

Abstract
The current unidimensional paradigm of kidney disease detection is incompatible with the complexity and heterogeneity of renal pathology. The diagnosis of kidney disease has largely focused on glomerular filtration, while assessment of kidney tubular health has notably been absent. Following insult, the kidney tubular cells undergo a cascade of cellular responses that result in the production and accumulation of low-molecular-weight proteins in the urine and systemic circulation. Modern advancements in molecular analysis and proteomics have allowed the identification and quantification of these proteins as biomarkers for assessing and characterizing kidney diseases. In this review, we highlight promising biomarkers of kidney tubular health that have strong underpinnings in the pathophysiology of kidney disease. These biomarkers have been applied to various specific clinical settings from the spectrum of acute to chronic kidney diseases, demonstrating the potential to improve patient care.
Keywords: biomarkers, acute kidney injury, AKI, chronic kidney disease, CKD, kidney
INTRODUCTION
Kidney diseases are complex and heterogeneous. Yet, clinical assessment of kidney disease largely relies largely on the glomerulus, the specialized filtering unit of the kidney. This unidimensional paradigm limits diagnosis and treatment of kidney diseases, the consequences of which are readily apparent: Both acute and chronic kidney diseases continue to outpace clinical management and are increasingly recognized as significant global health problems (1). And because these conditions are detected too late in the disease course, there have been no effective treatments developed to minimize kidney injury, alter the course of disease, or limit the associated morbidity and mortality.
Specifically, the diagnosis of kidney disease has relied on the serum creatinine, a breakdown product of creatine and phosphocreatine, that is largely freely filtered by the glomerulus. Accessible and affordable, serum creatinine has remained the gold standard for almost a century, despite its many well-recognized limitations as an indirect marker of kidney damage, including delayed detection of injury (Table 1) (2, 3). In addition, serum creatinine can increase in the absence of glomerular or tubular injury and can be unchanged under conditions of significant tubular injury, particularly when patients have good underlying kidney function and significant kidney reserve (4–6).
Table 1.
Limitations of serum creatinine in acute kidney injury (AKI) and chronic kidney disease (CKD)
| Disease | Limitations of serum creatinine |
|---|---|
| AKI and CKD | Nonspecific to disease etiology |
| Delayed marker of kidney damage | |
| Dependent on clinical characteristics (age, sex, muscle mass, etc.) | |
| Insensitive to small changes in GFR | |
| AKI | Dependent on hemodynamic steady state |
| Altered in hospitalized patients (i.e., by diuretics, IV fluids) | |
| Assay-related interference (i.e., by bilirubin) | |
| CKD | Unchanged despite kidney damage in tubulointerstitial and vascular disease |
| May be falsely low with significant proteinuria | |
| Provides imprecise eGFR estimations | |
| Requires special considerations for eGFR equations with changing muscle mass (i.e., in children, cirrhotics) |
Abbreviations: eGFR, estimated GFR; GFR, glomerular filtration rate.
To address these limitations, research employing novel technologies has focused on identifying structural markers of kidney tubular injury in the urine or systemic circulation that are directly produced by the kidney or build up as a result of the dysfunction of tubular cells following kidney damage (7–10). Linked to the pathophysiology of kidney injury, these biomarkers of tubular health may enable early detection, identification of the location of injury, etiologic discernment, and prognostic prediction of kidney diseases. In fact, the characterization of kidney tubules may be particularly important. The vast majority of the kidney’s energy expenditure is dedicated to the homeostatic maintenance of non-glomerular functions of the kidney, and recent studies have demonstrated that prognosis of kidney disease even if glomerular in origin (11) is dependent on the extent of tubulointerstitial fibrosis. In this review, we highlight promising kidney biomarkers that have strong underpinnings in the pathophysiology of kidney injury (12). These biomarkers have been investigated in various etiologies of acute kidney injury (AKI) and have more recently extended into various presentations of chronic kidney disease (CKD). We present the results of the applications of such biomarkers in various specific clinical settings from the spectrum of acute to chronic kidney diseases, demonstrating the breadth of clinical applications of these biomarkers.
BIOMARKERS OF KIDNEY TUBULAR HEALTH
Much of the research in the field of kidney biomarkers started with investigations of AKI, a condition in which serum creatinine may not be in steady state; thus, biomarkers that are closely correlated with true kidney tissue injury, akin to troponin for cardiac tissue, would be especially important for the detection of acute insults to the kidney. A variety of biomarkers of kidney tubular health have been identified in animal models and clinical studies that can localize to specific portions of the nephron and represent distinct mechanistic responses in the process of kidney injury. As direct indicators of kidney pathology, these biomarkers may address the limitations of serum creatinine. Several well-studied kidney biomarkers are presented with a broad physiologic overview, categorized by localization and mechanism of injury (Figures 1 and 2). These biomarkers provide a lens into the complexity of the pathogenesis of kidney injury and the heterogeneity that is not adequately captured by current approaches to characterize and treat kidney disease.
Figure 1.
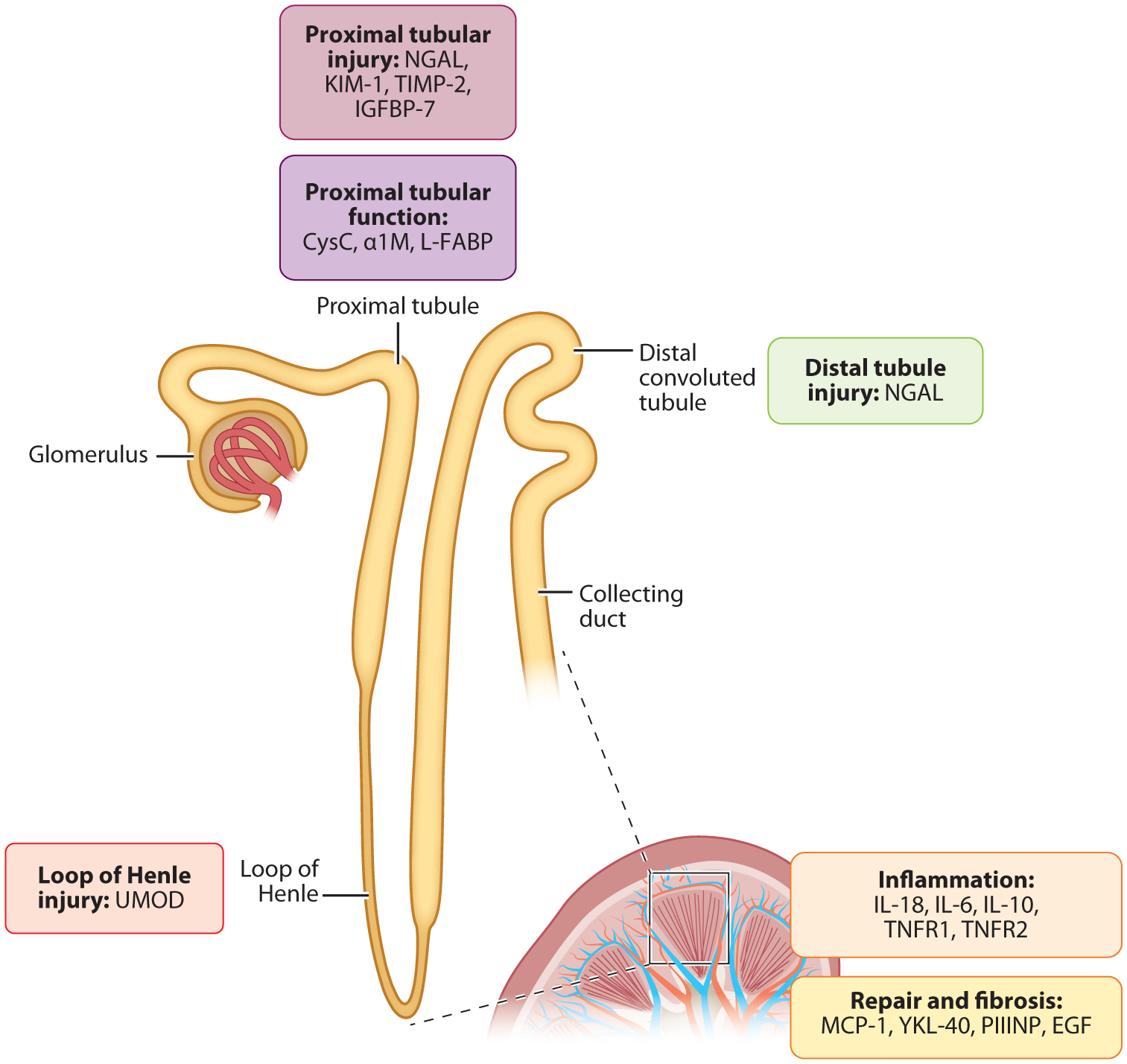
Biomarkers of kidney tubular health by anatomic localization and pathophysiology. Biomarkers of kidney tubular health are labeled corresponding to the anatomic site and/or mechanism of production. Abbreviations: EGF, epidermal growth factor; IGFBP-7, insulin-like growth factor-binding protein 7; IL, interleukin; KIM-1, kidney injury molecule-1; L-FABP, liver-type fatty acid–binding-protein; MCP-1, monocyte chemoattractant protein-1; NGAL, neutrophil gelatinase-associated lipocalin; PIIINP, procollagen type III N-terminal propeptide; TIMP-2, tissue inhibitor of metalloproteinases-2; UMOD, uromodulin; YKL-40, chitinase 3-like protein 1. Figure adapted with permission from Reference 82. Copyright 2013, American Society of Nephrology.
Figure 2.
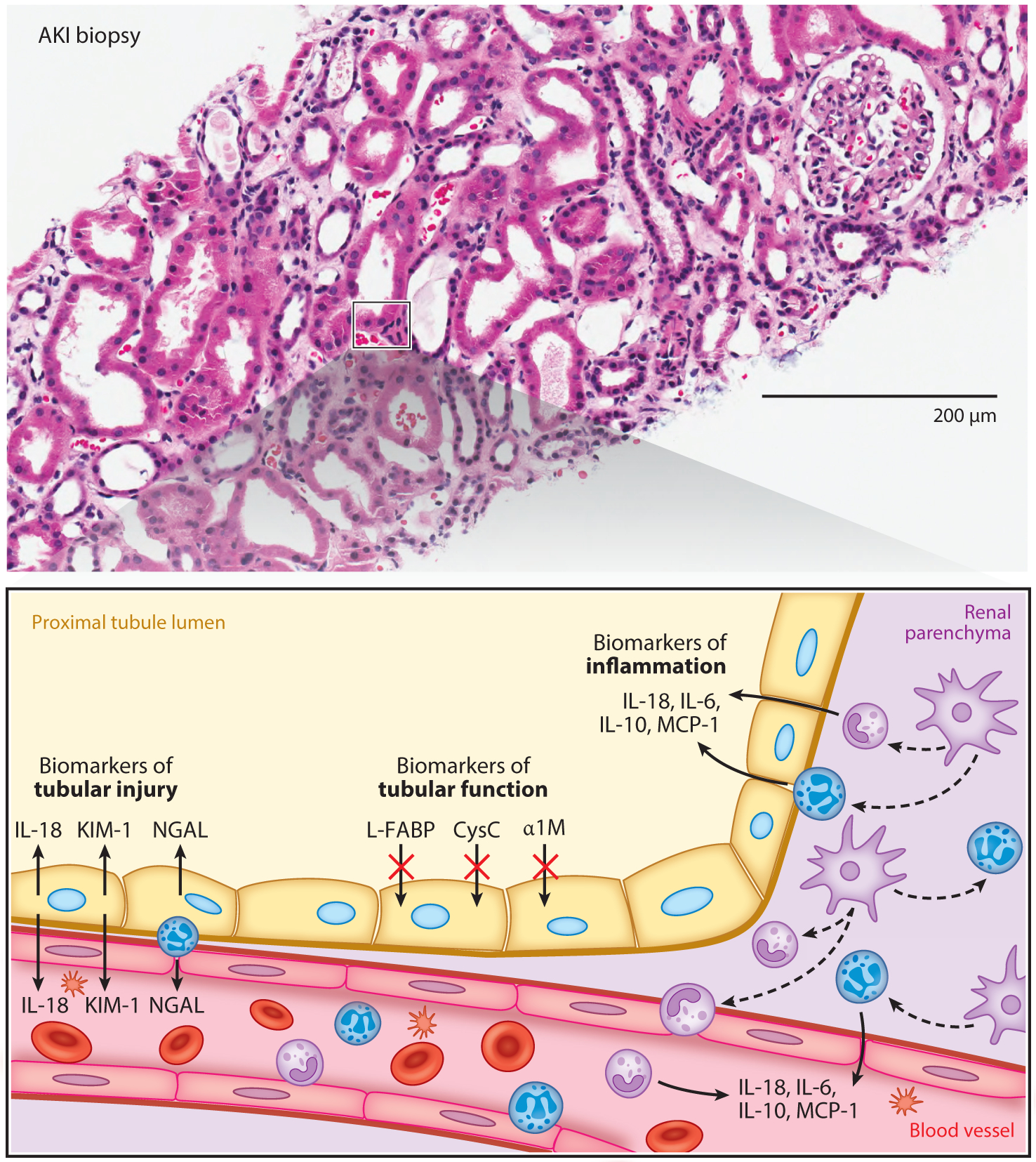
Biomarkers of AKI. The upper panel contains an AKI biopsy specimen, and the bottom panel is a close-up graphical representation of the portion of the AKI biopsy that is boxed in black and depicts three domains of kidney biomarkers. Biomarkers of tubular injury that are depicted include KIM-1 and NGAL, which are produced by proximal tubular cells early after kidney injury. Biomarkers of tubular function that are depicted include L-FABP, CysC, and α1M, which are proteins that build up in the urine due to tubular dysfunction. Biomarkers of inflammation that are depicted include IL-6, IL-10, IL-18, and MCP-1, which are produced as part of the inflammatory response to injury. Abbreviations: AKI, acute kidney injury; α1M, alpha-1 microglobulin; CysC, cystatin C; IL, interleukin; KIM-1, kidney injury molecule-1; L-FABP, liver-type fatty acid-binding-protein; MCP-1, monocyte chemoattractant protein-1; NGAL, neutrophil gelatinase-associated lipocalin.
Biomarkers of Tubular Injury
As a response to initial injury, tubular epithelial cells undergo subtle changes and release specific proteins into the urine and systemic circulation. Neutrophil gelatinase-associated lipocalin (NGAL), also known as lipocalin 2 (LCN2), is a glycoprotein bound to matrix metalloproteinase-9 in human neutrophils and is one of the most widely studied kidney biomarkers. It is a 25-kDa protein belonging to the lipocalin superfamily, which is involved in transport of hydrophilic substances through membranes to maintain cellular homeostasis (13). Through binding bacterial siderophores and sequestering iron, NGAL inhibits bacterial growth and is important in host defense against pathogens that require iron acquisition for survival. NGAL is expressed in various tissues in the body, such as the lung, gastrointestinal tract, liver, and kidney (14), and is markedly induced in injured epithelial cells in response to injury, inflammation, and neoplastic transformation (14). Thus, while both plasma and urinary NGAL have been investigated as promising biomarkers of kidney injury, urinary NGAL is more specific to that produced by the kidney following insult. Transcriptome profiling studies in rodent models identified NGAL as one of the most upregulated genes in the kidney very early after tubular injury, particularly in distal nephron segments, and evidence suggests that it may be the earliest known marker of kidney injury (15, 16). Urinary NGAL levels were significantly elevated within 2 h of renal ischemia-reperfusion injury in mouse models (17), and both urine and serum levels were elevated within 2 h following cardiac surgery in children who developed postoperative AKI (18). In addition, in CKD, urinary NGAL levels in humans have been demonstrated to be inversely correlated with estimated glomerular filtration rate (eGFR) and directly correlated with both interstitial fibrosis and tubular atrophy (19). Based on these promising findings, commercial NGAL tests have been approved for use in the detection of AKI in Europe and Asia with pending approval by the US Food and Drug Administration (FDA).
Kidney injury molecule-1 (KIM-1), also known as T cell immunoglobulin and mucin domain-containing protein-1 (TIM-1) and hepatitis A virus cellular receptor 1 (HAVCR-1), is a 90-kDa type 1 transmembrane glycoprotein that is significantly expressed in kidneys, specifically in proximal tubular cells, following ischemic injury, whereas it is virtually absent or present at low levels in healthy kidneys (20, 21). KIM-1 has evolved as a marker of proximal tubular injury, the hallmark of virtually all proteinuric, toxic, and ischemic renal diseases. KIM-1 has been shown to be a highly sensitive and specific marker of kidney injury in several animal models of kidney disease, including models of injury due to ischemia (20, 22) and various nephrotoxins (23–28). In addition to serving as a marker of tubular injury, preclinical data have also demonstrated that KIM-1 is upregulated in the later phases of AKI and is believed to play an important role in renal repair (29).
Tissue inhibitor of metalloproteinases-2 (TIMP-2) and insulin-like growth factor-binding protein 7 (IGFBP-7) are mediators of cell-cycle arrest, a commonly induced response to kidney injury. Both IGFBP-7 (through p523 and p21) and TIMP-2 (through p27) block the effect of cyclin-dependent protein kinase complexes and cause short periods of G1 cell-cycle arrest (30–32). These biomarkers were originally discovered in the clinical setting of critical illness and have been approved by the FDA for use in conjunction with clinical evaluation in intensive care unit patients who have acute cardiovascular and/or respiratory compromise (33).
Biomarkers of Tubular Function
The predominant function of the adenosine 5’-triphosphate (ATP)-rich proximal tubules is to reabsorb the majority of the filtrate. Thus, small molecules that are filtered by the glomerulus and processed or reabsorbed by the proximal tubules may serve as effective biomarkers of proximal tubular function. Increasing levels of these proteins in the urine may indicate reduced absorption by megalin, a multiligand endocytic receptor, in the proximal tubules prior to irreversible cell death. For example, cystatin C is a low-molecular-weight protein of 13 kDa produced at a constant rate by all nucleated cells and eliminated exclusively by glomerular filtration. Although it is neither secreted nor reabsorbed by renal tubules, it undergoes almost complete catabolism by proximal tubular cells and, thus, little if any appears in the urine under normal circumstances. Impairment of reabsorption in the proximal tubules can lead to marked increases in urinary levels of cystatin C in animals and humans (34, 35).
α1-Microglobulin (α1M) is another prototypical example of a marker of proximal tubular function. α1M is a low-molecular-weight glycoprotein of approximately 27 to 30 kDa and another member of the lipocalin superfamily. α1M is primarily synthesized by the liver and is available both in free form and as a complex with immunoglobulin A (IgA) (36). α1M is freely filtered at the glomerulus and completely reabsorbed via megalin mediation and catabolized by the normal proximal tubule. Therefore, an increase in the urinary concentration of α1M indicates proximal tubular injury or dysfunction, and patients with renal tubular diseases have been found to have elevated urinary α1M levels. Unlike β2-microglobulin and retinol-binding protein, which follow an analogous mechanism, α1M is more stable over a range of pH levels in the urine (37), which has currently made it a superior urinary biomarker of tubular dysfunction.
L-type or liver-type fatty acid-binding protein (L-FABP) is a 15-kDa protein that selectively binds to free fatty acids and transports them to mitochondria or peroxisomes, where free fatty acids are β-oxidized and participate in intracellular fatty acid homeostasis. First isolated in the liver as a binding protein for oleic acid and bilirubin, there are now known to be several different types of FABPs, which are expressed in a variety of tissues. Circulating L-FABP is thought to be filtered at the glomeruli and reabsorbed by proximal tubular cells. Although it is not synthesized in mouse models, L-FABP is expressed in the proximal tubules of humans following acute ischemic injury (38). Accordingly, elevated L-FABP levels have been shown to be a sensitive and specific marker of AKI in both adults and children (39, 40). Because L-FABP is also expressed by the liver, liver injury can be a potential contributor to increased urinary levels of this biomarker during AKI. However, previous clinical studies have shown that serum L-FABP levels do not have a significant influence on urinary levels and that urinary L-FABP levels are not significantly higher in patients with liver disease than in healthy subjects (39–42).
Uromodulin (UMOD), also known as Tamm-Horsfall protein, is an 85-kDa glycoprotein exclusively produced by cells of the thick ascending limb of Henle. It is the most abundant protein in physiologic urine and, with a large number of cysteine residues, has a tendency to aggregate and is the major constituent of hyaline casts (43). While UMOD’s physiologic function has yet to be elucidated, it has been implicated in regulation of salt homeostasis and conferring immunologic renal protection, including preventing infections and inhibiting nephrolithiasis. Studies in animal models and clinical settings have demonstrated its ability to serve as a biomarker for tubular mass and function and, accordingly, UMOD has been shown to be inversely associated with many kidney disease states. Evidence also shows that UMOD is a direct marker of the amount of intact tubular cells of the ascending limb of the loop of Henle and thus may represent a marker for the number of remaining functional tubules (44).
Biomarkers of Kidney Inflammation
The activation of inflammatory pathways within the kidneys and recruitment of inflammatory cells to the site of injury are early responses to kidney injury; such inflammatory mediators include interleukin-18 (IL-18), an 18-kDa proinflammatory cytokine, and a member of the IL-1 superfamily. As an early response to injury, the precursor of this inflammatory cytokine, pro-IL-18, is cleaved by caspase 1 within renal tubule cells and macrophages, and IL-18 is released into the tubular lumen and systemic circulation. Preclinical studies have demonstrated that IL-18 is a mediator of acute tubular injury, inducing both neutrophil and monocyte infiltration of the renal parenchyma (45, 46). In addition, IL-18 has been demonstrated to play a major role in macrophage activation, with mice engrafted with IL-18-deficient bone marrow experiencing less AKI than those with IL-18 replete marrow (47). Similarly, in IL-18 knockout mice with AKI, tumor necrosis factor-α (TNF-α), inducible nitric oxide synthase, macrophage inflammatory protein-2, and monocyte chemoattractant protein-1 (MCP-1) messenger RNA expression are all decreased, demonstrating the key inflammatory mediating effects of IL-18 on AKI. The immune response involves a variety of additional mediators, including IL-6 and IL-10. IL-6 is a major proinflammatory mediator that is well characterized in the orchestration of the inflammatory response following acute renal insult and has been shown to be a superior marker in renal patients compared with other proinflammatory candidates, such as the systemic inflammatory marker C-reactive protein. On the other hand, IL-10 is a prototypical anti-inflammatory cytokine that carries out the critical function of modulating and suppressing inflammation, antagonistic to the effects of IL-6.
In addition, kidney tubular cells also produce MCP-1 in response to proinflammatory cytokines, including TNF-α and IL-1β (48). MCP-1 is a chemotactic protein that attracts blood monocytes and tissue macrophages through interaction with the C-C motif cell-surface receptor chemokine receptor 2 (CCR2) (49, 50). In response to proinflammatory stimuli, MCP-1 is expressed in a variety of human cell types, including fibroblasts, endothelial cells, peripheral blood mononuclear cells, and epithelial cells (50–54). The lack of correlation between urinary and serum MCP-1 levels suggests that urinary MCP-1 is locally produced by the kidney rather than as a result of filtration of serum MCP-1 (55–57).
Soluble TNF receptors (TNFR1 and TNFR2), circulating markers of low-grade inflammation, have also recently been demonstrated as biomarkers of kidney disease. These soluble proteins are the circulating forms of the receptors shed from their membrane-bound counterparts, which are integral to TNF signaling pathways, and have been demonstrated to play important roles in the progression of atherosclerotic and kidney disease (58–60). Specifically, the TNF pathway has been involved in the development and progression of diabetic nephropathy in animal models (61), and TNF inhibition with the soluble TNFR2 fusion protein, etanercept, has improved albuminuria and tissue injury (62). TNF receptors belong to the TNF receptor superfamily, a group of type 1 single transmembrane glycoproteins. Binding of TNF-α to TNFRs regulates inflammatory responses and apoptosis via activation of nuclear factor kappa B (NF-κB) or activator protein 1 (AP-1). In humans, early studies have shown that increased levels of circulating TNFRs are strongly associated with the progression of diabetic nephropathy to CKD stage 3 and end-stage renal disease (ESRD), and with all-cause mortality (59, 60, 63). Although the literature has predominantly supported their use in diabetic nephropathy, subanalyses of nondiabetic patients in these studies have also confirmed their utility in other etiologies of kidney disease (64).
Biomarkers of Adaptive Repair and Fibrosis
Following inflammation, injury may be followed by reparative processes or ongoing progression of inflammation that eventually leads to fibrosis. These tightly regulated pathways of adaptive repair and fibrosis may be captured by urinary biomarkers involved in these pathways. YKL-40, also known as chitinase 3-Dlike protein 1 and BRP-39 in mice, is a 40-kDa inflammatory glycoprotein produced in a wide range of inflammatory cell types involved in modulating favorable responses to cellular damage (65). It has been hypothesized that this protein may signal the adaptive repair response following inflammation. For example, in hypoxic lung injury, BRP-39/YKL-40 has been demonstrated to limit lung injury, inflammation, and epithelial apoptosis (66). Studies of Brp39 knockout mice have revealed that macrophage-derived BRP-39 was critical in limiting renal tubular apoptosis via activation of Akt (also known as PKB, protein kinase B), and thus improved survival following renal ischemic-reperfusion injury (67).
In contrast, other biomarkers reflect the deposition of extracellular matrix that is the hallmark of fibrosis. Under physiologic conditions, kidneys have small amounts of collagen present in the intersitium, whereas kidneys having undergone progressive and sustained injury exhibit increased collagen production. Procollagen type III N-terminal propeptide (PIIINP) is the 42-kDa amino acid terminal peptide of type III procollagen, which is released during the synthesis and deposition of type III collagen. Urinary PIIINP levels are accordingly believed to be biomarkers of early stages of kidney fibrosis. Studies have found that urinary PIIINP levels did not correlate with proteinuria and thus likely represent intrarenal synthesis of this peptide (68). Ongoing work has focused on the evolving understanding of the pathophysiology of kidney injury in the modulation of renal repair. In contrast to the heart and brain, the kidney possesses intrinsic regenerative abilities following ischemic and toxic insults. Whether repair is promptly initiated or delayed may play an important role in outcomes following kidney injury. Thus, the process of and balance between adaptive and maladaptive repair may be an important junction for therapeutic intervention and has been the focus of active research efforts. Kidney biopsies are invasive and relatively difficult procedures and, thus, quantifying early fibrosis in CKD may become feasible with these noninvasive biomarkers.
For example, epidermal growth factor (EGF), which has been implicated in modulating the tubular response to injury (69, 70), was identified as a biomarker of chronic kidney disease via a renal biopsy transcriptome-driven discovery approach in a study of four diverse cohorts (71). Through an unbiased functional analysis of gene expression data, EGF was uniquely identified to be involved in kidney function decline. Along with intrarenal EGF mRNA, urinary EGF was found to be tightly correlated with eGFR at the time of biopsy and with longitudinal changes in eGFR, independent of traditional risk factors. Furthermore, EGF added predictive power to traditional clinical prognostic markers of CKD progression end points across the four diverse cohorts. As an especially promising biomarker, urinary EGF has been shown to be highly specific to the kidney and is normally minimally present in the plasma (72). Thus, it has been postulated that EGF may be a biomarker of the regenerative functional reserve and reflect the ability to respond to insults. Consistent with this understanding, exogenous EGF administration enhanced tubular repair and regeneration of kidney function in animal models of AKI; interestingly, however, in the presence of proinflammatory stimuli, EGF further exacerbated injury (73). In addition, urinary EGF has been shown to be inversely correlated with interstitial fibrosis (74), diabetic nephropathy (75), IgA nephropathy (69, 76), adult polycystic kidney disease (77), and pediatric CKD (78).
CLINICAL APPLICATIONS
Irrespective of etiology or clinical setting, a given loss of GFR due to kidney disease is detected by identical rises in serum creatinine levels. However, the meaning of these elevations in serum creatinine with respect to underlying mechanisms of injury and outcomes may significantly differ based on the physiologic context and the milieu in which the elevations occur. Biomarkers of tubular health have the ability to provide greater resolution into the nuances and complexities of these conditions, which have demonstrated the ability to improve detection, identify disease susceptibility, diagnose subclinical kidney disease, and predict prognosis of adverse events in a variety of clinical settings. It has become increasingly appreciated that any single biomarker may be insufficient to characterize the disease state. Rather, these biomarkers are context dependent. Accordingly, different biomarkers have demonstrated utility in different settings, reflecting the unique aspects of the underlying mechanisms of injury (Figure 3). Understanding the relationships between these different biomarker categories may improve the understanding of and ability to phenotype these disease processes and, in turn, inform the development of novel therapeutic compounds. We highlight observational data from several diverse clinical contexts in which these biomarkers have shown promise in advancing clinical care.
Figure 3.
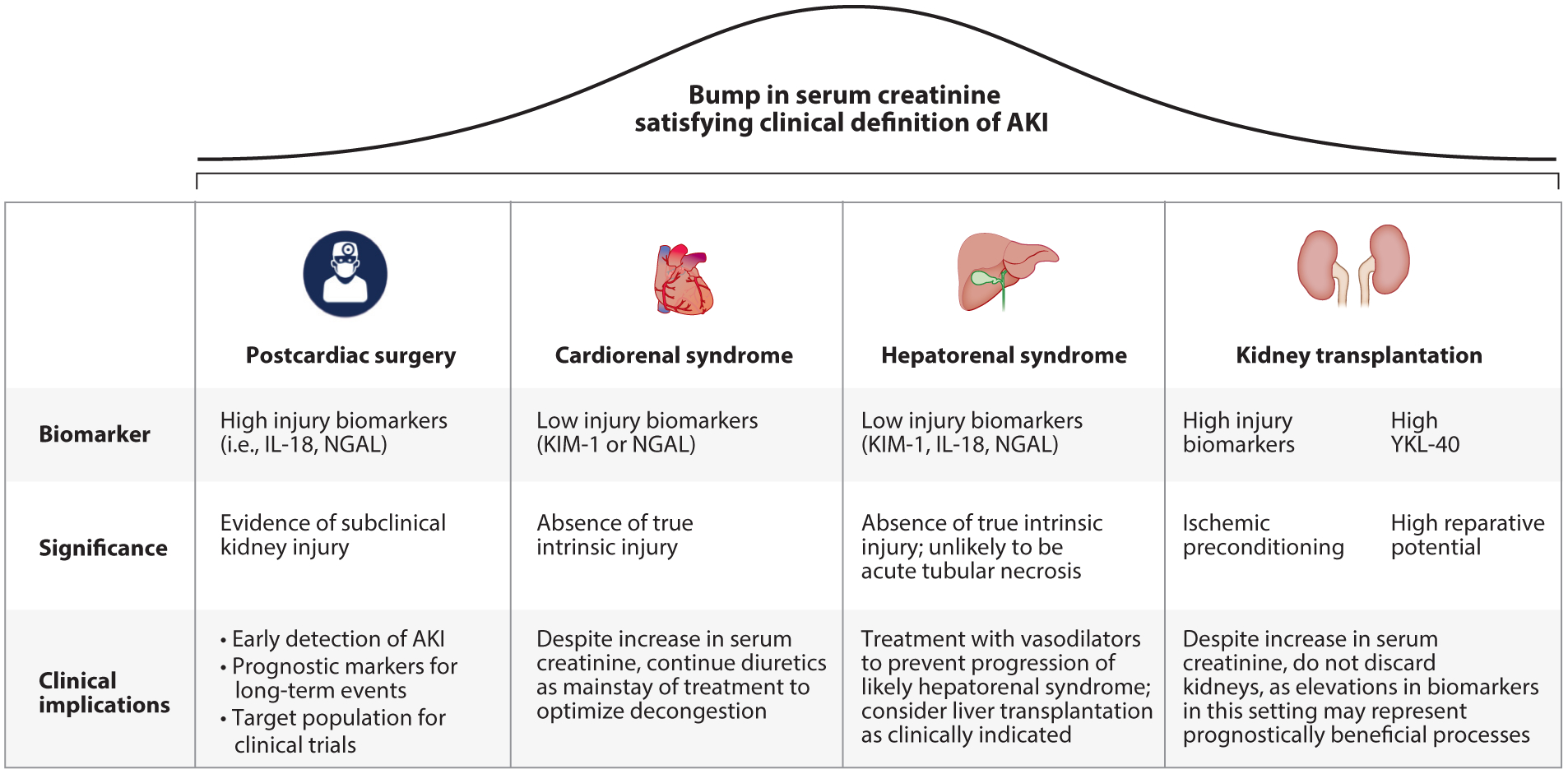
Distinguishing elevations of serum creatinine with biomarkers of kidney tubular health. Elevations in serum creatinine that meet the clinical definition of AKI do not necessarily represent true injury and have differing significance and clinical implications depending on the clinical milieu. Biomarker changes, their significance in elucidating the meaning of seemingly uniform elevations of serum creatinine, and clinical implications for patient management are detailed for the settings of postcardiac surgery, cardiorenal syndrome, hepatorenal syndrome, and kidney transplantation. Abbreviations: AKI, acute kidney injury; IL, interleukin; KIM-1, kidney injury molecule-1; NGAL, neutrophil gelatinase-associated lipocalin.
Acute Kidney Injury
The advances in the identification of these serum and urinary biomarkers representing tubular health have been investigated widely in AKI and can complement serum creatinine to improve patient management. Biomarkers can complement and provide mechanistic context to the rises in serum creatinine and improve clinical management of kidney disease in different settings (Figure 4).
Figure 4.
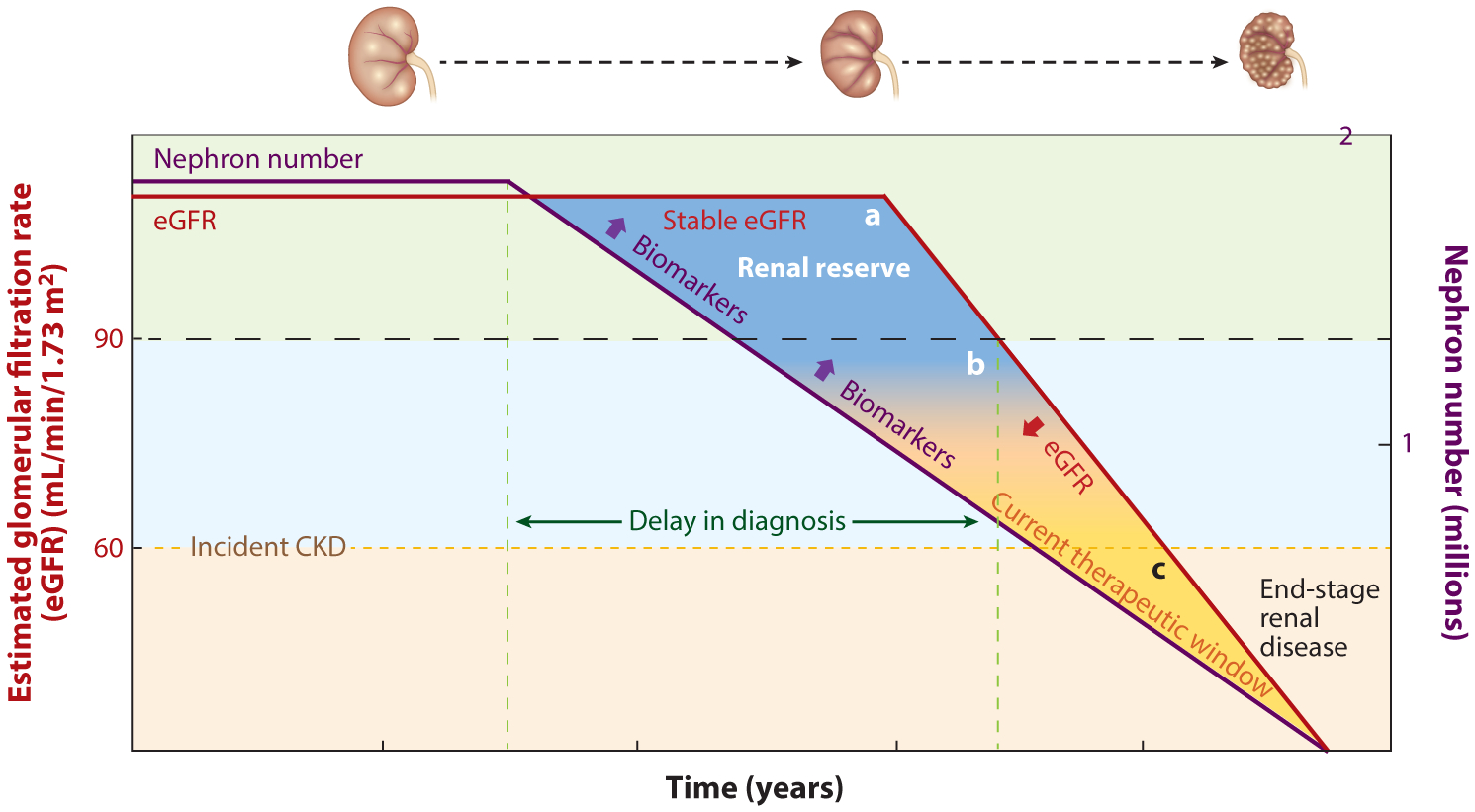
Opportunities for biomarkers of kidney tubular health to advance care in CKD. The left-hand y-axis depicts the eGFR, and the red line represents the eGFR trajectory with respect to time over the disease course. The right-hand y-axis depicts the number of remaining nephrons in millions, and the purple line depicts the nephron number with respect to time over the disease course. The delay in diagnosis (labeled as the distance between the dashed green vertical lines) may be reduced by use of biomarkers of kidney tubular health, which begin to increase as the nephron number begins to fall. In the earliest stages of CKD, the number of nephrons begins to decrease prior to being captured by clinically meaningful decreases in eGFR, as the kidney is able to compensate for loss of nephrons via the phenomenon of renal filtration reserve. Trapezoid A is labeled as renal reserve and encompasses the early course of disease when nephron numbers have begun to fall, yet eGFR is stable or only beginning to decline but is still above the threshold for clinical classification of CKD (dashed black horizontal line at eGFR of 90 mL/min/1.73 m2). In this range, novel biomarkers may address the critical limitations of serum creatinine and eGFR by detecting the earliest stages of kidney damage of incident CKD. Trapezoid B encompasses a region in which the effects of renal reserve may persist but are tapering out. In this range, decreases in nephron number may be paralleled by decreases in eGFR; thus, while biomarkers may be increasing and signaling intrinsic kidney damage, the kidney may also manifest loss of nephron number as commensurate eGFR declines because of the loss of renal reserve at this point. Thus, biomarkers may have less utility at this stage in the progression of CKD in discerning injury to the kidney; however, they may be able to address other limitations of serum creatinine. Triangle C represents advanced stages of CKD, in which the current therapeutic window for intervention lies. Utilizing biomarkers of tubular injury may allow for identification of therapeutic targets and shift this window to earlier points in the disease course. Abbreviations: CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate.
Early detection of subclinical AKI following cardiac surgery.
Cardiac surgery is a leading cause of ischemic AKI in hospitalized patients and strongly associated with significant morbidity and mortality. However, serum creatinine rises late in the disease course at 48–72 h postsurgery and limits the ability for early detection and intervention of these cases of AKI. The futility of attempting to treat patients following recognition of kidney injury based on serum creatinine has been compared to initiating treatment of myocardial infarction or stroke 48 h after the onset of ischemia. Preclinical models have demonstrated global changes in renal gene expression during early ischemic injury (79, 80), including production of proteins that have since been identified as biomarkers of AKI (81). Use of these biomarkers can augment the clinical ability to detect cases of kidney injury, improve risk stratification, and provide insight to therapeutic targets (82). Biomarkers of renal tubular injury, including IL-18, NGAL, and KIM-1, measured within 6 h following cardiac surgery, have been demonstrated to predict risk of AKI well before the rise in serum creatinine. For example, the highest quintiles of serum IL-18 within 6 h postsurgery were associated with a sevenfold increased risk of AKI in both adults and children, compared to the respective lowest quintiles (83, 84). Similarly, the highest quintiles of plasma NGAL and urinary KIM-1 were associated with increased risk of developing AKI, compared with the first quintile (adjusted OR 5.0, 95% CI: 1.6–15.3; adjusted OR 4.8, 95% CI: 1.6–14.6, respectively) (40, 83).
Although serum creatinine returns to normal levels in many of these cases of AKI, the burden on the kidneys may not be completely benign. For example, preclinical studies have demonstrated the development and persistence of inflammation, renal fibrosis, abnormal kidney gene expression profiles, and function deficits after ischemic kidney injury, despite a return of serum creatinine concentrations to normal values (85, 86). Accordingly, these biomarkers of tubular injury were also associated with severity and progression of AKI in the postcardiac surgery setting. The highest quintile of urinary IL-18 predicted those who progressed from early AKI (Acute Kidney Injury Network [AKIN] Stage 1) to more severe stages of AKI (AKIN Stage 2 or 3) (adjusted OR 3.00, 95% CI: 1.25–7.25) and was associated with duration of AKI >7 days (adjusted OR 2.90, 95% CI: 1.80–4.68) (87, 88). Similarly, those with the highest quintile of plasma NGAL were nearly eight times more likely to develop progressive AKI (OR 7.72, 95% CI: 2.65–22.49) compared with those in the first two quintiles. The highest quintile of urinary KIM-1 was also associated with longer duration of AKI (adjusted OR 2.3, 95% CI: 1.51–3.53) (88). L-FABP was associated with duration of AKI, and both the fourth and fifth quintiles were associated with a longer duration of AKI (adjusted OR 1.77, 95% CI: 1.17–2.67; adjusted OR 1.92, 95% CI: 1.26–2.93, respectively) (88).
Moreover, these biomarkers of tubular injury have even demonstrated potential to portend long-term mortality following cardiac surgery. Even in patients without AKI, the highest tertile of IL-18 was associated with increased risk of mortality over a median follow-up time of 3.0 years [interquartile range (IQR) 2.2 to 3.6] compared with the first tertile [adjusted hazard ratio (HR) 1.23, 95% CI: 1.02–1.48]. This effect was magnified in those subjects with perioperative AKI (adjusted HR 3.16, 95% CI: 1.53–6.53) (89). Similarly, the highest tertile of perioperative urinary KIM-1 was associated with increased mortality (adjusted HR 1.83, 95% CI: 1.44–2.33) in those who did not develop AKI as well as in those who developed AKI (adjusted HR 2.01, 95% CI: 1.31–3.1) (89). These findings may be especially critical because the association of troponin with mortality, in conjunction with its tissue specificity, was an important component of its incorporation into the diagnosis for myocardial infarction (90). However, given the long latency between an episode of kidney injury and mortality, in contrast to myocardial infarction, it may be difficult to determine causality. Nonetheless, these data suggest that recovery of serum creatinine levels may not capture the residual injury and fibrosis within the kidney that predisposes to increased morbidity and mortality. Recent evidence has demonstrated that elevated urinary kidney biomarkers of tubular damage, including α1M, PIIINP, and NGAL, are independent risk factors for both CVD and mortality over a median follow-up period of 12.4 years among elderly individuals with preserved baseline kidney function in a case-cohort study of the Health, Aging, and Body Composition (ABC) Study (91).
Distinguishing distinct etiologies of AKI in cardiorenal and hepatorenal syndromes.
Kidney injury in the settings of cardiorenal and hepatorenal syndromes (CRS, HRS) may have diverse underlying pathophysiology. Both cardiac dysfunction and cirrhosis can reduce renal perfusion through decreased forward flow and increased venous congestion, predisposing patients to kidney injury of prerenal physiology and intrinsic tubular injury. A wide variety of comorbidities and treatments may further complicate interpreting the meaning of rising serum creatinine concentrations. Biomarkers of tubular injury have demonstrated utility in distinguishing these distinct pathological processes.
For example, in the setting of CRS, aggressive diuresis is the mainstay of treatment to relieve congestion and restore cardiovascular hemodynamics but is also associated with elevations in serum creatinine, a phenomenon known as worsening renal function in the cardiology literature and often considered to be AKI (92, 93). Due to this perceived kidney damage, which is thought to be associated with adverse events (94, 95), diuresis and additional treatment are often halted (96). However, a rise in serum creatinine may not necessarily be uniformly associated with intrinsic kidney damage and adverse outcomes. For example, in a prospective cohort study of patients with acute decompensated heart failure, those who developed AKI following diuretic therapy did not have a significant rise in urinary NGAL levels (97). Furthermore, a recent ancillary study of the Renal Optimization Strategies Evaluation in Acute Heart Failure (ROSE-AHF) clinical trial using biomarkers of tubular health among 283 participants undergoing aggressive diuresis demonstrated that worsening renal function was not associated with elevations in biomarkers of kidney tubular injury, including NGAL and KIM-1, nor with adverse outcomes (98). The results demonstrated that the rise in creatinine may reflect clinically benign changes in filtration and suggest that such increases in creatinine may not warrant withdrawal of beneficial therapies.
Similarly, biomarkers have the ability to help narrow the differential diagnosis in the setting of cirrhosis between the two common etiologies of HRS and acute tubular necrosis. The ability to make the distinction between hepatorenal physiology and intrinsic tubular injury is critical, because treatments differ considerably. HRS may be reversed with restoration of renal perfusion through systemic vasoconstrictor therapy in addition to intravenous albumin treatment and subsequent liver transplantation. In contrast, patients with intrinsic tubular injury should be treated with dialysis and considered for combined liver-kidney transplantation (99–102). However, the current clinical paradigm to diagnose HRS is largely based on serum creatinine. Biomarkers of tubular health may distinguish causes of AKI and enable more rapid diagnoses and appropriate treatments. In a study of patients with cirrhosis who developed AKI, urinary levels of NGAL, IL-18, and L-FABP were elevated in patients with clinically adjudicated acute tubular necrosis but not in those with HRS or prerenal etiologies (103). In addition, the highest biomarker levels were found in patients with acute tubular injury, followed by HRS and prerenal azotemia, respectively, concordant with the physiology of the extent of tubular injury (100, 104). The combination of several biomarkers, including NGAL, L-FABP, and IL-18, was also assessed in diagnosing the type of AKI in patients with cirrhosis, with the likelihood of being diagnosed with acute tubular injury increasing in a stepwise fashion with the number of biomarkers that were above the optimal diagnostic cutoffs. For example, patients with cirrhosis who had at least one biomarker above the cutoff level were five times as likely to have acute tubular injury and those with all markers positive were 13 times as likely to have acute tubular injury (105).
Delineating prognosis of AKI in decreased donor kidney transplantation.
Ischemiareperfusion injury-mediated AKI is often seen in deceased donor kidneys, which subsequently have higher discard rates. Given the significant shortage of kidneys for transplantation, salvaging every viable kidney is imperative. However, serum creatinine may not accurately capture kidney injury and provide information about performance posttransplantation. In fact, an analysis of a biopsy series found that more than 20% of cases of biopsy-proven acute tubular injury did not meet the Kidney Disease: Improving Global Outcomes (KDIGO) serum creatinine-based AKI definition (106). Biomarkers of tubular injury can provide insight into the quality of deceased donor kidneys. For example, a study of deceased kidney donors demonstrated that serum creatinine had limited accuracy for diagnosing acute tubular necrosis, whereas urinary NGAL levels outperformed diagnosis of acute tubular injury (107). In addition, these biomarkers of tubular health may provide prognostic information. A large study of deceased donors of kidney injury demonstrated that increased urinary levels of NGAL, KIM-1, IL-18, and L-FABP were strongly associated with donor AKI but not with delayed graft function (107). Interestingly, the study reported that higher levels of kidney injury biomarkers were associated with higher 6-month eGFRs in recipients with delayed graft function. The protective effects of the intrinsic kidney injury from AKI are believed to be due to ischemic preconditioning, which involves the upregulation of anti-ischemic mediators. Consistent with these findings, ischemic preconditioning has been demonstrated to provide both acute and delayed protection against renal ischemia-reperfusion injury in mouse models via distinct signaling pathways (108).
In addition, YKL-40, a biomarker of adaptive repair after kidney injury, has been shown to be cytoprotective in the setting of kidney transplantation. Deceased kidney donors who had the highest YKL-40 levels had higher eGFRs at 6 months compared with patients whose donors had the lowest YKL-40 levels, after adjusting for donor and recipient factors, as well as the degree of kidney injury. Moreover, recipients with the highest donor YKL-40 levels also had 50% lower hazards of graft failure (adjusted HR 0.50, 95% CI: 0.27–0.94) (109).
Chronic Kidney Disease
In CKD, the timing and nature of the insult may be more difficult to estimate, as an accrual of kidney insults and injury over many years leads to the development of this condition. Thus, the early search for biomarkers of CKD lagged behind research of AKI. However, because AKI and CKD share similar underlying mechanisms of functional and structural injury and exist on the same pathophysiologic continuum, biomarkers of AKI have also been applied to CKD. The biomarkers have been especially promising in identifying susceptibility to and predicting the development of incident CKD; however, they have not been as robust in predicting CKD progression after adjustment for serum creatinine, and studies in different settings may be biased based on the etiology of underlying CKD and enrollment criteria. In participants without CKD at baseline, the “renal filtration reserve” (4, 5) is able to compensate for filtration deficits and, in effect, absorbs the insult (Figure 4). Biomarkers of tubular injury are critical to provide information about kidney tubular injury in these situations in which serum creatinine does not change appreciably. Accordingly, these biomarkers of tubular injury have been shown to be especially promising in prognosticating development of incident CKD. For example, in a nested-case control study of 686 participants from the Multi-Ethnic Study of Atherosclerosis (MESA), in which cases were defined as those with a baseline eGFR >60 ml/min who subsequently developed CKD Stage 3 and/or had a rapid drop in kidney function over the five-year study period, higher levels of KIM-1 were associated with increased odds of developing CKD Stage 3 or a rapid decline in eGFR (adjusted OR 1.15, 95% CI: 1.02–1.29). Similarly, at study entry, those in the highest decile of KIM-1 had a twofold increased risk of this same end point compared with the lower 90%. This ability to predict the development and progression of CKD was independent of the presence of albuminuria (110). Similarly, when investigated in a cohort of 149 persons with chronic congestive heart failure during 5 years of follow-up, urinary KIM-1 levels were strongly associated with the progression of CKD, defined as a >25% drop in eGFR from baseline (111).
However, when renal filtration reserve is lost, each insult to the kidney will correspond with an added decrease in glomerular filtration and, thus, serum creatinine may provide as much information as tubular injury biomarkers. For example, a recent study of participants in the Chronic Renal Insufficiency Cohort (CRIC) with baseline CKD demonstrated that urinary KIM-1, NGAL, and L-FABP levels were significantly associated with CKD progression in unadjusted analyses; however, once controlling for serum creatinine-based eGFR and urinary albumin/creatinine ratio, two traditional markers of kidney function, these biomarkers were no longer independently associated with CKD progression. In addition, none of the biomarkers improved risk stratification of the clinical model for CKD progression, suggesting that tubular injury biomarkers may have limited utility in patients who have diminished renal reserve (112). Additional studies of individuals with CKD have also demonstrated that tubular injury markers do not add to risk prediction of disease progression after accounting for traditional markers of kidney function (113–115).
Diabetic nephropathy.
Diabetes is major risk factor and leading cause of CKD and ESRD. However, 20–30% of individuals with diabetes have rapidly declining kidney function even when on optimal therapy with angiotensin-converting enzyme inhibition or angiotensin II receptor blockers. Kidney disease progression in diabetes is multifactorial and may be driven by ongoing inflammation, intrinsic tubular injury, or maladaptive repair, among other pathways (116, 117), and biomarkers may help to discern the dominant underlying etiologies (Figure 5). Addressing this key clinical question with biomarkers of tubular health may improve risk stratification and enable effective treatment of progressive diabetic kidney disease.
Figure 5.
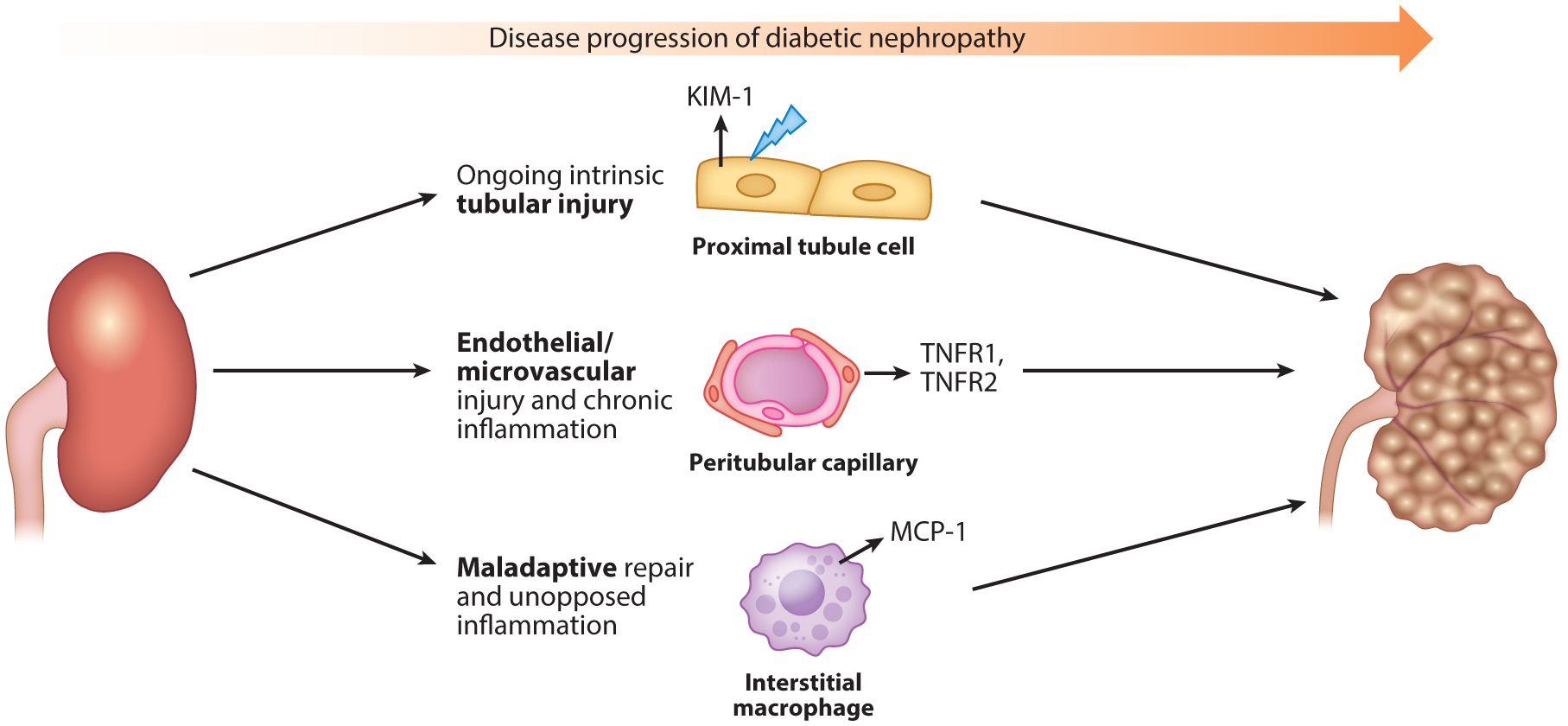
Distinct mechanisms of diabetic nephropathy progression. Diabetic nephropathy is a heterogeneous condition that may involve diverse mechanisms. This may explain why a portion of patients with diabetic kidney disease continue to experience disease progression, despite optimal therapy with the current standard of treatments, including angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers. These current therapies do not address the underlying mechanisms that drive the progression of diabetic kidney disease, which may include ongoing intrinsic tubular injury, manifested by elevated levels of KIM-1; endothelial/microvascular injury and chronic inflammation, manifested by elevated levels of TNFR1 and TNFR2; and maladaptive repair and unopposed inflammation, manifested by elevated levels of MCP-1. These biomarkers may discern distinct pathways of injury and inform therapeutic interventions for diabetic nephropathy. Abbreviations: KIM-1, kidney injury molecule-1; MCP-1, monocyte chemoattractant protein-1; TNFR, tumor necrosis factor receptor.
For example, biomarkers of tubular injury have been found to be associated with kidney disease in diabetic patients, indicating underlying intrinsic tubular injury. Plasma KIM-1 was measured in subjects from the Action to Control Cardiovascular Risk in Diabetes (ACCORD) and US Veterans Administration Nephropathy in Diabetes (VA-NEPHRON-D) cohorts. In the ACCORD cohort, those who went on to develop incident CKD had higher baseline KIM-1 levels. Similarly, in the VA-NEPHRON-D study, those who went on to develop progressive CKD had higher baseline levels (118). In both of these studies, those with the highest quartile of KIM-1 concentrations were more likely to experience adverse renal outcomes, compared with those in the lowest quartiles of KIM-1 concentrations (119, 120).
In these two cohorts, plasma TNFR1 and TNFR2 were also found to be higher among those with advanced diabetic kidney disease, compared with those with early stage disease at baseline, and higher levels of these two cytokine receptors predicted eGFR decline, even after adjustment for baseline eGFR and albuminuria (118). These biomarkers have been implicated in an inflammatory pathway representing ongoing endothelial inflammation, a known etiology of diabetic kidney disease (121). Consistent with this notion, the TNF pathway has been linked to diabetic kidney disease: Genome-wide association studies have demonstrated that transcripts inversely correlated with GFR clustered around TNF-α, which directly acts on podocytes to propagate the inflammatory cascade via TNFRs (122). In addition, several studies have found that plasma TNFR1 and TNFR2 are associated with incident diabetic kidney disease and ESRD in diabetic patients (59, 60, 123, 124). Interestingly, TNFR1 and TNFR2 may represent distinct pathways, but the nuances are still being elucidated (125).
Furthermore, urinary MCP-1 has also been found to be significantly elevated in patients with diabetic nephropathy, and these levels correlate significantly with albuminuria in humans as well as in experimental diabetic nephropathy (126–129). In a prospective observational study of patients with diabetic nephropathy, urinary MCP-1 levels were associated with macroalbuminuria and correlated with the subsequent rate of eGFR decline over a median follow-up of six years. These findings suggest that MCP-1 is associated with later-stage disease and potentially represents a pathway of maladaptive repair (130).
HIV-associated chronic kidney disease.
HIV-infected individuals are at substantial risk of kidney disease, as the virus is known to take host in and damage renal tubular cells, even in those with controlled viremia (131). In addition, the HIV-infected population has numerous metabolic and vascular comorbidities and treatment-related risk factors that increase the risk of kidney disease. Serum creatinine cannot distinguish between the many potential etiologies of kidney disease, which impedes clinical management. Biomarkers of tubular dysfunction and injury have been especially promising in this setting in detecting early injury attributable to the virus. For example, α1M levels in women infected with HIV were associated with both kidney function decline and mortality (132). Compared to those with the lowest α1M levels, those with the highest levels had an increased risk of developing CKD (adjusted OR 2.1, 95% CI: 1.3–3.4) and a 2.7-fold risk of a 10% decline in eGFR. This correlation in CKD development and progression was separate from the 1.6-fold adjusted risk of mortality, which accounted for baseline kidney function as well as the presence of albuminuria (132). In addition, urine IL-18 was associated with worsening renal function over time in a cohort of the Women’s Interagency HIV Study. IL-18 was higher in HIV-infected women compared to those who were not infected, as well as significantly associated with higher HIV RNA levels and lower CD4 counts, a lower proportion of hepatitis C infection, and lower HDL cholesterol levels (133, 134). Similar findings with IL-18 were also reported in a cross-sectional study of 1,144 men of the Multicenter AIDS Cohort Study (MACS), which found substantial elevations of IL-18, KIM-1, PIIINP, and albumin/creatinine ratio in HIV-infected men, compared with uninfected men (135).
These biomarkers can also identify kidney damage from tenofovir disoproxil fumarate (TDF), a widely used, first-line antiretroviral that is known to cause kidney disease via proximal tubular pathology (136, 137). Cross-sectional studies have demonstrated that cumulative TDF exposure in HIV-infected men enrolled in MACS was independently associated with elevations in concentrations of urinary α1M, IL-18, KIM-1, and PIIINP, but not with albuminuria, consistent with the notion that TDF is a proximal tubular toxin (138, 139). These tubular injury biomarkers may identify subclinical injury before the onset of overt, irreversible disease and distinguish kidney injury attributable to TDF use. Ongoing work is investigating the ability of biomarker patterns and signatures to discern diverse etiologies of kidney injury among HIV-infected individuals.
FUTURE DIRECTIONS
The use of biomarkers in nephrology has gained considerable traction; however, there are still strides to be made before they can cross the threshold of clinical application. As a variety of biomarkers of kidney health have demonstrated robust associations with adverse outcomes in diverse clinical settings, they have begun to shed light on unique biological pathways that mediate injury and repair. Studying biomarkers in clinical models has also highlighted the limitations of animal models that do not simulate the complexities of overlapping comorbidities, such as diabetes, hypertension, and aging. Future studies will need to develop animal models that can capture such complexity. This deeper understanding may lead to identification and individualization of effective therapeutic windows and targets that can maximally limit injury, promote repair, and prevent fibrosis without hindering an appropriate host defense.
These biomarkers may be applied as tools in clinical trials for the evaluation of novel therapeutics (Figure 6). Biomarkers of tubular injury can be used in clinical trials to enrich the population for true cases of intrinsic injury versus hemodynamic injury (as diagnostic biomarkers), select for participants at high risk for associated outcomes (as prognostic biomarkers), and identify those who may be more likely to respond to a particular intervention (as predictive biomarkers). These more sensitive and specific metrics for trial enrollment can increase statistical power, decrease the required sample size, and reduce trial costs. In a series of simulated clinical trials using diagnostic biomarkers to enroll patients at high risk for AKI following cardiac surgery in the TRIBE-AKI cohort, investigators demonstrated that trial costs could be substantially decreased from 29% to 64%, with the trade-off requiring a larger screening group of participants and limited generalizability (140). In addition, when IL-18 and NGAL were used as the outcomes for a simulated trial of statin use during the perioperative period, AKI defined by elevations in biomarkers could discern significant differences in the outcome and renoprotective effects of statins, whereas AKI defined by serum creatinine could not (141). These findings question whether the previously failed trials in nephrology were perhaps using the wrong target or suboptimal designs.
Figure 6.
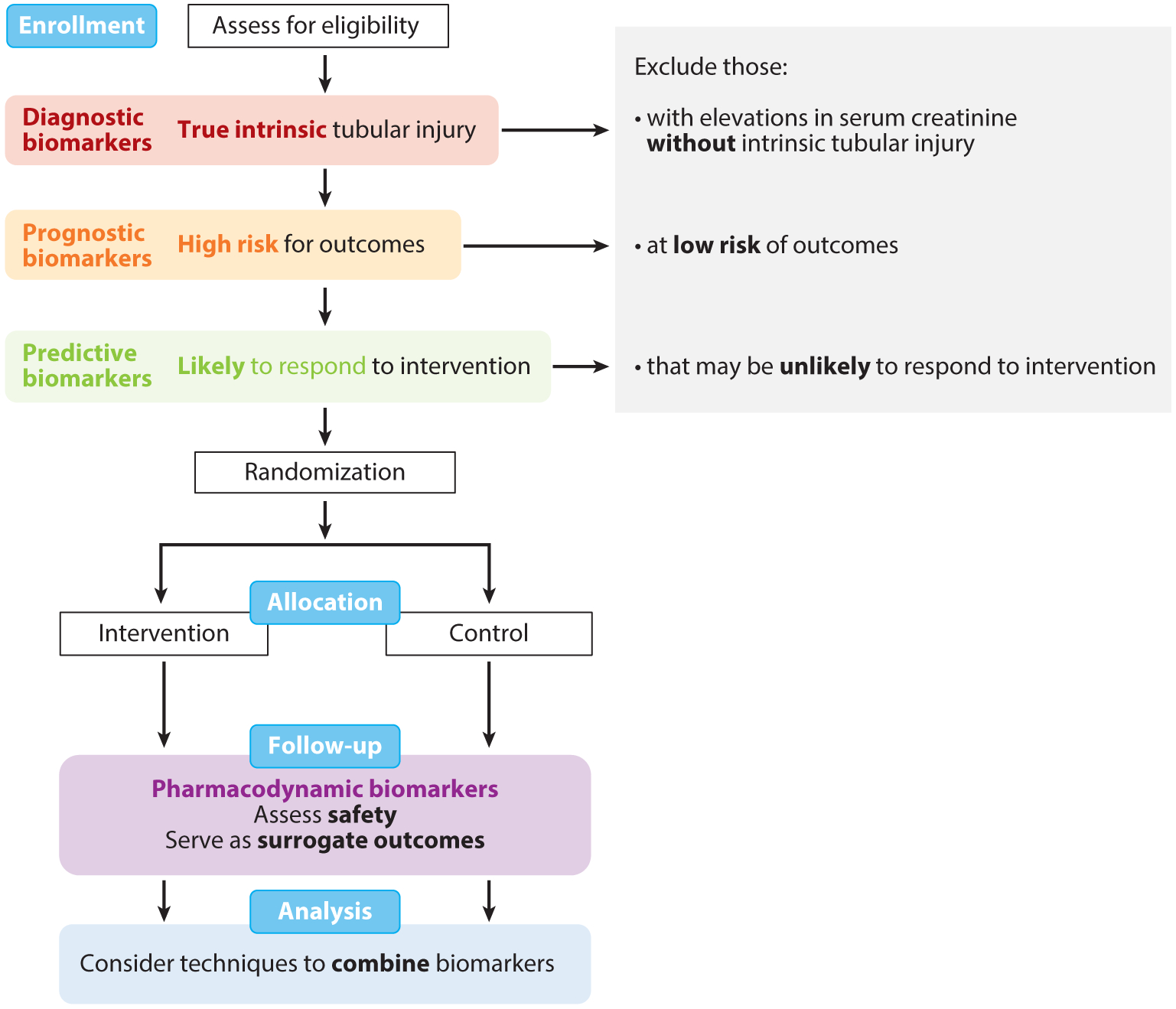
Application of kidney biomarkers in clinical trials. Biomarkers of tubular health may be applied in various stages of clinical trials. At enrollment, diagnostic biomarkers (red) can be used to identify participants with true intrinsic tubular injury; prognostic biomarkers (orange) can be used to identify participants at high risk for outcomes; and predictive biomarkers (green) can be used to identify participants likely to respond to an intervention of interest. By using these biomarkers to assess eligibility and exclude individuals who may not be truly suitable for the study despite an elevation in serum creatinine, investigators may be able to conduct more efficient, cost-effective trials. In the follow-up phase of trials, pharmacodynamics biomarkers (purple) may be used to assess safety of interventions and serve as surrogate outcomes for kidney damage. With the wide breath of biomarkers now available, biomarkers may be combined with novel statistical techniques in the analysis phase. Figure adapted with permission from Reference 140.
In addition, these biomarkers can be utilized as safety biomarkers for the monitoring of toxicity or harm in clinical trials. In fact, the FDA, along with its European and Japanese counterparts, has collaboratively approved a panel of urinary biomarkers of kidney injury that includes KIM-1, albumin, total protein, cystatin C, clusterin, trefoil factor 3, and α1M for use in preclinical models of nephrotoxicity (142–144).
Similar to the predictive biomarkers that have advanced the field of therapeutics in oncology, such as with the use of herceptin in human epidermal growth factor receptor 2 (HER2)-positive breast cancers, targeted interventions may play an important role in effective treatment of kidney disease. For example, a drug acting on the apoptosis pathway via the nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome complex may be effective in patients in whom this AKI pathway is active. A biomarker, such as urinary IL-18, that is released upon activation of the NLRP3 inflammasome pathway could be a predictive biomarker to select patients for enrollment in a trial of such a drug. In addition, individuals with diabetic nephropathy driven by high levels of TNFR1 and TNFR2 may indicate that anti-inflammatory agents could be repurposed to treat these conditions.
To achieve these goals, more work is needed to further phenotype these kidney disease syndromes with biomarkers of tubular injury and other biological data. In order to fully capture the diverse manifestations and apply them reliably to clinical trial design, new strategies that rely on a panel of several novel biomarkers will be necessary, perhaps as a composite biomarker score. Ongoing research is working toward contending with this complexity by using methods such as metabolomics profiling (145) and by investigating circulating microRNAs, which have been shown to specifically change in various kidney diseases (146). In addition, large-scale efforts are underway to elucidate and trace these very pathways of kidney disease. The ongoing National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)-funded Kidney Precision Medicine Project (147) aims to obtain kidney biopsies of AKI and CKD and construct a kidney tissue atlas to define disease subgroups with genetics, transcriptomics, and corresponding plasma and urine biomarkers. The availability of this tissue-based atlas will allow for comparisons of biomarkers to the true gold standard of kidney biopsies and discovery of novel biomarkers. These future studies may unmask more robust utility of biomarkers, which may have appeared to be suboptimal due to comparisons with the imperfect standard of serum creatinine. Similar to the field of oncology, the molecular phenotyping from this resource may lead to personalized treatments for kidney disease patients based on distinct disease signatures. Along with these developments, novel statistical approaches will be needed to evaluate the accuracy of biomarkers in risk prediction, especially as these markers are compared to the imperfect gold standard of serum creatinine. In addition, with the large number of biomarkers that are now available, biomarkers may need to be combined into composite scores to enhance predictive abilities and improve feasibility of implementation into clinical practice (148–150).
CONCLUSIONS
In conclusion, several promising biomarkers of kidney health that are involved in the pathophysiologic mechanism of kidney damage have demonstrated the potential to improve clinical treatment of kidney diseases. These biomarkers have demonstrated the ability to detect early damage, localize injury, and predict disease progression, severity, and associated long-term mortality. Future work is underway to characterize biological pathways that promote kidney repair and long-term survival, which may inform the development novel therapeutics in the field of nephrology.
ACKNOWLEDGMENTS
C. Parikh was supported by NIH grant RO1HL085757 to fund the TRIBE-AKI Consortium to study novel biomarkers of acute kidney injury in cardiac surgery. He is also a member of the NIH-sponsored Assessment, Serial Evaluation, and Subsequent Sequelae in Acute Kidney Injury (ASSESS-AKI) Consortium (U01DK082185), CKD Biomarker Consortium (UO1DK10696), and Kidney Precision Medicine Consortium (UG3DK114866).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, et al. 2005. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA 294:813–18 [DOI] [PubMed] [Google Scholar]
- 2.Kampmann J, Siersbaek-Nielsen K, Kristensen M, Hansen JM. 1974. Rapid evaluation of creatinine clearance. Acta Med. Scand 196:517–20 [DOI] [PubMed] [Google Scholar]
- 3.Nickolas TL, Barasch J, Devarajan P. 2008. Biomarkers in acute and chronic kidney disease. Curr. Opin. Nephrol. Hypertens 17:127–32 [DOI] [PubMed] [Google Scholar]
- 4.Bosch JP. 1995. Renal reserve: a functional view of glomerular filtration rate. Semin. Nephrol 15:381–85 [PubMed] [Google Scholar]
- 5.Herrera J, Rodríguez-Iturbe B. 1998. Stimulation of tubular secretion of creatinine in health and in conditions associated with reduced nephron mass. Evidence for a tubular functional reserve. Nephrol. Dial. Transplant 13:623–29 [DOI] [PubMed] [Google Scholar]
- 6.Wu I, Parikh CR. 2008. Screening for kidney diseases: older measures versus novel biomarkers. Clin. J. Am. Soc. Nephrol 3:1895–901 [DOI] [PubMed] [Google Scholar]
- 7.Zurbig P, Dihazi H, Metzger J, Thongboonkerd V, Vlahou A. 2011. Urine proteomics in kidney and urogenital diseases: moving towards clinical applications. Proteom. Clin. Appl 5:256–68 [DOI] [PubMed] [Google Scholar]
- 8.Shao C, Wang Y, Gao Y. 2011. Applications of urinary proteomics in biomarker discovery. Sci. China Live Sci 54:409–17 [DOI] [PubMed] [Google Scholar]
- 9.Boudonck KJ, Rose DJ, Karoly ED, Lee DP, Lawton KA, Lapinskas PJ. 2009. Metabolomics for early detection of drug-induced kidney injury: review of the current status. Bioanalysis 1:1645–63 [DOI] [PubMed] [Google Scholar]
- 10.Prunotto M, Ghiggeri GM, Candiano G, Lescuyer P, Hochstrasser D, Moll S. 2011. Urinary proteomics and drug discovery in chronic kidney disease: a new perspective. J. Proteome Res 10:126–32 [DOI] [PubMed] [Google Scholar]
- 11.Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, et al. 2016. Severity and frequency of proximal tubule injury determines renal prognosis. J. Am. Soc. Nephrol 27:2393–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woroniecki RP, Schnaper HW. 2009. Progression of glomerular and tubular disease. Semin. Nephrol 29:412–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flower DR. 1996. The lipocalin protein family: structure and function. Biochem. J 318(Part 1):1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soni SS, Cruz D, Bobek I, Chionh CY, Nalesso F, et al. 2010. NGAL: a biomarker of acute kidney injury and other systemic conditions. Int. Urol. Nephrol 42:141–50 [DOI] [PubMed] [Google Scholar]
- 15.Supavekin S, Zhang W, Kucherlapati R, Kaskel FJ, Moore LC, Devarajan P. 2003. Differential gene expression following early renal ischemia/reperfusion. Kidney Int. 63:1714–24 [DOI] [PubMed] [Google Scholar]
- 16.Yuen PST, Jo S-K, Holly MK, Hu X, Star RA. 2006. Ischemic and nephrotoxic acute renal failure are distinguished by their broad transcriptomic responses. Physiol. Genom 25:375–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, et al. 2003. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J. Am. Soc. Nephrol 14:2534–43 [DOI] [PubMed] [Google Scholar]
- 18.Mishra J, Dent C, Tarabishi R, Mitsnefes MM, Ma Q, et al. 2005. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 365:1231–38 [DOI] [PubMed] [Google Scholar]
- 19.Nickolas TL, Forster CS, Sise ME, Barasch N, Valle DS, et al. 2012. NGAL (Lcn2) monomer is associated with tubulointerstitial damage in chronic kidney disease. Kidney Int. 82:718–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, et al. 1998. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J. Biol. Chem 273:4135–42 [DOI] [PubMed] [Google Scholar]
- 21.Hubank M, Schatz DG. 1994. Identifying differences in mRNA expression by representational difference analysis of cDNA. Nucl. Acid Res 22:5640–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. 2002. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int. 62:237–44 [DOI] [PubMed] [Google Scholar]
- 23.Vaidya VS, Ramirez V, Ichimura T, Bobadilla NA, Bonventre JV. 2006. Urinary kidney injury molecule-1: a sensitive quantitative biomarker for early detection of kidney tubular injury. Am. J. Physiol. Ren. Physiol 290:517–29 [DOI] [PubMed] [Google Scholar]
- 24.Zhou Y, Vaidya VS, Brown RP, Zhang J, Rosenzweig BA, et al. 2008. Comparison of kidney injury molecule-1 and other nephrotoxicity biomarkers in urine and kidney following acute exposure to gentamicin, mercury, and chromium. Toxicol. Sci 101:159–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prozialeck WC, Vaidya VS, Liu J, Waalkes MP, Edwards JR, et al. 2007. Kidney injury molecule-1 is an early biomarker of cadmium nephrotoxicity. Kidney Int. 72:985–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jost G, Pietsch H, Sommer J, Sandner P, Lengsfeld P, et al. 2009. Retention of iodine and expression of biomarkers for renal damage in the kidney after application of iodinated contrast media in rats. Investig. Radiol 44:114–23 [DOI] [PubMed] [Google Scholar]
- 27.Pérez-Rojas J, Blanco JA, Cruz C, Trujillo J, Vaidya VS, et al. 2007. Mineralocorticoid receptor blockade confers renoprotection in preexisting chronic cyclosporine nephrotoxicity. Am. J. Physiol. Ren. Physiol 292:131–39 [DOI] [PubMed] [Google Scholar]
- 28.van Timmeren MM, Bakker SJL, Vaidya VS, Bailly V, Schuurs TA, et al. 2006. Tubular kidney injury molecule-1 in protein-overload nephropathy. Am. J. Physiol. Ren. Physiol 291:456–64 [DOI] [PubMed] [Google Scholar]
- 29.Ko GJ, Grigoryev DN, Linfert D, Jang HR, Watkins T, et al. 2010. Transcriptional analysis of kidneys during repair from AKI reveals possible roles for NGAL and KIM-1 as biomarkers of AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol 298:F1472–83 [DOI] [PubMed] [Google Scholar]
- 30.Price PM, Safirstein RL, Megyesi J. 2009. The cell cycle and acute kidney injury. Kidney Int. 76:604–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boonstra J, Post JA. 2004. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 337:1–13 [DOI] [PubMed] [Google Scholar]
- 32.Seo DW, Li H, Qu CK, Oh J, Kim YS, et al. 2006. Shp-1 mediates the antiproliferative activity of tissue inhibitor of metalloproteinase-2 in human microvascular endothelial cells. J. Biol. Chem 281:3711–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kashani K, Al-Khafaji A, Ardiles T, Artigas A, Bagshaw SM, et al. 2013. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit. Care 17:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Togashi Y, Sakaguchi Y, Miyamoto M, Miyamoto Y. 2012. Urinary cystatin C as a biomarker for acute kidney injury and its immunohistochemical localization in kidney in the CDDP-treated rats. Exp. Toxicol. Pathol 64:797–805 [DOI] [PubMed] [Google Scholar]
- 35.Koyner JL, Bennett MR, Worcester EM, Ma Q, Raman J, et al. 2008. Urinary cystatin C as an early biomarker of acute kidney injury following adult cardiothoracic surgery. Kidney Int. 74:1059–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Penders J, Delanghe JR. 2004. Alpha 1-microglobulin: clinical laboratory aspects and applications. Clin. Chim. Acta 346:107–18 [DOI] [PubMed] [Google Scholar]
- 37.Itoh Y, Kawai T. 1990. Human α1-microglobulin: its measurement and clinical significance. J. Clin. Lab. Anal 4:376–84 [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto T, Noiri E, Ono Y, Doi K, Negishi K, et al. 2007. Renal L-type fatty acid–binding protein in acute ischemic injury. J. Am. Soc. Nephrol 18:2894–902 [DOI] [PubMed] [Google Scholar]
- 39.Portilla D, Dent C, Sugaya T, Nagothu KK, Kundi I, et al. 2008. Liver fatty acid-binding protein as a biomarker of acute kidney injury after cardiac surgery. Kidney Int. 73:465–72 [DOI] [PubMed] [Google Scholar]
- 40.Parikh CR, Thiessen-Philbrook H, Garg AX, Kadiyala D, Shlipak MG, et al. 2013. Performance of kidney injury molecule-1 and liver fatty acid-binding protein and combined biomarkers of AKI after cardiac surgery. Clin. J. Am. Soc. Nephrol 8:1079–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakamura T, Sugaya T, Koide H. 2009. Urinary liver-type fatty acid-binding protein in septic shock: effect of polymyxin B-immobilized fiber hemoperfusion. Shock 31:454–59 [DOI] [PubMed] [Google Scholar]
- 42.Kamijo A, Sugaya T, Hikawa A, Yamanouchi M, Hirata Y, et al. 2006. Urinary liver-type fatty acid binding protein as a useful biomarker in chronic kidney disease. Mol. Cell. Biochem 284:175–82 [DOI] [PubMed] [Google Scholar]
- 43.Devuyst O, Olinger E, Rampoldi L. 2017. Uromodulin: from physiology to rare and complex kidney disorders. Nat. Rev. Nephrol 13:525. [DOI] [PubMed] [Google Scholar]
- 44.Steubl D, Block M, Herbst V, Nockher WA, Schlumberger W, et al. 2016. Plasma uromodulin correlates with kidney function and identifies early stages in chronic kidney disease patients. Medicine 95:e3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edelstein CL, Hoke TS, Somerset H, Fang W, Klein CL, et al. 2007. Proximal tubules from caspase-1-deficient mice are protected against hypoxia-induced membrane injury. Nephrol. Dial. Transpl 22:1052–61 [DOI] [PubMed] [Google Scholar]
- 46.Melnikov VY, Ecder T, Fantuzzi G, Siegmund B, Lucia MS, et al. 2001. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J. Clin. Investig 107:1145–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu H, Craft ML, Wang P, Wyburn KR, Chen G, et al. 2008. IL-18 contributes to renal damage after ischemia-reperfusion. J. Am. Soc. Nephrol 19:2331–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rovin BH, Yoshiumura T, Tan L. 1992. Cytokine-induced production of monocyte chemoattractant protein-1 by cultured human mesangial cells. J. Immunol 148:2148–53 [PubMed] [Google Scholar]
- 49.Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, et al. 1997. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Investig 100:2552–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leonard EJ, Yoshimura T. 1990. Human monocyte chemoattractant protein-1 (MCP-1). Immunol. Today 11:97–101 [DOI] [PubMed] [Google Scholar]
- 51.Yoshimura T, Leonard EJ. 1990. Secretion by human fibroblasts of monocyte chemoattractant protein-1, the product of gene JE. J. Immunol 144:2377–83 [PubMed] [Google Scholar]
- 52.Yoshimura T, Robinson EA, Tanaka S, Appella E, Leonard EJ. 1989. Purification and amino acid analysis of two human monocyte chemoattractants produced by phytohemagglutinin-stimulated human blood mononuclear leukocytes. J. Immunol 142:1956–62 [PubMed] [Google Scholar]
- 53.Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, et al. 1990. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. PNAS 87:5134–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elner SG, Strieter RM, Elner VM, Rollins BJ, Del Monte MA, Kunkel SL. 1991. Monocyte chemotactic protein gene expression by cytokine-treated human retinal pigment epithelial cells. Lab. Investig 64:819–25 [PubMed] [Google Scholar]
- 55.Wada T, Yokoyama H, Su SB, Mukaida N, Iwano M, et al. 1996. Monitoring urinary levels of monocyte chemotactic and activating factor reflects disease activity of lupus nephritis. Kidney Int. 49:761–67 [DOI] [PubMed] [Google Scholar]
- 56.Noris M, Bernasconi S, Casiraghi F, Sozzani S, Gotti E, et al. 1995. Monocyte chemoattractant protein-1 is excreted in excessive amounts in the urine of patients with lupus nephritis. Lab. Investig 73:804–9 [PubMed] [Google Scholar]
- 57.Kiyici S, Erturk E, Budak F, Ersoy C, Tuncel E, et al. 2006. Serum monocyte chemoattractant protein-1 and monocyte adhesion molecules in type 1 diabetic patients with nephropathy. Arch. Med. Res 37:998–1003 [DOI] [PubMed] [Google Scholar]
- 58.Bae E, Cha R-H, Kim YC, An JN, Kim DK, et al. 2017. Circulating TNF receptors predict cardiovascular disease in patients with chronic kidney disease. Medicine 96:e6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gohda T, Niewczas MA, Ficociello LH, Walker WH, Skupien J, et al. 2012. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J. Am. Soc. Nephrol 23:516–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Niewczas MA, Gohda T, Skupien J, Smiles AM, Walker WH, et al. 2012. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J. Am. Soc. Nephrol 23:507–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hasegawa G, Nakano K, Sawada M, Uno K, Shibayama Y, et al. 1991. Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int. 40:1007–12 [DOI] [PubMed] [Google Scholar]
- 62.Huang Y-S, Fu S-H, Lu K-C, Chen J-S, Hsieh H-Y, et al. 2017. Inhibition of tumor necrosis factor signaling attenuates renal immune cell infiltration in experimental membranous nephropathy. Oncotarget 8:111631–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fernández-Juárez G, Perez JV, Fernández JLL, Martinez-Martinez E, Cachofeiro V, et al. 2017. High levels of circulating TNFR1 increase the risk of all-cause mortality and progression of renal disease in type 2 diabetic nephropathy. Nephrology 22:354–60 [DOI] [PubMed] [Google Scholar]
- 64.Neirynck N, Glorieux G, Schepers E, Verbeke F, Vanholder R. 2015. Soluble tumor necrosis factor receptor 1 and 2 predict outcomes in advanced chronic kidney disease: a prospective cohort study. PLOS ONE 10:e0122073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee CG, Da Silva CA, Dela Cruz CS, Ahangari F, Ma B, et al. 2011. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. Physiol 73:479–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sohn MH, Kang M-J, Matsuura H, Bhandari V, Chen N-Y, et al. 2010. The chitinase-like proteins breast regression protein-39 and YKL-40 regulate hyperoxia-induced acute lung injury. Am. J. Respir. Crit. Care Med 182:918–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt IM, Hall IE, Kale S, Lee S, He C-H, et al. 2013. Chitinase-like protein Brp-39/YKL-40 modulates the renal response to ischemic injury and predicts delayed allograft function. J. Am. Soc. Nephrol 24:309–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ghoul BE, Squalli T, Servais A, Elie C, Meas-Yedid V, et al. 2010. Urinary procollagen III aminoterminal propeptide (PIIINP): a fibrotest for the nephrologist. Clin. J. Am. Soc. Nephrol 5:205–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Torres DD, Rossini M, Manno C, Mattace-Raso F, D’Altri C, et al. 2008. The ratio of epidermal growth factor to monocyte chemotactic peptide-1 in the urine predicts renal prognosis in IgA nephropathy. Kidney Int. 73:327–33 [DOI] [PubMed] [Google Scholar]
- 70.Grandaliano G, Gesualdo L, Bartoli F, Ranieri E, Monno R, et al. 2000. MCP-1 and EGF renal expression and urine excretion in human congenital obstructive nephropathy. Kidney Int. 58:182–92 [DOI] [PubMed] [Google Scholar]
- 71.Ju W, Nair V, Smith S, Zhu L, Shedden K, et al. 2015. Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci. Transl. Med 7:316ra193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mattila A-L, Viinikka L, Saario I, Perheentupa J. 1988. Human epidermal growth factor: renal production and absence from plasma. Regul. Pept 23:89–93 [DOI] [PubMed] [Google Scholar]
- 73.Lechner J, Malloth NA, Jennings P, Heckl D, Pfaller W, Seppi T. 2007. Opposing roles of EGF in IFN-α-induced epithelial barrier destabilization and tissue repair. Am. J. Physiol. Cell Physiol 293:C1843–50 [DOI] [PubMed] [Google Scholar]
- 74.Tang J, Liu N, Zhuang S. 2013. Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int. 83:804–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mathiesen ER, Nexø E, Hommel E, Parving HH. 1989. Reduced urinary excretion of epidermal growth factor in incipient and overt diabetic nephropathy. Diabetic Med. 6:121–26 [DOI] [PubMed] [Google Scholar]
- 76.Ranieri E, Gesualdo L, Petrarulo F, Schena FP. 1996. Urinary IL-6/EGF ratio: a useful prognostic marker for the progression of renal damage in IgA nephropathy. Kidney Int. 50:1990–2001 [DOI] [PubMed] [Google Scholar]
- 77.Weinstein T, Hwang D, Lev-Ran A, Ori Y, Korzets A, Levi J. 1997. Excretion of epidermal growth factor in human adult polycystic kidney disease. Israel J. Med. Sci 33:641–42 [PubMed] [Google Scholar]
- 78.Tsau YK, Chen CH. 1999. Urinary epidermal growth factor excretion in children with chronic renal failure. Am. J. Nephrol 19:400–4 [DOI] [PubMed] [Google Scholar]
- 79.Supavekin S, Zhang W, Kucherlapati R, Kaskel FJ, Moore LC, Devarajan P. 2003. Differential gene expression following early renal ischemia/reperfusion. Kidney Int. 63:1714–24 [DOI] [PubMed] [Google Scholar]
- 80.Villanueva S, Céspedes C, Vio CP. 2006. Ischemic acute renal failure induces the expression of a wide range of nephrogenic proteins. Am. J. Physiol. Regul. Integr. Comp. Physiol 290:R861–70 [DOI] [PubMed] [Google Scholar]
- 81.Bonventre JV, Yang L. 2011. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig 121:4210–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Koyner JL, Parikh CR. 2013. Clinical utility of biomarkers of AKI in cardiac surgery and critical illness. Clin. J. Am. Soc. Nephrol 8:1034–42 [DOI] [PubMed] [Google Scholar]
- 83.Parikh CR, Coca SG, Thiessen-Philbrook H, Shlipak MG, Koyner JL, et al. 2011. Postoperative biomarkers predict acute kidney injury and poor outcomes after adult cardiac surgery. J. Am. Soc. Nephrol 22:1748–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Parikh CR, Devarajan P, Zappitelli M, Sint K, Thiessen-Philbrook H, et al. 2011. Postoperative biomarkers predict acute kidney injury and poor outcomes after pediatric cardiac surgery. J. Am. Soc. Nephrol 22:1737–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Basile DP, Donohoe D, Roethe K, Osborn JL. 2001. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am. J. Physiol. Ren. Physiol 281:F887–99 [DOI] [PubMed] [Google Scholar]
- 86.Basile DP, Fredrich K, Alausa M, Vio CP, Liang M, et al. 2005. Identification of persistently altered gene expression in the kidney after functional recovery from ischemic acute renal failure. Am. J. Physiol. Ren. Physiol 288:F953–63 [DOI] [PubMed] [Google Scholar]
- 87.Koyner JL, Garg AX, Coca SG, Sint K, Thiessen-Philbrook H, et al. 2012. Biomarkers predict progression of acute kidney injury after cardiac surgery. J. Am. Soc. Nephrol 23:905–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Coca SG, Nadkarni GN, Garg AX, Koyner J, Thiessen-Philbrook H, et al. 2016. First post-operative urinary kidney injury biomarkers and association with the duration of AKI in the TRIBE-AKI cohort. PLOS ONE 11:e0161098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Coca SG, Garg AX, Thiessen-Philbrook H, Koyner JL, Patel UD, et al. 2014. Urinary biomarkers of AKI and mortality 3 years after cardiac surgery. J. Am. Soc. Nephrol 25:1063–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thygesen K, Alpert JS, White HD. 2007. Universal definition of myocardial infarction. Circulation 116:2634–53 [DOI] [PubMed] [Google Scholar]
- 91.Jotwani V, Katz R, Ix JH, Gutiérrez OM, Bennett M, et al. 2018. Urinary biomarkers of kidney tubular damage and risk of cardiovascular disease and mortality in elders. Am. J. Kidney Dis 72:205–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weinfeld MS, Chertow GM, Stevenson LW. 1999. Aggravated renal dysfunction during intensive therapy for advanced chronic heart failure. Am. Heart J 138:285–90 [DOI] [PubMed] [Google Scholar]
- 93.Knight EL, Glynn RJ, McIntyre KM, Mogun H, Avorn J. 1999. Predictors of decreased renal function in patients with heart failure during angiotensin-converting enzyme inhibitor therapy: results from the Studies of Left Ventricular Dysfunction (SOLVD). Am. Heart J 138:849–55 [DOI] [PubMed] [Google Scholar]
- 94.Krumholz HM, Chen Y-T, Vaccarino V, Wang Y, Radford MJ, et al. 2000. Correlates and impact on outcomes of worsening renal function in patients ≥65 years of age with heart failure. Am. J. Cardiol 85:1110–13 [DOI] [PubMed] [Google Scholar]
- 95.Smith GL, Vaccarino V, Kosiborod M, Lichtman JH, Cheng S, et al. 2003. Worsening renal function: What is a clinically meaningful change in creatinine during hospitalization with heart failure? J. Card. Failure 9:13–25 [DOI] [PubMed] [Google Scholar]
- 96.Testani JM, Brisco-Bacik MA. 2017. Worsening renal function and mortality in heart failure. Causality confounding? Circ. Heart Failure 10:e003835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dupont M, Shrestha K, Singh D, Awad A, Kovach C, et al. 2012. Lack of significant renal tubular injury despite acute kidney injury in acute decompensated heart failure. Eur. J. Heart Failure 14:597–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ahmad T, Jackson K, Rao VS, Tang WHW, Brisco-Bacik MA, et al. 2018. Worsening renal function in acute heart failure patients undergoing aggressive diuresis is not associated with tubular injury. Circulation 137:2016–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Velez JCQ, Kadian M, Taburyanskaya M, Bohm NM, Delay TA, et al. 2015. Hepatorenal acute kidney injury and the importance of raising mean arterial pressure. Nephron 131:191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dobre M, Demirjian S, Sehgal AR, Navaneethan SD. 2011. Terlipressin in hepatorenal syndrome: a systematic review and meta-analysis. Int. Urol. Nephrol 43:175–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Junior APN, Farias AQ, d’Albuquerque LAC, Carrilho FJ, Malbouisson LMS. 2014. Terlipressin versus norepinephrine in the treatment of hepatorenal syndrome: a systematic review and meta-analysis. PLOS ONE 9:e107466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nadim MK, Genyk YS, Tokin C, Fieber J, Ananthapanyasut W, et al. 2012. Impact of the etiology of acute kidney injury on outcomes following liver transplantation: acute tubular necrosis versus hepatorenal syndrome. Liver Transplant. 18:539–48 [DOI] [PubMed] [Google Scholar]
- 103.Belcher JM, Sanyal AJ, Peixoto AJ, Perazella MA, Lim J, et al. 2014. Kidney biomarkers and differential diagnosis of patients with cirrhosis and acute kidney injury. Hepatology 60:622–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fagundes C, Pépin M-N, Guevara M, Barreto R, Casals G, et al. 2012. Urinary neutrophil gelatinase-associated lipocalin as biomarker in the differential diagnosis of impairment of kidney function in cirrhosis. J. Hepatol 57:267–73 [DOI] [PubMed] [Google Scholar]
- 105.Belcher JM, Sanyal AJ, Peixoto AJ, Perazella MA, Lim J, et al. 2014. Kidney biomarkers and differential diagnosis of patients with cirrhosis and acute kidney injury. Hepatology 60:622–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chu R, Li C, Wang S, Zou W, Liu G, Yang L. 2014. Assessment of KDIGO definitions in patients with histopathologic evidence of acute renal disease. Clin. J. Am. Soc. Nephrol 9:1175–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reese PP, Hall IE, Weng FL, Schröppel B, Doshi MD, et al. 2015. Associations between deceased-donor urine injury biomarkers and kidney transplant outcomes. J. Am. Soc. Nephrol 27:1534–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Joo JD, Kim M, D’Agati VD, Lee HT. 2006. Ischemic preconditioning provides both acute and delayed protection against renal ischemia and reperfusion injury in mice. J. Am. Soc. Nephrol 17:3115–23 [DOI] [PubMed] [Google Scholar]
- 109.Puthumana J, Hall IE, Reese PP, Schröppel B, Weng FL, et al. 2017. YKL-40 associates with renal recovery in deceased donor kidney transplantation. J. Am. Soc. Nephrol 28:661–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Peralta CA, Katz R, Bonventre JV, Sabbisetti V, Siscovick D, et al. 2012. Associations of urinary levels of kidney injury molecule 1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) with kidney function decline in the Multi-Ethnic Study of Atherosclerosis (MESA). Am. J. Kidney Dis 60:904–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jungbauer CG, Uecer E, Stadler S, Birner C, Buchner S, et al. 2016. N-acteyl-β-D-glucosaminidase and kidney injury molecule-1: new predictors for long-term progression of chronic kidney disease in patients with heart failure. Nephrology 21:490–98 [DOI] [PubMed] [Google Scholar]
- 112.Hsu C, Xie D, Waikar SS, Bonventre JV, Zhang X, et al. 2017. Urine biomarkers of tubular injury do not improve on the clinical model predicting chronic kidney disease progression. Kidney Int. 91:196–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Foster MC, Coresh J, Bonventre JV, Sabbisetti VS, Waikar SS, et al. 2015. Urinary biomarkers and risk of ESRD in the atherosclerosis risk in communities study. Clin. J. Am. Soc. Nephrol 10:1956–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lin Hugo Y-H, Hwang D-Y, Lee Su C, Kuo H-T, Kuo M-C, et al. 2015. Urinary neutrophil gelatinase-associated lipocalin and clinical outcomes in chronic kidney disease patients. Clin. Chem. Lab. Med 53:73–83 [DOI] [PubMed] [Google Scholar]
- 115.Fufaa GD, Weil EJ, Nelson RG, Hanson RL, Bonventre JV, et al. 2015. Association of urinary KIM-1, L-FABP, NAG and NGAL with incident end-stage renal disease and mortality in American Indians with type 2 diabetes mellitus. Diabetologia 58:188–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Navarro-González JF, Mora-Fernández C. 2008. The role of inflammatory cytokines in diabetic nephropathy. J. Am. Soc. Nephrol 19:433–42 [DOI] [PubMed] [Google Scholar]
- 117.Mora C, Navarro JF. 2006. Inflammation and diabetic nephropathy. Curr. Diabetes Rep 6:463–68 [DOI] [PubMed] [Google Scholar]
- 118.Coca SG, Nadkarni GN, Huang Y, Moledina DG, Rao V, et al. 2017. Plasma biomarkers and kidney function decline in early and established diabetic kidney disease. J. Am. Soc. Nephrol 28:2786–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Conway BR, Manoharan D, Manoharan D, Jenks S, Dear JW, et al. 2012. Measuring urinary tubular biomarkers in type 2 diabetes does not add prognostic value beyond established risk factors. Kidney Int. 82:812–18 [DOI] [PubMed] [Google Scholar]
- 120.Vaidya VS, Niewczas MA, Ficociello LH, Johnson AC, Collings FB, et al. 2011. Regression of microalbuminuria in type 1 diabetes is associated with lower levels of urinary tubular injury biomarkers, kidney injury molecule-1, and N-acetyl-β-d-glucosaminidase. Kidney Int. 79:464–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Al-Lamki RS, Mayadas TN. 2015. TNF receptors: signaling pathways and contribution to renal dysfunction. Kidney Int. 87:281–96 [DOI] [PubMed] [Google Scholar]
- 122.Ledo N, Ko Y-A, Park A-SD, Kang H-M, Han S-Y, et al. 2015. Functional genomic annotation of genetic risk loci highlights inflammation and epithelial biology networks in CKD. J. Am. Soc. Nephrol 26:692–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pavkov ME, Nelson RG, Knowler WC, Cheng Y, Krolewski AS, Niewczas MA. 2015. Elevation of circulating TNF receptors 1 and 2 increases the risk of end-stage renal disease in American Indians with type 2 diabetes. Kidney Int. 87:812–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Krolewski AS, Niewczas MA, Skupien J, Gohda T, Smiles A, et al. 2014. Early progressive renal decline precedes the onset of microalbuminuria and its progression to macroalbuminuria. Diabetes Care 37:226–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Al-Lamki RS, Wang J, Vandenabeele P, Bradley JA, Thiru S, et al. 2005. TNFR1- and TNFR2-mediated signaling pathways in human kidney are cell type-specific and differentially contribute to renal injury. FASEB J. 19:1637–45 [DOI] [PubMed] [Google Scholar]
- 126.Banba N, Nakamura T, Matsumura M, Kuroda H, Hattori Y, Kasai K. 2000. Possible relationship of monocyte chemoattractant protein-1 with diabetic nephropathy. Kidney Int. 58:684–90 [DOI] [PubMed] [Google Scholar]
- 127.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH. 2006. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 69:73–80 [DOI] [PubMed] [Google Scholar]
- 128.Morii T, Fujita H, Narita T, Shimotomai T, Fujishima H, et al. 2003. Association of monocyte chemoattractant protein-1 with renal tubular damage in diabetic nephropathy. J. Diabetes Complic 17:11–15 [DOI] [PubMed] [Google Scholar]
- 129.Takebayashi K, Matsumoto S, Aso Y, Inukai T. 2006. Aldosterone blockade attenuates urinary monocyte chemoattractant protein-1 and oxidative stress in patients with type 2 diabetes complicated by diabetic nephropathy. J. Clin. Endocrinol. Metab 91:2214–17 [DOI] [PubMed] [Google Scholar]
- 130.Tam FWK, Riser BL, Meeran K, Rambow J, Pusey CD, Frankel AH. 2009. Urinary monocyte chemoattractant protein-1 (MCP-1) and connective tissue growth factor (CCN2) as prognostic markers for progression of diabetic nephropathy. Cytokine 47:37–42 [DOI] [PubMed] [Google Scholar]
- 131.Canaud G, Dejucq-Rainsford N, Avettand-Fenoel V, Viard JP, Anglicheau D, et al. 2014. The kidney as a reservoir for HIV-1 after renal transplantation. J. Am. Soc. Nephrol 25:407–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jotwani V, Scherzer R, Abraham A, Estrella MM, Bennett M, et al. 2015. Association of urine α1-microglobulin with kidney function decline and mortality in HIV-infected women. Clin. J. Am. Soc. Nephrol 10:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shlipak MG, Scherzer R, Abraham A, Tien PC, Grunfeld C, et al. 2012. Urinary markers of kidney injury and kidney function decline in HIV-infected women. J. Acquir. Immune Defic. Syndr 61:565–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jotwani V, Scherzer R, Abraham A, Estrella MM, Bennett M, et al. 2013. Does HIV infection promote early kidney injury in women? Antivir. Ther 19:79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jotwani V, Scherzer R, Estrella MM, Jacobson LP, Witt MD, et al. 2017. Association of HIV infection with biomarkers of kidney injury and fibrosis in the Multicenter AIDS Cohort Study. Antivir. Ther 22:421–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kohler JJ, Hosseini SH, Hoying-Brandt A, Green E, Johnson DM, et al. 2009. Tenofovir renal toxicity targets mitochondria of renal proximal tubules. Lab. Investig 89:513–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Herlitz LC, Mohan S, Stokes MB, Radhakrishnan J, D’Agati VD, Markowitz GS. 2010. Tenofovir nephrotoxicity: acute tubular necrosis with distinctive clinical, pathological, and mitochondrial abnormalities. Kidney Int. 78:1171–77 [DOI] [PubMed] [Google Scholar]
- 138.Jotwani V, Scherzer R, Estrella MM, Jacobson LP, Witt MD, et al. 2016. Brief report: cumulative tenofovir disoproxil fumarate exposure is associated with biomarkers of tubular injury and fibrosis in HIV-infected men. JAIDS J. Acquir. Immune Defic. Syndr 73:177–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jotwani V, Scherzer R, Estrella MM, Jacobson LP, Witt MD, et al. 2016. HIV infection, tenofovir, and urine α1-microglobulin: a cross-sectional analysis in the multicenter AIDS cohort study. Am. J. Kidney Dis 68:571–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Parikh CR, Moledina DG, Coca SG, Thiessen-Philbrook HR, Garg AX. 2016. Application of new acute kidney injury biomarkers in human randomized controlled trials. Kidney Int. 89:1372–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Molnar AO, Parikh CR, Coca SG, Thiessen-Philbrook H, Koyner JL, et al. 2014. Association between preoperative statin use and acute kidney injury biomarkers in cardiac surgery. Ann. Thoracic Surg 97:2081–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Goodsaid F, Frueh F. 2007. Biomarker qualification pilot process at the US Food and Drug Administration. AAPS J. 9:E105–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sistare FD, Dieterle F, Troth S, Holder DJ, Gerhold D, et al. 2010. Towards consensus practices to qualify safety biomarkers for use in early drug development. Nat. Biotechnol 28:446–54 [DOI] [PubMed] [Google Scholar]
- 144.Dieterle F, Sistare F, Goodsaid F, Papaluca M, Ozer JS, et al. 2010. Renal biomarker qualification submission: a dialog between the FDA-EMEA and Predictive Safety Testing Consortium. Nat. Biotechnol 28:455–62 [DOI] [PubMed] [Google Scholar]
- 145.Kimura T, Yasuda K, Yamamoto R, Soga T, Rakugi H, et al. 2016. Identification of biomarkers for development of end-stage kidney disease in chronic kidney disease by metabolomic profiling. Sci. Rep 6:26138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hocher B, Adamski J. 2017. Metabolomics for clinical use and research in chronic kidney disease. Nat. Rev. Nephrol 13:269–84 [DOI] [PubMed] [Google Scholar]
- 147.Kidney Precis. Med. Proj. 2018. Kidney Precision Medicine Project. Natl. Inst. Diabetes Dig. Kidney Dis., Bethesda, MD: https://www.niddk.nih.gov/research-funding/research-programs/kidney-precision-medicine-project-kpmp [Google Scholar]
- 148.Arthur JM, Hill EG, Alge JL, Lewis EC, Neely BA, et al. 2014. Evaluation of 32 urine biomarkers to predict the progression of acute kidney injury after cardiac surgery. Kidney Int. 85:431–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.McIlroy DR, Farkas D, Pan K, Pickering JW, Lee HT. 2018. Combining novel renal injury markers with delta serum creatinine early after cardiac surgery and risk-stratification for serious adverse outcomes: an exploratory analysis. J. Cardiothorac. Vasc. Anesth 32:2190–200 [DOI] [PubMed] [Google Scholar]
- 150.Basu RK, Wong HR, Krawczeski CD, Wheeler DS, Manning PB, et al. 2014. Combining functional and tubular damage biomarkers improves diagnostic precision for acute kidney injury after cardiac surgery. J. Am. Coll. Cardiol 64:2753–62 [DOI] [PMC free article] [PubMed] [Google Scholar]