

Abstract
T cells that are gene-modified with tumor-specific T cell receptors are a promising treatment for metastatic melanoma patients. In a clinical trial, we treated seven metastatic melanoma patients with autologous T cells transduced to express a tyrosinase-reactive T cell receptor (TCR) (TIL 1383I) and a truncated CD34 molecule as a selection marker. We followed transgene expression in the TCR-transduced T cells after infusion and observed that both lentiviral- and retroviral-transduced T cells lost transgene expression over time, so that by 4 weeks post-transfer, few T cells expressed either lentiviral or retroviral transgenes. Transgene expression was reactivated by stimulation with anti-CD3/anti-CD28 beads and cytokines. TCR-transduced T cell lentiviral and retroviral transgene expression was also downregulated in vitro when T cells were cultured without cytokines. Transduced T cells cultured with interleukin (IL)-15 maintained transgene expression. Culturing gene-modified T cells in the presence of histone deacetylase (HDAC) inhibitors maintained transgene expression and functional TCR-transduced T cell responses to tumor. These results implicate epigenetic processes in the loss of transgene expression in lentiviral- and retroviral-transduced T cells.
Keywords: TCR-transduced T cells, gene-modified T cells, HDAC inhibitors, sodium butyrate, vorinostat, gene silencing, transgene silencing, cancer immunotherapy, cell therapy, functional responses
Graphical Abstract
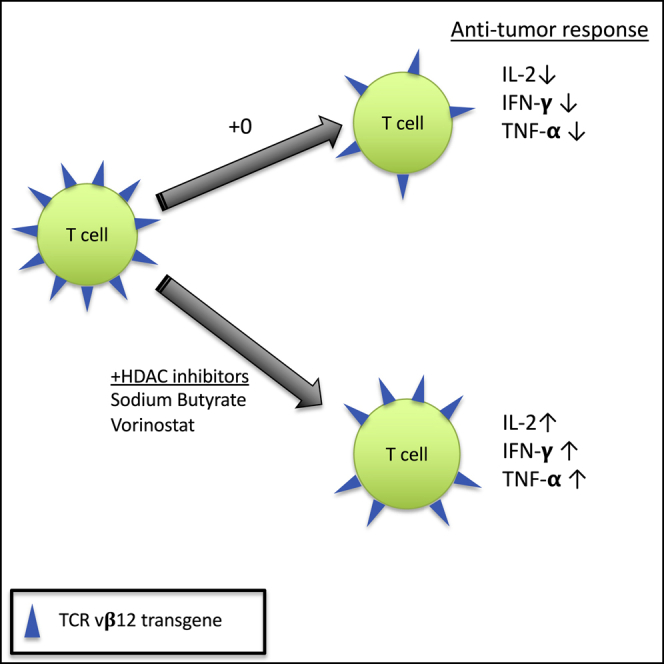
Nishimura and colleagues demonstrate that transgene expression in gene-modified anti-tumor T cells is reversibly downregulated over time in vivo and in vitro. Treatment with IL-15 or with HDAC inhibitors (sodium butyrate or vorinostat) reduced this downregulation and maintained T cell functional responses to tumor cells.
Introduction
Despite increased public health program-induced awareness of the risks of sun exposure, the incidence of melanoma has doubled in the last 40 years.1 This year alone, approximately 100,000 people will be diagnosed with melanoma in the United States, and 6,800 will die of their disease.2 Cancer immunotherapy has become a prevalent modality of treatment for metastatic melanoma. Forms of immunotherapy such as checkpoint inhibitors (e.g., anti-PD-1 and anti-CTLA-4)3 and tumor-infiltrating lymphocyte (TIL) therapy depend on activating and expanding pre-existing tumor-specific T cells.4 In contrast, genetic modification of autologous T cells to target the tumor does not depend on pre-existing anti-tumor T cells and has led to clinical responses in metastatic melanoma patients for over 14 years.5,6 Current testing of gene-modified T cell therapy is extensive, with over 100 clinical trials listed on ClinicalTrials.gov.
T cell receptor (TCR)-transduced T cells can induce partial or complete remission and can persist for years after transfer.7,8 While promising, many patients fail to respond to TCR-transduced T cells, and efforts are needed to improve the efficacy and durability of TCR-transduced T cell therapies.9, 10, 11, 12, 13, 14, 15, 16 Studies have found that clinical efficacy correlates with the expansion and persistence of adoptively transferred T cells.17 Unfortunately, most patients treated with TCR-transduced T cells have few detectable TCR-transduced T cells in the blood beyond a month post-transfer.8,9,11 Previous work demonstrated that TCR-transduced T cells may lose transgene transcription over time.13 Such loss of transgene transcription may be predicted to lead to loss of expression of the anti-tumor TCR, reducing long-term efficacy of gene-modified T cells.
Despite the importance of transgenic TCR expression for the anti-tumor function of transduced T cells, the processes regulating maintenance of transgene expression and effects of transgene expression on functional responses of transduced T cells are largely unexplored. While conducting a clinical trial, we had an opportunity to study transgene regulation in both lentiviral- and retroviral-transduced T cells. All patients were treated with autologous T cells transduced to express the TIL 1383I receptor, which responds to an HLA-A2-restricted tyrosinase peptide in a CD8-independent fashion, permitting treatment with both CD4+- and CD8+-transduced T cells. The first cohort of three patients were treated with lentiviral-transduced T cells, while the remaining four were treated with retroviral-transduced T cells. In this study, we found that both lentiviral- and retroviral-transduced T cells exhibit reversible downregulation of transgene expression in patients and in vitro. During in vitro culture, the presence of interleukin (IL)-15, but not antigen or IL-2, prevented downregulation of transgene expression. Culture with sodium butyrate, an HDAC inhibitor, reduced transgene expression downregulation and retained T cell function in the absence of cytokines. Collectively, these results suggest that transgene downregulation in T cells is epigenetically regulated by histone deacetylation. Although further testing is needed, these results suggest the possibility that histone deacetylase (HDAC) inhibitor treatment might enhance the long-term clinical efficacy of transduced T cell therapies in patients.
Results
Seven patients were treated with TIL 1383I TCR-transduced T cells, of which the first three were treated with lentiviral-transduced T cells (lentiviral transduced)8 and the remaining four with γ-retroviral-transduced T cells (retroviral transduced).18 We previously observed that the percentage of transduced T cells in the peripheral blood was highest in the first week post-transfer and then decreased thereafter.8 Closer investigation revealed a declining level of expression of the truncated CD34t marker gene (CD34) in CD34+ T cells in the weeks following adoptive transfer (Figure 1A). These decreases occurred in both CD34+ CD4+ (Figure 1B, top) and CD34+ CD8+ (Figure 1B, bottom) T cells in patients given retroviral- and lentiviral-transduced T cells. Decreasing transgene expression levels were observed in most patients, with the interesting exception of patient 2, who achieved a partial response after adoptive transfer, which eventually became a complete response after further immunotherapies.8 By 4 weeks post-transfer, a small percentage of patient T cells expressed low levels of the truncated CD34 marker gene (CD34) encoded in the lentiviral and retroviral vectors. We aimed to investigate whether TCR-transduced T cells might remain present but undetectable due to loss of transgene expression. To induce an activation state similar to that of cells undergoing transduction, T cells from patient samples taken 4 weeks post-adoptive transfer were activated with CD3- and CD28-crosslinking Human T-Activator beads (CD3/CD28 beads) in the presence of IL-2 and IL-15. Following stimulation, both the percent of transgene (CD34)-expressing T cells and the level of transgene expression were determined by flow cytometry (Figure 1C). Representative gating is shown in Figure S1. In all patients, activating the T cells increased the percent of detectable CD8+CD34+ T cells, although it did not consistently increase the percent of detectable CD4+CD34+ T cells (Figure 1D). However, the level of CD34 expression (the mean fluorescence intensity [MFI] of CD34 staining) on both CD4+CD34+ as well as CD8+CD34+ T cells was significantly increased, suggesting that activation did enhance transgene expression in both CD4+ and CD8+ T cells. Patients 4–7, treated with retrovirus-transduced T cells, appeared to have greater CD34 upregulation after restimulation than patients 1–3, treated with lentivirus-transduced T cells. This apparent difference might be due to the higher dose of cells in patients treated with retrovirus, to higher transgene expression in retrovirus-transduced T cells, or to differences in epigenetic regulation of the transgene expression between viral constructs. CD34 staining on CD34-negative CD4+ and CD8+ T cells was, as expected, low, and did not significantly change after restimulation (Figure S2, top). CD34 staining on total T cells from healthy donors also was low and showed only a very slight increase with stimulation, likely due to increased autofluorescence (Figure S2, bottom). These results indicate that retroviral- and lentiviral-transduced CD4+ and CD8+ T cells reversibly downregulate transgene expression in patients, potentially decreasing efficacy of transduced T cells over time.
Figure 1.

Transgene expression is reversibly downregulated in patients
(A and B) Cryopreserved patient PBMCs were thawed and analyzed by flow cytometry for expression of CD34 on live CD3+CD4+CD34+ or live CD3+CD8+CD34+ T cells. (A) Expression of CD34 versus CD4 on live CD3+ T cells for a patient given lentivirally transduced T cells (top, LV) and retrovirally transduced T cells (bottom, RV). (B) MFI of CD34 on live CD34+ CD4+ (top) and CD34+ CD8+ (bottom) T cells. (C and D) Patient blood samples were thawed then rested or cultured in complete media with IL-2 (300 IU/mL) and IL-15 (100 ng/mL) for 2 days, then restimulated with Human T-Activator CD3/CD28 beads at a 1:1 cell:bead ratio. (C) Expression of CD34 and gating on CD34+CD4+ and CD34+CD8+ are shown. (D, top) The percent CD34+CD4+ (left) and CD34+CD8+ (right) are shown for every patient; (bottom) the MFI of CD34 on CD34+CD4+ (left) and CD34+CD8+ (right) T cells is shown for every patient. Patients receiving lentiviral-transduced T cells are shown in blue, while those receiving retroviral-transduced T cells are shown in black. p values were calculated by a ratio paired t test and summarized with ∗∗p < 0.01 and ∗p < 0.05.
Prior to transfer into patients, T cells were maintained with high levels of IL-2 and IL-15 to promote T cell survival and growth. These levels of IL-2 and IL-15 are optimized for in vitro culture and are far higher than what has been observed in normal peripheral blood.19,20 In our trial, patients received low-dose IL-2 (72,000 IU/kg), for the first 7 days post-transfer, during which the percentage of transgene-expressing (CD34+) T cells increased in six of seven patients8 (Figure S3). After day seven, the percent of transduced T cells decreased. It was possible that the combination of low-dose IL-2 and increased bioavailability of IL-15 due to lymphopenia21,22 contributed to transgene expression in patients. We hypothesized that when levels of these cytokines naturally declined as patients stopped receiving IL-2 and recovered from lymphopenia, transgene expression was also declining. We cultured patient T cells transduced with lentiviral or retroviral vectors without adding IL-2 or IL-15 to determine whether transgene expression would downregulate in vitro in the absence of exogenous cytokines. During culture without cytokines, both the marker gene, CD34, and transgenic TCR expression (via Vβ12, the β chain of the TIL 1383I TCR) were monitored by flow cytometry over time. Both CD34 and Vβ12 expression decreased on transduced CD4+ and CD8+ T cells cultured without cytokines compared to cells cultured with IL-2 and IL-15 (Figure 2A). By day seven, a significant fraction of transgene expression was lost in T cells without cytokines compared to T cells maintained with IL-2 and IL-15 (Figure 2B). Interestingly, Vβ12 expression was more sensitive to downregulation than the CD34 marker, perhaps due to TCR cycling and competition for CD3 with the endogenous TCR.23 These results suggest that transgene expression is maintained by cytokines, and loss of transgene expression may be induced by cytokine withdrawal.
Figure 2.

Reversible transgene expression downregulation occurs in the absence of cytokines
(A and B) T cells from the apheresis of patients 1–3 were activated, separated into two groups, and transduced with lentivirus or retrovirus and expanded. Transduced T cells were cultured with and without cytokines for 23 days. CD34 and Vβ12 expression was assessed by flow cytometry. (A) Example of CD34 versus Vβ12 expression on CD4+ and CD8+ T cells on day 23. (B) Time course of CD34 (left) and Vβ12 (right) expression on CD4+ (top) and CD8+ (bottom) T cells. Data in (B) were analyzed by 2-way ANOVA and Tukey’s multiple comparisons test. (C) Transduced T cells were cultured for 4 weeks without cytokines, then restimulated. CD34 and Vβ12 expression was assessed on CD4+ and CD8+ T cells prior to and after restimulation for 2 days with IL-2 and IL-15 or with CD3/CD28 beads as well as IL-2 and IL-15. Data in (C) were analyzed by paired t test. p values are summarized as ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05.
Culturing T cells without cytokines causes some apoptosis (viability for cells cultured without cytokines for 23 days was 14.9% ± 4.2%, while viability for cells cultured with cytokines was 33.2% ± 4.2%). It was therefore possible that transduced T cells were selectively dying without cytokine support. To rule out the possibility that transgene expression loss in vitro might be due to selective death of transduced cells, we determined whether cells that had downregulated CD34 and Vβ12 expression in vitro were able to re-express transgene if restimulated. Lentiviral- and retroviral-transduced T cells cultured for 4 weeks in the absence of cytokines were restimulated for 2 days with cytokines (IL-2 and IL-15) with or without anti-CD3/anti-CD28 beads. Restimulation with both cytokines and anti-CD3/anti-CD28 significantly increased the percent of CD34+Vβ12+ cells in CD4+ and CD8+ populations for both lentiviral- and retroviral-transduced T cells (Figure 2C). Restimulation with cytokines alone significantly increased transgene expression in retroviral-transduced CD4+ and CD8+ T cells as well as lentiviral-transduced CD4+ T cells. There was a trend toward increase in transgene expression in lentiviral-transduced CD8+ T cells after cytokine restimulation, but it did not reach significance. Interestingly, although the percent of transgene downregulation was greater in retroviral-transduced T cells (Figures 2B and 2C), the retroviral-transduced T cells re-expressed transgene at substantially higher levels than lentiviral-transduced T cells as well. Although there are insufficient data to draw general conclusions about all retroviruses and lentiviruses, our results suggest that for this retrovirus and this lentivirus, the retroviral transgenes are more plastic—more susceptible to both downregulation and reversal of downregulation than the lentiviral transgenes. Overall, these results indicate that retroviral and lentiviral transgene expression is lost in vivo and during culture without cytokines due to a reversible process.
We next investigated whether IL-2, IL-15, or both were necessary for maintaining transgene expression. Culture of either lentiviral- or retroviral-transduced healthy donor CD4+ and CD8+ T cells with IL-15 alone maintained transgene expression equivalently to culture with both IL-2 and IL-15 (Figure 3A). Conversely, culture with IL-2 alone only maintained transgene expression slightly above culture with no cytokines. These results demonstrate that IL-15, but not IL-2, contributes to the maintenance of transgene expression over time in vitro.
Figure 3.

Culture in IL-15 reduces downregulation of transgene expression
Healthy donor T cells were activated, transduced with lentivirus (pLVX-1383I, left) or retrovirus (SAMEN-1383I, RV, right), then CD34-sorted and rapidly expanded. (A) Lentiviral- and retroviral-transduced T cells were then cultured in the presence or absence of IL-2 and IL-15. Expression of CD34 and Vβ12 was assessed by flow cytometry on CD4+ or CD8+ CD3+ T cells at indicated time points. (B) Lentiviral- and retroviral-transduced T cells were cultured for 14 days in the absence or presence of T2 cells, T2 cells pulsed with tyrosinase, 624 MEL cells, or IL-2 (300 IU/mL) and IL-15 (100 ng/mL). Tumor cells were added at a ratio of 1:4 tumor:T cell. On day 12, tumor cells were no longer detectable, and a second dose of tumor cells was added. Error bars represent standard deviation. Significance is shown by Student’s t test with p values summarized as ∗∗p < 0.01, and ∗p < 0.05.
Both TCR and cytokine stimuli were present during initial transduction. To investigate whether culturing with antigen would be sufficient to maintain transgene expression, transduced healthy donor T cells were cultured with either HLA-A2+ T2 cells pulsed with tyrosinase peptide (T2/tyrosinase), or an HLA-A2+ tyrosinase-expressing melanoma line, 624 MEL. Neither culture with T2/tyrosinase nor culture with 624 MEL cells prevented loss of transgene expression in T cells, whereas culture with IL-2 and IL-15 maintained significantly higher transgene expression than culture without cytokines (Figure 3B). These in vitro results, in addition to the loss of transgene expression in patients who have both tumors and melanocytes bearing antigen, suggest that antigen exposure alone is insufficient to maintain transgene expression. In the presence of IL-2 and IL-15, culturing T cells with antigen-expressing tumor cells did increase transgene expression on CD8+ T cells compared to CD8+ T cells cultured with cytokines alone (Figure S4). These results suggested that antigen stimulation of T cells enhances transgene expression in the presence of cytokines but is not sufficient to maintain transgene expression as a single agent.
Most of the DNA in resting T cells is bound up in transcriptionally inactive heterochromatin.24,25 T cell activation reduces heterochromatin and promotes upregulation of gene expression of many activation-associated genes.26 T cells were activated prior to transduction to enhance retroviral and lentiviral transgene integration. Most likely, retroviral and lentiviral transgenes then integrated into open chromatin near transcriptionally active sites. It is possible that as T cells enter resting states in patients or in vitro without cytokines, there is epigenetic downregulation of transgene expression along with the genes near which the transgenes inserted. Two methods of epigenetic downregulation of gene expression are through DNA methylation or histone deacetylation by histone deacetylases (HDACs). Therefore, we tested whether T cell culture with HDAC inhibitors, sodium butyrate or suberoylanilide hydroxamic acid (SAHA),27 or a DNA methyltransferase inhibitor, 5-aza-2-deoxycytidine (5a2d), prevented downregulation of transgene expression. Culture of transduced T cells with HDAC inhibitors prevented downregulation of transgene expression in the absence of cytokines (Figure 4). When cells were cultured with sodium butyrate and cytokines, the addition of both agents acted to maintain higher transgene expression than either alone, although SAHA treatment did not act additively with cytokines. DNA methyltransferase inhibition did not affect transgene expression in our model, a finding consistent with previous work showing that there is little methylation of transgenes in patients.13 These results indicate that when T cells are removed from high cytokine environments, histone deacetylation reduces transgene expression, and this process may be prevented by treatment with HDAC inhibitors. Further, T cells cultured with cytokines may also undergo some HDAC-mediated transgene downregulation, as treatment with sodium butyrate enhanced transgene expression in T cells cultured with cytokines.
Figure 4.

Culture with HDAC inhibitors reduces transgene expression downregulation
Transduced T cells (from healthy donors) were cultured for 12 days with no cytokines or with IL-2 and IL-15 in the absence or presence of HDAC inhibitors, sodium butyrate (SB) or suberoylanilide hydroxamic acid (SAHA), or a DNA methyltransferase inhibitor, 5-aza-2′-deoxycytidine (5a2d). (A) Representative fluorescence-activated cell sorting (FACS) plots of CD34 versus vβ12 on CD4+ (top) and CD8+ (bottom) T cells. (B) Comparison of percentages CD34+Vβ12+ of CD4+ (top) and CD8+ (bottom) T cells. Results are representative of at least four donors and three independent experiments. Significance was determined by paired t test (pairing on donor and virus type) with p values summarized as ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05
Culture in HDAC inhibitors prevented downregulation of transgene expression, leading us to question whether culture in HDAC inhibitors would further prevent loss of functional responses of transduced T cells to antigen. Transduced T cells from three donors were cultured with complete medium only, complete medium with cytokines, complete medium with sodium butyrate (1 mM), or complete medium with cytokines and sodium butyrate. After 10 days in culture, the T cells were then cocultured overnight (without cytokines or sodium butyrate) with T2/tyrosinase or with 624 MEL. CD4+ and CD8+ T cells were assessed for lytic granule production by staining for CD107a expression and for production of inflammatory cytokines IL-2, tumor necrosis factor alpha (TNF-α), and interferon (IFN)-γ by intracellular cytokine staining. The percentage of T cells producing one, two, three, or all four effector molecules is shown (Figure 5B). In both lentiviral- and retroviral-transduced T cells cultured without cytokines, culture in sodium butyrate increased the percent of cytokine-producing cells in both CD4+ and by CD8+ T cells. The addition of sodium butyrate to T cells cultured in IL-2 and IL-15 also appeared to increase cytokine production in both CD4+ and CD8+ T cells, though the difference did not reach significance in some cases due to substantial variability. A breakdown of the exact cytokine-producing profiles (Figure S5) of all groups suggests that culture in sodium butyrate increases the percentage of retroviral- and lentiviral-transduced T cells producing many cytokine profiles in both CD4+ and CD8+ T cells activated with 624 MEL or T2/tyrosinase. These results indicate that HDAC inhibition can retain antigen-specific T cell effector functions in TCR-transduced T cells cultured without cytokines and enhance effector functions in T cells cultured with cytokines. Further testing is required to determine whether HDAC inhibition can maintain T cell effector functions in vivo. If so, HDAC inhibition may permit more effective and long-lived transgenic T cell therapies.
Figure 5.

Transduced CD4+ and CD8+ T cell responses to antigen are protected in the absence of cytokines by sodium butyrate
Transduced T cells (from healthy donors) were cultured for 10 days with and without sodium butyrate and cytokines. After 10 days, cells were washed and cocultured with T2/tyrosinase or 624 MEL in the presence of Golgi blockers and a labeled anti-CD107a antibody. After the coculture, the cells were stained extracellularly for CD3, CD4, CD8, and CD34; stained with a viability dye; then fixed, permeabilized and stained intracellularly for TNF-α, IFN- γ, and IL-2. Statistics shown are Student’s t test of differences in total cells expressing 1 or more effector molecules (IL-2, IFN-γ, TNF-α, and CD107a). p values are summarized as ∗∗p < 0.01, and ∗p < 0.05.
Discussion
We found that transgene expression was downregulated in TCR-transduced CD4+ and CD8+ T cells adoptively transferred into metastatic melanoma patients. This phenomenon occurred regardless of whether patients received lentiviral- or retroviral-transduced T cells. Lentiviral and retroviral transgene expression downregulation was reproducible by in vitro culture without cytokines. In vitro culture in IL-15, but not IL-2 or antigen, maintained T cell transgene expression. Culturing T cells in the presence of an HDAC inhibitor prevented T cell transgene downregulation and maintained T cell functional responses in the absence of cytokines. Although we have previously observed that detectable (by flow cytometry) expression of transgene is not required for antigen-specific T cell responses of transduced T cells,28 here we observe that culture of transduced T cells with an HDAC inhibitor increased effector responses as well as transgene expression. Collectively, our results suggest that histone deacetylase activity reduces transgene expression and effector functions of transduced T cells.
Although both γ-retroviruses and lentivirus are being used in clinical trials, these vectors may not have identical epigenetic regulation. Both γ-retroviruses and lentiviruses integrate semi-randomly into the genome at sites associated with histone binding, which are also near transcriptionally active sites. However, γ-retrovirus and lentivirus integration patterns are non-identical, with γ-retroviruses favoring areas 5′ of genes, while lentiviruses favor introns.29 We compared transgene expression regulation across both types of viruses and found that both retroviral- and lentiviral-transduced T cells lost transgene expression, and transgene expression was restored after cytokine and TCR stimulation in both retroviral- and lentiviral-transduced T cells. Interestingly, downregulation and stimulation-induced reexpression appeared to be more robust in γ-retroviral-transduced T cells than in lentiviral-transduced T cells (Figures 1, 2, 3, and 4). It is possible that γ-retrovirus integration sites are more sensitive to epigenetic effects (both downregulation and upregulation) than lentiviral integration sites. It is also possible that the specific viruses used in this study are regulated differently due to uncontrolled factors, such as different promoters, regulatory elements, or copy number. Overall, however, neither retroviral- nor lentiviral-transduced T cells are immune to transgene expression loss. Further, similar mechanisms (IL-15, histone deacetylases) appear to regulate transgene expression in both lentiviral- and retroviral-transduced T cells.
Notably, the effects of HDAC inhibition were greater on expression of the T cell receptor (Vβ12) than on the CD34 gene. Both genes are co-transcribed as one mRNA, separated by a ribosomal skip sequence, so HDAC inhibition and prevention of chromatin condensation and gene transcription downregulation cannot account for this difference. It is likely that the TCR requires more mRNA for similar expression, as the TCR protein is constantly being cycled away from and back to the cell surface30 and must compete with endogenous TCR for CD3ζ chains.31 It is further possible that effects of HDAC inhibitors on other cellular pathways also contribute to the regulation of T cell receptor expression. For example, retroviruses have been shown to reduce expression of multiple CD3 components,32,33 and HDAC inhibition was shown to rescue CD3 expression. Therefore, HDAC inhibitors may be rescuing transgene expression and function via multiple mechanisms—both directly and by enhancing CD3 expression.
Culture of T cells in the presence of sodium butyrate maintained or increased the ability of TCR-transduced CD4+ and CD8+ T cells to make cytokines in response to antigen. This appears to be in contrast to past studies in which sodium butyrate was found to be suppressive and drive regulatory T cell (Treg) formation in the gut microenvironment34 and inhibit antigen-presenting cell (APC)-mediated activation of CD8+ T cells35,36 and induce anergy in naive T cells.37 However, recent papers have demonstrated that sodium butyrate and other HDAC inhibitors can enhance T helper 1 (Th1) responses,38,39 reduce T cell activation-induced cell death (AICD),40 and enhance the development of CD8+ T cell memory.41 Collectively, these results suggest that while sodium butyrate and other HDAC inhibitors can prevent or reduce naive cell activation, these inhibitors can also enhance existing effector T cell responses. In our transduced cells, is likely that HDAC inhibition results in maintaining expression of activation and/or growth-associated genes and maintaining expression of transgenes inserted into activation and growth-associated genetic loci as well.
Our results suggest that IL-15 treatment and/or HDAC inhibitors might enhance transduced T cell therapies in the clinic by enhancing TCR transgene expression. Preclinical studies have shown that HDAC inhibitors can synergize with other immunotherapies and enhance anti-tumor responses.42, 43, 44, 45 HDAC inhibitors may act to change the immune environment of tumors46 and are currently being tested in clinical trials in conjunction with immunotherapies.46 One caveat of using HDAC inhibitors (HDACi) that should be noted is that HDAC inhibitors such as vorinostat may be lymphotoxic. We observed a wide variety of toxicity from both sodium butyrate and vorinostat treatment in our assays, with the percent of viable cells reduced approximately 0%–50% with sodium butyrate and 25%–75% with vorinostat in the absence of cytokines. The presence of cytokines reduced the toxicity of the HDACi treatments. Interestingly, the toxicity of vorinostat in vitro was more dramatic than that observed in patients, in which less than 30% of patients had demonstrated any decrease in their white blood cell counts.47 Although IL-15 did not show dramatic results in the clinic as a single agent, and significant toxicity was observed,48 there is an IL-15 conjugate that is well tolerated by patients49 and has shown promising anti-tumortumor results when given with PD-1 blockade in an initial small-scale safety trial.50 It is possible that patients given transduced T cells would benefit from concurrent treatment with HDAC inhibitors or an IL-15 conjugate to enhance transgene expression and function. Overall, this study demonstrated that HDAC inhibitors can enhance T cell transgene expression and function in vitro. Further in vivo studies are necessary to determine the effects of HDAC inhibitors and/or IL-15 conjugates on transduced T cells in vivo in the tumor microenvironment, where effects on tumor, the presence of antigen, and suppressive mechanisms may enhance or detract from the effects of HDAC inhibitors or IL-15 treatment.
Materials and methods
Clinical trial design
These studies were performed at Loyola University Medical Center (Maywood, IL, USA) and the Earle A. Chiles Research Institute (Portland, OR, USA). The study was registered with ClinicalTrials.gov (NCT01586403 [lentivirus] and NCT02870244 [retrovirus]). Informed consent was obtained prior to enrolling patients. These studies were approved by the Institutional Review Board (LU 203732 [lentivirus] and LU 203729 [retrovirus]) and the Institutional Biosafety Committee (LU 203729 [retrovirus] and LU 203732 [lentivirus]) at Loyola University Medical Center as well as the Institutional Biosafety Committee and the Institutional Review Board (16-004B) at Providence Portland Medical Center. These studies were also approved by the Recombinant DNA Advisory Committee (RAC Protocol 1101-1086) at Loyola University Medical Center, the Cancer Therapy Evaluation Program (CTEP 9358), and the US Food and Drug Administration (IND 14971 [lentivirus] and 16315 [retrovirus]).
Patients 1–3 received 2.5 × 106 lentivirus-transduced T cells per kilogram. Patients 4–6 received 7.5 × 106 γ-retrovirus-transduced T cells per kilogram. Patient 7 received 2.5 × 107 γ-retrovirus-transduced T cells per kilogram. All patients received non-myeloablative lymphodepletion with intravenous once-daily administration of cyclophosphamide and fludarabine prior to T cell infusion (the day of infusion was designated as day 0). Cyclophosphamide was administered at 60 mg/kg on days −2 and −1 for patients 1–3, 60 mg/kg on days −3 and −2 for patients 4–6, and 1,000 mg/m2 on days −3 and −2 for patient 7. Fludarabine was administered at 25 mg/m2 on days −5, −4, −3, −2, and −1. All patients were further given low-dose IL-2 (72,000 IU/kg, Aldesleukin, Novartis, East Hanover, NJ, USA) for 1 week following the T cell infusion.
T cells for patient treatment were generated and patients were treated as follows. Briefly, patient peripheral blood mononuclear cells (PBMCs) were activated in AIM-V medium (Thermo Fisher) with 5% human AB serum (Valley Biomedical, Winchester, VA, USA) (AIM-V CM) with 50 ng/mL anti-CD3 antibody (OKT3, Miltenyi Biotec, Somerville, MA, USA), 300 IU/mL rhIL-2 (Aldesleukin, Novartis, East Hanover, NJ, USA) and 100 ng/mL rhIL-15 (Biological Resources Branch, NCI, Frederick, MD, USA). Three days after activation, T cells were spinoculated with lentivirus- or retrovirus-containing media on retronectin-coated (Takara Bio, Mountain View, CA, USA) plates. Three days after spinoculation, T cells were enriched for CD34-expressing cells (transduced T cells) using biodegradable CD34-binding immunomagnetic iron-dextrose particles (GMP-grade, Miltenyi Biotec) and a CliniMACS (Miltenyi Biotec). Enriched CD34+ T cells were expanded by a rapid expansion protocol (REP). For the REP, T cells were expanded by culture with irradiated pooled allogeneic PBMCs from at least three donors at a 1:200 T:PBMC ratio in AIM-V CM with 30 ng/mL anti-CD3 antibody, 300 IU/mL rhIL-2, and 100 ng/mL rhIL-15. At various points post-T cell transfer, patient blood samples were taken; white blood cells were isolated by density gradient centrifugation and then cryopreserved and analyzed at a later time point.
Viruses
The SAMEN-1383I γ-retroviral vector has been previously described.51 Briefly, expression is driven by a hybrid 5′ long terminal repeat (LTR), containing the human cytomegalovirus enhancer and promoter fused to the MMLV 5′ LTR followed by splice donor, packaging, and splice acceptor sites; then by the expression cassette of the cloned TIL 1383I TCR α gene; a P2A ribosomal skip site; the TIL 1383I TCR β gene; a T2A ribosomal skip site; and the truncated non-signaling CD34t expression marker. The lentiviral vector used to transduce T cells from patients 1–3 was a previously described proprietary vector (Lentigen Technology, Gaithersburg, MD, USA).8 For all in vitro experiments with healthy donor T cells, the pLVX-1383I lentiviral vector was used. The pLVX-1383I vector was produced from the pLVX-EF1alpha-mCherry-N1 vector (Clontech, Mountain View, CA, USA), which was modified to remove the mCherry and then include the same TIL 1383I TCR and CD34t expression cassette as the retroviral vector. Vector maps are shown in Figure S6.
Patient characteristics
Patients 1–3 were previously described.8 Patients 4–7 are described in Table S1.
Cell lines and primary cells
Healthy donor PBMCs were purchased as fully deidentified apheresis products (Key Biologics, Memphis, TN, USA). Cell lines used for T cell stimulation included T2 cells, a TAP-deficient HLA-A2+ cell line52 purchased from ATCC, and 624 MEL, a tyrosinase-producing, HLA-A2+ human melanoma line obtained from the Surgery Branch of the National Cancer Institute (NCI, Bethesda, MD, USA).53 Viral producer cell lines included GPRTG, a stable lentiviral producer line made from HEK293T cells transfected with the HIV gag-pol, rev, tat, and VSV-G env genes,54 provided by the National Gene Vector Biorepository (Indiana University, Indianapolis, IN, USA), and PG-13, a stable retroviral producer line made from NIH 3T3 cells transfected with Gibbon Ape Leukemia Virus (GALV) envelope and the Moloney murine leukemia virus (MMLV) gag-pol genes.55 GPRTG cells were transduced with the modified pLVX-1383I construct described above to generate a stable lentiviral producer line. PG-13 cells were transduced with the SAMEN-1383I construct described above to generate a stable retroviral producer line. T2 cells were maintained in RPMI (Thermo Fisher, Waltham, MA, USA) with 10% fetal calf serum (FCS) (RPMI CM). 624 MEL and GPRTG were maintained in DMEM (Thermo Fisher) with 10% FCS (DMEM CM). PG-13 cells were maintained in Iscove’s modified DMEM (Thermo Fisher) with 10% FCS (IMDMEM CM). After transduction, transduced T cells were maintained in culture in RPMI CM with 300 IU/mL rhIL-2 and 100 ng/mL rhIL-15 unless otherwise noted. When cultured without cytokines, T cells were cultured in RPMI CM alone. When cultured with HDAC inhibitors or methyltransferase inhibitors, T cells were cultured with 1 mM sodium butyrate, 1 μM SAHA, or 130 nM 5-aza-2-deoxycytidine.
Flow cytometry
Extracellular staining
T cells were stained with fluorescently labeled antibodies to human CD3, CD4, CD8, CD34, CD14, CD19 (Biolegend, San Diego, CA, USA), and TCR-Vβ12 (Beckman Coulter, Indianapolis, IN, USA), washed three times in PBS with 2% bovine serum albumin (2% PBSA), then resuspended in 2% PBSA for analysis on the flow cytometer. Live/dead differentiation was determined either using Live/Dead Fixable Aqua (Thermo Fisher Scientific, Waltham, MA, USA) after extracellular staining or by adding DAPI (Millipore Sigma, St. Louis, MO, USA) to a final concentration of 2 μg/mL immediately prior to analyzing samples on the flow cytometer. All samples were analyzed in the Loyola University Medical Center Flow Cytometry Core, on either an LSR Fortessa (BD Biosciences, San Jose, CA, USA) or a FACSCanto (BD Biosciences), and analyzed on FlowJo Software (BD Biosciences).
Intracellular cytokine staining
T cells were stimulated overnight in RPMI CM with either T2 cells pulsed with 10 μg/mL tyrosinase peptide (T2/tyrosinase) or a tyrosinase-expressing HLA-A2+ melanoma line (624 MEL) in the presence of Golgi blockers Brefeldin A (BFA, Biolegend) and Monensin (Biolegend). Fluorescently labeled anti-CD107a antibody (Biolegend) was also added to the coculture to stain for lytic granule release. After the culture, cells were washed and stained extracellularly with labeled antibodies to CD3, CD4, CD8, and CD34, then stained with Live/Dead Fixable Aqua as described above. After extracellular staining, cells were fixed and permeabilized using Fixation Buffer (Biolegend) and Intracellular Staining Permeabilization Wash Buffer (Perm/Wash Buffer, Biolegend) and stained intracellularly with fluorescently labeled antibodies specific for IL-2, IFNγ, and TNFα (Biolegend). Stained cells were then washed twice in Perm/Wash buffer and once in 2% PBSA prior to resuspension in 2% PBSA for analysis. Results of intracellular cytokine staining were assessed on an LSR Fortessa flow cytometer (BD Biosciences, San Jose, CA, USA), and analyzed using FlowJo software (BD Biosciences).
Transduction, CD34-isolation, and rapid expansion
Patient or healthy donor PBMCs were isolated by apheresis, and then red blood cells were removed using Ficoll (Cytiva Life Sciences, Marlborough, MA, USA) density gradient separation. T cells were activated by culturing PBMCs in RPMI CM with 50 ng/mL anti-CD3 antibody (OKT3, CD3 Pure, Miltenyi Biotec), 300 IU/mL rhIL-2 (Aldesleukin, Novartis), and 100 ng/mL rhIL-15 (Biological Resources Branch, NCI) for 3 days. Activated T cells were then transduced by spinoculation with lentiviral (pLVX-TIL 1383I-CD34t) or retroviral (SAMEN-TIL 1383I-CD34t) supernatants in the presence of 8 μg/mL polybrene (Millipore Sigma, St. Louis, MO, USA). Post-transduction, T cells were maintained in media containing rhIL-2 and rhIL-15 unless otherwise noted. After transduction, successfully transduced T cells were positively selected by CD34-binding magnetic bead isolation (Miltenyi Biotec) of CD34-expressing cells. Isolated CD34+ T cells were expanded using a rapid expansion protocol (REP). For the REP, T cells and pooled irradiated (5,000 Rads) feeder PBMC from at least three donors were cultured in RPMI CM with 30 ng/mL anti-CD3 antibody (OKT3, Miltenyi Biotec), 300 IU/mL rhIL-2 (Aldesleukin, Novartis) and 100 ng/mL rhIL-15 (Biological Resources Branch, NCI) for 10 days, after which T cells were used in experiments or cryopreserved for later use.
Acknowledgments
The authors would like to acknowledge their funding sources kindly provided by the National Cancer Institute (NCI) of the National Institutes of Health (NIH). These include grants R01CA90873 (M.I.N.), R01CA104947 (M.I.N.), R01CA104947-S1 (M.I.N.), P01CA154778 (M.I.N.), and R01AI129543-01 (M.I.N.). All flow cytometry was performed in the Loyola University Medical Center Flow Cytometry Core Facility with the support of Patricia Simms.
Author contributions
Conceptualization: T.V.M. and M.I.N.; methodology: T.V.M, G.M.S., and M.I.N.; investigation: T.V.M, G.M.S., M.D., S.Y.W., A.V.D., C.R.W., K.A.H., J.J.S., C.F., J.F., T.M., M.T., and D.M.; resources: C.V.G., B.D.C., J.I.C., B.A.F., and M.I.N.; writing – original draft: T.V.M. and M.I.N.; writing – review & editing: T.V.M., A.V.D., S.Y.W., and M.I.N.; supervision: M.I.N.; project administration: T.V.M., G.M.S., and M.I.N.; funding acquisition: M.I.N.
Declaration of interests
T.V.M. G.M.S., M.D., S.Y.W., A.V.D, C.R.W., K.A.H., J.J.S., C.V.G., C.F., J.F., T.M., M.T., D.M., B.D.C. B.A.F., and M.I.N. have no conflicts of interest to disclose. J.I.C. is on the speakers’ bureau for Bristol Meyers Squibb (BMS) and Merck; is a consultant for Clinigen; receives research support (to institution) from BMS, Prometheus, AVEO, and Roche/Genentech; and has a family member employed full time by BMS.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.01.014.
Supplemental information
References
- 1.Henley S.J., Ward E.M., Scott S., Ma J., Anderson R.N., Firth A.U., Thomas C.C., Islami F., Weir H.K., Lewis D.R. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer. 2020;126:2225–2249. doi: 10.1002/cncr.32802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Yi M., Jiao D., Xu H., Liu Q., Zhao W., Han X., Wu K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer. 2018;17:129. doi: 10.1186/s12943-018-0864-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg S.A., Packard B.S., Aebersold P.M., Solomon D., Topalian S.L., Toy S.T., Simon P., Lotze M.T., Yang J.C., Seipp C.A. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988;319:1676–1680. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 5.Duong C.P., Yong C.S., Kershaw M.H., Slaney C.Y., Darcy P.K. Cancer immunotherapy utilizing gene-modified T cells: From the bench to the clinic. Mol. Immunol. 2015;67(2 Pt A):46–57. doi: 10.1016/j.molimm.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 6.Bailey S.R., Maus M.V. Gene editing for immune cell therapies. Nat. Biotechnol. 2019;37:1425–1434. doi: 10.1038/s41587-019-0137-8. [DOI] [PubMed] [Google Scholar]
- 7.Scholler J., Brady T.L., Binder-Scholl G., Hwang W.T., Plesa G., Hege K.M., Vogel A.N., Kalos M., Riley J.L., Deeks S.G. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore T., Wagner C.R., Scurti G.M., Hutchens K.A., Godellas C., Clark A.L., Kolawole E.M., Hellman L.M., Singh N.K., Huyke F.A. Clinical and immunologic evaluation of three metastatic melanoma patients treated with autologous melanoma-reactive TCR-transduced T cells. Cancer Immunol. Immunother. 2018;67:311–325. doi: 10.1007/s00262-017-2073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan R.A., Dudley M.E., Wunderlich J.R., Hughes M.S., Yang J.C., Sherry R.M., Royal R.E., Topalian S.L., Kammula U.S., Restifo N.P. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis J.L., Theoret M.R., Zheng Z., Lamers C.H., Rosenberg S.A., Morgan R.A. Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin. Cancer Res. 2010;16:5852–5861. doi: 10.1158/1078-0432.CCR-10-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson L.A., Morgan R.A., Dudley M.E., Cassard L., Yang J.C., Hughes M.S., Kammula U.S., Royal R.E., Sherry R.M., Wunderlich J.R. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robbins P.F., Kassim S.H., Tran T.L., Crystal J.S., Morgan R.A., Feldman S.A., Yang J.C., Dudley M.E., Wunderlich J.R., Sherry R.M. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin. Cancer Res. 2015;21:1019–1027. doi: 10.1158/1078-0432.CCR-14-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burns W.R., Zheng Z., Rosenberg S.A., Morgan R.A. Lack of specific gamma-retroviral vector long terminal repeat promoter silencing in patients receiving genetically engineered lymphocytes and activation upon lymphocyte restimulation. Blood. 2009;114:2888–2899. doi: 10.1182/blood-2009-01-199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chodon T., Comin-Anduix B., Chmielowski B., Koya R.C., Wu Z., Auerbach M., Ng C., Avramis E., Seja E., Villanueva A. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin. Cancer Res. 2014;20:2457–2465. doi: 10.1158/1078-0432.CCR-13-3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abate-Daga D., Hanada K., Davis J.L., Yang J.C., Rosenberg S.A., Morgan R.A. Expression profiling of TCR-engineered T cells demonstrates overexpression of multiple inhibitory receptors in persisting lymphocytes. Blood. 2013;122:1399–1410. doi: 10.1182/blood-2013-04-495531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgan R.A., Chinnasamy N., Abate-Daga D., Gros A., Robbins P.F., Zheng Z., Dudley M.E., Feldman S.A., Yang J.C., Sherry R.M. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenberg S.A., Yang J.C., Sherry R.M., Kammula U.S., Hughes M.S., Phan G.Q., Citrin D.E., Restifo N.P., Robbins P.F., Wunderlich J.R. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lens M.B., Dawes M. Interferon alfa therapy for malignant melanoma: a systematic review of randomized controlled trials. J. Clin. Oncol. 2002;20:1818–1825. doi: 10.1200/JCO.2002.07.070. [DOI] [PubMed] [Google Scholar]
- 19.Kucera R., Topolcan O., Treskova I., Kinkorova J., Windrichova J., Fuchsova R., Svobodova S., Treska V., Babuska V., Novak J., Smejkal J. Evaluation of IL-2, IL-6, IL-8 and IL-10 in Malignant Melanoma Diagnostics. Anticancer Res. 2015;35:3537–3541. [PubMed] [Google Scholar]
- 20.Tallerico R., Cristiani C.M., Staaf E., Garofalo C., Sottile R., Capone M., Pico de Coaña Y., Madonna G., Palella E., Wolodarski M. IL-15, TIM-3 and NK cells subsets predict responsiveness to anti-CTLA-4 treatment in melanoma patients. OncoImmunology. 2016;6:e1261242. doi: 10.1080/2162402X.2016.1261242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dudley M.E., Yang J.C., Sherry R., Hughes M.S., Royal R., Kammula U., Robbins P.F., Huang J., Citrin D.E., Leitman S.F. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallen H., Thompson J.A., Reilly J.Z., Rodmyre R.M., Cao J., Yee C. Fludarabine modulates immune response and extends in vivo survival of adoptively transferred CD8 T cells in patients with metastatic melanoma. PLoS ONE. 2009;4:e4749. doi: 10.1371/journal.pone.0004749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klausner R.D., Lippincott-Schwartz J., Bonifacino J.S. The T cell antigen receptor: insights into organelle biology. Annu. Rev. Cell Biol. 1990;6:403–431. doi: 10.1146/annurev.cb.06.110190.002155. [DOI] [PubMed] [Google Scholar]
- 24.Grigoryev S.A., Nikitina T., Pehrson J.R., Singh P.B., Woodcock C.L. Dynamic relocation of epigenetic chromatin markers reveals an active role of constitutive heterochromatin in the transition from proliferation to quiescence. J. Cell Sci. 2004;117:6153–6162. doi: 10.1242/jcs.01537. [DOI] [PubMed] [Google Scholar]
- 25.Setterfield G., Hall R., Bladon T., Little J., Kaplan J.G. Changes in structure and composition of lymphocyte nuclei during mitogenic stimulation. J. Ultrastruct. Res. 1983;82:264–282. doi: 10.1016/s0022-5320(83)80014-8. [DOI] [PubMed] [Google Scholar]
- 26.Tu W.J., Hardy K., Sutton C.R., McCuaig R., Li J., Dunn J., Tan A., Brezar V., Morris M., Denyer G. Priming of transcriptional memory responses via the chromatin accessibility landscape in T cells. Sci. Rep. 2017;7:44825. doi: 10.1038/srep44825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siurala M., Havunen R., Saha D., Lumen D., Airaksinen A.J., Tähtinen S., Cervera-Carrascon V., Bramante S., Parviainen S., Vähä-Koskela M. Adenoviral Delivery of Tumor Necrosis Factor-α and Interleukin-2 Enables Successful Adoptive Cell Therapy of Immunosuppressive Melanoma. Mol. Ther. 2016;24:1435–1443. doi: 10.1038/mt.2016.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foley K.C., Spear T.T., Murray D.C., Nagato K., Garrett-Mayer E., Nishimura M.I. HCV T Cell Receptor Chain Modifications to Enhance Expression, Pairing, and Antigen Recognition in T Cells for Adoptive Transfer. Mol. Ther. Oncolytics. 2017;5:105–115. doi: 10.1016/j.omto.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Serrao E., Engelman A.N. Sites of retroviral DNA integration: From basic research to clinical applications. Crit. Rev. Biochem. Mol. Biol. 2016;51:26–42. doi: 10.3109/10409238.2015.1102859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu H., Rhodes M., Wiest D.L., Vignali D.A. On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity. 2000;13:665–675. doi: 10.1016/s1074-7613(00)00066-2. [DOI] [PubMed] [Google Scholar]
- 31.D’Oro U., Munitic I., Chacko G., Karpova T., McNally J., Ashwell J.D. Regulation of constitutive TCR internalization by the zeta-chain. J. Immunol. 2002;169:6269–6278. doi: 10.4049/jimmunol.169.11.6269. [DOI] [PubMed] [Google Scholar]
- 32.Akl H., Badran B., Dobirta G., Manfouo-Foutsop G., Moschitta M., Merimi M., Burny A., Martiat P., Willard-Gallo K.E. Progressive loss of CD3 expression after HTLV-I infection results from chromatin remodeling affecting all the CD3 genes and persists despite early viral genes silencing. Virol. J. 2007;4:85. doi: 10.1186/1743-422X-4-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Segura I., Delmelle-Wibaut C., Janssens M., Cleuter Y., van den Broeke A., Kettmann R., Willard-Gallo K.E. Human immunodeficiency virus type 2 produces a defect in CD3-gamma gene transcripts similar to that observed for human immunodeficiency virus type 1. J. Virol. 1999;73:5207–5213. doi: 10.1128/jvi.73.6.5207-5213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furusawa Y., Obata Y., Fukuda S., Endo T.A., Nakato G., Takahashi D., Nakanishi Y., Uetake C., Kato K., Kato T. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 35.Säemann M.D., Böhmig G.A., Osterreicher C.H., Burtscher H., Parolini O., Diakos C., Stöckl J., Hörl W.H., Zlabinger G.J. Anti-inflammatory effects of sodium butyrate on human monocytes: potent inhibition of IL-12 and up-regulation of IL-10 production. FASEB J. 2000;14:2380–2382. doi: 10.1096/fj.00-0359fje. [DOI] [PubMed] [Google Scholar]
- 36.Nastasi C., Fredholm S., Willerslev-Olsen A., Hansen M., Bonefeld C.M., Geisler C., Andersen M.H., Ødum N., Woetmann A. Butyrate and propionate inhibit antigen-specific CD8+ T cell activation by suppressing IL-12 production by antigen-presenting cells. Sci. Rep. 2017;7:14516. doi: 10.1038/s41598-017-15099-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson S.K., DeLoose A., Gilbert K.M. The ability of antigen, but not interleukin-2, to promote n-butyrate-induced T helper 1 cell anergy is associated with increased expression and altered association patterns of cyclin-dependent kinase inhibitors. Immunology. 2002;106:486–495. doi: 10.1046/j.1365-2567.2002.01457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kespohl M., Vachharajani N., Luu M., Harb H., Pautz S., Wolff S., Sillner N., Walker A., Schmitt-Kopplin P., Boettger T. The Microbial Metabolite Butyrate Induces Expression of Th1-Associated Factors in CD4+ T Cells. Front. Immunol. 2017;8:1036. doi: 10.3389/fimmu.2017.01036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luu M., Weigand K., Wedi F., Breidenbend C., Leister H., Pautz S., Adhikary T., Visekruna A. Regulation of the effector function of CD8+ T cells by gut microbiota-derived metabolite butyrate. Sci. Rep. 2018;8:14430. doi: 10.1038/s41598-018-32860-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao K., Wang G., Li W., Zhang L., Wang R., Huang Y., Du L., Jiang J., Wu C., He X. Histone deacetylase inhibitors prevent activation-induced cell death and promote anti-tumor immunity. Oncogene. 2015;34:5960–5970. doi: 10.1038/onc.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bachem A., Makhlouf C., Binger K.J., de Souza D.P., Tull D., Hochheiser K., Whitney P.G., Fernandez-Ruiz D., Dähling S., Kastenmüller W. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity. 2019;51:285–297.e5. doi: 10.1016/j.immuni.2019.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Hicks K.C., Knudson K.M., Lee K.L., Hamilton D.H., Hodge J.W., Figg W.D., Ordentlich P., Jones F.R., Rabizadeh S., Soon-Shjong P. Cooperative Immune-mediated Mechanisms of the HDAC Inhibitor Entinostat, an IL-15 Superagonist, and a Cancer Vaccine Effectively Synergize as a Novel Cancer Therapy. Clin. Cancer Res. 2020;26:704–716. doi: 10.1158/1078-0432.CCR-19-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laino A.S., Betts B.C., Veerapathran A., Dolgalev I., Sarnaik A., Quayle S.N., Jones S.S., Weber J.S., Woods D.M. HDAC6 selective inhibition of melanoma patient T-cells augments anti-tumor characteristics. J. Immunother. Cancer. 2019;7:33. doi: 10.1186/s40425-019-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Llopiz D., Ruiz M., Villanueva L., Iglesias T., Silva L., Egea J., Lasarte J.J., Pivette P., Trochon-Joseph V., Vasseur B. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 2019;68:379–393. doi: 10.1007/s00262-018-2283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng H., Zhao W., Yan C., Watson C.C., Massengill M., Xie M., Massengill C., Noyes D.R., Martinez G.V., Afzal R. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2016;22:4119–4132. doi: 10.1158/1078-0432.CCR-15-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mazzone R., Zwergel C., Mai A., Valente S. Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin. Epigenetics. 2017;9:59. doi: 10.1186/s13148-017-0358-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mann B.S., Johnson J.R., He K., Sridhara R., Abraham S., Booth B.P., Verbois L., Morse D.E., Jee J.M., Pope S. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin. Cancer Res. 2007;13:2318–2322. doi: 10.1158/1078-0432.CCR-06-2672. [DOI] [PubMed] [Google Scholar]
- 48.Conlon K.C., Potter E.L., Pittaluga S., Lee C.R., Miljkovic M.D., Fleisher T.A., Dubois S., Bryant B.R., Petrus M., Perera L.P. IL15 by Continuous Intravenous Infusion to Adult Patients with Solid Tumors in a Phase I Trial Induced Dramatic NK-Cell Subset Expansion. Clin. Cancer Res. 2019;25:4945–4954. doi: 10.1158/1078-0432.CCR-18-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Margolin K., Morishima C., Velcheti V., Miller J.S., Lee S.M., Silk A.W., Holtan S.G., Lacroix A.M., Fling S.P., Kaiser J.C. Phase I Trial of ALT-803, A Novel Recombinant IL15 Complex, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018;24:5552–5561. doi: 10.1158/1078-0432.CCR-18-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wrangle J.M., Velcheti V., Patel M.R., Garrett-Mayer E., Hill E.G., Ravenel J.G., Miller J.S., Farhad M., Anderton K., Lindsey K. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018;19:694–704. doi: 10.1016/S1470-2045(18)30148-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Norell H., Zhang Y., McCracken J., Martins da Palma T., Lesher A., Liu Y., Roszkowski J.J., Temple A., Callender G.G., Clay T. CD34-based enrichment of genetically engineered human T cells for clinical use results in dramatically enhanced tumor targeting. Cancer Immunol. Immunother. 2010;59:851–862. doi: 10.1007/s00262-009-0810-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salter R.D., Cresswell P. Impaired assembly and transport of HLA-A and -B antigens in a mutant TxB cell hybrid. EMBO J. 1986;5:943–949. doi: 10.1002/j.1460-2075.1986.tb04307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rivoltini L., Barracchini K.C., Viggiano V., Kawakami Y., Smith A., Mixon A., Restifo N.P., Topalian S.L., Simonis T.B., Rosenberg S.A. Quantitative correlation between HLA class I allele expression and recognition of melanoma cells by antigen-specific cytotoxic T lymphocytes. Cancer Res. 1995;55:3149–3157. [PMC free article] [PubMed] [Google Scholar]
- 54.Throm R.E., Ouma A.A., Zhou S., Chandrasekaran A., Lockey T., Greene M., De Ravin S.S., Moayeri M., Malech H.L., Sorrentino B.P., Gray J.T. Efficient construction of producer cell lines for a SIN lentiviral vector for SCID-X1 gene therapy by concatemeric array transfection. Blood. 2009;113:5104–5110. doi: 10.1182/blood-2008-11-191049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller A.D., Garcia J.V., von Suhr N., Lynch C.M., Wilson C., Eiden M.V. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J. Virol. 1991;65:2220–2224. doi: 10.1128/jvi.65.5.2220-2224.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.