

Abstract
Oligodendrocyte dysfunction has been implicated in the pathophysiology of amyotrophic lateral sclerosis (ALS), a neurodegenerative disorder characterized by progressive motor neuron loss. The failure of trophic support provided by oligodendrocytes is associated with a concomitant reduction in oligodendroglial monocarboxylate transporter 1 (MCT1) expression and is detrimental for the long-term survival of motor neuron axons. Therefore, we established an adeno-associated virus 9 (AAV9)-based platform by which MCT1 was targeted mostly to white matter oligodendrocytes to investigate whether this approach could provide a therapeutic benefit in the SOD1G93A mouse model of ALS. Despite good oligodendrocyte transduction and AAV-mediated MCT1 transgene expression, the disease outcome of SOD1G93A mice was not altered. Our study further increases our current understanding about the complex nature of oligodendrocyte pathology in ALS and provides valuable insights into the future development of therapeutic strategies to efficiently modulate these cells.
Keywords: amyotrophic lateral sclerosis, oligodendrocytes, monocarboxylate transporter 1, adeno-associated virus, gene therapy
Graphical Abstract
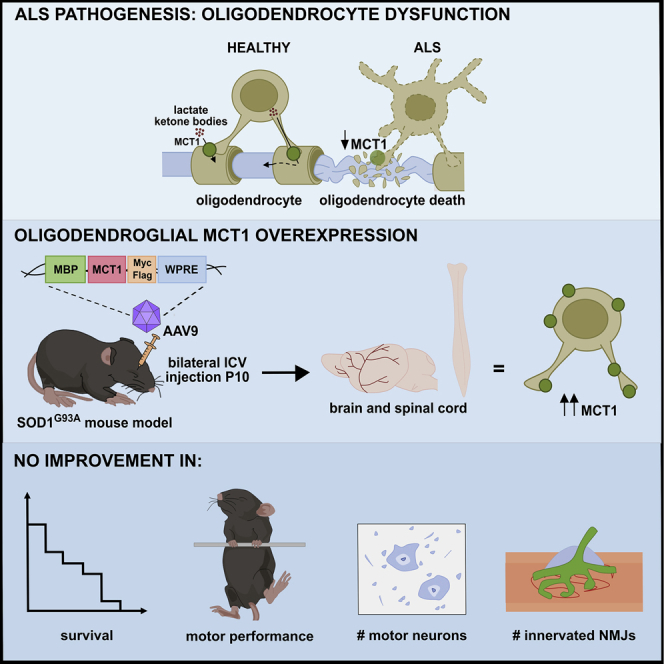
Dysfunctional oligodendrocytes contribute to motor neuron degeneration in ALS. Eykens et al. developed an AAV-based approach to stimulate oligodendrocyte protection in the SOD1G93A ALS mouse model. Using intracerebroventricular AAV9 delivery at postnatal day 10, they investigated the effect of restoring the expression of the oligodendroglial protein MCT1 on ALS pathology.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by the loss of both upper and lower motor neurons and typically strikes in midlife with an average age of onset in the mid to late 50s.1 Patients become wheelchair bound and generally succumb to the disease 3 to 5 years after diagnosis, mostly due to respiratory failure.2 Only in 10% of ALS patients a family history of disease is evident, with mutations in C9orf72, fused in sarcoma (FUS), TAR DNA-binding protein 43 (TARDBP), and superoxide dismutase 1 (SOD1) genes explaining about 70% of familial ALS.1,2 For the majority of sporadic patients (∼90%), the etiology remains unknown. Currently, no curative treatment is available and the two FDA-approved drugs, riluzole and edaravone, have demonstrated only a limited impact on the disease course or survival.1
For many years, it was thought that motor neurons were the main cell type affected in ALS. However, mutant SOD1-induced damage within glial cells was demonstrated to be instrumental in causing full-blown disease, qualifying ALS as a non-cell-autonomous disorder.3,4 Therefore, we focused on oligodendrocyte pathology as a component of the non-cell-autonomous disease mechanism of ALS. Several pathological studies reported oligodendrocyte abnormalities both in human ALS and ALS rodent models.5, 6, 7, 8 These include TDP-43 mislocalization and aggregation,5 changes in myelin protein composition,6 and increased oligodendrocyte progenitor cell (OPC) reactivity.9 Cell-fate tracing experiments in mice further confirmed the involvement of oligodendrocytes in ALS by demonstrating that OPCs proliferate alongside degenerating motor neurons and replace lost oligodendrocytes even before the manifestation of clinical symptoms.10 However, the newly formed oligodendrocytes are incapable of providing complete trophic support to the axons they enwrap. Both ALS patients and mutant SOD1 mice display reduced myelin basic protein (MBP) and monocarboxylate transporter 1 (MCT1) expression, reflecting oligodendrocyte dysfunction.11, 12, 13, 14 As a consequence, an important source of both trophic and structural support vanishes, thereby further weakening the already vulnerable motor neurons.
MCT1, along with MCT2 and MCT4, localizes to the central nervous system and is responsible for the proton-coupled transport of several monocarboxylate metabolites such as pyruvate, lactate, and ketone bodies.15,16 While MCT2 mainly localizes to neurons17,18 and MCT4 to astrocytes,17,18 MCT1 is primarily expressed in oligodendrocytes.11 This transporter is essential to meet the local energy requirements of neurons, since nutrients can only access motor neurons at the nodes of Ranvier or at the motor neuron cell body.13,17,19,20 Oligodendroglial MCT1 has recently been implicated in the astrocyte-neuron lactate shuttle, which serves as an important source of trophic support to neurons.13,21 Interestingly, removal of mutant SOD1 from the oligodendrocyte lineage in ALS mice preserved MCT1 expression and improved disease outcome.12 Similar observations were made in a recent study using induced pluripotent stem cell (iPSC)-derived co-cultures of oligodendrocytes and motor neurons.22 Mutant SOD1-expressing oligodendrocytes were able to induce motor neuron death, either in co-culture or upon incubation with oligodendrocyte-conditioned medium.22 These mutant oligodendrocytes produced lower amounts of lactate and expressed fewer MCT1 transporters. This means that the expression of mutant SOD1 in oligodendrocytes diminishes the ability of these cells to provide trophic support to motor neurons and strongly suggests that restoring oligodendroglial function might be beneficial for motor neuron survival.
Therefore, the aim of our study was to investigate whether enhancement of oligodendroglial MCT1 expression in ALS mice could increase the number of functional oligodendrocytes, leading to an improvement in disease outcome. To address this question, we established an adeno-associated virus 9 (AAV9)-based gene delivery platform by which MCT1 could be specifically targeted to oligodendrocytes in SOD1G93A mice. Despite having achieved efficient oligodendrocyte transduction and MCT1 transgene expression, the disease outcome in SOD1G93A mice was not altered.
Results
Efficient and specific GFP expression in oligodendrocytes throughout the CNS following i.c.v. MBP-GFP-AAV9 delivery
To study the role of specific oligodendroglial proteins such as MCT1 in ALS pathology, an efficient and selective oligodendrocyte-targeted gene delivery system is crucial. AAV-mediated central nervous system (CNS) targeted delivery of vectors that carry the MBP promoter confines transgene expression specifically to oligodendrocytes.23, 24, 25 To reach the most optimal gene delivery in both the brain and spinal cord, we modified the protocol described by Georgiou et al.24 to AAV9-mediated gene delivery via intracerebroventricular (i.c.v.) injections. The injections were performed at postnatal day 10, which was reported to improve the ratio of GFP-expressing oligodendrocytes throughout adult life.23
First, we evaluated the efficiency of our optimized delivery method using an MBP-GFP-AAV9 vector. The expression pattern of green fluorescent protein (GFP) was assessed 3 weeks following i.c.v. injection of C57BL/6 mice (1 × 1011 viral genomes [vg] per mouse). GFP-positive cells were detected alongside the sites of injection and also throughout different brain regions, including the corpus callosum, the forebrain, and hindbrain (Figures 1A–1D). Viral expression extended to gray and mostly white matter areas in the cervical, thoracic, and lumbar spinal cord (Figure 1E). Next, we determined the cell types expressing GFP using immunohistochemistry. Transverse sections of the lumbar spinal cord were double-stained with GFP and cell-type-specific markers for neurons (NeuN), astrocytes (GFAP), microglia (Iba1), OPCs (NG2), or oligodendrocytes (CC1). No GFP immunoreactivity was detected in neurons, astrocytes, microglia, or OPCs, while a strong GFP signal was found in CC1-positive oligodendrocytes (Figure 1F). We quantified the relative number of oligodendroglial, CC1-positive cells that were immunoreactive for GFP. Among all transduced cells in the lumbar spinal cord, quantification showed that nearly half of the oligodendrocytes were GFP-positive (Figure 1G). Western blot analysis further confirmed the presence of GFP in protein extracts from the brain, cervical, thoracic, and lumbar regions of the spinal cord, as well as in the sciatic nerve, but not in the gastrocnemius muscle (Figure 1H).
Figure 1.

Widespread and oligodendrocyte-specific AAV-mediated expression of GFP throughout the brain and spinal cord
(A–D) Images of various brain areas from a mouse that received an i.c.v. injection of the MBP-GFP-AAV9 vector examined around P30, as indicated including (A) the lateral ventricle, (B) the corpus callosum, (C) the forebrain, and (D) the hindbrain. Cell nuclei were counterstained with DAPI. Scale bar, 5 μm. (E) Representative transverse sections of the cervical, thoracic, and lumbar spinal cord were immunoreactive for GFP, whereas non-injected controls were not. (F) Immunostaining with cell-specific markers confirms that MBP-GFP-AAV9-induced GFP expression is not present in astrocytes (GFAP), OPCs (NG2), neurons (NeuN), and microglia (Iba1) but is localized in oligodendrocytes (CC1). Scale bar, 10 μm. (G) Following P10 delivery, GFP transgene expression was observed in 47% of all CC1+ oligodendrocytes in white matter regions of the lumber spinal cord. Data are presented as mean ± SEM, n = 3 mice per group. (H) Western blot analysis of GFP expression in protein extracts from spinal cord, brain, gastrocnemius, and sciatic nerve taken from non-transgenic (NTG) mice injected in both lateral ventricles with 1 × 1011 vg of MBP-GFP-AAV9. Non-injected (N.I.) mice were used as a control; SC C, cervical spinal cord; SC T, thoracic spinal cord; SC L, lumbar spinal cord; Gastrocn., Gastrocnemius muscle; SN, sciatic nerve.
Altogether, these results show that a single injection of the MBP-GFP-AAV9 vector in both lateral brain ventricles resulted in disperse CNS transduction. Highly efficient and oligodendrocyte-specific transgene expression was obtained in the brain and spinal cord. These data indicate that our adapted AAV9 system is a very good gene delivery platform that can be used for the modulation of oligodendrocyte function in mouse models with oligodendroglial pathology.
MBP-MCT1FLAG/Myc-AAV9 vector leads to oligodendrocyte-specific and efficient MCT1 overexpression
We next introduced a cDNA encoding mouse Mct1 into the MBP-AAV plasmid (MBP-MCT1FLAG/Myc-AAV), in order to obtain oligodendrocyte-specific MCT1 expression. The rationale for this was the observation that MCT1 expression is decreased in human ALS patients and rodent ALS models, which could contribute to the observed motor neuron death during ALS pathogenesis.11, 12, 13 Following i.c.v. injection of the MBP-MCT1FLAG/Myc-AAV9 vector in non-transgenic (NTG) mice at 10 days of age, we examined MCT1 transgene expression 3 weeks later in the CNS by immunostaining, qPCR, and western blot.
Oligodendrocytes were immunoreactive for the Myc tag, while Myc immunoreactivity was absent in the oligodendrocytes of non-injected animals (Figure 2A). Next, the mRNA expression level of MCT1 was determined in brain and spinal cord tissue of mice that were either non-injected or injected with MBP-GFP-AAV9, MBP-MCT1FLAG/Myc-AAV9, or MBP-empty-AAV9. The results showed a significant increase in MCT1 mRNA levels in brain and spinal cord tissues of mice injected with MBP-MCT1FLAG/Myc-AAV9, compared to non-injected controls (Figures 2B and 2C). No significant alteration in MCT1 mRNA expression was observed in mice injected with the control viral vectors. The expression of the MCT1 transgene was also confirmed by western blot analysis of brain and spinal cord lysates from MBP-MCT1FLAG/Myc-AAV9-injected mice using both anti-FLAG, as well as anti-MCT1 antibodies. The results confirmed the presence of a FLAG band, which was absent in non-injected controls (Figures 2D and 2E). Blotting with an MCT1 antibody revealed increased immunoreactivity in MCT1 overexpressing animals, with total MCT1 levels reaching about twice the amount of the non-injected mice (Figures 2D and 2E). Moreover, Myc immunoreactivity was observed in the sciatic nerve of mice injected with the MBP-MCT1FLAG/Myc-AAV vector and was absent in the sciatic nerve of non-injected controls (Figure S1). This observation strongly indicates that i.c.v. injection of our AAV vector resulted in transgene expression in the peripheral nervous system, more precisely in the sciatic nerve. Overall, these results show that AAV9-mediated MCT1 expression in the CNS and sciatic nerve was successful.
Figure 2.

i.c.v. MBP-MCT1FLAG/Myc-AAV9 injection leads to expression of the MCT1 transgene in NTG mice
(A) Double immunostaining for the oligodendrocyte marker CC1 (red) and the Myc tag (green) shows the presence of vector expressed MCT1 in spinal cord oligodendrocytes (left panel), while non-injected mice show no Myc immunoreactivity (right panel). Arrowheads indicate overlap between Myc and CC1. Scale bar, 20 μm (upper panel) and 10 μm (lower panels). (B and C) qPCR analysis of lumbar spinal cord and brain lysates from three MBP-MCT1FLAG/Myc-AAV9-injected mice shows upregulation of total MCT1 mRNA expression relative to Gapdh, Hprt, Ywaz, and Polr2a. Data are presented as mean ± SEM, n = 3 mice per group, One-way ANOVA, ∗p < 0.05, ∗∗∗∗p < 0.0001. (D and E) Western blot analysis of cervical spinal cord and brain tissue reveals the presence of a FLAG-positive band in injected mice, which is absent in non-injected mice. A non-specific band above the specific one is marked with an asterisk. Blotting with an MCT1 antibody and its quantification shows an increase of MCT1 protein following i.c.v. injection of the MBP-MCT1FLAG/Myc-AAV9 vector. GAPDH is used as a loading control. Data are presented as mean ± SEM, n = 3–5 mice per group, Student’s t test, ∗∗p < 0.01. NTG, non-transgenic mouse; EMPTY, MBP-Empty-AAV9-injected; GFP, MBP-GFP-AAV-injected; MCT1, MBP-MCT1FLAG/Myc-AAV9-injected.
AAV-mediated MCT1 overexpression does not improve key pathological features or survival in the SOD1G93A mouse model
MCT1 expression was shown to be decreased in human ALS patients and rodent ALS models.11, 12, 13 We confirmed the reduction of MCT1 protein levels at different stages of disease progression in the lumbar spinal cord of SOD1G93A mice compared to SOD1WT controls (Figure S2). Having established widespread CNS expression of our MBP-MCT1FLAG/Myc-AAV9 vector, including high expression levels in white matter oligodendrocytes, we next tested its effect in the SOD1G93A mouse model of ALS. ALS mice were injected with either the MBP-GFP-AAV9, MBP-MCT1FLAG/Myc-AAV9, or MBP-empty-AAV9 vector at postnatal day 10 and subjected to the hanging wire test to evaluate motor performance starting at the age of 60 days (Figure 3A), while survival was also monitored (Figure 3B).
Figure 3.

Overall survival rate, functional outcome, (motor) neuron survival, and neuromuscular junction (NMJ) innervation are not improved by AAV-mediated MCT1 overexpression in the CNS of ALS mice
(A) Disease onset determined by failure in hanging wire (HW) performance is not significantly altered in MCT1-overexpressing mice (median: EMPTY, 112 days; GFP, 113 days; MCT1, 114 days; log-rank test > 0.05). (B) Survival is not improved following i.c.v. MBP-MCT1FLAG/Myc-AAV9 injection in ALS mice (median: EMPTY, 154.5 days; GFP, 156 days; MCT1, 155 days; log-rank test > 0.05). (C) Disease duration, based on age at failure on the hanging wire test, is not significantly altered between groups. Data are presented as mean ± SEM, n = 23–26 mice per group, One-way ANOVA. Ns, non-significant. (D) Upper panel is the visualization of neurons by means of a thionin staining. The lower panel represents the quantification of neurons in the ventral horn of end-stage ALS mice categorized according to size and normalized to 300,000 μm2 of ventral horn. Neurons with an area above 250 μm2 were considered to be motor neurons. Data are presented as mean ± SEM, n = 4-5 mice per group, one-way ANOVA per size category, p > 0.05. (E) The upper panel represents an example of a denervated and innervated NMJ. The lower panel is the proportion of innervated and denervated NMJs in the gastrocnemius muscle at disease end-stage. Data are presented as mean ± SEM, n = 4-5 mice per group, one-way ANOVA p > 0.05p. Scale bar, 20 μm. SV2, synaptic vesicle protein; NFL, neurofilament; BTX, α-bungarotoxin; EMPTY, MBP-Empty-AAV9-injected; GFP, MBP-GFP-AAV9-injected; MCT1, MBP-MCT1FLAG/Myc-AAV9-injected.
No significant delay in age at disease onset, as determined by failure on the hanging wire test, could be observed for MBP-MCT1FLAG/Myc-AAV9-injected animals compared to the control groups (Figure 3A). Virally expressed MCT1 did not alter the median survival or disease duration of SOD1G93A mice, when compared to controls (Figures 3B and 3C).
At end-stage disease, the number of (motor) neurons present in the ventral horn of the spinal cord was not different between groups (Figure 3D). Longitudinal sections of the gastrocnemius muscle were used to monitor α-bungarotoxin (BTX)-positive motor endplates, while the axons were stained with neurofilament (NFL) and synaptic vesicle protein 2 (SV2; Figure 3E). At this time point, the majority of neuromuscular junctions (NMJs) were denervated in the ALS mice. MBP-MCT1FLAG/Myc-AAV9-injected mice displayed a non-significant trend toward a decrease in the number of innervated NMJs (Figure 3E). Overall, however, NMJ innervation or denervation were not altered between the different groups. Altogether, the treatment of ALS mice with a viral vector encoding MCT1 did not improve key pathological features or survival in the SOD1G93A mouse model.
Virally expressed MCT1 is still detectable at end-stage disease
As treatment with MBP-MCT1FLAG/Myc-AAV9 was not able to improve the disease outcome in the SOD1G93A mouse model, we next evaluated whether MCT1 was still present at end-stage disease. It was reported that AAV-mediated gene expression in the CNS was long-lasting.26,27 However, it might be that the increased turnover of oligodendrocytes in this ALS mouse model12,13 results in a subsequent loss of transgenic MCT1 expression. Therefore, we examined MCT1 transgene expression at disease end-stage by immunostaining, qPCR, and dot blot.
Lumbar spinal cord sections of end-stage ALS mice remained immunoreactive for Myc and showed a similar vector distribution as described for NTG mice (compare Figures 1E and 4A). qPCR results revealed a significant increase in MCT1 mRNA levels in the lumbar spinal cord of mice injected with MBP-MCT1FLAG/Myc-AAV9 compared to the controls, even at disease end-stage (Figure 4B). MCT1 protein expression was confirmed by dot blot analysis of brain lysates from mice injected with MBP-MCT1FLAG/Myc-AAV9, using an anti-FLAG antibody (Figure 4C). Altogether, these results show that MCT1 was still overexpressed at end-stage disease in the ALS mice and that a loss of MCT1 expression did not explain the lack of a therapeutic effect.
Figure 4.

Virally expressed MCT1 is still detectable at disease end-stage in ALS mice
(A) Double immunostaining for the oligodendrocyte marker CC1 (red) and the Myc tag (green) or GFP shows the presence of vector expressed MCT1 and GFP in spinal cord oligodendrocytes (upper and middle panel), while MBP-Empty-AAV-injected mice show no Myc or GFP immunoreactivity (lower panel). Scale bar, 50 μm. Right column represents a magnification of the left side images. Scale bar, 20 μm. (B) qPCR analysis of spinal cord lysates from MBP-MCT1FLAG/Myc-AAV9-injected mice shows upregulation of total MCT1 mRNA expression at end-stage disease. Data are presented as mean ± SEM, n = 4-9 mice per group, one-way ANOVA ∗∗∗p < 0.001. (C) Dot blot analysis of FLAG-tagged MCT1 protein levels in brain lysates. Ponceau staining was used as a loading control. EMPTY, MBP-Empty-AAV9-injected; GFP, MBP-GFP-AAV9-injected; MCT1, MBP-MCT1FLAG/Myc-AAV9-injected.
Discussion
Oligodendrocytes provide both structural and trophic support to axons and are pivotal for the integrity and long-term survival of axons.28,29 Failure of oligodendrocytes has already been implicated in several disorders such as leukodystrophies,30 Alzheimer’s disease,31,32 multiple sclerosis,33 and more recently in ALS.11, 12, 13 Previous work revealed that oligodendrocytes are dysfunctional in ALS and that this contributes to the observed motor neuron death. To this end, impairment of trophic support, as a consequence of reduced oligodendroglial MCT1 expression, has been proposed as one of the pathogenic mechanisms of ALS.11, 12, 13 Therefore, the main goal of this study was to develop a gene therapy approach to restore MCT1 levels, specifically in oligodendrocytes. We achieved widespread oligodendroglial transduction in brain and spinal cord, following a single injection of the MBP-MCT1FLAG/Myc-AAV9 vector into the lateral ventricles. Using this approach in the SOD1G93A mouse model of ALS, we did not observe a functional rescue of the disease phenotype, nor a survival benefit. As a consequence, we conclude that upregulation of the MCT1 lactate transporter in white matter oligodendrocytes does not ameliorate the disease outcome in these ALS mice, at least with the overexpression we were able to achieve.
To date, several studies have tried to establish an AAV-mediated gene delivery platform for oligodendrocytes with varying levels of success in terms of specificity and efficacy.23 We were able to overcome this issue via i.c.v. administration of AAV9 at the age of 10 days in mice. At this age, a significant number of mature oligodendrocytes is already present, which was previously reported to improve the ratio of GFP-expressing oligodendrocytes throughout adult life.23 As the focal injection of AAV in brain tissue only allows for local vector spread over a range of a few millimeters,23,24 we decided to deliver the constructs via i.c.v. injections. With this approach, we took advantage of the ventricular system that interconnects the brain and spinal cord via the central canal and facilitates cerebrospinal fluid (CSF) flow.34 We indeed succeeded in achieving widespread and oligodendrocyte-selective AAV-mediated transgene expression throughout the brain and spinal cord, with extremely high transduction levels within white matter regions. Our transduction profile was comparable to an earlier study that reported a transduction efficiency of 41% of spinal cord oligodendrocytes, following injection of the same MBP-GFP-AAV1/2 construct in the internal capsule of neonatal mice.24
Next, we used our AAV platform as a tool to express genes specifically in oligodendrocytes in the context of ALS. To this end, we investigated whether restoration of oligodendroglial MCT1 expression and hence trophic support provided by oligodendrocytes could be beneficial in the SOD1G93A mouse model of ALS. We established oligodendrocyte-specific AAV9-mediated MCT1 expression in vivo. This approach significantly increased MCT1 mRNA and protein expression levels in brain and spinal cord tissue. In addition, virally expressed MCT1 was still detectable at end-stage disease.
Despite these results, we were not able to improve disease outcome in the SOD1G93A mouse model of ALS. This was unexpected, particularly because the deletion of mutant SOD1 in oligodendrocytes was previously associated with an approximately 2-fold increase in MCT1 protein at disease onset and was beneficial in the SOD1G37R mouse model of ALS.12 Given our comparable levels of MCT1 protein overexpression, one possibility for the absence of a therapeutic effect might be the difference in disease severity between the SOD1G37R and SOD1G93A mouse models. Both models are characterized by dramatic loss of ventral horn motor neurons, accompanied by progressive hind- and forelimb weakness and ultimately death.35 However, it must indeed be noted that these animals phenotypically differ in the ages at which these symptoms occur and their overall time of survival. SOD1G93A mice display a faster and more aggressive disease progression, which might contribute to the fact that a 2-fold increase of MCT1 expression might not be enough to prolong survival and slow down disease progression as seen in the SOD1G37R mouse model.
Another possible explanation can be found in the complex nature of oligodendrocyte pathology in ALS. It has been demonstrated that numerous oligodendrocyte functions are disrupted in ALS, including trophic support, myelination, and oligodendrocyte differentiation. These observations raise the possibility that multiple injuries occur in oligodendrocytes during ALS pathogenesis, which could make it very challenging to establish improved oligodendroglial health. For instance, previous efforts to restore proper oligodendrocyte differentiation in ALS mice, by inhibiting factors that negatively influence oligodendrocyte maturation, were unsuccessful.36 What we can learn from this study is that intervening at only one level of the problem, whether it be oligodendrocyte differentiation, trophic support, or myelination, might not be sufficient to elicit a disease-relevant improvement in oligodendrocyte function. Therefore, it is of translational relevance to explore strategies by which initial oligodendrocyte degeneration can be prevented or move to combination therapies tackling multiple aspects of the pathological processes. For instance, the replacement of defective oligodendrocytes with healthy ones can potentially also promote the protection of neurons. To this end, transplanted OPCs have been shown to integrate and differentiate into mature oligodendrocytes in vivo,37, 38, 39 and this procedure has already demonstrated significant improvements in recovery of locomotor function in a rat model of spinal cord injury.37,39 Despite the fact that OPC transplantation is still in its infancy, it might hold great promise to restore oligodendrocyte function in ALS and other neurodegenerative disorders.
In the current study, we mainly targeted oligodendrocytes residing in white matter regions, which are less affected than gray matter ones during ALS pathogenesis.12 However, the importance of white matter oligodendrocytes in supporting motor neurons should not be underestimated. Motor neuronal cell bodies and axon terminals are localized in the gray matter, while the white matter is made out of myelinated fibers that also possess a significant energy requirement.40,41 The axons of motor neurons arising from the motor cortex pass through these white matter columns before synapsing with lower motor neurons. Therefore, local MCT1-mediated lactate delivery to these extremely long axons in the white matter is a very important source of energy, which is critical to maintain the integrity of the motor axons initiating in the motor cortex. On this note, one has to keep in mind that properly functioning MCT1 transporters might be compromised by defects in downstream lactate transport, such as axonal MCT2 expression or function. This could result in lactate being exported by MCT1, as expected, but merely accumulating in the periaxonal space due to lack of axonal uptake. In addition, it must be noted that lower motor neurons also have a significant association with Schwann cells42,43 and that we detected the MCT1 transgene in the sciatic nerve of mice that were injected with the MBP-MCT1FLAG/Myc-AAV9 vector (Figure S1). Since it has been demonstrated that Schwann cells contribute to disease progression in ALS mice,42,44 we must bear in mind that our approach might also have induced alterations within the sciatic nerves of the SOD1G93A mice.
In conclusion, our study highlights that many aspects of oligodendrocyte pathology in ALS need to be further investigated to gain a better insight into strategies to efficiently modulate these cells during the disease. Once we have a better mechanistic insight, our highly effective AAV9-based gene delivery platform will be a valuable tool to correct the function of these cells in the context of ALS, as well as in other neurodegenerative disorders with oligodendroglial involvement.
Materials and methods
Animal housing and study approval
Mice were housed in the KU Leuven animal facilities on a 12-h light-dark cycle at a temperature of 20°C. Animals were given free access to standard rodent chow and water and were helped with their fluid intake at later disease stages by provision of an extended spout. Protocols were designed to minimize animal discomfort, and all experiments were approved by the local Ethical Committee of the University of Leuven, Belgium (P120/2015).
Mouse models
Human wild-type SOD1 mice (B6SJL-Tg [SOD1]2Gur/J; stock number: 002297) overexpress the normal allele of the human SOD1 gene. This strain serves as a control for human mutant SOD1 overexpressing mice (B6SJL-Tg [SOD1∗G93A] 1Gur/J; stock number: 002726). Both mouse strains were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and were maintained on a C57BL/6 background after backcrossing to C57BL/6 for more than 20 generations. C57BL/6 NTG littermates were used as additional controls.
Disease onset and survival
The hanging wire test was used to assess hind- and forelimb muscular strength. Mice were placed on a wire grid, which was inverted, and the latency to fall was recorded over a maximum period of 60 s. The age of disease onset was taken to be the point at which a mouse was no longer able to hold on for 60 s. Survival was determined by the loss of the righting reflex, defined as the ability to regain normal posture within 20 s of being placed on its side. This time point was considered as the end-stage of the disease, whereupon the mouse was sacrificed. The investigator was blinded to the genotype and/or the treatment when assessing this parameter.
AAV expression construct cloning
The AAV plasmid entailing the MBP promoter-driven GFP, woodchuck hepatitis virus post-transcriptional regulatory element (WPRE), and the bovine growth hormone polyadenylation sequence (bGHpA), flanked by inverted terminal repeats (pAAV.MBP-GFP) was described previously23 and was used to generate the MBP-driven MCT1FLAG/Myc (pAAV.MBP-MCT1FLAG/Myc) and empty promoter (pAAV.MBP-Empty) plasmids. Briefly, a geneblock fragment containing the cDNA sequence of the mouse Mct1 gene (NM_009196) with a Myc- and FLAG-tag added to the ORF, was digested with AgeI/SacII and inserted into the pAAV.MBP-GFP plasmid that had also been digested with AgeI/SacII, to obtain the pAAV.MBP-MCT1FLAG/Myc construct. The latter was digested with AgeI/EcoRI and subsequently blunted, resulting in pAAV.MBP-Empty.
AAV production
The production of AAV9 viral stocks was performed according to the helper virus-free three-plasmid transfection of HEK293T cells.45,46 Briefly, HEK293T cells (20 plates of Ø15 cm) were transfected when reaching 80% confluence with a mixture of polyethylenimine (PEI; 1 μg/μL, Polyscience, Niles, IL, USA, MW 25,000, 23966-2), the vector plasmid (10 μg/plate, containing the transgene cassette under control of the MBP promoter and AAV2 inverted terminal repeats), the adenovirus helper plasmid (ΔF6, 20 μg/plate), and the AAV packaging plasmid carrying the rep2 and cap9 genes (10 μg/plate) dissolved in NaCl (150 mM) and 25 mL of pre-warmed Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies, Carlsbad, CA, USA, 41965039) supplemented with 5% of fetal bovine serum (FBS). 5 h post-transfection, 12 mL of DMEM supplemented with 10% FBS was added to the plates. Cells were harvested 72 h after transfection and pelleted for 10 min at 1,500 rpm at 4°C with a 420R Hettich centrifuge (Sigma-Aldrich, St. Louis, MO, USA). The supernatant was discarded, and cell pellets were lysed in 2 mL/plate of lysis buffer (150 mM NaCl and 1 M Tris-HCl pH 8.5 in 500 mL of endotoxin free H2O). This mixture underwent three freeze-thaw cycles of 10 min using dry ice/ethanol and a 37°C water bath, after which the samples were centrifuged for 15 min at 2,500 rpm and 4°C. Benzonase (250 U/mL, Sigma-Aldrich, E1014) was added to the supernatant at a final concentration of 50 U/mL and incubated for 30 min at 37°C. The clarified lysates were centrifuged at 8,500 rpm at room temperature for 20 min and passed through a 0.45 μm filter. Next, a discontinuous iodixanol (Optiprep Density Gradient Medium [60% (w/v) solution of iodixanol in water], Sigma-Aldrich, D1556) gradient was prepared in a Beckman OptiSeal 25 × 77 mm tube (Beckman, 361625) by careful layering of iodixanol solutions in the following order: 60%, 40%, 25%, and 15%. The crude lysate was then layered carefully onto the gradient with a Pasteur pipette. Gradients were then centrifuged at 50,000 rpm for 1 h and 40 min at 12°C using a 50.2Ti rotor (Beckman). Afterward, the fraction between the 40% and 60% interface was collected using a MicroFil needle (28G, 67 mm; WPI, Euskirchen, Germany, MF28G67-5) attached to a 5 mL syringe with tubing system (Hospithera, Brussels, Belgium, 126.100). Purified vectors were desalted and concentrated with Amicon Ultra-cell 100K filter units (Merck-Millipore, Burlington, USA, UFC910024) by adding 1X PBS-MK on top of the filter (5X stock; 250 mL 10X PBS, 2.5 mL 1 M MgCl2, 6.25 mL 1 M KCl, and endotoxin-free H2O in 500 mL). When the total volume was reduced to about 150 μL, the vector solution was collected in PBS with 0.01% of pluronic F68. Aliquots were made and stored at −80°C until further use. Vector purity was determined using a silver stain (Pierce, Rockford, IL, USA) according to the manufacturer’s instructions. Vector genome titers were determined by RT-PCR using Taqman Universal PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) with primers and probe targeting the WPRE or GFP. Titers were expressed as vg/mL.
Stereotactic injections
Before animals reached the age of 10 days (day of injection), a DNA sample was obtained for genotyping (SOD1G93A-positive versus wild-type mice). For each litter, the investigator made sure that for each experimental condition (MCT1, GFP, or empty vector) at least one mouse was represented to always have littermate controls. Postnatal day 10 mice (C57BL/6 mice for transduction efficiency study and SOD1G93A for the treatment study) were anesthetized with isoflurane for both induction (4%, 1l/min oxygen) and maintenance (2%, 1l/min oxygen) of anesthesia. Both male and female mice were used for experiments. Before surgery, mice received a subcutaneous injection of Vetergesic (0.05 mg/kg) and topical administration of Xylocaine (6 mg/kg). Mice were then placed onto a stereotaxic frame (Kopf Instruments, Tujunga, CA, USA) using an adaptor for neonatal mice. 4 μL of the vector solution was injected into the lateral ventricles (x = 0.8 mm, y = −0.2 mm, z = 2 mm), equivalent to a dose of 5 × 1010 vg of the MBP-MCT1Myc/FLAG-AAV9 and MBP-GFP-AAV9 vector stock, or 4 × 1010 vg of the MBP-Empty-AAV9 vector stock. Delivery was performed at a rate of 500 nL/min, using a microprocessor-controlled mini-pump (World Precision Instruments, Sarasota, FL, USA) connected to a 33G Hamilton syringe. The needle was left in place for 5 min prior to slowly retracting it from the ventricles. Afterward the pups were left underneath an infrared light to recover their mobility before returning as a group to their mother. Mice were observed until postnatal day 31 or death.
RNA isolation and RT-PCR
Mice were anesthetized with 10% Nembutal (Ceva Chemicals, Hornsby, NSW, Australia) and transcardially perfused with 1X PBS (Sigma-Aldrich). Tissues were harvested and immediately stored at −80°C until further processing. Total RNA was extracted using TriPure (Roche, Basel, Switzerland) and isopropanol purification. The RNA containing fraction was further isolated using the RNeasy Mini Kit (QIAGEN, Valencia, CA, USA), according to the manufacturer’s instructions with the inclusion of an optional DNase digestion step. cDNA was synthesized from 1 μg RNA using the Superscript III First-Strand Synthesis Mastermix kit (Invitrogen, Carlsbad, CA, USA). Quantitative real-time PCR reactions were performed on 5 μL cDNA (of a 1/10 dilution) using Taqman Fast Universal PCR Master Mix 2X (Applied Biosystems, Foster City, CA, USA) and following Taqman gene expression assays as follows: Ywhaz (Mm.PT.58.8991239), Gapdh (Mm.PT.39a.1), Hprt (Mm00446968_m1 or Mm.PT.39a.22214828), Polr2a (Mm.PT.39a.22214849), and Mct1 (Mm.PT.58.7462799). Samples were run in triplicate in a 96-well plate and thermal cycling was performed on a StepOne-Plus Real-Time PCR system (Applied Biosystems) using a standard (∼40 min) amplification protocol. Data were analyzed with qBase Plus software (Biogazelle, Ghent, Belgium) using the geNorm analysis method, which allows to determine the most stable reference genes from a set of tested candidate reference genes.
Western blot
Fresh tissue samples were collected and lysed in radioimmunoprecipitation (RIPA) buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholic acid, 0.5% SDS), supplemented with cOmplete, EDTA-free protease inhibitor cocktail (Roche, Basel, Germany). Tissues were homogenized with the MagNA lyser oscillator (Roche, Germany) at 6,500 rpm for 30 s in a 5427R centrifuge (Eppendorf, Hamburg, Germany). Protein concentrations were measured with the micro BCA kit (Pierce). 30–50 μg of protein from each tissue lysate was loaded on a 10% SDS-polyacrylamide-gel electrophoresis (SDS-PAGE) gel. Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore) by a semi-dry transfer apparatus (Hoefer, San Francisco, CA, USA, TE70XP) at 180 mA for 1 h and 45 min. The membrane was then blocked with 5% skimmed milk in Tris-buffered saline/Tween (TBS-T: 10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Tween 20) for 1 h at room temperature. Immunoblots were then incubated with primary antibodies against MCT1 (chicken, 1/1,000; Millipore, AB1286), FLAG (mouse, 1/1,000; Sigma, F3165), GFP (chicken, 1/1,000; Abcam, ab13970), and GAPDH (mouse, 1/2,000; Ambion, AM4300) in TBS-T at 4°C overnight. After washing the membranes with TBS-T and incubating them for 1 h with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (1/5,000; Santa Cruz Biotechnology, Dallas, TX, USA), protein bands were visualized using enhanced chemiluminescence (ECL substrate; Pierce). Blots were scanned using the ImageQuant LAS 4000 Biomolecular Imager (GE Healthcare, Chicago, IL, USA) and band intensities were quantified using ImageQuant software (GE Healthcare). A mild reblotting buffer (Merck-Millipore) was applied to strip the blots between incubation with the MCT1 and FLAG antibody.
Dot blot
Fresh tissue samples were collected and lysed in RIPA buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholic acid, 0.5% SDS), supplemented with cOmplete, EDTA-free protease inhibitor cocktail (Roche). Tissues were homogenized with the MagNA lyser oscillator (Roche) at 6,500 rpm for 30 s in a 5427R centrifuge (Eppendorf). Protein concentrations were measured with the micro BCA kit (Pierce). 30 μg of protein was applied to a nitrocellulose membrane with a pore size of 0.2 μm (GE Healthcare) and air-dried for 25 min. The membrane was then blocked with 5% skimmed milk in TBS-T for 1 h at room temperature and incubated with a primary anti-FLAG antibody (mouse, 1/1,000; Sigma, F3165) for 1 h at room temperature. After washing the membranes with TBS-T and incubating them for 1 h with the anti-mouse HRP-conjugated secondary antibody (1/5,000: Santa Cruz Biotechnology), protein dots were visualized using ECL substrate (Pierce). Blots were scanned using the ImageQuant LAS 4000 Biomolecular Imager (GE Healthcare).
Immunostainings
Mice were anaesthetized with 10% Nembutal (Ceva) and transcardially perfused with 1X PBS (Sigma-Aldrich) followed by perfusion with 4% paraformaldehyde (PFA). Lumbar spinal cord or brain tissues were harvested, post-fixed with 4% PFA for 2 h on ice and dehydrated overnight in a 30% sucrose solution at 4°C. Following embedding in optimal cutting temperature (OCT) compound mounting medium for cryotomy (VWR, Radnor, PA, USA), the samples were stored at −80°C until further processing. 20 μm transversal cryosections were obtained and processed for immunostaining. Sections were blocked for 1 h with 10% normal donkey serum (Sigma-Aldrich) in PBS-T (0.1% Triton X-100, Sigma-Aldrich) and incubated for 1 h at room temperature with primary antibodies dissolved in PBS-T supplemented with 10% normal donkey serum (Table 1). Sections were then washed with PBS-T and incubated with the appropriate secondary antibodies (Thermo Fisher Scientific) for 1 h at room temperature in PBS-T supplemented with 10% normal donkey serum. Slides were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA) and images were acquired with a Leica TCS SP8 confocal laser scanning microscope using a 20X or 40X objective (Leica Microsystems). The percentage of GFP-expressing oligodendrocytes was determined by counting CC1- and GFP-positive cells from at least four non-adjacent sections from three different animals using the NIS software.
Table 1.
Overview of the antibodies used for immunofluorescence
| Target | Antibody | Species reactivity | Supplier | Catalogue no. | Dilution |
|---|---|---|---|---|---|
| Oligodendrocytes | anti-CC1 | Mouse | Abcam | AB16794 | 1/100 |
| MCT1 | anti-MCT1 | Chicken | Millipore | AB1286 | 1/200 |
| Myc | anti-Myc | Rat | Biorad | MCA1929 | 1/200 |
| OPCs | anti-NG2 | Mouse | Millipore | MAB5384 | 1/200 |
| Neurons | anti-NeuN | Mouse | Millipore | MAB377 | 1/500 |
| Astrocytes | anti-GFAP | Mouse | Sigma | G3893 | 1/200 |
| Microglia | anti-CD11b | Rat | AbD Serotec | MCA74G | 1/200 |
Motor neuron count
Mice were anesthetized with 10% Nembutal (Ceva) and transcardially perfused with 1X PBS (Sigma-Aldrich) followed by perfusion with 4% PFA. Lumbar spinal cords were harvested, post-fixed with 4% PFA for 2 h on ice, and dehydrated overnight in a 30% sucrose solution at 4°C. Following embedding in OCT compound (VWR, Radnor, USA), the samples were stored at −80°C until further processing. 20 μm cryosections were obtained and every tenth section was stained with 2X thionin solution to visualize the motor neuron cell bodies in the ventral horn. Briefly, cryosections were fixed for 1 min in 100% ethanol in a cryochamber and subsequently washed for 15 s in distilled H2O, stained with 2X thionin for 45 s, washed twice in distilled H2O, and dehydrated in 70%, 90%, and 100% ethanol (30 s each step). Histological sections were cleared with Histoclear solution for 1 min and mounted with PerTex medium (Histolab, Västra Frölunda, Sweden). A total of 10 spinal cord sections for each mouse were analyzed, covering a range of approximately 1.2 mm as described before. Images were acquired with a Zeiss Imager M1 at 10X magnification. The number and size of normal-appearing neurons were quantified using AxioVision software (version 4.8; Carl Zeiss AG, Oberkochen, Germany) and were normalized to the size of the ventral horn. To this end, the values obtained for each spinal cord section were divided by the ventral horn size of the section in question and were then normalized by correcting to the average ventral horn size in a NTG mouse (300,000 μm2).
Histological examination of NMJs
Prior to dissection, mice were euthanized with 10% Nembutal and subsequent cervical dislocation. The gastrocnemius muscle was dissected, snap-frozen in liquid-nitrogen-cooled isopentane and stored at −80°C. On the day of the experiment, longitudinal cryosections of 20 μm were fixed in 4% PFA for 10 min before rinsing with 1X PBS. Tissue was subsequently permeabilized with PBS-T for 5 min and blocked in PBS-T supplemented with 10% normal donkey serum (NDS, Sigma-Aldrich) for 1 h. Neuronal axons (anti-NFL light chain antibody (Alexa Fluor 488 conjugated, Cell Signaling, 8024S, 1/500), presynaptic terminals (anti-SV2A, Developmental Studies Hybridoma Bank, 1/100), and motor endplates (Alexa Fluor 555-conjugated α-bungarotoxin, Life Technologies, B35451, 1/5,000) were stained with primary antibodies dissolved in PBS-T supplemented with 10% NDS and incubated overnight. Slides were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA) and images were acquired with a Leica TCS SP8 confocal laser scanning microscope (Leica Microsystems). For each experimental condition, at least 100 NMJs were analyzed for innervation on a Zeiss Imager M1 microscope system using a 40X objective (Carl Zeiss, AG). Assessments were undertaken by an investigator blinded to the different groups.
Statistical analysis
Statistical analysis was done with GraphPad Prism 8 software (Graphad Software, La Jolla, CA, USA) and the statistical tests used are specified in the figure legends. Non-parametric testing was performed when the data obtained were not normally distributed (Shapiro-Wilk normality test).
Acknowledgments
This work was supported by VIB, the Fund for Scientific Research Flanders (FWO), the University of Leuven (GOA/11/014 and C14/17/107), the Interuniversity Attraction Poles Programme of the Belgian Federal Science Policy Office (P7/16), the European Research Council (ERC grant agreement number 340429), and the ALS Liga (Belgium). L.V.D.B., P.V.D., and W.R. are supported by the “Opening the Future” Fund (KU Leuven). W.R. and P.V.D. are supported through the Emil von Behring Chair for Neuromuscular and Neurodegenerative Disorders, the Laevers Fund for ALS Research, and the “Een Hart voor ALS” and the “Valéry Perrier race against ALS” funds of the University of Leuven. P.V.D. holds a senior clinical investigatorship of FWO-Vlaanderen. M.G.H. is supported by the ERC Starting Grant 281961 (AstroFunc) and ERC Proof-of-Concept Grant 713755 (AD-VIP), FWO (1513616N), Thierry Latran Foundation (SOD-VIP), and Stichting Alzheimer Onderzoek (Pilot grant P#14006 and Standard grant 2018-0029).
Author contributions
C.E. designed and performed all the experiments and wrote the paper with input from all authors. E.R., L.R., A.B.-.A. and N.H. participated in scientific discussions and provided technical support. A.L. executed the analysis of the motor neurons and NMJ counts. A.K., N.C., and S.M. provided technical support for imaging acquiring and analysis. S.D. and M.G.H. shared the AAV vector production protocol, provided C.E. with their AAV production/purification equipment, and participated in scientific discussions. M.K. and G.v.J. developed and shared the MBP-GFP-AAV plasmid used throughout the study and provided intellectual input. P.V.D., L.V.D.B., and W.R. supervised the project and contributed to the conception and design of the study.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.01.006.
Contributor Information
Ludo Van Den Bosch, Email: ludo.vandenbosch@kuleuven.vib.be.
Wim Robberecht, Email: wim.robberecht@kuleuven.be.
Supplemental information
References
- 1.Brown R.H., Al-Chalabi A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017;377:162–172. doi: 10.1056/NEJMra1603471. [DOI] [PubMed] [Google Scholar]
- 2.Swinnen B., Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014;10:661–670. doi: 10.1038/nrneurol.2014.184. [DOI] [PubMed] [Google Scholar]
- 3.Boillée S., Vande Velde C., Cleveland D.W. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Ilieva H., Polymenidou M., Cleveland D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neumann M., Kwong L.K., Truax A.C., Vanmassenhove B., Kretzschmar H.A., Van Deerlin V.M., Clark C.M., Grossman M., Miller B.L., Trojanowski J.Q., Lee V.M. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J. Neuropathol. Exp. Neurol. 2007;66:177–183. doi: 10.1097/01.jnen.0000248554.45456.58. [DOI] [PubMed] [Google Scholar]
- 6.Niebroj-Dobosz I., Rafałowska J., Fidziańska A., Gadamski R., Grieb P. Myelin composition of spinal cord in a model of amyotrophic lateral sclerosis (ALS) in SOD1G93A transgenic rats. Folia Neuropathol. 2007;45:236–241. [PubMed] [Google Scholar]
- 7.Seilhean D., Cazeneuve C., Thuriès V., Russaouen O., Millecamps S., Salachas F., Meininger V., Leguern E., Duyckaerts C. Accumulation of TDP-43 and alpha-actin in an amyotrophic lateral sclerosis patient with the K17I ANG mutation. Acta Neuropathol. 2009;118:561–573. doi: 10.1007/s00401-009-0545-9. [DOI] [PubMed] [Google Scholar]
- 8.Mackenzie I.R., Ansorge O., Strong M., Bilbao J., Zinman L., Ang L.C., Baker M., Stewart H., Eisen A., Rademakers R., Neumann M. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 2011;122:87–98. doi: 10.1007/s00401-011-0838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Magnus T., Carmen J., Deleon J., Xue H., Pardo A.C., Lepore A.C. Adult glial precursor proliferation in mutant SOD1G93A mice. Glia. 2008;56:200–208. doi: 10.1002/glia.20604. [DOI] [PubMed] [Google Scholar]
- 10.Kang S.H., Fukaya M., Yang J.K., Rothstein J.D., Bergles D.E. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668–681. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee Y., Morrison B.M., Li Y., Lengacher S., Farah M.H., Hoffman P.N., Liu Y., Tsingalia A., Jin L., Zhang P.W. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang S.H., Li Y., Fukaya M., Lorenzini I., Cleveland D.W., Ostrow L.W., Rothstein J.D., Bergles D.E. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 2013;16:571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Philips T., Bento-Abreu A., Nonneman A., Haeck W., Staats K., Geelen V., Hersmus N., Küsters B., Van Den Bosch L., Van Damme P. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain. 2013;136:471–482. doi: 10.1093/brain/aws339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nonneman A., Robberecht W., Van Den Bosch L. The role of oligodendroglial dysfunction in amyotrophic lateral sclerosis. Neurodegener. Dis. Manag. 2014;4:223–239. doi: 10.2217/nmt.14.21. [DOI] [PubMed] [Google Scholar]
- 15.Garcia C.K., Goldstein J.L., Pathak R.K., Anderson R.G., Brown M.S. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: implications for the Cori cycle. Cell. 1994;76:865–873. doi: 10.1016/0092-8674(94)90361-1. [DOI] [PubMed] [Google Scholar]
- 16.Halestrap A.P., Price N.T. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem. J. 1999;343:281–299. [PMC free article] [PubMed] [Google Scholar]
- 17.Pellerin L., Pellegri G., Bittar P.G., Charnay Y., Bouras C., Martin J.L., Stella N., Magistretti P.J. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci. 1998;20:291–299. doi: 10.1159/000017324. [DOI] [PubMed] [Google Scholar]
- 18.Rafiki A., Boulland J.L., Halestrap A.P., Ottersen O.P., Bergersen L. Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience. 2003;122:677–688. doi: 10.1016/j.neuroscience.2003.08.040. [DOI] [PubMed] [Google Scholar]
- 19.Morrison B.M., Lee Y., Rothstein J.D. Oligodendroglia: metabolic supporters of axons. Trends Cell Biol. 2013;23:644–651. doi: 10.1016/j.tcb.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasparov S. Are Astrocytes the Pressure-Reservoirs of Lactate in the Brain? Cell Metab. 2016;23:1–2. doi: 10.1016/j.cmet.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Jha M.K., Morrison B.M. Glia-neuron energy metabolism in health and diseases: New insights into the role of nervous system metabolic transporters. Exp. Neurol. 2018;309:23–31. doi: 10.1016/j.expneurol.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferraiuolo L., Meyer K., Sherwood T.W., Vick J., Likhite S., Frakes A., Miranda C.J., Braun L., Heath P.R., Pineda R. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc. Natl. Acad. Sci. USA. 2016;113:E6496–E6505. doi: 10.1073/pnas.1607496113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Jonquieres G., Mersmann N., Klugmann C.B., Harasta A.E., Lutz B., Teahan O., Housley G.D., Fröhlich D., Krämer-Albers E.M., Klugmann M. Glial promoter selectivity following AAV-delivery to the immature brain. PLoS ONE. 2013;8:e65646. doi: 10.1371/journal.pone.0065646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Georgiou E., Sidiropoulou K., Richter J., Papaneophytou C., Sargiannidou I., Kagiava A., von Jonquieres G., Christodoulou C., Klugmann M., Kleopa K.A. Gene therapy targeting oligodendrocytes provides therapeutic benefit in a leukodystrophy model. Brain. 2017;140:599–616. doi: 10.1093/brain/aww351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Jonquieres G., Spencer Z.H.T., Rowlands B.D., Klugmann C.B., Bongers A., Harasta A.E., Parley K.E., Cederholm J., Teahan O., Pickford R. Uncoupling N-acetylaspartate from brain pathology: implications for Canavan disease gene therapy. Acta Neuropathol. 2018;135:95–113. doi: 10.1007/s00401-017-1784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu R., Janson C.G., Mastakov M., Lawlor P., Young D., Mouravlev A., Fitzsimons H., Choi K.L., Ma H., Dragunow M. Quantitative comparison of expression with adeno-associated virus (AAV-2) brain-specific gene cassettes. Gene Ther. 2001;8:1323–1332. doi: 10.1038/sj.gt.3301529. [DOI] [PubMed] [Google Scholar]
- 27.Sehara Y., Fujimoto K.I., Ikeguchi K., Katakai Y., Ono F., Takino N., Ito M., Ozawa K., Muramatsu S.I. Persistent Expression of Dopamine-Synthesizing Enzymes 15 Years After Gene Transfer in a Primate Model of Parkinson’s Disease. Hum. Gene Ther. Clin. Dev. 2017;28:74–79. doi: 10.1089/humc.2017.010. [DOI] [PubMed] [Google Scholar]
- 28.Nave K.A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 2010;11:275–283. doi: 10.1038/nrn2797. [DOI] [PubMed] [Google Scholar]
- 29.Simons M., Nave K.A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2015;8:a020479. doi: 10.1101/cshperspect.a020479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saugier-Veber P., Munnich A., Bonneau D., Rozet J.M., Le Merrer M., Gil R., Boespflug-Tanguy O. X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat. Genet. 1994;6:257–262. doi: 10.1038/ng0394-257. [DOI] [PubMed] [Google Scholar]
- 31.Roher A.E., Weiss N., Kokjohn T.A., Kuo Y.M., Kalback W., Anthony J., Watson D., Luehrs D.C., Sue L., Walker D. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry. 2002;41:11080–11090. doi: 10.1021/bi026173d. [DOI] [PubMed] [Google Scholar]
- 32.Mitew S., Kirkcaldie M.T., Halliday G.M., Shepherd C.E., Vickers J.C., Dickson T.C. Focal demyelination in Alzheimer’s disease and transgenic mouse models. Acta Neuropathol. 2010;119:567–577. doi: 10.1007/s00401-010-0657-2. [DOI] [PubMed] [Google Scholar]
- 33.John G.R., Shankar S.L., Shafit-Zagardo B., Massimi A., Lee S.C., Raine C.S., Brosnan C.F. Multiple sclerosis: re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat. Med. 2002;8:1115–1121. doi: 10.1038/nm781. [DOI] [PubMed] [Google Scholar]
- 34.DeVos S.L., Miller T.M. Direct intraventricular delivery of drugs to the rodent central nervous system. J. Vis. Exp. 2013;12:e50326. doi: 10.3791/50326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maragakis N.J. Motor neuron disease: progressive muscular atrophy in the ALS spectrum. Nat. Rev. Neurol. 2010;6:187–188. doi: 10.1038/nrneurol.2010.29. [DOI] [PubMed] [Google Scholar]
- 36.Eykens C., Nonneman A., Jensen C., Iavarone A., Van Damme P., Van Den Bosch L., Robberecht W. Conditional deletion of Id2 or Notch1 in oligodendrocyte progenitor cells does not ameliorate disease outcome in SOD1G93A mice. Neurobiol. Aging. 2018;68:1–4. doi: 10.1016/j.neurobiolaging.2018.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun Y., Xu C.C., Li J., Guan X.Y., Gao L., Ma L.X., Li R.X., Peng Y.W., Zhu G.P. Transplantation of oligodendrocyte precursor cells improves locomotion deficits in rats with spinal cord irradiation injury. PLoS ONE. 2013;8:e57534. doi: 10.1371/journal.pone.0057534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu L., Ryu J., Hiel H., Menon A., Aggarwal A., Rha E., Mahairaki V., Cummings B.J., Koliatsos V.E. Transplantation of human oligodendrocyte progenitor cells in an animal model of diffuse traumatic axonal injury: survival and differentiation. Stem Cell Res. Ther. 2015;6:93. doi: 10.1186/s13287-015-0087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J., Xiong L.L., Wang Y.C., He X., Jiang L., Fu S.J., Han X.F., Liu J., Wang T.H. Oligodendrocyte precursor cell transplantation promotes functional recovery following contusive spinal cord injury in rats and is associated with altered microRNA expression. Mol. Med. Rep. 2018;17:771–782. doi: 10.3892/mmr.2017.7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matute C., Ransom B.R. Roles of white matter in central nervous system pathophysiologies. ASN Neuro. 2012;4:e00079. doi: 10.1042/AN20110060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou T., Ahmad T.K., Gozda K., Truong J., Kong J., Namaka M. Implications of white matter damage in amyotrophic lateral sclerosis (Review) Mol. Med. Rep. 2017;16:4379–4392. doi: 10.3892/mmr.2017.7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lobsiger C.S., Boillee S., McAlonis-Downes M., Khan A.M., Feltri M.L., Yamanaka K., Cleveland D.W. Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc. Natl. Acad. Sci. USA. 2009;106:4465–4470. doi: 10.1073/pnas.0813339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L., Sharma K., Grisotti G., Roos R.P. The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiol. Dis. 2009;35:234–240. doi: 10.1016/j.nbd.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turner B.J., Ackerley S., Davies K.E., Talbot K. Dismutase-competent SOD1 mutant accumulation in myelinating Schwann cells is not detrimental to normal or transgenic ALS model mice. Hum. Mol. Genet. 2010;19:815–824. doi: 10.1093/hmg/ddp550. [DOI] [PubMed] [Google Scholar]
- 45.Rincon M.Y., de Vin F., Duqué S.I., Fripont S., Castaldo S.A., Bouhuijzen-Wenger J., Holt M.G. Widespread transduction of astrocytes and neurons in the mouse central nervous system after systemic delivery of a self-complementary AAV-PHP.B vector. Gene Ther. 2018;25:83–92. doi: 10.1038/s41434-018-0005-z. [DOI] [PubMed] [Google Scholar]
- 46.Fripont S., Marneffe C., Marino M., Rincon M.Y., Holt M.G. Production, Purification, and Quality Control for Adeno-associated Virus-based Vectors. J. Vis. Exp. 2019;(143) doi: 10.3791/58960. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.