

Introduction
Development of therapeutic strategies for immune-mediated tumor destruction, previously an elusive goal, has been accelerated by understanding the molecular basis of immune recognition and regulation of cancer cells. It is now well known that the immune system plays a pivotal role in monitoring cancer development (1–4). This concept of “cancer immunoediting” (2, 5, 6) holds that the immune system not only protects the host against primary cancer development, but also sculpts tumor immunogenicity (6). Cancer immunoediting is a dynamic process composed of three phases: elimination, equilibrium, and escape (6). Elimination represents the classical concept of cancer immunosurveillance (7, 8), equilibrium is the period of immune-mediated latency after incomplete tumor destruction (9), and escape refers to the final outgrowth of tumors that have outstripped immunological restraints (10). In support of this concept, presence of tumor-infiltrating lymphocytes (TILs) is associated with improved clinical outcome in epithelial ovarian cancer (EOC) (11–14).
Significant progress has been made in the development of antitumor immunity by initiating de-novo or boosting pre-existing immune responses; some have gained regulatory approval (Table 1). These interventions include vaccines, cell-based therapy, checkpoint blockade, and oncolytic virotherapy. Dramatic clinical responses in certain solid cancers treated with monoclonal antibodies targeting checkpoint pathways have spurred the popularity of utilizing the immune system to control unchecked tumor growth. FDA approval of checkpoint inhibitors, anti-cytotoxic T lymphocyte antigen-4 (anti-CTLA-4) and anti-programmed death-1 (anti-PD-1) for several solid tumors and sipuleucel-T for metastatic prostate cancer (15, 16) suggest these promising results may be expanded to EOC.
Table 1:
Available Anti-cancer Immunotherapies
| Approach | Licensed |
|---|---|
| Tumor targeting antibodies | Yes |
| DC vaccination | Yes |
| Peptide vaccines | Yes |
| Immunostimulatory cytokines | Yes |
| Immunomodulatory antibodies | Yes |
| Oncolytic virotherapy | Yes |
| TLR agonists | Yes |
| DNA and recombinant viral vaccines | No |
| Inhibitors of IDO, arginase | No |
| Adoptive cell therapy | No |
EOC tumor antigens and vaccine therapy
The development of approaches to analyze humoral (17) and cellular (18) immune reactivity to cancer led to the molecular characterization of tumor antigens recognized by autologous CD8+ T-cells (19) and/or antibodies (20) including serological analysis of recombinant cDNA expression libraries (SEREX) (21), differential gene expression analysis, T-cell epitope cloning (TEPIC) (22, 23), and bioinformatics (24, 25). As a consequence, human tumor antigens (TA) can be broadly classified into one or more of the following categories:
differentiation antigens [e.g. tyrosinase (26), Melan-A/MART-1 (27), gp100 (28)];
mutational antigens [e.g. CDK4 (29), β-catenin (30), caspase-8 (31), P53 (32)];
splice variant antigens [e.g. NY-CO-37/PDZ-45 (32), ING1 (35)];
glycolipid antigens;
cancer-testis antigens (CTA) [e.g. MAGE (22), NY-ESO-1 (21), LAGE-1 (38)].
These TAs give rise to epitopes presented on tumor cells in the context of major histocompatibility complex (MHC) molecules, thereby stimulating CD8+ or CD4+ T-cells.
Although there are several options in deciding which antigen to target, fundamental requirements of ideal TA include:
limited or no expression in normal tissues, but aberrant expression at high frequencies in tumor;
immunogenicity; and
a role in tumor progression.
Self-antigens
While no current self-TA completely meets all criteria, the CTA family is closest. Criteria for placing antigens in this category are (39, 40):
predominant expression in testis germ cells and generally not in other normal tissues;
expression in malignant tumors of different histological types;
expression in malignancies in a lineage non-specific fashion;
often mapping on the X-chromosome;
often members of multigene families.
Despite their poorly characterized biologic function, their expression is known to be restricted in immune privileged sites such as testes, placenta, and fetal ovary, but not in other normal tissues. Abnormal expression of these germ-line genes in malignant tumors may reflect activation of a silenced “gametogenic program,” which ultimately leads to tumor progression and broad immunogenicity (41). CTA immunogenicity has led to development of cancer vaccines targeting these antigens in many solid tumors.
Expression of 162 CTA across 53 normal samples from GTeX and 31 tumor samples from TCGA revealed a strikingly contrasting profile. Tumor samples had heterogeneous expression of CTA, while normal tissues (beside testis) had minimal or absent CTA expression (Figure 1). These results strongly suggest CTA can be exquisite candidates for immune targeting in EOC. Furthermore, as their transcription is epigenetically regulated (42–44), there are opportunities to reinstate CTA expression with DNA methyltransferase inhibitors.
Figure 1.
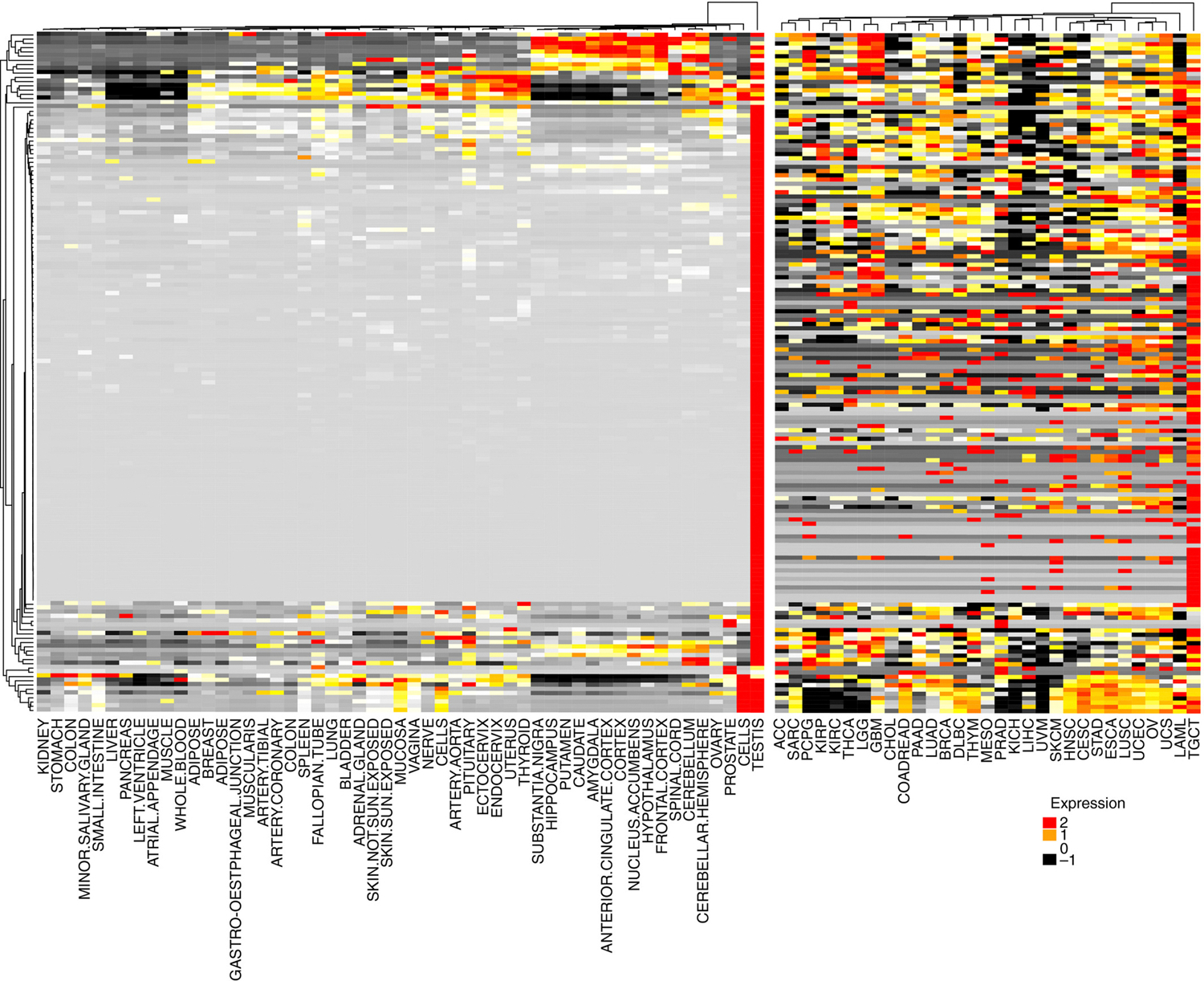
Expression pattern of 162 CT genes across normal tissues from GTeX and patient tumor samples from TCGA. RNASeq data were obtained from GTeX (normal tissues) or TCGA PANCancer study (tumors). The median expression per each CT gene was calculated across all patients in a specific tissue. Each cell in the heatmap indicates the median expression of a CT gene in the tissue indicated at the bottom of the figure. Red cells indicate high expression and black cells low expression levels.
From Want MY, Lugade AA, Battaglia S, et al. Nature of tumour rejection antigens in ovarian cancer. Immunology 2018; with permission.
The identification and characterization of peptide epitopes from TAs, along with the relative ease of cGMP-grade peptide production, led to a large number of vaccine studies utilizing these peptide epitopes in EOC (Table 2). The most common cancer vaccine strategy is to administer full-length recombinant protein or peptides, most often via intramuscular, subcutaneous, or intradermal route, together with one or more immunostimulatory adjuvants. While short peptides (8–12 a.a.) directly bind to surface MHC, synthetic long peptides (25–30 a.a.) are endocytosed, processed, and presented to elicit an immune response (45). Several reports indicate therapeutic activity of synthetic long peptides is superior to that of their shorter counterparts, especially when they include epitopes recognized by both cytotoxic and helper T-cells or when conjugated to adjuvants (45, 46).
Table 2.
Selected cancer vaccine studies
| Antigen | Phase | Disease | Technology | Co-therapy | Sponsor | Reference |
|---|---|---|---|---|---|---|
| NY-ESO-1 | I | Metastatic cancer | Recombinant protein | GLA -SE | Immune Design | NCT02015416 |
| I | Ovarian, fallopian tube cancer | DEC-205 Fusion protein | Poly-ICLC, IDO1 inhibitor | Roswell Park Cancer Institute | NCT02166905 | |
| I | NY-ESO-1 expressing solid tumors | DEC-205 Fusion protein/Dendritic ell | Rapamycin | Roswell Park Cancer Institute | NCT01522820 | |
| I/II | NY-ESO-1 expressing tumors | DEC-205 Fusion protein | Resiquimod, Poly-ICLC | Celldex Therapeutics | NCT00948961 | |
| I | NY-ESO-1 expressing tumors | Full length protein | Montanide, Resiquimod | Mount Sinai School of Medicine | NCT00821652 | |
| I | Ovarian, fallopian, primary peritoneal cancer | Peptide | Decitabine, Doxorubicin, Montanide | Roswell Park Cancer Institute | NCT01673217 | |
| I | NY-ESO-1/LAGE-1 expressing tumors | Peptide | CpG7909, Montanide | Ludwig Institute for Cancer Research | NCT00199836 | |
| I | Ovarian, fallopian, primary peritoneal cancer | Peptide | Montanide | Memorial Sloan Kettering Cancer Center | NCT00066729 | |
| I | Prostate cancer | Peptide | Baylor College of Medicine | NCT00616291 | ||
| I | Ovarian, fallopian, primary peritoneal cancer | Overlapping Long peptides (OLP4) | Montanide, Poly-ICLC | Ludwig Institute for Cancer Research | NCT00616941 | |
| I | Ovarian, fallopian, primary peritoneal cancer | Vector (ALVAC(2)-NY-ESO-1(M) TRICOM) | GM-CSF, Rapamycin | Roswell Park Cancer Institute | NCT01536054 | |
| I | Ovarian, fallopian, primary peritoneal cancer | Vector (ALVAC(2)-NY-ESO-1(M) TRICOM) | GM-CSF | Ludwig Institute for Cancer Research | NCT00803569 | |
| II | Ovarian, fallopian, primary peritoneal cancer | Vector (Fowlpox-NY-ESO-1) | Recombinant Vaccinia-NY-ESO_1 | Ludwig Institute for Cancer Research | NCT00112957 |
Several NY-ESO-1 vaccine clinical trials have demonstrated clinical activity, but these studies were small and not definitive (47, 48). At present, no peptide- or DNA-based anticancer vaccine is currently FDA approved. Nevertheless, a recent retrospective analysis of EOC patients with NY-ESO-1 positive tumors indicated that vaccination targeting the antigen led to improvement in overall survival (OS) by >2 years (49).
Several forms of DC-based vaccine approaches have been developed, most involving isolation of circulating monocytes and their differentiation ex-vivo, in the presence of agents that promote DC maturation. The autologous DCs are injected into patients upon exposure to tumor antigen and thus elicit tumor-specific immune responses in-vivo. Another strategy is fusion of TA with mAbs that bind endocytosis receptors (e.g., CD206, DEC-205) on the surface of DCs (50).
Non-self neoantigens
Advances in next-generation sequencing (NGS) and epitope prediction now permit rapid identification of mutational neoantigens. This has led to efforts in utilizing neoantigens for personalized cancer immunotherapies. Indirect support for this approach comes from studies demonstrating:
infusion of autologous ex-vivo expanded TILs can induce objective clinical responses in melanoma (51), and
the relationship between pretherapy CD8+ T-cell infiltrates and response to checkpoint blockade in melanoma (52).
NGS permit identification of mutations present within the tumor exome allowing for neoantigen prediction. Several pre-clinical and clinical studies have now confirmed the possibility of identifying neoantigens on this basis (53–57). Although there are limitations of probing the mutational profile of tumor in a single biopsy (58, 59), it is evident the vast majority of neoantigens occur within exonic sequence and do not lead to formation of neoantigens recognized by autologous T-cells (59, 60). Consequently, a robust pipeline for filtering cancer exome data is essential. Stimulation of neoantigen-specific T-cell responses in cancer patients can be accomplished using two possible approaches. The first is to synthesize long peptide vaccines that encode a set of predicted neoantigens. The second approach is to identify and expand pre-existing neoantigen-specific T-cell populations to create either bulk neoantigen-specific T-cell products or TCR-engineered T-cells for adoptive therapy. This latter approach was recently tested in a pilot clinical trial of autologous DCs pulsed with oxidized autologous whole-tumor cell lysate (OCDC), which was injected intranodally in platinum-treated, recurrent EOC patients (61).
Immune inhibitory network and immune checkpoint inhibitors in EOC
A major barrier to successful cancer immunotherapy is an immunosuppressive TME. Even if large numbers of tumor-specific T-cells are generated in patients by active immunization or adoptive transfer, these T-cells may not readily destroy tumor targets. In EOC, some of the major mechanisms that subvert anti-tumor immunity in the TME include Tregs (11, 62), MDSC (63–65), inhibitory cytokines such as transforming growth factor-β (TGFβ) (66), immune checkpoint receptors (67–70), and indoleamine-2,3-dioxygenase (IDO) (71–73). This redundant immunosuppressive network may pose an impediment to immunotherapy, thus facilitating tumor progression.
Emerging evidence suggests that inhibitory receptor expression on TA-specific T-cells is one mechanism by which tumors evade immunosurveillance (74). Although inhibitory receptor blockade has shown significant promise (75–77), recent studies indicate that multiple inhibitory receptors are often co-expressed on TA-specific T-cells (78). In human EOC, TA-specific CD8+ T-cells co-expressing PD-1 and LAG-3 exhibit significantly impaired IFN-γ and TNF-α production compared with single positive cells (70). Simultaneous blockade of both receptors restored effector function of these TA-specific T-cells to a level above single receptor blockade (70). In an EOC mouse model, synergistic blockade of LAG-3 and PD-1 enhanced CD8+ TIL function and promoted tumor control, while single-agent blockade had little or no effect (79).
Immune modulation is designed to reinstate an existing anticancer immune response or elicit novel responses as a result of antigen spreading. This has been achieved through four general strategies:
inhibition of immunosuppressive receptors expressed by activated T-cells;
inhibition of the principal ligands of these receptors;
activation of co-stimulatory receptors expressed by effector T-cells; and
neutralization of immunosuppressive mediators in the TME.
The first published data supporting checkpoint blockade as a potentially valuable therapeutic approach in EOC were trials of nivolumab (67), and BMS-93655 (anti-PD-L1) (75). In 20 nivolumab-treated patients in whom responses could be evaluated, the best overall response was 15% and the disease control rate was 45%. Two additional trials using avelumab and pembrolizumab were presented at the annual ASCO meeting in 2015. Of 75 heavily pre-treated avelumab-treated EOC patients, 8 patients experienced partial responses, 33 patients had stable disease, and there were no complete responses, with a disease control rate of 54.7% (80). In another Phase-1b study, 26 heavily pre-treated EOC patients with PD-L1 expression ≥1% on tumor cells were treated with pembrolizumab. The results showed one complete response, two partial responses and six patients with stable disease, corresponding to a disease control rate of 34.6% (81).
While these results are promising, the mechanism(s) of resistance to immune checkpoints in EOC are unclear. It is possible redundant immunosuppressive mechanisms counteract the beneficial effects of checkpoint blockade. Interestingly, a recent study in a murine EOC model showed anti-PD-1 monotherapy resulted in compensatory induction of other checkpoints, a feedback loop further contributing to immunosuppression (82). Additional checkpoint blockade agents are in various phases of clinical development, including anti-LAG-3 and anti-TIM3. Finally, emerging evidence suggests that the clinical efficacy of checkpoint blockade may be profoundly influenced by the mutational burden and “neoantigens” specific to the neoplasm (83), as higher neoantigen load leads to recruitment of a diverse repertoire of neoantigen-specific T-cells, leading to more effective tumor control.
Another critical tolerogenic mechanism in EOC is mediated by IDO, an immunoregulatory enzyme that contributes to profound immunosuppression (72). IDO catalyzes the rate-limiting step of tryptophan degradation. Reduction in local tryptophan levels and the production of tryptophan catabolites both contribute to immunosuppression (84), culminating in negative effects on T-cell proliferation, function, and survival. IDO activity also promotes the differentiation of naïve T-cells to Tregs (85). Since increased Treg activity has been shown to promote tumor growth and Treg depletion has been shown to allow an otherwise ineffectual anti-tumor immune response to occur (62), IDO expansion of Tregs provides an additional immunosuppressive mechanism.
In addition to directly inhibiting IDO enzymatic activity, second-generation IDO1 inhibitors such as epacadostat and navoximod have entered clinical trials due to their favorable pharmacokinetic profile. Phase I clinical trials with these orally available compounds have demonstrated safety and biological efficacy based on reversal of tryptophan depletion (86). A recently completed trial (NCT02042430) sought to determine the magnitude by which epacadostat alters CD8+ TIL frequency when administrated prior to surgery in newly diagnosed stage III-IV EOC patients. Another approach tests whether concomitant IDO inhibition and NY-ESO-1 vaccination will enhance the generation of durable antitumor CD8+ T-cells in EOC patients (NCT02166905).
Adoptive Cellular Therapy
Among various immunotherapeutic approaches, adoptive T cell therapy (ACT) has resulted in objective responses in the majority of treated patients (87). ACT approaches involve:
the collection of circulating T-cells or TILs (88);
modification and/or expansion ex-vivo; and
their re-infusion to patients after lymphodepleting chemotherapy.
Initial studies demonstrating the potential of T-cell immunotherapy to eradicate solid tumors came from the NCI in adoptive transfer studies of TILs (89, 90). Unfortunately, methods of isolating and manufacturing TILs are labor intensive and only successful in a subset of patients (91, 92). In order to improve the therapeutic potential of ACT, peripheral blood lymphocytes can be genetically modified to express: (i) a TA-specific T-cell receptor (TCR) (87), or (ii) “chimeric antigen receptor” (CAR) expressing the TA-binding domain of an immunoglobulin linked to costimulatory molecules (93). In EOC, targets for the TCR approach include NY-ESO-1, MAGE-A4, and WT1. Targets for CAR-T-cells include MUC16, mesothelin, and folate receptor. Several studies are ongoing or completed testing CD8TCR-engineered T-cells in patients. Although spectacular responses have been observed, the majority of clinical responses are short-lived with ultimate tumor relapse. One explanation for this sub-optimal outcome is the relatively limited long-term survival and effector function due to suppression or exhaustion of infused T-cells.
Previous ACT trials have focused on CD8TCR but not CD4TCR. Because CD4+ T-cells maintain CD8+ T-cell responses (94, 95) and rescue exhausted T-cells (96), long-lasting anti-tumor responses are expected by the synergy of CD8TCR− and CD4TCR-engineered T-cells. Recently, two types of TA-specific CD4+ T-cells, tumor-recognizing or non-tumor-recognizing, have been identified that play distinct roles in the TME (97). Though both recognize NY-ESO-1 presented by APCs, only tumor-recognizing CD4 directly recognize cancer cells in an antigen/MHC-restricted manner (97, 98).
Oncolytic Virus-based therapy
Oncolytic viruses (OV) are non-pathogenic viral strains that specifically infect cancer cells, triggering their demise. The anti-neoplastic potential of OV can be innate via a cytopathic effect or by mediating oncolysis due to expression of gene products that are potentially lethal for the host cell. Increasing preclinical and clinical evidence indicate that the therapeutic activity of oncolytic viruses is also related to their ability to elicit immune responses as they (i) reprogram the inflammatory TME to be more immunogenic and (ii) promote the release of TA. OVs can be genetically engineered to endow them with additional attributes, such as antagonism of chemokine receptors (99).
Results from a study testing talimogene laherparepvec (T-VEC), a modified herpes simplex virus type-1, were recently reported (100). Researchers randomized 436 patients with aggressive, inoperable melanoma to receive either T-VEC or a control immunotherapy. 16.3% of the group given T-VEC showed a durable response of >6 months, with some responses extending past three years.
Combination of ACT and OV may have beneficial synergistic effect. In mouse models, it has been shown that T-cells loaded with oncolytic vesicular stomatitis virus efficiently delivered the virus to metastatic lymph nodes leading to tumor clearance (101). The loading of antigen-specific T-cells with vesicular stomatitis virus enhanced the delivery of the virus to lung tumors (102) and the associated pro-inflammatory TME enhanced antigen-specific T-cell proliferation and survival within the tumor.
Conclusions and Future Directions
Cancer immunotherapy is evolving quickly and understanding the dynamics of the antitumor immune response, especially in regards to immunosuppression and counter-regulation, will lead to development of effective personalized targeted approaches. A future direction for EOC is to develop approaches based on shared antigens and the patient’s neo-antigenome. This will require a pipeline for rapid and reliable neoantigen identification, consisting of a multidisciplinary team of clinicians and bioinformaticians. Recent examples of “off-the-shelf” tumor antigens and engineered T-cells are limited to the KRAS G12D TCR (103–105). Identifying other pairs of antigen moieties and TCR sequences could offer great benefits for ACT and vaccination strategies in EOC.
Immunotherapy mediates tumor destruction, but also triggers coordinated induction of counter-regulatory and suppressive pathways. Concomitant blockade of suppressive pathways at the time of vaccination or T-cell transfer will allow inflammation-induced transformation of the TME from a tolerogenic to an immunogenic milieu. Based on promising results of PD-1/PD-L1 pathway blockade, it is important to consider opportunities for combination therapies. These include PD-1/PD-L1 blockade with anti-CTLA-4 or anti-LAG-3, an approach that has demonstrated excellent results in pre-clinical models of ovarian cancer and melanoma (70, 79, 106–110). Additional potential combinations include targeted agents (e.g. BRAF and EGFR targeted agents) (111), blocking IDO, chemotherapies with potential to cause immunogenic cell death, and vaccine combinations.
Key points:
Harnessing the immune system to eradicate established tumors is emerging as a viable and efficacious therapy for advanced ovarian cancer.
Shared tumor antigens, such as NY-ESO-1, or patient-specific mutational neoantigens are attractive targets for initiation of T-cell responses.
The presence of redundant and compensatory immune checkpoint pathways indicate that combinatorial checkpoint blockade may be required for effective tumor control in ovarian cancer.
Addressing the molecular mechanisms governing poor in-vivo persistence of engineered T-cells will enhance the therapeutic potential of adoptive T-cell therapy.
Synopsis.
Clinical progress in the field of cancer immunotherapy has been slow for many years but within the last 5 years, breakthrough successes have brought immunotherapy to the forefront in cancer therapy. Promising results have been observed in solid tumors and hematological malignancies with adoptive cell therapy using tumor-infiltrating lymphocytes (TILs), host cells genetically engineered with antitumor T-cell receptors (TCRs) or chimeric antigen receptors (CARs), immune checkpoint inhibitors, and oncolytic virotherapy. However, most treatment modalities have shown limited efficacy when utilized as monotherapy. The complex nature of cancer with intra- and inter-tumor antigen and genomic heterogeneity, coupled with the immunosuppressive microenvironment, emphasizes the potential need to personalize immunotherapy by manipulating the patient’s own immune system against cancer. For successful and long-lasting cure of cancer, a multi-modal approach is essential, combining antitumor cell therapy with manipulation of multiple pathways in the tumor microenvironment (TME) to ameliorate tumor-induced immunosuppression.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Doll R, Kinlen L. Immunosurveillance and cancer: epidemiological evidence. British medical journal. 1970;4:420–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8. [DOI] [PubMed] [Google Scholar]
- 3.Galon J, Angell HK, Bedognetti D, et al. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39:11–26. [DOI] [PubMed] [Google Scholar]
- 4.Slaney CY, Rautela J, Parker BS. The emerging role of immunosurveillance in dictating metastatic spread in breast cancer. Cancer research. 2013;73:5852–7. [DOI] [PubMed] [Google Scholar]
- 5.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60. [DOI] [PubMed] [Google Scholar]
- 6.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–48. [DOI] [PubMed] [Google Scholar]
- 7.Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. [DOI] [PubMed] [Google Scholar]
- 8.Street SE, Hayakawa Y, Zhan Y, et al. Innate immune surveillance of spontaneous B cell lymphomas by natural killer cells and gammadelta T cells. J Exp Med. 2004;199:879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacKie RM, Reid R, Junor B. Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N Engl J Med. 2003;348:567–8. [DOI] [PubMed] [Google Scholar]
- 10.Marincola FM, Jaffee EM, Hicklin DJ, et al. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000;74:181–273. [DOI] [PubMed] [Google Scholar]
- 11.Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci. 2005;102:18538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13. [DOI] [PubMed] [Google Scholar]
- 13.Ovarian Tumor Tissue Analysis C. Dose-Response Association of CD8+ Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol. 2017;3:e173290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang WT, Adams SF, Tahirovic E, et al. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol. 2012;124:192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carey TE, Takahashi T, Resnick LA, et al. Cell surface antigens of human malignant melanoma: mixed hemadsorption assays for humoral immunity to cultured autologous melanoma cells. Proc Natl Acad Sci. 1976;73:3278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knuth A, Danowski B, Oettgen HF, et al. T-cell-mediated cytotoxicity against autologous malignant melanoma: analysis with interleukin 2-dependent T-cell cultures. Proc Natl Acad Sci. 1984;81:3511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–7. [DOI] [PubMed] [Google Scholar]
- 20.Sahin U, Tureci O, Schmitt H, et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc Natl Acad Sci. 1995;92:11810–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen YT, Scanlan MJ, Sahin U, et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci. 1997;94:1914–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boon T, van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med. 1996;183:725–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van den Eynde BJ, Boon T. Tumor antigens recognized by T lymphocytes. Int J Clin Lab Res. 1997;27:81–6. [DOI] [PubMed] [Google Scholar]
- 24.Scanlan MJ, Altorki NK, Gure AO, et al. Expression of cancer-testis antigens in lung cancer: definition of bromodomain testis-specific gene (BRDT) as a new CT gene, CT9. Cancer Lett. 2000;150:155–64. [DOI] [PubMed] [Google Scholar]
- 25.Alpen B, Gure AO, Scanlan MJ, et al. A new member of the NY-ESO-1 gene family is ubiquitously expressed in somatic tissues and evolutionarily conserved. Gene. 2002;297:141–9. [DOI] [PubMed] [Google Scholar]
- 26.Brichard V, Van Pel A, Wolfel T, et al. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med. 1993;178:489–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coulie PG, Brichard V, Van Pel A, et al. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med. 1994;180:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawakami Y, Eliyahu S, Jennings C, et al. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol. 1995;154:3961–8. [PubMed] [Google Scholar]
- 29.Wolfel T, Hauer M, Schneider J, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–4. [DOI] [PubMed] [Google Scholar]
- 30.Robbins PF, El-Gamil M, Li YF, et al. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med. 1996;183:1185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandruzzato S, Brasseur F, Andry G, et al. A CASP-8 mutation recognized by cytolytic T lymphocytes on a human head and neck carcinoma. J Exp Med. 1997;186:785–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scanlan MJ, Chen YT, Williamson B, et al. Characterization of human colon cancer antigens recognized by autologous antibodies. Int J Cancer. 1998;76:652–8. [DOI] [PubMed] [Google Scholar]
- 33.Cheever MA, Disis ML, Bernhard H, et al. Immunity to oncogenic proteins. Immunol Rev. 1995;145:33–59. [DOI] [PubMed] [Google Scholar]
- 34.Gnjatic S, Cai Z, Viguier M, et al. Accumulation of the p53 protein allows recognition by human CTL of a wild-type p53 epitope presented by breast carcinomas and melanomas. J Immunol. 1998;160:328–33. [PubMed] [Google Scholar]
- 35.Jager E, Jager D, Knuth A. CTL-defined cancer vaccines: perspectives for active immunotherapeutic interventions in minimal residual disease. Cancer Metastasis Rev. 1999;18:143–50. [DOI] [PubMed] [Google Scholar]
- 36.Tindle RW. Human papillomavirus vaccines for cervical cancer. Curr Opin Immunol. 1996;8:643–50. [DOI] [PubMed] [Google Scholar]
- 37.Lennette ET, Winberg G, Yadav M, et al. Antibodies to LMP2A/2B in EBV-carrying malignancies. Eur J Cancer. 1995;31A:1875–8. [DOI] [PubMed] [Google Scholar]
- 38.Lethe B, Lucas S, Michaux L, et al. LAGE-1, a new gene with tumor specificity. Int J Cancer. 1998;76:903–8. [DOI] [PubMed] [Google Scholar]
- 39.Tureci O, Sahin U, Zwick C, et al. Identification of a meiosis-specific protein as a member of the class of cancer/testis antigens. Proc Natl Acad Sci. 1998;95:5211–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Old LJ. Cancer/testis (CT) antigens - a new link between gametogenesis and cancer. Cancer Immunity. 2001;1:1. [PubMed] [Google Scholar]
- 41.Simpson AJ, Caballero OL, Jungbluth A, et al. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–25. [DOI] [PubMed] [Google Scholar]
- 42.Akers SN, Odunsi K, Karpf AR. Regulation of cancer germline antigen gene expression: implications for cancer immunotherapy. Future Oncol. 2010;6:717–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woloszynska-Read A, James SR, Link PA, et al. DNA methylation-dependent regulation of BORIS/CTCFL expression in ovarian cancer. Cancer Immun. 2007;7:21. [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang W, Barger CJ, Link PA, et al. DNA hypomethylation-mediated activation of Cancer/Testis Antigen 45 (CT45) genes is associated with disease progression and reduced survival in epithelial ovarian cancer. Epigenetics. 2015;10:736–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. 2008;8:351–60. [DOI] [PubMed] [Google Scholar]
- 46.Odunsi K, Qian F, Matsuzaki J, et al. Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T cell responses in ovarian cancer. Proc Natl Acad Sci. 2007;104:12837–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Odunsi K, Matsuzaki J, Karbach J, et al. Efficacy of vaccination with recombinant vaccinia and fowlpox vectors expressing NY-ESO-1 antigen in ovarian cancer and melanoma patients. Proc Natl Acad Sci. 2012;109:5797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davis ID, Chen W, Jackson H, et al. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4+ and CD8+ T cell responses in humans. Proc Natl Acad Sci. 2004;101:10697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szender JB, Papanicolau-Sengos A, Eng KH, et al. NY-ESO-1 expression predicts an aggressive phenotype of ovarian cancer. Gynecol Oncol. 2017;145:420–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsuji T, Matsuzaki J, Kelly MP, et al. Antibody-targeted NY-ESO-1 to mannose receptor or DEC-205 in vitro elicits dual human CD8+ and CD4+ T cell responses with broad antigen specificity. J Immunol. 2011;186:1218–27. [DOI] [PubMed] [Google Scholar]
- 51.Dudley ME, Gross CA, Somerville RP, et al. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. Jour clin oncol. 2013;31:2152–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castle JC, Kreiter S, Diekmann J, et al. Exploiting the mutanome for tumor vaccination. Cancer research. 2012;72:1081–91. [DOI] [PubMed] [Google Scholar]
- 54.Duan F, Duitama J, Al Seesi S, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med. 2014;211:2231–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wick DA, Webb JR, Nielsen JS, et al. Surveillance of the tumor mutanome by T cells during progression from primary to recurrent ovarian cancer. Clin canc res. 2014;20:1125–34. [DOI] [PubMed] [Google Scholar]
- 57.Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Linnemann C, vanBuuren MM, Bies L, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat med. 2015;21:81–5. [DOI] [PubMed] [Google Scholar]
- 60.Lu YC, Yao X, Crystal JS, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin canc res. 2014;20:3401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanyi JL, Bobisse S, Ophir E, et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med. 2018;10. [DOI] [PubMed] [Google Scholar]
- 62.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat med. 2004;10:942–9. [DOI] [PubMed] [Google Scholar]
- 63.Godoy HE, Khan AN, Vethanayagam RR, et al. Myeloid-derived suppressor cells modulate immune responses independently of NADPH oxidase in the ovarian tumor microenvironment in mice. PloS one. 2013;8:e69631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Obermajer N, Muthuswamy R, Odunsi K, et al. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011;71:7463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horikawa N, Abiko K, Matsumura N, et al. Expression of Vascular Endothelial Growth Factor in Ovarian Cancer Inhibits Tumor Immunity through the Accumulation of Myeloid-Derived Suppressor Cells. Clin canc res. 2016; [DOI] [PubMed]
- 66.Govindaraj C, Scalzo-Inguanti K, Madondo M, et al. Impaired Th1 immunity in ovarian cancer patients is mediated by TNFR2+ Tregs within the tumor microenvironment. Clin immunol. 2013;149:97–110. [DOI] [PubMed] [Google Scholar]
- 67.Hamanishi J, Mandai M, Ikeda T, et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol. 2015;33:4015–22. [DOI] [PubMed] [Google Scholar]
- 68.Hamanishi J, Mandai M, Iwasaki M, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci. 2007;104:3360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamanishi J, Mandai M, Konishi I. Immune checkpoint inhibition in ovarian cancer. International immunology. 2016;28:339–48. [DOI] [PubMed] [Google Scholar]
- 70.Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci. 2010;107:7875–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Inaba T, Ino K, Kajiyama H, et al. Role of the immunosuppressive enzyme indoleamine-2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol Oncol. 2009;115:185–92. [DOI] [PubMed] [Google Scholar]
- 72.Qian F, Villella J, Wallace PK, et al. Efficacy of levo-1-methyl tryptophan and dextro-1-methyl tryptophan in reversing indoleamine-2,3-dioxygenase-mediated arrest of T-cell proliferation in human epithelial ovarian cancer. Cancer Res. 2009;69:5498–504. [DOI] [PubMed] [Google Scholar]
- 73.Takao M, Okamoto A, Nikaido T, et al. Increased synthesis of indoleamine-2,3-dioxygenase protein is positively associated with impaired survival in patients with serous-type, but not with other types of, ovarian cancer. Oncology reports. 2007;17:1333–9. [PubMed] [Google Scholar]
- 74.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Callahan MK, Wolchok JD, Allison JP. Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy. Semin oncol. 2010;37:473–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baitsch L, Legat A, Barba L, et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PloS one. 2012;7:e30852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang RY, Eppolito C, Lele S, et al. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. 2015;6:27359–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Disis ML, Patel MR, Pant S, et al. Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with previously treated, recurrent or refractory ovarian cancer: A phase Ib, open-label expansion trial. Jour clin oncol. 2015;33:5509 [Google Scholar]
- 81.Varga A, Piha-Paul SA, Ott PA, et al. Antitumor activity and safety of pembrolizumab in patients with PD-L1 positive advanced ovarian cancer: Interim results from a phase Ib study. Jour clin oncol. 2015;33:5510 [Google Scholar]
- 82.Huang R, Francois A, McGray AJR, et al. Compensatory upregulation of PD-1, LAG-3 and CTLA-4 limits the efficacy of single agent checkpoint blockade in metastatic ovarian cancer. OncoImmunology. 2017; [DOI] [PMC free article] [PubMed]
- 83.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Munn DH. Indoleamine 2,3-dioxygenase, tumor-induced tolerance and counter-regulation. Curr Opin Immunol. 2006;18:220–5. [DOI] [PubMed] [Google Scholar]
- 85.Fallarino F, Grohmann U, You S, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176:6752–61. [DOI] [PubMed] [Google Scholar]
- 86.Liu X, Shin N, Koblish HK, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115:3520–30. [DOI] [PubMed] [Google Scholar]
- 87.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rosenberg SA, Restifo NP, Yang JC, et al. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. Jour clin oncol. 2005;23:2346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dudley ME, Wunderlich JR, Shelton TE, et al. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. Jour immunother. 2003;26:332–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dudley ME, Yang JC, Sherry R, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. Jour clin oncol. 2008;26:5233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. Jour virol. 1994;68:8056–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schoenberger SP, Toes RE, vanderVoort EI, et al. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–3. [DOI] [PubMed] [Google Scholar]
- 96.Aubert RD, Kamphorst AO, Sarkar S, et al. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci. 2011;108:21182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matsuzaki J, Tsuji T, Luescher I, et al. Nonclassical antigen-processing pathways are required for MHC class II-restricted direct tumor recognition by NY-ESO-1-specific CD4(+) T cells. Canc immunol res. 2014;2:341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tsuji T, Matsuzaki J, Caballero OL, et al. Heat shock protein 90-mediated peptide-selective presentation of cytosolic tumor antigen for direct recognition of tumors by CD4(+) T cells. J Immunol. 2012;188:3851–8. [DOI] [PubMed] [Google Scholar]
- 99.Gil M, Komorowski MP, Seshadri M, et al. CXCL12/CXCR4 blockade by oncolytic virotherapy inhibits ovarian cancer growth by decreasing immunosuppression and targeting cancer-initiating cells. J Immunol. 2014;193:5327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. Jour clin oncol. 2015; [DOI] [PubMed]
- 101.Qiao J, Kottke T, Willmon C, et al. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat med. 2008;14:37–44. [DOI] [PubMed] [Google Scholar]
- 102.Qiao J, Wang H, Kottke T, et al. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene therapy. 2008;15:604–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med. 2016;22:26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tran E, Robbins PF, Lu YC, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016;375:2255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Woo SR, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer research. 2012;72:917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kelleher RJ, Balu-Iyer S, Loyall JL, et al. Extracellular Vesicles Present in Human Ovarian Tumor Microenvironments Induce a Phosphatidylserine Dependent Arrest in the T Cell Signaling Cascade. Canc immunol res. 2015; [DOI] [PMC free article] [PubMed]
- 109.Mikucki ME, Fisher DT, Matsuzaki J, et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun. 2015;6:7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zsiros E, Tsuji T, Odunsi K. Adoptive T-cell therapy is a promising salvage approach for advanced or recurrent metastatic cervical cancer. J Clin Oncol. 2015;33:1521–2. [DOI] [PubMed] [Google Scholar]
- 111.Larkin J, Lao CD, Urba WJ, et al. Efficacy and Safety of Nivolumab in Patients With BRAF V600 Mutant and BRAF Wild-Type Advanced Melanoma: A Pooled Analysis of 4 Clinical Trials. JAMA Oncol. 2015;1:433–40. [DOI] [PubMed] [Google Scholar]