

Abstract
Modification of endogenous proteins by drugs and drug metabolites are thought to be a cause of idiosyncratic adverse drug reactions (IADRs). Trimethoprim (TMP) is a commonly prescribed antibiotic that has been implicated in IADRs; however, there is no known mechanism by which this drug or its metabolites modify proteins. This study describes the results of screening trimethoprim and its primary metabolites for the ability to covalently modify human serum albumin (HSA). The first step of the screen was in vitro reactions of the compounds with HSA followed by western blotting with antisera specific to drug-modified proteins. Compounds with positive signal in the western blot were then screened using an untargeted peptide profiling method to discover modified peptides. This strategy identified two sites in HSA that are modified by incubation with a TMP metabolite, α-hydroxy trimethoprim (Cα-OH-TMP).
Keywords: Trimethoprim, metabolism, protein adduct, adverse drug reaction
Introduction
Trimethoprim (TMP), in combination with sulfamethoxazole, is a commonly prescribed antibiotic that is associated with idiosyncratic adverse drug reactions (IADRs). Until recently, TMP was assumed to be an unreactive, innocent bystander as sulfamethoxazole alone has been implicated in causing IADRs, given its ability to undergo bioactivation and protein adduct formation (Sanderson et al. 2007). However, more recent in vitro data support the bioactivation of TMP to reactive iminoquinone methide and para-quinone methide TMP intermediates (Damsten et al. 2008; Lai et al. 1999). Human liver microsomal (HLM) incubation and inhibition studies in the presence of NADPH identified CYP3A4 as the primary enzyme responsible for TMP bioactivation, resulting in TMP-N-acetylcysteine (TMP-NAC) adduct formation (Goldman et al. 2015). Incubation of [14C]TMP with HLMs and NADPH resulted in estimated covalent binding comparable to other drugs associated with covalent binding-induced adverse reactions (Goldman et al. 2016). TMP-NAC adducts have been identified in the urine of patients exposed to TMP which is further evidence of TMP bioactivation in vivo (van Haandel et al. 2014).
IADRs have in part been attributed to the ability of a small molecules to undergo bioactivation resulting in an electrophile that can subsequently form an adduct with an endogenous protein (Evans et al. 2004). A drug-modified protein (hapten) may then trigger an immune response resulting in an IADR (Cho and Uetrecht 2017). Greater daily doses and covalent binding capability are associated with increased idiosyncratic drug toxicity (Nakayama et al. 2009). TMP is typically prescribed at high daily doses (>300mg/day) and has evidence of significant covalent protein binding, further supporting a potential role for TMP induced IADRs. Parent drug bioactivation or subsequent reactive metabolites can both result in covalent binding leading to drug-modified proteins (Cho and Uetrecht 2017). Even some relatively stable drugs or circulating metabolites can directly generate protein adducts. For example, isoniazid-modified human serum albumin (HSA) peptides were detected following incubation of isoniazid directly with HSA without oxidative enzymes or redox-active metals (Meng et al. 2015). Carbamazepine 10,11-epoxide (CBZE), initially presumed to be a stable circulating plasma metabolite, can spontaneously form CBZE-glutathione adducts, and covalently bind to HSA (Yip et al. 2017).
TMP’s primary metabolites, trimethoprim 1-N-oxide (1-NO-TMP), trimethoprim 3-N-oxide (3-NO-TMP), 3´-desmethyl-TMP (3-DM-TMP), 4´-desmethyl-TMP (4-DM-TMP), and α-hydroxy trimethoprim (Cα-OH-TMP), do not appear overtly reactive (Figure 1) (Sigel et al. 1973). Remarkably, preliminary in-house data indicated that Cα-OH-TMP was able to modify proteins directly, without enzymatic activity. Given these findings and the limited understanding of TMP adduct formation capabilities, we set out to systematically screen TMP primary metabolites for the capacity to modify HSA.
Fig. 1.
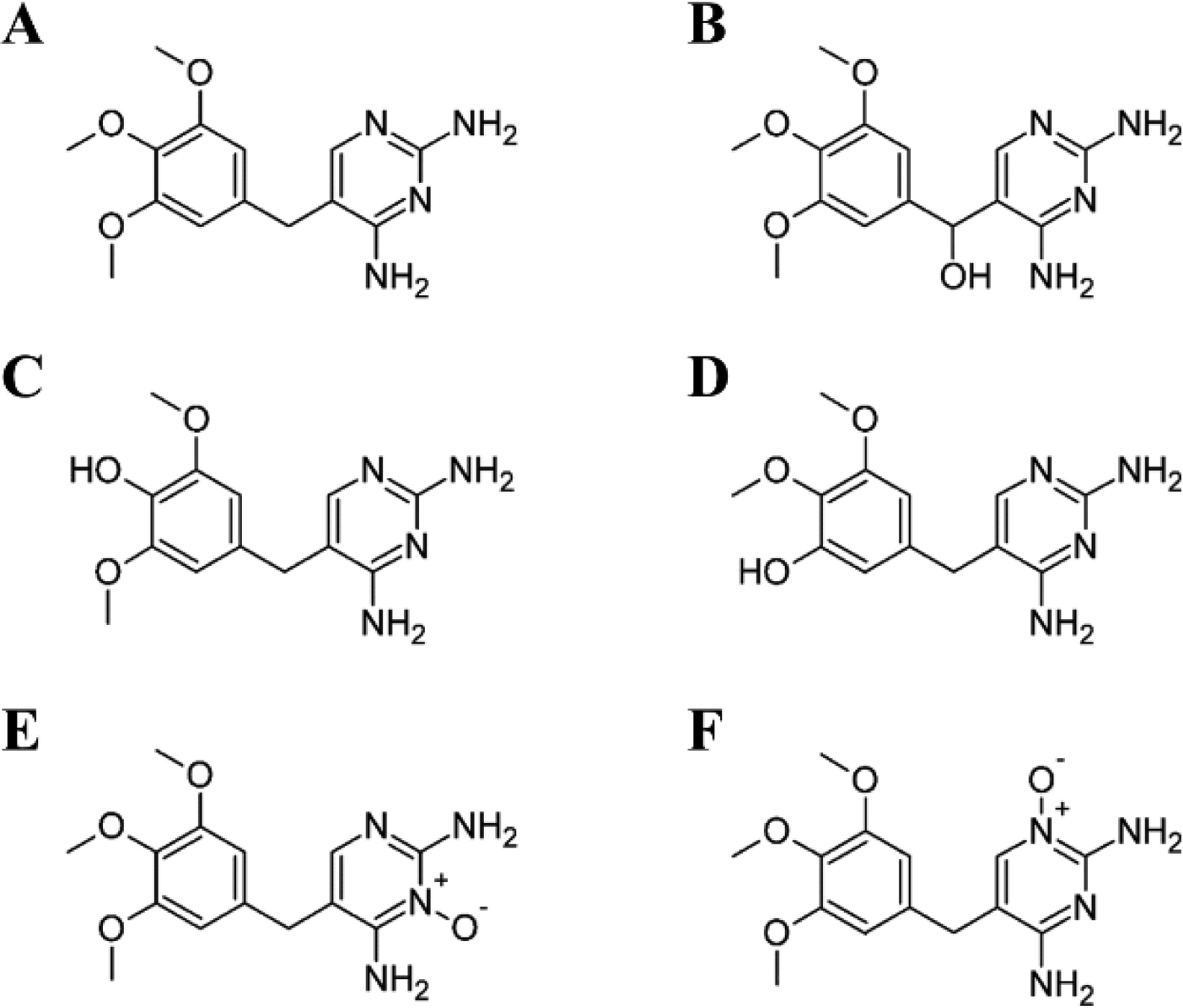
Primary trimethoprim metabolites a, TMP b, Cα-OH-TMP c, 4-DM-TMP d, 3-DM-TMP, e, 1-NO-TMP f, 3-NO-TMP
Materials and Methods
Chemicals and Reagents
1-NO-TMP, 3-NO-TMP, 3´-DM-TMP, 4´-DM-TMP, Cα-OH-TMP, and Cα-NAC-TMP were custom synthesized by Artis-Chem Co. Ltd. (Shanghai, China). Purity of the standards was determined to be ≥98% by H-NMR, LC-MS and LC-UV at 214nm. LC-MS Optima® grade acetonitrile, methanol, and water were purchased from Fisher Scientific (Fairlawn, NJ, USA). All reagents were reagent grade quality or higher. EDTA, formic acid (LC-MS grade), glucose-6-phosphate dehydrogenase, magnesium chloride, N-acetyl-L-cysteine (NAC), NADP, potassium phosphate dibasic, potassium phosphate monobasic, keyhole limpet hemocyanin (KLH), RNase, N-Hydroxysuccinimde, N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC) were purchased from Sigma Aldrich Chemical Co. (St. Louis, MO). Normal goat serum (Sigma), poly(vinyl alcohol) (Sigma) Tween 20 (Bio-Rad), Goat anti-Rabbit IgG (H+L) (Thermo Scientific), Poly-Horseradish peroxidase (HRP) Secondary Antibody (Thermo Scientific), HRP conjugate (Thermo Scientific), TMB Substrate Kit (Thermo Scientific).
Synthesis of Immunogen and Immunization Protocol
The TMP antigen was prepared by coupling TMP-NAC to KLH and RNase by EDC conjugation. Two rabbits were immunized with TMP antigen per the standard 70-day rabbit immunization protocol for rabbit polyclonal antibody production (Thermo Scientific). Primary injection occurred on day 1 with 0.50 mg of antigen, followed by boost injections with 0.25 mg of antigen on days 14, 28, and 42. Sera was collected on days 35, 56, 58 and terminal bleed. Antisera was characterized by enzyme-linked immunosorbent assay (ELISA). A dilution of 1:102400 detected antigen and inhibition of 16% antisera binding was observed with 500 μM TMP.
Incubation of human serum albumin with trimethoprim metabolites
Trimethoprim and metabolites (1-NO-TMP, 3-NO-TMP, 3-DM-TMP, 4-DM-TMP and Cα-OH-TMP) were solubilized from powder in phosphate buffer saline containing 1mg/ml HSA. Solutions were held at 37°C in microcentrifuge tubes. Samples were diluted in 4X LDS buffer to achieve a protein concentration of 0.5 μg/μL in preparation for western blot analysis. Sample preparation for LC-MS analysis was as follows: 500uL ice cold acetone was added to 100uL incubation mixture (100 μg HSA protein equivalent) and kept at −20°C until samples were processed for digestion and analysis.
Western blotting for detection of trimethoprim modified proteins
Incubations at a 33:1 drug: albumin molar ratio were used to compare adduct formation of trimethoprim and metabolites. Drug: albumin molar ratios from 33:1 to 1:1 were used for concentration-dependent experiments. Protein adducts were visualized using SDS-PAGE and western blotting using 4uG HSA/well. After blocking, membranes were incubated in 5% milk containing 1:500 primary antibody for 12–16 hours with rocking at 4°C then washed in Tris buffered saline with 0.1% Tween 20 (TBST) 4X for 5 minutes each. This was followed by a 1-hour incubation with a 1:2000 dilution of IgG Goat/Anti-rabbit HRP secondary (Thermo Fisher 31460) in TBST and washed 4X in TBST for 5 minutes each. Images were developed using SuperSignal West Pico Plus chemiluminescent solution (Thermo Fisher 34580) and visualized via a Thermo Fisher iBright FL1000 imager.
Proteomic sample preparation
The acetone quenched samples were spun at 10,000 g for ten minutes to precipitate the protein. The supernatant was removed, and the samples were dried under nitrogen flow. The precipitated protein samples were digested using Sequencing Grade Modified Trypsin (Promega, Madison, WI). Briefly, the samples were resuspended in 50uL 6 M urea/50 mM ammonium bicarbonate pH 8. For reduction, dithiothreitol was added to a final concentration of 5 mM and incubated for thirty minutes at 37°C. For cysteine capping, iodoacetamide was added to a final concentration of 15 mM and incubated for thirty minutes in the dark at room temperature. The digest was diluted to 300 μL with 50 mM ammonium bicarbonate pH 8, and trypsin was added (1:50, trypsin: protein) for an overnight incubation at 37°C. Formic acid was added to 1% prior to analysis.
Liquid chromatography mass spectrometry
The tryptic digests were analyzed on a Waters Xevo G2-XS quadrupole time-of-flight (QTOF) mass spectrometer coupled to a Waters Acquity M-class UPLC system with an ion-key ionization source. Samples were trapped on an M-class symmetry C18 column (300μM × 50mm packed with a 5μM particle), followed by separation on a peptide BEH (1.7 μM particle) ion-key column (150μM × 50mm). The liquid chromatography gradient proceeded from 3–50% acetonitrile/water (0.1% formic acid) over 20 minutes at a flowrate of 1.5 μL/min. Samples were ionized in positive mode with ESI parameters as follows: capillary voltage, 3 kV ; sampling cone voltage, 40 V; source temperature, 120°C; desolvation temperature, 600°C; cone gas flow, 25.0 L/hr. Full scan LC-MS data was acquired over a range of 400–2000 m/z in sensitivity mode with leucine enkephalin as a lockspray analyte. Fragmentation analysis for peptide identification was performed using MSMS mode for individual target ions and fast data dependent acquisition (fastDDA) over the range of 150–1400 m/z and 250–1500 m/z, respectively.
LC-MS Data analysis
Data files were centroided and converted to NetCDF files by Mass Lynx 4.1 software and the DataBridge utility. The converted files were uploaded to XCMS Online for data processing. A pairwise analysis was created to compare each metabolite/drug treated group to the vehicle group. Each analysis consisted of four LC-MS runs for each group. The predefined parameters for HPLC/Q-TOF were used with the following modifications: m/z deviation of 15 ppm, signal to noise threshold of 10, and a prefilter intensity of 1000. Feature detection used the centWave algorithm, and the retention time correction used the obiwarp method. Features were considered significant with fold changes of at least three-fold and p-values less than 0.05 by a Welch’s t-test. Manual peak inspection was performed using Mass Lynx software.
Spectra for unidentified peptides were searched against the human Uniprot database (downloaded 10/31/2019) using Peaks Studio 10 (Bioinformatics Solutions Inc., Waterloo, CA). The following search parameters were used: mass precursor tolerance of 15 ppm, fragment mass error tolerance of 0.1 Da, trypsin as the enzyme, fixed modification of carbamidomethylation (C), variable modification of TMP (288.122, on Cys, His, and Lys). The false discovery rate (FDR), calculated using a decoy fusion method, were used to filter the peptide hits; the FDR was set to 0.1% (-logP 28.7).
Results and Discussion
An initial screening of TMP and its five primary metabolites for their ability to covalently bind protein was performed. Metabolites were incubated with HSA and subjected to western blot analysis with an antibody that recognizes TMP metabolite-modified proteins (Fig. 2a). Cα-OH-TMP clearly had a robust signal by western blotting with TMP and 1-NO-TMP having much weaker signals. All other metabolites had signals equivalent to untreated HSA. These data indicated Cα-OH-TMP, 1-NO-TMP, and TMP may potentially form covalent adducts with albumin. Because of the robust western blotting signal, dose-dependent incubations of Cα-OH-TMP and HSA were performed (Fig. 2b). The western signal observed increased with dose, indicating that the signal was specific to Cα-OH-TMP-mediated covalent binding.
Fig. 2.
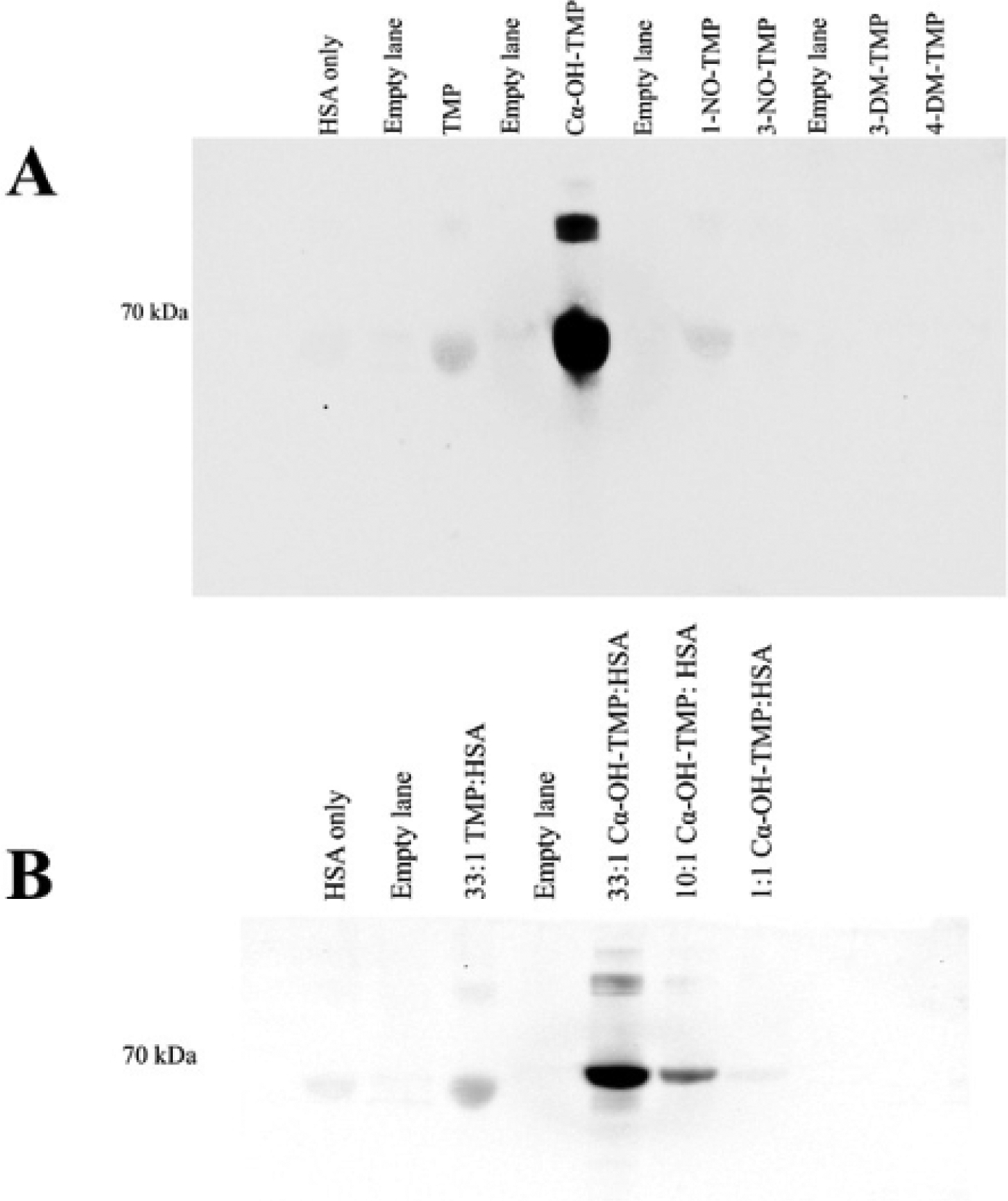
Western blot analysis a, TMP and TMP metabolites incubated with HSA b, dose- dependent HSA treatment with Cα-OH-TMP
Given that TMP is known to be highly protein bound (Wijkstrom and Westerlund 1983), the possibility that some of the western blot signal for TMP or its metabolites is due to noncovalent binding was considered. Therefore, an untargeted peptide profiling strategy was used as a secondary screen for covalent protein binding. XCMS has been widely used to identify changing metabolites, including peptides, between different sample sets (Nolte et al. 2009; Tautenhahn et al. 2012). XCMS aligns chromatograms from liquid chromatography mass spectrometry (LC-MS) experiments, quantifies changing species, and generates reports with ratios and statistics. This tool allowed identification of potentially modified peptides in a relatively unbiased manner.
In the secondary screen for covalent binding, HSA was treated with the compounds, which had positive signal in the first screen: TMP, Cα-OH-TMP, and 1-NO-TMP. The samples were then precipitated and digested with trypsin prior to LC-MS analysis. The compound-treated versus untreated samples were then compared using XCMS. The results of the XCMS analyses for each compound are summarized in Table 1. The data was quite dense with approximately 20,000 features being detected in each comparative analysis. To identify only potentially modified peptides, the XCMS data was filtered to only look for significant features elevated in the treated samples. Cα-OH-TMP-treated samples had 30 significant features. TMP itself had no significant features, and 1-NO-TMP had three. The data was then manually curated to look for features that had reasonable peak quality and were present only in treated samples. The species that passed the manual verification process were consolidated by merging different charge states and deisotoping the data. After data curation, four candidate modified peptides from Cα-OH-TMP-treated samples were identified (Table 1). For clarity, these peptides were named Peptides 1–4. None of the changing features from 1-NO-TMP treatment passed manual data curation. Representative extracted ion chromatograms for Peptide 1 and Peptide 2 illustrate that they are only present in the Cα-OH-TMP-treated samples (Fig. 3a–b). The differences in the areas calculated by XCMS for these two modified peptides are illustrated by boxplots (Fig. 3c–d).
Table 1.
Changing features obtained during XCMS processing
| Metabolite | Features | Filtered Featuresa | Manually verified potential peptidesb |
|---|---|---|---|
| TMP | 19603 | 0 | 0 |
| Cα-OH-TMP | 20033 | 30 | 4 |
| 1-NO-TMP | 19393 | 3 | 0 |
Elevated in treated sample, not deisotoped, fold>3, pvalue<0.05
Deisotoped, consolidated charge states, no overlapping spectra
Fig. 3.
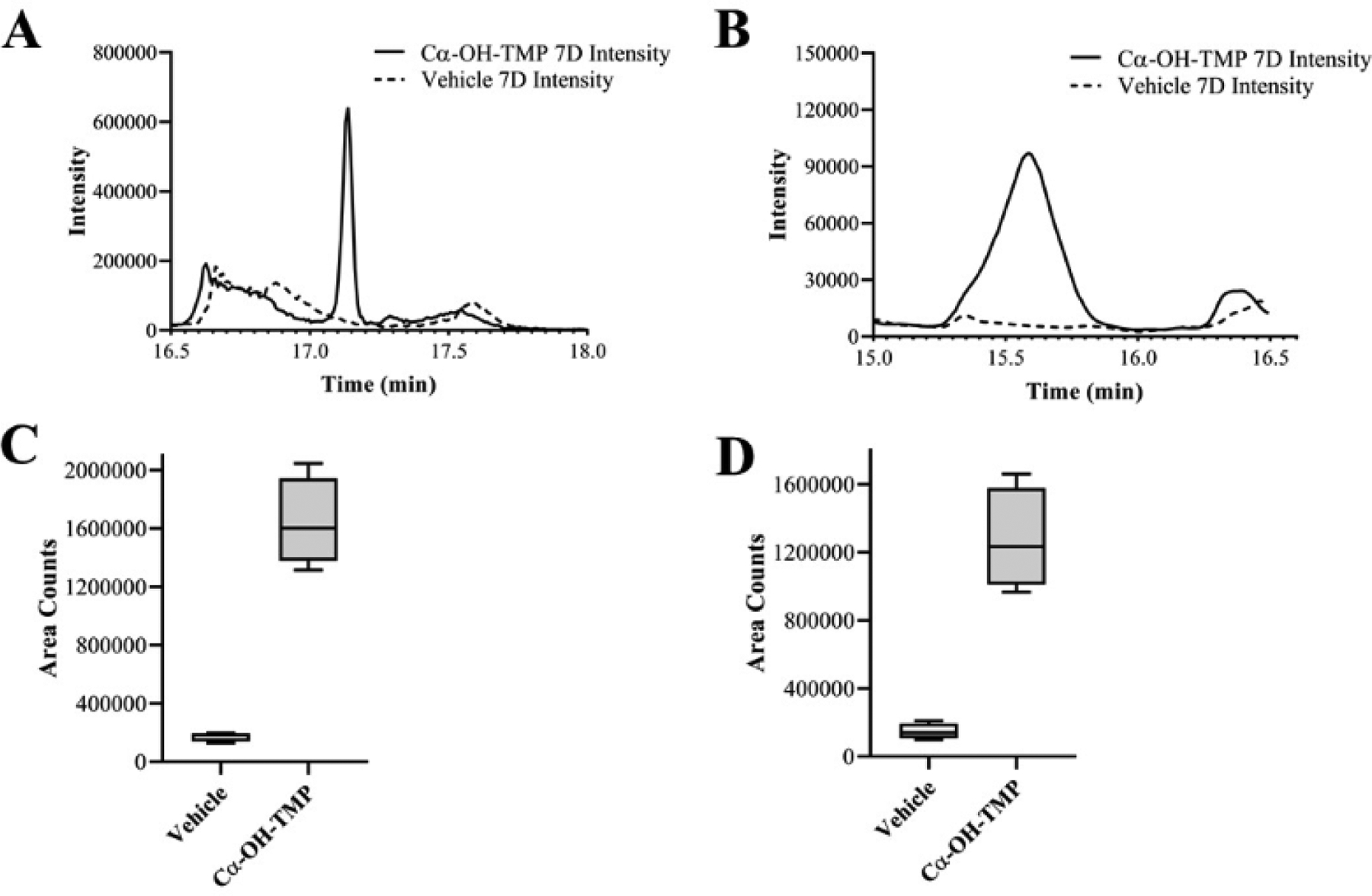
Representative extracted ion chromatograms (EICs) and area boxplots from XCMS data a, EIC for Peptide 1 (681.1) b, EIC for Peptide 2 (816.37) c, boxplot for Peptide 1 (681.1) d, boxplot for Peptide 2 (816.37). Boxes depict the 25–75th percentiles with the whiskers depicting the minimum and maximum values
The four candidate modified peptides from Cα-OH-TMP treatment were analyzed using tandem mass spectrometry (MS2) approaches, the Peaks database searching tool, and manual spectrum inspection (Tran et al. 2019). The 288.12 Dalton modification used for the database search was based upon the structure of the NAC adduct of TMP, which has been observed in patient samples and human liver microsome incubations (Goldman et al. 2015; van Haandel et al. 2014). Two peptides of the four candidate peptides, Peptides 1 and 2, were confidently identified with TMP modifications (Table 2). The tandem spectra used to identify these peptides show that both of these peptides had a common fragment at m/z 289.13 (Fig. 4), which is the same fragment reported for the NAC adduct of TMP (Fig. 5) (van Haandel et al. 2014). This common fragment ion further supports that these peptides are modified by treatment with Cα-OH-TMP. The modification on Peptide 1 is likely on the cysteine residue, since the mass of the peptide is consistent with no cysteine carbamidomethylation and a modification of 288.12 Daltons. The Peaks algorithm calculates an A-Score for each modification to express the probability that a modification is on an individual amino acid (Chalkley and Clauser 2012). The A-score for the modification on the cysteine of Peptide 1 is 25.58, which is equivalent to a p-value of 0.003. The strong y ions from y11-y19 further support this modification site; however, without stronger y1-y8 ions, the specific modification site is not unambiguous. The cysteine residue of Peptide 1 is Cys34, which is a residue in HSA that is frequently modified by electrophiles, electrophilic drugs, and metabolites (Sabbioni and Turesky 2017). This is attributed to the fact that Cys34 exists as a free thiol and has a low pKa relative to other cysteine residues (Stewart et al. 2005). The modification on Peptide 2 is predicted to be on the C-terminal lysine, Lys313, residue with an A-score of 43.05, which is equivalent to a p-value of 0.00005. However, the absence of a b or y ion with the intact modification, leaves open the possibility that the histidine residue, His288, is modified. His288 is a site that has previously been reported as a site for modification by 4-hydroxy-trans-2-nonenal (Aldini et al. 2006). The lack of a modified b or y ion could be due to fragmentation conditions, which destroy the modification at relatively low energies, generating an intense 289.13 reporter ion signal. This indicates that, like glycosylation or phosphorylation, the tendency to lose the modification during fragmentation might present challenges in accurately localizing the modification within a peptide (Hinneburg et al. 2016).
Table 2.
Modified peptides identified after Cα-OH-TMP treatment
| Peptide | m/za | Charge State | RTa | Peptide identificationb | Residues of mature HSA | Ratioa | p-valuea | Filtered Featuresa | Mass | −10logPc | A-scored |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide 1 | 907.80 | 3 | 17.1 | ALVLIAFAQYLQQC#PFEDHVK | 21–41 | 4.0 | 1.33E-05 | 5 | 2720.38 | ||
| 681.10* | 4 | 17.1 | ALVLIAFAQYLQQC#PFEDHVK | 21–41 | 9.5 | 2.18E-03 | 5 | 2720.38 | 57.31 | 25.58 | |
| 545.08 | 5 | 17.1 | ALVLIAFAQYLQQC#PFEDHVK | 21–41 | 4.5 | 7.39E-03 | 4 | 2720.38 | |||
| Peptide 2 | 1088.16 | 3 | 15.5 | SHCÎAEVENDEMPADLPSLAADFVESK# | 287–313 | 40.6 | 1.65E-03 | 4 | 3261.46 | ||
| 816.37* | 4 | 15.5 | SHCÎAEVENDEMPADLPSLAADFVESK# | 287–313 | 8.7 | 4.16E-03 | 7 | 3261.46 | 45.79 | 43.05 | |
| Peptide 3 | 588.48 | 5 | 13.7 | --- | --- | 12.4 | 3.76E-02 | 2 | 2937.38 | ||
| Peptide 4 | 511.92 | 6 | 13.3 | --- | --- | 33.6 | 4.14E-02 | 1 | 3065.48 |
m/z targeted for peptide identification
Modification due to treatment (+288.12 daltons)
Carbamidomethylation modification due to cysteine capping
Data from XCMS processing (monoisotopic m/z, retention time, ratio, p-value from Welch’s t-test, feature count)
Modified HSA sequences identified by Peaks database search
Peptide score from Peaks database search
Score localizing modification to specific amino acid, A-score=−10*log(p-value)
Fig. 4.
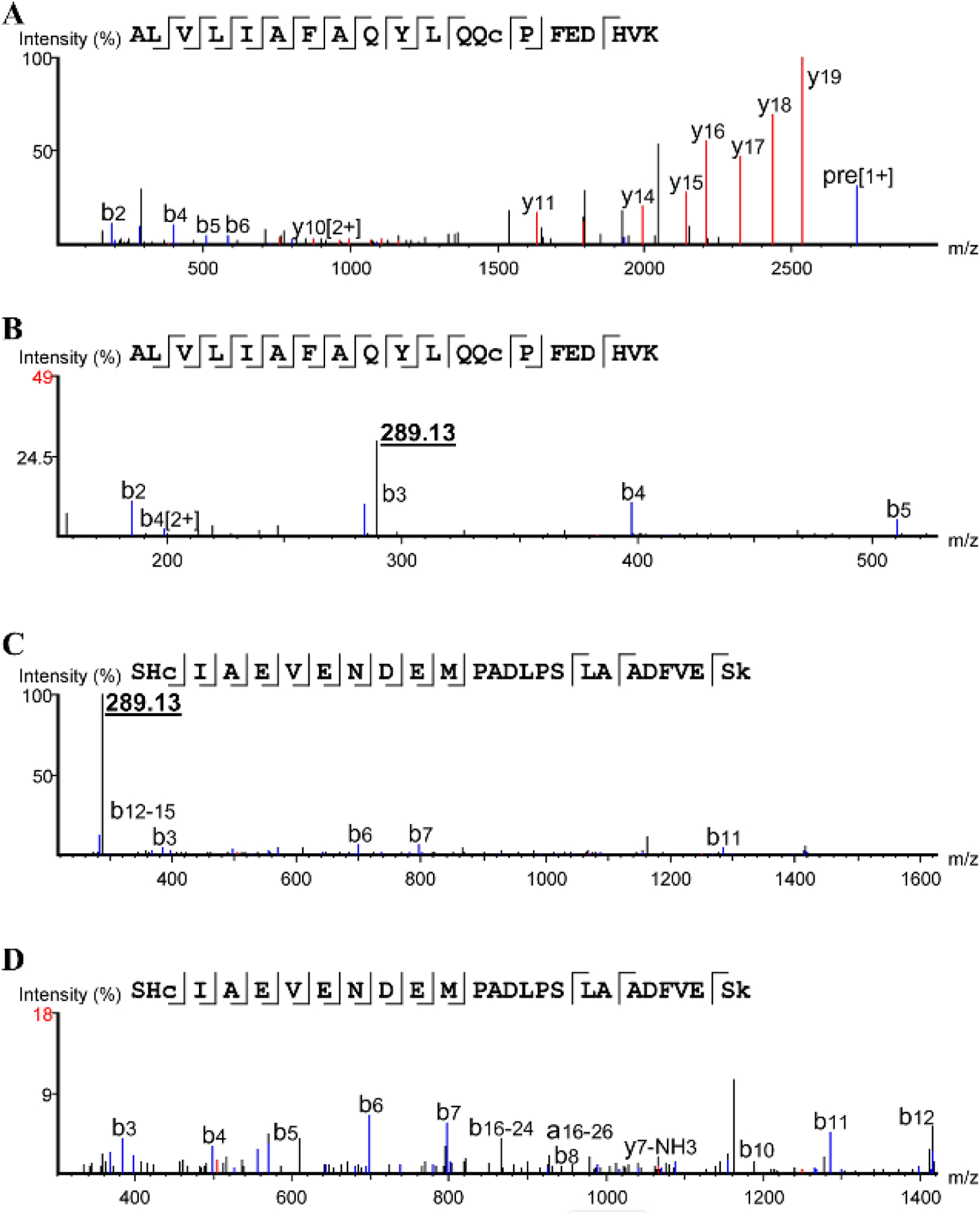
Fragmentation spectra a, spectrum of Peptide 1 (m/z 681.1) showing b and y ions b, zoomed in spectrum of Peptide 1 (m/z 681.1) showing 289.13 reporter c, zoomed out spectrum of Peptide 2 (m/z 816.37) showing 289.13 reporter d, spectrum of Peptide 2 (m/z 816.37) showing b and y ions
Fig. 5.
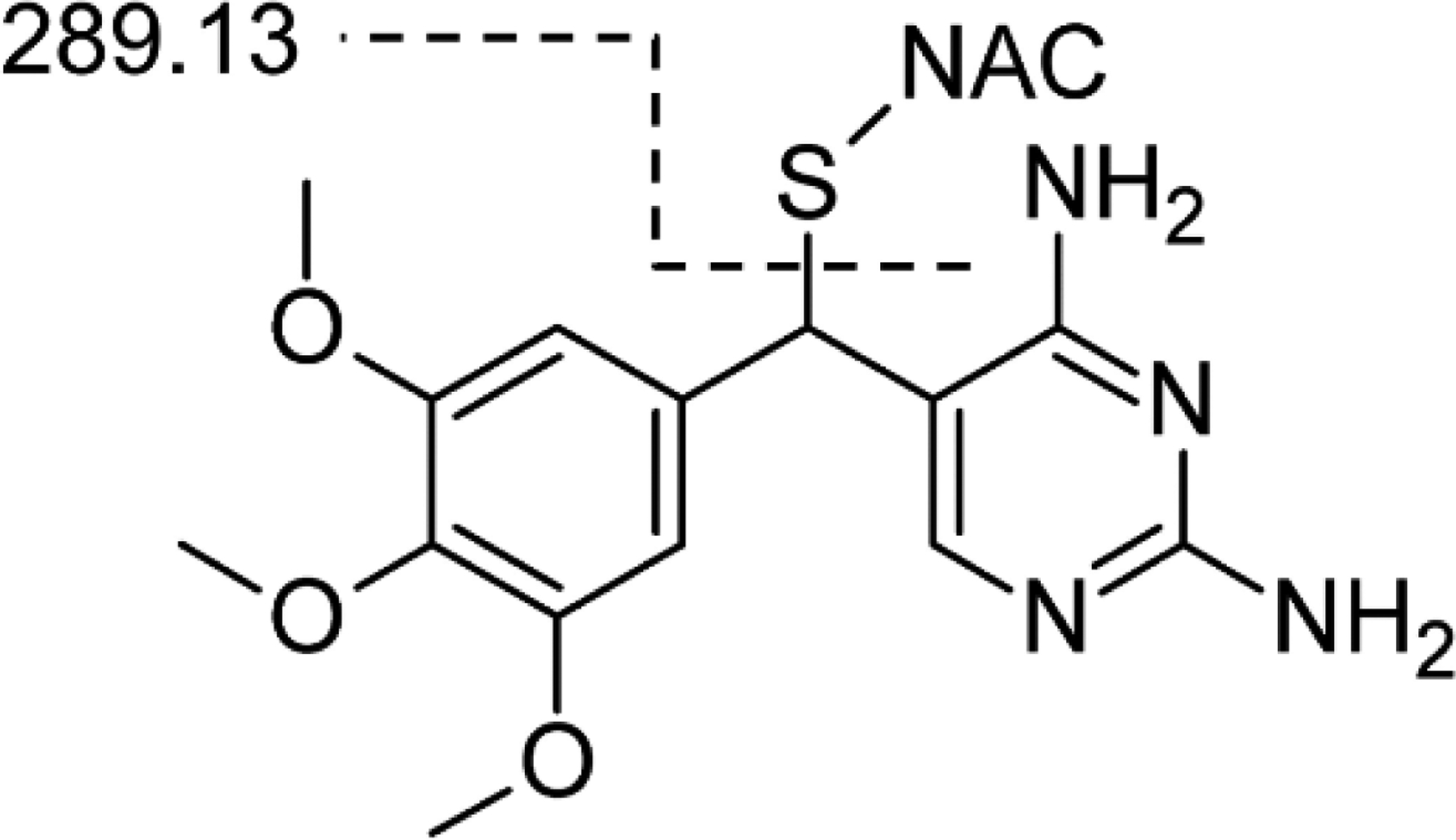
Cα-NAC-TMP structure showing fragmentation to form 289.13 fragment
The LC-MS signal of the modified peptides was compared at one and seven days to determine whether the peptides were detectable at shorter timepoints (Fig. 6). The signals of Peptides 1 and 2 were much lower at one day than at seven days, indicating the extremely slow kinetics of this reaction. These slow reactions are not completely implausible given that IADRs, including those caused by TMP-sulfamethoxazole, characteristically occur at least 1–2 weeks after treatment initiation (Uetrecht and Naisbitt 2013). While Cα-OH-TMP does not appear overtly reactive, the data indicates it can react to form adducts with nucleophilic amino acids. We speculate that this might occur via an SN1-type reaction with a stabilized carbocation.
Fig. 6.
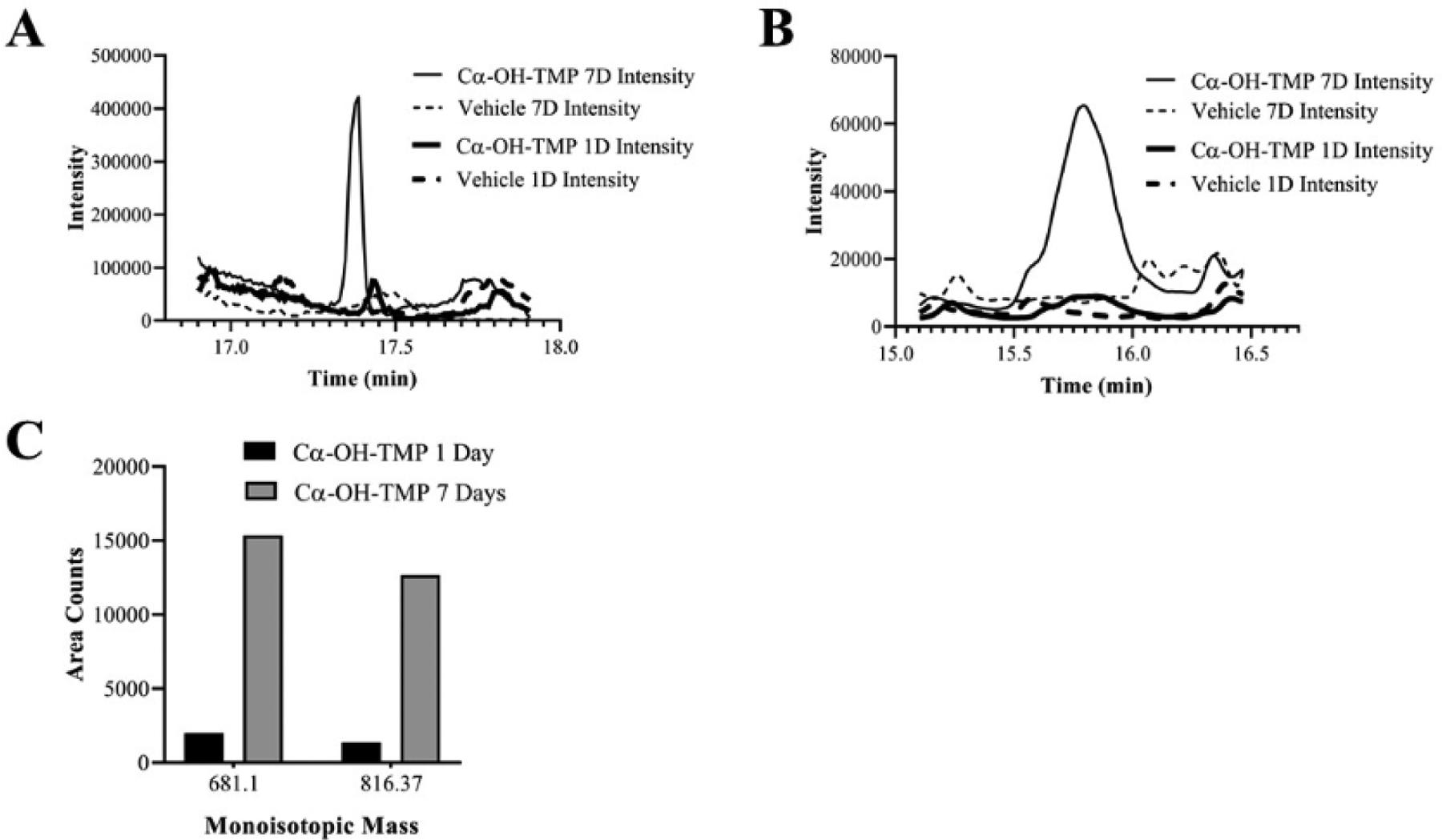
Measurements of peptides during a time course treatment of HSA with Cα-OH-TMP a, time course EIC for Peptide 1 (681.1) b, time course EIC for Peptide 2 (816.37) c, Mass Lynx area count time course for Peptide 1 (681.1) and Peptide 2 (816.37)
Conclusions
This work identified two HSA peptides that can be modified by treatment with the TMP primary metabolite, Cα-OH-TMP. However, Cα-OH-TMP-dependent modification of proteins may only be part of the story. Further work is required to identify the role of secondary and phase II metabolism in the bioactivation of TMP and formation of protein adducts. The role of TMP protein adducts in the development of IADRs also remains to be established. Immunological studies, such as proliferation of patient T cells, are potential routes to test the mechanistic role of individual protein adducts in patient IADRs (Meng et al. 2017). Future work will also include developing methodologies to detect TMP-adducted proteins in patient samples, which could increase our understanding of IADRs and/or become potential biomarkers in this patient population.
Acknowledgements
We would like to thank Robert Hanzlik and Yakov Koen at the University of Kansas, Department of Medicinal Chemistry, for their assistance in TMP antigen generation and antisera dilution and inhibition work. This work was supported by the National Institutes of Health R01GM129783 (J.L.G).
Abbreviations Used
- TMP
Trimethoprim
- IADR
idiosyncratic adverse drug reactions
- HLM
human liver microsomal
- NAC
N-acetylcysteine
- 1-NO-TMP
1-N-oxide
- 3-NO-TMP
trimethoprim 3-N-oxide
- 3-DM-TMP
3´-desmethyl-TMP
- 4-DM-TMP
4´-desmethyl-TMP
- Cα-OH-TMP
α-hydroxy trimethoprim
- CBZE
Carbamazepine 10,11-epoxide
- HAS
human serum albumin
- KLH
keyhole limpet hemocyanin
- LC-MS
liquid chromatography mass spectrometry
- EIC
extracted ion chromatogram
Footnotes
Conflict of Interest:
The authors declare they have no conflict of interest.
References
- Aldini G, Gamberoni L, Orioli M, Beretta G, Regazzoni L, Maffei Facino R, Carini M (2006) Mass spectrometric characterization of covalent modification of human serum albumin by 4-hydroxy-trans-2-nonenal. Journal of mass spectrometry : JMS 41:1149–1161 [DOI] [PubMed] [Google Scholar]
- Chalkley RJ, Clauser KR (2012) Modification site localization scoring: strategies and performance. Mol Cell Proteomics 11:3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho T, Uetrecht J (2017) How Reactive Metabolites Induce an Immune Response That Sometimes Leads to an Idiosyncratic Drug Reaction. Chem Res Toxicol 30:295–314 [DOI] [PubMed] [Google Scholar]
- Damsten MC, de Vlieger JS, Niessen WM, Irth H, Vermeulen NP, Commandeur JN (2008) Trimethoprim: novel reactive intermediates and bioactivation pathways by cytochrome p450s. Chem Res Toxicol 21:2181–2187 [DOI] [PubMed] [Google Scholar]
- Evans DC, Watt AP, Nicoll-Griffith DA, Baillie TA (2004) Drug-protein adducts: an industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem Res Toxicol 17:3–16 [DOI] [PubMed] [Google Scholar]
- Goldman JL, Koen YM, Rogers SA, Li K, Leeder JS, Hanzlik RP (2016) Bioactivation of Trimethoprim to Protein-Reactive Metabolites in Human Liver Microsomes. Drug Metab Dispos 44:1603–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JL, Leeder JS, Van Haandel L, Pearce RE (2015) In Vitro Hepatic Oxidative Biotransformation of Trimethoprim. Drug Metab Dispos 43:1372–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinneburg H, Stavenhagen K, Schweiger-Hufnagel U, Pengelley S, Jabs W, Seeberger PH, Silva DV, Wuhrer M, Kolarich D (2016) The Art of Destruction: Optimizing Collision Energies in Quadrupole-Time of Flight (Q-TOF) Instruments for Glycopeptide-Based Glycoproteomics. Journal of the American Society for Mass Spectrometry 27:507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WG, Zahid N, Uetrecht JP (1999) Metabolism of trimethoprim to a reactive iminoquinone methide by activated human neutrophils and hepatic microsomes. J Pharmacol Exp Ther 291:292–299 [PubMed] [Google Scholar]
- Meng X, Al-Attar Z, Yaseen FS, Jenkins R, Earnshaw C, Whitaker P, Peckham D, French NS, Naisbitt DJ, Park BK (2017) Definition of the Nature and Hapten Threshold of the beta-Lactam Antigen Required for T Cell Activation In Vitro and in Patients. J Immunol 198:4217–4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Maggs JL, Usui T, Whitaker P, French NS, Naisbitt DJ, Park BK (2015) Auto-oxidation of Isoniazid Leads to Isonicotinic-Lysine Adducts on Human Serum Albumin. Chem Res Toxicol 28:51–58 [DOI] [PubMed] [Google Scholar]
- Nakayama S, Atsumi R, Takakusa H, Kobayashi Y, Kurihara A, Nagai Y, Nakai D, Okazaki O (2009) A zone classification system for risk assessment of idiosyncratic drug toxicity using daily dose and covalent binding. Drug Metab Dispos 37:1970–1977 [DOI] [PubMed] [Google Scholar]
- Nolte WM, Tagore DM, Lane WS, Saghatelian A (2009) Peptidomics of prolyl endopeptidase in the central nervous system. Biochemistry 48:11971–11981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbioni G, Turesky RJ (2017) Biomonitoring Human Albumin Adducts: The Past, the Present, and the Future. Chem Res Toxicol 30:332–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JP, Naisbitt DJ, Farrell J, Ashby CA, Tucker MJ, Rieder MJ, Pirmohamed M, Clarke SE, Park BK (2007) Sulfamethoxazole and its metabolite nitroso sulfamethoxazole stimulate dendritic cell costimulatory signaling. J Immunol 178:5533–5542 [DOI] [PubMed] [Google Scholar]
- Sigel CW, Grace ME, Nichol CA (1973) Metabolism of trimethoprim in man and measurement of a new metabolite: a new fluorescence assay. J Infect Dis 128:Suppl:580–583 p [DOI] [PubMed] [Google Scholar]
- Stewart AJ, Blindauer CA, Berezenko S, Sleep D, Tooth D, Sadler PJ (2005) Role of Tyr84 in controlling the reactivity of Cys34 of human albumin. The FEBS journal 272:353–362 [DOI] [PubMed] [Google Scholar]
- Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G (2012) XCMS Online: a web-based platform to process untargeted metabolomic data. Analytical chemistry 84:5035–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A, Li M (2019) Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nature methods 16:63–66 [DOI] [PubMed] [Google Scholar]
- Uetrecht J, Naisbitt DJ (2013) Idiosyncratic adverse drug reactions: current concepts. Pharmacological reviews 65:779–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haandel L, Goldman JL, Pearce RE, Leeder JS (2014) Urinary biomarkers of trimethoprim bioactivation in vivo following therapeutic dosing in children. Chem Res Toxicol 27:211–218 [DOI] [PubMed] [Google Scholar]
- Wijkstrom A, Westerlund D (1983) Plasma protein binding of sulphadiazine, sulphamethoxazole and trimethoprim determined by ultrafiltration. Journal of pharmaceutical and biomedical analysis 1:293–299 [DOI] [PubMed] [Google Scholar]
- Yip VLM, Meng X, Maggs JL, Jenkins RE, Marlot PT, Marson AG, Park BK, Pirmohamed M (2017) Mass Spectrometric Characterization of Circulating Covalent Protein Adducts Derived from Epoxide Metabolites of Carbamazepine in Patients. Chem Res Toxicol 30:1419–1435 [DOI] [PubMed] [Google Scholar]