

Abstract
Objectives
KPC-producing Klebsiella pneumoniae (KPC-Kp) isolates commonly co-harbour the aminoglycoside-modifying enzyme (AME) gene aac(6’)-Ib, which encodes an AME that can confer resistance to some of the commercially available aminoglycosides. We sought to determine the influence of AAC(6’)-Ib in KPC-Kp on the pharmacodynamic activity of aminoglycosides.
Methods
Six KPC-Kp clinical isolates, three with and three without aac(6’)-Ib, were analysed. Using these isolates, the bacterial killing of amikacin, gentamicin and tobramycin was assessed in static time–kill experiments. The pharmacodynamic activity of the aminoglycosides was then assessed in a dynamic one-compartment infection model over 72 h using simulated human pharmacokinetics of once-daily dosing with amikacin (15 mg/kg), gentamicin (5 mg/kg) and tobramycin (5 mg/kg).
Results
At clinically relevant aminoglycoside concentrations in time–kill experiments and the dynamic one-compartment model, gentamicin was more active than amikacin or tobramycin against the isolates harbouring aac(6’)-Ib. Amikacin, gentamicin and tobramycin all showed progressively reduced bacterial killing with exposure to repeated doses against most isolates in the dynamic one-compartment model. MIC values were generally not a good predictor of gentamicin pharmacodynamic activity against KPC-Kp, but were more reliable for amikacin and tobramycin.
Conclusions
Gentamicin may be preferred over amikacin or tobramycin for treatment of KPC-Kp infections. However, gentamicin MICs are not a consistent predictor of its pharmacodynamic activity and unexpected treatment failures are possible.
Introduction
Carbapenem-resistant Enterobacterales (CRE) are a serious public health threat, since they are associated with substantial morbidity and mortality.1 Among CRE, the KPC enzyme is the most common cause of carbapenem resistance in the USA and is often acquired through horizontal transmission on mobile genetic elements, such as plasmids.2–4 Although new antimicrobials with activity against KPC-producing Klebsiella pneumoniae (KPC-Kp) are available, infections caused by KPC-Kp are still associated with mortality rates up to 45% and may require treatment with a combination antimicrobial regimen.5–9
Aminoglycoside-based combinations remain a potentially effective treatment option, but the role of aminoglycosides for KPC-Kp infections has not been fully determined.9–12 One major concern is that KPC plasmids often co-harbour the genes that encode aminoglycoside-modifying enzymes (AMEs), such that KPC-Kp may be resistant to both carbapenems and certain aminoglycosides.13 AMEs, which are the most common and clinically relevant determinant of aminoglycoside resistance in K. pneumoniae, elicit resistance by acetylating, phosphorylating or adenylating vulnerable amino- or hydroxyl- groups of aminoglycoside antibiotics.14 The most common clinically relevant AME gene co-harboured by KPC-Kp is aac(6’)-Ib, which encodes an enzyme that fully inactivates amikacin and tobramycin and only partially inactivates gentamicin formulations (inactivates gentamicin components C1a and C2, but not gentamicin C1) by acetylation.
Although >90% of KPC-Kp isolates possess aac(6’)-Ib, the clinical impact of this AME on aminoglycoside therapy has not been clearly defined.15 Among isolates with aac(6’)-Ib, there is often discordance between predicted aminoglycoside resistance based on genotype and observed resistance based on phenotypic MICs. For example, although AAC(6’)-Ib inactivates amikacin in vitro, it is still found in the majority of carbapenem-resistant K. pneumoniae that are considered susceptible to amikacin.15,16 Clinically, aminoglycoside dosing generates high peak and low trough concentrations that may also induce expression of AMEs or enable aminoglycoside tolerance to form. There are no studies currently available that compare the pharmacodynamic activity of aminoglycosides against clinical KPC-Kp isolates with aac(6’)-Ib. Thus, the preferred aminoglycoside for KPC-Kp with aac(6’)-Ib remains unclear and may not be well predicted based on aminoglycoside MICs alone. Currently, there is minimal evidence to guide selection of an aminoglycoside based on genotype alone, but an improved understanding of the influence of each AME on aminoglycoside pharmacodynamics may help to facilitate implementation of rapid diagnostic tests that detect AMEs. The purpose of this study was to define the impact of aac(6’)-Ib on the pharmacodynamic activity of amikacin, gentamicin and tobramycin in clinical KPC-Kp isolates.
Methods
Bacterial isolates, WGS and antibiotic susceptibility testing
Three clinical KPC-Kp isolates with aac(6’)-Ib (NU-CRE055, 085 and 213) and three isolates without aac(6’)-Ib (NU-CRE193, 195 and 236) were used in each experiment. ATCC BAA-1705, which harbours blaKPC-2 and aac(6’)-Ib, was used as a control isolate for time–kill experiments. KPC-Kp isolates were selected that had MICs of amikacin, gentamicin and tobramycin that represent the susceptibilities of the majority of clinical isolates.15,16 Each isolate underwent WGS as previously described.17 Briefly, DNA was extracted using the Promega Maxwell 16 instrument (Madison, WI, USA). Sequencing libraries were prepared using the Illumina Nextera XT kit (Illumina, Inc., San Diego, CA, USA) and sequenced on the Illumina HiSeq platform to generate 150 bp paired-end reads. Reads were quality trimmed and Illumina adapter sequences were removed using Trimmomatic v0.32.18De novo assembly was performed using SPAdes v3.9.1.19 Quality control was performed by aligning trimmed reads to assembly contigs using the Burrows-Wheeler Alignment (‘BWA’) tool (v0.7.15).20 All contigs shorter than 200 bp or with an average fold coverage of <5× per base were removed. Genome sequences were aligned against the NCBI Bacterial Antimicrobial Resistance Reference Gene Database (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA313047) and ResFinder (http://www.genomicepidemiology.org) using BLAST, with other resistance genes identified by alignment to K. pneumoniae strain MGH78578 (CP000647.1) using CLC Sequence Viewer Version 7.8.1.
Amikacin (Lot# SLBT0718), gentamicin (Lot# SLBT5354) and tobramycin (Lot# SLBS8814) were purchased from Sigma–Aldrich (St Louis, MO, USA). MICs were determined by broth microdilution in triplicate according to CLSI guidelines.21 MICs and relevant resistance genes are shown in Table 1.
Table 1.
KPC-Kp isolates with and without the AME gene aac(6’)-Ib and their aminoglycoside MICs
| Isolate | aac(6’)-Ib genea | KPC gene | Amikacin MIC (mg/L) | Gentamicin MIC (mg/L) | Tobramycin MIC (mg/L) |
|---|---|---|---|---|---|
| NU-CRE193 | no | bla KPC-3 | 1 | 0.25 | 0.5 |
| NU-CRE195 | no | bla KPC-3 | 0.5 | 0.25 | 0.25 |
| NU-CRE236 | no | bla KPC-3 | 2 | 0.5 | 1 |
| NU-CRE055 | yes | bla KPC-3 | 32 | 0.5 | 32 |
| NU-CRE085 | yes | bla KPC-3 | 8 | 0.25 | 8 |
| NU-CRE213 | yes | bla KPC-3 | 8 | 0.25 | 8 |
Some isolates co-harboured additional AME genes not predicted to inactivate amikacin, gentamicin or tobramycin as described in Table S3.
Time–kill experiments
Static concentration time–kill experiments were conducted to initially compare the pharmacodynamic activity of amikacin, gentamicin and tobramycin against isolates of KPC-Kp with and without aac(6’)-Ib. Time–kill experiments were performed in duplicate at an inoculum of ∼108 cfu/mL, as previously described.22 In order to thoroughly quantify the concentration–response relationship in each isolate, multiplicative concentrations of amikacin (1.5, 3, 6, 12, 24, 48, 96, 192, 384 and 768 mg/L), gentamicin (0.5, 1, 2, 4, 8, 16, 32, 64, 128 and 256 mg/L) and tobramycin (0.5, 1, 2, 4, 8, 16, 32, 64, 128 and 256 mg/L) were used. Amikacin concentrations were 3× higher than gentamicin and tobramycin to account for potency differences between aminoglycosides and the higher amikacin concentrations observed in humans.23,24 Viable bacterial cell counts were performed at 0, 1, 2, 4, 6, 8 and 24 h. The lower limit of quantification (LLQ) for viable colony counting in time–kill experiments and the one-compartment model was 250 cfu/mL. Data below the LLQ are included to provide the greatest amount of information, but should be interpreted with caution.
One-compartment pharmacokinetic/pharmacodynamic models
Pharmacokinetic/pharmacodynamic studies using a one-compartment model examined the effect of simulated human drug exposures over 72 h on microbiological response. Experiments were performed in duplicate.25 Briefly, CAMHB (Becton Dickinson) was continuously pumped into sealed central reservoirs housed in a 37°C incubator. The central reservoirs contained the bacteria at an inoculum of ∼108 cfu/mL and a magnetic stir bar to ensure constant mixing and homogeneity. Viable bacterial cell counts were obtained at 0, 1, 2, 4, 6, 24, 25, 26, 28, 30, 48, 49, 50, 52, 54 and 72 h after the start of drug administration. Aminoglycosides were infused over 30 min into the central reservoir via a syringe pump (NE-1000X2; New Era Pumps Systems). Human simulated pharmacokinetic profiles of amikacin, gentamicin and tobramycin were tested against each KPC-Kp isolate using maximal FDA-approved daily doses26–28 (Table S1, available as Supplementary data at JAC Online). LC-MS/MS was used to validate the aminoglycoside concentrations in the one-compartment pharmacokinetic/pharmacodynamic models, as outlined in the Supplementary data.
Data analysis
Time–kill experiments were used to compare the dose–response effect between KPC-Kp isolates with and without aac(6’)-Ib. A Student’s t-test was used to compare mean bacterial killing at 24 h between isolates with and without aac(6’)-Ib. Data were also plotted as aminoglycoside concentration against 24 h log10 cfu/mL reduction and then fit to Hill-type models (Equation 1 to estimate the four parameters of the concentration–response relationship. To determine if variation in aminoglycoside activity was due to the difference in MIC or the presence/absence of aac(6’)-Ib, aminoglycoside concentrations were also normalized to the MIC for each respective isolate by dividing the aminoglycoside concentration by the MIC. The dependent variable, E, is the pharmacodynamic effect of the aminoglycoside concentration (C). E0 is the effect in the absence of drug, Emax is the maximal drug effect and EC50 is the concentration or concentration: MIC required to achieve 50% of maximal drug effect. H is the Hill or sigmoidicity constant.
Analysis of the pharmacodynamic data from the one-compartment model was performed using the log ratio area (LRA) to integrate bacterial killing across all timepoints, as described previously (Equation 2).29 The area under the cfu/mL versus time curve from 0 to 72 h (AUCFU) was calculated by the linear trapezoidal rule.
Results
WGS and assembly
Sequencing statistics and assembly characteristics are summarized in Table S2. Briefly, assembled genomes ranged in size between 5.45 and 5.87 Mb (average = 5.66 Mb) with an average fold coverage of 164× (range = 94–439×). The average number of contigs per assembly was 122 (range = 86–142) and the average N50, or the length of the shortest contig at 50% of the total genome length, was 162 kb (range = 137–222 kb). The complete antibiotic resistance gene profiles for each isolate are outlined in Table S3. The aac(6’)-Ib gene sequences in NU-CRE055, 085 and 213 were identical.
Time–kill experiments
Amikacin, gentamicin and tobramycin displayed a clear concentration–response relationship for KPC-Kp with and without aac(6’)-Ib and few instances of regrowth were observed (Figure 1). Based on the ATCC BAA-1705 control results, the time–kill model was well established in our laboratory (Figure S1).30 Mean bacterial killing at 24 h for the fCmax concentration of amikacin (48 mg/L) was significantly higher against KPC-Kp without aac(6’)-Ib (−5.89 log10 cfu/mL) compared with KPC-Kp with aac(6’)-Ib (−0.99 log10 cfu/mL) (P < 0.05). Tobramycin also had a significant difference in killing at the fCmax concentration (16 mg/L) for isolates without aac(6’)-Ib (−8.24 log10 cfu/mL) compared with isolates with aac(6’)-Ib, where it failed to kill (+0.46 log10 cfu/mL) (P < 0.05). However, there was no significant difference in bacterial killing at the gentamicin fCmax concentration (16 mg/L) between isolates without (−6.86 log10 cfu/mL) and with (−5.63 log10 cfu/mL) aac(6’)-Ib (P > 0.05). When compared with the MIC, the lowest aminoglycoside concentration to achieve bactericidal activity for KPC-Kp isolates without aac(6’)-Ib was between 12× and 24× MIC, between 4× and 16× MIC and between 8× and 32× MIC for amikacin, gentamicin and tobramycin, respectively. However, for KPC-Kp isolates with aac(6’)-Ib, bactericidal activity for amikacin, gentamicin and tobramycin initially occurred between ≥48× and 96× MIC, between 8× and 64× MIC and at ≥16× MIC, respectively.
Figure 1.

Activity of amikacin, gentamicin and tobramycin against KPC-Kp isolates NU-CRE193, 195 and 236 without aac(6’)-Ib (a–i) and isolates NU-CRE055, 085 and 213 with aac(6’)-Ib (j–r) in time–kill experiments over 24 h. Each line represents the mean of two duplicate runs. The LLQ for bacterial density was 250 cfu/mL.
The pharmacodynamic relationships between concentration and effect for each isolate were well described by Hill-type models (Figure 2a–c). In general, higher concentrations of amikacin, gentamicin and tobramycin were required to reach maximal bacterial killing (Emax) for isolates with aac(6’)-Ib. Therapeutically relevant concentrations of amikacin and tobramycin would only be capable of achieving half-maximal activity against KPC-Kp without aac(6’)-Ib, as the EC50 values for isolates with aac(6’)-Ib exceeded physiological concentrations. However, the gentamicin EC50 values for all isolates were within a clinically relevant range (gentamicin EC50 range across all isolates = 1.1–16.8 mg/L). The difference in pharmacodynamic activity between KPC-Kp isolates without and with aac(6’)-Ib was in large part explained by the MIC of amikacin (Figure 2d) and tobramycin (Figure 2f), where EC50 values for each genotype were not significantly different (P > 0.05) when the aminoglycoside concentration was normalized by the MIC. However, amikacin concentrations up to 768 mg/L were unable to eradicate isolates with aac(6’)-Ib, so Emax values were different. For gentamicin, the differences in MIC did not entirely explain the differences observed in bacterial killing (Figure 2e). KPC-Kp isolates without and with aac(6’)-Ib required concentrations of 9.7× and 32.1× MIC to achieve 50% of maximal drug effect, respectively, which were significantly different (P < 0.001).
Figure 2.

Analysis of the pharmacokinetic/pharmacodynamic relationship between aminoglycoside concentration and bacterial killing for KPC-Kp with (green) and without (blue) aac(6’)-Ib in time–kill experiments. Bacterial reduction at 24 h versus aminoglycoside concentration (a–c) and concentrations normalized by aminoglycoside MICs (d–f) were fit to Hill-type functions (lines). EC50 and r2 values corresponding to parameters from the Hill-type functions from the MIC normalized fits (d–f) appear below each panel.
One-compartment pharmacokinetic/pharmacodynamic models
Amikacin, gentamicin and tobramycin all displayed similar general patterns of killing against KPC-Kp in the one-compartment model (Figure 3). After each aminoglycoside dose, there was a reduction in bacterial viability for approximately 4–6 h, followed by a period of regrowth prior to the next dose. However, the extent of killing varied between aminoglycosides and was also dependent on the presence of aac(6’)-Ib. The first doses of amikacin, gentamicin and tobramycin caused mean bacterial reductions of −2.58, −3.88 and −2.18 log10 cfu/mL, respectively, across all isolates.
Figure 3.

Pharmacodynamic effects of human simulated pharmacokinetics for amikacin (a), gentamicin (b) and tobramycin (c) against KPC-Kp with aac(6’)-Ib (green) and without aac(6’)-Ib (blue) in the one-compartment pharmacokinetic/pharmacodynamic model over 72 h. Each line represents the mean of two duplicate experiments. The LLQ for bacterial density was 250 cfu/mL. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
The presence of aac(6’)-Ib diminished the activity of amikacin and tobramycin more than it impacted gentamicin. The first amikacin dose caused a mean bacterial reduction of −3.94 log10 cfu/mL for KPC-Kp without aac(6’)-Ib, whereas it only killed the KPC-Kp with aac(6’)-Ib −1.22 log10 cfu/mL. Average killing for the first tobramycin dose was −4.21 log10 cfu/mL against KPC-Kp without aac(6’)-Ib, while tobramycin had only a marginal effect on isolates with aac(6’)-Ib (−0.14 log10 cfu/mL maximum reduction). In contrast to amikacin and tobramycin, gentamicin activity was similar for isolates with (−3.33 log10 cfu/mL maximum reduction) and without (−4.44 log10 cfu/mL maximum reduction) aac(6’)-Ib.
The LRA confirmed that, on average, amikacin and tobramycin activity was diminished against KPC-Kp isolates with aac(6’)-Ib (Figure 4). However, despite this general trend, amikacin demonstrated the least overall killing against NU-CRE236 (LRA = −0.27), which was driven by amikacin’s inactivity after the second and third doses (Figure 5a). In contrast to amikacin and tobramycin, the mean LRA following exposure to gentamicin was similar for KPC-Kp isolates without aac(6’)-Ib (LRA = −0.51) and with aac(6’)-Ib (LRA = −0.56). Despite the similar mean LRA between genotypes following gentamicin exposure, there was still a large degree of inter-isolate variability in response to gentamicin even though the isolates had similar MICs (0.25–0.5 mg/L across all isolates). Elevated amikacin and tobramycin MICs were generally predictive of poorer bacterial killing.
Figure 4.
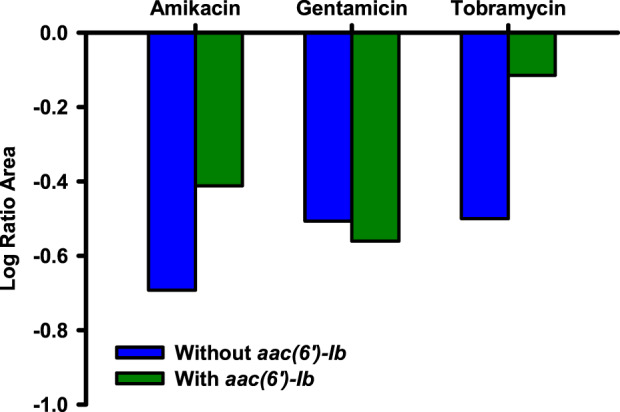
Pharmacodynamic relationship between aminoglycoside and KPC-Kp genotype. Mean LRA integrated killing over 72 h in the one-compartment model for KPC-Kp isolates without aac(6’)-Ib (blue bars) and with aac(6’)-Ib (green bars). This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Figure 5.

Maximum reduction in viable bacterial counts following the first (black bars), second (red bars) and third (grey bars) aminoglycoside doses in the one-compartment model. Amikacin (a), gentamicin (b) and tobramycin (c) were administered to KPC-Kp isolates without aac(6’)-Ib (NU-CRE193, 195 and 236) and with aac(6’)-Ib (NU-CRE055, 085 and 213). This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Interestingly, attenuated bacterial killing after the first dose of each aminoglycoside was observed for the majority of the isolates with and without aac(6’)-Ib (Figure 5). For example, the first amikacin dose against NU-CRE236 caused mean killing of −1.97 log10 cfu/mL, but the second and third doses only reduced viable bacterial counts by −0.36 and −0.29 log10 cfu/mL, respectively. Attenuation of bacterial killing was observed for all three KPC-Kp isolates without aac(6’)-Ib for all three aminoglycosides. For some KPC-Kp isolates with aac(6’)-Ib, diminished killing was also noted for amikacin (two out of three isolates) and gentamicin (one out of three isolates). The limited activity of tobramycin against any of the KPC-Kp isolates with aac(6’)-Ib prohibits comparison of killing across doses one to three.
Discussion
The AAC(6’)-Ib AME is expressed by >90% of KPC-Kp, highlighting the need to define its impact on the pharmacodynamics of aminoglycosides.15 In the current study, we found that gentamicin may be preferred over amikacin or tobramycin for treatment of KPC-Kp infections when an aminoglycoside is required, since it displayed the greatest activity against isolates with and without aac(6’)-Ib. This is consistent with a previous study in which we showed that amikacin failed to kill an amikacin-susceptible (MIC = 4 mg/L) carbapenem-resistant Escherichia coli with aac(6’)-Ib in the hollow-fibre infection model.22 In agreement with our study, previous time–kill experiments by Bremmer et al.31 also showed that amikacin activity against KPC-Kp isolates with aac(6’)-Ib was proportional to the MIC. Further, they also found that regrowth was common in the presence of clinically relevant concentrations of amikacin for susceptible KPC-Kp isolates. In a previous translational model, Caulin et al.32 demonstrated that the level of AAC(6’)-Ib expression among isogenic strains of K. pneumoniae appeared to be well correlated with MICs of aminoglycosides and the activity of amikacin or isepamicin in a time–kill model, but not in a rabbit endocarditis model. In the 9 day rabbit endocarditis model, significant reduction in bacterial density was seen in the pan-susceptible control strain lacking AAC(6’)-Ib, but the bacterial reduction in strains with AAC(6’)-Ib was more modest, suggesting that in vitro amikacin activity (MIC = 4 mg/L) may not translate to in vivo activity for infections with high bacterial burden and AAC(6’)-Ib. Plazomicin is another aminoglycoside that maintains a high degree of in vitro activity in the presence of most AMEs found within KPC-Kp isolates and may be more efficacious than amikacin, gentamicin or tobramycin against isolates with aac(6’)-Ib.33
Calls to lower the aminoglycoside susceptibility threshold have been made for over a decade, citing the unacceptably low probabilities of good clinical outcome when treating isolates with higher MICs,34,35 concerns about reaching pharmacokinetic/pharmacodynamic targets in critically ill patients36 and a dearth of safety data for the doses required to treat all isolates included in the current CLSI susceptibility breakpoints (amikacin ≤16 mg/L, gentamicin/tobramycin ≤4 mg/L).37 Other reports have shown that, among K. pneumoniae, the presence of aac(6’)-Ib with or without other AMEs may not raise the MIC of traditional aminoglycosides above the current CLSI susceptibility thresholds, thereby declaring many isolates with AMEs as aminoglycoside susceptible.38 For these reasons, the National Antimicrobial Susceptibility Testing Committee for the United States (USCAST) has recently updated its report for recommended aminoglycoside in vitro susceptibility breakpoints on the basis of preclinical efficacy data, Monte Carlo simulations and MIC distributions [susceptibility breakpoints: amikacin ≤4 mg/L, gentamicin ≤2 mg/L (gentamicin-pneumonia ≤1 mg/L), tobramycin ≤2 mg/L (tobramycin-pneumonia ≤1 mg/L)].39 EUCAST has similar breakpoints to USCAST for gentamicin and tobramycin (susceptibility breakpoint: ≤2 mg/L), but the amikacin breakpoint is higher (susceptibility breakpoint: ≤8 mg/L).40
The most important difference between USCAST and CLSI/EUCAST MIC interpretations for isolates in our study was for amikacin. Applying the USCAST breakpoints to the KPC-Kp isolates used in our study, all three of the isolates without aac(6’)-Ib would be considered susceptible to amikacin, whereas the three isolates with aac(6’)-Ib would be considered resistant to amikacin. However, according to current CLSI breakpoints, none of the isolates was resistant to amikacin (five susceptible, one intermediate). Applying EUCAST breakpoints, five isolates would also be considered susceptible to amikacin, including two of three KPC-Kp that harboured aac(6’)-Ib and were minimally killed by amikacin. Amikacin activity correlated well with the isolate’s MIC regardless of the presence of aac(6’)-Ib. In agreement with USCAST breakpoints, the time–kill analyses revealed that the EC50 values for the susceptible KPC-Kp isolates (EC50 = 3.4–45.1 mg/L) were clinically achievable and are below fCmax concentrations attained following extended-interval amikacin dosing.39 Amikacin EC50 values for the resistant KPC-Kp isolates, with aac(6’)-Ib, were much higher (EC50 = 100–637 mg/L). However, against an inoculum of 108 cfu/mL in the dynamic model, the maximum FDA-approved amikacin dose (15 mg/kg every 24 h) was only bactericidal against a single isolate. Therefore, doses of amikacin >15 mg/kg may be necessary to overcome high bacterial density infections, even for isolates with amikacin MICs ≤4 mg/L. The current study enunciates the disparity between the current aminoglycoside FDA-approved dosing and susceptibility breakpoints and provides further evidence that revisions to the CLSI and EUCAST breakpoints, particularly for amikacin, may be necessary.
Extended-interval aminoglycoside dosing, which utilizes high doses administered less frequently, can reduce nephrotoxicity by providing intervals of low drug concentrations. To the best of our knowledge, this is the first study to show diminished killing with extended-interval dosing in K. pneumoniae. Adaptive resistance to repeated aminoglycoside doses has primarily been studied in Pseudomonas aeruginosa,41–46 where it has been observed in vitro, in animal models and in patients. Additional studies have also found diminished killing by repeated exposure to aminoglycosides in E. coli43,47 and Enterobacter cloacae.43 The mechanism(s) by which the isolates in the present study adapted to aminoglycoside exposure may at least in part differ from these previous studies, since K. pneumoniae does not express the efflux pumps that contribute to adaptive resistance in P. aeruginosa.48
Attenuation of aminoglycoside activity in KPC-Kp may be caused by the formation of an aminoglycoside-resistant and/or an aminoglycoside-tolerant bacterial population. However, aminoglycoside resistance in K. pneumoniae is primarily driven by the acquisition of additional AMEs, which is not possible in our closed in vitro model. Unlike the previous studies that observed aminoglycoside adaptive resistance in other species, our study included isolates with pre-existing AMEs. Aminoglycoside exposure can induce expression of pre-existing AMEs through the presence of RNA riboswitches that can bind specific aminoglycosides and modulate downstream expression of the AME.49,50 Though future studies are required to fully investigate the mechanism, the specificity of RNA riboswitches for certain aminoglycosides may in part explain the inter-isolate variability in gentamicin activity observed for KPC-Kp with aac(6’)-Ib. For example, the KPC-Kp isolate with aac(6’)-Ib that did not respond as well to gentamicin despite susceptibility (i.e. NU-CRE055) may possess a riboswitch that is induced in the presence of gentamicin and increases expression of AAC(6’)-Ib over time. It is also possible that aminoglycoside tolerance mechanisms are responsible for the attenuated activity of repeated doses. Aminoglycoside tolerance can be caused by reduced aminoglycoside uptake by the bacterial cell.51 Bacterial cells can also activate amino acid biosynthesis to reduce aminoglycoside uptake.52 Since the isolates in the present study were not isogenic, inter-isolate variations in metabolic response to aminoglycosides may have also contributed to the isolate differences in response to gentamicin. Small-colony variants (SCVs) can also develop during aminoglycoside exposure and display aminoglycoside resistance; however, no SCVs were observed in our dynamic one-compartment experiments.
There are a few limitations to note about the present study. The first is that experiments were performed in a relatively limited number of KPC-Kp isolates. Although the isolate number we used is consistent with similar studies, future studies should evaluate additional isolates with diverse backgrounds to solidify our observations. Another limitation is that the development of a biofilm on the one-compartment pharmacokinetic/pharmacodynamic model could not be ruled out and may have also contributed to regrowth. However, we did not detect biofilm growth during any experiment.
In conclusion, gentamicin may be preferred over amikacin or tobramycin for treatment of infections caused by KPC-Kp with and without aac(6’)-Ib. For amikacin and tobramycin, the degree of bacterial killing was correlated with the aminoglycoside MIC, which is likely linked to the presence of aac(6’)-Ib. Worryingly though, the pharmacodynamic activity of gentamicin was not consistent for all isolates and its use may still lead to unexpected treatment failures, encouraging the need to optimize aminoglycoside combination regimens for KPC-Kp. Our data also suggest that USCAST breakpoints for amikacin may better separate isolates with and without aac(6’)-Ib than CLSI or EUCAST breakpoints. However, doses exceeding the approved amikacin dose may still be necessary to achieve bactericidal killing, even for isolates with MICs ≤4 mg/L. We are also the first (to the best of our knowledge) to show diminishing activity of aminoglycosides with repeat once-daily dosing in K. pneumoniae. Improved understanding of the influence of AME genes, such as aac(6’)-Ib, on aminoglycoside pharmacodynamics may help optimize the use of aminoglycosides for KPC-Kp infections.
Data availability
This Whole Genome Shotgun project has been deposited at GenBank under the accession numbers JAAUWY000000000 (K. pneumoniae NU-CRE055), JAAUWZ000000000 (K. pneumoniae NU-CRE085), JAAUXA000000000 (K. pneumoniae NU-CRE193), JAAUXB000000000 (K. pneumoniae NU-CRE195), JAAUXC000000000 (K. pneumoniae NU-CRE213) and JAAUXD000000000 (K. pneumoniae NU-CRE236).
Supplementary Material
Funding
The project was funded by the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust (to Z.P.B. and A.R.H.). This project has been funded in part by the National Institutes of Health under Grants R01AI118257, U19AI135964, K24AI04831 and R21AI129167 (all to A.R.H.). Z.P.B. was supported in part by the National Center for Advancing Translational Sciences, National Institutes of Health, under Grant KL2TR002002.
Transparency declarations
None to declare.
Disclaimer
The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Supplementary data
Supplementary data, including Tables S1 to S3 and Figure S1, are available at JAC Online.
References
- 1.CDC. Antibiotic Resistance Threats in the United States. 2013. http://www.cdc.gov/drugresistance/threat-report-2013/index.html.
- 2. Munoz-Price LS, Poirel L, Bonomo RA. et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis 2013; 13: 785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Logan LK, Weinstein RA.. The epidemiology of carbapenem-resistant Enterobacteriaceae: the impact and evolution of a global menace. J Infect Dis 2017; 215: Suppl 1: S28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramirez MS, Iriarte A, Reyes-Lamothe R. et al. Small Klebsiella pneumoniae plasmids: neglected contributors to antibiotic resistance. Front Microbiol 2019; 10: 2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ramos-Castaneda JA, Ruano-Ravina A, Barbosa-Lorenzo R. et al. Mortality due to KPC carbapenemase-producing Klebsiella pneumoniae infections: systematic review and meta-analysis: mortality due to KPC Klebsiella pneumoniae infections. J Infect 2018; 76: 438–48. [DOI] [PubMed] [Google Scholar]
- 6. Wunderink RG, Giamarellos-Bourboulis EJ, Rahav G. et al. Effect and safety of meropenem-vaborbactam versus best-available therapy in patients with carbapenem-resistant Enterobacteriaceae infections: the TANGO II randomized clinical trial. Infect Dis Ther 2018; 7: 439–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tumbarello M, Trecarichi EM, Corona A. et al. Efficacy of ceftazidime-avibactam salvage therapy in patients with infections caused by Klebsiella pneumoniae carbapenemase-producing K. pneumoniae. Clin Infect Dis 2019; 68: 355–64. [DOI] [PubMed] [Google Scholar]
- 8. Rodriguez-Bano J, Gutierrez-Gutierrez B, Machuca I. et al. Treatment of infections caused by extended-spectrum-β-lactamase-, AmpC-, and carbapenemase-producing Enterobacteriaceae. Clin Microbiol Rev 2018; 31: e00079-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hawkey PM, Warren RE, Livermore DM. et al. Treatment of infections caused by multidrug-resistant Gram-negative bacteria: report of the British Society for Antimicrobial Chemotherapy/Healthcare Infection Society/British Infection Association Joint Working Party. J Antimicrob Chemother 2018; 73 Suppl 3: iii2–78. [DOI] [PubMed] [Google Scholar]
- 10. Falagas ME, Lourida P, Poulikakos P. et al. Antibiotic treatment of infections due to carbapenem-resistant Enterobacteriaceae: systematic evaluation of the available evidence. Antimicrob Agents Chemother 2014; 58: 654–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shields RK, Clancy CJ, Press EG. et al. Aminoglycosides for treatment of bacteremia due to carbapenem-resistant Klebsiella pneumoniae. Antimicrob Agents Chemother 2016; 60: 3187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rafailidis PI, Falagas ME.. Options for treating carbapenem-resistant Enterobacteriaceae. Curr Opin Infect Dis 2014; 27: 479–83. [DOI] [PubMed] [Google Scholar]
- 13. Bush K. Carbapenemases: partners in crime. J Glob Antimicrob Resist 2013; 1: 7–16. [DOI] [PubMed] [Google Scholar]
- 14. Ramirez MS, Tolmasky ME.. Aminoglycoside modifying enzymes. Drug Resist Updat 2010; 13: 151–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Almaghrabi R, Clancy CJ, Doi Y. et al. Carbapenem-resistant Klebsiella pneumoniae strains exhibit diversity in aminoglycoside-modifying enzymes, which exert differing effects on plazomicin and other agents. Antimicrob Agents Chemother 2014; 58: 4443–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Galani I, Nafplioti K, Adamou P. et al. Nationwide epidemiology of carbapenem resistant Klebsiella pneumoniae isolates from Greek hospitals, with regards to plazomicin and aminoglycoside resistance. BMC Infect Dis 2019; 19: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bachta KER, Ozer EA, Pandit A. et al. Draft genome sequence of Pseudomonas aeruginosa strain BWH047, a sequence type 235 multidrug-resistant clinical isolate expressing high levels of colistin resistance. Microbiol Resour Announc 2019; 8: e00623-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bolger AM, Lohse M, Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30: 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bankevich A, Nurk S, Antipov D. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012; 19: 455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H, Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.CLSI. Performance Standards for Antimicrobial Susceptibility Testing—Twenty-Ninth Edition: M100 2019.
- 22. Bulman ZP, Chen L, Walsh TJ. et al. Polymyxin combinations combat Escherichia coli harboring mcr-1 and blaNDM-5: preparation for a postantibiotic era. MBio 2017; 8: e00540-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bailey TC, Little JR, Littenberg B. et al. A meta-analysis of extended-interval dosing versus multiple daily dosing of aminoglycosides. Clin Infect Dis 1997; 24: 786–95. [DOI] [PubMed] [Google Scholar]
- 24. Yu PK, Washington JA 2nd. Comparative in vitro activity of three aminoglycosidic antibiotics: BB-K8, kanamycin, and gentamicin. Antimicrob Agents Chemother 1973; 4: 133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bergen PJ, Tsuji BT, Bulitta JB. et al. Synergistic killing of multidrug-resistant Pseudomonas aeruginosa at multiple inocula by colistin combined with doripenem in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 2011; 55: 5685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Urban AW, Craig WA.. Daily dosage of aminoglycosides. Curr Clin Top Infect Dis 1997; 17: 236–55. [PubMed] [Google Scholar]
- 27. Cobussen M, Stassen PM, Posthouwer D. et al. Improving peak concentrations of a single dose regime of gentamicin in patients with sepsis in the emergency department. PLoS One 2019; 14: e0210012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blackburn LM, Tverdek FP, Hernandez M. et al. First-dose pharmacokinetics of aminoglycosides in critically ill haematological malignancy patients. Int J Antimicrob Agents 2015; 45: 46–53. [DOI] [PubMed] [Google Scholar]
- 29. Bulman ZP, Ly NS, Lenhard JR. et al. Influence of rhlR and lasR on polymyxin pharmacodynamics in Pseudomonas aeruginosa and implications for quorum sensing inhibition with azithromycin. Antimicrob Agents Chemother 2017; 61: e00096-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu W, Shen P, Bao Z. et al. In vitro antibacterial activity of fosfomycin combined with other antimicrobials against KPC-producing Klebsiella pneumoniae. Int J Antimicrob Agents 2017; 50: 237–41. [DOI] [PubMed] [Google Scholar]
- 31. Bremmer DN, Clancy CJ, Press EG. et al. KPC-producing Klebsiella pneumoniae strains that harbor AAC(6’)-Ib exhibit intermediate resistance to amikacin. Antimicrob Agents Chemother 2014; 58: 7597–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Caulin E, Coutrot A, Carbon C. et al. Resistance to amikacin and isepamicin in rabbits with experimental endocarditis of an aac(6')-Ib-bearing strain of Klebsiella pneumoniae susceptible in vitro. Antimicrob Agents Chemother 1996; 40: 2848–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Castanheira M, Davis AP, Serio AW. et al. In vitro activity of Plazomicin against Enterobacteriaceae isolates carrying genes encoding aminoglycoside-modifying enzymes most common in US Census divisions. Diagn Microbiol Infect Dis 2019; 94: 73–7. [DOI] [PubMed] [Google Scholar]
- 34. Drusano GL, Ambrose PG, Bhavnani SM. et al. Back to the future: using aminoglycosides again and how to dose them optimally. Clin Infect Dis 2007; 45: 753–60. [DOI] [PubMed] [Google Scholar]
- 35. Drusano GL, Louie A.. Optimization of aminoglycoside therapy. Antimicrob Agents Chemother 2011; 55: 2528–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zavascki AP, Klee BO, Bulitta JB.. Aminoglycosides against carbapenem-resistant Enterobacteriaceae in the critically ill: the pitfalls of aminoglycoside susceptibility. Expert Rev anti Infect Ther 2017; 15: 519–26. [DOI] [PubMed] [Google Scholar]
- 37. Bland CM, Pai MP, Lodise TP.. Reappraisal of contemporary pharmacokinetic and pharmacodynamic principles for informing aminoglycoside dosing. Pharmacotherapy 2018; 38: 1229–38. [DOI] [PubMed] [Google Scholar]
- 38. Landman D, Babu E, Shah N. et al. Activity of a novel aminoglycoside, ACHN-490, against clinical isolates of Escherichia coli and Klebsiella pneumoniae from New York City. J Antimicrob Chemother 2010; 65: 2123–7. [DOI] [PubMed] [Google Scholar]
- 39.National Antimicrobial Susceptibility Testing Committee for the United States (USCAST). Aminoglycoside In Vitro Susceptibility Test Interpretive Criteria Evaluations. Version 1.3. 2019. http://www.uscast.org.
- 40.EUCAST. Breakpoint Tables for Interpretation of MICs and Zone Diameters. 2020.
- 41. Barclay ML, Begg EJ, Chambers ST.. Adaptive resistance following single doses of gentamicin in a dynamic in vitro model. Antimicrob Agents Chemother 1992; 36: 1951–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Daikos GL, Jackson GG, Lolans VT. et al. Adaptive resistance to aminoglycoside antibiotics from first-exposure down-regulation. J Infect Dis 1990; 162: 414–20. [DOI] [PubMed] [Google Scholar]
- 43. Gould IM, Milne K, Harvey G. et al. Ionic binding, adaptive resistance and post-antibiotic effect of netilmicin and ciprofloxacin. J Antimicrob Chemother 1991; 27: 741–8. [DOI] [PubMed] [Google Scholar]
- 44. Daikos GL, Lolans VT, Jackson GG.. First-exposure adaptive resistance to aminoglycoside antibiotics in vivo with meaning for optimal clinical use. Antimicrob Agents Chemother 1991; 35: 117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xiong YQ, Caillon J, Kergueris MF. et al. Adaptive resistance of Pseudomonas aeruginosa induced by aminoglycosides and killing kinetics in a rabbit endocarditis model. Antimicrob Agents Chemother 1997; 41: 823–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barclay ML, Begg EJ, Chambers ST. et al. Adaptive resistance to tobramycin in Pseudomonas aeruginosa lung infection in cystic fibrosis. J Antimicrob Chemother 1996; 37: 1155–64. [DOI] [PubMed] [Google Scholar]
- 47. Mohamed AF, Nielsen EI, Cars O. et al. Pharmacokinetic-pharmacodynamic model for gentamicin and its adaptive resistance with predictions of dosing schedules in newborn infants. Antimicrob Agents Chemother 2012; 56: 179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hocquet D, Vogne C, El Garch F. et al. MexXY-OprM efflux pump is necessary for a adaptive resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob Agents Chemother 2003; 47: 1371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang S, He W, Sun W. et al. Integron-derived aminoglycoside-sensing riboswitches control aminoglycoside acetyltransferase resistance gene expression. Antimicrob Agents Chemother 2019; 63: e00236-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jia X, Zhang J, Sun W. et al. Riboswitch control of aminoglycoside antibiotic resistance. Cell 2013; 152: 68–81. [DOI] [PubMed] [Google Scholar]
- 51. Allison KR, Brynildsen MP, Collins JJ.. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 2011; 473: 216–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shan Y, Lazinski D, Rowe S. et al. Genetic basis of persister tolerance to aminoglycosides in Escherichia coli. MBio 2015; 6: e00078-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This Whole Genome Shotgun project has been deposited at GenBank under the accession numbers JAAUWY000000000 (K. pneumoniae NU-CRE055), JAAUWZ000000000 (K. pneumoniae NU-CRE085), JAAUXA000000000 (K. pneumoniae NU-CRE193), JAAUXB000000000 (K. pneumoniae NU-CRE195), JAAUXC000000000 (K. pneumoniae NU-CRE213) and JAAUXD000000000 (K. pneumoniae NU-CRE236).