-regulated after exercise within the hippocampus, as well as other regions such as the frontal cortex and medial septum/diagonal band (MS/DB; Fabel et al. 2003; Griesbach et al. 2009; Neeper et al. 1996; Tong et al. 2012; Vivar et al. 2012). However, BDNF emerged as the key modulator leading to memory improvements following exercise and environmental enrichment (Cotman et al. 2007; Gomez-Pinilla et al. 2008; Vaynman et al. 2004). Exercise-induced changes in BDNF drive improvements in hippocampal neurogenesis (van Praag 2009; van Praag et al. 2000), and are correlated with improvements spatial memory (see Berchtold et al. 2010; Christie et al. 2005; O’Callaghan et al. 2007). However, NGF is also a potent driver of neural recovery, but its role in exercise-induced brain and behavioral recovery is less studied (see Hall et al. 2018). NGF infusions into the intraventricular space has been shown to recover spatial memory deficits, revive cholinergic neurons within the basal forebrain, and increase the expression of vesicular ACh transporter within the hippocampus in rodents exposed to long-term (6–9 months) ethanol (20%) in the drinking water (Cadete-Leite et al. 2003; Lukoyanov et al. 2003; Pereira et al. 2016).
Although the hallmark neuropathology in the PTD model, and KS, is neuronal loss in the anterior, medial, and midline thalamus, as well as the mammillary bodies, there is significant, loss (30%) loss of MS/DB cholinergic neurons with concomitant reductions in cholinergic innervation of the hippocampus and frontal cortex that leads to blunted hippocampal and cortical behaviorally-stimulated ACh efflux (Anzalone et al. 2010; Hall and Savage. 2016; Hall et al. 2018; Savage et al. 2007). Nevertheless, the PTD model is responsive to cholinergic modulation: increasing cholinergic tone, with cholinergic agonists (physostigmine, tacrine) within the septohippocampal circuit reduces or eliminates the spatial memory impairment following thiamine deficiency (Roland et al. 2008; 2010). Similar to other models of memory dysfunction, the forebrain cholinergic system is critical in modulating the recovery of learning and memory.
We have shown that the effects of exercise go beyond enhanced BDNF and hippocampal neurogenesis: Exercise induces cortical cytogenesis (Hall et al. 2014), as well as rescues cholinergic neurons in the MS/DB (Hall and Savage 2016, Hall et al. 2018; Vetreno and Crews 2018; Vetreno et al. 2019), following PTD-induced pathology or adolescent ethanol exposure. Furthermore, our data demonstrate that exercise-induced improvements in NGF, not BDNF, drive both recovery spatial memory and cholinergic phenotype rescue in the PTD model of KS (Hall et al. 2018).
The effects of exercise following binge ethanol exposure during adulthood and adolescents have also been explored. Intense ethanol exposure (oral ethanol gavage every 8-hrs for 4 days straight) during adulthood led to a decrease in the expression of BDNF and a reduction of granular cells within the dentate gyrus of the hippocampus, which was associated with spatial navigation impairments, in binged female rats, but not male rats (Maynard et al. 2018). Post-binge exercise increased BNDF levels and recovered granule cell loss in the female hippocampus (Maynard et al. 2018). In male rats exposed to intermittent ethanol throughout adolescents (oral gavage, 2 days on 2 days off, from post-natal day 25–55), there is a reduction in cortical and hippocampal NGF, BDNF, a loss of hippocampal neurogenesis, and a reduction of neurons in the basal forebrain that express the cholinergic phenotype, all of which were recovered with post-binge exercise (Vetreno and Crews. 2018; Vetreno et al. 2019). Interestingly, the adolescent binge-induced reduction of cholinergic neurons was associated with epigenetic silencing of cholinergic phenotype genes, which were reversed by exercise (Vetreno et al. 2019). Thus, there can be behavioral recovery following thiamine deficiency or extensive ethanol exposure is primarily driven by enhancement within the septohippocampal circuit.
6.2.2. Environment enrichment can rescue some behavioral deficits related to thalamic pathology
Several papers have examined whether EE recovered cognitive behaviors and circuit function following experimental-induced lesions of the anterior thalamus or the midline thalamus. First, it should be noted that like alcohol-related brain damage, in standard-housed laboratory rats, there is little spontaneous recovery of behavioral deficits after thalamic lesions. EE for rodents includes group housing, running wheels, and the replacement of toy objects throughout the cage. It, therefore, provides multisensory cognitive stimulation, increased physical activity, and broader social and emotional experiences. Rat models suggest that the deficits caused by ATN damage, which is associated with “diencephalic amnesia”, can be ameliorated by EE.
Spatial impairment in the water maze, both flexible strategy adaptation and reference memory, following lesion to the ATN, were recovered when rats were housed in an EE following thalamic pathology (Wolff et al. 2008). EE also recovered spatial working memory in rats with anterior thalamic lesions (Loukavenko et al. 2007). Although the damaged thalamus remained unchanged, enrichment increased CA1 spine density in both sham rats and those with anterior thalamic damage (Dalrymple-Alford et al. 2015). Administered neurotrophic drug cerebrolysin reduced spatial working memory deficits in rats with ATN lesions, and when given along with EE the benefit was synergistic (Loukavenko et al. 2016). However, neither the drug treatment nor EE recovered suppressed c-fos activity with the retrosplenial cortex of rats with ATN lesions. These data suggest that the recovery observed involved other brain regions, not the retrosplenial cortex. Most recently, EE prevented the decrease in prefrontal theta power in rats with ATN lesions and appeared to increases functional connectivity between the prefrontal cortex and hippocampus through by passing the anterior thalamus (Ulrich et al. 2019).
There are also data that demonstrates that EE also improves spatial navigation in the water maze following lesions to the ventral midline thalamus (Re/Rh; Ali et al. 2017). Furthermore, following a long 25-day retention interval, c-Fos immunoreactivity in the mPFC was suppressed in rats with ventral midline thalamic lesions, but exposure to EE corrected the retrieval-triggered c-Fos expression within the mPFC. The reinstatement of memory and activation of the mPFC following EE suggests that functional connectivity within the hippocampus and prefrontal cortex, possibly through the amygdala, is a driver of behavioral EE-induced recovery following thalamic pathology (Ali et al. 2017).
Thus, although adult alcohol-related thalamic pathology is not recovered by pharmacological or behavioral interventions, improvements in the extended hippocampal circuit that include the basal forebrain and frontocortical restoration appear to revive circuit functioning, leading to restored cognitive functioning. The nervous system retains an impressive capacity to recover and adapt following alcohol-related brain damage, irrelevant whether the pathology was induced during development or adulthood.
7. Conclusions
All the forebrain regions involved in cognition receive input from the thalamus, and cortical regions are typically innervated by more than one thalamic nucleus; in addition, 30–50% of cortical regions project back to the thalamus (Varela 2014). The midline thalamus, specifically the anterior, intralaminar and ventral midline nuclei, have emerged as significant contributors to cognitive processing, including attention, spatial navigation, working memory, remote memory, and behavioral flexibility. However, determining the individual nucleus-specific contributions within the thalamus to behavior can be difficult due to the heterogeneity of neuronal function that exists within each nucleus. For instance, the intralaminar nuclei have multiple distinct subregions that govern different anatomical connectivity and behavioral correlates (Saalmann 2014). Similarly, the Re/Rh nuclei have differences in connectivity across its rostrocaudal extent (Dolleman-van der Weel et al. 2019), and recent indications of a functionally relevant dorsal-ventral division as well (Angulo-Garcia et al. 2018). Thus, there is difficulty in pin-pointing the distinct contributions of relatively small subnuclei within the thalamus through either experimental approached or observation of disease states. Although there are technical limitations to uncovering these specific contributions, the concurrent approach of multiple techniques to provide convergent evidence is the best solution. Several recent technical advances in viral tracing, optogenetics and chemogenetics, allows for more selectivity in targeting of different thalamic regions and circuits, which has opened up new possibilities in the integration of specific thalamic cortical and subcortical pathway. Many midline thalamic nuclei have the capacity to influence prefrontal cortex (directly and indirectly) and this provides reason to expect overlapping as well as unique behavioral contributions. This is both the case between nuclei, as well as within individual nuclei, as functional circuits exist both between nuclei, and within them (Dolleman-van der Weel et al. 2019).
Despite such limitations, the field has moved away from the conception of the thalamus as a simple relay station. Damage to the thalamus can affect the initial acquisition, directing attention, short-term representation, monitoring, and updating current information within a changing environment, to long-term consolidation (Wolff and Vann 2019). Thus, a more current view is that the midline thalamus integrates information from midbrain, diencephalic, hippocampal, and cortical regions to directly influence cognitive functioning. Research regarding the function of the midline thalamus in cognition has revealed a complex contribution of the thalamus for cognition, as well as a role for the thalamus in developmental and neurodegenerative disorders.
Alcohol is neurotoxic, and it functions as teratogen during development and evokes neurodegenerative cascades in adulthood. We selectively reviewed the damaging effects of alcohol during early development and adulthood on the thalamus. As stated, alcohol is not highly regionally selective as a neurotoxin, but the midline thalamus is a key target. As reviewed, specific nuclei within the thalamus display unique vulnerability to alcohol-related neurotoxicity. Exposure to high levels of alcohol during brain developmental brain damage the ATN (Wozniak et al. 2004) and selectively the Re (Gursky et al. 2019). Long-term ethanol exposure (over 6 months in the rodent) in adulthood becomes confounded with thiamine deficiency (Toledo Nunes et al. 2019). However, extensive thiamine deficiency is linked to midline pathology (ATN, IML, gelatinosus and posterior nuclei), buts spares ventral midline cell loss (Bobal and Savage 2015). However, given that prefrontal cortex receives afferent projections from a large number of thalamic nuclei, including the anterior, intralaminar, midline nuclei (Barbas et al. 1991), a range of frontocortical dysfunction (disinhibition, poor working memory, cognitive and behavioral inflexibility) is observed following developmental or adult ethanol exposure. There is also hippocampal dysfunction (impaired spatial navigation and spatial memory, configural dysfunction) following developmental and adult alcohol exposure that is due to hippocampal pathology, as well as damage to the ATN. Understanding the specificity of thalamic influence on corticothalamic and thalamic-hippocampal circuitry provides a long-overdue foundation for understanding the consequences of alcohol exposure paradigms and alcohol use disorders. In summary, understanding of thalamic dysfunction and circuit adaptation will further aid in the development of successful therapeutics.
Acknowledgements:
Supported by NIH grant numbers R21AA026613 and R01AA027269 to A.Y.K., and P50AA017823-Main 2 and R01AA021775 to L.M.S. and the Developmental Exposure Alcohol Research Center at Binghamton University.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the above stated funding agencies. The authors have no conflicts of interest to declare.
Abbreviations:
- AC
Anterior cingulate cortex
- Ach
Acetylcholine
- AD
Anterodorsal thalamic nucleus
- AM
Anteromedial thalamic nucleus
- ATN
Anterior thalamic nucleus
- AV
Anteroventral thalamic nucleus
- AVVL
Anteroventral ventrolateral nucleus
- BAC
Blood alcohol concentration
- BDNF
Brain derived neurotrophin factor
- CE
Complex environment
- CET
Chronic ethanol treatment
- CL
Central lateral thalamic nucleus
- CM
Central medial thalamic nucleus
- DREADDs
Designer Receptors Exclusively Activated by Designer Drugs
- E
Embryonic day
- EAS
Early acute stage
- EC
Entorhinal cortex
- EE
Enriched environment
- FAS
Fetal Alcohol Syndrome
- FASD
Fetal Alcohol Spectrum Disorders
- HD
Head direction
- IAM
Interoanteromedial thalamic nucleus
- IkBa
Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- IL
Infralimbic cortex
- IL-1β
Interleukin-1β
- IL-6
Interleukin-6
- IMD
Intermediodorsal thalamic nuclei
- KS
Korsakoff Syndrome
- LAS
Late acute stage
- MAS
Middle acute stage
- MO
Medial orbital cortex
- mPFC
Medial prefrontal cortex
- MRI
Magnetic resonance imaging
- MS/DB
Medial septum/diagonal band
- NGF
Nerve growth factor
- NMTP
Non-matching-to-position
- PC
Paracentral thalamic nucleus
- PD
Postnatal day
- PF
Parafascicular thalamic nucleus
- PL
Prelimbic cortex
- PTD
Pyrithiamine-induced thiamine deficiency
- Re
Nucleus reuniens
- Rh
Rhomboid nucleus
- SPF
Subparafascicular thalamic nucleus
- TNF-ɑ
Tumor necrosis factor-ɑ
- VEGF
Vascular endothelial growth factor
- WE
Wernicke’s Encephalopathy
- WKS
Wernicke-Korsakoff Syndrome
References:
- Aggleton JP, Nelson AJ. (2015). Why do lesions in the rodent anterior thalamic nuclei cause such severe spatial deficits? Neurosci Biobehav Rev. 4: 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggleton JP, Hunt PR, Nagle S, Neave N. (1996). The effects of selective lesions within the anterior thalamic nuclei on spatial memory in the rat. Behav Brain Res. 81:189–198. [DOI] [PubMed] [Google Scholar]
- Aghaie CI, Hausknecht KA, Wang R, Dezfuli PH, Haj-Dahmane S, Kane CJ. et al. (2019). Prenatal Ethanol Exposure and Postnatal Environmental Intervention Alter Dopaminergic Neuron and Microglia Morphology in the Ventral Tegmental Area During Adulthood. Alcohol Clin Exp Res. 10.1111/acer.14275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akers KG, Kushner SA, Leslie AT, Clarke L, van der Kooy D, Lerch JP, Frankland PW (2011). Fetal alcohol exposure leads to abnormal olfactory bulb development and impaired odor discrimination in adult mice. Mol Brain. 4. 10.1186/1756-6606-4-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali M, Cholvin T, Muller MA, Cosquer B, Kelche C, Cassel JC, Pereira de Vasconcelos A. (2017). Environmental enrichment enhances system level consolidation of a spatial memory after lesions of the ventral midline thalamus. Neurobiol Learn Mem. 141:108–123. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. (1979). Development of the diencephalon in the rat. IV. Quantitative study of the time of origin of neurons and the internuclear chronological gradients in the thalamus. J Comp Neurol, 188(3), 455–471, doi: 10.1002/cne.901880308. [DOI] [PubMed] [Google Scholar]
- Angulo-Garcia D, Ferraris M, Ghestem A, Bernard C, Quilichini PP (2018). Spatio-temporal organization of cell assemblies in Nucleus Reuniens during slow oscillations. BiorXiv. doi: 10.1101/474973 [DOI] [Google Scholar]
- Anzalone S, Vetreno RP, Ramos RL, Savage LM. (2010). Cortical cholinergic abnormalities contribute to the amnesic state induced by pyrithiamine-induced thiamine deficiency in the rat. Eur J Neurosci. 32: 847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archibald SL, Fennema-Notestine C, Gamst A, Riley EP, Mattson SN, Jernigan TL. (2007). Brain dysmorphology in individuals with severe prenatal alcohol exposure. Dev. Med. Child Neurol 43(3): 148–154. 10.1111/j.1469-8749.2001.tb00179.x [DOI] [PubMed] [Google Scholar]
- Armstrong-James M, Ross DT, Chen F, Ebner FF. (1988). The effect of thiamine deficiency on the structure and physiology of the rat forebrain. Metab Brain Dis. 3: 91–124. [DOI] [PubMed] [Google Scholar]
- Aronne MP, Guadagnoli T, Fontanet P, Evrard SG, Brusco A. (2011). Effects of prenatal ethanol exposure on rat brain radial glia and neuroblast migration. Experimental Neurology, 229(2), 364–371, doi: 10.1016/j.expneurol.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Bailey KR, Mair RG. (2005). Lesions of specific and nonspecific thalamic nuclei affect prefrontal cortex-dependent aspects of spatial working memory. Behav Neurosci. 119: 410–419. [DOI] [PubMed] [Google Scholar]
- Barbas H, Henion TH, Dermon CR. (1991). Diverse thalamic projections to the prefrontal cortex in the rhesus monkey. J Comp Neurol. 313:65–94. [DOI] [PubMed] [Google Scholar]
- Bayer SA (1980). Development of the hippocampal region in the rat. I. Neurogenesis examined with 3H-thymidine autoradiography. J Comp Neurol, 190(1), 87–114, doi: 10.1002/cne.901900107. [DOI] [PubMed] [Google Scholar]
- Beracochea D, Lescaudron L, Verna A, Jaffard R. (1987). Neuroanatomical effects of chronic ethanol consumption on dorsomedial and anterior thalamic nuclei and on substantia innominata in mice. Neurosci Lett. 73: 81–84. [DOI] [PubMed] [Google Scholar]
- Berchtold NC, Castello N, Cotman CW. (2010). Exercise and time-dependent benefits to learning and memory. Neurosci, 167: 588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendse HW, Groenewegen HJ. (1991). Restricted cortical termination fields of the midline and intralaminar thalamic nuclei in the rat. Neurosci. 42:73–102. [DOI] [PubMed] [Google Scholar]
- Bertram EH, Zhang DX. (1999). Thalamic excitation of hippocampal CA1 neurons: a comparison with the effects of CA3 stimulation. Neurosci. 92: 15–26. [DOI] [PubMed] [Google Scholar]
- Bielawski DM, Abel EL (2002). The effect of administering ethanol as single vs. divided doses on blood alcohol levels in the rat. Neurotoxicol Teratol 24:559–562. 10.1016/S0892-0362(02)00207-6 [DOI] [PubMed] [Google Scholar]
- Bobal MG, Savage LM. (2015). The role of ventral midline thalamus in cholinergic-based recovery in the amnestic rat. Neuroscience. 285: 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonthius DJ, West JR (1990). Alcohol‐Induced Neuronal Loss in Developing Rats: Increased Brain Damage with Binge Exposure. Alcohol Clin Exp Res 14:107–118. 10.1111/j.1530-0277.1990.tb00455.x [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, West JR (1991). Permanent neuronal deficits in rats exposed to alcohol during the brain growth spurt. Teratology 44:147–163. 10.1002/tera.1420440203 [DOI] [PubMed] [Google Scholar]
- Boschen KE, McKeown SE, Roth TL, Klintsova AY (2017). Impact of exercise and a complex environment on hippocampal dendritic morphology, Bdnf gene expression, and DNA methylation in male rat pups neonatally exposed to alcohol. Dev Neurobiol 77:708–725. 10.1002/dneu.22448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer JF, Tranter KM, Sarkar S, Hanig JP. (2018). Microglial activation and vascular responses that are associated with early thalamic neurodegeneration resulting from thiamine deficiency. Neurotox. 65: 98–110. [DOI] [PubMed] [Google Scholar]
- Brown HD, Baker PM, Ragozzino ME. (2010). The parafascicular thalamic nucleus concomitantly influences behavioral flexibility and dorsomedial striatal acetylcholine output in rats. J Neurosci. 30: 14390–14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk JA, Mair RG. (1998). Thalamic amnesia reconsidered: excitotoxic lesions of the intralaminar nuclei, but not the mediodorsal nucleus, disrupt place delayed matching-to-sample performance in rats. Behav Neurosci. 112: 54–67. [PubMed] [Google Scholar]
- Byatt G, Dalrymple-Alford JC. (1996). Both anteromedial and anteroventral thalamic lesions impair radial-maze learning in rats. Behav Neurosci. 110: 1335–1348. [DOI] [PubMed] [Google Scholar]
- Cassel JC. (2013). The reuniens and rhomboid nuclei: neuroanatomy, electrophysiological characteristics and behavioral implications. Pro in Neurobio. 111: 34–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassel JC, Pereira de Vasconcelos A. (2015). Importance of the ventral midline thalamus in driving hippocampal functions. Prog Brain Res. 219: 145–161. [DOI] [PubMed] [Google Scholar]
- Caudill MA, Strupp BJ, Muscalu L, Nevins J., Canfield RL (2018). Maternal choline supplementation during the third trimester of pregnancy improves infant information processing speed: A randomized, double-blind, controlled feeding study. FASEB J 32:2172–2180. 10.1096/fj.201700692RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell TD, Margret CP, Li CX, Waters RS (2007). Long-term effects of prenatal alcohol exposure on the size of the whisker representation in juvenile and adult rat barrel cortex. Alcohol 41:239–251. 10.1016/J.Alcohol.2007.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng DT, Jacobson SW, Jacobson JL, Molteno CD, Stanton ME, Desmond JE (2015). Eyeblink classical conditioning in alcoholism and fetal alcohol spectrum disorders. Front. Psychiatry [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IY, Allan AM, Cunningham LA (2005). Moderate fetal alcohol exposure impairs the neurogenic response to an enriched environment in adult mice. Alcohol Clin Exp Res 29:2053–2062. 10.1097/01.alc.0000187037.02670.59 [DOI] [PubMed] [Google Scholar]
- Cholvin T, Hok V, Giorgi L, Chaillan F, Poucet B. (2018). Ventral midline thalamus is necessary for hippocampal place field stability and cell firing modulation. J Neurosci. 38: 158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholvin T, Loureiro M, Cassel R, Cosquer B, Geiger K, De Sa Nogueira D, Raingard H, Robelin L, Kelche C, Pereira de Vasconcelos A, Cassel JC. (2013). The ventral midline thalamus contributes to strategy shifting in a memory task requiring both prefrontal cortical and hippocampal functions. J Neurosci. 33: 8772–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie BR, Swann SE, Fox CJ, Froc D, Lieblich SE, Redila V, Webber A. (2005). Voluntary exercise rescues deficits in spatial memory and long-term potentiation in prenatal ethanol-exposed male rats. Eur J Neurosci. 21: 1719–1726. [DOI] [PubMed] [Google Scholar]
- Clark BJ, Taube JS. (2012). Vestibular and attractor network basis of the head direction cell signal in subcortical circuits. Front Neural Circuits. 6:7. doi: 10.3389/fncir.2012.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner JM, Chiba AA, Tuszynski MH (2005). The basal forebrain cholinergic system is essential for cortical plasticity and functional recovery following brain injury. Neuron 46:173–179. 10.1016/j.neuron.2005.03.003 [DOI] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC, Christie LA. (2007). Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends in Neurosci. 30: 464–472. [DOI] [PubMed] [Google Scholar]
- Cudd TA (2005). Animal model systems for the study of alcohol teratology. Exp Biol Med 230:389–393. 10.1177/15353702-0323006-06 [DOI] [PubMed] [Google Scholar]
- Dalrymple-Alford JC, Harland B, Loukavenko EA, Perry B, Mercer S, Collings DA, Ulrich K, Abraham WC, McNaughton N, Wolff M. (2015). Anterior thalamic nuclei lesions and recovery of function: Relevance to cognitive thalamus. Neurosci Biobehav Rev. 54:145–160. [DOI] [PubMed] [Google Scholar]
- Davoodi FG, Motamedi F, Naghdi N, Akbari E. (2009). Effect of reversible inactivation of the reuniens nucleus on spatial learning and memory in rats using Morris water maze task. Behav. Brain Res 198: 130–135. [DOI] [PubMed] [Google Scholar]
- Dias GP, Cocks G, Do Nascimento Bevilaqua MC, Egidio Nardi A, Thuret S. (2016). Resveratrol: A Potential Hippocampal Plasticity Enhancer. Oxid Med Cell Longev 10.1155/2016/9651236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, Sands J (1979). Comparative aspects of the brain growth spurt. Early Hum Dev 311:79–83. 10.1016/0378-3782(79)90022-7 [DOI] [PubMed] [Google Scholar]
- Dolleman-van der Weel MJ, Griffin AL, Ito HT, Shapiro ML, Witter MP, Vertes RP, Allen TA. (2019). The nucleus reuniens of the thalamus sits at the nexus of a hippocampus and medial prefrontal cortex circuit enabling memory and behavior. Learn Mem. 26: 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolleman-Van der Weel MJ, Lopes da Silva FH, Witter MP. (1997). Nucleus reuniens thalami modulates activity in hippocampal field CA1 through excitatory and inhibitory mechanisms. J Neurosci. 17: 5640–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolleman-van der Weel MJ, Lopes da Silva FH, Witter MP. (2017). Interaction of nucleus reuniens and entorhinal cortex projections in hippocampal field CA1 of the rat. Brain Struct Funct. 222: 2421–2438. [DOI] [PubMed] [Google Scholar]
- Dolleman-van der Weel MJ, Morris RG, Witter MP. (2009). Neurotoxic lesions of the thalamic reuniens or mediodorsal nucleus in rats affect non-mnemonic aspects of watermaze learning. Brain Struct Funct. 213: 329–342. [DOI] [PubMed] [Google Scholar]
- Dror V, Eliash S, Rehavi M, Assaf Y, Biton IE, Fattal-Valevski A. (2010). Neurodegeneration in thiamine deficient rats-A longitudinal MRI study. Brain Res. 1308: 176–184. [DOI] [PubMed] [Google Scholar]
- Duan AR, Varela C, Zhang Y, Shen Y, Xiong L, Wilson MA, Lisman J. (2015). Delta frequency optogenetic stimulation of the thalamic nucleus reuniens is sufficient to produce working memory deficits: relevance to schizophrenia. Biol Psychiatry. 77: 1098–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudchenko PA, Wood ER, Smith A. (2019). A new perspective on the head direction cell system and spatial behavior. Neurosci Biobehav Rev. 105: 24–33. [DOI] [PubMed] [Google Scholar]
- Dumont JR, Amin E, Aggleton JP. (2014). Selective importance of the rat anterior thalamic nuclei for configural learning involving distal spatial cues. Euro J Neurosci. 39: 241–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunty WC Jr, Zucker RM, & Sulik KK (2002). Hindbrain and Cranial Nerve Dysmorphogenesis Result from Acute Maternal Ethanol Administration. Developmental Neuroscience, 24(4), 328–342 [DOI] [PubMed] [Google Scholar]
- El Shawa H, Abbott CW, Huffman KJ (2013). Prenatal ethanol exposure disrupts intraneocortical circuitry, cortical gene expression, and behavior in a mouse model of FASD. J Neurosci 33:18893–18905. 10.1523/JNEUROSCI.3721-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerlund Å, Heikkinen S, Autti-Rämö I, Korkman M, Timonen M, Kuusi T., et al. (2006). Brain metabolic alterations in adolescents and young adults with fetal alcohol spectrum disorders. Alcohol Clin Exp Res 30:2097–2104. 10.1111/j.1530-0277.2006.00257.x [DOI] [PubMed] [Google Scholar]
- Farber NB, Creeley CE, Olney JW. (2010). Alcohol-induced neuroapoptosis in the fetal macaque brain. Neurobiol. Dis 40: 200–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis DD, Champagne FA, Liu D, Meaney MJ (1999). Maternal Care, Gene Expression, and the Development of Individual Differences in Stress Reactivity. Acad Sci 896:66–84. 10.1111/j.1749-6632.1999.tb08106.x [DOI] [PubMed] [Google Scholar]
- Fuglestad AJ, Fink BA, Eckerle JK, Boys CJ, Hoecker HL, Kroupina MG, et al. (2013). Inadequate intake of nutrients essential for neurodevelopment in children with fetal alcohol spectrum disorders (FASD). Neurotoxicol Teratol 39:128–132. 10.1016/j.ntt.2013.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel KI, Johnston S, Weinberg J. (2002). Prenatal ethanol exposure and spatial navigation: Effects of postnatal handling and aging. Developmental Psychobiology, 40(4), 345–357. [DOI] [PubMed] [Google Scholar]
- Gao C, Leng Y, Ma J, Rooke V, Rodriguez-Gonzalaez S, Ramakrishman C, et al. (2020). Two genetically, anatomically and functionally distinct cell types segregate across anteroposterior axis of paraventricular thalamus. Nature Neuroscience doi: 10.1038/s41593-019-0572-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin EA, O’Leary-Moore SK, Khan AA, Parnell SE, Ament JJ, Dehart DB, et al. (2010). Magnetic resonance microscopy defines ethanol-induced brain abnormalities in prenatal mice: Effects of acute insult on gestational day 7. Alcohol Clin Exp Res 34:98–111. 10.1111/j.1530-0277.2009.01071.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Vaynman S, Ying Z. (2008). Brain-derived neurotrophic factor functions as a metabotrophin to mediate the effects of exercise on cognition. Eur J Neurosci. 28: 2278–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodlett CR, Kelly SJ, West JR (1987). Early postnatal alcohol exposure that produces high blood alcohol levels impairs development of spatial navigation learning. Psychobiology 15:64–74. 10.3758/BF03327265 [DOI] [Google Scholar]
- Goodlett CR, Lundahl KR (1996). Temporal determinants of neonatal alcohol-induced cerebellar damage and motor performance deficits. Pharmacol Biochem Behav 55:531–540. 10.1016/S0091-3057(96)00248-1 [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Gomez-Pinilla F. (2009). Exercise-induced improvement in cognitive performance after traumatic brain injury in rats is dependent on BDNF activation. Brain Res. 1288: 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granato A, Santarelli M, Sbriccoli A, Minciacchi D. (1995). Multifaceted alterations of the thalamo-cortico-thalamic loop in adult rats prenatally exposed to ethanol. Anatomy and Embryology, 191(1), 11–23, doi: 10.1007/bf00215293. [DOI] [PubMed] [Google Scholar]
- Groenewegen HJ (1988). Organization of the afferent connections of the mediodorsal thalamic nucleus in the rat, related to the mediodorsal-prefrontal topography. Neuroscience 24:379–431 doi: 10.1016/0306-4522(88)90339-9 [DOI] [PubMed] [Google Scholar]
- Gursky ZH, Klintsova AY (2017). Wheel Running and Environmental Complexity as a Therapeutic Intervention in an Animal Model of FASD. J Vis Exp 54947. 10.3791/54947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gursky ZH, Savage LM, Klintsova AY. (2019). Nucleus reuniens of the midline thalamus of a rat is specifically damaged after postnatal alcohol exposure, NeuroRep. 30: 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gursky ZH, Spillman EC, Klintsova AY. (2020). Gursky, Z. H., Spillman, E. C., & Klintsova, A. Y. (2020).Single-day postnatal alcohol exposure induces apoptotic cell death and causes long-term neuron loss in rodent thalamic nucleus reuniens. Neuroscience. 435: 124–134. 10.1016/j.neuroscience.2020.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JM, Vetreno RP, Savage LM. (2014). Differential cortical neurotrophin and cytogenetic adaptation after voluntary exercise in normal and amnestic rats. Neurosci. 258: 131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JM, Savage LM. (2016). Exercise leads to the re-emergence of the cholinergic/nestin neuronal phenotype within the medial septum/diagonal band and subsequent rescue of both hippocampal Ach efflux and spatial behavior. Exp Neurol. 278: 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JM, Gomez-Pinilla F, Savage LM. (2018). Nerve growth factor is responsible for exercise-induced recovery of septohippocampal cholinergic structure and function. Front Neurosci. 12:773. doi: 10.3389/fnins.2018.00773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallock HL, Wang A, Griffin AL. (2016). Ventral midline thalamus is critical for hippocampal-prefrontal synchrony and spatial working memory. J Neurosci. 36: 8372–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallock HL, Wang A, Shaw CL, Griffin AL. (2013). Transient inactivation of the thalamic nucleus reuniens and rhomboid nucleus produces deficits of a working-memory dependent tactile-visual conditional discrimination task. Behav Neurosci. 127: 860–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton DA, Candelaria-Cook FT, Akers KG, Rice JP, Maes LI, Rosenberg M., et al. (2010). Patterns of social-experience-related c-fos and Arc expression in the frontal cortices of rats exposed to saccharin or moderate levels of ethanol during prenatal brain development. Behavioural Brain Research, 214(1), 66–74, doi: 10.1016/j.bbr.2010.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton GF, Boschen KE, Goodlett CR, Greenough WT, Klintsova AY (2012). Housing in Environmental Complexity Following Wheel Running Augments Survival of Newly Generated Hippocampal Neurons in a Rat Model of Binge Alcohol Exposure During the Third Trimester Equivalent. Alcohol Clin Exp Res 36:1196–1204. 10.1111/j.1530-0277.2011.01726.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton GF, Criss KJ, Klintsova AY (2015). Voluntary exercise partially reverses neonatal alcohol-induced deficits in mPFC layer II/III dendritic morphology of male adolescent rats. Synapse 69:405–415. 10.1002/syn.21827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton GF, Murawski NJ, St.Cyr SA, Jablonski SA, Schiffino FL, Stanton ME, et al. (2011). Neonatal alcohol exposure disrupts hippocampal neurogenesis and contextual fear conditioning in adult rats. Brain Res 1412:88–101. 10.1016/J.BRAINRES.2011.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton GF, Whitcher LT, Klintsova AY (2010). Postnatal binge-like alcohol exposure decreases dendritic complexity while increasing the density of mature spines in mPFC Layer II/III pyramidal neurons. Synapse 64:127–135. 10.1002/syn.20711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan AJ (2014). Review: Environmental enrichment and brain repair: Harnessing the therapeutic effects of cognitive stimulation and physical activity to enhance experience-dependent plasticity. Neuropathol Appl Neurobiol 40:13–25. 10.1111/nan.12102 [DOI] [PubMed] [Google Scholar]
- Harding A, Halliday G, Caine D, Kril J. (2000). Degeneration of anterior thalamic nuclei differentiates alcoholics with amnesia. Brain, 123: 141–154. [DOI] [PubMed] [Google Scholar]
- Hauer BE, Pagliardini S, Dickson CT. (2019). The reuniens nucleus of the thalamus has an essential role in coordinating slow-wave activity between neocortex and hippocampus. eNeuro. 6: pii: ENEURO.0365–19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazell AS, Butterworth RF. (2009). Update of cell damage mechanisms in thiamine deficiency: focus on oxidative stress, excitotoxicity and inflammation. Alcohol Alcohol. 44: 141–147. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Butterworth RF, Hakim AM. (1993). Cerebral vulnerability is associated with selective increase in extracellular glutamate concentration in experimental thiamine deficiency. J Neurochem. 61: 1155–1158. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Faim S, Wertheimer G, Silva VR, Marques CS. (2013). The impact of oxidative stress in thiamine deficiency: a multifactorial targeting issue. Neurochem Int. 62: 796–802. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Sheedy D, Oanea R, Aghourian M, Sun S, Jung JY, Wang D, Wang C. (2010). Loss of astrocytic glutamate transporters in Wernicke encephalopathy. Glia. 58: 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton MB, Mitchell JJ, Paiva M, Walker DW (2000). Ethanol-induced alterations in the expression of neurotrophic factors in the developing rat central nervous system. Dev Brain Res 121:97–107. 10.1016/S0165-3806(00)00032-8 [DOI] [PubMed] [Google Scholar]
- Hembrook JR, and Mair RG (2011). Lesions of reuniens and rhomboid thalamic nuclei impair radial maze win-shift performance. Hippo. 21: 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hembrook JR, Onos KD, and Mair RG (2012). Inactivation of ventral midline thalamus produces selective spatial delayed conditional discrimination impairment in the rat. Hippo. 22: 853–860. [DOI] [PubMed] [Google Scholar]
- Henriques JF, Portugal CC, Canedo T, Relvas JB, Summavielle T, Socodato R. (2018). Microglia and alcohol meet at the crossroads: Microglia as critical modulators of alcohol neurotoxicity. Toxicol Lett. 283: 21–31. [DOI] [PubMed] [Google Scholar]
- Hoover WB, Vertes RP. (2012). Collateral projections from nucleus reuniens of thalamus to hippocampus and medial prefrontal cortex in the rat: a single and double retrograde fluorescent labeling study. Brain Struct Funct. 217: 191–209. [DOI] [PubMed] [Google Scholar]
- Hoyme HE, Kalberg WO, Elliott AJ, Blankenship J, Buckley D, Marais A-S., et al. (2016). Updated Clinical Guidelines for Diagnosing Fetal Alcohol Spectrum Disorders. Pediatrics 138:. 10.1542/peds.2015-4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idrus NM, Breit KR, Thomas JD (2017). Dietary choline levels modify the effects of prenatal alcohol exposure in rats. Neurotoxicol Teratol 59:43–52. 10.1016/J.NTT.2016.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, et al. (2000). Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science (80-) 287:1056–1060. 10.1126/science.287.5455.1056 [DOI] [PubMed] [Google Scholar]
- Ito HT, Moser EI, Moser MB. (2018). Supramammillary nucleus modulates spike-time coordination in the prefrontal-thalamo-hippocampal circuit during navigation. Neuron. 99: 576–587. [DOI] [PubMed] [Google Scholar]
- Ito HT, Zhang SJ, Witter MP, Moser EI, Moser MB. (2015). A prefrontal-thalamo-hippocampal circuit for goal-directed spatial navigation. Nature. 522: 50–55. [DOI] [PubMed] [Google Scholar]
- Jankowski MM, Islam MN, Wright NF, Vann SD, Erichsen JT, Aggleton JP, O’Mara SM. (2014). Nucleus reuniens of the thalamus contains head direction cells. Elife. 14:3. doi: 10.7554/eLife.03075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski MM, Ronnqvist KC, Tsanov M, Vann SD, Wright NF, Erichsen JT, Aggleton JP, O’Mara SM. (2013). The anterior thalamus provides a subcortical circuit supporting memory and spatial navigation. Front Syst Neurosci. 7:45. doi: 10.3389/fnsys.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TM, Witter MP. (1991). Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 313: 574–586. [DOI] [PubMed] [Google Scholar]
- Jayachandran M, Linley SB, Schlecht M, Mahler SV, Vertes RP, Allen TA. (2019). Prefrontal Pathways Provide Top-Down Control of Memory for Sequences of Events. Cell Rep. 28: 640–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirikowic TL, McCoy SW, Lubetzky-Vilnai A, Price R, Ciol MA, Kartin D, et al. (2013). Sensory control of balance: A comparison of children with fetal alcohol spectrum disorders to children with typical development. J Popul Ther Clin Pharmacol [PMC free article] [PubMed] [Google Scholar]
- Jones K, Smith D (1973). Recognition of the Fetal Alcohol Syndrome in Early Infancy. Lancet 302:999–1001. 10.1016/S0140-6736(73)91092-1 [DOI] [PubMed] [Google Scholar]
- Karuppagounder SS, Shi Q, Xu H, Gibson GE. (2007). Changes in inflammatory processes associated with selective vulnerability following mild impairment of oxidative metabolism. Neurobiol Dis. 26: 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Fukabori R, Nishizawa K, Okada K, Yoshioka N, Sugawara M, Maejima Y, Shimomura K, Okamoto M, Eifuku S, Kobayashi K. (2018). Action selection and flexible switching controlled by the intralaminar thalamic neurons. Cell Rep. 22: 2370–2382. [DOI] [PubMed] [Google Scholar]
- Kelly SJ, Lawrence CR (2008). Intragastric intubation of alcohol during the perinatal period. Methods Mol Biol 447:101–110. 10.1007/978-1-59745-242-7_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G (2019). Environmental enrichment, new neurons and the neurobiology of individuality. Nat Rev Neurosci. 4: 235–245. [DOI] [PubMed] [Google Scholar]
- Klein MM, Cholvin T, Cosquer B, Salvadori A, Le Mero J, Kourouma L, et al. (2019) Ventral midline thalamus lesion prevents persistence of new (learning-triggered) hippocampal spines, delayed neocortical spinogenesis, and spatial memory durability. Brain Struct Funct 224:1659–1676 doi: 10.1007/s00429-019-01865-1 [DOI] [PubMed] [Google Scholar]
- Klintsova AY, Cowell RM, Swain RA, Napper RM, Goodlett CR, Greenough WT (1998). Therapeutic effects of complex motor training on motor performance deficits induced by neonatal binge-like alcohol exposure in rats: I. Behavioral results. Brain Res 800:48–61. 10.1016/S0006-8993(98)00495-8 [DOI] [PubMed] [Google Scholar]
- Klintsova AY, Hamilton GF, Boschen KE (2013). Long-term consequences of developmental alcohol exposure on brain structure and function: Therapeutic benefits of physical activity. Brain Sciences 3(1) 1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klintsova AY, Scamra C, Hoffman M, Napper RM, Goodlett CR, Greenough WT (2002). Therapeutic effects of complex motor training on motor performance deficits induced by neonatal binge-like alcohol exposure in rats:: II. A quantitative stereological study of synaptic plasticity in female rat cerebellum. Brain Res 937:83–93. 10.1016/S0006-8993(02)02492-7 [DOI] [PubMed] [Google Scholar]
- Knierim JJ, Kudrimoti HS, McNaughton BL. (1995). Place cells, head direction cells, and the learning of landmark stability. J Neurosci. 15: 1648–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koger SM, Mair RG. (1994). Comparison of the effects of frontal cortical and thalamic lesions on measures of olfactory learning and memory in the rat. Behav Neurosci. 108: 1088–1100. [DOI] [PubMed] [Google Scholar]
- Kotkoskie LA, Norton S. (1989a). Cerebral cortical morphology and behavior in rats following acute prenatal ethanol exposure. Alcohol Clin Exp Res, 13(6), 776–781. [DOI] [PubMed] [Google Scholar]
- Kotkoskie LA, Norton S. (1989b). Morphometric analysis of developing rat cerebral cortex following acute prenatal ethanol exposure. Exp Neurol, 106(3), 283–288. [DOI] [PubMed] [Google Scholar]
- Krout KE, Belzer RE, Loewy AD. (2002). Brainstem projections to midline and intralaminar thalamic nuclei of the rat. J Comp Neurol. 448: 53–101. [DOI] [PubMed] [Google Scholar]
- Kumar A, Singh CK, LaVoie HA, DiPette HA, Singh DJ, Singh US. (2011). Resveratrol Restores Nrf2 Level and Prevents Ethanol-Induced Toxic Effects in the Cerebellum of a Rodent Model of Fetal Alcohol Spectrum Disorders. Mol Pharmacol 80:446 LP–457. 10.1124/mol.111.071126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais PJ, Mair RG, Anderson CD, McEntee WJ. (1988). Long-lasting changes in regional brain amino acids and monoamines in recovered pyrithiamine treated rats. Neurochem Res. 13: 1199–1206. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Savage LM. (1995). Thiamine deficiency in rats produces cognitive and memory deficits on spatial tasks that correlate with tissue loss in diencephalon, cortex and white matter. Behav Brain Res. 68: 75–89. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX. (1993). Extracellular glutamate is increased in thalamus during thiamine deficiency-induced lesions and is blocked by MK-801. J Neurochem. 61: 2175–2182. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX, Savage LM. (1996). Neuropathology of thiamine deficiency: an update on the comparative analysis of human disorders and experimental models. Metab Brain Dis. 11: 19–37. [DOI] [PubMed] [Google Scholar]
- Lara-Vásquez A, Espinosa N, Durán E, Stockle M, Fuentealba P. (2016). Midline thalamic neurons are differentially engaged during hippocampus network oscillations. Scientific Reports, 6, 29807, doi: 10.1038/srep29807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law LM, Smith DM. (2012). The anterior thalamus is critical for overcoming interference in a context-dependent odor discrimination task. Behav Neurosci. 126: 710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layfield DM, Patel M, Hallock H, Griffin AL. (2015). Inactivation of the nucleus reuniens/rhomboid causes a delay-dependent impairment of spatial working memory. Neurobiol Learn Mem. 125:163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel C, Rasmussen C, Wyper K, Walker L, Andrew G, Yager J. (2008). Brain Diffusion Abnormalities in Children With Fetal Alcohol Spectrum Disorder. Alcohol Clin Exp Res. 32(10): 1732–1740. 10.1111/j.1530-0277.2008.00750.x [DOI] [PubMed] [Google Scholar]
- Lee MH, Rabe A. (1999). Infantile Handling Eliminates Reversal Learning Deficit in Rats Prenatally Exposed to Alcohol. Alcohol, 18(1), 49–53, doi: 10.1016/S0741-8329(98)00067-6. [DOI] [PubMed] [Google Scholar]
- Light KE, Serbus DC, Santiago M (1989). Exposure of Rats to Ethanol from Postnatal Days 4 to 8: Alterations of Cholinergic Neurochemistry in the Cerebral Cortex and Corpus Striatum at Day 20. Alcohol Clin Exp Res 13:29–35. 10.1111/j.1530-0277.1989.tb00279.x [DOI] [PubMed] [Google Scholar]
- Linley SB, Gallo MM, Vertes RP. (2016). Lesions of the ventral midline thalamus produce deficits in reversal learning and attention on an odor texture set shifting task. Brain Res. 1649: 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski RJ, Hammond P, O’Leary-Moore SK, Ament JJ, Pecevich SJ, Jiang Y, et al. (2012). Ethanol-Induced Face-Brain Dysmorphology Patterns Are Correlative and Exposure-Stage Dependent. PLoS One 7:e43067. 10.1371/journal.pone.0043067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez J, Wolff M, Lecourtier L, Cosquer B, Bontempi B, Dalrymple-Alford J, Cassel JC. (2009). The intralaminar thalamic nuclei contribute to remote spatial memory. J Neurosci. 29:3302–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukavenko EA, Ottley MC, Moran JP, Wolff M, Dalrymple-Alford JC. (2007). Towards therapy to relieve memory impairment after anterior thalamic lesions: improved spatial working memory after immediate and delayed postoperative enrichment. Eur J Neurosci. 26: 3267–3276. [DOI] [PubMed] [Google Scholar]
- Loukavenko EA, Wolff M, Poirier GL, Dalrymple-Alford JC. (2016). Impaired spatial working memory after anterior thalamic lesions: recovery with cerebrolysin and enrichment. Brain Struct Funct. 221: 1955–1970. [DOI] [PubMed] [Google Scholar]
- Luo J (2015). Effects of ethanol on the cerebellum: Advances and prospects. Cerebellum 14:383–385. 10.1007/s12311-015-0674-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden JT, Thompson SM, Magcalas CM, Wagner JL, Hamilton DA, Savage DD., et al. (2020). Moderate prenatal alcohol exposure reduces parvalbumin expressing GABAergic interneurons in the dorsal hippocampus of adult male and female rat offspring. Neurosci Lett 718:134700. 10.1016/J.NEULET.2019.134700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair RG, Anderson CD, Langlais PJ, McEntee WJ. (1985). Thiamine deficiency depletes cortical norepinephrine and impairs learning processes in the rat. Brain Res. 360: 273–284. [DOI] [PubMed] [Google Scholar]
- Mair RG, Anderson CD, Langlais PJ, McEntee WJ. (1988). Behavioral impairments, brain lesions and monoaminergic activity in the rat following recovery from a bout of thiamine deficiency. Behav Brain Res. 27: 223–239. [DOI] [PubMed] [Google Scholar]
- Mair RG, Burk JA, Porter MC. (1998). Lesions of the frontal cortex, hippocampus, and intralaminar thalamic nuclei have distinct effects on remembering in rats. Behav Neurosci. 112: 772–792. [DOI] [PubMed] [Google Scholar]
- Mair RG, Burk JA, Porter MC. (2003). Impairment of radial armmaze delayed nonmatching after lesions of anterior thalamus and parahippocampal cortex. Behav Neurosci. 117: 96–605. [DOI] [PubMed] [Google Scholar]
- Mair RG, Koch JK, Newman JB, Howard JR, Burk JA. (2002). A double dissociation within striatum between serial reaction time and radial maze delayed nonmatching performance in rats. J Neurosci. 22: 6756–6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair RG, Miller RL, Wormwood BA, Francoeur MJ, Onos KD, Gibson BM. (2015). The neurobiology of thalamic amnesia: Contributions of medial thalamus and prefrontal cortex to delayed conditional discrimination. Neurosci Biobehav Rev. 54: 161–74. [DOI] [PubMed] [Google Scholar]
- Maisson DJ, Gemzik ZM, Griffin AL. (2018). Optogenetic suppression of the nucleus reuniens selectively impairs encoding during spatial working memory. Neurobiol Learn Mem. 155: 78–85. [DOI] [PubMed] [Google Scholar]
- Marchand A, Faugère A, Coutureau E, Wolff M. (2014). A role for anterior thalamic nuclei in contextual fear memory. Brain Struct Funct. 219:1575–1586. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Rilep EP, Jernigan TL, Ehlers CL, Delis DC, Jones KL, Stern C, Johnson KA, Hesselink JR, Bellugi U. (1992). Fetal Alcohol Syndrome: A Case Report of Neuropsychological, MRI, and EEG Assessment of Two Children. Alcohol Clin Exp Res. 16(5): 1001–1003. 10.1111/j.1530-0277.1992.tb01909.x [DOI] [PubMed] [Google Scholar]
- Matsumoto N, Minamimoto T, Graybiel AM, Kimura M. (2001). Neurons in the thalamic CM-Pf complex supply striatal neurons with information about behaviorally significant sensory events. J Neurophysiol. 85: 960–976. [DOI] [PubMed] [Google Scholar]
- Maynard ME, Barton EA, Robinson CR, Wooden JI, Leasure JL. (2018). Sex differences in hippocampal damage, cognitive impairment, and trophic factor expression in an animal model of an alcohol use disorder. Brain Struct Funct. 223:195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna JT, Vertes RP. (2004). Afferent projections to nucleus reuniens of the thalamus. J Comp Neurol. 480: 115–142. [DOI] [PubMed] [Google Scholar]
- Mei H, Logothetis NK, Eschenko O. (2018). The activity of thalamic nucleus reuniens is critical for memory retrieval, but not essential for the early phase of “off-line” consolidation. Learn Mem. 25: 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meintjes EM, Narr KL, van der Kouwe AJW, Molteno CD, Pirnia T, Gutman B, Woods RP, Thompson PM, Jacobson JL, Jacobson SW. (2014). A tensor-based morphometry analysis of regional differences in brain volume in relation to prenatal alcohol exposure. Neuroimage Clin. 5: 152–160. 10.1016/j.nicl.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalick SM, Crandall JE, Langlois JC, Krienke JD, Dube WV. (2001). Prenatal ethanol exposure, generalized learning impairment, and medial prefrontal cortical deficits in rats. Neurotoxicology and Teratology, 23(5), 453–462, doi: 10.1016/S0892-0362(01)00168-4. [DOI] [PubMed] [Google Scholar]
- Minciacchi D, Granato A. (1989). Development of the thalamocortical system: Transient‐crossed projections to the frontal cortex in neonatal rats. Journal of Comparative Neurology, 281(1), 1–12, doi: 10.1002/cne.902810102. [DOI] [PubMed] [Google Scholar]
- Mohammed AH, Zhu SW, Darmopil S, Hjerling-Leffler J, Ernfors P, Winblad B, et al. (2002). Environmental enrichment and the brain. Prog Brain Res 138:109–133. 10.1016/S0079-6123(02)38074-9 [DOI] [PubMed] [Google Scholar]
- Mooney SM, Miller MW (2010). Prenatal exposure to ethanol affects postnatal neurogenesis in thalamus. Exp Neurol 223:566–573. 10.1016/J.EXPNEUROL.2010.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney SM, Miller MW (2001). Effects of prenatal exposure to ethanol on the expression of bcl-2, bax and caspase 3 in the developing rat cerebral cortex and thalamus. Brain Res 911:71–81. 10.1016/S0006-8993(01)02718-4 [DOI] [PubMed] [Google Scholar]
- Morton RA, Diaz MR, Topper LA, Valenzuela CF (2014). Construction of Vapor Chambers Used to Expose Mice to Alcohol During the Equivalent of all Three Trimesters of Human Development. JoVE e51839. 10.3791/51839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser MB, Rowland DC, Moser EI. (2015). Place cells, grid cells, and memory. Cold Spring Harb Perspect Biol. 7(2):a021808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, & Handa RJ (1997). Fetal Alcohol Exposure Produces Delay-Dependent Memory Deficits in Juvenile and Adult Rats. Alcoholism: Clinical and Experimental Research, 21(4), 710–715, doi: 10.1111/j.1530-0277.1997.tb03826.x. [DOI] [PubMed] [Google Scholar]
- Nardelli A, Lebel C, Rasmussen C, Andrew G, Beaulieu C. (2011). Extensive Deep Gray Matter Volume Reductions in Children and Adolescents with Fetal Alcohol Spectrum Disorders. Alcohol Clin Exp Res. 35(8): 1404–1417. 10.1111/j.1530-0277.2011.01476.x [DOI] [PubMed] [Google Scholar]
- Neeper SA, Gomez-Pinilla F, Choi J, Cotman CW. (1996). Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res. 726: 49–56. [PubMed] [Google Scholar]
- Nelson AJD, Powell AL, Kinnavane L, Aggleton JP. (2018). Anterior thalamic nuclei, but not retrosplenial cortex, lesions abolish latent inhibition in rats. Behav Neurosci. 132: 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes PT, Gómez-Mendoza DP, Rezende CP, Figueiredo HCP, Ribeiro AM. (2018). Thalamic proteome changes and behavioral impairments in thiamine-deficient rats. Neurosci. 385: 181–197. [DOI] [PubMed] [Google Scholar]
- Nunes PT, Kipp BT, Reitz NL, Savage LM. (2019). Aging with alcohol-related brain damage: Critical brain circuits associated with cognitive dysfunction. Int Rev Neurobiol. 148: 101–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan RM, Ohle R, Kelly AM. (2007). The effects of forced exercise on hippocampal plasticity in the rat: A comparison of LTP, spatial- and non-spatial learning. Behav Brain Res. 176:362–366. [DOI] [PubMed] [Google Scholar]
- Olson AK, Eadie BD, Ernst C, Christie BR (2006). Environmental enrichment and voluntary exercise massively increase neurogenesis in the adult hippocampus via dissociable pathways. Hippocampus 16:250–260. 10.1002/hipo.20157 [DOI] [PubMed] [Google Scholar]
- Otero NKH, Thomas JD, Saski CA, Xia X, Kelly SJ. (2012). Choline Supplementation and DNA Methylation in the Hippocampus and Prefrontal Cortex of Rats Exposed to Alcohol During Development. Alcohol Clin Exp Res 36:1701–1709. 10.1111/j.1530-0277.2012.01784.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent A, Mackey A, De Bellefeuille L. (1983). The subcortical afferents to caudate nucleus and putamen in primate: a fluorescence retrograde double labeling study. Neurosci. 10: 1137–1150. [DOI] [PubMed] [Google Scholar]
- Perkins AE, Fadel JR, Kelly SJ (2015). The effects of postnatal alcohol exposure and galantamine on the context pre-exposure facilitation effect and acetylcholine efflux using invivo microdialysis. Alcohol 49:193–205. 10.1016/j.alcohol.2015.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry BAL, Mitchell AS. (2019). Considering the evidence for anterior and laterodorsal thalamic nuclei as higher order relays to cortex. Front Mol Neurosci. 12:167. doi: 10.3389/fnmol.2019.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli B, Weinberg J, Hicks GG. (2018). Effects of prenatal alcohol exposure (PAE): insights into FASD using mouse models of PAE. Biochem. Cell Biol 69(2): 131–147. 10.1139/bcb-2017-0280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitel AL, Chételat G, Le Berre AP, Desgranges B, Eustache F, Beaunieux H. (2012). Macrostructural abnormalities in Korsakoff syndrome compared with uncomplicated alcoholism. Neurology. 78: 1330–1333. [DOI] [PubMed] [Google Scholar]
- Pitel AL, Segobin SH, Ritz L, Eustache F, Beaunieux H. (2015). Thalamic abnormalities are a cardinal feature of alcohol-related brain dysfunction. Neurosci Biobehav Rev. 54: 38–45. [DOI] [PubMed] [Google Scholar]
- Preston-Ferrer P, Coletta S, Frey M, Burgalossi A. (2016). Anatomical organization of presubicular head-direction circuits. Elife. e14592. doi: 10.7554/eLife.14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rema V, Ebner FF (1999). Effect of enriched environment rearing on impairments in cortical excitability and plasticity after prenatal alcohol exposure. J Neurosci 19:10993–11006. 10.1523/jneurosci.19-24-10993.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice D, Barone S. (2000). Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environmental Health Perspectives, 108(Suppl 3), 511–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley EP, Barron S, Driscoll CD, Hamlin RT. (1986). The Effects of Physostigmine on Open-Field Behavior in Rats Exposed to Alcohol Prenatally. Alcoholism: Clinical and Experimental Research, 10(1), 50–53, doi: 10.1111/j.1530-0277.1986.tb05613.x. [DOI] [PubMed] [Google Scholar]
- Robinson JK, Mair RG. (1992). MK-801 prevents brain lesions and delayed-nonmatching-to-sample deficits produced by pyrithiamine-induced encephalopathy in rats. Behav Neurosci. 106: 623–633. [PubMed] [Google Scholar]
- Rodriguez CI, Davies S, Calhoun V, Savage DD, Hamilton DA (2016). Moderate Prenatal Alcohol Exposure Alters Functional Connectivity in the Adult Rat Brain. Alcohol Clin Exp Res 40:2134–2146 doi: 10.1111/acer.13175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Mark K, Vetreno RP, and Savage LM (2008). Increasing hippocampal acetylcholine levels enhance behavioral performance in an animal model of diencephalic amnesia. Brain Res. 1234: 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Levinson M, Vetreno RP, Savage LM. (2010). Differential effects of systemic and intraseptal administration of the acetylcholinesterase inhibitor tacrine on the recovery of spatial behavior in an animal model of diencephalic amnesia. Eur J Pharm. 629: 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Svensson FP, Mazeh A, Kocsis B. (2017). Prefrontal-hippocampal coupling by theta rhythm and by 2–5 Hz oscillation in the delta band: The role of the nucleus reuniens of the thalamus. Brain Struct Funct. 222: 2819–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saalmann YB. (2014). Intralaminar and medial thalamic influence on cortical synchrony, information transmission and cognition. Front Syst Neurosci. 8:83. doi: 10.3389/fnsys.2014.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli M, Granato A, Sbriccoli A, Gobbi G, Janiri L, Minciacchi D. (1995). Alterations of the thalamo-cortical system in rats prenatally exposed to ethanol are prevented by concurrent administration of acetyl- l-carnitine. Brain Research, 698(1), 241–247, doi: 10.1016/0006-8993(95)00997-5. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Liachenko S, Paule MG, Bowyer J, Hanig JP. (2016). Brain endothelial dysfunction following pyrithiamine induced thiamine deficiency in the rat. Neurotox. 57: 298–309. [DOI] [PubMed] [Google Scholar]
- Savage LM, Castillo R, Langlais PJ. (1998). Effects of lesions of thalamic intralaminar and midline nuclei and internal medullary lamina on spatial memory and object discrimination. Behav Neurosci. 112: 1339–1352. [DOI] [PubMed] [Google Scholar]
- Savage LM, Hall JM, Vetreno RP. (2011). Anterior thalamic lesions alter both hippocampal-dependent behavior and hippocampal acetylcholine release in the rat. Learn Mem. 18: 751–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Sweet AJ, Castillo R, Langlais PJ. (1997). The effects of lesions to thalamic lateral internal medullary lamina and posterior nuclei on learning, memory and habituation in the rat. Behav Brain Res. 82: 133–147. [DOI] [PubMed] [Google Scholar]
- Savage LM, Hall J, Resende LS (2012). Translational rodent models of Korsakoff syndrome reveal the critical neuroanatomical substrates of memory dysfunction and recovery. Neuropsych Rev. 22: 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider RD, Thomas JD (2016). Adolescent Choline Supplementation Attenuates Working Memory Deficits in Rats Exposed to Alcohol During the Third Trimester Equivalent. Alcohol Clin Exp Res 40:897–905. 10.1111/acer.13021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber WB, St. Cyr SA, Jablonski SA, Hunt PS, Klintsova AY, Stanton ME. (2013). Effects of exercise and environmental complexity on deficits in trace and contextual fear conditioning produced by neonatal alcohol exposure in rats. Dev Psychobiol 55:483–495. 10.1002/dev.21052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segobin S, Laniepce A, Ritz L, Lannuzel C, Boudehent C, Cabé N, Urso L, Vabret F, Eustache F, Beaunieux H, Pitel AL. (2019). Dissociating thalamic alterations in alcohol use disorder defines specificity of Korsakoff’s syndrome. Brain. 142: 1458–1470. [DOI] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. (2013). Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Progress in Neurobiology, 106–107, 1–16, doi: 10.1016/j.pneurobio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SM. (2016). Thalamus plays a central role in ongoing cortical functioning. Nat Neurosci. 19: 533–541. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Guillery RW. (1996). Functional organization of thalamocortical relays. J Neurophysiol. 76: 1367–1395. [DOI] [PubMed] [Google Scholar]
- Sidibé M, Bevan MD, Bolam JP, Smith Y. (1997). Efferent connections of the internal globus pallidus in the squirrel monkey: I. Topography and synaptic organization of the pallidothalamic projection. J Comp Neurol. 382: 323–347. [PubMed] [Google Scholar]
- Stanton ME, Goodlett CR (1998). Neonatal ethanol exposure impairs eyeblink conditioning in weanling rats. Alcohol Clin Exp Res 22:270–275. 10.1111/j.1530-0277.1998.tb03649.x [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A. (2008). Neuroimaging of the Wernicke-Korsakoff syndrome. Alcohol Alcohol: 44, 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sziklas V, Petrides M. (1999). The effects of lesions to the anterior thalamic nuclei on object-place associations in rats. Eur J Neurosci. 11: 559–566. [DOI] [PubMed] [Google Scholar]
- Taggart TC, Simmons RW, Thomas JD, Riley EP (2017). Children with Heavy Prenatal Alcohol Exposure Exhibit Atypical Gait Characteristics. Alcohol Clin Exp Res 41:1648–1655. 10.1111/acer.13450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JS (2007). The head direction signal: origins and sensory-motor integration. Ann Rev of Neurosci. 30:181–207. [DOI] [PubMed] [Google Scholar]
- Terasaki LS, Schwarz JM (2016). Effects of Moderate Prenatal Alcohol Exposure during Early Gestation in Rats on Inflammation across the Maternal-Fetal-Immune Interface and Later-Life Immune Function in the Offspring. J Neuroimmune Pharmacol 11:680–692. 10.1007/s11481-016-9691-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J, Wasserman E, West J, Goodlett C. (1996). Behavioral deficits induced by bingelike exposure to alcohol in neonatal rats: importance of developmental timing and number of episodes. Dev Psychobiol., 29(5), 433–452. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Goodlett CR, West JR (1998). Alcohol-induced Purkinje cell loss depends on developmental timing of alcohol exposure and correlates with motor performance. Dev Brain Res 105:159–166. 10.1016/S0165-3806(97)00164-8 [DOI] [PubMed] [Google Scholar]
- Thomas JD, Garcia GG, Dominguez HD, Riley EP (2004). Administration of eliprodil during ethanol withdrawal in the neonatal rat attenuates ethanol-induced learning deficits Psychopharmacology 175:189–195 doi: 10.1007/s00213-004-1806-x [DOI] [PubMed] [Google Scholar]
- Thomas JD, Sather TM, Whinery LA (2008). Voluntary Exercise Influences Behavioral Development in Rats Exposed to Alcohol During the Neonatal Brain Growth Spurt. Behav Neurosci 122:1264–1273. 10.1037/a0013271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V, Chopra K (2011). Resveratrol prevents alcohol-induced cognitive deficits and brain damage by blocking inflammatory signaling and cell death cascade in neonatal rat brain. J Neurochem 117:678–690. 10.1111/j.1471-4159.2011.07236.x [DOI] [PubMed] [Google Scholar]
- Todd KG, Butterworth RF. (1998). Evaluation of the role of NMDA-mediated excitotoxicity in the selective neuronal loss in experimental Wernicke encephalopathy. Exp Neurol. 149: 130–138. [DOI] [PubMed] [Google Scholar]
- Toledo Nunes P, Vedder LC, Deak T, Savage LM. (2019). A Pivotal Role for Thiamine Deficiency in the Expression of Neuroinflammation Markers in Models of Alcohol-Related Brain Damage. Alcohol Clin Exp Res. 43: 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson D, Wilce P, Bedi K. (1998). Spatial Learning Ability of Rats Following Differing Levels of Exposure to Alcohol During Early Postnatal Life. Physiol Behav 63:205–211. 10.1016/S0031-9384(97)00424-1 [DOI] [PubMed] [Google Scholar]
- Tong L, Prieto GA, Kramar EA, Smith ED, Cribbs DH, Lynch G, Cotman CW. (2012). Brain-Derived Neurotrophic Factor-Dependent Synaptic Plasticity Is Suppressed by Interleukin-1 via p38 Mitogen-Activated Protein Kinase. J Neurosci. 32: 17714–17724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treit S, Lebel C, Baugh L, Rasmussen C, Andrew G, Beaulieu C. (2013). Longitudinal MRI Reveals Altered Trajectory of Brain Development during Childhood and Adolescence in Fetal Alcohol Spectrum Disorders. J Neurosci. 33(24): 10098–10109. 10.1523/JNEUROSCI.5004-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsanov M, Chah E, Vann SD, Reilly RB, Erichsen JT, Aggleton JP, O’Mara SM. (2011) Theta-modulated head direction cells in the rat anterior thalamus. J Neurosci. 31: 9489–9502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich K, Spriggs MJ, Abraham WC, Dalrymple-Alford JC, McNaughton N. (2019). Environmental enrichment increases prefrontal EEG power and synchrony with the hippocampus in rats with anterior thalamus lesions. Hippo. 29: 128–140. [DOI] [PubMed] [Google Scholar]
- Uylings HBM, Kuypers K, Diamond MC, Veltman WAM (1978). Effects of differential environments on plasticity of dendrites of cortical pyramidal neurons in adult rats. Exp Neurol 62:658–677. 10.1016/0014-4886(78)90276-5 [DOI] [PubMed] [Google Scholar]
- Valenzuela CF, Morton RA, Diaz MR, Topper L (2012). Does moderate drinking harm the fetal brain? Insights from animal models. Trends Neurosci 35:284–292. 10.1016/J.TINS.2012.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Werf YD, Witter MP, Groenewegen HJ. (2002). The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Brain Res Rev. 39: 107–140. [DOI] [PubMed] [Google Scholar]
- van Eden C, Kros J, & HB U (1990). The development of the rat prefrontal cortex. Its size and development of connections with thalamus, spinal cord and other cortical areas. Prog Brain Res, 85, 169–183. [DOI] [PubMed] [Google Scholar]
- van Praag H, Kempermann G, Gage FH. (2000). Neural consequences of environmental enrichment. Nat Rev Neurosci. 1: 191–198. [DOI] [PubMed] [Google Scholar]
- van Praag H. (2009). Exercise and the brain: something to chew on. Trends Neurosci. 32 :283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vann SD, Brown MW, Aggleton JP (2000). Fos expression in the rostral thalamic nuclei and associated cortical regions in response to different spatial memory tests. Neuroscience 101:983–991 [DOI] [PubMed] [Google Scholar]
- Varela C. (2014). Thalamic neuromodulation and its implications for executive networks. Front Neural Circuits. 8:69. doi: 10.3389/fncir.2014.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela C, Kumar S, Yang JY, Wilson MA. (2014). Anatomical substrates for direct interactions between hippocampus, medial prefrontal cortex, and the thalamic nucleus reuniens. Brain Struct Funct. 219: 911–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaynman S, Ying Z, Gomez-Pinilla F. (2004). Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur Jour Neurosci. 20: 2580–2590. [DOI] [PubMed] [Google Scholar]
- Vedder LC, Hall JM, Jabrouin KR, Savage LM. (2015). Interactions between chronic ethanol consumption and thiamine deficiency on neural plasticity, spatial memory, and cognitive flexibility. Alcohol Clin Exp Res. 39: 2143–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuganti R, Kalluri H, Yi JH, Bowen KK, Hazell AS. (2006). Gene expression changes in thalamus and inferior colliculus associated with inflammation, cellular stress, metabolism and structural damage in thiamine deficiency. Eur J Neurosci. 23: 1172–1188. [DOI] [PubMed] [Google Scholar]
- Vertes RP, Albo Z, Viana Di Prisco G. (2001). Theta-rhythmically firing neurons in the anterior thalamus: implications for mnemonic functions of Papez’s circuit. Neurosci, 104: 619–625 [DOI] [PubMed] [Google Scholar]
- Vertes RP. (2006). Interactions among the medial prefrontal cortex, hippocampus and midline thalamus in emotional and cognitive processing in the rat. Neurosci. 142:1–20. [DOI] [PubMed] [Google Scholar]
- Vertes RP, Hoover WB, Do Valle AC, Sherman A, Rodriguez JJ. (2006). Efferent projections of reuniens and rhomboid nuclei of the thalamus in the rat. J Comp Neurol. 499: 768–796. [DOI] [PubMed] [Google Scholar]
- Vertes RP, Linley SB, Groenewegen HJ, Witter MP. (2015a). Thalamus. In Paxinos G (Ed.), The Rat Nervous System (4th ed.): Academic Press. [Google Scholar]
- Vertes RP, Linley SB, Hoover WB. (2015b) Limbic circuitry of the midline thalamus. Neurosci Biobehav Rev. 54: 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Bohnsack JP, Kusumo H, Liu W, Pandey SC, Crews FT. (2019). Neuroimmune and epigenetic involvement in adolescent binge ethanol-induced loss of basal forebrain cholinergic neurons: Restoration with voluntary exercise. Addict Biol. doi: 10.1111/adb.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Crews FT. (2018). Adolescent binge ethanol-induced loss of basal forebrain cholinergic neurons and neuroimmune activation are prevented by exercise and indomethacin. PLoS One. 13:e0204500. doi: 10.1371/j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Hall JM, Savage LM. (2011). Alcohol-related amnesia and dementia: animal models have revealed the contributions of different etiological factors on neuropathology, neurochemical dysfunction and cognitive impairment. Neurobiol Learn Mem. 96: 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor M, Adams RD, Collins GH. (1971). The Wernicke-Korsakoff syndrome Philadelphia: F.A.Davis Co. [PubMed] [Google Scholar]
- Vivar C, Potter MC, van Praag H. (2012). All About Running: Synaptic Plasticity, Growth Factors and Adult Hippocampal Neurogenesis. Neurogenesis and Neural Plasticity, 15: 189–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivar C, van Praag H (2017). Running Changes the Brain: the Long and the Short of It. Physiology 32:410–424. 10.1152/physiol.00017.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss MW, Vivar C, Kramer AF, van Praag H (2013). Bridging animal and human models of exercise-induced brain plasticity. Trends Cogn Sci 17:525–544. 10.1016/J.TICS.2013.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JR, Hamre KM, Pierce DR (1984). Delay in brain growth induced by alcohol in artificially reared rat pups. Alcohol 1:213–222. 10.1016/0741-8329(84)90101-0 [DOI] [PubMed] [Google Scholar]
- Whitcher LT, Klintsova AY (2008). Postnatal binge-like alcohol exposure reduces spine density without affecting dendritic morphology in rat mPFC. Synapse 62:566–573. 10.1002/syn.20532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LD, & Barone S Jr (2001). Qualitative and quantitative estimates of apoptosis from birth to senescence in the rat brain. Cell Death And Differentiation, 8, 345, doi: 10.1038/sj.cdd.4400816. [DOI] [PubMed] [Google Scholar]
- Winter SS, Clark BJ, Taube JS. (2015). Spatial navigation. Disruption of the head direction cell network impairs the parahippocampal grid cell signal. Science. 347: 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff M, Loukavenko EA, Will BE, Dalrymple-Alford JC. (2008). The extended hippocampal-diencephalic memory system: enriched housing promotes recovery of the flexible use of spatial representations after anterior thalamic lesions. Hippo. 18: 996–1007. [DOI] [PubMed] [Google Scholar]
- Wolff M, Vann SD. (2019). The Cognitive Thalamus as a Gateway to Mental Representations. J Neurosci. 39: 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouterlood FG, Saldana E, Witter MP. (1990). Projection from the nucleus reuniens thalami to the hippocampal region: light and electron microscopic tracing study in the rat with the anterograde tracer Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 296:179–203. [DOI] [PubMed] [Google Scholar]
- Wozniak JR, Fuglestad AJ, Eckerle JK, Fink BA, Hoecker HL, Boys CJ, et al. (2015). Choline supplementation in children with fetal alcohol spectrum disorders: A randomized, double-blind, placebo-controlled trial. Am J Clin Nutr 102:1113–1125. 10.3945/ajcn.114.099168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ. (2004). Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis. 17: 403–414. [DOI] [PubMed] [Google Scholar]
- Wozniak JR, Muetzel RL (2011). What does diffusion tensor imaging reveal about the brain and cognition in fetal alcohol spectrum disorders? Neuropsychol Rev 21:133–147. 10.1007/s11065-011-9162-1 [DOI] [PubMed] [Google Scholar]
- Yasukawa T, Kita T, Xue Y, Kita H (2004). Rat intralaminar thalamic nuclei projections to the globus pallidus: a biotinylated dextran amine anterograde tracing study. J Comp Neurol. 471: 153–67. [DOI] [PubMed] [Google Scholar]
- Ye X, Kapeller-Libermann D, Travaglia A, Inda MC, Alberini CM (2017). Direct dorsal hippocampal-prelimbic cortex connections strengthen fear memories. Nat Neurosci 20:52–61 doi: 10.1038/nn.4443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Yang Y, Gao L, Zhao J, Cai Y, Huang J, et al. (2015). Protective effects of resveratrol on the inhibition of hippocampal neurogenesis induced by ethanol during early postnatal life. Biochim Biophys Acta - Mol Basis Dis 1852:1298–1310. 10.1016/J.BBADIS.2015.03.009 [DOI] [PubMed] [Google Scholar]
- Zeisel SH (2011). What choline metabolism can tell us about the underlying mechanisms of fetal alcohol spectrum disorders. Mol Neurobiol 44:185–191. 10.1007/s12035-011-8165-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Burk JA, Glode BM, Mair RG. (1998). Effects of thalamic and olfactory cortical lesions on continuous olfactory delayed nonmatching-to-sample and olfactory discrimination in rats (Rattus norvegicus). Behav Neurosci. 112: 39–53. [DOI] [PubMed] [Google Scholar]
- Zhang SX, Weilersbacher GS, Henderson SW, Corso T, Olney JW, Langlais PJ. (1995). Excitotoxic cytopathology, progression, and reversibility of thiamine deficiency-induced diencephalic lesions. J Neuropathol Exp Neurol. 54: 255–267. [DOI] [PubMed] [Google Scholar]