

Abstract
Metastatic medullary thyroid cancer (MTC) is a rare but often aggressive thyroid malignancy with a 5-year survival rate of less than 40% and few effective therapeutic options. Adoptive T cell immunotherapy using chimeric antigen receptor (CAR)-modified T cells (CAR Ts) is showing encouraging results in the treatment of cancer, but development is challenged by the availability of suitable target antigens. We identified glial-derived neurotrophic factor (GDNF) family receptor alpha 4 (GFRα4) as a putative antigen target for CAR-based therapy of MTC. We show that GFRα4 is highly expressed in MTC, in parafollicular cells within the thyroid from which MTC originates, and in normal thymus. We isolated two single-chain variable fragments (scFvs) targeting GFRα4 isoforms a and b by antibody phage display. CARs bearing the CD3ζ and the CD137 costimulatory domains were constructed using these GFRα4-specific scFvs. GFRα4-specific CAR Ts trigger antigen-dependent cytotoxicity and cytokine production in vitro, and they are able to eliminate tumors derived from the MTC TT cell line in an immunodeficient mouse xenograft model of MTC. These data demonstrate the feasibility of targeting GFRα4 by CAR T and support this antigen as a promising target for adoptive T cell immunotherapy and other antibody-based therapies for MTC.
Keywords: Medullary thyroid carcinoma, immunotherapy, GFRa4, CAR T cells, RETMC
Graphical Abstract
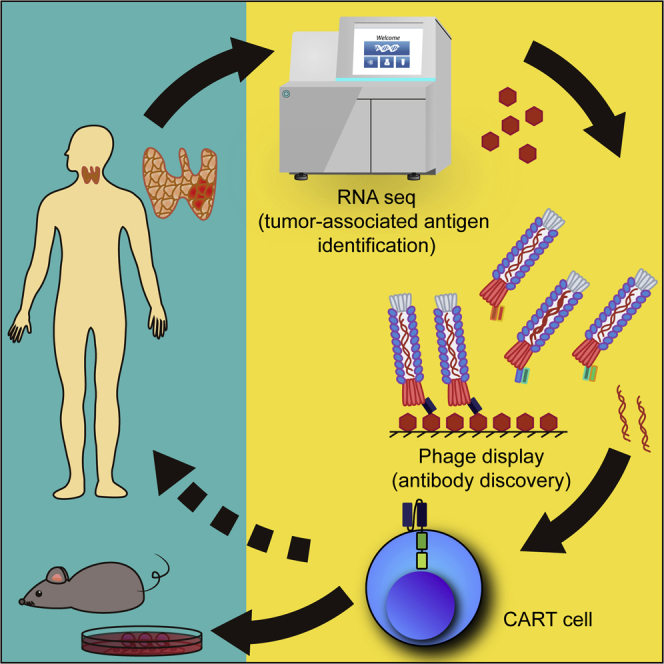
Metastatic medullary thyroid carcinoma (MTC) is a rare but aggressive malignancy with no curative options. By sequencing mRNA from a patient’s tumor, Siegel and colleagues identified and characterized GFRα4 as a promising tumor target antigen and demonstrated that CAR T immunotherapy targeting this antigen controlled MTC in tumor xenograft models.
Introduction
Medullary thyroid carcinoma (MTC) is the third most common thyroid malignancy, with both sporadic and hereditary forms.1,2 Unlike the majority of thyroid cancers that arise from the follicular epithelium of the thyroid, MTC arises from the malignant transformation of parafollicular cells (also known as C cells) within the thyroid, which are neuroendocrine cells that secrete the polypeptide hormone calcitonin.1,2 The hereditary form of MTC, which comprises approximately 25% of cases, is associated with germline mutations in the RET proto-oncogene, and this form can be associated with tumors in the adrenal or parathyroid glands as part of the multiple endocrine neoplasia type 2 (MEN2) syndrome.1,2 Sporadic cases of MTC, which typically present at a more advanced stage at diagnosis compared with hereditary forms, are also frequently associated with somatic mutations within the RET proto-oncogene, supporting the underlying importance of this gene in MTC.3 While early-stage disease can be cured by total thyroidectomy, therapeutic options are limited for patients with metastatic disease, where the 5-year survival is less than 40%.2,3 Unlike in other forms of thyroid cancer of follicular origin, there is no role for therapeutic radioactive iodine in MTC. There is also a limited role for cytotoxic chemotherapy in this disease, and a role for immunotherapy is unknown; in general, the prognosis for metastatic MTC remains poor.4 Recently, several small-molecule tyrosine kinase inhibitors (TKIs) have been introduced for the treatment of metastatic MTC.5 These agents result in objective responses in only a subset of patients (27%–40%); however, the long-term durability of these responses is unclear.6,7 Thus, there remains a strong need for new therapies for MTC.
Chimeric antigen receptor (CAR) technology, in which an extracellular antigen-binding domain from an antibody is fused to cytoplasmic signaling domains of the T cell receptor and costimulatory receptors, has shown notable promise for the treatment of advanced B cell malignancies.8 When expressed on a patient’s own T cells, a CD19-specific CAR directs those T cells to specifically kill CD19 antigen-expressing cancer cells and normal B cells, leading to complete and durable remission of disease even in late-stage cancer patients.9, 10, 11 While highly effective in B cell malignancies, applications of CAR T-based therapies to other malignancies, especially solid tumors, has been hampered by the availability of suitable target antigens with expression that is limited to tumors and to tissues with replaceable functions.12,13 Early-phase studies with several CARs for the treatment of solid tumors have led to severe adverse events due to target expression on critical normal tissues.12,14
Through RNaseq analysis of MTC, we identified the RET-associated receptor of the glial-derived neurotrophic factor (GDNF) receptor (GFR) family, GFRα4, as an MTC-associated antigen with highly restricted expression in humans. There are four known GDNF receptors: GFRα1, GFRα2, GFRα3, and GFRα4.15 Unlike the other family members, which display widespread expression within the brain and central nervous system of mammals, GFRα4 expression appears restricted to normal parafollicular cells within the thyroid.16 GFRα4 is predicted to produce 3 isoforms in humans, two GPI-linked membrane-bound forms (a and b), differing by 47 amino acids in the extracellular region, and a putative secreted form, GFRα4c.16 Mice bearing deletions of GFRα4 appear healthy, with no obvious developmental abnormalities.17 GFRα4 knockout mice exhibit defects in calcitonin regulation, suggesting that GFRα4 is critical for normal parafollicular cell function; however, the role of calcitonin in normal human physiology is unknown.18 Although calcitonin infusion can induce some hypocalcemia, patients with extremely low or absent calcitonin secretion following total thyroidectomy or MTC patients with extremely high calcitonin concentrations fail to show any obvious signs of calcium or bone abnormalities.19
Based on the expression of GFRα4 by MTC and otherwise restricted expression to normal parafollicular cells, along with the observation that elimination of normal parafollicular cells through thyroidectomy does not lead to adverse clinical effects, we hypothesize that GFRα4 is a useful target antigen for CAR-based T cell immunotherapy for MTC. We therefore developed single-chain variable fragments that permit specific targeting of GFRα4 by CAR T therapy for MTC. In this report, we present the results of preclinical studies that include the feasibility of targeting this receptor via CAR along with an in-depth evaluation of GFRα4 expression in normal human tissues to support further investigation of GFRα4-specific immunotherapy in the treatment of MTC.
Results
Target identification and validation
RNA-seq analysis was performed on a tumor sample from a 49-year-old male with metastatic MTC; the patient’s tumor harbored an acquired RET-activating mutation, Glu632-Leu633 deletion, in one allele of RET20 in addition to a germline (Tyr791Phe) variant in the other allele. Gene expression of his tumor compared to publicly available normal tissue expression data generated by GTEx revealed that the gene GFRA4 was highly expressed in his MTC (Figure 1A) and that this gene is one of the highest differentially expressed genes, relative to normal tissues. Consistent with its expression in MTC, which arises from thyroid parafollicular cells, GFRα4 RNA expression also showed relative thyroid specificity by RNA in situ hybridization and qRT-PCR of normal tissues (Figures 1B–1D; Table S1). Although GFRα4 RNA was detected in testis by qPCR, this was not observed by in situ hybridization. Interestingly, a GFRα4 expression signal is also identified in testis according to the Human Protein Atlas, although the level is far lower than in normal thyroid.21 Lindahl et al.16 did not detect GFRA4 mRNA in human testis by endpoint RT-PCR. Thus, the discrepancy in our results may reflect an artifact of the qPCR assay or a level that is below the level of sensitivity of some assays (i.e., ISH, RT-PCR). Consistent with Lindahl et al.,16 we found GFRα4 mRNA expression in 7 out of 7 cases of MTC evaluated (Figure 1C; Figure S1) as well as the MTC-derived TT and MZ-CRC1 cell lines, although the level was 100-fold lower in MZ-CRC1. As expected, GFRα4 expression was seen in scattered cells within normal thyroid parenchyma but not the follicular epithelium. GFRα4 mRNA expression examined by ISH in samples from cynomolgus macaques also showed thyroid specificity and is summarized in Table S2.
Figure 1.

GFRA4 mRNA expression in normal human tissues and MTC
(A) Log-transformed gene expression of MTC sample of index patient (y axis) is plotted against log-transformed maximum expression of genes across normal tissues (x axis). (B) Relative expression of GFRA4 (isoforms a and b) across various human cell lines and normal tissues by qPCR with expression in thyroid set to 100. Representative data from two experiments are shown. Error bars indicate 1 standard deviation of the mean. (C) RNA in situ hybridization using probes against GFRA4 (isoforms a and b), DAPB (negative control), and PPIB (positive control) in the indicated FFPE samples. Two representative examples of 7 tested MTC samples are shown. MTC 1 is from index patient. (D) RNA in situ hybridization with GFRα4 probes, as in (C), in the following normal human tissues or MTC: (i) cerebellum; (ii) frontal lobe; (iii) temporal lobe; (iv) occipital lobe; (v) parietal lobe; (vi) insula; (vii) hippocampus; (viii) pons; (ix) medullar; (x) pituitary; (xi) thyroid; (xii) MTC; (xiii) thymus; (xiv) testis; (xv) kidney.
RET-driven signaling is thought to play a central role in both hereditary and sporadic forms of MTC. Indeed, TKIs are thought to control MTC growth by inhibiting RET signaling, and RET knockdown in TT cells was shown to inhibit growth in culture.22,23 Since RET is known to complex with GFR family members at the cell surface, we sought to determine if GFRα4 may be required to maintain stable RET expression, and consequent signaling, in MTC cells. If GFRα4 is required, we reasoned that the risk of tumor escape due to antigen loss following GFRα4-directed immunotherapy would be lower. However, while knockdown of RET inhibited TT cell proliferation as previously published, knockdown of GFRα4 had no effect (Figure S2).
Isolation and validation of GFRα4-specific antibody
As no antibodies were available for GFRα4 that were shown to bind well to both of its cell-surface GPI-linked isoforms, we screened a naive rabbit-human chimeric Fab library to isolate GFRα4-specific binders that could be used to generate scFv for incorporation into a CAR. After 4 rounds of panning against microplate-bound human GFRα4 isoform a or b in the presence of a 10-fold mass excess of soluble GFRα1, GFRα2, and GFRα3 proteins to avoid isolating cross-reactive antibodies, we isolated 2 unique clones, termed P4-6 and P4-10, that each bound to both isoforms of GFRα4 but not to GFRα1, GFRα2, and GFRα3 (Figure S3).
P4-6 and P4-10 Fabs were assessed for cross-reactivity to other human membrane proteins using a membrane proteome array consisting of over 4,500 unique human membrane proteins (performed by Integral Molecular, Philadelphia, PA, USA). P4-10 demonstrated reactivity against only GFRα4, whereas P4-6 showed reactivity against GFRα4 as well as SYT2, a synaptic vesicle membrane protein involved in vesicle exocytosis (Figure S4). Given its specificity, we conducted pre-clinical CAR T cell studies using P4-10.
In vitro evaluation of GFRα4-targeted CAR
A CAR consisting of the P4-10 variable domains with 4-1BB and CD3ζ signaling domains (P4-10bbz) (Figure 2A) was expressed on primary human T cells (Figure 2B) and a Jurkat cell line expressing an NFAT-driven GFP reporter construct. As expected, P4-10bbz-expressing Jurkat cells showed activation (i.e., expressed GFP) over basal levels in response to wells coated with GFRα4a recombinant protein but not wells coated with the other GFRα recombinant proteins (Figure 2C). We next investigated the potential for CAR activation by soluble GFRα4, given the potential existence of soluble GFRα4, either produced as secreted isoform, GFRα4c,16 or due to shedding of the GPI-linked forms (GFRα4a and GFRα4b). Since the serum concentration of a putative soluble GFRα4 by MTC is unknown, we used recombinant human GFRα4a-Fc at concentrations above those described for other membrane-shed tumor-associated proteins that have been targeted by CAR T cells, namely mesothelin and B cell maturation antigen (BCMA), which have been detected at ng/mL concentrations in serum.24,25 We detected no activation of P4-10bbz in reporter Jurkat cells or primary T cells by soluble GFRα4 at a concentration of 1 μg/mL and only activation of primary P4-10bbz T cells at 3 μg/mL, which is likely well above physiologic levels (Figures 2D and 2E).
Figure 2.

P4-10 CAR T cells specifically respond to GFRα4 protein in vitro
(A) Schematic of CAR construct inserted downstream of an EF1a-derived promoter within a 3rd-generation lentiviral vector plasmid. (B) Primary human T cells were activated using anti-CD3/anti-CD28-coated beads followed by transduction with lentiviral vectors encoding the indicated CAR genes or were left non-transduced (NTD). Seven days later, T cells were stained with biotinylated F(ab′)2-specifc goat anti-mouse or donkey anti-rabbit followed by streptavidin-AF647 and analysis by flow cytometry. (C) NFAT-GFP reporter Jurkat cells expressing no CAR (NTD), 19bbz, or the P4-10bbz CAR were cultured in wells that had been coated overnight with GFRα1, GFRα2, GFRα3, GFRα4, and OKT3 proteins. Following overnight culture, cells were analyzed by flow cytometry for GFP expression. A representative of 3 independent experiments is shown. (D and E) NFAT-GFP reporter Jurkat cells (D) or primary human T cells (E) expressing no CAR (NTD), or the P4-10bbz CAR were cultured in wells that had been coated overnight with OKT3 or GFRα4 (bound) or in media with soluble GFRα4 (soluble). Following overnight culture, cells were analyzed by flow cytometry for GFP expression (*p < 0.001 compared to media control, ANOVA, Dunnett’s multiple comparisons test). Representatives of 2 independent experiments each are shown. Error bars indicate 1 standard deviation of the mean.
P4-10bbz Jurkat cells were activated by both MTC cell lines, TT and MZ-CRC1, albeit less so by the latter, consistent with reduced GFRA4 mRNA levels in these cells (Figure 1B). P4-10bbz CAR T cells lysed TT and MZ-CRC1 cells (Figure 3B; Figure S5) as well as K562 cells engineered to express either membrane-bound isoform of GFRα4, but not K562 cells that express other GFR family members (Figure 3B; Figures S6A, S6B, and S6D) and produced interleukin-2 (IL-2) and interferon (IFN) γ in response to cells expressing GFRα4 (Figure 3C; Figure S6C). Interestingly, P4-10bbz Jurkat cells were also activated by the neuroblastoma cell line SY5Y, which has been described to express GFRα4,26 but were not activated by 293T cells (Figure S7).
Figure 3.

P4-10 CAR T cells specifically respond to GFRα4-expressing cells in vitro
(A) NFAT-GFP reporter Jurkat cells expressing no CAR (NTD), 19bbz, or the P4-10bbz CAR were co-cultured with the indicated stimuli overnight, followed by assessment of GFP expression in the Jurkat cells by flow cytometry (∗p < 0.05, ANOVA, Dunnett’s multiple comparisons test compared to media control). A representative of 3 independent experiments is shown. (B) Primary human T cells expressing no CAR (NTD), 19bbz, or P4-10bbz were co-cultured for approximately 18 h with 51Cr-loaded TT cells or K562 cells expressing the indicated antigens, at the indicated effector:target ratios. Following co-culture, supernatant was assessed for released radioactivity. Percentage of the lysis was calculated according to the formula: 100 × (experimental − spontaneous)/(maximum − spontaneous), where spontaneous represents radioactivity released from cultures of target cells alone and maximum represents radioactivity released by target cells lysed with 5% SDS. Error bars represent 1 standard deviation of three technical replicates, and results are representative of at least 3 independent experiments. (C) Primary human T cells expressing no CAR (NTD), 19bbz, or P4-10bbz were co-cultured for 24 h with media, Nalm6 cells, TT cells, or beads coated with anti-CD3/anti-CD28 antibodies. IL-2 and IFNγ in co-culture supernatants were measured by ELISA. All error bars indicate 1 standard deviation of three technical replicates, and results are representative of at least 3 independent experiments.
In vivo evaluation of efficacy of GFRα4 CAR
To test P4-10bbz CAR T cells in vivo, we used an MTC xenograft mouse model. Click-beetle green (CBG) luciferase was expressed in TT cells, and these cells were subcutaneously implanted into non-obese diabetic severe combined immunodeficiency-gamma (NSG) mice. Mice then developed localized solid tumors that were measured by luminescence and size. P4-10bbz CAR T cells effectively eradicated tumors in this xenograft model (Figures 4A and 4B), and antitumor activity was accompanied by robust T cell expansion (Figure 4C). To compare P4-10bbz CAR T cells with an established CD19-directed CAR (19bbz) standard,27 we ectopically expressed GFRα4b in Nalm6 cells, an acute lymphoblastic leukemia cell line that expresses CD19 (Figure S5A). 2 × 106 Nalm6-GFRα4b cells expressing CBG were purified by fluorescence-activated cell sorting (FACS), and cells were injected intravenously (i.v.) into NSG mice. Five days later, 2 × 106 CAR+ or non-CAR (NTD) T cells were injected i.v., and tumor burden was followed by luminescence imaging. Both 19bbz and P4-10bbz CAR T cells eliminated the Nalm6 signal to the limit of detection with comparable kinetics (Figure 4D). T cells in P4-10bbz-treated mice expanded more robustly compared to 19bbz-treated mice (Figure 4E), which may be related to an off-target antigen-driven stimulation (see below).
Figure 4.

P4-10bbz CAR T cells control GFRα4-expressing tumors in vivo
(A and B) NSG mice were implanted subcutaneously with 5 × 106 TT cells engineered to express click beetle green luciferase (CBG). On day 9 post-TT cell implantation, mice received 1 × 107 T cells expressing no CAR (NTD, n = 7) or P4-10bbz (n = 8). Tumor burden was assessed by bioluminescence imaging (A) and by caliper measurement (B). Each line in (A) represents an individual mouse, and curves in (B) represent mean volumes; error bars indicate 1 standard deviation. (C) On day 21 after TT injection, peripheral blood human T cells were counted by flow cytometry (*p < 0.05, Student’s t test). (D) NSG mice were injected intravenously with 2 × 106 Nalm6 cells engineered to express CBG and GFRα4. Five days later, mice were injected intravenously with 2 × 106 control T cells (NTD, n = 10), 19bbz (n = 10), or P4-10bbz (n = 10) CAR T cells, and tumor burden was assessed by bioluminescent imaging (*p < 0.05, 1-way ANOVA, Dunnett’s multiple comparisons test compared to NTD). (E) On days 13 and 20 after T cell injection, peripheral blood human T cells were counted by flow cytometry (*p < 0.05, ANOVA, Tukey’s multiple comparisons test). All error bars indicate 1 standard deviation of the mean.
Investigation of in vivo toxicity
Unexpectedly, in both TT and Nalm6 xenograft models, P4-10bbz-treated mice developed evidence of skin toxicity, which grossly involved the skin of the tail, footpads, and occasionally the ears (Figures 5A and 5B). Gross signs of toxicity typically became evident after tumor eradication, beginning with erythema and focal desquamation in the tail progressing to marked ulceration associated with purulent exudate. The footpads similarly developed marked ulceration and, in some mice, the outer ears became erythematous. Microscopic evaluation showed a progression from basal lymphocytic infiltration of the epidermis to marked lymphocytic interface dermatitis with basal epithelial cell apoptosis and satellitosis (Figure 5C). Lesions progressed to ulceration with epithelial necrosis covered by serocellular exudate containing coccoid bacteria (Figure 5D). Mild lymphocytic infiltration and epithelial cell apoptosis, although much less pronounced and without progression to ulceration, was also observed in stratified squamous epithelium of the middle ear, the squamous portions of the esophagus and stomach, and skin overlying other regions of the body. Other organs were normal or variably showed lymphocytic infiltration that was also seen in control T cell-treated mice (Table S3).
Figure 5.

Skin toxicity is directed against murine but not human keratinocytes
(A) NSG mice as shown in Figure 3A implanted subcutaneously with TT cells were injected with non-CAR T cells (top) or P4-10bbz CAR T cells (bottom). (B) Hematoxylin and eosin-stained sections of tail skin from NSG mice bearing TT cell tumors followed by treatment with P4-10bbz CAR T cells prior to gross evidence of skin toxicity (top) and after ulceration (bottom). (C) Sections of skin from an NSG mouse and from a human donor stained with P4-10 antibody. (D) NFAT-GFP reporter Jurkat cells expressing 19bbz, P4-10bbz, or PX44bbz CARs were co-cultured overnight with the indicated target cells or in wells coated with the CD3-specific antibody, OKT3. Cells were then stained with a CD69-specific antibody, and GFP and CD69 expression were analyzed by flow cytometry. A representative of 2 independent experiments is shown. (∗∗p < 0.005, ∗∗∗p < 0.0005, 2-way ANOVA with Tukey’s multiple comparisons test [statistics shown for Mu. KC groups only]). Mu. KC, murine keratinocytes; Hu. KC, human keratinocytes. All error bars indicate 1 standard deviation of the mean.
Many of the histologic features were reminiscent of graft-versus-host disease. Thus, although 19bbz and NTD cells were produced in parallel to P4-10bbz cells from the same healthy donor, we repeated the in vivo experiment using CAR T cells in which the TCR was deleted by CRISPR-Cas9 targeting TRBC (TRBCko). Additionally, we engineered TT cells to express CD19 in order to stimulate 19bbz CAR Ts in vivo. In contrast to continued tumor growth in mice treated with NTD T cells, TT-CD19+ tumors were eradicated with similar kinetics by 19bbz, P4-10bbz, and P4-10bbz+TRBCko CAR T cells (Figure S9A). However, only mice that received P4-10bbz or P4-10bbz+TRBCko CAR T cells developed the toxicity, with similar kinetics (Figure S9B). In P4-10bbz+TRBCko CAR-treated mice, T cells in the blood and infiltrating the skin remained negative for surface CD3 (Figure S10) and TCRβ (Figure S11), respectively, indicating the toxicity was TCR independent.
Thus, we focused on CAR-mediated mechanisms. Since soluble GFR family members have been reported to bind to their co-receptor, RET, in trans,28,29 we hypothesized that GFRα4 released by TT cells or Nalm6-GFRα4 cells could potentially bind to RET on mouse tissue and serve as CAR targets. However, this is unlikely, since RET is expressed widely, and such a mechanism would be expected to cause more widespread tissue toxicity; additionally, we found that P4-10bbz CAR T cells caused toxicity in NSG mice even in the absence of tumors (Figure S12). To evaluate the possibility of an “on-target off-tumor” effect, we first determined whether the P4-10bbz CAR could recognize murine GFRα4. Human P4-10bbz CAR T cells did lyse K562 cells expressing mouse GFRα4 (Figure S13). However, both qPCR (Figure S14A) and RNA ISH (Figure S16) showed no evidence of GFRA4 transcripts in mouse skin from various body regions. These data suggested that the toxicity is due to off-target reactivity. Human skin samples also demonstrated no expression of GFRA4 mRNA by qPCR despite detection of ACTB indicating adequate RNA quality (Figures S14B and S14C).
To determine if the reactivity in mouse skin is relevant to human skin, we performed IHC using P4-10 expressed as a soluble full-length immunoglobulin G (IgG). Mouse skin from the tail, flank, and ears showed diffuse staining of keratinocytes (KCs) by P4-10. In contrast, human skin from 3 of 3 tested individuals showed no staining with P4-10 antibody (Figures 5E and 5F). As expected, P4-10 stained MTC and TT cells (Figure S15). Next, we functionally tested P4-10 reactivity against murine and human KCs. Jurkat cells expressing an NFAT-driven GFP reporter were transduced to express 19bbz, P4-10bbz, or PX44bbz, a positive-control CAR directed against desmoglein 1 and desmoglein 3 on murine and human KCs.30 These CAR-reporter cells were co-cultured with K562-CD19, K562-GFRα4, and murine or human KCs cultured under in low- (0.06 mM) or high- (0.2 mM) calcium media, the latter stimulating terminal KC differentiation. After overnight co-culture, GFP and CD69 expression was measured by flow cytometry to assess CAR activation. As expected, 19bbz was activated by K562-CD19, and PX44bbz was activated by both murine and human KCs. Consistent with findings by IHC, P4-10bbz was activated by murine KCs but not by human KCs. Additionally, primary human P4-10bbz CAR T cells also did not react to human keratinocytes whereas PX44bbz CAR T cells did (Figure S17). Lastly, cytotoxicity of P4-10bbz CAR T cells was tested against a panel of primary human cells, including human keratinocytes, and showed specific lysis of only the control TT cell targets (Figure S18). Although the target antigen in mouse KCs remains unknown, the aggregate data suggest that the cross-reactive target underlying the observed toxicity in mice is likely not relevant to humans.
P4-10 humanization for clinical CAR evaluation
In order to decrease the potential immunogenicity of the rabbit-derived P4-10 scFv for use in the clinic, we humanized both the rabbit variable heavy chain (VH) and variable light chain (VL) domains by CDR grafting based on published methods.31 Two new scFvs were designed and incorporated into CAR constructs, termed CAR25 and CAR29 (Figure S19). The VL domains of both are identical and are composed of framework regions derived from the human lambda germline gene IGLV4-69∗01. The VH domains of CAR25 and CAR29 are based on human heavy chain germline framework regions of IGHV4-38-2∗02 and IGHV3-48∗03, respectively. Each of the 2 humanized VH domains as well as their common VL domain were found to have 85% amino acid sequence identity to human VH and VL germline genes, a level of humanization that is comparable to or greater than many humanized antibodies currently in clinical use.32 We measured the affinity of the rabbit P4-10 and CAR29 scFvs against human GFRα4 by surface plasmon resonance (SPR); the dissociation constant (KD) of P4-10 was 9.5 × 10−9 M and 1.14 × 10−9 M for GFRα4a and GFRα4b, respectively, and that of CAR29 was 4.87 × 10−8 M and 1.19 × 10−8 M (Figure S20). Despite a 5- to 10-fold decrease in affinity of the CAR29 scFv, we found that each of the CARs utilizing humanized P4-10 scFv was indistinguishable from the parental rabbit P4-10 CAR in terms of efficacy and specificity in vitro and in terms of in vivo efficacy and dermal toxicity (Figure S21).
Discussion
We have shown that GFRα4 is a promising target for immunotherapy to treat MTC. Consistent with findings of Lindahl et al.,16 we show, using a highly specific and sensitive RNA-ISH assay, that GFRα4 expression in humans and cynomolgus macaques is limited to parafollicular cells of the thyroid, strongly suggesting absence of protein expression. Also confirming their findings, we found GFRα4 expression in all tested cases of MTC, which derive from parafollicular cells. Others have reported GFRα4 expression in human tissues apart from the thyroid.26,33 Using confocal imaging and sub-cellular fractionation with western blot, Lee et al.33 reported plasma membrane expression of GFRα4 in normal human brain and mislocalization to the ER in glioma samples. However, using ISH, we were unable to detect GFRα4 mRNA in human and cynomolgus macaque brain samples from various brain regions. Using qPCR, we found GFRα4 signals from brain regions were 10- to 1,000-fold lower compared to normal thyroid and MTC cell lines. Their study did not specify the location of brain tissue tested or the source of anti-GFRα4 antibody used.33 The discrepancy between our findings may be due to differential sensitivity of the assays but could also be due to GFRα4 expression in an infrequent population of cells that was missed due to sampling or dilution among negative cells. Wang et al.,26,34 investigating a potential role of GFRα4 in the pathogenesis of Hirschsprung’s disease, showed decreased GFRα4 mRNA and GFRα4 protein in colonic tissue from affected subjects compared to normal individuals. However, they did not compare expression to that found in thyroid tissue.26,34 We found no evidence of GFRα4 mRNA expression in colon, including enteric ganglia using ISH and qPCR. Additionally, the antibody used in their studies reportedly detected GFRα4 protein in 293T cells. In contrast, we found no evidence of GFRα4 expression in 293T cells when tested using our sensitive P4-10bbz CAR reporter cells (Figure 3D). Another study also investigating the potential role of GFRα4 in Hirschsprung’s disease found no mutations or variants of GFRα4 associated with disease.35 Thus, it remains unclear if GFRα4 is expressed in the colon. If it is, its expression may be at low levels or in infrequent cells.
GFRα4 is predicted to occur as two different GPI-linked membrane-bound isoforms, GFRα4a and GFRα4b, and a third soluble form, GFRα4c, although secretion of the soluble protein has yet to be demonstrated convincingly.16 Using phage display, we constructed P4-10bbz, a CAR that targets both surface GPI-linked isoforms. We showed in vitro efficacy of T cells expressing P4-10bbz against target cells expressing GFRα4a or GFRα4b, but not other GFRα family members. Using a tumor xenograft model, we showed in vivo efficacy of P4-10bbz CAR T cells against MTC. In a model using Nalm6 cells expressing both CD19 and GFRα4, P4-10bbz CAR T cells eradicated antigen-positive tumor cells as effectively as the CD19-targeted “gold-standard” CAR T cell.
Tumor-escape due to antigen loss has been described following CAR T cell therapy in B cell malignancies.36,37 To address the possibility of antigen loss as a mechanism of relapse in MTC, we performed small interfering RNA (siRNA)-mediated silencing of GFRα4 in TT cells and found no defect in TT cell proliferation. Thus, antigen-loss tumor escape could occur following GFRα4-targeted therapy. However, whether GFRα4-independent growth is specific to TT cells or also a feature of primary MTC cells remains to be determined. Interestingly, we found reduced expression of RET protein, but not mRNA, following GFRα4 knockdown using two different siRNAs. The reason for this is not clear but may indicate that RET is more stable when in complex with its co-receptor GFRs; loss of GFRα4 in TT cells could result in more rapid degradation of RET protein. One limitation of our study is the relatively few MTC lines available for evaluation. We tested TT and MZ-CRC1 cells, the only two MTC cell lines of which we are aware, and found that both express GFRα4 but at very different levels, for unknown reasons. Each harbors a different RET-activating mutation, and it is unknown whether this contributes to the expression level of GFRα4, a binding partner of RET.
In vivo murine studies revealed P4-10bbz CAR-mediated toxicity affecting squamous epithelia, most prominently in the skin of the tail and foot pads. The combined results of several experiments presented suggest that the toxicity is the result of off-target reactivity of the CAR to an unknown antigen found in murine but not human keratinocytes. Although a limitation of the present study is the lack of identification of the off-target antigen, we provide multiple lines of evidence to suggest its lack of relevance to humans. This underscores limitations inherent in the in vivo models available for evaluating CAR therapies. Future work to further evaluate the safety of GFRα4-targeted immunotherapies could employ a syngeneic CAR T model targeting murine or knocked-in human GFRα4, keeping in mind, however, that murine and human GFRα4 appear to undergo differential splicing.
In summary, we have identified and validated GFRα4 as a promising therapeutic target for treatment of MTC. We have developed a GFRα4-directed CAR T cell immunotherapy that could be an effective treatment for patients with MTC, who currently have no curative options. A clinical trial to test GFRα4-directed CAR T cells for MTC is currently under development.
Materials and methods
Antibodies and flow cytometry
Expression of the FMC63-based 19bbz CAR was assessed using biotin-conjugated goat anti-mouse antibody (Jackson ImmunoResearch, West Grove, PA, USA) followed by streptavidin-PE (BD Biosciences, San Jose, CA, USA) or streptavidin-APC (BD Biosciences). P4-10bbz expression was assessed by staining with biotin-conjugated donkey anti-rabbit antibody (Jackson ImmunoResearch) followed by streptavidin-PE or streptavidin-APC. Expression of GFRα4 was assessed using recombinant IgG comprising the P4-10 rabbit variable region fused to either a rabbit, mouse, or human constant region. The rabbit construct (a kind gift from Novartis) was detected by AF647-conjugated donkey anti-rabbit (Jackson ImmunoResearch), and the mouse/human constructs (constructed by WuXi AppTec, Philadelphia, PA, USA) were detected by AF647-conjugated goat anti-mouse or -human antibodies, respectively (Jackson ImmunoResearch). Expression of GFRα1, GFRα2, and GFRα3 was assessed using polyclonal rabbit anti-GFRα1, anti-GFRα2, and anti-GFRα3 (Sino Biologicals, Wayne, PA, USA) followed by AF647-conjugated goat anti-rabbit antibody (Thermo Fisher Scientific). Human T cells in mouse blood were enumerated using anti-CD3-APC-Cy7 (BD Biosciences), anti-CD4-V450 (BD Biosciences), anti-CD8-PE (BD Biosciences), anti-CD45-APC (BD Biosciences), and CountBright Absolute Counting Beads (Thermo Fisher Scientific). After a 20-min incubation, samples were fixed using BD Pharm Lyse. For analysis of Nalm6 cells in mouse blood, red blood cells were first lysed with a 5-min incubation in ACK lysis buffer (Thermo Fisher Scientific). Samples were then washed once and stained with P4-10 antibody, anti-CD3-APC-Cy7, anti-CD45-PE (BD Biosciences), and anti-CD19 (Beckman Coulter, Atlanta, GA, USA). Following a 30-min incubation and two washes, samples were stained with AF647-conjugated goat anti-rabbit antibody for 30 min. After two washes, samples were fixed with buffered 1% paraformaldehyde. All samples were analyzed on a LSRFortessa (BD Biosciences), and data analysis was performed using FlowJo software (FlowJo).
Phage display
Antibody fragments to human GFRα4 isoform a (GFRα4a) and isoform b (GFRα4b) were isolated by phage display using a naive chimeric rabbit-human Fab M13 phage display library.38 The library comprised >10 billion independent rabbit variable regions displayed on phage as Fab fragments with human CH1 heavy chain constant regions and human light chain Cκ or Cλ constant regions. Library selections against human Fc-fusion constructs of microplate-immobilized GFRα4a and GFRα4b were performed in separate experiments and carried out as described39 in the presence of 10-fold excess soluble Fc-GFRα1, Fc-GFRα2, and Fc-GFRα3 to prevent cross-reactivity with related GDNF family alpha receptors or human Fc constant region fragments. Human GFRα1, 2, 3, and 4a were purchased from R&D Systems (Minneapolis, MN, USA), and GFRα4b was purchased from LakePharma (Belmont, CA, USA).
After four rounds of panning, individual phage-displayed Fab clones were prepared, and phage ELISA and nucleotide sequencing39 revealed 2 unique GFRα4a- and GFRα4b-binding Fabs designated P4-6 and P4-10. Immunoglobulin sequence analysis of recombinant antibodies were performed using the IMGT tools V-Quest40 and DomainGapAlign,41 available at http://www.imgt.org.
SPR analysis
Kinetic and thermodynamic parameters for scFv binding to antigen were measured on a Biacore X100 instrument (Cytiva) at 25°C using 1× HBS-EP+ (Cytiva) as running buffer. A mouse anti-human IgG CH2 monoclonal antibody (Cytiva) was immobilized on a CM5 sensor chip via standard NHS/EDC coupling methods (Cytiva) to capture the antigens hFc-GFRα4a or hFc-GFRα4b. Rabbit scFv or humanized scFv serially two-fold diluted in running buffer were then injected respectively at five different concentrations with a replicate of the lowest concentration to confirm regeneration of the sensor chip. Biacore X100 Control Software (version 2.0.1) was used to collect data and Biacore X100 Evaluation Software (version 2.0.1) to analyze data. Calculation of association (kon) and dissociation (koff) rate constants was based on a 1:1 Langmuir binding model. The equilibrium KD was calculated from koff/kon.
CAR construction
A scFv construct was synthesized from the VH and VL fragments of the P4-10 Fab nucleotide sequence in VH-VL orientation with the 15 amino acid glycine-serine linker, GGGGSGGGGSSGGGS, between the variable domains, flanked by BamH1 and Nhe1 restriction sites (Genewiz, South Plainfield, NJ, USA). These were cloned into a lentiviral plasmid vector containing a glycine-serine hinge-domain, GGGGSGGGGS, followed by a CD8 transmembrane domain and the cytoplasmic domains of 4-1BB and CD3ζ driven by the EF1α promoter.27 The PX44 CAR, directed to the keratinocyte adhesion molecules desmogleins 1 and 3, was similarly constructed by synthesis of the PX44 scFv42 flanked by BamH1 and Nhe1 followed by cloning into the CAR lentivirus vector in place of the P4-10 scFv. The human CD19 directed FMC63-based CAR has been described previously.27
CAR T cell production
T cells were activated using anti-CD3/anti-CD28 coated Dynabeads (Thermo Fisher Scientific, Waltham, MA, USA) at a bead:cell ratio of 3:1 and cultured in RPMI1640 with 10% fetal bovine serum (FBS), 10 mM HEPES, and 1% penicillin/streptomycin. Twenty-four hours after activation, the cells were transduced with lentiviruses at a MOI of 2 to 3. T cells were then expanded in media containing 100 IU/mL IL-2 (Proleukin) for 10–14 days prior to cryopreservation or use in functional studies.
T cell receptor-deficient CAR T cells were generated by T cell activation and viral transduction as described.43 On day 3 of culture, T cells were de-beaded and electroporated (360 V, 1 ms pulse, 2 mm gap cuvette) using an ECM 830 Square Wave Electroporation system (BTX, Holliston, MA, USA) at a concentration of 5–10 × 107 cells/mL in Opti-MEM with in vitro-transcribed Cas9 mRNA at a final concentration of 20 μg per 100 μL. Cas9 mRNA was generated from a pGEM vector encoding the Cas9 gene using mScript (Cellscript, Madison, WI, USA) following the manufacturer’s recommendations. Following electroporation, T cells were returned to complete media with IL-2. On day 4, T cells were electroporated under the same conditions with in vitro-transcribed gRNA targeting TRBC43 at a final concentration of 8–10 μg per 100 μL. T cells were then returned to complete media. Between days 9 and 11 of culture, TCR-positive cells were depleted using an anti-CD3 depletion kit (Miltenyi Biotec, Auburn, CA, USA). Labeled cells were serially passed through two columns over the magnet, which consistently yielded >95% surface CD3-negative cells as assessed by flow cytometry. Cells were expanded for an additional 5 days prior to cryopreservation.
Primary cells and cell lines
TT, MZ-CRC1, K562, HaCaT, and Nalm6 cells were obtained from ATCC. Jurkat cells stably expressing an NFAT-driven GFP reporter were a kind gift of Arthur Weiss (University of California, San Francisco). K562-CD19 and TT-CD19 cells were generated by transduction with a lentivirus encoding human CD19.27 K562 cells expressing GFRα1, GFRα2, and GFRα3 were constructed by transduction with lentiviruses containing GFP followed by a T2A sequence, followed by the GFRα1/2/3 coding sequences. Nalm6 cells were transduced with a lentivirus encoding CBG luciferase and, where indicated, a lentiviral vector encoding GFRα4a or GFRα4b. All cell lines were cultured in RPMI1640 containing 10% fetal bovine serum, 10 mM HEPES, and 1% penicillin/streptomycin.
Primary human T cells, collected from healthy volunteer donors, were obtained from the Human Immunology Core at the University of Pennsylvania. Bulk T cells were isolated by negative selection using RosetteSep (StemCell Technologies, Vancouver, BC, Canada).
Human primary keratinocytes isolated from human foreskin were obtained from the University of Pennsylvania Dermatology Core. Murine keratinocytes were isolated according to a published method.44
NFAT-GFP Jurkat cell reporter assay
NFAT-GFP reporter Jurkat cells were engineered to express CARs by transduction with CAR-encoding lentiviral vectors. Jurkat cells were incubated overnight in the presence of indicated target cells at a 2:1 (target:Jurkat cell) ratio or in the presence of the indicated plate-bound proteins. OKT3 (10 μg/mL, Biolegend) was adsorbed onto culture wells by overnight incubation at 4°C. GFR proteins (10 μg/mL) were absorbed overnight to wells precoated with anti-human Fc (10 μg/mL).
Tumor xenograft studies
NSG (NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ) mice were housed in the Stem Cell Xenograft Core Facility at the University of Pennsylvania under pathogen-free conditions. The experimental protocols were approved by the University of Pennsylvania IACUC.
In the Nalm6-GFRα4 model, 6- to 8-week-old NSG mice were injected with 2 × 106 Nalm6 cells transduced with GFRα4b and co-transduced with CBG luciferase emitting in the green spectrum (Chrom-Luc Green, Promega, Madison, WI, USA). These cells had been sorted to approximately 98% GFRα4b positivity. Cells were injected in a volume of 100 μL PBS via tail vein. On day 5, the mice were randomly assigned to study groups and injected with NTD, 19bbz CAR, or P4-10bbz CAR T cells at 2 × 106 cells in 100 μL PBS per mouse through the tail vein. Tumor burden was assessed by imaging anesthetized mice using a Xenogen Spectrum system and Living Image v4.2 software following intraperitoneal injection of 150 mg/kg D-luciferin (Caliper Life Sciences, Hopkinton, MA, USA). Each animal was imaged alone (for photon quantification) or in groups of up to 5 mice (for display purposes) in the anterior-posterior prone position at the same relative time point after luciferin injection (6 min). Data were collected until the midrange of the linear scale was reached (600–60,000 counts) or maximal exposure settings reached (f-stop 1, large binning, and 120 s), and then converted to photons/s/cm2/steradian to normalize each image for exposure time, f-stop, binning, and animal size.
For the TT tumor model, 6- to 8-week-old mice were subcutaneously implanted with TT cells or TT cells engineered to express CD19. Tumor volume was assessed by external caliper measurement of the greatest longitudinal diameter (length) and the greatest transverse diameter (width). Tumor volume was calculated by the formula: tumor volume = 1/2(length × width2). When tumors reached at least 100 mm3, mice were randomly assigned to study groups and received NTD T cells or indicated CAR T cells (5 × 106) through tail vein injection.
In some experiments, TT cells transduced with CBG were injected into adult NSG mice, and tumors were assessed following CAR T cell injections by bioluminescent imaging as described above for the Nalm6 model.
Chromium release assay
Target cells were loaded with 51Cr and combined with differing amounts of transduced T cells in U-bottom plates. After a 4-h incubation at 37°C, the release of free 51Cr was measured using a COBRA II automated gamma-counter (Packard Instrument Company). The percent specific lysis was calculated using the formula: % specific lysis = 100 × (experimental cpm release − spontaneous cpm release)/(total cpm release − spontaneous cpm release). All data are presented as a mean ± standard deviation of triplicate wells.
Cytokine analysis
T cells were co-incubated with target cells at a 1:1 ratio or with anti-CD3/anti-CD28 beads at a 3:1 (bead:cell) ratio for 24 h. Culture supernatant was then harvested and frozen at −80°C. IFNγ and IL-2 in supernatants were measured by ELISA (R&D Systems) following the manufacturer’s instructions. Data are presented as a mean ± standard deviation of triplicate wells.
qRT-PCR
RNA from a normal human tissue panel was obtained from Takara Clontech (Mountain View, CA, USA). Additionally, RNA was isolated from TT, MZ-CRC1, and K562 using the RNeasy Mini Kit (QIAGEN, Germantown, MD, USA). RNA from human skin samples was isolated using the RNeasy Fibrous Tissue Mini Kit (QIAGEN). cDNA was synthesized using the High-Capacity RNA-to-cDNA kit from Applied Biosystems (Foster City, CA, USA). qPCR was performed using 100 ng of the cDNA per well in 20-μL reactions. Primers for GFRα4, ACTB, and RET were purchased from Applied Biosystems. Relative expression of GFRα4 and RET were calculated using the 2−ΔΔCt method using ACTB as a reference gene.
RNA in situ hybridization
Chromogenic RNA in situ hybridization (RNAscope LS 2.5 Detection System; Advanced Cell Diagnostics, Newark, CA, USA) was performed on formalin-fixed paraffin-embedded tissue using a Leica Bond III instrument and probes to human GFRα4 (Probe-Hs-GFRA4 #417428; Advanced Cell Diagnostics). Positive (Hs-PPIB #313908; Advanced Cell Diagnostics) and negative (Hs-DapB #312038; Advanced Cell Diagnostics) control probes were used to assess RNA and tissue quality.
Immunohistochemistry
Representative samples of normal NSG mouse and human skin were investigated by immunohistochemistry using a P4-10 rabbit variable region/mouse constant region (IgG1-κ) chimeric antibody. Immunostaining was performed using a Leica BOND RXm automated platform combined with the Bond Polymer Refine Detection kit (Leica #DS9800), which allows detection of both mouse and rabbit tissue-bound IgG-based primary antibodies. This approach allowed the simultaneous detection of both the mouse and the rabbit components of the P4-10 chimeric primary antibody. For the mouse component of P4-10 antibody, isotype-matched controls were generated using the same protocol with an irrelevant mouse monoclonal primary antibody (i.e., MUM1P/IRF4, DAKO/Agilent, catalog #M7259) incubated at the same concentration of the P4-10 antibody (15 μg/mL). For the rabbit component of P4-10 chimeric antibody, the rabbit anti-mouse IgG antibody linker present in the kit served as isotype control. Subcutaneous xenografts generated in NSG mice transplanted with TT cells (ATCC CRL-1803) were used as positive controls.
For immunohistochemistry, 5-μm-thick paraffin sections were mounted on ProbeOn slides (Thermo Fisher Scientific). Briefly, after dewaxing and rehydration, sections were pretreated with the epitope retrieval BOND ER1 low pH buffer (Leica #AR9961) for 20 min at 98°C. Endogenous peroxidase was inactivated with 3% H2O2 for 10 min at room temperature (RT). Nonspecific tissue-antibody interactions were blocked with Leica PowerVision IHC/ISH Super Blocking solution (PV6122) for 30 min at RT. The same blocking solution also served as diluent for the primary antibody. Immunoreactivity was revealed with the diaminobenzidine (DAB) chromogen reaction. Slides were finally counterstained in hematoxylin, dehydrated in an ethanol series, cleared in xylene, and permanently mounted with a resinous mounting medium (Thermo Fisher Scientific ClearVue coverslipper).
RET and GFRα4 knockdown
TT cells that were engineered to express GFP were electroporated with siRNA oligonucleotides (Integrated DNA Technologies, Coralville, IA, USA) targeting GFP (positive control), RET, GFRα4, or with a non-targeting siRNA (negative control). Twenty-four hours later, cells were lysed, and mRNA was isolated for measurement of RET and GFRα4 by qPCR. Cells were also maintained in culture and counted 7 days later to assess proliferation.
Statistical analysis
Statistical analyses, as indicated in figure legends, were performed using Prism (GraphPad Software, San Diego, CA, USA).
Data and materials availability
Plasmids and lentiviral vectors encoding the scFvs and CARs as well as cell lines described in this manuscript are available to interested investigators, but their availability will depend upon the execution of a material transfer agreement with the University of Pennsylvania.
Acknowledgments
The authors thank Roger B. Cohen, MD, for critical review of this manuscript, Xiaowei Xu, MD, PhD, for advice related to toxicity evaluation, Daniel Martinez and Amy Ziober for assistance with immunohistochemistry and RNAscope studies, and Jiangtao Ren, PhD, and Yangbing Zhao, PhD, for assistance with TRBC deletion using CRISPR/Cas9. The work presented was funded through the REACT Thyroid Foundation and Tmunity Therapeutics, with additional support from Novartis. V.G.B. is funded by the Burroughs Wellcome Fund.
Author contributions
V.G.B. conceived, designed, and performed experiments; analyzed data; and wrote the manuscript. L.L., S.K., D.A., K.Z., Z.Z., S.N.C., G.G., E.R., K.M., and M.D.F. assisted with experiments and data analysis. R.S.G., H.P., and C.R. supported antibody discovery and development. D.L.S. conceived, designed, and performed experiments; analyzed data; and edited the manuscript. M.C.M. conceived and designed experiments, analyzed data, and wrote the manuscript.
Declaration of interests
D.L.S., V.G.B., C.R., R.S.G., and M.C.M. have filed a patent application on technology presented in this manuscript.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.01.012.
Contributor Information
Vijay G. Bhoj, Email: vbhoj@pennmedicine.upenn.edu.
Don L. Siegel, Email: siegeld@pennmedicine.upenn.edu.
Supplemental information
References
- 1.Pacini F., Castagna M.G., Cipri C., Schlumberger M. Medullary thyroid carcinoma. Clin. Oncol. (R. Coll. Radiol.) 2010;22:475–485. doi: 10.1016/j.clon.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Roy M., Chen H., Sippel R.S. Current understanding and management of medullary thyroid cancer. Oncologist. 2013;18:1093–1100. doi: 10.1634/theoncologist.2013-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ernani V., Kumar M., Chen A.Y., Owonikoko T.K. Systemic treatment and management approaches for medullary thyroid cancer. Cancer Treat. Rev. 2016;50:89–98. doi: 10.1016/j.ctrv.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Schlumberger M., Carlomagno F., Baudin E., Bidart J.M., Santoro M. New therapeutic approaches to treat medullary thyroid carcinoma. Nat. Clin. Pract. Endocrinol. Metab. 2008;4:22–32. doi: 10.1038/ncpendmet0717. [DOI] [PubMed] [Google Scholar]
- 5.Klein Hesselink E.N., Steenvoorden D., Kapiteijn E., Corssmit E.P., van der Horst-Schrivers A.N., Lefrandt J.D., Links T.P., Dekkers O.M. Therapy of endocrine disease: response and toxicity of small-molecule tyrosine kinase inhibitors in patients with thyroid carcinoma: a systematic review and meta-analysis. Eur. J. Endocrinol. 2015;172:R215–R225. doi: 10.1530/EJE-14-0788. [DOI] [PubMed] [Google Scholar]
- 6.Elisei R., Schlumberger M.J., Müller S.P., Schöffski P., Brose M.S., Shah M.H., Licitra L., Jarzab B., Medvedev V., Kreissl M.C. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013;31:3639–3646. doi: 10.1200/JCO.2012.48.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wells S.A., Jr., Robinson B.G., Gagel R.F., Dralle H., Fagin J.A., Santoro M., Baudin E., Elisei R., Jarzab B., Vasselli J.R. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J. Clin. Oncol. 2012;30:134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milone M.C., Bhoj V.G. The Pharmacology of T Cell Therapies. Mol. Ther. Methods Clin. Dev. 2018;8:210–221. doi: 10.1016/j.omtm.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhoj V.G., Arhontoulis D., Wertheim G., Capobianchi J., Callahan C.A., Ellebrecht C.T., Obstfeld A.E., Lacey S.F., Melenhorst J.J., Nazimuddin F. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood. 2016;128:360–370. doi: 10.1182/blood-2016-01-694356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., JULIET Investigators Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 12.Knochelmann H.M., Smith A.S., Dwyer C.J., Wyatt M.M., Mehrotra S., Paulos C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018;9:1740. doi: 10.3389/fimmu.2018.01740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei J., Han X., Bo J., Han W. Target selection for CAR-T therapy. J. Hematol. Oncol. 2019;12:62. doi: 10.1186/s13045-019-0758-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamers C.H., Sleijfer S., van Steenbergen S., van Elzakker P., van Krimpen B., Groot C., Vulto A., den Bakker M., Oosterwijk E., Debets R., Gratama J.W. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol. Ther. 2013;21:904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Airaksinen M.S., Titievsky A., Saarma M. GDNF family neurotrophic factor signaling: four masters, one servant? Mol. Cell. Neurosci. 1999;13:313–325. doi: 10.1006/mcne.1999.0754. [DOI] [PubMed] [Google Scholar]
- 16.Lindahl M., Poteryaev D., Yu L., Arumae U., Timmusk T., Bongarzone I., Aiello A., Pierotti M.A., Airaksinen M.S., Saarma M. Human glial cell line-derived neurotrophic factor receptor alpha 4 is the receptor for persephin and is predominantly expressed in normal and malignant thyroid medullary cells. J. Biol. Chem. 2001;276:9344–9351. doi: 10.1074/jbc.M008279200. [DOI] [PubMed] [Google Scholar]
- 17.Lindfors P.H., Lindahl M., Rossi J., Saarma M., Airaksinen M.S. Ablation of persephin receptor glial cell line-derived neurotrophic factor family receptor alpha4 impairs thyroid calcitonin production in young mice. Endocrinology. 2006;147:2237–2244. doi: 10.1210/en.2005-1620. [DOI] [PubMed] [Google Scholar]
- 18.Felsenfeld A.J., Levine B.S. Calcitonin, the forgotten hormone: does it deserve to be forgotten? Clin. Kidney J. 2015;8:180–187. doi: 10.1093/ckj/sfv011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mundy G.R., Guise T.A. Hormonal control of calcium homeostasis. Clin. Chem. 1999;45:1347–1352. [PubMed] [Google Scholar]
- 20.Bongarzone I., Vigano E., Alberti L., Mondellini P., Uggeri M., Pasini B., Borrello M.G., Pierotti M.A. The Glu632-Leu633 deletion in cysteine rich domain of Ret induces constitutive dimerization and alters the processing of the receptor protein. Oncogene. 1999;18:4833–4838. doi: 10.1038/sj.onc.1202848. [DOI] [PubMed] [Google Scholar]
- 21.The Human Protein Atlas. GFRA4. https://www.proteinatlas.org/search/gfra4.
- 22.Lian E.Y., Maritan S.M., Cockburn J.G., Kasaian K., Crupi M.J., Hurlbut D., Jones S.J., Wiseman S.M., Mulligan L.M. Differential roles of RET isoforms in medullary and papillary thyroid carcinomas. Endocr. Relat. Cancer. 2017;24:53–69. doi: 10.1530/ERC-16-0393. [DOI] [PubMed] [Google Scholar]
- 23.Koga K., Hattori Y., Komori M., Narishima R., Yamasaki M., Hakoshima M., Fukui T., Maitani Y. Combination of RET siRNA and irinotecan inhibited the growth of medullary thyroid carcinoma TT cells and xenografts via apoptosis. Cancer Sci. 2010;101:941–947. doi: 10.1111/j.1349-7006.2009.01484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hassan R., Remaley A.T., Sampson M.L., Zhang J., Cox D.D., Pingpank J., Alexander R., Willingham M., Pastan I., Onda M. Detection and quantitation of serum mesothelin, a tumor marker for patients with mesothelioma and ovarian cancer. Clin. Cancer Res. 2006;12:447–453. doi: 10.1158/1078-0432.CCR-05-1477. [DOI] [PubMed] [Google Scholar]
- 25.Chen H., Li M., Xu N., Ng N., Sanchez E., Soof C.M., Patil S., Udd K., Bujarski S., Cao J. Serum B-cell maturation antigen (BCMA) reduces binding of anti-BCMA antibody to multiple myeloma cells. Leuk. Res. 2019;81:62–66. doi: 10.1016/j.leukres.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 26.Wang G., Guo F., Wang H., Liu W., Zhang L., Cui M., Wu X. Downregulation of microRNA-483-5p Promotes Cell Proliferation and Invasion by Targeting GFRA4 in Hirschsprung’s Disease. DNA Cell Biol. 2017;36:930–937. doi: 10.1089/dna.2017.3821. [DOI] [PubMed] [Google Scholar]
- 27.Milone M.C., Fish J.D., Carpenito C., Carroll R.G., Binder G.K., Teachey D., Samanta M., Lakhal M., Gloss B., Danet-Desnoyers G. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paratcha G., Ledda F., Baars L., Coulpier M., Besset V., Anders J., Scott R., Ibáñez C.F. Released GFRalpha1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron. 2001;29:171–184. doi: 10.1016/s0896-6273(01)00188-x. [DOI] [PubMed] [Google Scholar]
- 29.Yang J., Runeberg-Roos P., Leppänen V.M., Saarma M. The mouse soluble GFRalpha4 receptor activates RET independently of its ligand persephin. Oncogene. 2007;26:3892–3898. doi: 10.1038/sj.onc.1210161. [DOI] [PubMed] [Google Scholar]
- 30.Ellebrecht C.T., Bhoj V.G., Nace A., Choi E.J., Mao X., Cho M.J., Di Zenzo G., Lanzavecchia A., Seykora J.T., Cotsarelis G. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353:179–184. doi: 10.1126/science.aaf6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goydel RS, Weber J, Peng H, Qi J, Soden J, Freeth J, Park H, Rader C.J Biol Chem. 2020 May 1;295(18):5995-6006. doi: 10.1074/jbc.RA120.012791. [DOI] [PMC free article] [PubMed]
- 32.Mayrhofer P., Kunert R. Nomenclature of humanized mAbs: Early concepts, current challenges and future perspectives. Hum. Antibodies. 2019;27:37–51. doi: 10.3233/HAB-180347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee K., Byun K., Hong W., Chuang H.Y., Pack C.G., Bayarsaikhan E., Paek S.H., Kim H., Shin H.Y., Ideker T., Lee B. Proteome-wide discovery of mislocated proteins in cancer. Genome Res. 2013;23:1283–1294. doi: 10.1101/gr.155499.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang G., Zhang L., Wang H., Cui M., Liu W., Liu Y., Wu X. Demethylation of GFRA4 Promotes Cell Proliferation and Invasion in Hirschsprung Disease. DNA Cell Biol. 2018;37:316–324. doi: 10.1089/dna.2017.3928. [DOI] [PubMed] [Google Scholar]
- 35.Borrego S., Fernández R.M., Dziema H., Niess A., López-Alonso M., Antiñolo G., Eng C. Investigation of germline GFRA4 mutations and evaluation of the involvement of GFRA1, GFRA2, GFRA3, and GFRA4 sequence variants in Hirschsprung disease. J. Med. Genet. 2003;40:e18. doi: 10.1136/jmg.40.3.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orlando E.J., Han X., Tribouley C., Wood P.A., Leary R.J., Riester M., Levine J.E., Qayed M., Grupp S.A., Boyer M. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018;24:1504–1506. doi: 10.1038/s41591-018-0146-z. [DOI] [PubMed] [Google Scholar]
- 37.Pillai V., Muralidharan K., Meng W., Bagashev A., Oldridge D.A., Rosenthal J., Van Arnam J., Melenhorst J.J., Mohan D., DiNofia A.M. CAR T-cell therapy is effective for CD19-dim B-lymphoblastic leukemia but is impacted by prior blinatumomab therapy. Blood Adv. 2019;3:3539–3549. doi: 10.1182/bloodadvances.2019000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng H., Nerreter T., Chang J., Qi J., Li X., Karunadharma P., Martinez G.J., Fallahi M., Soden J., Freeth J. Mining Naïve Rabbit Antibody Repertoires by Phage Display for Monoclonal Antibodies of Therapeutic Utility. J. Mol. Biol. 2017;429:2954–2973. doi: 10.1016/j.jmb.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rader C., Steinberger P., Barbas C.F., III . Chapter 10. Selection from Antibody Libraries; Chapter 11. Analysis of Selected Antibodies. In: Barbas C.F. III, Burton D.R., Scott J.K., Silverman G.J., editors. Phage Display: A Laboratory Manual. Cold Spring Harbor Laboratory Press; 2001. pp. 10.11–10.20; 11.11–11.24. [Google Scholar]
- 40.Brochet X., Lefranc M.P., Giudicelli V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008;36:W503–W508. doi: 10.1093/nar/gkn316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ehrenmann F., Kaas Q., Lefranc M.P. IMGT/3Dstructure-DB and IMGT/DomainGapAlign: a database and a tool for immunoglobulins or antibodies, T cell receptors, MHC, IgSF and MhcSF. Nucleic Acids Res. 2010;38:D301–D307. doi: 10.1093/nar/gkp946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Payne A.S., Ishii K., Kacir S., Lin C., Li H., Hanakawa Y., Tsunoda K., Amagai M., Stanley J.R., Siegel D.L. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J. Clin. Invest. 2005;115:888–899. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ren J., Zhang X., Liu X., Fang C., Jiang S., June C.H., Zhao Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. 2017;8:17002–17011. doi: 10.18632/oncotarget.15218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li F., Adase C.A., Zhang L.J. Isolation and Culture of Primary Mouse Keratinocytes from Neonatal and Adult Mouse Skin. J. Vis. Exp. 2017;125:56027. doi: 10.3791/56027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.