

Purpose of review
Primary biliary cholangitis (PBC) is characterized by autoimmune damage of intrahepatic bile ducts associated with a loss of tolerance to mitochondrial antigens. PBC etiopathogenesis is intriguing because of different perplexing features, namely: a) although mitochondria are present in all cell types and tissues, the damage is mainly restricted to biliary epithelial cells (BECs); b) despite being an autoimmune disorder, it does not respond to immunosuppressive drugs but rather to ursodeoxycholic acid, a bile salt that induces HCO3− rich choleresis; c) the overwhelming female preponderance of the disease remains unexplained. Here we present an etiopathogenic view of PBC which sheds light on these puzzling facts of the disease.
Recent findings
PBC develops in patients with genetic predisposition to autoimmunity in whom epigenetic mechanisms silence the Cl−/HCO3− exchanger AE2 in both cholangiocytes and lymphoid cells. Defective AE2 function can produce BECs damage as a result of decreased biliary HCO3− secretion with disruption of the protective alkaline umbrella that normally prevents the penetration of toxic apolar bile salts into cholangiocytes. AE2 dysfunction also causes increased intracellular pH (pHi) in cholangiocytes, leading to the activation of soluble adenylyl cyclase, which sensitizes BECs to bile salt-induced apoptosis. Recently, mitophagy was found to be inhibited by cytosolic alkalization and stimulated by acidification. Accordingly, we propose that AE2 deficiency may disturb mitophagy in BECs, thus, promoting the accumulation of defective mitochondria, oxidative stress and presentation of mitochondrial antigens to the immune cells. As women possess a more acidic endolysosomal milieu than men, mitophagy might be more affected in women in an AE2-defective background. Apart from affecting BECs function, AE2 downregulation in lymphocytes may also contribute to alter immunoregulation facilitating autoreactive T-cell responses.
Summary
PBC can be considered as a disorder of Cl−/HCO3− exchange in individuals with genetic predisposition to autoimmunity.
Keywords: Ae2 KO mice, antimitochondrial antibodies, biliary bicarbonate umbrella, epigenetic mechanisms, female predominance, miR-506, mitophagy, Na+–independent Cl−/HCO3− anion exchanger 2, pHi disturbance, promoter hypermethylation, soluble adenylyl cyclase
INTRODUCTION
Primary biliary cholangitis (PBC) is an autoimmune disease that affects interlobular bile ducts causing ductopenia and progressive cholestasis. It is characterized by the presence (in 95% of the patients) of antimitochondrial antibodies (AMAs), overwhelming female preponderance (10 : 1) and frequent association with sicca syndrome and other autoimmune diseases, such as Hashimoto's thyroiditis, reumathoid arthritis and scleroderma [1–7]. PBC-specific AMAs are directed against components of the 2-oxo dehydrogenase complexes, principally the inner lipoyl domain in the E2 component of the pyruvate dehydrogenase complex (PDC-E2), but also the branched chain 2-oxo-acid dehydrogenase (BCOADC-E2) and 2-oxoglutarate dehydrogenase complex (OGDC-E2) [8,9]. Additionally, and regardless of AMA status, 30% of patients may develop different PBC-specific antinuclear autoantibodies presenting multiple nuclear-dot or nuclear-membrane staining patterns [10]. These autoantibodies include the highly specific anti-gp210 and anti-p62 (with 95% specificity for PBC each) as well as anti-sp100, anti-PML and anti-sp140 [5,10].
PBC has variable presentation. Although there are patients who evolve rapidly to advanced forms of liver damage, in most cases, the progression is slow with long lasting asymptomatic evolution in some individuals [1]. However, if the disease is left untreated, the persisting damage of bile ducts leads to increasing cholestasis with jaundice, pruritus and hypercholesterinemia, ending up in biliary cirrhosis requiring liver transplantation [11,12].
Nowadays, growing clinical awareness and widespread use of AMA test in the community allow diagnosis of PBC at an early stage when daily administration of ursodeoxycholic acid (UDCA) is particularly efficient at improving biochemical markers, attenuating bile duct injury and halting the evolution of the disease to advanced cholestasis and liver failure [13–15]. Indeed, PBC patients who respond favorably to UDCA (more than 60%) have a life expectancy similar to that of age and sex-matched normal population [16–18]. With ongoing development of novel therapies, combination regimes are expected to achieve adequate control of the disease in those patients who do not respond to UDCA alone [11,19,20]. In fact, sufficiently powered phase III clinical trials have shown that two drugs, the FXR agonist obeticholic acid and bezafibrate (a PPAR-α agonist that reduces bile salt synthesis and increases biliary phospholipid secretion), provide benefit in combination with UDCA to patients with incomplete response to UDCA monotherapy [11,19].
Box 1.
no caption available
PATHOLOGIC FINDINGS AND IMMUNOLOGICAL MECHANISMS
The distinctive pathological finding in PBC is the presence of dense portal and periportal inflammatory infiltrates surrounding the small and medium-sized intrahepatic bile ducts. Inflammatory cells permeate the biliary epithelium, which appears disrupted, with irregularities of the biliary lumen. The inflammatory infiltrate is composed mainly of CD4+ and CD8+ T cells, macrophages, B lymphocytes, plasma cells, NK and NKT cells, and variable presence of eosinophils [21]. Infiltrating CD8+ T cells exert a direct cytotoxic attack on bile duct epithelium, whereas CD4+ cells are helper T (Th) cells that act by producing inflammatory cytokines, which stimulate autoreactive cytotoxic CD8+ T cells and autoantibody production by B cells [22,23▪▪]. As a reflection of the loss of tolerance to PDC-E2, CD4+ and CD8+ T cells reacting against this mitochondrial protein are abundantly found in the liver of patients with PBC and, interestingly, both B and T-cell epitopes include the lipoyl domain of E2 [24]. Regulatory T cells (Treg) are reduced in the liver and peripheral blood of patients with PBC [25], a defect which likely facilitates autoimmune responses.
In PBC, B-cell follicles are frequently observed in the portal tract in the vicinity of biliary ducts. These lymphocytic aggregates – which are termed tertiary lymphoid structures (TLSs) when present outside secondary lymphoid organs – can be found in the target tissues of other autoimmune diseases. They are characterized by the presence of high endothelial cell venules and an organized disposition of B and T cells in connection with follicular dendritic cells. TLSs provide a cytokine and chemokine-rich milieu, where B cells, follicular dendritic cells and T cells interact enabling affinity maturation of effector immune cells fostering humoral and cellular autoimmunity [26]. It should be mentioned that early PBC is characterized by IL-12-driven Th1 type of immune response, whereas IL-23-mediated Th17 type of response prevails in later stages promoting fibrosis in advanced disease [27].
Although PBC-specific AMA are not directly pathogenic, immunocomplexes formed by AMA and the cognate antigen can activate local and regional dendritic cells boosting immunity against E2 subunit. In fact, it has been shown that macrophages from PBC patients, but not from normal individuals, produce high amounts of proinflammatory cytokines, including IL-6, TNFα and IL-12, when incubated with apoptotic bodies from BECs and AMA, likely reflecting M1 polarization of the phagocytic cells [23▪▪,28].
Notably, PDC-E2 is upregulated in injured BECs and, because of its lack of glutathiolation, it may remain intact in apoptotic bodies from these cells [29,30,31▪]. The apoptotic blebs of damaged BECs can be engulfed by antigen-presenting cells to stimulate adaptive immunity against this mitochondrial antigen particularly when the individual has a genetic background predisposing to autoimmunity.
The question arises as to what is the basic alteration of BECs that causes cell stress, PDC-E2 enrichment and apoptosis.
BILIARY EPITHELIAL CELL DYSFUNCTION IN PRIMARY BILIARY CHOLANGITIS: AN ESSENTIAL DETERMINANT OF BILIARY EPITHELIAL CELL STRESS LEADING TO IMMUNE-MEDIATED BILE DUCT INJURY
BECs are polarized cells, which play a central role in bile formation by secreting HCO3− and water to the ductular lumen, thus, fluidizing and alkalizing canalicular bile [32▪▪,33,34]. Although cholangiocytes represent about 3–5% of total liver cell population, they generate 25–40% of total bile flow in humans, indicating how active are these cells at transporting water and HCO3− to bile [32▪▪,33,35].
Ductular secretion of HCO3− takes place via the Cl−/HCO3− exchanger AE2 (also known as SLC4A2) located at the apical membrane of BECs [34,36,37▪▪,38▪▪]. Notably, AE2 mRNA levels are much higher in cholangiocytes than in hepatocyes [39]. During the postprandial period, the gastrointestinal hormone secretin binds the secretin receptor at the basolateral membrane of BECs resulting in increased intracellular cAMP levels with subsequent PKA activation and transfer of cytoplasmic vesicles containing AE2, the Cl− channel cystic fibrosis transmembrane regulator (CFTR) and the water channel aquaporin 1 (AQP1) to the luminal membrane [40]. This event promotes CFTR-mediated exit of Cl− to the lumen, generating a Cl− gradient, which stimulates Cl−/HCO3− exchange via AE2, leading to increased biliary HCO3− and water secretion [32▪▪,33,41]. CFTR also mediates ATP release to bile [42]. Luminal ATP stimulates apical P2Y receptors followed by activation of the apical, type III inositol 1,4,5-triphosphate receptor (InsP3R, a Ca2+ release channel present at the endoplasmic reticulum) with subsequent rise of intracellular Ca2+ levels. Increased intracellular Ca2+ enhances luminal Cl− gradient by activating the apical Ca2+-dependent Cl− channel TMEM16A, thus, promoting AE2-mediated Cl−/HCO3− exchange and biliary HCO3− secretion [42]. Acetylcholine can also stimulate biliary HCO3− secretion via activation of basolateral InsP3Rs (types I and II) and elevation of intracellular Ca2+[42].
AE2 deficiency in primary biliary cholangitis
In the early 1990s, it was shown that PBC patients exhibit reduced AE2 mRNA levels in the liver and in peripheral blood mononuclear cells [43▪▪]. Defective hepatic AE2 expression in PBC was corroborated by immunohistochemistry studies demonstrating reduced AE2 staining at the apical membrane of BECs of interlobular bile ducts [38▪▪]. Notably, it was found that UDCA therapy improved hepatic AE2 expression in parallel with amelioration of cholestasis [38▪▪,43▪▪]. Consistent with the concept that PBC is associated with deficient HCO3− transport to bile, PET imaging revealed that secretin-induced biliary HCO3− secretion was impaired in PBC patients and that UDCA therapy corrected this defect [44▪▪]. In-vitro studies using isolated cholangiocytes further confirmed diminished AE2 activity in specimens from individuals with PBC [45].
The notion that AE2 deficiency is central in the pathogenesis of PBC was supported by findings in Ae2a,b knockout mice. These animals recapitulate most features observed in PBC patients, including development of AMA, increased IgM levels, disturbed immunoregulation, progressive cholestasis and nonsuppurative cholangitis [46–48]. In fact, all aged Ae2a,b knockout mice (15–25 months) exhibit portal mononuclear infiltration with abundant cytotoxic lymphocytes, bile duct damage and ductopenia [47]. Moreover, Ae2a,b-deficient mice manifest an altered cholangiocyte gene expression profile consistent with the existence of oxidative stress and enhanced antigen presentation [46].
Upregulation of miR-506 in primary biliary cholangitis cholangiocytes
In the last years, it was found that miR-506 (has-miR-506–3p/5p), a micro RNA that targets both AE2 and InsP3R3, is upregulated in BECs from PBC patients compared with either patients with primary sclerosing cholangitis or healthy controls [49,50]. Noteworthy, transfection of PBC cultured cholangiocytes with antimiR-506 oligonucleotides increases AE2 activity [49], whereas experimental overexpression of miR-506 in cholangiocytes diminishes AE2 expression and activity, causes PDC-E2 overexpression, impairs mitochondrial energy metabolism and promotes oxidative stress, endoplasmic reticulum stress and bile acid induced-apoptosis [31▪]. Moreover, lymphocytes from PBC patients are activated and proliferate when cocultured with miR-506-overexpressing cholangiocytes [31▪]. Accordingly, in PBC patients, the upregulation of miR-506 in cholangiocytes might impair biliary HCO3− secretion while promoting a proinflammatory and immunogenic phenotype in BECs. Of interest, the MiR-506 gene is located in the X chromosome, a fact that might be related to the female predominance observed in PBC [49]. Of note, proinflammatory cytokines stimulate MiR-506 promoter activity [31▪], suggesting that inflammatory processes generated by environmental factors might contribute to AE2 downregulation in the biliary epithelium of PBC patients via increased miR-506 expression.
AE2 deficiency increases pHi, enhances the sensitivity to proapoptotic signaling and may potentially affect mitophagy in BECs
In addition to be a crucial mediator of biliary HCO3− secretion, AE2 is an acid loader involved in regulating intracellular pH (pHi) [41]. Accordingly, AE2 dysfunction in BECs results in increased pHi [41,48,49]. Notably, it was found that intracellular alkalization activates the HCO3− sensor soluble adenylyl cyclase (sAC), which sensitizes BECs to bile salt-induced apoptosis [51,52▪▪].
In addition, deficient AE2 activity might likely impinge on a variety of cell functions, including autophagy and mitophagy, which are processes critically dependent on pHi [53▪▪]. In particular, mitophagy, the specific autophagic elimination of mitochondria, is induced by cytosolic acidification, whereas intracellular alkalization impairs this process, ultimately leading to the accumulation of defective mitochondria, production of reactive oxygen species and activation of cell death pathways [53▪▪,54]. When dysfunctional mitochondria are not properly eliminated, the activation of intracellular innate immunity sensors, such as the NLRP3 inflammasome, stimulates the production of proinflammatory cytokines, such as IL-1β and IL-18 [55▪,56]. Thus, mitophagy represents a critical cell-intrinsic, quality-control and anti-inflammatory mechanism, which is likely disturbed in PBC BECs. Defective mitophagy would make these cells to overexpress mitochondrial antigens and to acquire a proinflammatory and immunogenic phenotype. Importantly, Matheoud et al.[57▪▪] showed that the blockade of mitophagy was accompanied by presentation of high levels of mitochondrial antigens on major histocompatibility complex class I, an observation which is clearly relevant to the pathogenesis of PBC.
The potential involvement of dysregulated mitophagy in the pathogenesis of PBC is attractive as it might explain the enrichment of BECs in mitochondrial proteins and their increased presentation to the immune system. Interestingly, it has been recently shown that there are sex differences in the pH of intraphagolysosomal milieu, which is more acidic in women than in men [58▪▪]. Accordingly, it would be expected that defects in acidifiers like AE2 might affect mitophagy to a greater extent in women. Moreover, mitochondrial dynamics have been found to be markedly influenced by sex hormones [59]. PBC is characterized by an overwhelming female preponderance and although autoimmune diseases are in general more common in women than in men, it is tempting to hypothesize that a preferential disturbance of mitophagy in women in a context of defective AE2 function might contribute to explain the strong female bias of the disease.
Defective ‘biliary bicarbonate umbrella’ in primary biliary cholangitis
As indicated above, in addition to disturbing the intracellular milieu of cholangiocytes, impaired AE2 expression results in reduced alkalization of bile. Ten years ago, Beuers et al.[60] proposed that the bile duct epithelium is protected against the toxic effects of bile salts by the presence of a HCO3−–rich glycocalyx covering the luminal pole of cholangiocytes (a protective barrier designated as the ‘biliary bicarbonate umbrella’). This alkaline protective barrier is generated by the active AE2-mediated secretion of HCO3− to bile. Thanks to this alkaline shield, glycine-conjugated bile salts with a pKa of 4, like glycochenodeoxycholic acid, are maintained in a polar membrane-impermeant form and therefore are not able to enter BECs [60,61▪▪]. When AE2 activity is impaired, glycine-conjugated bile salts remain in an apolar state and penetrate into cholangiocytes promoting oxidative stress, cell senescence and apoptosis [51,62,63▪].
The therapeutic effect of ursodeoxycholic acid
UDCA promotes biliary HCO3− secretion and therefore strengthens the defensive alkaline barrier, which protects BECs [61▪▪]. The fact that most PBC patients have a positive response to UDCA while immunosuppressive drugs (including newer biologics used to modify the immune system) do not provide a clear benefit [19,64–68] supports the notion that AE2 deficiency and a defective biliary HCO3− umbrella are key determinants of bile duct damage in PBC. The mechanisms by which UDCA increases biliary HCO3− secretion involve the activation of PI3K/Akt pathway, which stimulates the release of ATP leading to the extrusion of Cl− to the lumen through the Ca2+-dependent Cl− channel TMEM16A and subsequent Cl−/HCO3− exchange [61▪▪]. In addition, UDCA upregulates AE2 gene expression when acting in conjunction with glucocorticoids [69]. Moreover, UDCA induces nitric oxide synthase in liver tissue and promotes Mrp2-mediated canalicular secretion of nitric oxide in the form of S-nitrosoglutathione (GSNO), a compound that displays potent antiapoptotic effects on BECs [70]. Additionally, GSNO was shown to enhance UDCA-mediated ATP release by cholangiocytes further boosting biliary bicarbonate secretion [70].
GENETIC, EPIGENETIC AND ENVIRONMENTAL FACTORS
Genetic factors
Abundant evidence has been accumulated supporting the view that PBC occurs as a result of a dysfunction of the biliary epithelium in individuals with genetic predisposition to autoimmunity. Genome-wide association studies [71,72] and dense fine-mapping association analysis [73] have related susceptibility to PBC with variations in genes regulating innate and adaptive immunity, similarly to what occurs in autoimmune diseases responsive to immunosuppressive drugs [11,23▪▪]. However, studies in discordant monozygotic twins showing differences in gene expression profile and DNA methylation [74,75] indicate that besides genetics, epigenetic factors contribute to the development of the disease.
Concerning AE2 gene variations, conventional genotyping of selected single nucleotide polymorphisms (rs2069443, rs2303933, rs2303937 and rs2303941) in Japanese PBC patients revealed associations with disease susceptibility and/or anticentromere antibody production [76]. In whites, however, no association of AE2 single nucleotide polymorphisms with PBC susceptibility was found, though two variants were reported to influence AMA status [77], whereas the synonymous variation rs2303932 was associated with reduced PBC progression in French patients receiving UDCA [78].
Environmental and epigenetic factors
Environmental elements may affect the phenotype of patients, particularly via epigenetic changes (DNA methylation patterns, noncoding RNAs like microRNAs and noncoding long RNAs, histone modifications, such as acetylation, methylation, phosphorylation, ubiquitination and sumoylation) leading to disturbed gene expression. So far, a significant number of epigenetic changes have been reported in PBC, which include altered expression of microRNAs (thoroughly reviewed by Rodrigues et al.[4]) as well as methylation abnormalities of genes (either hyper or hypomethylation) on the X chromosome and autosomal chromosomes [4,23▪▪,74,79]. When affecting promoter regions, hypermethylation is consistently related to silencing of gene expression, whereas hypermethylation in intragenic regions is usually associated with enhanced expression [80].
Recently, we reported that there is a significant increase in CpG-cytosine methylation affecting selective promoter regions of the AE2 gene both in liver and PBMCs from PBC individuals [81▪]. Moreover, mean methylation rates inversely correlate with mRNA levels [81▪]. Of note, selective CpG-hypermethylation clusters in PBC affected relevant nuclear factor sites/motifs [81▪,82–84].
Among environmental exposures affecting DNA methylation, smoking has been associated with high risk of PBC [85–87], being also related to increased DNA-methyltransferase expression [88]. Therefore, DNA methylation affecting AE2 and additional genes may constitute a link between genetic and environmental risk factors in PBC pathogenesis.
THE ROLE OF AE2 DEFICIENCY IN THE DYSREGULATION OF IMMUNE RESPONSE
As mentioned, in PBC, AE2 mRNA levels are decreased not only in the liver but also in peripheral lymphoid cells [43▪▪]. Thus, altered pHi because of defective AE2 activity may affect the function of immune cells in PBC patients [48,89]. Indeed, we found that Ae2a,b-deficient mice exhibit reduced Treg population, progressive expansion of CD8+ T cells and increased production of IL-12 and IFNγ [46,47]. Notably, we observed that CD4+ T cells express several transporters that mediate cytosolic acidification, including AE2, AE1 and Na+–HCO3− cotransporter, whereas the last two are poorly expressed in CD8+ T cells. Thus, CD8 T cells rely heavily on AE2 for intracellular acidification. Accordingly, AE2-deficient CD8+ T cells have high pHi and, upon stimulation, become more alkaline, produce more IL-2 and exhibit increased proliferative response [48]. These findings suggest that AE2 deficiency in lymphoid cells might contribute to dysregulated T-cell responses in PBC patients.
PBC is characteristically a disease affecting middle-aged women. Data from Ae2a,b-knockout mice illuminate the effect of age in the development of the disease [47]. In young Ae2a,b-knockout mice, portal tracts were infiltrated with activated T cells expressing PD1 and producing high amounts of IFNγ (this cytokine induces PD-L1 expression in cholangiocytes and in other cell types of the portal tract). In young knockout animals, the clonal expansion of autoreactive lymphocytes within the liver was inhibited by PD1/PD-L1 interaction. In contrast, in old Ae2a,b-knockout mice, PD1 gene expression was epigenetically silenced by DNA methylation in CD8+ T cells. This age-related epigenetic downregulation of PD1 allowed autoreactive CD8+ T cells to escape PD1/PD-L1-induced apoptosis, favoring clonal expansion and enhanced lymphocytic infiltration of portal tracts with aggravation of bile duct lesion. Similar epigenetic changes in patients with PBC would explain why this disease is commonly manifested later in life, between 40 and 60 years of age.
CONCLUSION
PBC results from autoimmune attack to the epithelium of interlobular bile ducts. PBC develops in patients with a genetic background predisposing to autoimmunity. The target of the autoimmune reaction is the mitochondrial antigen E2 component of the 2-oxo dehydrogenase complexes, in particular PDC-E2, which is overexpressed by stressed BECs. The immune-mediated cholangiocellular injury appears to be secondary to the failure of BEC to secrete HCO3− to bile due to AE2 deficiency. This defect is likely because of epigenetic factors involving miR-506 upregulation and AE2 promoter methylation. Impaired biliary HCO3− secretion disrupts the defensive bicarbonate umbrella allowing the penetration of hydrophobic bile acids into BECs resulting in senescence and apoptosis of these cells. In addition, AE2 deficiency alters cholangiolar pHi leading to sAC activation and increased sensitivity to bile salt-induced apoptosis. Increased pHi likely disturbs mitophagy, with resulting accumulation of faulty mitochondria, and this would trigger oxidative stress while promoting the presentation of mitochondrial antigens to the immune system. It could be hypothesized that as women exhibit a more acidic intraendolysosomal milieu than men, AE2 deficiency may disturb mitophagy more intensely in women, thus, contributing to explain the marked predominance of the disease in women. In PBC, AE2 deficiency also affects lymphocytes likely cooperating to the immunological derangement present in this condition. In summary, PBC could be considered as a disease that occurs in patients with genetic predisposition to autoimmunity in which AE2 is epigenetically downregulated both in liver and lymphoid cells. These concepts are illustrated in Fig. 1.
FIGURE 1.
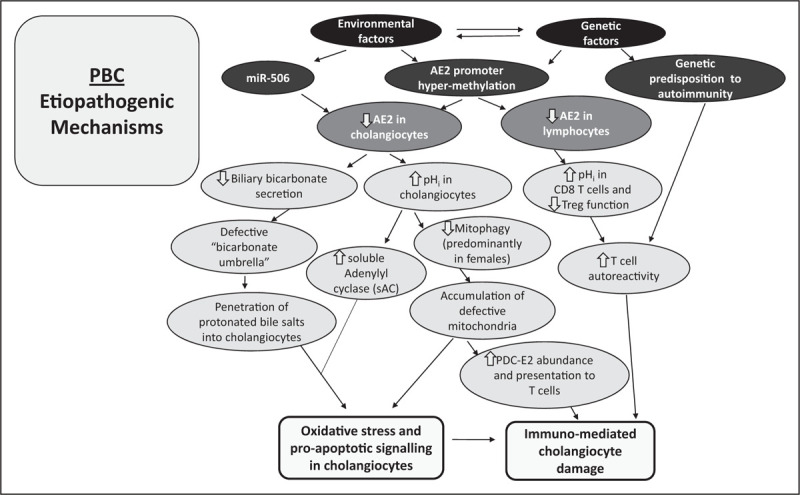
PBC: Etiopathogenic mechanisms. Epigenetic, genetic and environmental factors concur in causing PBC. Epigenetic mechanisms (including microRNAs and promoter hypermethylation) induce downregulation of AE2 in both cholangiocytes and lymphocytes. Defective AE2 function in cholangiocytes decreases biliary bicarbonate secretion while increasing intracellular pH (pHi). The disruption of the protective bicarbonate umbrella allows penetration of apolar bile salts into hepatocytes promoting cell apoptosis, which is favored by the activation of soluble adenylyl cyclase (sAC) because of the elevation of pHi. This alteration may also affect mitophagy, especially in women (in whom the endolysosomal milieu is more acidic than in men). Impaired mitophagy would lead to oxidative stress, accumulation of defective mitochondria, PDC-E2 overexpression and presentation of mitochondrial antigens to the immune system. These changes lead to immuno-mediated cell damage specially in individuals with genetic predisposition to autoimmunity. On the other hand, AE2 deficiency in lymphocytes, particularly in CD8 T cells, may contribute to enhance autoreactive T-cell responses.
Acknowledgements
None.
Financial support and sponsorship
None.
Conflicts of interest
JP and JFM none.
JMB: INCYTE: grant for research; ROCHE: grant for research; BAYER: fee for lectures; INTERCEPT: fee for lectures; OWL METABOLOMICS and QED Therapeutics: fee for consulting.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1.Lindor KD, Bowlus CL, Boyer J, et al. Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2019; 69:394–419. [DOI] [PubMed] [Google Scholar]
- 2.Hirschfield GM, Beuers U, Corpechot C, et al. EASL Clinical Practice Guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol 2017; 67:145–172. [DOI] [PubMed] [Google Scholar]
- 3.Terziroli Beretta-Piccoli B, Mieli-Vergani G, Vergani D, et al. The challenges of primary biliary cholangitis: what is new and what needs to be done. J Autoimmun 2019; 105:102328. [DOI] [PubMed] [Google Scholar]
- 4.Rodrigues PM, Perugorria MJ, Santos-Laso A, et al. Primary biliary cholangitis: a tale of epigenetically-induced secretory failure? J Hepatol 2018; 69:1371–1383. [DOI] [PubMed] [Google Scholar]
- 5.Trivedi PJ, Cullen S. Etiopathogenesis of primary biliary cirrhosis: an overview of recent developments. Hepatol Int 2013; 7:28–47. [DOI] [PubMed] [Google Scholar]
- 6.Poupon R. Primary biliary cirrhosis: a 2010 update. J Hepatol 2010; 52:745–758. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura M. Clinical significance of autoantibodies in primary biliary cirrhosis. Semin Liver Dis 2014; 34:334–340. [DOI] [PubMed] [Google Scholar]
- 8.He XS, Ansari AA, Ridgway WM, et al. New insights to the immunopathology and autoimmune responses in primary biliary cirrhosis. Cell Immunol 2006; 239:1–13. [DOI] [PubMed] [Google Scholar]
- 9.Hirschfield GM, Gershwin ME. The immunobiology and pathophysiology of primary biliary cirrhosis. Annu Rev Pathol 2013; 8:303–330. [DOI] [PubMed] [Google Scholar]
- 10.Bogdanos DP, Komorowski L. Disease-specific autoantibodies in primary biliary cirrhosis. Clin Chim Acta 2011; 412:502–512. [DOI] [PubMed] [Google Scholar]
- 11.Gulamhusein AF, Hirschfield GM. Primary biliary cholangitis: pathogenesis and therapeutic opportunities. Nat Rev Gastroenterol Hepatol 2020; 17:93–110. [DOI] [PubMed] [Google Scholar]
- 12.Younossi ZM, Bernstein D, Shiffman ML, et al. Diagnosis and management of primary biliary cholangitis. Am J Gastroenterol 2019; 114:48–63. [DOI] [PubMed] [Google Scholar]
- 13.Lindor KD, Poupon R, Poupon R, et al. Ursodeoxycholic acid for primary biliary cirrhosis. Lancet 2000; 355:657–658. [DOI] [PubMed] [Google Scholar]
- 14.Poupon RE, Poupon R, Balkau B. Ursodiol for the long-term treatment of primary biliary cirrhosis. The UDCA-PBC Study Group. N Engl J Med 1994; 330:1342–1347. [DOI] [PubMed] [Google Scholar]
- 15.Harms MH, de Veer RC, Lammers WJ, et al. Number needed to treat with ursodeoxycholic acid therapy to prevent liver transplantation or death in primary biliary cholangitis. Gut 2020; 69:1502–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corpechot C, Carrat F, Bahr A, et al. The effect of ursodeoxycholic acid therapy on the natural course of primary biliary cirrhosis. Gastroenterology 2005; 128:297–303. [DOI] [PubMed] [Google Scholar]
- 17.Parés A, Caballería L, Rodés J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology 2006; 130:715–720. [DOI] [PubMed] [Google Scholar]
- 18.Lindor K. Ursodeoxycholic acid for the treatment of primary biliary cirrhosis. N Engl J Med 2007; 357:1524–1529. [DOI] [PubMed] [Google Scholar]
- 19.Corpechot C, Poupon R, Chazouilleres O. New treatment/targets for primary biliary cholangitis. JHEP Rep 2019; 1:203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carbone M, Nardi A, Flack S, et al. Pretreatment prediction of response to ursodeoxycholic acid in primary biliary cholangitis: development and validation of the UDCA Response Score. Lancet Gastroenterol Hepatol 2018; 3:626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsuneyama K, Baba H, Morimoto Y, et al. Primary biliary cholangitis: its pathological characteristics and immunopathological mechanisms. J Med Invest 2017; 64:7–13. [DOI] [PubMed] [Google Scholar]
- 22.Tsuda M, Ambrosini YM, Zhang W, et al. Fine phenotypic and functional characterization of effector cluster of differentiation 8 positive T cells in human patients with primary biliary cirrhosis. Hepatology 2011; 54:1293–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23▪▪.Lleo A, Leung PS, Hirschfield GM, Gershwin EM. The pathogenesis of primary biliary cholangitis: a comprehensive review. Semin Liver Dis 2020; 40:34–48. [DOI] [PubMed] [Google Scholar]; An updated review of the pathogenic mechanims causing PBC.
- 24.Kita H, Matsumura S, He XS, et al. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest 2002; 109:1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lan RY, Cheng C, Lian ZX, et al. Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology 2006; 43:729–737. [DOI] [PubMed] [Google Scholar]
- 26.Pipi E, Nayar S, Gardner DH, et al. Tertiary lymphoid structures: autoimmunity goes local. Front Immunol 2018; 9:1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang CY, Ma X, Tsuneyama K, et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology 2014; 59:1944–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lleo A, Bowlus CL, Yang GX, et al. Biliary apotopes and antimitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology 2010; 52:987–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Odin JA, Huebert RC, Casciola-Rosen L, et al. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J Clin Invest 2001; 108:223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lleo A, Selmi C, Invernizzi P, et al. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology 2009; 49:871–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31▪.Erice O, Munoz-Garrido P, Vaquero J, et al. MicroRNA-506 promotes primary biliary cholangitis-like features in cholangiocytes and immune activation. Hepatology 2018; 67:1420–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]; An important article showing that upregulation of miR-506 sensitizes cholangiocytes to proapoptotic stimuli, induces PDC-E2 overexpression and causes impaired energy metabolism in mitochondria leading to oxidative and ER stress in these cells.
- 32▪▪.Banales JM, Huebert RC, Karlsen T, et al. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol 2019; 16:269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive review on cholangiocyte biology and diseases affecting bile duct epithelium.
- 33.Banales JM, Prieto J, Medina JF. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol 2006; 12:3496–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strazzabosco M. Transport systems in cholangiocytes: their role in bile formation and cholestasis. Yale J Biol Med 1997; 70:427–434. [PMC free article] [PubMed] [Google Scholar]
- 35.Boyer JL. Andreoli TE, Hoffman JF, Fanestil DD, Schultz SG. Mechanisms of bile secretion and hepatic transport. Physiology of membrane disorders. New York: Plenum Publishing Corporation; 1986. 609–636. [Google Scholar]
- 36.Banales JM, Arenas F, Rodríguez-Ortigosa CM, et al. Bicarbonate-rich choleresis induced by secretin in normal rat is taurocholate-dependent and involves AE2 anion exchanger. Hepatology 2006; 43:266–275. [DOI] [PubMed] [Google Scholar]
- 37▪▪.Martínez-Ansó E, Castillo JE, Díez J, et al. Immunohistochemical detection of chloride/bicarbonate anion exchangers in human liver. Hepatology 1994; 19:1400–1406. [PubMed] [Google Scholar]; This article revealed for the first time the presence of AE2 protein at the luminal pole of intrahepatic bile duct epithelial cells.
- 38▪▪.Medina JF, Martínez-Ansó E, Vázquez JJ, Prieto J. Decreased anion exchanger 2 immunoreactivity in the liver of patients with primary biliary cirrhosis. Hepatology 1997; 25:12–17. [DOI] [PubMed] [Google Scholar]; This article revealed that AE2 protein was poorly expressed in intrahepatic BECs in PBC patients.
- 39.García C, Montuenga LM, Medina JF, Prieto J. In situ detection of AE2 anion-exchanger mRNA in the human liver. Cell Tissue Res 1998; 291:481–488. [DOI] [PubMed] [Google Scholar]
- 40.Tietz PS, Marinelli RA, Chen XM, et al. Agonist-induced coordinated trafficking of functionally-related transport proteins for water and ions in cholangiocytes. J Biol Chem 2003; 278:20413–20419. [DOI] [PubMed] [Google Scholar]
- 41.Concepcion AR, Lopez M, Ardura-Fabregat A, Medina JF. Role of AE2 for pHi regulation in biliary epithelial cells. Front Physiol 2014; 4:413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minagawa N, Nagata J, Shibao K, et al. Cyclic AMP regulates bicarbonate secretion in cholangiocytes throug the release of ATP into bile. Gastroenterology 2007; 133:1592–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43▪▪.Prieto J, Qian C, García N, et al. Abnormal expression of anion exchanger genes in primary biliary cirrhosis. Gastroenterology 1993; 105:572–578. [DOI] [PubMed] [Google Scholar]; This is the first article showing defective AE2 expression in PBC.
- 44▪▪.Prieto J, García N, Martí-Climent JM, et al. Assessment of biliary bicarbonate secretion in humans by positron emission tomography. Gastroenterology 1999; 117:167–172. [DOI] [PubMed] [Google Scholar]; This work is the first in-vivo imaging of biliary bicarbonate secretion in humans and the demonstration that this process is impaired in PBC and is ameliorated by UDCA therapy.
- 45.Melero S, Spirlì C, Zsembery A, et al. Defective regulation of cholangiocyte Cl−/HCO3− and Na+/H+ exchanger activities in primary biliary cirrhosis. Hepatology 2002; 35:1513–1521. [DOI] [PubMed] [Google Scholar]
- 46.Salas JT, Banales JM, Sarvide S, et al. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology 2008; 134:1482–1493. [DOI] [PubMed] [Google Scholar]
- 47.Concepcion AR, Salas JT, Saez E, et al. −/−CD8+ T cells undergo activation and programmed death-1 repression in the liver of aged Ae2a,b−/− mice favoring autoimmune cholangitis. Oncotarget 2015; 6:28588–28606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Concepcion AR, Salas JT, Sarvide S, et al. Anion exchanger 2 is critical for CD8+ T cells to maintain pHi homeostasis and modulate immune responses. Eur J Immunol 2014; 44:1341–1351. [DOI] [PubMed] [Google Scholar]
- 49.Banales JM, Saez E, Uriz M, et al. Up-regulation of microRNA 506 leads to decreased Cl−/HCO3− anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology 2012; 56:687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ananthanarayanan M, Banales JM, Guerra MT, et al. Posttranslational regulation of the type III inositol 1,4,5-trisphosphate receptor by miRNA-506. J Biol Chem 2015; 290:184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang JC, Go S, de Waart DR, et al. Soluble adenylyl cyclase regulates bile salt-induced apoptosis in human cholangiocytes. Hepatology 2016; 64:522–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52▪▪.Chang JC, Go S, Verhoeven AJ, et al. Role of the bicarbonate-responsive soluble adenylyl cyclase in cholangiocyte apoptosis in primary biliary cholangitis; a new hypothesis. Biochim Biophys Acta 2018; 1864:1232–1239. [DOI] [PubMed] [Google Scholar]; This article shows that the elevation of pHi in cholangiocytes resulting from defective AE2 expression sensitizes these cells to proapoptotic stimuli via activation of sAC.
- 53▪▪.Berezhnov AV, Soutar MP, Fedotova EI, et al. Intracellular pH modulates autophagy and mitophagy. J Biol Chem 2016; 291:8701–8708. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article conveys the important message that cytosolic acidification stimulates mitophagy.
- 54.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011; 333:1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55▪.Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020; 16:3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]; This comprehensive review illuminates the role of defective mitophagy in tissue inflammation and autoimmunity.
- 56.Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016; 164:896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57▪▪.Matheoud D, Sugiura A, Bellemare-Pelletier A, et al. Parkinson's disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell 2016; 166:314–327. [DOI] [PubMed] [Google Scholar]; This is a relevant article showing that a defect in mitophagy promotes the presentation of mitochondrial antigens to the immune system, a fact that might shed light on the pathogenesis of PBC.
- 58▪▪.Harris VM, Harley ITW, Kurien BT, et al. Lysosomal pH is regulated in a sex dependent manner in immune cells expressing CXorf21. Front Immunol 2019; 10:578. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows that the pH of the endolysomal compartment is more acidic in women than in men.
- 59.Ventura-Clapier R, Moulin M, Piquereau J, et al. Mitochondria: a central target for sex differences in pathologies. Clin Sci (Lond) 2017; 131:803–822. [DOI] [PubMed] [Google Scholar]
- 60.Beuers U, Hohenester S, de Buy Wenniger LJ, et al. The biliary Cl−/HCO3− umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 2010; 52:1334–1340. [DOI] [PubMed] [Google Scholar]
- 61▪▪.van Niekerk J, Kersten R, Beuers U. Role of bile acids and the biliary HCO3− umbrella in the pathogenesis of primary biliary cholangitis. Clin Liver Dis 2018; 22:457–479. [DOI] [PubMed] [Google Scholar]; This report describes in detail the implication of defective biliary bicarbonate umbrella in cholangiocyte damage in PBC.
- 62.Hisamoto S, Shimoda S, Harada K, et al. Hydrophobic bile acids suppress expression of AE2 in biliary epithelial cells and induce bile duct inflammation in primary biliary cholangitis. J Autoimmun 2016; 75:150–160. [DOI] [PubMed] [Google Scholar]
- 63▪.Sasaki M, Sato Y, Nakanuma Y. An impaired biliary bicarbonate umbrella may be involved in dysregulated autophagy in primary biliary cholangitis. Lab Invest 2018; 98:745–754. [DOI] [PubMed] [Google Scholar]; This article links defective bicarbonate umbrella with impairment of autophagy in PBC, thus, establishing a connection between autophagia and changes in pHi.
- 64.Molinaro A, Marschall HU. Why doesn’t primary biliary cholangitis respond to immunosuppressive medications? Curr Hepatology Rep 2017; 16:119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bowlus CL, Yang GX, Liu CH, et al. Therapeutic trials of biologics in primary biliary cholangitis: an open label study of abatacept and review of the literature. J Autoimmun 2019; 101:26–34. [DOI] [PubMed] [Google Scholar]
- 66.Floreani A, Franceschet I, Perini L, et al. New therapies for primary biliary cirrhosis. Clin Rev Allergy Immunol 2015; 48:263–272. [DOI] [PubMed] [Google Scholar]
- 67.Hirschfield GM, Dyson JK, Alexander GJM, et al. The British Society of Gastroenterology/UK-PBC primary biliary cholangitis treatment and management guidelines. Gut 2018; 67:1568–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hirschfield GM, Gershwin ME, Strauss R, et al. PURIFI Study Group. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: a proof-of-concept study. Hepatology 2016; 64:189–199. [DOI] [PubMed] [Google Scholar]
- 69.Arenas F, Hervías I, Úriz M, et al. Combination of ursodeoxycholic acid and glucocorticoids upregulates the AE2 alternate promoter in human liver cells. J Clin Invest 2008; 118:695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rodríguez-Ortigosa CM, Banales JM, Olivas I, et al. Biliary secretion of S-nitrosoglutathione is involved in the hypercholeresis induced by ursodeoxycholic acid in the normal rat. Hepatology 2010; 52:667–677. [DOI] [PubMed] [Google Scholar]
- 71.Hirschfield GM, Invernizzi P. Progress in the genetics of primary biliary cirrhosis. Semin Liver Dis 2011; 31:147–156. [DOI] [PubMed] [Google Scholar]
- 72.Nakamura M, Nishida N, Kawashima M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet 2012; 91:721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu JZ, Almarri MA, Gaffney DJ, et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat Genet 2012; 44:1137–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Selmi C, Cavaciocchi F, Lleo A, et al. Genome-wide analysis of DNA methylation, copy number variation, and gene expression in monozygotic twins discordant for primary biliary cirrhosis. Front Immunol 2014; 5:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xie YQ, Ma HD, Lian ZX. Epigenetics and primary biliary cirrhosis: a comprehensive review and implications for autoimmunity. Clin Rev Allergy Immunol 2016; 50:390–403. [DOI] [PubMed] [Google Scholar]
- 76.Aiba Y, Nakamura M, Joshita S, et al. Genetic polymorphisms in CTLA4 and SLC4A2 are differentially associated with the pathogenesis of primary biliary cirrhosis in Japanese patients. J Gastroenterol 2011; 46:1203–1212. [DOI] [PubMed] [Google Scholar]
- 77.Juran BD, Atkinson EJ, Larson JJ, et al. Common genetic variation and haplotypes of the anion exchanger SLC4A2 in primary biliary cirrhosis. Am J Gastroenterol 2009; 104:1406–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Poupon R, Ping C, Chretien Y, et al. Genetic factors of susceptibility and of severity in primary biliary cirrhosis. J Hepatol 2008; 49:1038–1045. [DOI] [PubMed] [Google Scholar]
- 79.Lleo A, Zhang W, Zhao M, et al. DNA methylation profiling of the X chromosome reveals an aberrant demethylation on CXCR3 promoter in primary biliary cirrhosis. Clin Epigenetics 2015; 7:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aran D, Toperoff G, Rosenberg M, Hellman A. Replication timing related and gene body-specific methylation of active human genes. Hum Mol Genet 2011; 20:670–680. [DOI] [PubMed] [Google Scholar]
- 81▪.Arenas F, Hervías I, Sáez E, et al. Promoter hypermethylation of the AE2/SLC4A2 gene in PBC. JHEP Rep 2019; 1:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article revealed that promoter hypermethylation is one of the mechanisms involved in AE2 downregulation both in liver and peripheral mononuclear cells from PBC patients.
- 82.Malumbres R, Lecanda J, Melero S, et al. HNF1α upregulates the human AE2 anion exchanger gene (SLC4A2) from an alternate promoter. Biochem Biophys Res Commun 2003; 311:233–240. [DOI] [PubMed] [Google Scholar]
- 83.Medina JF, Acín A, Prieto J. Molecular cloning and characterization of the human AE2 anion exchanger (SLC4A2) gene. Genomics 1997; 39:74–85. [DOI] [PubMed] [Google Scholar]
- 84.Medina JF, Lecanda J, Acín A, et al. Tissue-specific N-terminal isoforms from overlapping alternate promoters of the human AE2 anion exchanger gene. Biochem Biophys Res Commun 2000; 267:228–235. [DOI] [PubMed] [Google Scholar]
- 85.Kim KA, Kim YS, Park SH, et al. Environmental risk factors and comorbidities of primary biliary cholangitis in Korea: a case-control study. Korean J Intern Med 2020; doi: 10.3904/kjim.2019.234. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Prince MI, Ducker SJ, James OF. Case-control studies of risk factors for primary biliary cirrhosis in two United Kingdom populations. Gut 2010; 59:508–512. [DOI] [PubMed] [Google Scholar]
- 87.Gershwin ME, Selmi C, Worman HJ, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology 2005; 42:1194–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hammons GJ, Yan Y, Lopatina NG, et al. Increased expression of hepatic DNA methyltransferase in smokers. Cell Biol Toxicol 1999; 15:389–394. [DOI] [PubMed] [Google Scholar]
- 89.Celay J, Lozano T, Concepcion AR, et al. Targeting the anion exchanger 2 with specific peptides as a new therapeutic approach in B lymphoid neoplasms. Haematologica 2018; 103:1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]