

Graphical abstract
Keywords: Deep learning, Classification, Single cell data
Abstract
Cell type classification is an important problem in cancer research, especially with the advent of single cell technologies. Correctly identifying cells within the tumor microenvironment can provide oncologists with a snapshot of how a patient’s immune system reacts to the tumor. Wide and deep learning (WDL) is an approach to construct a cell-classification prediction model that can learn patterns within high-dimensional data (deep) and ensure that biologically relevant features (wide) remain in the final model. In this paper, we demonstrate that regularization can prevent overfitting and adding a wide component to a neural network can result in a model with better predictive performance. In particular, we observed that a combination of dropout and regularization can lead to a validation loss function that does not depend on the number of training iterations and does not experience a significant decrease in prediction accuracy compared to models with , dropout, or no regularization. Additionally, we show WDL can have superior classification accuracy when the training and testing of a model are completed data on that arise from the same cancer type but different platforms. More specifically, WDL compared to traditional deep learning models can substantially increase the overall cell type prediction accuracy (36.5 to 86.9%) and T cell subtypes (CD4: 2.4 to 59.1%, and CD8: 19.5 to 96.1%) when the models were trained using melanoma data obtained from the 10X platform and tested on basal cell carcinoma data obtained using SMART-seq. WDL obtains higher accuracy when compared to state-of-the-art cell classification algorithms CHETAH (70.36%) and SingleR (70.59%).
1. Introduction
Immunology is quickly becoming a popular area of study in cancer research and offers an opportunity to expand our understanding and ability to treat patients. Estimating the immune composition of an individual’s tumor has been the focus of several studies which have developed deconvolution methods [1], [2] to estimate the cellular composition of the tumor micro-environment with bulk RNA expression data. Recently with the advent of single cell sequencing researchers are now able to measure gene expression in individual cells within the tumor-microenvironment and classify cells using hierarchical clustering and correlation-based methods [3], [4], [5]. Cell type classification can be conducted by constructing visualizations such as t-Distributed Stochastic Neighbor Embedding (t-SNE) [6] or Uniform Manifold Approximation and Projection (UMAP) [7] plots to define clusters and assign these clusters to different cell types based on enriched canonical markers. However, a major drawback of this canonical process is that it heavily relies on the researchers’ knowledge on the cell-type-specific signature genes, and it can become arbitrary when making conclusions based on only a handful of genes. Also, the cell type marker genes are cancer type-specific and may not generalize to other datasets [8]. In addition, discriminating between fine immune cell subtypes, such as exhausted CD8 T cells vs. activated CD8 T cells, effector CD4 T cells vs. naive CD4 T cells, is a much more challenging task due to the lack of universal marker genes.
Identification of highly specific cell types is now possible with the development of single cell RNA-sequencing technology. However, a challenge in cell annotation in single cell RNA-sequencing is that transcription profiles are difficult to transfer between different platforms. Multiple platforms have been developed for single cell RNA-sequencing including SMART-seq [9], CEL-seq [10], Fluidigm C1 [11], SMART-seq2 [12], and more advanced droplet-based platforms including Drop-seq [13] and 10X Genomics Chromium system [14], etc. The two most commonly used platforms are SMART-seq/SMART-seq2 and 10X. The 10X platform is a droplet-based approach that generates a unique molecular identifier (UMI) at or ends to diminish the sequencing reads representation biases due to library amplification. On the other hand, SMART-seq and SMART-seq2 are designed to generate full-length cDNA. Droplet-based or -tag methods like 10X can capture much more cells which in turn can give better overview of the heterogeneity within population; while a full-length proposal like SMART-seq is better suited for studies concerned with isoforms, splicing, or gene fusion. Due to the differences in how they amplify the mRNA transcripts, the data generated from these platforms are not directly comparable, which presents a great challenge to the integrated cell type identification in cross-platform datasets [15], [16], [17], [18]. Therefore, there is a great need for automatic cell identification methods that be used across studies, single cell platforms, and cancer types.
While there are many different single cell RNA-sequencing platforms whose results are on different scales and not directly comparable, the underlying gene to gene relationships should be consistent and navigating these relationships may allow for borrowing of information from different technologies. Deep learning brings us the possibility to explore and summarize complex highly non-linear relationships into high-level features from high throughput data sources. Deep learning is a powerful machine learning technique that is often used in visual recognition[19], [20], natural language processing [21], [22], and starting to infiltrate the realm of cancer research [23], [24], [25]. Deep learning detects patterns in data by using neural networks with many layers of nodes by transforming the output model of the nodes from the previous layer with non-linear functions. The coefficients output from each node are augmented using gradient descent in order to optimize the prediction error of the network.
Wide and deep learning (WDL) combines a deep neural network with a generalized linear model (GLM) based on a small set of features. Deep learning tends to generalize patterns in the data, while in contrast GLMs may only memorize the patterns in the data. WDL has been shown to be an effective tool in recommender systems [26]. Specifically, we propose utilizing a deep learning model which can leverage large dimensional data (deep), as well as incorporate a few known biologically relevant genes in the last hidden layer of a neural network to emphasize their biological importance (wide). The wide part of the model allows us to make cell type classification more precise and fine-tuned to classify more specific immune cell subtypes such as distinguishing activated from exhausted CD8 T cells.
This paper serves three purposes. First, we illustrate a deep learning framework to automatically classify cells, which can greatly reduce the burden of manually identifying clusters of cells and then annotating them based on cluster-specific genes. This is particularly useful for more specialized cell subtypes which lack the universal canonical marker genes that can distinguish them from similar cell types. For example, well-known markers of exhausted T cells, such as LAG3, PDCD1, and HAVCR2, are also highly expressed in activated T cells. Therefore, it is hard to discriminate these similar cell types using a single or a handful of markers. Second, it provides some background information about deep learning, specifically focusing on regularization methods to avoid overfitting the model. Models are trained, validated, and subsequently used to classify cells from the same dataset. This scenario is realistic since it is possible that some hospitals may not have the resources needed for generating large amounts of data to build their own model. In addition, in many clinical studies the patients’ tumor samples are collected over a fairly long period of time (years) in several batches. Waiting till sample collection is finished before single cell RNA-sequencing data analysis is not realistic. It will be extremely helpful to train a deep learning model using samples collected at earlier time points and subsequently apply it to later samples. Lastly, this paper provides an illustration of how incorporating known biologically relevant biomarkers can be used to transfer knowledge. In this scenario, we explore the possibility to transfer cell type annotations across different single cell RNA-sequencing platforms, which can help make full use of the enormous publicly available single cell RNA-seq data that are generated by different technologies.
In the methods section, we will describe the data single cell RNA-seq datasets used in study, provide background about deep learning, and wide and deep learning. Then in the results section, we will present results from training and testing the models in the two scenarios. Finally, we make some concluding comments and discussion in the discussion section.
2. Methods
2.1. Yost data
Yost et al. [8] conducted droplet-based 10X 5’ single-cell RNA-sequencing on 79,046 cells from primary tumors of 11 patients with advanced basal cell carcinoma before and after anti-PD-1 treatment. In total, RNA profiles from 53,030 malignant, immune and stromal cells, and 33,106 T cells were obtained from single cell RNA-sequencing. The cell types of interest were T cells, B cells, nature killer (NK) cells, macrophages, cancer-associated fibroblasts (CAFs), endothelial cells, plasma cells, melanocytes, and tumor cells. The T cells were further classified into regulatory (Tregs), follicular helpers (), T helper 17 (), naive T cells, activated CD8, exhausted CD8, effector CD8, and memory CD8 T cells (Supplementary Fig. 1). We downloaded the raw gene count for single cells from Gene Expression Omnibus (GEO) under accession GSE123814, and normalized the data following Methods in Yost et al. using Seurat[27]. Specifically, the raw counts were normalized by the total expression of each cell, with the cell cycle effects accounted for. The resulting normalized gene expressions were used subsequently in the Wide and Deep Learning models. The UMAP projections and cell type classification provided by Yost et al. were used to visualize the data. The cell type classification was also used as cell type for each cell when assessing the Wide and Deep Learning models.
2.2. Tirosh data
Tirosh et al. [28] applied SMART-seq to 4,645 single cells isolated from 19 freshly procured human melanoma tumors, profiling T cells, B cells, NK cells, macrophages, endothelial cells, CAFs, and melanoma cells. To further analyze the T cell subtypes, we downloaded the log-transformed TPM (Transcripts per Million reads) gene expression values provided by the study and imported them to Seurat [27]. S and G2/M cell cycle phase scores were assigned to cells based on previously defined gene sets using CellCycleScoring function to qualitatively evaluate the cell cycle variations in each cell. Scaled z-scores for each gene were then calculated using ScaleData function by regressing against the S and G2/M phases scores to remove the cell-specific cell cycle variations. Shared nearest neighbor (SNN) based clustering method was used to identify clusters based on the first 30 principle components computed from scaled data with resolution = 1. UMAPs were generated using the same principle components with perplexity = 30 and used for all visualization. A total of 15 distinctive clusters were identified and annotated by identifying differentially expressed marker genes for each cluster and comparing them to known cell type markers and markers reported by Tirosh et al. From this analysis, we confirmed the cell annotation provided by Tirosh et al. and were able to further identify CD4 T cells and CD8 T cells. The normalized and scaled z-scores were used as gene expression values in the deep learning models.
2.3. Background for classification problems with deep learning
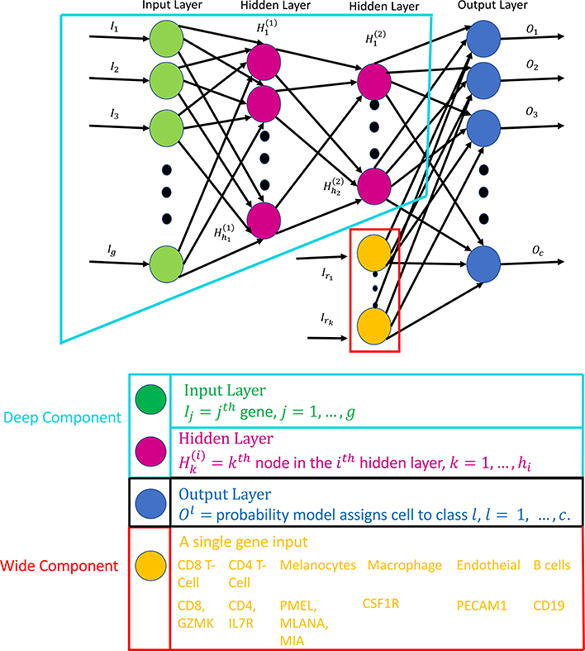
Deep neural networks (DNN) are able to learn and condense highly non-linear features (genes) into a high level summary through the use of composition of functions. These functions are dot products that undergo a non-linear transformation and are then passed into another function in the next layer. There are three types of layers in a DNN which are input, hidden, and output layers. Each node in the input layer corresponds to the expression of a single gene. Information from the input layer is passed to each node in a hidden layer which optimizes the weights in a dot product to minimize the cell type classification loss. The nodes in the output layer produce the probability that a cell is classified as a specific type. The architecture of a generic DNN with two hidden layers is seen in Fig. 1. For multi-class classification the objective function to be minimized is so called cross-entropy function, ,
| (1) |
where m is the number of cell types, n is the sample size, is 1 if the cell is type j and 0 otherwise, and Q is the predicted cell type probability distribution for cell i. Note that each cell can have a different predicted probability distribution. This function is minimized by gradient descent which is computed by iterating the chain rule over all layers of the model. A DNN is updated using stochastic gradient descent (SGD), which at every iteration computes an gradient with a random subset of cells instead of using all of the data and then updates the model weights. SGD greatly reduces the computational burden but requires many iterations due random sampling. The architecture of a DNN is complex and requires careful tuning. Some examples of tuning parameters are batch size, iterations to train the model, and nodes in each layer [29], [30].
Fig. 1.

Depiction of a generic wide and deep learning neural network where the wide component is surrounded by the red lines and the deep component is encompassed by the turquoise lines. The deep component is equivalent to a traditional deep learning model described in Sections 2.2, 3.1. The gene names in yellow correspond to the genes that are used in the wide part of the WDL model in Section 3.2.
The metrics we used to analyze the predictive capability of a particular model were accuracy and area under the curve (AUC). Accuracy was defined as the percentage of cells that were classified as the correct type of cell by the DNN. AUC, for binary classification, is computed by determining the area under of receiver operator curve, which is the curve generated by plotting the false positive rate (x-axis) versus the true positive rate (y-axis) at varying probability thresholds. There are two ways to compute AUC for multi-class classification, the first is one versus the rest [31], [32], and the second is one versus one, which computes all pairwise AUCs [33]. The average over all computed AUCs is then reported as the final AUC. One versus rest AUC is not reliable when there is an imbalance in the classes, which is often the case in cell classification, therefore in this study we report one versus one AUC to access the overall predictive capability of models.
A challenge to training a deep learning model is to ensure that the results can be generalized to new data sets. One of the simplest ways to prevent overfitting is to reduce the number of hidden layers or nodes which in turn decreases the number of parameters estimated by the model. Another technique is dropout which randomly deletes a specified proportion of nodes from each layer in the neural network. By deleting different sets of nodes in each iteration the model is trained on different sub-networks and becomes less sensitive to the specific weights of nodes. Dropout can speed up the training of a DNN, however, it may require more iterations to train the network. It is recommended that the percentage of nodes to delete from each layer should be between 20–50% [34] Lastly, constraints can be imposed on the weight vector of each node, requiring the norm to be small. The regularizers work in a similar way to lasso or ridge penalization in a regression setting where there is an additional parameter which changes the influence of the penalization term. The aim is either to keep the value of the weights small or push as many as possible to zero (lasso). Elastic net penalization has also been used which allows for a balance between the (lasso) and (ridge) penalty [30], [35], [36].
Understanding which genes were influential to a successful cell type classification model is important for validating the results and can lead to the detection of novel genes for future research. Despite many machine learning techniques being seen as ‘black boxes’, there have been efforts to interpret the results. One simple approach to evaluate the importance of a feature is calculating the dot product of consecutive nodes [37]. This was originally proposed for neural networks with a single hidden layer, but we extend this work to a neural work with two hidden layers.
| (2) |
where corresponds to the matrix that contains the coefficients. By ranking these weights of each gene we can gain some notion of variable importance in the final classification.
2.4. Emphasizing important genes for improved cell type classifiers
Wide and deep learning (WDL) involves merging a set of features, a wide component, with the last hidden layer in a DNN, a deep component. Adding these features in the final step will ensure that they are emphasized in the model, since they may be lost due to dropout or assigned with small weights. The wide component is a generalized linear model where the input is a set of original features. Wide components tend to memorize the patterns of the data, while deep components can generalize non-linear patterns. The architecture of a WDL model is shown in Fig. 1. In this study, specific genes exclusively expressed by a particular cell type are added to the last hidden layer forcing the model to emphasize them more. This may allow a DNN to produce a more accurate model than a model constructed with only a deep part, especially in scenarios where the data are obtained from different platforms or cancer types. Markers were selected based on literature and prior knowledge of surface markers for each cell type.
3. Results
3.1. Neural network tuning
In this section, we want to describe how the hyperparameters (number of nodes, type of regularization) were selected. Traditional deep learning models with two hidden layers were constructed with no regularization (No Dropout), dropout for both hidden layers (Dropout Only), dropout and an regularizer for both hidden layers (Dropout + ), and lastly dropout and an regularizer for both hidden layers (Dropout + ). A grid search was employed where the first hidden layer could have 1, 5, 10, 25, 50, 75, 100, 500, 1000 nodes and the second hidden layer could have 1, 5, 10, 25, 50, 75, 100, 500 nodes with a batch size of 128. Each neural network was optimized with the Adam optimizer [38] with a learning rate of 0.01. The activation function for each hidden layer was the rectified linear units (ReLU), , and the softmax activation function was used for the output layer. To enforce the and a kernel regularizer was used with a 0.01 regularization factor to reduce the weights, W, of the activation function, . The weights of each layer were initialized using Glorot uniform initializer which random selects values between and , where n and m are the number of input and output nodes, respectively.
In order to determine which DNN architecture leads to good results, we split the data into training, testing, and validation sets. This was conducted by randomly splitting the total dataset into a training and testing (75%) and testing (25%) set, and then further splitting the first set into a training (80% of the first set or 60% of the total dataset) and validation (20% of the first set or 15% of the total dataset) set. A z-transformation was applied to each feature in the combined training and validation sets, and then these summary statistics were used to conduct the analogous transformation on the testing set. In this section, the training, testing, and validation sets were all from the Yost data.
Fig. 2 shows the training and validation accuracy (left) and loss (right) using the same architecture for each model, and a numerical summary is provided in Table 1 for the architecture leading to the best validation accuracy. Notice that with No Dropout, Dropout Only, and Dropout + the validation loss increased as the model is trained. On the other hand, the validation accuracy and validation loss remained consistent with the training loss as epochs increase for Dropout + , suggesting that even if the model is trained with an excessive number of iterations, the model performance will not suffer heavily from overtraining. While the No Dropout and Dropout only models had the lowest validation loss and nearly 100% training accuracy, they are undoubtedly overtrained and will likely not generalize well for future data.
Fig. 2.

Comparison of the accuracy (A) and categorical cross-entropy loss (B) defined in Eq. (1) for a single replicate of four models from Section 3.1 with varying methods of regularization and drop out. The red and turquoise lines correspond to the performance on the training and validation set, respectively.
Table 1.
Summary of the number of nodes in each layer, validation, and testing accuracy. The architecture was selected based on the model that highest accuracy when classifying cells in the validation data set. The testing accuracy arises from training a model with the specified architecture with training + validation datasets and testing on previously unused test set. The mean (standard deviation) of 10 replications of each model is presented for accuracy and area under curve (AUC).
| Validation | First | Second | Testing | Testing | |
|---|---|---|---|---|---|
| Accuracy | Layer | Layer | Accuracy | AUC | |
| No | 94.8 | 100 | 50 | 94.9 | 0.999 |
| Dropout | (0.457) | (0.248) | (1.28e−4) | ||
| Dropout | 93.5 | 100 | 75 | 94.1 | 0.996 |
| Only | (0.259) | (0.312) | (3.74e−4) | ||
| Dropout + | 92.6 | 75 | 100 | 92.4 | 0.998 |
| (0.461) | (0.517) | (2.13e−4) | |||
| Dropout + | 93.9 | 100 | 75 | 93.4 | 0.999 |
| (0.450) | (0.244) | (1.28e−4) |
The overall accuracy of the Dropout + model is 93.8%, with the prediction accuracy of individual cell types ranging from 85% to 100% (Fig. 3A). T cell subtypes are similar in gene expression profiles and are difficult to distinguish. T cell subtype classification is commonly done as a second stage of classification where only the T cells are considered [8]. Fig. 3A shows that using a deep learning framework, each T cell subtype is classified with at least 85% accuracy, and 5 out of the 7 T cell subtypes had greater than 90% percent accuracy, and the misclassified cells were classified as another type of T cell. In single cell RNA-sequencing, the separation between activated and exhausted CD8 T cells is particularly difficult. The exhausted cells are considered chronically activated, and they also highly express the activation markers such as TNF and IFNG. The subtle difference between activated and exhausted CD8 T cells is the overexpression of exhaustion markers such as TIGIT and HAVCR2. Our deep learning model with Dropout+ setting was able to capture these genes and ranked their importance as and in a total of 22,890 genes (Fig. 3B, Supplementary Table 1). In addition, the model puts high emphasis on the genes that are typically over-expressed in tissue-resident memory cells, such as LAYN and CXCL13 (Fig. 3B), which is consistent with Yost et al. original findings. The cell type classification accuracies for No Dropout, Dropout only, and Dropout + are included in Supplementary Fig. 2.
Fig. 3.

Heatmap of accuracy by cell type for a deep learning model (A), trained and tested on the Yost dataset, with two hidden layers with 100 and 75 nodes respectively, and 20% dropout and regularization. Average expression of the top 20 most influential genes by cell type (B) where the size of the dot corresponds to the proportion of cells that express this gene and color ranging from red to red, indicating low to high average expression. The accuracy for the heatmap represents the average accuracy for 10 replications.
We conducted a series of experiments to explore the impact of the number of hidden layers of deep learning models. First, we constructed DNNs with one hidden layer with 100 nodes for Dropout + model, and 25 nodes for No Dropout, Dropout Only, and Dropout + models. These configurations were selected by conducting a grid search using the values 1, 10, 25, 50, 100, 150, 200, 250, 500, and 1000 (10 replications for each value was conducted). We found that the number of nodes that yield the best validation accuracy was 25 nodes for No Dropout, Dropout only, and Dropout + , and 100 nodes for Dropout + . Second, we constructed DNNs with three hidden layers with 100, 50, and 25 nodes, respectively. The accuracy and AUC of these models are shown in Supplementary Table 2. These experiments are displayed in Supplementary Table 2 and are very similar to those in Table 1. Also, note that a single layer DNN is only exploring the correlation structure, while a DNN with more layers can leverage more complex relationships in the data. The overall accuracy for a DNN with three hidden layers is only 0.3% more than the overall accuracy of a model with two hidden layers which is not practically different. Thus we will proceed with the model with two layers since it is more simple and maybe easier to generalize to different datasets.
There are many heuristics for selecting the number of nodes in two-layer models. One heuristic is the so-called pyramid rule heuristic, which uses following formula:
where n is the number of inputs, m is the number of outputs, and [39]. There were 22,890 genes input into the model and 18 classes, hence we used 2,113 and 196 nodes. The accuracy of the pyramid rule models is dramatically decreased (Supplementary Table 2) when compared to the two-layer models (Table 1) for all models except Dropout + . We illustrated that a model resulting from combining Dropout and regularization is robust to overfitting, as this model has many more parameters than the models presented in Table 1. In each set of experiments in the section, we see that Dropout + may not always have the highest accuracy, but based on the training plots it is the least sensitive to poorly specified models. Based on these findings, all models discussed in the remainder of this paper are constructed with dropout and regularization, and is referred to as the naive model, especially since there is not a significant difference between the testing accuracy of the four methods.
3.2. Testing on different datasets
Both naive and WDL learning models were constructed using the basal cell carcinoma data generated by Yost et al. and tested using melanoma data produced by Tirosh et al. Both models were constructed with 100 and 75 nodes for the first and second layers respectively. A comparison of the true cells types and the predicted cell types from the WDL model are shown in Fig. 4A and B. The naive model had an overall accuracy of 33% and did poorly on T cell subtypes, B cells, and NK cells (Fig. 4C). Another discrepancy is that a large portion of cells were misclassified as CAFs. With the addition of the 11 markers listed in Fig. 1, the WDL model can better discriminate subtypes of T cells, CD8 T cells and CD4 T cells, with an accuracy of 96% and 59% respectively (Fig. 4B right) and obtained an overall accuracy of 86.8% accuracy (Supplementary Fig. 5). The classification accuracy for each cell types ranges from 55% for NK cells to 97% for melanoma and macrophages, with 6 of the 8 cell types having accuracy higher than 93% (Fig. 4B right). A majority of misclassified CD4 T cells were classified as CD8 T cells within the same major type. The largest increase of accuracy was observed for B cells from 25% in the naive model to 93% in the WDL model.
Fig. 4.

Comparison of the true cell types (A) and predicted cell types from a WDL model (B). Side-by-side comparison of the average accuracy by cell type for 10 replications of the naive and WDL models (C).
To understand why the models perform differently, we focus on the differences between specific markers that were highly influential in each model. The importance of the top 20 markers and their importance are displayed in the average expression profiles for each cell type in Fig. 5A and B. A total of 11 markers were included in the wide part of the WDL model, and these have by far the largest importance in the model. Five well-known T cell markers (CD8A, CD8B, GZMK, CD4, and IL7R) were given large weights by the model, which explains why the WDL model was able to better classify CD4 and CD8 T cells.
Fig. 5.

Dot plots for the Tirosh data using both naive (A) and WDL (B) with average, based on 10 replications, gene importance weights increasing with brown color scale. The gene names highlighted in red correspond to gene that were included in the wide part of the WDL model, and the red gene names correspond to the genes that were not in the wide part yet were influential in both the naive and WDL models. Full list of genes and weights is included in Supplementary Table 3.
To determine the impact of the location of the wide component, we further appended the wide component onto the first hidden layer; thus the number of nodes in the first hidden layer is , using notation from Fig. 1. The mean accuracy and AUC for the 10 replicates were 79.74 and 4.85 respectively, which were significantly lower than the proposed WDL model with wide components on second layer (p-0.000472, t-test). The summary statistics are shown in Supplementary Table 4.
Additionally, to better understand the applicability of the WDL model across training sets from different platforms, we constructed 10 models by swapping the training and testing datasets. The models were trained on Tirosh et al. data generated from SMART-seq and tested on Yost el al. data generated from 10X. For each model, there were 100 nodes in the first layer. 75 nodes in the second layer, and the same genes shown in Fig. 1 are used in the wide component. The mean overall accuracy for these replications was 40.53 and 81.04 for the naive and WDL. The summary statistics are shown in Supplementary Table 4. WDL model dramatically improves the classification of all cell types except macrophages (Supplemental Fig. 5). Overall, the WDL model performed well across cell types by using the SMART-seq data as training. However, the accuracy was lower than WDL model using 10X as testing. This can be explained by the fact that 10X is better at discriminating cell types while SMART-seq is more suitable to detect lowly expressed genes.
Lastly, to study the impact of the number and the genes on the accuracy of a wide and deep learning model, we constructed models using different sets of genes (Supplementary Table 5) as the wide component with Yost et al. as training and Tirosh et al. as testing. Gene set 1, which focused mostly on CD8 T cells, did a poor job identifying CD4 T cells and melanocytes cells. By specifically adding genes exclusively expressed in melanocytes, such as MIA, PMEL, and MLANA, the accuracy was dramatically increased for melanocytes. In addition, CSF1R (Gene Set 3) and PECAM1 (Gene set 4) increased the accuracy of macrophages and endothelial cells respectively when compared to Gene Set 2. Gene Set 6, which consists of all genes in Gene Sets 1–5, led to a lower overall and B cell accuracy. Additionally, CAF and CD4 T cells had lower accuracy than Gene Sets 3–5. Finally, with CD19 to target B cells, Gene Set 7 obtained higher accuracy for B cells, CAFs, and CD4 T cells. We also noticed that targeting a single cell type may decrease the accuracy of another cell type. For example, the B cell classification accuracy dropped in Gene Set 4 when adding marker for endothelial cells compared to Gene Set 2. In addition, although the overall accuracies of Gene Sets 2–5 were similar, the accuracy of certain cell types may vary depending on whether and how many of their markers were specified as the wide components. The decision of markers to include should be experiment specific and depends on the focus of the study. In general, we have observed that the accuracy of a cell type can be improved by adding markers exclusively expressed by that cell type.
3.3. Comparison to other cell classification methods
To further assess the deep learning framework, we compared the Dropout + model to the state-of-art single cell type identification methods, including (1) Garnett, a prior-knowledge method that trains the classifier based on prior-known markers [40]; (2) CHETAH, a hierarchical classifier which is based on correlation calculated using the set of genes that best discriminates between cell types [3]; (3) SingleR, a method that assigns cell types based on correlation to annotated reference data using selected genes provided by users [4]. We evaluated the performance with two experimental settings: (1) intra-dataset in which the training and testing data were split from Yost et al. data as described in Results 3.1; (2) inter-dataset in which the models were trained on Yost et al. data generated with 10X and tested on Tirosh et al. data generated with SMART-seq. All methods were run under their default settings. For the prior-knowledge method Garnett that requires a marker gene file as input to guide the training, we used the marker genes provided by Yost et al., and also specified parent–child relationships among the cell types based on prior knowledge (Supplementary Table 6). The cells classified as the parent type of their true cell types by Garnett were considered as intermediate, for example, a CD8 exhausted T cell was classified as a CD8 T cell. For the hierarchical classifier CHETAH, the algorithm automatically selected genes that were distinctive enough between cell types, learned the hierarchical structures, and assigned cells into cell types with intermediate categories. For the correlation-based method SingleR, the genes used to calculate correlation were defined by differential expression analysis. The top 100 most differentially expressed genes for each cell type were selected. In all methods, cells classified as their true cell types were considered correct, while cells classified other than their true cell types or intermediate types are considered incorrect. Overall our naive DL model in the intra-dataset and WDL model in the inter-dataset outperformed other methods (Fig. 6). In general, all methods performed better in intra-dataset compared to inter-dataset experiment, which is consistent with the previous observation that the performance of classifiers is low when they are trained or tested on SMART-seq data [41]. In intra-dataset setting, naive DL shows much higher accuracy for CD8 T cell subtypes than the other three methods (Fig. 3A, Supplementary Fig. 6 left). The WDL model in inter-dataset setting also performed significantly better to discriminate CD8 and CD4 cells than all other methods (Fig. 4C, Supplementary Fig. 6 right).
Fig. 6.

Comparing deep learning models to other methods using intra-dataset (left) and inter-dataset (right) experiments. Percentages of cells classified into each category were shown in different colors. Note that naive DL and WDL are the average accuracy for 10 models.
4. Discussion
WDL presents an opportunity to use a small set of commonly known biological markers for cell type classification to allow models to be slightly less data-driven. We have illustrated a substantial increase in overall accuracy (36.5 to 86.9%) and for T cell subtypes (CD4 increased from 2.4 to 59.1% and CD8 increased from 19.5 to 96.1%). We have demonstrated that this can allow for training and testing models from data obtained from different platforms and types of skin cancer. Further refinement for the classification of fine T cell subtypes is needed to address questions such as ‘how strong is the CD8 T cell response to a tumor?’, i.e., determine the proportion of exhausted CD8 T cells, which are very relevant in cancer research. Additionally, there is a great need to develop systems to transfer knowledge across cancer types. WDL allows the opportunity to address this by including general set of genes for cell type classification and avoiding adding data/cancer specific markers as shown in Section 3.2.
In addition to adding a wide component to a DNN there is a need for careful consideration for how a model is trained to avoid the memorization of data. While dropout is a great tool for making deep learning models more generalizable there are many applications where there is a need for additional steps to avoid overfitting. Regularization is computationally intensive but makes deep learning models generalizable to test datasets. Models can very easily be overtrained, but a combination of dropout and regularization can provide a loss function that is stable across the training iterations. Another challenge for deep learning in general is that the randomness in the initialization of node weights, dropout, and batches can lead to dramatically different performances for models that are tuned in the same manner and data. Studying an ensemble of deep neural networks could help study the stability of the models and comparing the most important biomarkers in each model can provide further confidence that the markers that are highly influential. Identifying these genes can help clinicians understand the commonality between immune cells behavior across cancer types providing better insight and treatment of the cancers themselves.
There are significant challenges when applying models to data from different platforms. In particular, SMART-seq and 10X are fundamentally different in the way the RNA is processed to cDNA. In addition, the 10X is a UMI-based method which largely reduces the effects of PCR application bias. A previous study has shown the cell classification methods generally had low performance on SMART-seq data [41]. Despite the differences in training and testing data structures, the WDL models performed well on both 10X and SMART-seq platforms. Another challenge of processing single-cell RNA sequencing data is normalization. Due to the high prevalence of zero counts, cautions need to be taken when adjusting for unwanted batch effects in single-cell RNA sequencing data. A few deep learning-based methods have been developed to deal with this problem, such as DESC [42]. The models proposed in this study only take the processed single-cell RNA sequencing data, which has been normalized and batch corrected. It will be of interest to extend our work into a cell type classification method with batch effect removal as a future direction.
An advantage of deep learning to correlation-based methods such as CHETAH and SingleR is the use of multiple layers which allows for the exploration of nonlinear relationships in the data. Additionally, deep learning was employed using all genes, even those that were not expressed in many cells. For instance, in the intra-dataset experiment, Yost et al. identified EOMES to discriminate CD8 memory T cells, and TIGIT and CXCL13 to differentiate CD8 T exhausted cells (Supplementary Fig. 4 and Supplementary Table 3). However, these markers were not included in CHETAH or SingleR correlation calculation, which led to poorer accuracy for CD8 memory T cells and CD8 exhausted T cells (Supplementary Fig. 6 left).
One reason that WDL, CHETAH, and SingleR are successful at cell type classification in inter-datasets is that they are supervised by training data, unlike Garnett where the classifier relies on a handful of prior-knowledge genes. In particular, WDL learns the weights of genes automatically from the data, while allowing important genes to have higher weights; CHETAH and SingleR calculate correlation on genes that are most discriminative between cell populations in the training data, instead of using only a few prior-known genes. The naive deep learning models, on the other hand, performed well in the intra-data/platform experiment but poorly in the inter-data/platform experiment. This is because it does not utilize any prior knowledge and the model is left finding patterns from the full set of genes. Also, the overall gene expression patterns differ even for the same cell type due to different amplification protocols and technologies. WDL model was able to obtain a higher accuracy than the naive model for every cell type. However, CHETAH and SingleR are able to classify some cell types better than WDL. No markers were included for CAFs, myofibroblasts, or DCs in the WDL model, because no markers have been shown exclusively in these cell types across different platforms. Supplementary Fig. 4 illustrates that CST3 was given a large weight and highly expressed in CAFs, macrophages, melanocytes, and myofibroblasts. This explains why WDL had difficulty discriminating CAFs, macrophages, melanocytes, and myofibroblasts. With further research, it will be possible to improve the WDL model by identifying better markers for these cell types.
The true type of a cell is usually determined by in vivo study to test the cell’s function, which is a laborious process and not applicable in single-cell RNA sequencing experiments. Training of a DNN can reduce this time and lead to a model that is able to be applied to new samples directly. The training a DNN with the Yost data took approximately 1.5 h, while it took 20 min to train a model with the Tirosh data. From single-cell RNA sequencing data, the researchers can only infer the cell type by intensively interrogating the gene expression profile and comparing it to known cell types. Also, for groups of cells that do not match any known cell types, it is difficult to annotate them. Therefore, in most single-cell RNA sequencing studies, the term “cell clusters” is used to refer to groups of cell with a distinctive expression profile, instead of “cell types”. Here, we use the “true cell type” to refer to the cell annotation inferred by the original studies in Yost et al. and Tirosh et al. just for the model assessment. Despite using “cell clusters” as a surrogate for the true cell type it is still important to have algorithms that can quickly analyze the cells for a patient to get an idea of how the immune system is reacting to an illness such as cancer.
5. Funding
This work was supported in part by Institutional Research Grant No. 14-189-19 (to XW, XY) from the American Cancer Society, and a Department Pilot Project Award from Moffitt Cancer Center (to XY). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
6. Authors’ contributions
All authors read and approved the final manuscript. CW, BLF, JRC, XW, and XY conceived the study. CW, XW and XY designed the algorithm, performed the analyses, interpreted the results and wrote the manuscript.
7. Code availability
The code, documentation, and subsets of both the Tirosh and Yost datasets, in order to make the repository self complete, are provided at https://github.com/cwilso6/WDL_CellType_Class.
CRediT authorship contribution statement
Christopher M. Wilson: Formal analysis, Investigation, Visualization, Writing - review & editing. Brooke L. Fridley: Conceptualization, Methodology, Supervision, Writing - review & editing. José R. Conejo-Garcia: Conceptualization, Supervision, Writing - review & editing. Xuefeng Wang: Conceptualization, Supervision, Writing - review & editing. Xiaoqing Yu: Conceptualization, Visualization, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors would like to thank Colleagues at Department of Biostatistics and Bioinformatics at Moffitt Cancer Center for providing feedback.
Footnotes
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.csbj.2021.01.027.
Contributor Information
Xuefeng Wang, Email: xuefeng.wang@moffitt.org.
Xiaoqing Yu, Email: xiaoqing.yu@moffitt.org.
Supplementary data
The following are the Supplementary data to this article:
References
- 1.Newman A., Liu C., Green M., Gentles A., Feng W., Xu Y., Hoang C., Diehn M., Alizadeh A. Robust enumeration of cell subsets from tissue expression profiles. PLoS Med. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sturm G, Finotello F, Petitprez F, Zhang JD, Baumbach J, Fridman WH, List M, Aneichyk T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019;35(14):i436–45. arXiv:https://academic.oup.com/bioinformatics/article-pdf/35/14/i436/28913288/btz363.pdf, doi:10.1093/bioinformatics/btz363. URL: https://doi.org/10.1093/bioinformatics/btz363. [DOI] [PMC free article] [PubMed]
- 3.de Kanter JK, Lijnzaad P, Candelli T, Margaritis T, Holstege FCP. CHETAH: a selective, hierarchical cell type identification method for single-cell RNA sequencing. Nucl Acids Res 2019;47(16):e95–e95. arXiv:https://academic.oup.com/nar/article-pdf/47/16/e95/31234637/gkz543.pdf, doi:10.1093/nar/gkz543. URL: https://doi.org/10.1093/nar/gkz543. [DOI] [PMC free article] [PubMed]
- 4.Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R., Butte A.J., Bhattacharya M. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdelaal T., Michielsen L., Cats D., Hoogduin D., Mei H., Reinders M.J.T., Mahfouz A. A comparison of automatic cell identification methods for single-cell rna sequencing data. Gen Biol. 2019;20(1):194. doi: 10.1186/s13059-019-1795-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res 2008;9:2579–605. URL: http://www.jmlr.org/papers/v9/vandermaaten08a.html.
- 7.McInnes L, Healy J, Melville J. Umap: Uniform manifold approximation and projection for dimension reduction, cite arxiv:1802.03426Comment: Reference implementation available at http://github.com/lmcinnes/umap; 2018. URL: http://arxiv.org/abs/1802.03426.
- 8.Yost K.E., Satpathy A.T., Wells D.K., Qi Y., Wang C., Kageyama R., McNamara K.L., Granja J.M., Sarin K.Y., Brown R.A., Gupta R.K., Curtis C., Bucktrout S.L., Davis M.M., Chang A.L.S., Chang H.Y. Clonal replacement of tumor-specific t cells following pd-1 blockade. Nat Med. 2019;25(8):1251–1259. doi: 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Picelli S, Björklund rK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 2013;10(11):1096–8. doi:10.1038/nmeth.2639. URL: https://doi.org/10.1038/nmeth.2639 [DOI] [PubMed]
- 10.Hashimshony T., Wagner F., Sher N., Yanai I. Cel-seq: single-cell rna-seq by multiplexed linear amplification. Cell Rep. 2012;2(3):666–673. doi: 10.1016/j.celrep.2012.08.003. URL: https://doi.org/10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Islam S., Zeisel A., Joost S., La Manno G., Zajac P., Kasper M., Lönnerberg P., Linnarsson S. Quantitative single-cell rna-seq with unique molecular identifiers. Nature methods. 2014;11(2):163–166. doi: 10.1038/nmeth.2772. URL:https://doi.org/10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- 12.Picelli S., Faridani O.R., Björklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length rna-seq from single cells using smart-seq2. Nat Protocols. 2014;9(1):171–181. doi: 10.1038/nprot.2014.006. URL: https://doi.org/10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 13.Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M., Trombetta J.J., Weitz D.A., Sanes J.R., Shalek A.K., Regev A., McCarroll S.A. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202–1214. doi: 10.1016/j.cell.2015.05.002. URL: https://europepmc.org/articles/PMC4481139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., Gregory M.T., Shuga J., Montesclaros L., Underwood J.G., Masquelier D.A., Nishimura S.Y., Schnall-Levin M., Wyatt P.W., Hindson C.M., Bharadwaj R., Wong A., Ness K.D., Beppu L.W., Deeg H.J., McFarland C., Loeb K.R., Valente W.J., Ericson N.G., Stevens E.A., Radich J.P., Mikkelsen T.S., Hindson B.J., Bielas J.H. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. doi: 10.1038/ncomms14049. URL: https://europepmc.org/articles/PMC5241818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X., Li T., Liu F., Chen Y., Yao J., Li Z., Huang Y., Wang J. Comparative analysis of droplet-based ultra-high-throughput single-cell rna-seq systems. Mol Cell. 2019;73(1):130–142.e5. doi: 10.1016/j.molcel.2018.10.020. URL: http://www.sciencedirect.com/science/article/pii/S1097276518308803. [DOI] [PubMed] [Google Scholar]
- 16.See P., Lum J., Chen J., Ginhoux F. A single-cell sequencing guide for immunologists. Front Immunol. 2018;9:2425. doi: 10.3389/fimmu.2018.02425. URL: https://www.frontiersin.org/article/10.3389/fimmu.2018.02425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vieth B., Parekh S., Ziegenhain C., Enard W., Hellmann I. A systematic evaluation of single cell rna-seq analysis pipelines. Nat Commun. 2019;10(1):4667. doi: 10.1038/s41467-019-12266-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen G., Ning B., Shi T. Single-cell rna-seq technologies and related computational data analysis. Front Genet. 2019;10:317. doi: 10.3389/fgene.2019.00317. URL: https://www.frontiersin.org/article/10.3389/fgene.2019.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Islam MT, Karim Siddique BMN, Rahman S, Jabid T. Image recognition with deep learning. In: 2018 International conference on intelligent informatics and biomedical sciences (ICIIBMS), vol. 3; 2018. p. 106–10.
- 20.Jia X. Image recognition method based on deep learning. In: 2017 29th Chinese control and decision conference (CCDC); 2017. p. 4730–5.
- 21.Young T., Hazarika D., Poria S., Cambria E. Recent trends in deep learning based natural language processing [review article] IEEE Comput Intell Mag. 2018;13(3):55–75. [Google Scholar]
- 22.Sharma A.R., Kaushik P. 2017 International conference on computing, communication and automation (ICCCA) 2017. Literature survey of statistical, deep and reinforcement learning in natural language processing; pp. 350–354. [Google Scholar]
- 23.Cheerla A, Gevaert O. Deep learning with multimodal representation for pancancer prognosis prediction. Bioinformatics 2019;35(14):i446–54. arXiv:https://academic.oup.com/bioinformatics/article-pdf/35/14/i446/28913346/btz342.pdf, doi:10.1093/bioinformatics/btz342. URL: https://doi.org/10.1093/bioinformatics/btz342. [DOI] [PMC free article] [PubMed]
- 24.Tang B., Pan Z., Yin K., Khateeb A. Recent advances of deep learning in bioinformatics and computational biology. Front Genet. 2019;10:214. doi: 10.3389/fgene.2019.00214. URL: https://www.frontiersin.org/article/10.3389/fgene.2019.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen H., Engkvist O., Wang Y., Olivecrona M., Blaschke T. The rise of deep learning in drug discovery. Drug Discov Today. 2018;23(6):1241–1250. doi: 10.1016/j.drudis.2018.01.039. URL: http://www.sciencedirect.com/science/article/pii/S1359644617303598. [DOI] [PubMed] [Google Scholar]
- 26.Cheng H-T, Ispir M, Anil R, Haque Z, Hong L, Jain V, Liu X, Shah H, Koc L, Harmsen J, et al. Wide & deep learning for recommender systems. In: Proceedings of the 1st workshop on deep learning for recommender systems – DLRS 2016. doi:10.1145/2988450.2988454. URL: https://doi.org/10.1145/2988450.2988454.
- 27.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. URL: https://europepmc.org/articles/PMC6687398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., Fallahi-Sichani M., Dutton-Regester K., Lin J.-R., Cohen O., Shah P., Lu D., Genshaft A.S., Hughes T.K., Ziegler C.G.K., Kazer S.W., Gaillard A., Kolb K.E., Villani A.-C., Johannessen C.M., Andreev A.Y., Van Allen E.M., Bertagnolli M., Sorger P.K., Sullivan R.J., Flaherty K.T., Frederick D.T., Jané-Valbuena J., Yoon C.H., Rozenblatt-Rosen O., Shalek A.K., Regev A., Garraway L.A. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell rna-seq. Science. 2016;352(6282):189–196. doi: 10.1126/science.aad0501. arXiv:https://science.sciencemag.org/content/352/6282/189.full.pdf, URL: https://science.sciencemag.org/content/352/6282/189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gulli A., Pal S. Deep learning with Keras. Packt Publishing Ltd. 2017 [Google Scholar]
- 30.Goodfellow I, Bengio Y, Courville A. Deep learning. The MIT Press; 2016.
- 31.Fawcett T. An introduction to roc analysis. Pattern Recognit Lett 2006;27(8):861–74. rOC Analysis in Pattern Recognition. doi: 10.1016/j.patrec.2005.10.010. URL: http://www.sciencedirect.com/science/article/pii/S016786550500303X.
- 32.Provost F, Domingos P. Well-trained pets: improving probability estimation trees; 2000.
- 33.Hand D.J., Till R.J. A simple generalisation of the area under the roc curve for multiple class classification problems. Mach Learn. 2001;45(2):171–186. [Google Scholar]
- 34.Srivastava N., Hinton G., Krizhevsky A., Sutskever I., Salakhutdinov R. Dropout: A simple way to prevent neural networks from overfitting. J Mach Learn Res. 2014;15(56):1929–1958. URL: http://jmlr.org/papers/v15/srivastava14a.html. [Google Scholar]
- 35.Wu C., Gales M.J.F., Ragni A., Karanasou P., Sim K.C. Improving interpretability and regularization in deep learning. IEEE/ACM Trans Audio Speech Lang Process. 2018;26(2):256–265. [Google Scholar]
- 36.Yu K, Xu W, Gong Y. Deep learning with kernel regularization for visual recognition. In: Koller D, Schuurmans D, Bengio Y, Bottou L, editors. Advances in neural information processing systems 21. Curran Associates Inc; 2009. p. 1889–96. arXiv:https://academic.oup.com/bioinformatics/article-pdf/26/10/1340/16892402/btq134.pdf, doi:10.1093/bioinformatics/btq134. URL: https://doi.org/10.1093/bioinformatics/btq134.
- 37.Garson GD. Interpreting neural-network connection weights; 1991.
- 38.Kingma DP, Ba J. Adam: A method for stochastic optimization; 2017. arXiv:1412.6980.
- 39.Masters T. Academic Press Professional Inc.; USA: 1993. Practical neural network recipes in C++ [Google Scholar]
- 40.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., Gregory M.T., Shuga J., Montesclaros L., Underwood J.G., Masquelier D.A., Nishimura S.Y., Schnall-Levin M., Wyatt P.W., Hindson C.M., Bharadwaj R., Wong A., Ness K.D., Beppu L.W., Deeg H.J., McFarland C., Loeb K.R., Valente W.J., Ericson N.G., Stevens E.A., Radich J.P., Mikkelsen T.S., Hindson B.J., Bielas J.H. Massively parallel digital transcriptional profiling of single cells. Nature communications. 2017;8:14049. doi: 10.1038/ncomms14049. URL:https://europepmc.org/articles/PMC5241818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdelaal T., Michielsen L., Cats D., Hoogduin D., Mei H., Reinders M.J.T., Mahfouz A. A comparison of automatic cell identification methods for single-cell rna sequencing data. Gen Biol. 2019;20(1):194. doi: 10.1186/s13059-019-1795-z. URL: https://europepmc.org/articles/PMC6734286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X., Wang K., Lyu Y., Pan H., Zhang J., Stambolian D., Susztak K., Reilly M.P., Hu G., Li M. Deep learning enables accurate clustering with batch effect removal in single-cell rna-seq analysis. Nat Commun. 2020;11(1):2338. doi: 10.1038/s41467-020-15851-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.