

Abstract
Myelomeningocele (MMC) affects one in 1000 newborns annually worldwide and each surviving child faces tremendous lifetime medical and caregiving burdens. Both genetic and environmental factors contribute to disease risk but the mechanism is unclear. This study examined 506 MMC subjects for ultra-rare deleterious variants (URDVs, absent in gnomAD v2.1.1 controls that have Combined Annotation Dependent Depletion score ≥ 20) in candidate genes either known to cause abnormal neural tube closure in animals or previously associated with human MMC in the current study cohort. Approximately 70% of the study subjects carried one to nine URDVs among 302 candidate genes. Half of the study subjects carried heterozygous URDVs in multiple genes involved in the structure and/or function of cilium, cytoskeleton, extracellular matrix, WNT signaling, and/or cell migration. Another 20% of the study subjects carried heterozygous URDVs in candidate genes associated with gene transcription regulation, folate metabolism, or glucose metabolism. Presence of URDVs in the candidate genes involving these biological function groups may elevate the risk of developing myelomeningocele in the study cohort.
Subject terms: Developmental biology, Genetics, Neuroscience, Diseases, Molecular medicine, Neurology, Risk factors
Introduction
Successful neural tube formation relies on a series of orchestrated biological processes to facilitate convergent extension of neural ectodermal cells (NE) on the neural plate. NE cells at the midline and the dorsolateral regions form the medial hinge point (MHP) and the dorsolateral hinge point (DLHP) through mechanisms including apical construction and localized cell proliferation, and proliferation of non-neural ectodermal cells (NNE) that facilitate closing of the neural tube1 (Fig. 1). These biological processes involve sensing signals in the extracellular matrix by signal receptors on the cell surface and sub-organelles (e.g. mechanochemical sensors on primary cilia) which subsequently trigger remodeling of cytoskeletal structures and orchestrate directional migration of NE and NNE cells2. The neural tube can fail to close due to impaired convergent extension, MHP or DLHP formation, structural integrity of NE, or midline fusion of NE or NNE3. Maintaining NNE integrity and controlling epithelial-to-mesenchymal-transition of neural crest cells4, and balancing proliferation of hindgut cells5 (of endodermal origin) may also play important roles in the development of neural tube. Synchrony of all these processes together transform the neural plate into neural folds that meet and merge to form the neural tube. Factors interrupting the structure and/or function and spatial temporal integrity of these synchronous processes could result in failure of neural tube closure1,3. More than 400 genes have been identified in animal models that are required to maintain normal neural tube closure6–8. Many of the neural tube defect (NTD) animal model genes constitute structural and/or functional components of cilium, extracellular matrix, the WNT signaling pathway, actomyosin cytoskeleton remodeling and cell migration3,9.
Figure 1.
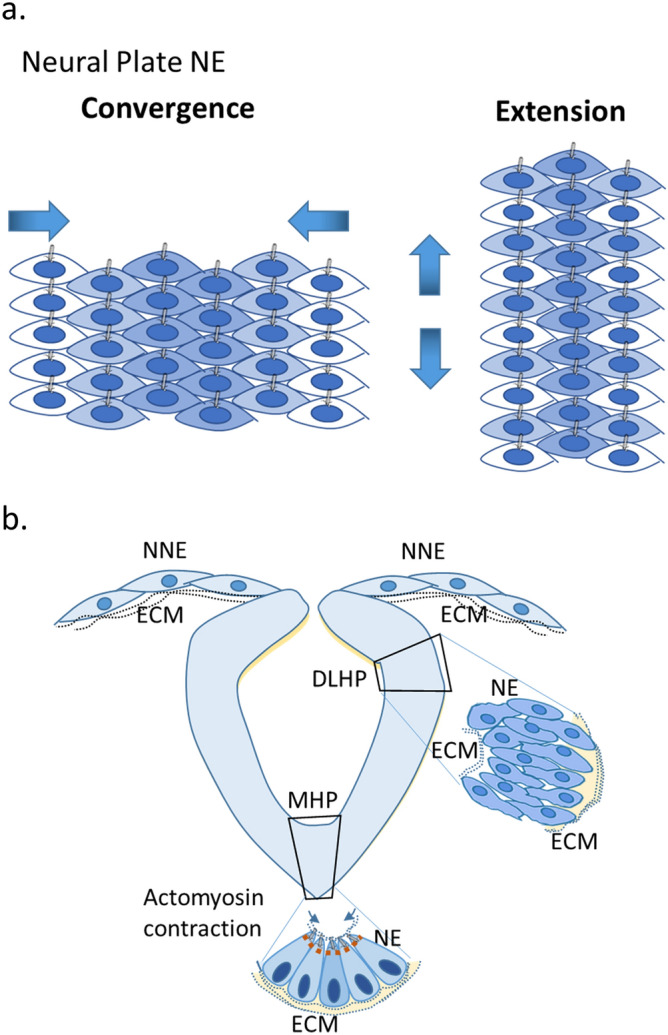
Neural tube formation involves a series of biological processes including mechanochemical signal sensing from the environment to orchestrate cell proliferation, cytoskeleton remodeling, and migration of neural and non-neural ectodermal cells. (a) Neural plate cells undergo convergent-extension to facilitate posterior-anterior elongation of the neural tube. Neuroectodermal cells (NE) migrate to the midline in response to extracellular signals received by cilia in NE cells to establish and maintain polarity. (b) Midline NE cells undergo apical constriction via a biological process involving actomyosin cytoskeleton remodeling to form the midline hinge point (MHP). NE cell and non-neuroectodermal (NNE) cells continue proliferation and migration during the neural tube closing process upon sensing signals in the extracellular matrix (ECM). NE cells at the dorsolateral hinge point (DLHP) undergo proliferation, migration dorsally, and reshaping to form the DLHP bringing the NE/NNE edges at the dorsal midline together. Advancing migration and proliferation of NNE and mesenchymal cells facilitate ectodermal layer extension when dorsal midline cells’ protrusions meet and join at the closing point. The process also brings NE cells together to establish cell junctions and close the NT.
An increasing number of heterozygous deleterious genetic variants in genes regulating planar cell polarity (PCP) have been revealed in human NTD cases and suggest that multi-genic heterozygous deleterious variants may contribute to NTD development10–13 . Knowledge is lacking regarding the extent of deleterious variants in genes involving various biological processes contributing to human myelomeningocele (MMC). It is possible that defective genes not known to interact with PCP genes in combination with defective PCP genes together can confer risk of MMC. Genes of interest for this study included variants of 551 candidate genes (537 mouse genes and 14 human NTD associated genes) previously shown to cause NTDs in mouse embryos or previously associated with human NTD (see Supplementary Table 1). In a complex trait study, the effect size of genetic variants has been shown to be inversely related to the frequency of the alternate allele of variants14, and new and private variants could constitute an important reservoir of disease risk alleles15. Single nucleotide variants (SNVs) in 551 candidate genes that were absent in non-Finnish European (NFE) or Ad-mixed American (AMR) in gnomAD v2.1.1 (controls) and which have top 1% deleteriousness (Combined Annotation Dependent Depletion phred score, C-score ≥ 20)16 were catalogued and defined as ultra-rare deleterious variants (URDVs) for analysis in the study. The majority of the homozygous defective mouse genes caused open neural tubes in the cranial regions whereas heterozygous mice had normal neural tube development9. Interestingly, some mice with a subset of these genes in the digenic heterozygote condition developed spina bifida, suggesting that oligogenic heterozygosity is a possible mechanism for spina bifida development17. Recent human spina bifida studies revealed cases carrying only heterozygous damaging variants of PCP genes and examples of subjects with digenic heterozygote rare variants in some PCP genes were observed13. In addition, some cases had damaging variants in two PCP genes each inherited from one of the parents12. Oligogenic inheritance is a demonstrated disease mechanism in many human diseases including the birth defect with holoprosencephaly18. Representation of oligogenic heterozygous URDVs in two or more NTD candidate genes found in MMC subjects in the study will be reported. Ontology analysis suggested URDV-containing NTD candidate genes were enriched for the same ontology group or in multiple categories including cilium, cytoskeleton, extracellular matrix, WNT signaling, and cell migration. The potential contribution of the observed results to MMC development in humans will be discussed.
Results
Global landscape of single nucleotide variants of MMC exomes
Three European (EA) and two Mexican (MA) MMC subjects had variant counts less than 80% of the median variant counts of the study subjects and were removed from further analysis, leaving 254 EA and 252 MA MMC exomes. A total of 597,401 high confidence SNVs that passed filtering steps described in the Materials and Method section were selected for further analysis. Using dbNSFP v4.0 to annotate high quality SNVs, 112,369 variants predicted to be functionally significant in 16,135 genes were selected for further analysis that included 109,727 missense, 1814 stop_gained, 84 stop_lost and 771 splice site variants. Non-coding SNVs and synonymous SNVs were not analyzed in this study.
Using AMR and NFE datasets from gnomAD v2.1.1 (controls) as references, approximately 15.1% and 13.2% of the SNVs in EA MMC and MA MMC respectively were ultra-rare variants (URVs) with alternate allele frequency (aAF) in the gnomAD v2.1.1 (controls) equal to zero or with an absence of the known variant. URVs found only in MMC subjects may constitute an important enriched pool of risk alleles for further analysis15. An average of 40 URVs per subject were identified. Over 98% of URVs occurred only once. Approximately 7400 URVs had C-score ≥ 20 in each ethnic group (see Supplementary Table 2). These variants were defined as ultra-rare functionally deleterious SNVs (URDVs) and used for further analysis. Each study subject had approximately 29 URDVs with range between 9 and 90.
Eighty-eight subjects were re-sequenced using the Illumina NGS platforms. Of the 30,789 SNVs in 33 EA and 37,924 SNVs in 55 MA identified from the Ion Proton platform in this study, over 98.5% were also identified by the Illumina NGS platforms.
Landscape of URVs in NTD candidate genes in human MMC exomes
There are several hundred genes known to cause NTDs in mice under monogenic and oligogenic conditions3,9. We sought insight through examining SNVs in 551 NTD candidate genes (see Supplementary Table 1) known to cause abnormal neural tube closure in animal models (537 genes), and 14 genes associated with human MMC in our study cohort. A total of 2134 SNVs in 440 genes were identified from EA MMC exomes and 2496 SNV in 447 genes were identified from MA MMC exomes.
Ultra-rare functional deleterious SNVs (URDVs) in candidate genes
Many studies have suggested the impact of variants is inversely proportionate to the alternate allele frequencies (aAF)14. Using aAF of variants in NFE/AMR gnomAD (controls) exomes as a filter, all 506 MMC subjects carried 31–69 rare deleterious variants (RDVs, aAF ≤ 0.01) in NTD candidate genes whereas the number of URDVs per subject varied between zero and eight (see Fig. 2 & Supplementary Table 3). With increased findings of novel and very rare variants in PCP genes associated with human NTDs in many previous studies3,19, we chose to focus on the novel URDVs, which are more likely to have higher effect size, for subsequent analyses.
Figure 2.

Distributions of URDVs Identified by WES. (a) Chart shows count of URDVs in NTD candidate gene(s) identified per exome of MM subjects varied from 0 to 8. In this study, the number of EA MMC subjects is 254 and the number of MA MMC subjects is 252. Approximately 25–30% subjects had no URDVs in NTD candidate genes, around 35% had one URDV, and the remaining had more than one. (b) Shows counts of URDVs discovered per NTD candidate gene ranged from one to nine with nearly 65% genes containing only one. Total number of URDV-containing NTD candidate genes found in EA and MA MMC subjects are 202 and 203 respectively.
Among 254 EA MMC subjects there were 307 URDVs in 202 genes. On average, each URDV-containing gene in EA MMC group had 1.52 URDVs. The range of URDVs per gene found in EA MMC was zero to six. Among 252 MA MMC subjects, there were 331 URDVs found in 203 genes. An average of 1.63 URDVs in a URDV-containing NTD candidate gene was identified in MA MMC group. The range of URDVs per gene found in MA MMC subjects was zero to nine. Over 97% of URDVs had an allele count (AC) of one and a few had two to three. The majority (~ 95%) of URDVs were missense changes while the remaining ~ 3.4% were stop_gained or splice site changes (1.3% and 2.4% in EA and MA groups respectively). Around 70% of MMC subjects in both ethnic groups had at least one URDV in an NTD candidate gene, with the highest URDV counts being eight and five in EA MMC and MA MMC subjects, respectively (Fig. 2). There were 78 EA MMC subjects and 67 MA MMC subjects who had no URDVs in NTD candidate genes.
URDV-containing NTD candidate genes were either common to both ethnicities or found uniquely in EA MMC and MA MMC exomes. There were 99 genes unique to EA MMC, 100 genes unique to MA MMC and 103 genes were common to both ethnic groups. Examining EA MMC and MA MMC respectively, most (66.7% and 63.4%) of the genes had one URDVs; 21.3% and 22.1% had two; 10.9% and 7.4% had three and the remaining few had four to nine URDVs. For the entire study cohort, 636 URDVs with 603 missense, 21 stop_gained and 12 splice site changes in 302 NTD candidate genes were discovered (Supplementary Table 1). These URDVs were manually verified using the online Integrative Genomics Viewer (IGV) Web App (https://igv.org/app). Seven subjects carried two heterozygous URDVs in their candidate genes (Supplementary Table 3). Visual inspection using IGV revealed the two heterozygous URDVs were in cis positions in the candidate genes (i.e. FREM2, FOLR2, RPGRIP1L, BMP2, and CGN) of five subjects respectively. One subject (i.e. C35-874, Supplementary Table 3) had two heterozygous URDVs in trans position in the KIF7 gene. The remaining subject (i.e. F57-264, Supplementary Table 3) carried two URDVs 1400 bp apart in two different exons of PNPLA6 therefore, we were unable to determine whether the URDVs were in cis or in trans.
The number of URDVs identified was not directly proportionate to the length of coding sequence (CDS) of genes in the two subject groups (Supplementary Table 3). Genes with longer transcript length (CDS length > 10 Kb) had a higher number of URDVs as expected (e.g. FREM2, HSPG2, APOB, DYNC2H1 and HTT) although their URDVs/Kb CDS was less than one. Overall, 96 genes had URDV densities of over one per Kb of CDS and the 64 genes had ≥ 1.25 URDVs/Kb CDS (Fig. 3, Supplementary Table 3). These 96 genes harboring 272 URDVs accounted for over 40% of the total 636 URDVs identified in 302 candidate genes. The median size of the CDS for the 96 genes was 1.39 Kb.
Figure 3.
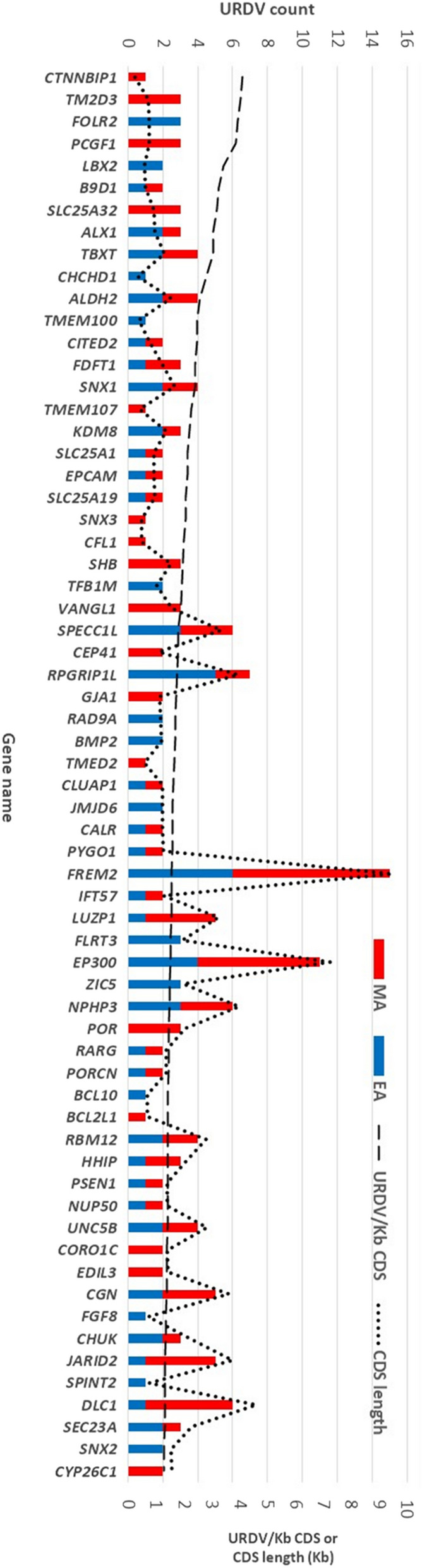
URDV density per Kb coding sequence of NTD candidate genes. The majority of 302 NTD candidate genes show URDV/KbCDS density less than one. The x-axis showed the gene names ordered by the URDV/Kb CDS from highest (4.1 for CTNNBP1) to lowest (1.28, CYP26C1). Dashed line (- - - -) showed the URDV/Kb CDS ratio of the gene and dotted line (…….) showed the length of CDS in Kb. Count of URDV in EA showed in blue bar and MA showed in red bar. Approximately half of the 64 genes had URDVs found in both EA and MA. One-third of the 64 genes have ≥ 2 URDVs/Kbp CDS.
Nine genes in MA MMC subjects had a higher number of URDVs, ranging from four to nine, with the most in FREM2. In EA MMC subjects, eight genes had four to six URDVs with six in APOB and in FREM2. Three genes (EP300, HSPG2, and FREM2) found in both EA and MA MMC subjects had more than ten URDVs with FREM2 and EP300 had an URDV density around 1.5.
Ontology Enrichment of NTD candidate genes with URDVs in MMC exomes
Among the 551 NTD candidate genes, enrichment of components in cilium, cytoskeleton, extracellular matrix and cell migration/adhesion, and WNT signaling were suggested (Supplementary Table 4). Using the ToppCluster20 online program, genes between EA and MA groups consisting of URDVs were compared to the group of genes lacking URDVs using Bonferroni correction for multiple testing. Analysis results showed genes in EA and MA groups had higher folds of enrichment in ontology classes than those of the overall 551 candidate genes. However, there was very distinct overrepresentation of subclasses of ontology for URDV-containing candidate genes in the subjects and these subclasses were absent among the candidate genes without URDVs (Fig. 4 and Supplementary Table 4). Enrichment of cellular component genes containing URDVs was seen mostly among ciliary components and intraciliary transport, actin filament, and microtubule cytoskeleton organization in both ethnic groups (Fig. 4a, and Supplementary Table 4). URDV-containing genes in EA MMC subjects were associated with components found in neuronal cell body, perinuclear region of cytoplasm, ciliary transition fiber, and WNT/SHH signaling. URDV-containing genes in MA MMC subjects were associated with components of ciliary basal body actin-based cytoskeleton and cell projection and cell–cell junctions. Differences in cellular component genes with URDV between EA and MA subjects were observed.
Figure 4.


Results of ontology and pathway enrichment analysis of candidate genes with and without URDVs identified. Enrichment analysis of (a) cellular components, (b) molecular functions, (c) pathways, and (d) biological processes, were performed using online tools ToppFun and ToppCluster. Count of URDV-containing genes in ontology class for EA (blue) and MA (red) MMC subjects and genes without URDVs (green) were shown with bar charts. Enrichment folds were represented with dashed lines for URDV-containing genes in EA (blue), URDV-containing genes in MA (red) and genes without URDVs (green). Enrichment comparisons passed Bonferroni correction. Detail descriptions of ontology, gene names, fold enrichment, and Bonferroni corrected P values can be found in Supplementary Table 4.
Molecular function analysis showed enrichment of URDV-containing candidate genes with molecular functions in transcription modulation, beta-catenin binding, cell adhesion, and cytoskeleton binding in both EA and MA MMC exomes (Fig. 4b, and Supplementary Table 4). EA MMC cases had enriched candidate genes related to hedgehog family protein and WNT signaling molecule binding. MA MMC cases had enriched candidate genes involved in DNA binding with transcription factors and nuclear hormone receptors. Together, nearly half of the MMC subjects in the study cohort had one or more URDVs in the candidate genes belonging to the ontological groups associated with ciliary structures and functions.
Both EA and MA MMC exomes consistently had URDV-containing genes disproportionately represented in the Hedgehog/WNT signaling and cancer signaling pathways (Fig. 4c, and Supplementary Table 4). Of note, EA MMC exomes uniquely enriched with URDVs in genes from WNT/PCP, SHH and TGFβ signaling pathways. MA MMC exomes were uniquely enriched with URDVs in genes related to thyroid hormone signaling, NOTCH signaling, and anchoring ciliary basal body.
Most of the NTD candidate genes involve biological processes of embryo development of organs particularly for the central nervous system and other organ systems (Supplementary Table 4). Ontology terms for biological processes are also consistent with those described in the cellular components and molecular functions sections (Fig. 4d).
Ciliary structure and functions are closely associated with GO terms for cytoskeleton/microtubules, extracellular matrix, WNT signaling, and migration of cells and cellular components. To further dissect the properties of URDV-containing NTD candidate genes and to examine the URDV distribution in MMC exomes, we performed analysis of genes assigned to these five GO terms (Supplementary Table 5). Candidate genes in each of these ontological groups consisted a range of 12–23.2% of the total URDVs (Table 1). Together, nearly half of the MMC subjects in the study cohort have one or more URDVs in the candidate genes belonging to the five ontological groups. There were 113 and 110 NTD candidate genes with URDVs respectively in the EA and MA subject groups that were associated with functions for cilium, ECM, cytoskeleton and WNT signaling and cell migration. Of these genes, some belong in only one GO term and many in two to four GO terms with cross-interacting roles (Fig. 5, and Supplementary Table 6). Of 364 subjects in the study cohort who bore at least one URDV in an NTD candidate gene, nearly 75% had URDVs affecting genes associated with cilium, WNT signaling, ECM, cytoskeleton and cell migration remodeling suggesting they may confer increased genetic risk to MMC development. The remaining 25% of MMC subjects had URDV-containing NTD candidate genes that were associated with other gene functions such as transcription modulation, folate one carbon metabolism network and glucose oxidative stress21 not related to the ontologies examined in this study.
Table 1.
Distribution of URDVs and genes in five biological ontology groups among MMC subjects.
| Categories | EA | MA | ||||||
|---|---|---|---|---|---|---|---|---|
| #URDV | #gene | #URDV/ gene | #subjects | #URDV | #gene | #URDV/ gene | Ð#subjects | |
| Cilium | 63 (20.3%) | 35 (17.2%) | 1.80 | 54 (21.3%) | 53 (16.1%) | 33 (16.3%) | 1.61 | 49 (19.4%) |
| Cytoskeleton | 72 (23.2% | 42 (20.6%) | 1.71 | 61 (24.0%) | 76 (23.0%) | 48 (23.6%) | 1.58 | 65 (25.8%) |
| ECM | 46 (14.8%) | 22 (10.8%) | 2.09 | 40 (15.7%) | 38 (11.5%) | 16 (7.9%) | 2.38 | 35 (13.9%) |
| WNT-signaling | 44 (14.1%) | 31 (15.2%) | 1.42 | 41 (16.1%) | 51 (15.5%) | 32 (15.8%) | 1.59 | 46 (18.3%) |
| Cell migration | 77 (24.8%) | 52 (25.5%) | 1.48 | 70 (27.6%) | 66 (20.0%) | 44 (21.7%) | 1.50 | 55 (21.8%) |
| Unique subtotal | 185 (59.4%) | 113 (55.4%) | 1.63 | 128 (50.4%) | 181 (54.8%) | 110 (54.2%) | 1.65 | 125 (49.6%) |
| Other ontologies | 125 (40.6%) | 89 (44.1%) | 1.36 | 48 (18.9%) | 150 (45.2%) | 93 (45.8%) | 1.61 | 60 (23.8%) |
| No URDVs | – | – | – | 78 (30.7%) | – | – | – | 67 (26.6%) |
| Total | 310 | 202 | 1.56 | 254 | 331 | 203 | 1.63 | 252 |
Note. Count of URDV, or neural tube defect candidate gene, or subjects with myelomeningocele are shown with the percentage to the total count of each column in bracket. Some genes are classified to more than one category. Count of URDVs, or gene or individual are presented follow by the percentage of the count presented in bracket. Some genes can be assigned to more than one ontology categories. A unique subtotal count shows the subtotal number of the URDV, or gene, or subject without double counting when a gene has multiple ontology groups assigned. Not in above – represent URDVs, genes or subjects outside the five ontologies. URDV—ultra-rare deleterious variant, EA—European American subject, MA—Mexican American subject. ECM – extracellular matrix.
Figure 5.

URDV containing candidate genes involved in multiple ontology groups. The Venn diagrams showed the relation and distribution of UTDV count and NTD candidate genes count to five ontology groups for EA (a) and MA (b). The number of URDVs and number of genes classified to cilium, WNT signaling, cytoskeleton, extracellular matrix (ECM) and cell migration are shown as fraction (count of URDV/count of gene). Genes classified to multiple categories were shown in the overlapped subset compartments between categories. Details on genes within each segment can be found in Supplementary Table 5.
Subjects had multiple URDV-containing genes affecting one or multiple ontologies
Recently published articles demonstrated the genetic contribution in individuals affected with NTDs that were potentially digenic heterozygote with de novo and/or rare deleterious variants inherited from each parent12. In this study, nearly 30% of MMC subjects had two or more URDVs in different NTD candidate genes belonging to the same and/or different ontologies. Overall, 85 EA MMC and 91 MA MMC exomes had multiple URDV-containing genes classified with different ontologies (Supplementary Table 3). Approximately 40% of these subjects had URDV-containing NTD candidate genes representing various combinations of the five ontologies presented above.
Oligogenic heterozygote URDVs involving the same ontology
A subset of MMC subjects including 25 EA and 21 MA were carrying two to three URDV-containing candidate genes that belong to the same ontology group (Table 2). Nine EA MMC and four MA MMC subjects had URDVs found in two candidate genes associated with cilia structure/function (Table 2). Of note, many cilium gene products also play roles in one or more of the other four ontologies examined here. Two EA MMC and five MA MMC subjects had URDVs in two genes with functions associated with WNT signaling (Table 2). Eleven EA MMC and nine MA MMC subjects had URDVs found in two genes with functions associated with cytoskeleton structures and functions (Table 2). Three EA MMC and one MA MMC subjects had URDVs found in two or three genes with functions associated with ECM structure and functions genes (Table 2). Finally, seven EA MMC and 10 MA MMC subjects had URDVs found in two or more candidate genes regulating cell migration (Table 2). Fifteen EA MMC subjects and 20 MA MMC subjects were carrying multiple URDV-containing candidate genes in different ontological groups.
Table 2.
Subjects with multiple URDVs in candidate genes classified with one or more ontological groups.






Note: EA—European American subjects, MA—Mexican American subjects. URDV—ultra-rare deleterious single nucleotide variant defined in Materials and Method section. Gene name—is the symbol of gene recommended by the Human Genome Organization Gene Nomenclature Committee (HGNC). Gene assigned into cilium, WNT-signaling, extracellular matrix (ECM), cytoskeleton and cell migration was described in Materials and Methods, and Supplementary Table 3. C-Score—Combined Annotation Dependent Depletion (CADD) score is a broadly applicable metric that objectively weights and integrates diverse information that integrates multiple functional annotations into one metric by contrasting variants that survived natural selection with simulated mutations.
URDV-containing genes and human neural tube expression and mouse phenotypes
The expression profiles of genes for four or more human neural tubes at Carnegie Stages (CS) 12 and 13 by SAGE were available for use to annotate NTD candidate genes with URDVs22. Expression of nearly 40% of the 302 URDV-containing NTD candidate genes was consistently detectable in CS12 and/or CS13 human neural tubes compared to less than 15% of 249 genes with no URDV identified (Supplementary Table 2). A Chi-square test comparing NTD candidate genes with and without URDVs showed significantly more URDV-containing genes expressed in human CS12 and CS13 (p < 0.0001, Supplementary Table 2). In addition, over 80% of the 302 URDV-containing NTD candidate genes caused embryonic lethality on or before neural tube closure in knockout mice of these genes9.
Discussion
This study revealed that 70% of the 506 MMC subjects consisting of the two ethnic groups with the highest prevalence of myelomeningocele in North America carry ultra-rare deleterious variants (URDVs) in 302 genes previously demonstrated to cause NTD phenotypes in animal models9 or associated with human NTDs (Supplementary Table 2). Ontology enrichment analyses showed around 50% of subjects in the cohort have one or more URDV-containing NTD candidate genes potentially impairing the structure and/or function of one or more ontological groups including cilium, SHH and WNT signaling, remodeling ECM and cytoskeleton, and cell migration. Normal structure and function of these genes are necessary for successful closure of the neural tube in animals1,3. The study results shown here may have a high impact on our understanding of the genetic mechanisms of human MMC.
One-fifth of MMC subjects in the study had URDVs in NTD candidate genes affecting cilium structure and/or function, suggesting human myelomeningocele risk is very likely associated with ciliopathy. The extent of URDV-containing ciliopathy genes associated with human NTD cases is not known. A review article by Vogel et al. (2012) noted that some human syndromes such as Meckel-Gruber syndrome and Joubert Syndrome that present with NTDs were associated with ciliopathies and recommended genetic screening of ciliopathy genes for human NTD cases23. Both NE and NNE involve the primary cilium playing roles in transducing extracellular signals to regulate cell growth, proliferation, directional migration and adhesion vital to neural tube development1,2,24–29. Many mutant mouse models with loss of function in the cilia-associated genes developed NTDs9. A high proportion of copy number variations in cilia genes have also been found in subjects affected by NTDs30. In this study, heterozygous URDVs identified in B9D1, CC2D2A, INPP5E, KIAA0586, KIF7, MKS1, NPHP3, PIBF1, RPGRIP1L, and TMEM67 belong to ciliopathy genes known to contribute to autosomal recessive syndromes MKS and JBTS in humans. In addition, there were cilium dependent Hedgehog signaling genes: DYNC2H1, GLI2, GLI3, GPR161, HHIP, IFT172, IFT57, IFT88, KIF7, MKS1, PRKACA, PTCH1, RPGRIP1L, SMO, and TULP3 present in MMC exomes. This study also found digenic heterozygous URDVs in two cilia genes in 13 MMC subjects. Several subjects had two URDV-containing genes classified to sub-functional groups such as Smoothened signal regulation (i.e. C2CD3-KIF7), Hedgehog off state (i.e. DYNC2H1-RPGRIP1L), Hedgehog signaling (i.e. HHIP-DYNC2H1), and dynein intermediate chain binding (i.e. DYNC2H1-HTT). It is likely that two genes within the same sub-functional group could contribute to digenic impairment of ciliary structure and/or functions critical to neural tube closure increasing the risk of MMC in these cases.
The PCP pathway modulates cellular functions such as ciliogenesis, actomyosin cytoskeleton and microtubules remodeling that are critical to neural tube formation1,31–36 . Normal ciliogenesis is important for non-canonical WNT PCP signaling37. Many studies showed defects in cilia can lead to canonical WNT signaling over-activation29. Furthermore, some evidence suggests cilium is a potential regulator (molecular switch) between canonical and non-canonical WNT signaling38. Neural tube formation involves biomechanical mechanisms resulting in mediolateral convergence and rostro-caudal extension of neural plate and axial tissues1. The convergent extension process depends on normal non-canonical WNT/PCP pathway activity. WNT/PCP activity influences remodeling of the cytoskeleton that drives events such as reshaping cells and bending the neural plate. Deleterious genetic variants disrupting normal function of core PCP maintenance genes (e.g. CELSR1, DACT1, SCRIB and VANGL2) has been associated with ~ 20% of craniorachischisis cases and 8% of spina bifida cases3,39. The extent of non-canonical WNT/PCP gene variants identified from subjects affected by various types of NTDs was recently published19. This study identified 7% of total MMC subjects with one or more URDVs affecting the WNT/PCP signaling pathway and 3–4% affecting core PCP pathway genes involved in neural tube closure. A handful of WNT signaling genes were shown to have higher mutational burden in the current study cohort40. Two URDVs [i.e. VANGL1 p.(V239I) and LRP6 p.(Y544C)] in two Mexican American subjects in the study cohort had been shown to cause functional loss of the proteins41,42. Discovery of heterozygous digenic deleterious variants in human NTD cases with spina bifida (i.e. PTK7/ SCRIB) and anencephaly (i.e. CELSR1/SCRIB; CELSR1/DVL3) strongly support the strategy of screening PCP genes to discover risk alleles contributing to human NTDs3,12. Here, we identified seven new digenic combinations of WNT/PCP signaling genes with three of these combinations (i.e. CELSR1-LRP6, SMURF2-LRP6, and PTK7-VANGL1) involving the core PCP pathway. Of these, three subjects had URDVs potentially interfering with regulation of planar polarity establishment (GO:0090175). These new gene combinations suggest previously unknown gene–gene interaction candidates for validation with animal studies in the future.
Knowledge of the extent of genetic risk of cytoskeleton structure function components to human MMC is limited. A quarter of the study subjects have one or more URDVs in 65 NTD candidate genes coding for cytoskeleton components to make microtubule structures: actomyosin filaments forming cell projections such as filopodia, lamellipodia, axoneme and cilia. Myosin motors along actin filaments facilitate vesicular cargo and biomolecule transport throughout the cell in response to the cues of signaling molecules transmitted from cilia37. In a study of 43 NTD trios, Lemay et al. (2015)43 identified heterozygote de novo deleterious variants in the SHROOM3 gene of one MMC case and one anencephaly case. Only one MMC subject in this study had a SHROOM3 URDV. Interestingly, half of the 65 cytoskeleton associated genes in MA and two-third in EA are associated with cytoskeleton and cilium, and the remaining genes belonging to actin cytoskeleton only. Digenic heterozygous URDV combinations found in MMC subject included C2CD3/KIF7, CC2D2A/WD11, DYNC2H1/RPGRIP1L and HHT/DYNC2H1 affecting cytoskeleton and cilium; and ABL1/CGN, KIF20B/KATNAL2 and MYH10/CGN affecting actin cytoskeleton function.
Nearly 15% of the subjects in this study had URDVs in NTD candidate genes coding for ECM components. The ECM compartment serves as a microenvironment retaining growth factors and signaling molecules (e.g. SHH, BMP, WNT, EGFs and FGFs) to recruit or influence neuroepithelial cells to determine shape, size, polarity and migration status for neural tube closing28. The roles of ECM components on regulating the position of the nucleus, cell shape, proliferation, migration, and morphogenesis of neural tube has been well-discussed1. Loss of function in some ECM components (e.g. Frem2 and Hspg2) caused NTDs in animal models9. This study is the first to reveal URDVs in FREM2, which were present in around 3% of study subjects, and URDVs in HSPG2, which were present in 2% of study subjects. Pathogenic variants in FREM2 have been associated with Cryptophthalmos (#123,570) and Fraser syndrome 2 (#617,666) with skull abnormalities or encephaloceles44. Digenic heterozygotes in two to four ECM genes were present in eight subjects in this study.
URDVs in cell migration genes were present in over 20% of the study subjects with many of these genes also involved in cilia, cytoskeleton, ECM, and WNT signaling. Ventral-dorsal migration of cells into the neural fold and rapid cell proliferation contribute to bending of the dorsolateral spinal neural plate, facilitating neural tube closure45. The ct mouse mutant has increased hindgut cell proliferation due to abnormal Grhl3 expression at the caudal region which surpasses NE and NNE migration and proliferation and prevents spinal neural tube closure5,46. Knockdown of the cell migration molecule Itgb1 prevents closure of the neuropore47. Mice with ablation of Rac1, a molecule modulating actin cytoskeleton remodeling and cell migration in NNE, developed open spina bifida, exencephaly or anencephaly48. In addition, impaired mesodermal cell migration due to diabetic pregnancy led to NTDs in mouse49. Seventeen MMC subjects in this study had oligogenic heterozygous URDVs in genes involving in cell migration providing examples for testing oligogenic heterozygote effects on cell migration as a mechanism of MMC development.
Knowledge of oligogenic gene–gene interaction between genes of different ontologies contributing to risk of myelomeningocele is lacking. Oligogenic inheritance in holoprosencephaly, a rare birth defect of the brain, was recently demonstrated18. Evidence of mouse embryos with oligogenic heterozygous PCP related genes knock-out developed spina bifida suggesting a subject having heterozygous URDVs in two NTD candidate genes belonging to the same ontology group may increase risk of MMC development9,17. So far, no non-syndromic human spina bifida cases with homozygous deleterious variants in one gene has been found, and only cases with digenic heterozygous rare deleterious variants in two PCP genes had been reported12,13. This study cohort showed examples of MMC subjects carrying multiple NTD candidate genes containing URDVs and one subject has two compound heterozygous URDVs in the KIF7 gene. Figure 6 summarized the potential impact of URDVs in NTD candidate genes identified from 506 MMC subjects. Hypothesis-free testing of random combinations of gene–gene interaction in animal models is too labor intensive and cost-prohibitive. While MMC risk for some subjects might be rationalized by disrupted genetic interaction between URDV-containing genes within the same ontology, subjects with URDV disrupted genes in different ontologies will be challenging. Here, the study results provide examples of new highly probable genetic interacting partners elevating MMC risk through an oligogenic heterozygote mechanism. For example, subject E52-879 has URDVs p.(D358N) in AGO2 and p.(Y544C) in LRP6. While interaction of AGO2 and LRP6 has not yet been demonstrated, it has been shown that inactivated canonical WNT signaling facilitated nuclear entry of miR-133a and complexed with AGO2 to suppress DnmT3b expression in mouse HL-1 cells50. Also, the presence of p.Y544C impaired LRP6 localization to plasma membrane for facilitating WNT signaling42. Subject D22-469 carries URDVs p.(N35K) and p.(H31R) respectively in SNX3 and CITED2, known components for WNT signaling and cell migration respectively. SNX3 binds WLS to regulate recycling of WLS thereby regulating the level of WNT excretion. Furthermore, it has been demonstrated that SNX3 with the subject’s URDV p.(N35K) failed to interact with WLS affecting WNT sorting and secretion51. A recent report demonstrated the developmental defects of Cited2 morphants in Zebrafish could be rescued with exogenous WNT5A and WNT1152.
Figure 6.

Summary of proportion of study subjects with URDV-containing NTD candidate genes constituting the components of cilium structure and function, WNT-signaling, cytoskeleton remodeling, extracellular matrix (ECM) remodeling and cell migration. URDVs were identified in the DNAs extracted from blood lymphocytes and expected to be present in all body cell types including both neural and non-neural ectodermal cells (NE and NNE respectively). Identifying the URDVs is the first of many steps leading to the understanding of the genetic mechanisms of human MMC development. Cross interaction of genes within and between the five groups is anticipated with many genes assigned to more than one of these groups. Presence of multiple genes with URDVs in one subject provide basis for testing potential gene–gene interaction that may interrupt cell proliferation, or convergent-extension, or apical constriction, or midline fusion or a combination of these processes leading to MMC.
This study identified URDVs in NTD candidate genes in 70% of MMC subjects lending strong support to the approach of screening NTD candidate genes to identify high impact genetic risk factors associated with human MMC development. Results of oligogenic URDV-containing cases present in this study provide a rational basis to test oligo-genic combinations in animal models to elucidate the mechanism(s) of MMC in humans. New oligogenic disease mechanism(s) for MMC development can be discovered from the new combinations of genes presented in this study and not previously known to cause NTDs. Testing genetic interactions between URDV-containing NTD candidate genes discovered from MMC subjects to understand human MMC mechanism is sound and promising.
In the study, we observed the presence of URDV-containing candidate genes that were common to EA and MA, and unique to one or the other ethnic population. Many of the genes unique to EA or MA belongs to the same ontology subclasses and a small number of genes belong to distinct specific subclasses. Differences in subclass information may help to reveal the disease mechanisms for the subjects who carried these variants. The potential of utilizing the observed differences in these genetic variants to predict differential risks in different ethnic populations may require replication with additional studies.
The study results were limited to examining the impact of ultra-rare SNVs in the coding region and the splice sites of NTD candidate genes. The impact of URDVs in genes not known to cause NTDs in mice has not been evaluated. Yet there are a significant portion of genes which have important biological function and are detected in the developmental stages when the neural tube closes that can affect neural tube development. Importance of rare deleterious variants, out of frame indel variants, deleterious non-coding variants and copy number variants should be recognized. Previous genetic association studies demonstrated some impact of common and rare deleterious variants to NTD risk in humans but generally lack replication due to various limitations of association study designs, ethnic background, and sample size. This study cohort has similar limitations. Separate analysis using all the common variants identified from the subject exomes did not yield significant association with the selected NTD candidate genes after correcting for multiple testing.
Materials and methods
Study population
A total of 511 subjects were selected for whole exome sequencing (WES) from a myelomeningocele study cohort enrolled from spina bifida clinics in five locations of North America between 1997 and 2010 (Au et al., 2008)53. All subjects provided an informed consent and enrolled in accordance with an institutional IRB at the University of Texas Health Science Center (UTHealth) at Houston. WES was performed on DNAs of 257 EA MMC including 140 females and 117 males, and DNAs of 254 MA MMC including 134 females and 120 males. Subjects in the study are sporadic cases and reported no family history of MMC. The majority (81%) of the study subjects were born before January 1998, the date North American countries mandated fortification of food crops with folic acid54. Fifty-two subjects were born in 1998 and 46 were born after 1998. Approximately 24% of the study subjects have MMC lesions at or above vertebrae L1 up to T10, 66.7% at or below vertebrae L2 down to sacrum, 1.7% had lesion spanning L4 to T10, and 8.2% had no specific lesion level information. Around 60% of the study subjects had hydrocephaly, 5.5% had arrested hydrocephaly, 3.7% had no hydrocephaly and 31.1% had no specific information. The majority subjects (74.8%) did not have information on Chiari Malformation, 0.6% had type I and 22.5% had type I Chiari Malformation, and 2.2% were normal. Blood samples were collected from subjects and parents where possible and genomic DNA was extracted for the study.
Exome sequencing and variant annotation
Exome library probes were made from an in-house design based on TargetSeq (Life Technologies, Inc.) with addition of splice sites, UTRs, small non-coding RNAs (e.g., microRNAs) and a selection of miRNA binding sites, and 200 bp promoter regions. High quality genomic DNA samples were processed using the exome library probes and the captured DNA products were sequenced following the manufacturer’s standard protocol for multiplexed sequencing using the P1 chip on the Ion Proton platform (Life Technologies, Inc.). Quality of sequencing was maintained at 40–60 million reads with read length between 120 and 150 bases, and over 75% reads were on-target for all successfully sequenced samples. Other quality control measures were implemented to map 45–60,000 SNP per sample with ~ 50% heterozygote variants and the Ts/Tv ratio average of 2.5. Samples that failed to meet the above quality criteria were re-sequenced. If re-sequencing failed, another subject’s DNA will be used to reach the goal of sequencing at least 500 subjects.
For sequence data that passed variant level and sample level quality filters, variant calling was conducted using Genome Analysis Toolkit GATK HaplotypeCaller version 3.x following best practice guidelines55. Additional variant filtering steps had been described previously21,40. Briefly, only variants designated a “PASS” by Variant Quality Score Recalibration (VQSR), met map quality score < 20, and having inbreeding coefficient < -0.3 were retained for further analysis. Individual sample filters were used to ensure that only high-fidelity variants with alternate allele depth > 25%, a read depth > 10, and genotype quality score > 20 were analyzed. Allele count (AC), allele number (AN), and allele frequency (AF) were recalculated for individual ethnicities after the filtering processes. Filtered high quality single-nucleotide variants (SNVs) were annotated using the non-synonymous single-nucleotide variant functional predictions (dbNSFP)56 version 4.0 database for all functional prediction information publicly available. Further analysis was focused on SNVs leading to stop_gained, stop_lost, non-synonymous, splice donor, and splice acceptor site changes in the canonical transcripts.
Thirty-three EA subjects and 55 MA subjects in the study were also re-sequenced using Illumina NGS platform (i.e. NovaSeq). Raw sequence reads of WES were mapped to GRCh38 (hg38) and read for WGS were mapped to GRCh37 (hg19). Variants were called following the GATK best practice guidelines. Then the variant data generated was filtered using the same filtering criteria described above for the Ion Proton generated data. Filtered variant data of the 88 subjects generated by the Ion Proton platform used in the study were extracted and compared to filtered variant data generated by the Illumina Platform to verify variants with concordance.
Ultra-rare functional deleterious SNVs (URDVs) analysis
The alternate allele frequencies (aAFs) of variants reported for the exomes of the non-Finnish European (NFE) and Ad Mixed American (AMR) control populations in the genome aggregation database gnomAD v.2.1.1 (https://gnomad.broadinstitute.org/) were used to annotate the single nucleotide variants (SNVs) in the study57. Variants having ethnic aAF = 0 or absent in gnomAD v2.1.1 (controls) despite high location coverage in gnomAD were defined as ultra-rare SNVs (URVs). Variants having aAFs in NFE or AMR gnomAD v2.1.1 (controls) less than 0.01 were defined as rare, whereas aAF ≥ 0.01 were defined as common. Combined Annotation Dependent Depletion16 (CADD; https://cadd.gs.washington.edu/) phred scores (C-score) of variants were used as the model to predict deleteriousness and variants with a C-score ≥ 20 (top 1% most deleterious) cutoff were defined as ultra-rare deleterious variants (URDVs) and retained for further comparison in the study. Genes with URDVs were further evaluated for potential association with MMC in the study cohort using the in silico function analysis tools. In addition, all URDVs identified in the NTD candidate genes in Supplementary Table 3 had been manually examined and confirmed using IGV Web App (https://igv.org/app/)58.
Online variant analysis tools
Annotation of gene and function with Gene Ontology59 (GO; http://geneontology.org) terms were used to estimate the extent of enrichment for particular ontology in molecular function, biological process, cell components and pathways in each group of genes found with URDVs to evaluate their potential relevance to development of neural tube defects. Several web based tools were used including PubMed and Mouse Genome Database9 ( http://www.informatics.jax.org/phenotypes.shtml) to compile a list of genes known to associate with neural tube defects (Supplementary Table 2) searching for terms: neural tube defects, open neural tube, spina bifida, anencephaly, exencephaly, craniorachischisis, and abnormal neural tube morphology. ToppFun (https://toppgene.cchmc.org/enrichment.jsp) and ToppCluster20 (https://toppcluster.cchmc.org/) were used to evaluate ontology enrichment and for comparing multiple groups of genes (Chen et al. 2009). Genes in the GO terms were extracted using g:Profiler60 (https://biit.cs.ut.ee/gprofiler/convert). Gene tolerant to loss-of-function mutation (pLi score) and haploinsufficiency were annotated using resources from DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources(DECIPHER; http://decipher.sanger.ac.uk)61.
To annotate expression of the genes of interest in animal neural tube, a database for human neural tubes during Carnegie stages CS12 and CS1322 (Krupp et al., 2008) was used. Genes expressed in mouse future spinal cord between Theiler Stages TS11-19 and early embryonic lethality mouse genes (MP:0008762) were extracted from MGI (http://www.informatics.jax.org/) for function annotation.
NTD candidate genes
Increasing numbers of published reports showed compound heterozygote deleterious variants of mouse NTD genes in human NTD subjects suggesting these variants as genetic risk factors for human NTDs. To facilitate variant containing gene prioritization, a list of 551 genes (Supplementary Table 2) was assembled from Mouse Genome Database9 (http://www.informatics.jax.org/) using search terms: open neural tube (MP:00000929), exencephaly (MP:0000914), anencephaly (MP:0001890), craniorachischisis (MP:0008784), and abnormal neural tube closure (MP:0003720). Also included were RARA, RARG, KIAA0586 and KATNAL2 with evidence of open neural tube defects in mice, chicken, frog and human62–64. Mouse NTD mutants not assigned to a gene and genes assigned only to spina bifida occulta were not included in this study. In addition, a list of 14 genes were added because association has been established in our study cohort (Supplementary Table 2). Together, a total of 551 candidate genes were selected for the study.
Ethical compliance
Affected subjects and/or their parents were recruited for the research study with a consenting protocol approved by the University of Texas Health Science Center (UTHealth) at Houston Institutional Review Board.
Supplementary Information
Acknowledgements
We gratefully acknowledge the subjects of the study cohort for participation supported by program project 5P01HD035946-02, and the studies and participants who provide biological samples and data to gnomAD. Also, we acknowledge Sarah Riosa for technical support, and Tina Findley, Laura Farach and Parker Laville for their help to review and comment on the manuscript. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Institute of Child Health and Human Development; the National Institutes of Health; or the U.S. Department of Health and Human Services. Grant support: NIH NICHD R01HD073434.
Author contributions
K.S.A., H.N., and J.E.H. were responsible for study design, analyzing and reviewing data. K.S.A., H.N., A.C.M. and J.E.H. were responsible for monitoring experimental progress. A.A-K., K.S., and M.G. provided list of genes expressed in human neural tubes at CS12 and CS13. D-K.K. performed WES sequencing experiments and raw data QC. P.H., C.B., L.H., M.R.B. provided bioinformatics support on exome sequence data QC, filtering, and annotating variants. S.L. and J.G. were responsible for resequencing a subset of 88 subjects in study cohort and provided variant data for comparison. All authors were responsible for manuscript review.
Funding
NIH/NICHD R01HD073434.
Data availability
Variants information is present in Supplementary Table 3. Also, variants will be available for review upon publication at dbSNP (https://www.ncbi.nlm.nih.gov/snp/) under BioProject ID: PRJNA611755.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material availlable at 10.1038/s41598-021-83058-7.
References
- 1.Nikolopoulou E, Galea GL, Rolo A, Greene NDE, Copp AJ. Neural tube closure: Cellular, molecular and biomechanical mechanisms. Development. 2017;144:552–566. doi: 10.1242/dev.145904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Veland IR, Lindbæk L, Christensen ST. Linking the primary cilium to cell migration in tissue repair and brain development. Bioscience. 2014;64:1115–1125. doi: 10.1093/biosci/biu179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Juriloff D, Harris M. Insights into the etiology of mammalian neural tube closure defects from developmental, genetic and evolutionary studies. J. Dev. Biol. 2018;6:22. doi: 10.3390/jdb6030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ray HJ, Niswander LA. Grainyhead-like 2 downstream targets act to suppress epithelial-to-mesenchymal transition during neural tube closure. Development. 2016;143:1192–1204. doi: 10.1242/dev.129825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gustavsson P, et al. Increased expression of Grainyhead-like-3 rescues spina bifida in a folate-resistant mouse model. Hum. Mol. Genet. 2007;16:2640–2646. doi: 10.1093/hmg/ddm221. [DOI] [PubMed] [Google Scholar]
- 6.Harris MJ, Juriloff DM. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res. Part A Clin. Mol. Teratol. 2010;88:653–669. doi: 10.1002/bdra.20676. [DOI] [PubMed] [Google Scholar]
- 7.Salbaum JM, Kappen C. Neural tube defect genes and maternal diabetes during pregnancy. Birth Defects Res. Part A Clin. Mol. Teratol. 2010;88:601–611. doi: 10.1002/bdra.20680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eppig JT, et al. The Mouse Genome Database (MGD): Comprehensive resource for genetics and genomics of the laboratory mouse. Nucleic Acids Res. 2012 doi: 10.1093/nar/gkr974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blake JA, Bult CJ, Kadin JA, Richardson JE, Eppig JT. The Mouse Genome Database (MGD): Premier model organism resource for mammalian genomics and genetics. Nucleic Acids Res. 2011;39:D842–D848. doi: 10.1093/nar/gkq1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Z, et al. Genetic analysis of Wnt/PCP genes in neural tube defects. BMC Med. Genomics. 2018;11:38. doi: 10.1186/s12920-018-0355-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishida M, et al. A targeted sequencing panel identifies rare damaging variants in multiple genes in the cranial neural tube defect, anencephaly. Clin. Genet. 2018;93:870–879. doi: 10.1111/cge.13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, et al. Digenic variants of planar cell polarity genes in human neural tube defect patients. Mol. Genet. Metab. 2018;124:94–100. doi: 10.1016/j.ymgme.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemay P, et al. Whole exome sequencing identifies novel predisposing genes in neural tube defects. Mol. Genet. Genomic Med. 2019;7:e00467. doi: 10.1002/mgg3.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bao EL, Cheng AN, Sankaran VG. The genetics of human hematopoiesis and its disruption in disease. EMBO Mol. Med. 2019;11:e10316. doi: 10.15252/emmm.201910316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katsanis N, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science (80-.) 2001 doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- 16.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris MJ, Juriloff DM. Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Res. Part A Clin. Mol. Teratol. 2007;79:187–210. doi: 10.1002/bdra.20333. [DOI] [PubMed] [Google Scholar]
- 18.Kim A, et al. Integrated clinical and omics approach to rare diseases: Novel genes and oligogenic inheritance in holoprosencephaly. Brain. 2019 doi: 10.1093/brain/awy290. [DOI] [PubMed] [Google Scholar]
- 19.Wang M, Capra, Kibar Update on the role of the non-canonical Wnt/planar cell polarity pathway in neural tube defects. Cells. 2019;8:1198. doi: 10.3390/cells8101198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaimal V, Bardes EE, Tabar SC, Jegga AG, Aronow BJ. ToppCluster: A multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. 2010;38:W96–W102. doi: 10.1093/nar/gkq418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hillman P, et al. Identification of novel candidate risk genes for myelomeningocele within the glucose homeostasis/oxidative stress and folate/one-carbon metabolism networks. Mol. Genet. Genomic Med. 2020;8(11):e1495. doi: 10.1002/mgg3.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krupp DR, et al. Transcriptome profiling of genes involved in neural tube closure during human embryonic development using long serial analysis of gene expression (long-SAGE) Birth Defects Res. Part A Clin. Mol. Teratol. 2012;94:683–692. doi: 10.1002/bdra.23040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogel TW, Carter CS, Abode-Iyamah K, Zhang Q, Robinson S. The role of primary cilia in the pathophysiology of neural tube defects. Neurosurg. Focus. 2012;33:E2. doi: 10.3171/2012.6.FOCUS12222. [DOI] [PubMed] [Google Scholar]
- 24.Christensen, S. T., Pedersen, S. F., Satir, P., Veland, I. R. & Schneider, L. Chapter 10 the primary cilium coordinates signaling pathways in cell cycle control and migration during development and tissue repair. In Current Topics in Developmental Biology 261–301 (2008). 10.1016/S0070-2153(08)00810-7. [DOI] [PubMed]
- 25.Murdoch JN, Copp AJ. The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res. Part A Clin. Mol. Teratol. 2010;88:633–652. doi: 10.1002/bdra.20686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ybot-Gonzalez P, Cogram P, Gerrelli D, Copp AJ. Sonic hedgehog and the molecular regulation of mouse neural tube closure. Development. 2002;129:2507–2517. doi: 10.1242/dev.129.10.2507. [DOI] [PubMed] [Google Scholar]
- 27.Ybot-Gonzalez P, et al. Convergent extension, planar-cell-polarity signalling and initiation of mouse neural tube closure. Development. 2007;134:789–799. doi: 10.1242/dev.000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB, Christensen ST. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019;15:199–219. doi: 10.1038/s41581-019-0116-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wheway, G., Nazlamova, L. & Hancock, J. T. Signaling through the primary cilium. Front. Cell Dev. Biol.6, 8. 10.3389/fcell.2018.00008 (2018). [DOI] [PMC free article] [PubMed]
- 30.Chen X, et al. Detection of Copy Number Variants Reveals Association of Cilia Genes with Neural Tube Defects. PLoS ONE. 2013;8:e54492. doi: 10.1371/journal.pone.0054492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rolo A, Escuin S, Greene NDE, Copp AJ. Rho GTPases in mammalian spinal neural tube closure. Small GTPases. 2018;9:283–289. doi: 10.1080/21541248.2016.1235388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malicki JJ, Johnson CA. The cilium: Cellular antenna and central processing unit. Trends Cell Biol. 2017;27:126–140. doi: 10.1016/j.tcb.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gurniak CB, Perlas E, Witke W. The actin depolymerizing factor n-cofilin is essential for neural tube morphogenesis and neural crest cell migration. Dev. Biol. 2005;278:231–241. doi: 10.1016/j.ydbio.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 34.Wallingford JB. Planar cell polarity, ciliogenesis and neural tube defects. Hum. Mol. Genet. 2006;15:R227–R234. doi: 10.1093/hmg/ddl216. [DOI] [PubMed] [Google Scholar]
- 35.May-Simera HL, Kelley MW. Cilia, Wnt signaling, and the cytoskeleton. Cilia. 2012;1:7. doi: 10.1186/2046-2530-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oteiza P, et al. Planar cell polarity signalling regulates cell adhesion properties in progenitors of the zebrafish laterality organ. Development. 2010;137:3459–3468. doi: 10.1242/dev.049981. [DOI] [PubMed] [Google Scholar]
- 37.Mirvis M, Stearns T, James Nelson W. Cilium structure, assembly, and disassembly regulated by the cytoskeleton. Biochem. J. 2018;475:2329–2353. doi: 10.1042/BCJ20170453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simons M, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beaumont M, et al. Targeted panel sequencing establishes the implication of planar cell polarity pathway and involves new candidate genes in neural tube defect disorders. Hum. Genet. 2019;138:363–374. doi: 10.1007/s00439-019-01993-y. [DOI] [PubMed] [Google Scholar]
- 40.Hebert H, et al. Burden of rare deleterious variants in WNT signaling genes among 511 myelomeningocele patients. PLoS One. 2020;15(9):83. doi: 10.22541/au.158983195.57483136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reynolds A, et al. VANGL1 rare variants associated with neural tube defects affect convergent extension in zebrafish. Mech. Dev. 2010;127:385–392. doi: 10.1016/j.mod.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lei Y, et al. Rare LRP6 Variants Identified in Spina Bifida Patients. Hum. Mutat. 2015;36:342–349. doi: 10.1002/humu.22750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lemay P, et al. Loss-of-function de novo mutations play an important role in severe human neural tube defects. J. Med. Genet. 2015;52:493–497. doi: 10.1136/jmedgenet-2015-103027. [DOI] [PubMed] [Google Scholar]
- 44.van Haelst MM, Scambler PJ, Hennekam RCM. Fraser syndrome: A clinical study of 59 cases and evaluation of diagnostic criteria. Am. J. Med. Genet. Part A. 2007;143A:3194–3203. doi: 10.1002/ajmg.a.31951. [DOI] [PubMed] [Google Scholar]
- 45.McShane SG, et al. Cellular basis of neuroepithelial bending during mouse spinal neural tube closure. Dev. Biol. 2015;404:113–124. doi: 10.1016/j.ydbio.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brook FA, Shum ASW, Van Straaten HWM, Copp AJ. Curvature of the caudal region is responsible for failure of neural tube closure in the curly tail (ct) mouse embryo. Development. 1991;113:671–678. doi: 10.1242/dev.113.2.671. [DOI] [PubMed] [Google Scholar]
- 47.Morita H, et al. Cell movements of the deep layer of non-neural ectoderm underlie complete neural tube closure in Xenopus. Development. 2012;139:1417–1426. doi: 10.1242/dev.073239. [DOI] [PubMed] [Google Scholar]
- 48.Rolo A, Galea GL, Savery D, Greene NDE, Copp AJ. Novel mouse model of encephalocele: Post-neurulation origin and relationship to open neural tube defects. Dis. Model. Mech. 2019;12:dmm04083. doi: 10.1242/dmm.040683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salbaum JM, et al. Novel mode of defective neural tube closure in the non-obese diabetic (NOD) mouse strain. Sci. Rep. 2015;5:16917. doi: 10.1038/srep16917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Mauro V, Crasto S, Colombo FS, Di Pasquale E, Catalucci D. Wnt signalling mediates miR-133a nuclear re-localization for the transcriptional control of Dnmt3b in cardiac cells. Sci. Rep. 2019;9:9320. doi: 10.1038/s41598-019-45818-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown HM, Murray S, Northrup H, Au KS, Niswander LA. Snx3 is important for mammalian neural tube closure via its role in canonical and non-canonical WNT signaling. Development. 2020;147(22):dev192518. doi: 10.1242/dev.192518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Santos JMA, et al. Exogenous WNT5A and WNT11 proteins rescue CITED2 dysfunction in mouse embryonic stem cells and zebrafish morphants. Cell Death Dis. 2019 doi: 10.1038/s41419-019-1816-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Au KS, et al. Characteristics of a spina bifida population including North American Caucasian and Hispanic individuals. Birth Defects Res. Part A Clin. Mol. Teratol. 2008;82:692–700. doi: 10.1002/bdra.20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.FDA-DHHS. Food Additives Permitted for Direct Addition to Food for Human Consumption; Folic Acid. vol. 81 https://www.govinfo.gov/content/pkg/FR-2016-04-15/pdf/2016-08792.pdf (2016). [PubMed]
- 55.Auwera, G. A. et al. From FastQ data to high‐confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinforma.43 (2013). [DOI] [PMC free article] [PubMed]
- 56.Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 2016;37:235–241. doi: 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karczewski KJ, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013 doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carbon S, et al. The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 2019;47:D330–D338. doi: 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raudvere U, et al. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Firth HV, et al. DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lohnes D, et al. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development. 1994;120:2723–2748. doi: 10.1242/dev.120.10.2723. [DOI] [PubMed] [Google Scholar]
- 63.Willsey HR, et al. Katanin-like protein Katnal2 is required for ciliogenesis and brain development in Xenopus embryos. Dev. Biol. 2018;442:276–287. doi: 10.1016/j.ydbio.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alby C, et al. Mutations in KIAA0586 cause lethal ciliopathies ranging from a hydrolethalus phenotype to short-rib polydactyly syndrome. Am. J. Hum. Genet. 2015;97:311–318. doi: 10.1016/j.ajhg.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Variants information is present in Supplementary Table 3. Also, variants will be available for review upon publication at dbSNP (https://www.ncbi.nlm.nih.gov/snp/) under BioProject ID: PRJNA611755.