

Abstract
Background & Aims:
Chronic colonic inflammation leads to dysplasia and cancer in patients with inflammatory bowel disease (IBD). We have described the critical role of innate immune signaling via toll-like receptor 4 (TLR4) in the pathogenesis of dysplasia and cancer. In the current study, we interrogate the intersection of TLR4 signaling, epithelial redox activity, and the microbiota in colitis-associated neoplasia.
Methods:
IBD and colorectal cancer datasets were analyzed for expression of TLR4, dual oxidase 2 (DUOX2), and NADPH oxidase 1 (NOX1). Epithelial production of hydrogen peroxide (H2O2) was analyzed in murine colonic epithelial cells and colonoid cultures. Colorectal cancer models were carried out in villin-TLR4 mice, carrying a constitutively active form of TLR4, their littermates, and villin-TLR4 mice backcrossed to DUOXA-KO mice. The role of the TLR4-shaped microbiota in tumor development was tested in wild-type germ-free mice.
Results:
Activation of epithelial TLR4 was associated with upregulation of DUOX2 and NOX1 in IBD and colorectal cancer. DUOX2 was exquisitely dependent on TLR4 signaling and mediated the production of epithelial H2O2. Epithelial H2O2 was significantly increased in villin-TLR4 mice; TLR4-dependent tumorigenesis required the presence of DUOX2 and a microbiota. Mucosa-associated microbiota transferred from villin-TLR4 mice to wild-type germ-free mice caused increased H2O2 production and tumorigenesis.
Conclusions:
Increased TLR4 signaling in colitis drives expression of DUOX2 and epithelial production of H2O2. The local milieu imprints the mucosal microbiota and imbues it with pathogenic properties demonstrated by enhanced epithelial ROS and increased development of colitis-associated tumors. The interrelationship between epithelial ROS and tumor-promoting microbiota requires a two-pronged strategy to reduce the risk of dysplasia in colitis patients.
Keywords: Ulcerative colitis, NADPH oxidases, colitis-associated cancer, microbiome
Graphical Abstract
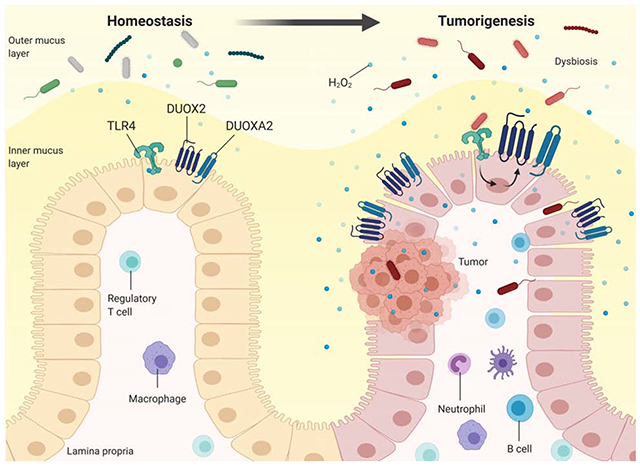
Lay Summary
Innate immune signaling in response to the mucosal microbiota drives epithelial reactive oxygen species to cause colitis-associated cancer.
Background
Inflammatory bowel diseases (IBD) are common and are increasing in incidence as more parts of the world are becoming industrialized1. Patients with long-standing ulcerative colitis (UC) are at increased risk of developing colorectal cancer (CRC) when compared to the general population2. As prevalence of IBD increases, this raises concern for an increase in world-wide cases of colitis-associated CRC (CAC)3, 4. The intensity, extent, and duration of inflammation all contribute to an increased risk of cancer5. The most recent population-based studies estimate that 15-20% of IBD patients with pancolitis develop CAC after 10 years of disease3, 4. CAC has a poorer survival than sporadic CRC in advanced and metastatic stages, and accounts for 15% of the mortality in IBD patients6, 7. Yet, progression from dysplasia to cancer is poorly understood.
Two hallmarks of IBD and CAC are the abnormal host response towards the microbiota8, 9 and the alterations in the microbial composition, termed dysbiosis10, 11. In humans it is difficult to tease out whether inflammation or dysbiosis comes first12. One host-dependent mechanism proposed to induce alterations in the microbiota at a compositional and functional level is the production of reactive oxygen species (ROS)13, 14. Whereas ROS production during inflammation has been largely attributed to infiltrating immune cells, the release of reactive oxygen intermediates by the epithelium has not been thoroughly explored. Epithelial ROS are produced as byproducts of mitochondrial activity or via NADPH oxidases, which are essential for the protection of the host against microbial tissue colonization14–16. Indeed, mutations in the NADPH oxidase dual oxidase 2 (DUOX2) have been associated with increased pathogen invasiveness and development of IBD17 and very early onset IBD18, 19. On the other hand, a characteristic of IBD is recapitulation of an immune response against commensal microbes in the absence of a known pathogen. Thus, in established UC, the mucosa is characterized by an upregulation in DUOX211, 20. We and others have demonstrated that this enzyme is induced by microbial stimuli21, 22 and drives epithelial hydrogen peroxide (H2O2) release in response to interferon-γ (IFNγ) and flagellin22. However, the consequences of DUOX2 overexpression during inflammation have not been investigated.
Given the link between inflammation, CAC, and the microbiota, we have been interested in the contribution of innate immune responses in the progression from inflammation to CAC. Toll-like receptor 4 (TLR4) is an innate immune receptor that recognizes Gram negative lipopolysaccharide (LPS), triggering antimicrobial and proinflammatory responses23, 24. We have reported the overexpression of TLR4 in the colonic epithelial cells (CECs) of UC and CRC patients25, 26. Transgenic mice expressing a constitutively active TLR4 in the intestinal epithelium, villin-TLR4, are more susceptible to inflammation, CRC, and CAC and have a mucosal microbiome that is shaped by TLR4 signaling27, 28. Notably, the microbiota of villin-TLR4 mice can transmit susceptibility to colitis in wild-type mice27.
In the current study, we interrogate the interplay between upregulated epithelial TLR4 signaling, the microbiota, and the development of tumorigenesis. In an unbiased search for genes regulated by TLR4 in the setting of CAC, the most highly expressed genes are involved in ROS generation by CECs. We demonstrate that epithelial TLR4 drives the expression of DUOX2 and the production of H2O2 by CECs. We also show that TLR4-shaped microbiota and DUOX2 participate in tumor initiation and that TLR4-shaped microbiota, which induces epithelial ROS production, is sufficient to transmit susceptibility to CAC. We believe that our study sheds light on a mechanism through which innate immune responses to the microbiota, even in the absence of overt inflammation, accelerate the transition from inflammation to dysplasia in IBD. This situation mimics what is observed in patients whose colitis is controlled but progress to dysplasia and cancer.
Methods
Animals
Villin-TLR4 mice expressing the CD4-TLR4 transgene23 under the villin promoter were generated as described previously29. TLR4 knock-out (TLR4-KO; 10ScNJ), NOX1-KO (Nox1tm1Kkr) and C57BI/6 mice were purchased from the Jackson Laboratory. Epithelial DUOXA1/A2-KO mice (DUOXA-KO) were obtained by crossing the DuoxA1/A2-floxed mice generated at Dr. Kaunitz’s laboratory (UCLA) with villin-cre (Tg(Vil1-cre)997Gum) mice purchased from the Jackson Laboratory. These mice are widely accepted as a model to investigate the role of DUOX2 in the colon given that the expression of DUOX1 in the gut is exceedingly low21. Villin-TLR4 DUOXA-KO mice were generated by backcrossing both strains. All mice, generated on a C57Bl/6 background, were housed in specific pathogen-free (SPF) conditions with a controlled temperature of 20±2°C and free access to food and water. All experiments were performed using mice between 8 to 16 weeks of age, of both sexes, with the approval of the Institutional Animal Care and Use Committee (IACUC) at the University of Miami (Protocols 17-196 and 18-169). The University of Miami is internationally accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Germ-free mice and microbial engraftment
Wild-type and villin-TLR4 germ-free (GF) mice were generated at the University of Miami Gnotobiotic Facility. Microbial engraftment was performed by transferring the mucosa-associated microbiota from C57Bl/6J or villin-TLR4 donor mice to wild-type GF recipient mice. Briefly, the mucosa-associated microbiota was extracted by homogenizing flushed colons in Hank’s balanced salt solution using a BeadBlaster-24 (Benchmark) in a vinyl anaerobic chamber (Coy Laboratory Products). One mL of Hank’s balanced salt solution was used for every 50 mg of colon. Wild-type GF recipient mice were orally gavaged with 200 μL of the resulting slurry and housed in separate biocontainment unit isocages depending on the donor mouse microbiome (vilin-TLR4 versus C57Bl/6J). After three weeks of engraftment, 2 recipient mice per each donor mouse were euthanized and a piece of mucosa was analyzed by 16s rRNA sequencing to verify engraftment. The remaining mice underwent the azoxymethane / dextran sulfate sodium (AOM-DSS) model of tumorigenesis.
Induction of tumorigenesis
To investigate tumorigenesis, two models were utilized: the AOM-DSS model of CAC and the AOM model of CRC. In the AOM-DSS model, mice were injected intraperitoneally once with 7.4 mg/kg AOM (Sigma-Aldrich) and then administered two or three cycles of DSS (Affymetrix USB, MW 40,000-50,000) in their drinking water for 5-7 consecutive days, depending on the experiment. Since villin-TLR4 mice are more susceptible to DSS28, they were treated with 1.5% DSS, while engrafted mice and wild-type littermates received 3% DSS. To analyze the various stages of tumor development, mice were euthanized on day 35 (dysplastic phase), 56, or 72 (tumor phases). During the DSS cycles and the first recovery week, mice were monitored daily for weight loss. Fluid supplementation was applied to mice losing more than 25% of their initial body weight30, whereas endpoint criteria were applied to mice losing more than 30% of their initial body weight or displaying lack of exploratory behavior.
In the AOM model of CRC, mice were injected intraperitoneally with 14.8 mg/kg AOM once a week for 6 consecutive weeks and were euthanized at weeks 12 or 17, as previously described25. GF status at the end of the experiment was corroborated by 16s rRNA PCR from stool collected directly from the colon during euthanasia (Supplementary Figure 4).
Collection of tissue samples
Mice were euthanized by cervical dislocation under isoflurane (Piramal Critical Care) anesthesia and the colon was removed, flushed, cut wide open, and pinned flat on a Sylgard-coated Petri dish. Tumor lesions imaged under a Nikon SMZ800 stereomicroscope were stitched using Adobe Photoshop CC (Adobe Systems Inc) and quantified for number and size using ImageJ (National Institutes of Health). A longitudinal section of the colon was prepared as a Swiss roll and fixed in 4% paraformaldehyde for histology, whereas the rest of the colon was used for CEC isolation. Tumor surrounding areas were homogenized in TRIzol reagent (Thermo-Fisher Scientific) for RNA isolation.
Isolation of CECs and colonoid preparation
CECs were isolated by chelation in 20 mM EDTA in Hank’s balanced salt solution for 1 hour at room temperature followed by gentle shaking. CECs were either lysed in TRIzol reagent for qPCR or pelleted for determination of H2O2 production and preparation of colonoids. Prior to seeding the colonoids, the crypts were digested with Dispase (StemCell Technologies; 1 U/L) in the presence of 2.5 μM Thiazovivin (Cayman Chemical) to obtain single cell suspension and then resuspended in ice-cold Cultrex reduced growth factor basement membrane, type R1 (R&D systems). Colonoids were initially grown in 50% conditioned medium containing wnt3a, R-spondin-3, noggin, and 20% fetal bovine serum supplemented with glycogen synthase kinase 3β inhibitor Chir99021 (5 μM), 2.5 μM Thiazovivin, and 100 μg/mL Primocin. After 2 days of expansion, colonoids were cultured for an additional 5 days in medium containing DMEM/F12, 10% R-spondin-2 and 10% noggin-conditioned media, 10% fetal bovine serum, Primocin and Chir00921. Colonoids were challenged with ultrapure LPS (1 μg/mL), IFNγ (100 ng/mL), heat killed Faecalibacterium prausnitzii (106 cells/mL, strain A2-165), or adherent invasive E. coli (106 cells/mL, AIEC strain LF82) for 24 hours before determinations.
Measurement of hydrogen peroxide production
Amplex red (Thermo-Fisher Scientific) was used to measure the real-time kinetics of H2O2 production by live cells. CECs or colon organoids seeded in a 96 well plate were incubated in Dulbecco’s phosphate-buffered saline solution containing Ca2+, Mg2+, 0.1 U/mL horseradish peroxidase, and 30 μM Amplex red (as per manufacturer’s instructions) with modifications22. Dimethyl sulfoxide (DMSO; vehicle) or 10 μM of the NADPH oxidase inhibitor diphenyleneiodonium (DPI) were added to assess H2O2 production induced by NADPH oxidases. Fluorescence was read at 40-60 second intervals for 10 minutes at 37°C (Ex 530 nm/Em 590 nm) in a Synergy H1 fluorometer (BioTek). H2O2 production was normalized to cell viability via MTT assay (ATCC) that was performed per manufacturer’s instructions. All samples were assayed in triplicates.
Statistical analysis
All data analysis and plots were performed using Prism8 (GraphPad Software, Inc.) and compared using Chi-square, t-test, or two-way ANOVA, as indicated. Results are presented as mean values and standard deviation (SD), and a P value of <0.05 was considered significant.
Results
Dysregulation of TLR4 is associated with overexpression of DUOX2 and NOX1
We have previously reported that UC patients with dysplasia have increased intestinal epithelial expression of TLR431 and that activation of TLR4 in CECs renders mice more susceptible to tumorigenesis25, 28. To take an unbiased view of the molecular signature of TLR4-dependent tumorigenesis, we used gene expression arrays (Mouse Exonic Evidence-Based Oligonucleotide microarray) to look for differentially expressed genes. Villin-TLR4 mice or wild-type littermate controls were treated with AOM-DSS28 and tissue from areas surrounding tumors was collected to analyze the gene expression changes in non-dysplastic, at risk epithelium. Several pathways related to immune activation were upregulated in villin-TLR4 mice when compared to their littermates (Supplementary Datasheet 1; GSE141767). Enzymes associated with ROS production, including inducible nitric oxide synthase (Nos2), Duox2, and Nox1, were among the most differentially upregulated genes in villin-TLR4 mucosa (Figure 1A).
Figure 1 |. Activation of TLR4 is associated with upregulation of NADPH oxidases in colonic tissue.
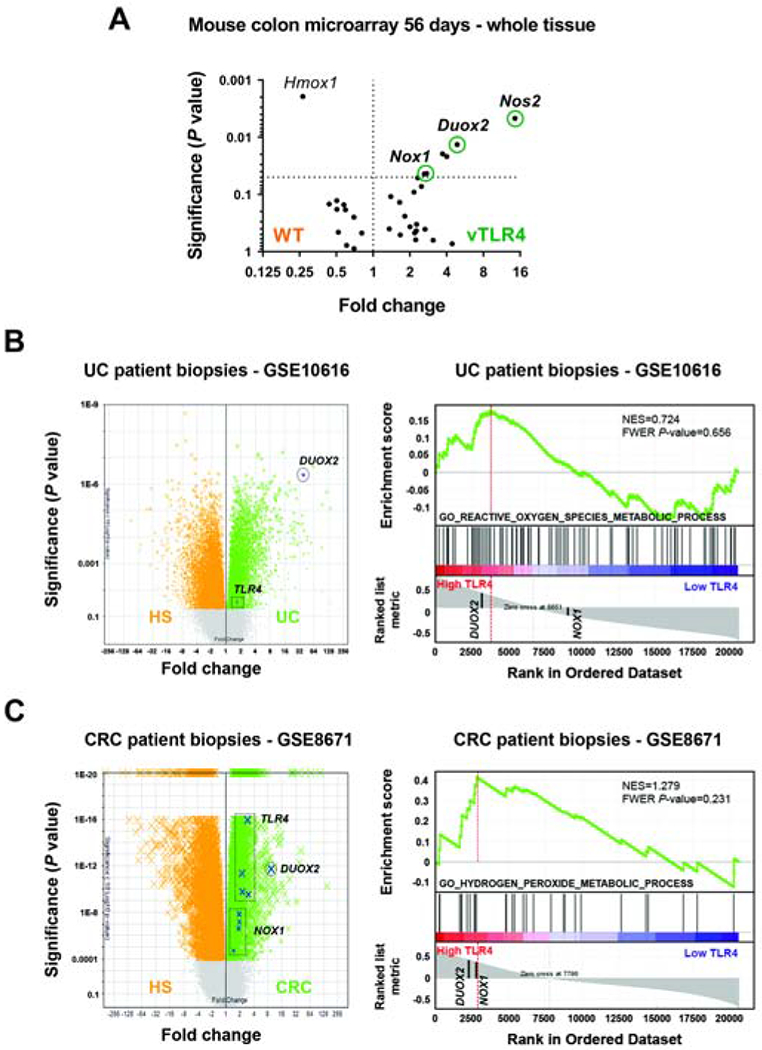
(A) Non-involved areas surrounding tumors were assayed on a microarray. Volcano plot shows the most representative upregulated genes in villin-TLR4 (vTLR4) mice and their wild-type littermates (WT) for ROS-associated pathways. (B) Expression of TLR4, DUOX2 and NOX1 in the dataset GSE10616 of UC patient biopsies. Volcano plot shows TLR4 and DUOX2 in UC patients vs healthy subjects (HS). Boxes highlight the location of different probes for the same transcript. GSEA plot shows that DUOX2 is in the leading-edge genes upregulated in association with TLR4 overexpression. (C) Expression of TLR4, DUOX2 and NOX1 in the dataset GSE8671 of CRC patient biopsies. Volcano plot shows gene expression in CRC adenoma vs healthy tissue. GSEA plot shows that DUOX2 and NOX1 are in the leading-edge genes upregulated in association with TLR4 overexpression.
The role of the NADPH oxidases DUOX2 and NOX1 in IBD or CAC is poorly understood. To identify how TLR4 expression correlates with expression of NADPH oxidases in human disease, we used a bioinformatics approach. We interrogated available gene expression datasets of Crohn’s disease, UC (GSE1061632) and CRC patients (GSE867133) for the expression of TLR4, DUOX2 and NOX1. We found that TLR4 and DUOX2 transcripts were significantly increased in UC, whereas TLR4, DUOX2 and NOX1 were all upregulated in Crohn’s disease and CRC patients (Figure 1B and C, Supplementary Figure 1A and B; left panels). Furthermore, we generated gene set enrichment analysis (GSEA) plots for these datasets for pathways related to ROS metabolism stratified by TLR4 expression and observed that DUOX2, but not NOX1, was consistently found in the leading-edge genes associated with increased expression of TLR4 (Figure 1B and C, Supplementary Figure 1A and B; right panels). These data suggest that the NADPH oxidases DUOX2 and NOX1 are linked to TLR4 upregulation in UC, Crohn’s disease, and CRC samples, as well as in our TLR4-dependent murine model of CAC.
Bacterial signaling through TLR4 regulates expression of Duox2 and Nox1 in CECs in vivo and in vitro
Our data in a murine model of TLR4-dependent CAC and patients with UC or CRC suggest that DUOX2 and NOX1 are regulated by TLR4. We next wished to determine whether TLR4 and the microbiota regulate Duox2 and Nox1 in CECs. To address this question, we used CECs or colon tissues from villin-TLR4 and wild-type littermates raised in specific pathogen-free (SPF) versus germ-free (GF) conditions. We compared mRNA expression of Duox2 and Nox1 by in situ hybridization and qRT-PCR, and their respective regulator proteins Duoxa2, Noxo1, Noxa1, and Cyba34, 35 by qRT-PCR. We found that wild-type GF mice had reduced expression of Duox2 and Duoxa2 when compared to wild-type SPF mice (Supplementary Figure 2B). Consistently, Duox2 transcripts in wild-type GF mice were clearly reduced and expressed at the bottom of the crypt when compared to SPF mice, which essentially expressed Duox2 at the tips of the crypts (Figure 2A). Constitutive activation of TLR4 in villin-TLR4 mice induced the expression of NADPH oxidases in SPF mice and restored expression of these genes under GF conditions (Figure 2B). Duox2 was induced all along the crypt axis and more markedly at the tips of the crypts, whereas Nox1 transcripts accumulated at the lower 1/3 of the crypt (Figure 2A and Supplementary Figure 2A). Only Noxo1 was TLR4-independent, as indicated by two-way ANOVA (P<0.05 in Noxa1, and P<0.001 in Duox2, Duoxa2, Nox1, and Cyba transcripts). These data demonstrate that Duox2 and Nox1 are regulated by the microbiota and that TLR4 signaling is sufficient to induce their expression.
Figure 2 |. TLR4 controls the expression of epithelial Duox2 and Nox1.
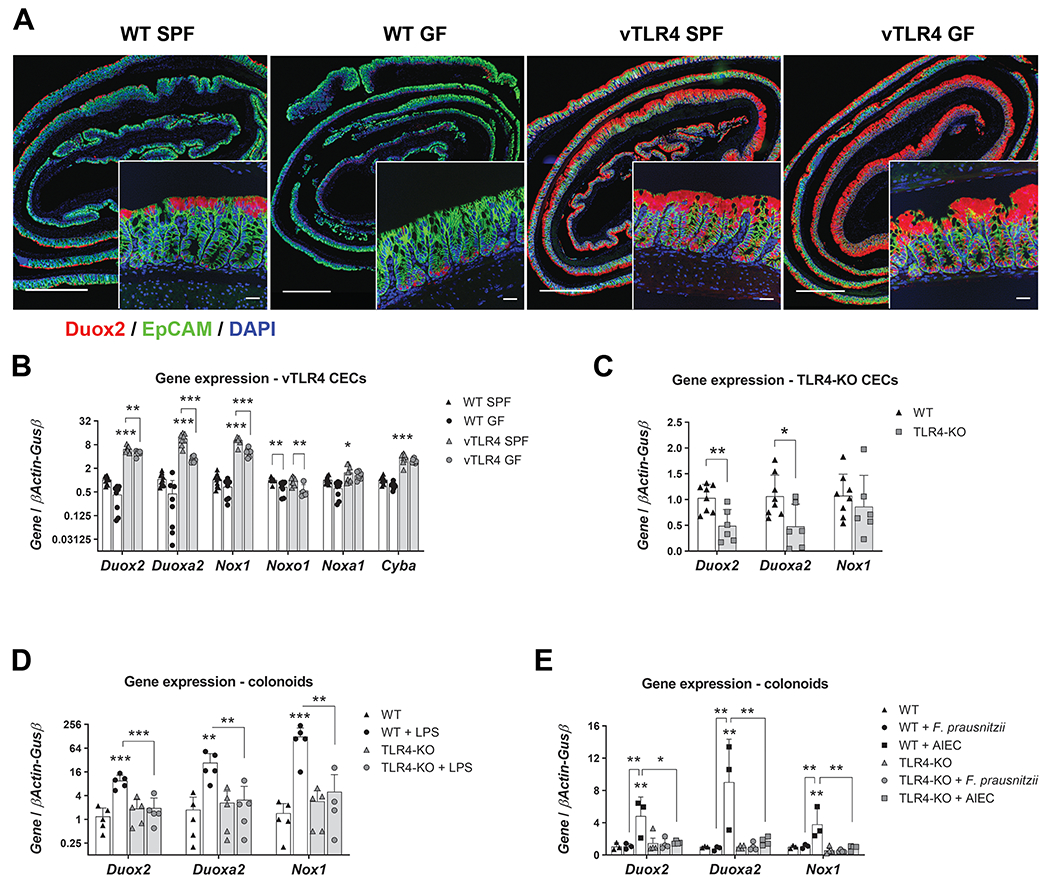
(A) Representative micrographs show Duox2 transcripts (red) counterstained with EpCAM (green) and DAPI (blue). Micrograph scale bar = 1 mm; inset scale bar = 25 μm. (B) Freshly isolated CECs were analyzed by qRT-PCR for the expression of selected transcripts. Villin-TLR4 SPF CECs showed increased expression of Duox2, Duoxa2, Nox1, Cyba (***P<0.001), and Noxa1 (*P<0.05) when compared to WT SPF CECs (n=8-10 mice). Data were analyzed by two-way ANOVA followed by Sidak’s post-hoc test for each gene. “Microbiota” (SPF vs GF) was identified as a significant source of variation in the expression of Duox2, Duoxa2, Nox1, Noxo1 (all P<0.001), Noxa1, and Cyba (both P<0.05); n=6-10 mice. (C) Gene expression in freshly isolated CECs of TLR4-KO and WT mice. Duox2 (**P<0.01) and Duoxa2 (*P<0.05) were significantly downregulated in TLR4-KO CECs (n=6-8 mice). Data were analyzed by unpaired t-test for each gene. (D) Cultured WT and TLR4-KO colonoids were stimulated with LPS for 24 hours and their gene expression was determined by qPCR. Duox2, Nox1 (***P<0.001), and Duoxa2 (**P<0.01) transcripts were significantly upregulated by LPS in WT colonoids (n=5 cultures). Data were analyzed by two-way ANOVA followed by Sidak’s post-hoc test for each gene. (E) Cultured WT and TLR4-KO colonoids were stimulated with heat killed F. prausnitzii or AIEC for 24 hours and their gene expression was determined by qPCR. Duox2, Duoxa2, and Nox1 (**P<0.01) transcripts were significantly upregulated by AIEC in WT colonoids (n=3-4 cultures). Data were analyzed by two-way ANOVA followed by Sidak’s post-hoc test for each gene.
To further investigate whether Duox2 and Nox1 expression depends strictly on TLR4, we isolated CECs from TLR4-KO and wild-type mice raised in SPF conditions and compared the gene expression for Duox2, Nox1, and related genes. Steady state levels of Duox2 and Duoxa2, but not NOX1-associated genes, were highly dependent on TLR4, as TLR4-KO CECs had significantly reduced transcript levels for both genes (Figure 2C and Supplementary Figure 2C). These results indicate that DUOX2 is tightly regulated by TLR4, whereas redundant mechanisms control expression of NOX1.
To corroborate that TLR4-mediated induction of NADPH oxidase expression occurs primarily in CECs and is not dependent on interactions with other cell types, we cultured wild-type and TLR4-KO colonoids and stimulated them with LPS for 24 hours. In contrast to TLR4-KO cells, wild-type colonoids upregulated Duox2, Duoxa2, and Nox1 upon stimulation with LPS, corroborating that epithelial TLR4 activation is sufficient to upregulate Duox2 and Nox1 in CECs (Figure 2D and Supplementary Figure 2D). Furthermore, we tested the requirement of TLR4 to respond to IBD-relevant bacteria, namely Faecalibacterium prausnitzii and adherent invasive E. coli (AIEC). F. prausnitzii is a Gram positive bacteria with a recognized protective role in IBD36, whereas AIEC is a Gram negative pathobiont often associated with development and aggravation of IBD37. Interestingly, whereas heat killed F. prausnitzii did not induce the upregulation of NADPH oxidases, AIEC induced the expression of Duox2, Duoxa2, Nox1 and Cyba in a TLR4-dependent manner (Figure 2E and Supplementary Figure 2E). These results suggest that CECs upregulate expression of NADPH oxidases in response to bacterial recognition via TLR4 and that DUOX2 is highly dependent on TLR4 signaling.
Epithelial TLR4 activation induces H2O2 production in a DUOX2-dependent manner
We next sought to determine the functional correlate of TLR4-induced NADPH oxidase expression, namely production of H2O2. To measure epithelial H2O2 production, we recently described a modified Amplex Red assay that can be used in CECs22. Freshly isolated CECs from villin-TLR4 mice had a marked increase in the production rate of H2O2 when compared to wild-type CECs (Figure 3A). Furthermore, whereas CECs isolated from wild-type GF mice produced less H2O2 than those isolated from wild-type SPF mice (Supplementary Figure 3A), constitutively active TLR4 in villin-TLR4 GF mice sustained high levels of H2O2 production, demonstrating that TLR4 signaling is sufficient to induce epithelial release of H2O2 (Figure 3A). Wild-type and TLR4-KO CECs showed similar rates of steady state H2O2 production in SPF conditions (Supplementary Figure 3B). To determine the involvement of NADPH oxidases in TLR4-induced epithelial H2O2 production, freshly isolated CECs were assayed for H2O2 production and treated with the NADPH oxidase inhibitor DPI or vehicle (DMSO). DPI significantly reduced the production rates of H2O2 in both wild-type and villin-TLR4 CECs when compared to DMSO (Figure 3B). Similarly, wild-type colonoids stimulated with LPS for 24 hours showed an increased release of H2O2 that was blocked by incubation with DPI (Figure 3C). These experiments demonstrate that TLR4-mediated H2O2 production is dependent on NADPH oxidase activity. Specificity of TLR4 activation by LPS was confirmed in TLR4-KO colonoids, which did not respond to LPS challenge (Figure 3C) but did respond to stimulation with IFN-γ (Supplementary Figure 3C).
Figure 3 |. Activation of TLR4 induces epithelial production of H2O2 via DUOX2.
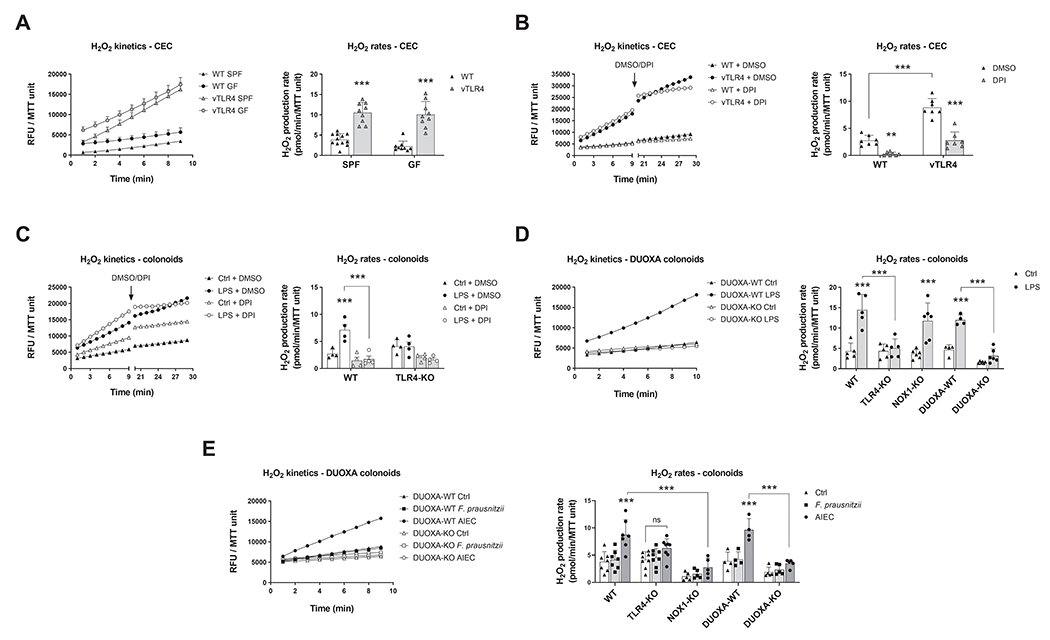
Left panels show kinetic production of H2O2 in one representative experiment expressed in relative fluorescence units (RFU) normalized to MTT viability values. Right panels show the H2O2 production rate in all experiments. (A) Freshly isolated CECs were assayed for the kinetic production of H2O2 in SPF and GF-raised villin-TLR4 mice and littermates. Villin-TLR4 mice had increased H2O2 production rates (***P<0.001) when compared to WT littermates (n=9-12 mice). (B) We tested the effects of NADPH oxidase inhibition in freshly isolated CECs by adding DPI or DMSO after 9 minutes of kinetic determination and measuring for 10 additional minutes. DPI markedly reduced H2O2 production in WT (**P<0.01) and villin-TLR4 (***P<0.001) CECs (n=7 mice). (C) Production of H2O2 was determined in WT and TLR4-KO colonoids stimulated for 24 hours with LPS. Left panel shows H2O2 production kinetics in WT organoids. LPS stimulation induced a release of H2O2 in WT colonoids (***P<0.001) that was completely abrogated by DPI (***P<0.001; n=4 cultures). (D) and (E) TLR4-KO, DUOXA-KO, and NOX1-KO colonoids were stimulated with LPS (n=4-6 cultures), F. prausnitzii, or AIEC (n=4-9 cultures) and their release of H2O2 was determined (***P<0.001). Data in all figures were analyzed by two-way ANOVA followed by Sidak’s post-hoc test.
Next, to assess the relative contribution of DUOX2 and NOX1 to H2O2 production downstream of TLR4, we generated colonoids from DUOXA-KO and NOX1-KO mice. In the absence of NOX1, LPS-induced H2O2 production was not reduced compared with wild-type colonoids (Figure 3D). Conversely, absence of DUOXA resulted in a total lack of response to LPS when compared to that of their control colonoids (DUOXA-wild-type floxed organoids not expressing the villin-cre recombinase) (Figure 3D). These data suggest that DUOX2 is the main NADPH oxidase involved in TLR4-dependent induction of H2O2. Finally, we investigated the requirement for TLR4, NOX1, and DUOX2 to produce epithelial H2O2 in response to IBD-relevant bacteria. Whereas stimulation with heat-killed F. prausnitzii did not induce release of H2O2, AIEC caused a marked increase in the production of epithelial ROS that was abrogated in TLR4, NOX1, and DUOXA-KO colonoids (Figure 3E). These observations indicate that CECs respond to Gram negative bacterial challenge by inducing NADPH oxidase-mediated release of ROS in a TLR4-dependent fashion.
Colitis-associated dysplasia and tumorigenesis are associated with increased H2O2 synthesis by CECs
We have shown that villin-TLR4 mice develop more and larger tumors when challenged with AOM-DSS28. Above we show that NADPH oxidase-mediated production of H2O2 is elevated in CECs from villin-TLR4 mice. To examine the dynamic production of H2O2 by CECs and the relationship to dysplasia and tumor development in wild-type versus villin-TLR4 mice, we performed the AOM-DSS model. To look at the relationship of epithelial H2O2 production to the inflammation-dysplasia cascade, we euthanized mice during the second cycle of DSS, before tumor development, or two weeks after the second cycle, when tumors are established (Figure 4A). Our results show that administration of 1.5% DSS in villin-TLR4 mice and 3% DSS in their wild-type littermates led to the development of high grade dysplasia in 100% of villin-TLR4 mice, whereas only 43% of wild-type littermates developed any degree of dysplasia (wild-type=3/7 vs villin-TLR4=7/7 mice, P<0.05; Figure 4B). By day 56, villin-TLR4 mice had extensive tumor development compared with wild-type mice (Figure 4C). Villin-TLR4 mice lesions were characterized by marked nuclear crowding, with stratification and hyperchromasia, as well as the presence of cribriform patterns reminiscent of that seen in patients with high-grade dysplasia (Figure 4D). In contrast, wild-type littermates showed a higher proportion of regenerative crypts, with clear differentiation towards the epithelial surface, as well as a lower inflammation score (Figure 4D). Coincident with this, we observed increased production of CEC H2O2 after subsequent DSS challenges, which was significantly higher in villin-TLR4 mice CECs when compared to those of wild-type littermates (Figure 4E). Two-way ANOVA identified an interaction effect between “time” and “TLR4 activation” factors (P<0.05), demonstrating that the release of H2O2 increased at higher rates in villin-TLR4 mice during the CAC model. The positive correlation between H2O2 production and histologic score (r=0.837, P<0.0001; Figure 4F) suggests that CEC-mediated H2O2 production is linked to the severity of dysplasia and tumorigenesis.
Figure 4 |. Epithelial release of H2O2 is sequentially increased in the different phases of tumor development.
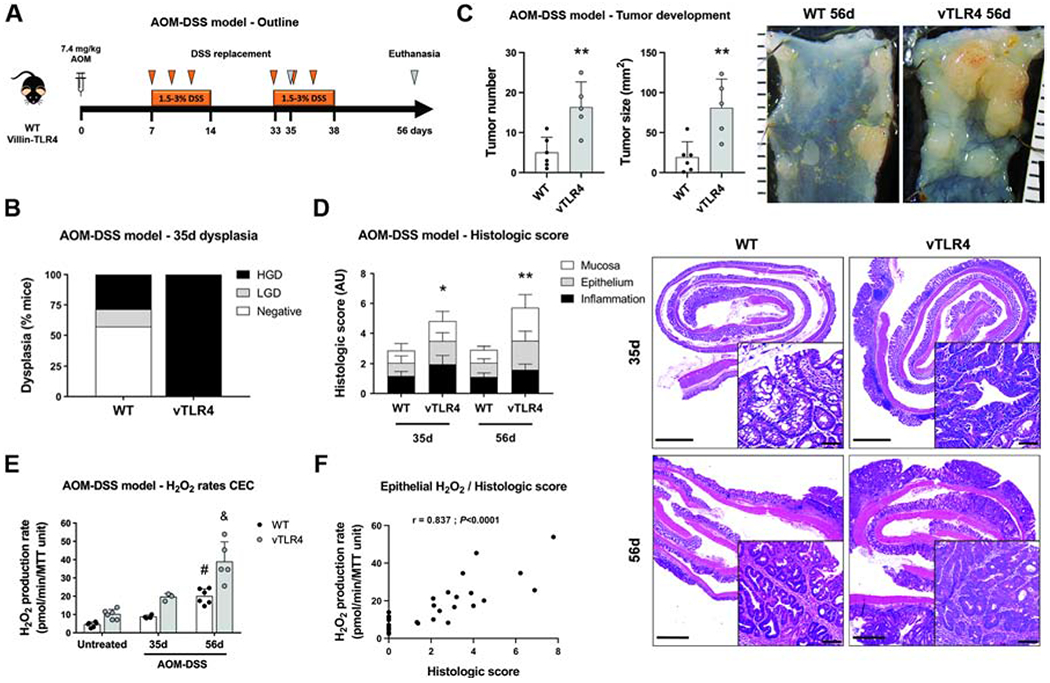
(A) Villin-TLR4 mice and their WT littermates underwent the AOM-DSS model of CAC for 35 or 56 days. (B) Percentage of mice that developed no dysplasia (negative), low-grade dysplasia (LGD), or high-grade dysplasia (HGD) at 35 days (n=7 mice). Dysplasia (LGD+HGD) was compared between groups by Chi-square test. (C) Tumor number and size (**P<0.01) were increased in villin-TLR4 when compared to WT mice after a 56d-CAC model (n=5-6 mice). Micrographs show representative distal colons of each group of mice; scale bar in mm. (D) Histologic score was increased in villin-TLR4 mice when compared to WT littermates on days 35 (*P<0.05; n=7 mice) and 56 (**P<0.01; n=5-6 mice). Right panels show representative micrographs for WT and villin-TLR4 mice colon at both times. Micrograph scale bar = 1 mm; inset scale bar = 50 μm. (E) Epithelial H2O2 production rate in freshly isolated CECs throughout the CAC model. (#) WT-56d vs WT-untreated (***P<0.001) and WT-56d vs WT-35d (*P<0.05); (&) vTLR4-56d vs vTLR4-untreated (***P<0.001), vTLR4-56d vs vTLR4-35d (***P<0.001); and vTLR4-56d vs WT-56d (***P<0.001; n=3-6 mice). “Time” (P<0.001), “TLR4 activation” (P<0.001), and the interaction between these (P<0.05) were also identified as significant variation factors in epithelial release of H2O2. (F) Spearman correlation between histologic score and CEC H2O2 production (r=0.837; P<0.0001; n=30 mice).
Both DUOX2 signaling and the microbiome are required for TLR4-dependent colonic tumorigenesis
The correlation between epithelial H2O2 production and histologic score led us to hypothesize that epithelial ROS participate in tumor initiation and progression. To test this hypothesis, we used repeated administration of AOM, without DSS, as a model of CRC to mitigate the confounding effect of ROS from mucosal phagocytic cells. This model is analogous to patients with dysplasia in the absence of active inflammation. We have previously shown that villin-TLR4 mice develop tumors with AOM alone by 17 weeks, whereas wild-type C57/Bl6 mice require more than 30 weeks25, 38. To investigate the involvement of epithelial ROS and particularly DUOX2 in this model, we backcrossed villin-TLR4 mice to DUOXA-KO mice and compared adenoma formation (Figure 5A). Genetic deletion of DUOXA led to a significant reduction in Duox2 expression (Figure 5B), which was found essentially at the tips of the crypts (Figure 5D, upper panels) and caused a decrease in the production of H2O2 by CECs (Figure 5C). Villin-TLR4 DUOXA-KO mice developed fewer tumors, of smaller size, when compared to villin-TLR4 littermates (Figure 5D, H&E panel, and E). These findings demonstrate that activation of DUOX2 and production of epithelial ROS in response to microbial stimuli are required for the development of colonic neoplasia.
Figure 5 |. Epithelial DUOX2 and TLR4-shaped microbiota induce tumor initiation.
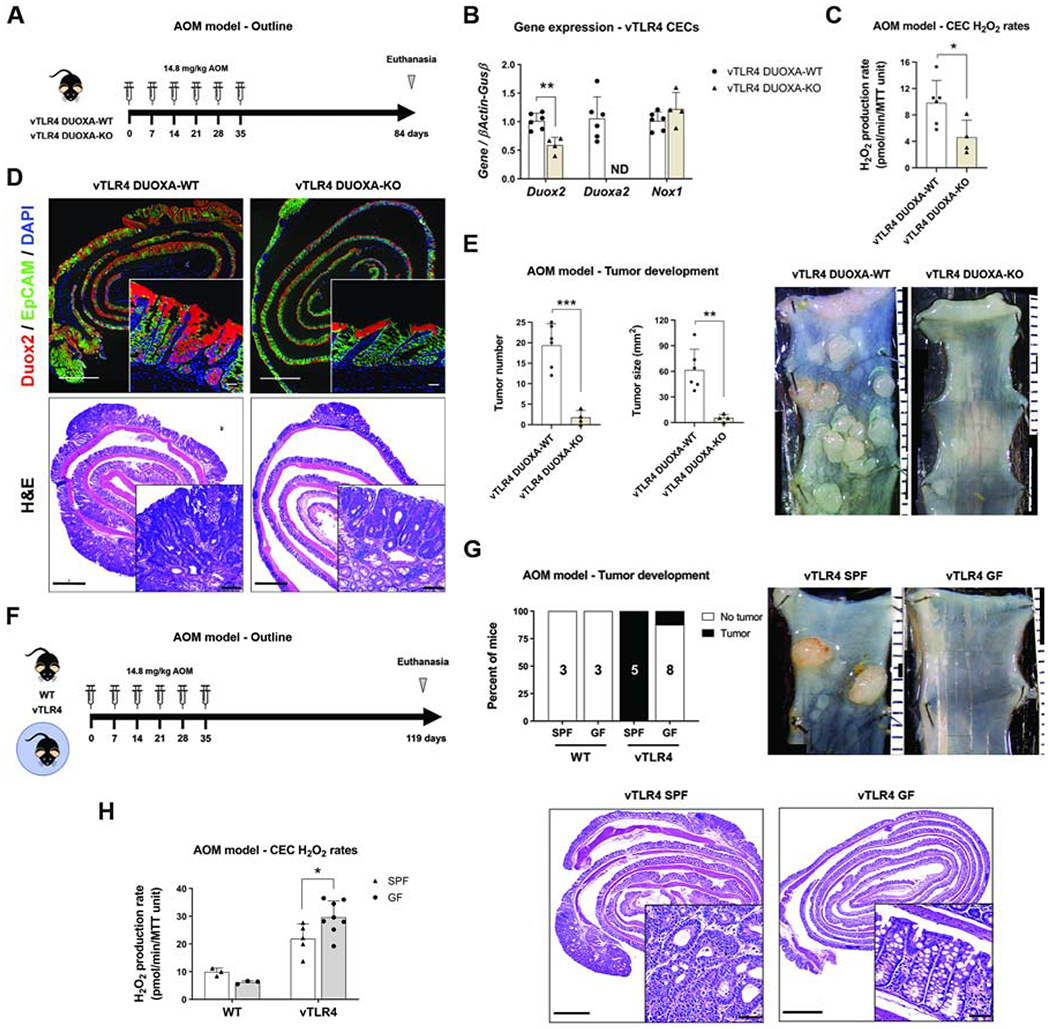
(A) Villin-TLR4 DUOXA-KO and their villin-TLR4 littermates underwent the AOM model of CRC for 12 weeks. (B) Gene expression in CECs. Deletion of Duoxa (non-detectable, ND) caused a significant downregulation of Duox2 (**P<0.01; n=4-6 mice). (C) CEC production of H2O2 in villin-TLR4 mice was reduced by deletion of DUOXA (*P<0.05; n=4-6 mice). (D) Representative micrographs of Duox2 in situ hybridization and H&E for AOM-treated villin-TLR4 mice. Micrograph scale bar = 1 mm; inset scale bar = 25 and 50 μm, respectively. (E) Tumor number (***P<0.001) and size (**P<0.01) were decreased in villin-TLR4 lacking DUOXA (n=4-6 mice). Micrographs show representative distal colons of each group of mice; scale bar in mm. (F) Villin-TLR4 mice and their WT littermates raised in SPF and GF conditions underwent the AOM model of CRC for 17 weeks. (G) Percentage of mice developing tumors in each condition is shown in black (n=3-8 mice). Tumor development was compared between groups by Chi-square test. Representative micrographs of AOM-treated villin-TLR4 mice grown in SPF and GF conditions. Micrograph scale bar = 1 mm; inset scale bar = 50 μm. (H) Epithelial H2O2 production rate at the time of euthanasia (n=3-8 mice). Villin-TLR4-SPF vs villin-TLR4-GF, *P<0.05, as determined by two-way ANOVA followed by Sidak’s post-hoc test.
We next sought to determine whether epithelial TLR4-induced ROS are sufficient to induce tumorigenesis or whether there is a requirement for the microbiota. To answer this question, we compared the development of tumors in villin-TLR4 mice raised in GF versus SPF conditions (Figure 5F). Consistent with our published findings25, neither wild-type SPF nor wild-type GF mice undergoing this model developed colon adenomas (Figure 5G). Villin-TLR4 mice raised in SPF conditions developed tumors in 100% of cases (5/5 mice). However, villin-TLR4 mice under GF conditions were significantly protected from AOM-induced tumors; only one tumor developed in one mouse out of eight (12.5%) (P<0.01; Figure 5G), demonstrating that TLR4-mediated susceptibility to tumorigenesis is dependent on the microbiota. To address the involvement of epithelial ROS in tumor initiation in this CRC model, we analyzed the H2O2 production in CECs isolated from non-tumor areas. Although H2O2 production in CECs from villin-TLR4 kept in GF conditions was higher than in villin-TLR4 SPF mice (Figure 5H), villin-TLR4 GF mice did not develop adenomas (Figure 5G). These findings demonstrate that TLR4 signaling drives epithelial ROS even under GF conditions but is not sufficient to initiate tumorigenesis in the absence of the microbiota.
TLR4-shaped microbiota transfers epithelial redox activity and susceptibility to tumorigenesis
We show above that both epithelial ROS and the microbiota interact to accelerate tumorigenesis in villin-TLR4 mice. We have previously shown that TLR4 expression in the epithelium shapes the mucosa-associated microbiota and can transfer susceptibility to colitis27. We next asked whether the microbiota of villin-TLR4 mice could transfer the susceptibility to tumors. To determine whether TLR4-shaped microbiota promotes tumorigenesis, wild-type GF mice were orally gavaged with the mucosa-associated microbiota of wild-type versus villin-TLR4 mice and underwent the AOM-DSS model of CAC (Figure 6A). Three weeks after the initial engraftment, a small group of mice were euthanized to analyze the engraftment stability using 16s rRNA sequencing of donor and recipient mucosa-associated microbiota samples. Wild-type donor microbiota had an ≥88% engraftment at the phylum, class, and genus-level while villin-TLR4 microbiota had a ≥70% engraftment (Supplementary Table 2). We did not find significant differences between wild-type and villin-TLR4 mice with respect to the overall community structure of the microbiota (Supplementary Figure 5).
Figure 6 |. TLR4-shaped microbiota transfers tumor susceptibility to WT mice.
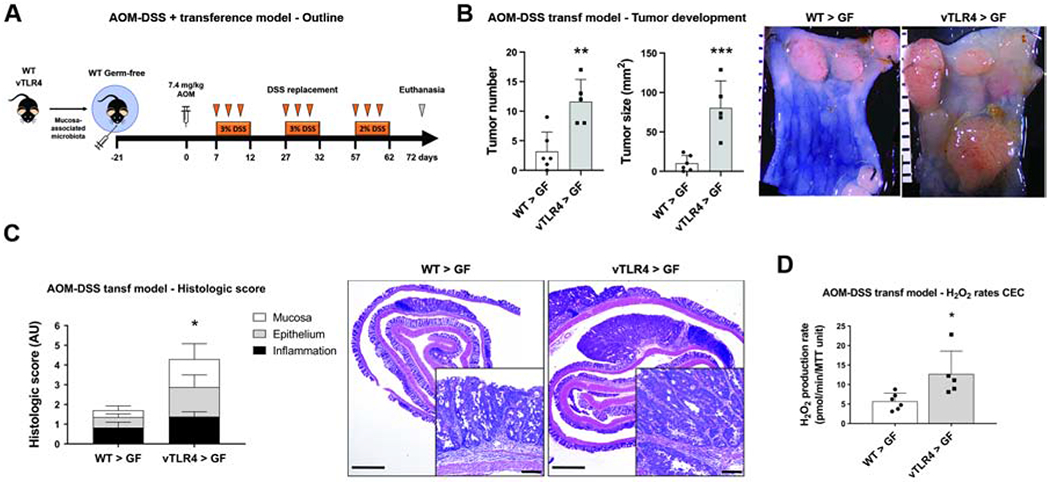
(A) The mucosa-associated microbiota of villin-TLR4 and WT mice were used to colonize WT GF mice that subsequently underwent the AOM-DSS CAC model. (B) Tumor number (**P<0.01) and size (***P<0.001) in GF mice receiving WT (WT > GF) or villin-TLR4 (vTLR4 > GF) microbiota (n=5-6 recipient GF mice). Micrographs show representative distal colons of each group of mice; scale bar in mm. (C) Histologic score was increased in vTLR4 > GF mice when compared to WT > GF littermates (*P<0.05; n=5-6 mice). Representative micrographs show tumor development in mice. Micrograph scale bar = 1 mm; inset scale bar = 50 μm. (D) Epithelial H2O2 production rate at the time of euthanasia (*P<0.05; n=5-6 mice).
Wild-type GF mice receiving the mucosa-associated microbiota of villin-TLR4 donors developed more and larger tumors than wild-type recipients of wild-type microbiota (Figure 6B), as well as more severe histologic lesions (Figure 6C), emulating the villin-TLR4 phenotype. Moreover, transfer of villin-TLR4-derived microbiota was also associated with a higher rate of H2O2 synthesis by CECs (Figure 6D), suggesting that TLR4-shaped microbiota enhances susceptibility to tumorigenesis and induces redox activity in the mucosa that may provide a feed forward loop for tumorigenesis. These results demonstrate that TLR4 signaling affects the functional properties of the microbiota, imbuing the ability to enhance H2O2 production and tumorigenesis.
Discussion
The incidence of colon cancer in IBD continues to be higher than the general population, and death from colon cancer is also two-fold higher in spite of earlier staged cancer at diagnosis2. These observations compel us to identify targetable pathways that can alter the natural history of the disease. Here, we show that TLR4 upregulation is a common feature in tissues from IBD and CRC patients and is accompanied by increases in expression of DUOX2. We mimicked this observation in diverse experimental models to understand the mechanistic implications. We demonstrate that activation of TLR4 and DUOX2 increases the production of H2O2 by CECs, which in turn promotes tumor initiation (a greater number) and progression (larger, higher grades of dysplasia). Using mucosa-associated microbiota from tumor-prone villin-TLR4 mice, we further show that the microbiota functionally transmit susceptibility to tumorigenesis and production of epithelial ROS. In clinical terms, the implication is that host innate immune responses and the mucosa-associated microbiota are necessary co-conspirators in the development of dysplasia and ultimately cancer.
Our laboratory has carefully characterized the involvement of TLR4 in colon cancer. In human tissue microarrays, we described that TLR4 expression increases specifically in CECs as tissues progress from normal to neoplastic stages25, 26. Subsequently, we demonstrated that epithelial TLR4 deficiency protected mice from CAC39 and that overactivation of this receptor led to increased susceptibility to CAC and CRC25, 28. Here, we show that epithelial TLR4 signaling triggers an oxidative program characterized by a marked increase in DUOX2 expression in human samples and mice. Functionally, the upregulation of DUOX2 translates into enhanced epithelial redox activity that, in the presence of a microbiota, promotes the development of tumors. Given the proximity between DUOX2 (top of the crypts) and the mucosa-associated microbiota and our findings in villin-TLR4 GF mice treated with AOM, we believe that DUOX2 shapes the microbiota to confer pro-tumorigenic properties. Previous studies have proposed that epithelial ROS can alter the gut microbiota either at a compositional40 or functional level14, 15. We did not find significant differences in the community structure of the mucosa-associated microbiota between wild-type and villin-TLR4 mice, suggesting that functional alterations in the microbiota are responsible for the increased pro-tumorigenic activity. Traditionally, ROS has been implicated in activating redox signaling pathways in stem cells41 and it is possible that epithelial TLR4-dependent ROS contributes to this mechanism as well. Deeper studies will be necessary to identify potential modifications and culprits in the host and the microbiota with tumor promoting properties. Ultimately, these findings have important implications in the design of new therapeutic approaches to prevent dysplastic progression and suggest that therapeutic strategies need to address the host’s antimicrobial response to mitigate the risk of CRC42, 43.
Our study corroborates that DUOX2 is upregulated in IBD11, 20 and extends it to CRC. We show a direct link between recognition of Gram negative bacteria via TLR4, regulation of DUOX2 activity, and subsequent production of epithelial H2O2. Although we also observed an upregulation of NOX1, we focused on DUOX2 since we and others have previously shown that TLR4 and IBD dysbiosis upregulate Duox221 and increase the production of epithelial H2O2 in GF mice22. Now, we show that dysregulation of this NADPH oxidase is necessary but not sufficient to cause tumors, and that the presence of the microbiota is also required to cause dysplasia. Taken together, our previous and current data support an interactive feedback loop: TLR4 activation leading to increased ROS, ROS-mediated modification of the microbiome that in turn further enhances DUOX2 activity and H2O2 production. Interestingly, in vitro, IBD-protective bacteria such as F. prausnitzii do not induce H2O2 production and may constitute an approach to ameliorating the functional tumorigenic properties of the microbiome. Our experiments also indicate that overt inflammation is not necessary to generate local H2O2 or cause tumors. Indeed, villin-TLR4 mice do not display signs of inflammation in steady state conditions25. These findings suggest that increased epithelial TLR4 signaling even during remission could participate in perpetuating local H2O2, dysbiosis13, 40, and therefore increase the chances of developing dysplasia.
In summary, we show the mechanistic link between TLR4 activation, microbiota, and tumorigenesis. Although our mouse model and in vitro system focused on TLR4, we believe this is a model for increased TLR signaling more broadly. Our results suggest that we must develop strategies to target common pathways in TLR signaling and at the same time address the tumor-promoting microbiota. Because the process is driven by epithelial TLR signaling, it is theoretically possible that localized inhibitors of TLR signaling can be developed without incurring the potential risk of systemic TLR inhibition. Our studies also help to shed light on why certain patients develop dysplasia long after the active inflammation has been treated. We believe that selective targeting of ROS pathways and microbiome-based approaches need to be delivered simultaneously to mitigate dysplasia in IBD patients, even in those in endoscopic and histologic remission.
Supplementary Material
What you need to know.
Background and context: Epithelial NADPH oxidase DUOX2 is upregulated in biopsies from patients with IBD and colorectal cancer but the functional implications are unknown. New Findings: The microbiota activate TLR4 which in turn stimulates epithelial ROS through DUOX2. Increased epithelial ROS production is associated with pro-tumorigenic microbiota. Both an altered microbiota and epithelial ROS are needed for colonic tumorigenesis. Limitations: Our experiments require corroboration in IBD patients. Impact: Our data show that microbial-induced epithelial ROS participate in tumorigenesis even in the absence of overt inflammation, shedding light on why certain patients develop dysplasia long after the active inflammation has been treated.
Acknowledgements
The authors acknowledge Dr. Kaunitz and Dr. Akiba at UCLA for generating and providing the original DUOXA epithelial knockout mice. We also acknowledge Dr. Sokol, Dr. Rolhion, and Ms. Straube at Sorbonne Université (INSERM) for providing heat killed F. prausnitzii and AIEC. Graphical abstract was created with BioRender.com.
Grant support
This work was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK099076), the Micky & Madeleine Arison Family Foundation Crohn’s & Colitis Discovery Laboratory, and Martin Kalser Chair to MTA.
Abbreviations
- AOM
azoxymethane
- CAC
colitis-associated cancer
- CRC
colorectal cancer
- DSS
dextran sulfate sodium
- DMSO
dimethyl sulfoxide
- DPI
diphenyleneiodonium
- DUOX2
dual oxidase 2
- DUOXA2
dual oxidase maturation factor 2
- GSEA
gene set enrichment analysis
- GF
germ-free
- HGD
high-grade dysplasia
- H2O2
hydrogen peroxide
- IBD
inflammatory bowel disease
- IFNγ
interferon-γ
- CEC
colonic epithelial cell
- KO
knockout
- LPS
lipopolysaccharide
- LGD
low-grade dysplasia
- NOS2
inducible nitric oxide synthase
- NOX1
NADPH oxidase 1
- NOXA1
Nox activator 1
- NOXO1
Nox organizer 1
- ROS
reactive oxygen species
- SPF
specific pathogen free
- SD
standard deviation
- TLR4
toll-like receptor 4
- UC
ulcerative colitis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
We have read the journal’s policy and the authors of this manuscript have the following competing interests: MTA has served as a consultant to Boehringer Ingelheim Pharmaceuticals, Gilead, Janssen, Abbvie, Eli Lilly and Landos Biopharma. MTA serves as a trainer or lecturer for Imedex, Focus Medical Communications and Cornerstones Health, Inc. MTA has funded investigator-initiated projects by Pfizer, Prometheus Laboratories and Takeda Pharmaceuticals. This does not alter the authors’ adherence to the journal’s policies on sharing data and materials. All other authors declare no conflict of interest.
Transcript profiling: GSE141767 microarray data are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141767
References
- 1.Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012;142:46–54 e42; quiz e30. [DOI] [PubMed] [Google Scholar]
- 2.Olen O, Erichsen R, Sachs MC, et al. Colorectal cancer in ulcerative colitis: a Scandinavian population-based cohort study. The Lancet 2019. [DOI] [PubMed] [Google Scholar]
- 3.Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med 1990;323:1228–33. [DOI] [PubMed] [Google Scholar]
- 4.Rubio CA, Befrits R. Colorectal adenocarcinoma in Crohn’s disease: a retrospective histologic study. Dis Colon Rectum 1997;40:1072–8. [DOI] [PubMed] [Google Scholar]
- 5.Nowacki TM, Bruckner M, Eveslage M, et al. The risk of colorectal cancer in patients with ulcerative colitis. Dig Dis Sci 2015;60:492–501. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe T, Konishi T, Kishimoto J, et al. Ulcerative colitis-associated colorectal cancer shows a poorer survival than sporadic colorectal cancer: a nationwide Japanese study. Inflamm Bowel Dis 2011;17:802–8. [DOI] [PubMed] [Google Scholar]
- 7.Munkholm P Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment Pharmacol Ther 2003;18 Suppl 2:1–5. [DOI] [PubMed] [Google Scholar]
- 8.Chu H, Khosravi A, Kusumawardhani IP, et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 2016;352:1116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schirmer M, Garner A, Vlamakis H, et al. Microbial genes and pathways in inflammatory bowel disease. Nat Rev Microbiol 2019;17:497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richard ML, Liguori G, Lamas B, et al. Mucosa-associated microbiota dysbiosis in colitis associated cancer. Gut Microbes 2018;9:131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lloyd-Price J, Arze C, Ananthakrishnan AN, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019;569:655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fritsch J, Abreu MT. The Microbiota and the Immune Response: What Is the Chicken and What Is the Egg? Gastrointest Endosc Clin N Am 2019;29:381–393. [DOI] [PubMed] [Google Scholar]
- 13.Zhu W, Winter MG, Byndloss MX, et al. Precision editing of the gut microbiota ameliorates colitis. Nature 2018;553:208–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alvarez LA, Kovacic L, Rodriguez J, et al. NADPH oxidase-derived H2O2 subverts pathogen signaling by oxidative phosphotyrosine conversion to PB-DOPA. Proc Natl Acad Sci U S A 2016;113:10406–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corcionivoschi N, Alvarez LA, Sharp TH, et al. Mucosal reactive oxygen species decrease virulence by disrupting Campylobacter jejuni phosphotyrosine signaling. Cell Host Microbe 2012;12:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grasberger H, El-Zaatari M, Dang DT, et al. Dual oxidases control release of hydrogen peroxide by the gastric epithelium to prevent Helicobacter felis infection and inflammation in mice. Gastroenterology 2013;145:1045–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levine AP, Pontikos N, Schiff ER, et al. Genetic Complexity of Crohn’s Disease in Two Large Ashkenazi Jewish Families. Gastroenterology 2016;151:698–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayes P, Dhillon S, O’Neill K, et al. Defects in NADPH Oxidase Genes NOX1 and DUOX2 in Very Early Onset Inflammatory Bowel Disease. Cell Mol Gastroenterol Hepatol 2015;1:489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parlato M, Charbit-Henrion F, Hayes P, et al. First Identification of Biallelic Inherited DUOX2 Inactivating Mutations as a Cause of Very Early Onset Inflammatory Bowel Disease. Gastroenterology 2017;153:609–611 e3. [DOI] [PubMed] [Google Scholar]
- 20.Haberman Y, Tickle TL, Dexheimer PJ, et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest 2014;124:3617–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grasberger H, Gao J, Nagao-Kitamoto H, et al. Increased Expression of DUOX2 Is an Epithelial Response to Mucosal Dysbiosis Required for Immune Homeostasis in Mouse Intestine. Gastroenterology 2015;149:1849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burgueno JF, Fritsch J, Santander AM, et al. Intestinal epithelial cells respond to chronic inflammation and dysbiosis by synthesizinf H2O2. Front Physiol 2019;10:1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997;388:394–7. [DOI] [PubMed] [Google Scholar]
- 24.Burgueno JF, Abreu MT. Epithelial Toll-like receptors and their role in gut homeostasis and disease. Nat Rev Gastroenterol Hepatol 2020;17:263–278. [DOI] [PubMed] [Google Scholar]
- 25.Santaolalla R, Sussman DA, Ruiz JR, et al. TLR4 activates the beta-catenin pathway to cause intestinal neoplasia. PLoS One 2013;8:e63298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sussman DA, Santaolalla R, Bejarano PA, et al. In silico and Ex vivo approaches identify a role for toll-like receptor 4 in colorectal cancer. J Exp Clin Cancer Res 2014;33:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dheer R, Santaolalla R, Davies JM, et al. Intestinal Epithelial Toll-Like Receptor 4 Signaling Affects Epithelial Function and Colonic Microbiota and Promotes a Risk for Transmissible Colitis. Infect Immun 2016;84:798–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fukata M, Shang L, Santaolalla R, et al. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm Bowel Dis 2011;17:1464–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shang L, Fukata M, Thirunarayanan N, et al. Toll-like receptor signaling in small intestinal epithelium promotes B-cell recruitment and IgA production in lamina propria. Gastroenterology 2008;135:529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burgueno JF, Lang JK, Santander AM, et al. Fluid supplementation accelerates epithelial repair during chemical colitis. PLoS One 2019;14:e0215387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukata M, Chen A, Vamadevan AS, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007;133:1869–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kugathasan S, Baldassano RN, Bradfield JP, et al. Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nat Genet 2008;40:1211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sabates-Bellver J, Van der Flier LG, de Palo M, et al. Transcriptome profile of human colorectal adenomas. Mol Cancer Res 2007;5:1263–75. [DOI] [PubMed] [Google Scholar]
- 34.Grasberger H, Refetoff S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem 2006;281:18269–72. [DOI] [PubMed] [Google Scholar]
- 35.Panday A, Sahoo MK, Osorio D, et al. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol 2015;12:5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 2008;105:16731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palmela C, Chevarin C, Xu Z, et al. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 2018;67:574–587. [DOI] [PubMed] [Google Scholar]
- 38.Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc 2007;2:1998–2004. [DOI] [PubMed] [Google Scholar]
- 39.Fukata M, Hernandez Y, Conduah D, et al. Innate immune signaling by Toll-like receptor-4 (TLR4) shapes the inflammatory microenvironment in colitis-associated tumors. Inflamm Bowel Dis 2009;15:997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winter SE, Lopez CA, Baumler AJ. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep 2013;14:319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 2014;15:411–21. [DOI] [PubMed] [Google Scholar]
- 42.Coleman OI, Lobner EM, Bierwirth S, et al. Activated ATF6 Induces Intestinal Dysbiosis and Innate Immune Response to Promote Colorectal Tumorigenesis. Gastroenterology 2018;155:1539–1552 e12. [DOI] [PubMed] [Google Scholar]
- 43.Zhu W, Miyata N, Winter MG, et al. Editing of the gut microbiota reduces carcinogenesis in mouse models of colitis-associated colorectal cancer. J Exp Med 2019;216:2378–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.