

Abstract
Lack of routine fresh or frozen tissue is a barrier to widespread transcriptomic analysis of adrenal cortical tumors and an impediment to translational research in endocrinology and endocrine oncology. Our group has previously pioneered the use of targeted amplicon-based next-generation sequencing for archival formalin-fixed paraffin-embedded (FFPE) adrenal tissue specimens to characterize the spectrum of somatic mutations in various forms of primary aldosteronism. Herein, we developed and validated a novel 194-amplicon targeted next-generation RNA sequencing (RNAseq) assay for transcriptomic analysis of adrenal tumors using clinical-grade FFPE specimens. Targeted RNAseq-derived expression values for 27 adrenal cortical tumors, including aldosterone-producing adenomas (APA; N = 8), cortisol-producing adenomas (CPA; N = 11), and adrenal cortical carcinomas (ACC; N = 8), highlighted known differentially-expressed genes (DEGs; i.e., CYP11B2, IGF2, etc.) and tumor type-specific transcriptional modules (i.e., high cell cycle/proliferation transcript expression in ACC, etc.), and a subset of DEGs was validated orthogonally using quantitative reverse transcription PCR (qRT-PCR). Finally, unsupervised hierarchical clustering using a subset of high-confidence DEGs revealed three discrete clusters representing APA, CPA, and ACC tumors with corresponding unique gene expression signatures, suggesting potential clinical utility for a transcriptomic-based approach to tumor classification. Overall, these data support the use of targeted amplicon-based RNAseq for comprehensive transcriptomic profiling of archival FFPE adrenal tumor material and indicate that this approach may facilitate important translational research opportunities for the study of these tumors.
Keywords: Next-generation sequencing (NGS), quantitative reverse-transcription polymerase chain reaction (qRT-PCR), aldosterone-producing adenoma (APA), cortisol-producing adenoma (CPA), adrenal cortical carcinoma (ACC), FFPE
INTRODUCTION
The adrenal cortex plays an important role in maintenance of metabolic homeostasis through the production of steroid hormones, which help regulate mineral and carbohydrate balance [1]. Inappropriate adrenal production of these hormones leads to primary aldosteronism (PA) in the case of aldosterone [2] and Cushing’s syndrome (CS) in the case of cortisol [3]. Somatic gene mutations resulting in the formation of benign, hormone-secreting adrenal cortical tumors [i.e., aldosterone-producing adenomas (APA) or cortisol-producing adenomas (CPA)] are a frequent underlying cause of PA [4] and CS [5]. Somatic mutations and other genomic alterations are also common in adrenal cortical carcinomas (ACC) – rare malignant adrenal cortical tumors, which may be associated with excess steroid hormone production [6]. While the distinction between adenoma and carcinoma is critical for the clinical management of adrenal cortical tumors, these entities may demonstrate overlapping clinical and pathologic features, suggesting a potential need for ancillary molecular tools to facilitate accurate diagnosis.
Prior genome-wide gene expression studies suggest that APA, CPA, and ACC have distinct transcriptomic profiles [6–8]; however, the relative rarity of adrenal cortical tumors (ACC, in particular) in surgical cohorts has been a limiting factor for the study of these tumors. Furthermore, until recently, comprehensive molecular profiling was essentially limited to high-quality fresh or frozen tissue samples, which are not routinely available for most clinical adrenal cortical tumor specimens. Our group has pioneered the use of targeted amplicon-based next-generation DNA sequencing (DNAseq) for formalin-fixed paraffin-embedded (FFPE) tissue to characterize the spectrum of somatic mutations in APA [9] and aldosterone-producing cell clusters (APCC) [10] in routine clinical adrenal specimens. We have also recently developed and validated targeted amplicon-based next-generation RNA sequencing (RNAseq) approaches for transcriptomic profiling of FFPE tissue and applied these methods to explore transcriptomic heterogeneity in clinical bladder and prostate cancer specimens [11,12].
In this study, we describe the development and validation of a targeted RNAseq assay for transcriptomic analysis of adrenal tumors using clinical-grade FFPE specimens and demonstrate that this approach has potential utility for differentiating among benign from malignant adrenal cortical tumors.
MATERIALS AND METHODS
This study was approved by the Institutional Review Board at the University of Michigan (HUM00083056) with a waiver of informed consent. The experimental approach is outlined in Figure 1 and described in detail below.
Figure 1. Immunohistochemistry (IHC)-guided capture of formalin-fixed paraffin-embedded (FFPE) adrenal tissue for targeted next-generation RNA sequencing (RNAseq) analysis.
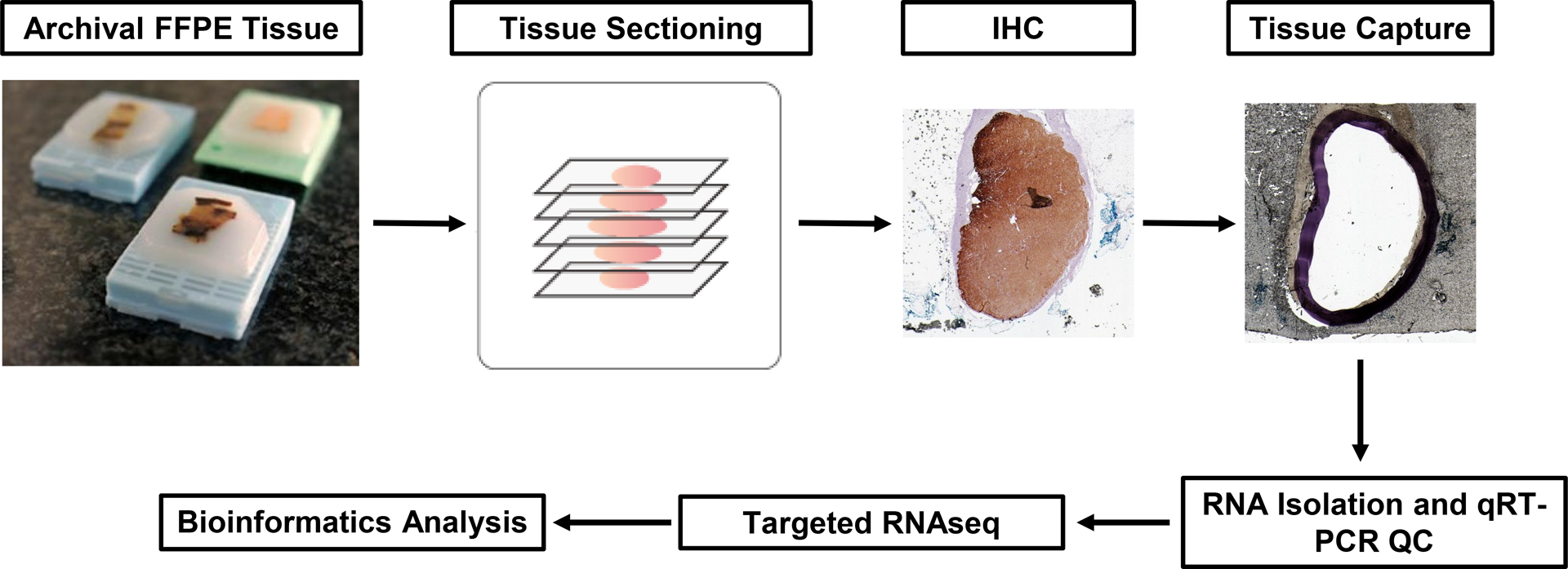
Serial 5 μm sections were generated from archival FFPE adrenal specimens, and IHC for CYP11B2 (aldosterone-producing adenomas) or CYP17A1 (cortisol-producing adenomas) was performed on the first slide to identify the area for transcriptomic profiling. Using the IHC-stained slide as a guide, the outlined area was captured from multiple unstained slides using a scalpel and a dissecting microscope. [For adrenal cortical carcinoma (ACC) specimens, all FFPE tissue on unstained slides was captured (see Materials and Methods for details).] RNA was extracted from pooled captured FFPE tissue, and after sample-level quality control (QC) with quantitative reverse transcription PCR (qRT-PCR), the FFPE-extracted RNA was utilized as input for targeted amplicon-based RNAseq using Ion Torrent™ technology. Log2-transformed read-level data was normalized to a set of housekeeping genes for bioinformatics analysis.
Cohort Selection
Clinical adrenal specimens from patients who underwent adrenalectomy at Michigan Medicine were identified from surgical pathology databases and/or prospectively-maintained clinical adrenal tumor databases. The clinical diagnosis of PA and CS was made according to the Endocrine Society Clinical Practice Guidelines or institutional consensus available at the time. For all cases, available H&E slides and ancillary material was reviewed by an experienced endocrine pathologist (T.J.G.) to confirm the diagnosis and select material for analysis, and the corresponding FFPE tissue blocks were retrieved for subsequent molecular studies. Using 5 μm FFPE sections, CYP11B2 (aldosterone synthase) and CYP17A1 (17α-hydroxylase) immunohistochemistry (IHC) was performed for APA and CPA specimens, respectively, as described previously [9]. Relative to the adjacent normal adrenal cortical tissue, APA were required to be CYP11B2 IHC positive, while CPA were required to be CYP17A1 IHC positive. ACC specimens were included based on histopathologic review alone, without relative steroidogenic enzyme expression analysis. Clinical information for the cohort and available mutational data for APA and CPA samples is available in Table S1.
Sample Preparation and RNA Extraction
For APA and CPA specimens, CYP11B2 and CYP17A1 IHC staining, respectively, was used to guide tissue sampling; IHC positive areas were outlined in ink on eight serial unstained slides and FFPE tissue was scraped for RNA extraction using a scalpel under a dissecting microscope. In contrast, for ACC specimens, all FFPE tissue on unstained slides was scraped for RNA extraction. Scraped FFPE tissue was stored at 4° Celsius in microcentrifuge tubes prior to RNA extraction using the AllPrep DNA/RNA FFPE Kit (Qiagen, Hilden, Germany).
Quantitative Reverse Transcription PCR (qRT-PCR)
For each sample, 200 ng of FFPE-extracted RNA was reverse transcribed using the Applied Biosystems High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA). Quantitative PCR (qPCR) was performed using an Applied Biosystems StepOnePlus Real-Time PCR System (Thermo Fisher Scientific) with commercially-available or custom-designed TaqMan primers and the TaqMan Fast Universal PCR Master Mix II (Thermo Fisher Scientific) using standard conditions (see Table S2 for qPCR assay information). Threshold cycle (Ct) values were calculated from the resulting fluorescence curves. PPIA was utilized for sample quality control (QC), and samples with highly-degraded RNA [based on a Ct (PPIA) > 31] were excluded from subsequent analyses. ACTB was used as a housekeeping gene for normalization. For each sample, log2-normalized expression values were calculated as follows: Ct (ACTB) – Ct (gene of interest). [Samples with a Ct (gene of interest) ≥ 40 were assigned a Ct value of 40.]
Targeted RNAseq
FFPE-extracted RNA was quantitated using a Qubit fluorometer (Thermo Fisher Scientific), and for each sample, up to 15 ng of FFPE-extracted RNA was reverse transcribed using SuperScript VILO (Thermo Fisher Scientific). Amplicon libraries were then generated for each sample from the resulting cDNA using the Ion AmpliSeq Library Kit Plus (Thermo Fisher Scientific) and a custom adrenal-specific AmpliSeq RNA panel – which was designed using the Ion AmpliSeq Designer (Thermo Fisher Scientific). This custom AmpliSeq RNA panel targets 194 genes, including adrenal and adrenal tumor-related transcripts associated with development, differentiation, tumorigenesis, and steroidogenesis; the panel also targets a number of housekeeping genes for sample QC and normalization. Target gene and amplicon information is available in Table S3. Amplicon libraries were quantitated on a QuantStudio 12K Flex (Thermo Fisher Scientific) using the Ion Library TaqMan Quantitation Kit (Thermo Fisher Scientific) prior to templating with an Ion Chef System (Thermo Fisher Scientific) and next-generation sequencing with an Ion S5 or Ion GeneStudio S5 Prime System (Thermo Fisher Scientific).
Next-Generation Sequencing (NGS) Data Analysis
NGS read data was processed and aligned using Torrent Suite (Thermo Fisher Scientific), and for each sample, amplicon-level read count data was generated using the coverageAnalysis plugin. Samples with less than 500,000 total reads and/or less than 55% end-to-end (E2E) reads were excluded from subsequent analyses. Amplicon-level E2E read counts were utilized to generate gene expression estimates, and amplicons without at least 100 E2E reads in at least two samples were excluded from subsequent analyses. For initial analysis of housekeeping genes, log2-transformed amplicon-level E2E read counts were normalized to sample-level total E2E read counts. Potential housekeeping genes with high variance across samples (σ > 1), high relative average expression levels (Z-score > 1.5), and/or low correlation with other housekeeping genes (average Pearson correlation coefficient < 0.4) were excluded, leaving a final set of six housekeeping genes for normalization: BANF1, CFL1, NUDFA2, PSMB4, RPN1, and SUMO2. For all subsequent analyses, log2-transformed amplicon-level E2E read counts were normalized to the mean of these six housekeeping genes. For each sample, a “Proliferation Score” was calculated as:
The following cell cycle/proliferation transcripts were included in the Proliferation Score calculation: ASPM, BUB1B, CDK1, DLGAP5, KIAA0101, MKI67, PBK, PRC1, RRM2, TOP2A, and UBE2C. Raw Proliferation Scores were converted to percentile rank for subsequent visualization and analysis.
Statistical Analyses
Exploratory pairwise differential expression analysis of log2-normalized expression values derived from targeted RNAseq data was performed using a standard parametric approach (i.e., Student’s t-test) with Benjamini-Hochberg post-hoc correction for multiple hypothesis testing [false discovery rate (FDR) < 5%]. Pairwise differentially expressed genes (DEGs) were explored using the Gene List Analysis tool from PANTHER [13]. Subsequently, DEGs with the potential for distinguishing among tumor types were identified from targeted RNAseq data using differential expression analysis with a standard parametric approach (i.e., one-way ANOVA), Bonferroni post-hoc correction for multiple hypothesis testing, and Tukey’s range test for significance among pairwise comparisons. Log2-normalized expression values derived from qRT-PCR data and Proliferation Scores were compared using one-way ANOVA, and single-gene correlation between paired RNAseq- and qRT-PCR-derived log2-normalized expression values was assessed using the Pearson correlation coefficient and Student’s t-test. Differences in clinical variables across tumor types within the study cohort were examined using standard statistical methods. Adjusted or unadjusted p-values < 0.05 were considered statistically significant (as applicable). All statistical analyses were performed in Excel (Microsoft, Redmond, WA) using the XLSTAT add-on (Addinsoft, Paris, France).
RESULTS
A Novel Targeted RNAseq Assay Generates Biologically Relevant Transcriptomic Information from FFPE Adrenal Tumor Specimens
A total of 32 adrenal cortical tumor samples (10 APA, 11 CPA, and 11 ACC) were selected for initial validation of the RNAseq assay. Of these, 31 (96.8%) passed the PPIA qRT-PCR QC threshold, and 27 (87.1%) passed subsequent RNAseq QC criteria (see Materials and Methods for details). These 27 samples formed the final study cohort, and included 8 APA [mean age = 50.6 years (range = 41–64 years); male-to-female ratio (M:F) = 3:1], 11 CPA [mean age = 46.7 years (range = 33–62 years); M:F = 1:10], and 8 ACC [mean age = 55.8 years (range = 37–83 years); M:F = 1:1] (see Table S1 for details). While a statistically significant difference in sex distribution across the tumor types was detected (P < 0.05), there was no significant difference in age distribution (P > 0.05). The majority of the included FFPE specimens were less than 5 years old; however, a subset of the specimens was more than 10 years old. Median E2E RNAseq reads were 3,082,034 [interquartile range (IQR) = 2,128,180–3,742,391], and median %E2E RNAseq reads were 90.81% (IQR = 89.61–91.56%) (see Table S4 for details). Of the 194 amplicons that comprise our targeted adrenal-focused RNAseq panel, 185 passed initial QC criteria and were included for analysis in this study (see Materials and Methods for details). Raw read counts and log2-normalized expression values are available in Table S5 and Table S6, respectively. Amplicon-level analysis across all samples demonstrated expected patterns of gene expression correlation (i.e., cell cycle/proliferation transcripts, adrenal medulla transcripts, etc.; see Figure S1). Similarly, sample-level principal component analysis (PCA) utilizing all gene expression values highlighted discrete clustering of tumor types using the first two principal components (see Figure S2). Finally, unsupervised clustering of median-centered gene expression values revealed distinct transcriptional modules associated with specific tumor subtypes (i.e., high expression of cell cycle/proliferation genes in ACC, high expression of subsets of metabolic enzymes in APA and CPA, etc.; see Figure S3).
Targeted RNAseq Identifies DEGs Among Adrenal Cortical Tumors
These data support the ability of our targeted RNAseq panel to generate biologically relevant transcriptomic information from FFPE adrenal tumor specimens. To further characterize the transcriptional differences across our cohort, we utilized pairwise differential expression analysis to identify potential DEGs among different types of adrenal cortical tumors. Comparison of APA and CPA highlighted 49 potential DEGs, including CYP11B2 (>2,000-fold higher in APA) and GSTA1 (19.4-fold higher in CPA) (see Table S7 and Figure S4A); APA-high genes were associated with multiple G-protein coupled receptor (GPCR) signaling pathways (i.e., 5HT, angiotensin, etc.), while CPA-high genes were associated with the insulin growth factor (IGF) signaling pathway (see Table S8). Comparison of ACC and APA revealed 60 potential DEGs, including insulin-like growth factor 2 (IGF2) (>100-fold higher in ACC) and CYP11B2 (>4,000-fold higher in APA) (see Table S7 and Figure S4B); ACC-high genes were associated with the IGF signaling pathway, nucleotide biosynthesis, DNA replication, and the p53 and ubiquitin proteasome pathways, while APA-high genes were again associated with GPCR signaling pathways (see Table S8). Comparison of ACC and CPA showed 46 potential DEGs, including PBK (>100-fold higher in ACC) and HSD3B2 (37.7-fold higher in CPA) (see Table S7 and Figure S4C); CPA-high genes were associated with sex hormone biosynthesis, while ACC-high genes were again associated with the IGF signaling pathway, nucleotide biosynthesis, DNA replication, and the p53 and ubiquitin proteasome pathways (see Table S8). Finally, we utilized an orthogonal approach (qRT-PCR) to validate a subset of the DEGs identified by our targeted RNAseq assay, including CYP11B2 and IGF2 (see Figures 2A and 2C). As expected, CYP11B2 expression was significantly higher in APA than CPA and ACC (P < 0.001), and IGF2 expression was significantly higher in ACC than APA and CPA (P < 0.001). Significant differential expression of ASPM, ATP9A, CDO1, GSTA1, NDRG4, RELN, and WISP2 (P < 0.005) across tumor types was also confirmed by qRT-PCR (see Table S9 for raw Ct values and Table S10 for expression differences), and there was high correlation between RNAseq and qRT-PCR-derived expression values (see Figures 2B and 2D; Pearson correlation coefficient = 0.55–0.93; P < 0.005).
Figure 2. Orthogonal validation of differential gene expression identified by targeted next-generation RNA sequencing (RNAseq) of archival formalin-fixed paraffin-embedded (FFPE) adrenal tumor tissue.
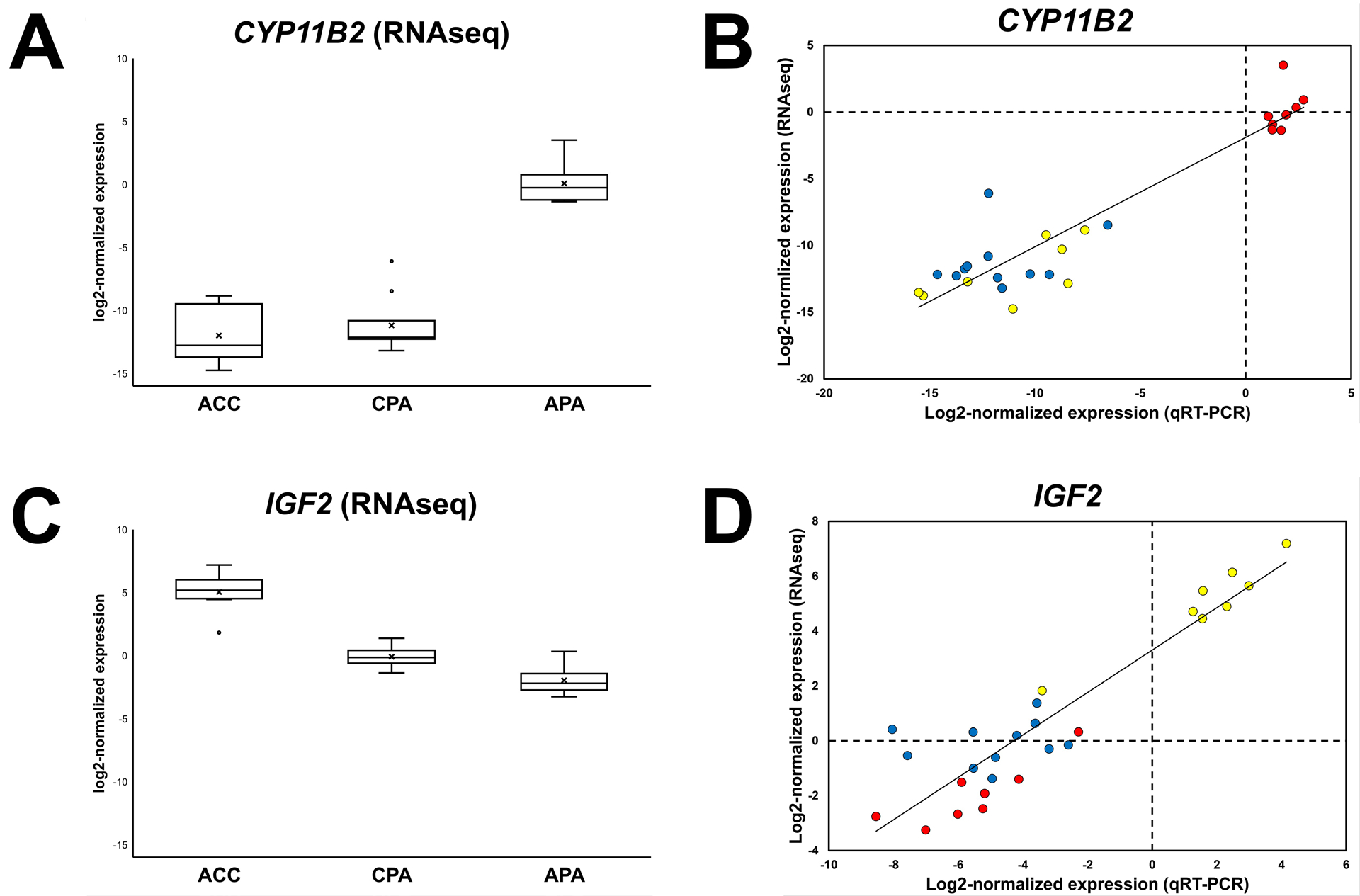
(A and C) Box plots of log2-normalized CYP11B2 and IGF2 expression values generated from targeted RNAseq data highlight differential gene expression across adrenal tumor types [adrenal cortical carcinoma (ACC), cortisol-producing adenoma (CPA), aldosterone-producing adenoma (APA)] (P < 0.001), which is confirmed by quantitative reverse transcription PCR (qRT-PCR) analysis (P < 0.001). (B and D) Scatter plots of paired RNAseq- and qRT-PCR-derived log2-normalized CYP11B2 and IGF2 expression values demonstrate high correlation across adrenal tumor types (Pearson correlation coefficient > 0.92; P < 0.001). ACC = yellow, CPA = blue, and APA = red.
Targeted RNAseq Can Differentiate Among Benign and Malignant Adrenal Cortical Tumors
These data support the ability of our targeted RNAseq panel to robustly detect differential gene expression among FFPE adrenal cortical tumor specimens. Given the potential overlap of clinical and pathologic findings in benign and malignant adrenal cortical tumors, we hypothesized that the ability to detect unique transcriptomic signatures may facilitate accurate classification of these tumors. To identify DEGs from our RNAseq data that may be informative for tumor type classification, we utilized a separate differential expression analysis approach with conservative post-hoc corrections for multiple hypothesis testing and multiple comparisons. This approach identified 40 DEGs across all tumor types, including CYP11B2, IGF2, and AVPR1A (see Table S11), and unsupervised hierarchical clustering of all samples with this subset of 40 DEGs revealed three discrete sample-level clusters representing APA, CPA, and ACC tumors with corresponding unique amplicon-level gene expression clusters (see Figure 3A). Importantly, despite a significant difference in sex distribution among the tumor types in our cohort, no sex-related differential gene expression was detected, suggesting that the observed transcriptomic differences are intrinsic to tumor types. Finally, as many of the ACC-high DEGs correspond to cell cycle/proliferation transcripts (e.g., PBK, TOP2A, etc.), a median-weighted expression average of these transcripts [i.e., “Proliferation Score”] was calculated for each sample (see Materials and Methods for details), and as expected, ACC showed significantly higher Proliferation Scores than APA and CPA (P < 0.001) (see Figure 3B). Indeed, sample-level integration of IGF2 expression and Proliferation Scores revealed a discrete ACC sample cluster with high IGF2 expression values and Proliferation Scores (see Figure 3C). Interestingly, one sample (“ACC1”) – corresponding to a low-grade ACC specimen – showed intermediate IGF2 expression but a comparatively high Proliferation Score, highlighting the potential utility of this integrated transcriptome-based approach to tumor classification.
Figure 3. Targeted next-generation RNA sequencing (RNAseq) of archival formalin-fixed paraffin-embedded (FFPE) adrenal tumor tissue generates biologically and clinically relevant transcriptomic data.
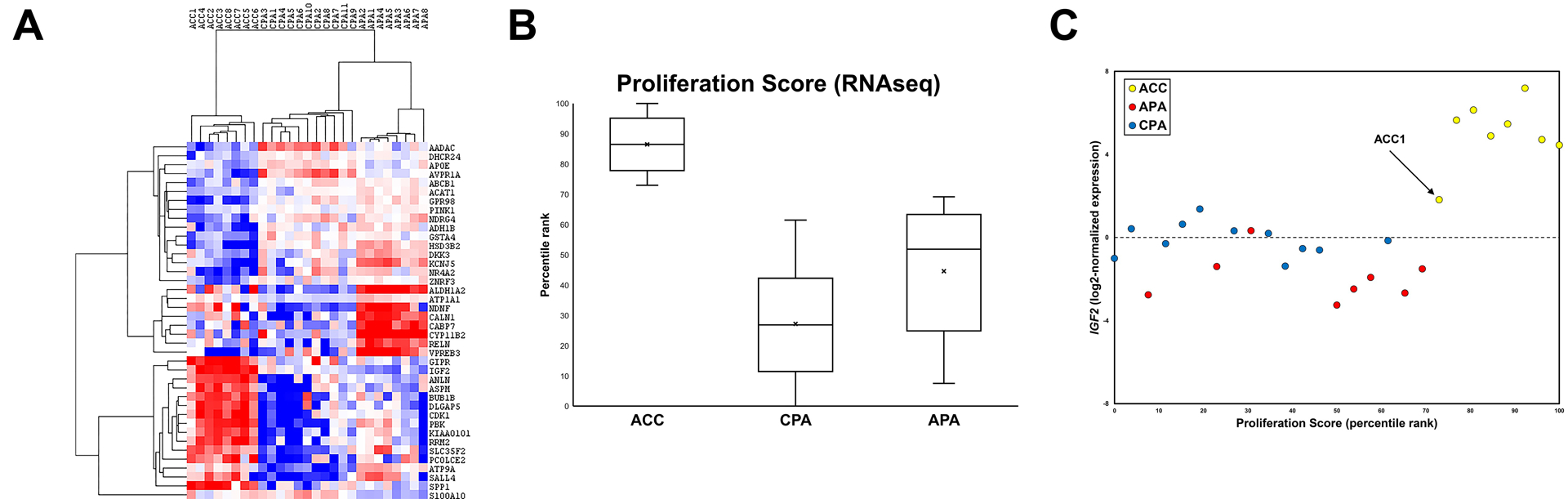
(A) Unsupervised hierarchical clustering of median-centered log2-normalized RNAseq expression values for 40 differentially-expressed genes reveals three discrete sample-level clusters representing adrenal cortical carcinoma (ACC), cortisol-producing adenoma (CPA), and aldosterone-producing adenoma (APA) with corresponding unique gene-level expression clusters. Range = −5 to 5 (blue to red). (B) Box plots of calculated Proliferation Score (percentile rank) generated from targeted RNAseq data confirm the high level of cell cycle/proliferation transcript expression in ACC compared to CPA and APA (P < 0.001). (C) Integrated visualization of calculated Proliferation Score (percentile rank; x-axis) and log2-normalized IGF2 expression values (y-axis) generated from targeted RNAseq data demonstrate a discrete ACC sample cluster with high IGF2 and cell cycle/proliferation transcript expression; a low-grade ACC sample (ACC1) showed intermediate IGF2 expression with a comparatively high Proliferation Score.
DISCUSSION AND CONCLUSIONS
The development and application of robust NGS approaches to study molecular alterations in archival FFPE tissue specimens has been integral to our understanding of the molecular basis of human disease, as the ability to characterize increasing numbers of rare tumors (e.g., ACC) and/or larger, more diverse cohorts of common tumors (e.g., APA and CPA) has shed new light on genomic heterogeneity across human neoplasia. Indeed, over the past decade, our group has pioneered the use of IHC-guided tissue macrodissection and targeted amplicon-based DNAseq with clinical FFPE adrenal specimens to characterize the genetic causes of PA, including APA and APCC [9,10,14–17]. It was just under ten years ago that the first somatic aldosterone-driver mutations (in KCNJ5) were described in APA; however, with the recent identification of somatic CACNA1H and CLCN2 mutations in small subsets of APA [14,18], aldosterone-driver mutations can now be detected in over 90% of APA.
Similar to the progress made in our understanding of the genomic basis for adrenal cortical tumors over the past decade, the ability to obtain robust transcriptomic data in a high-throughput manner from archival FFPE specimens has the potential to facilitate breakthroughs for translational research of these tumors. In this study, we validated a novel adrenal-focused targeted RNAseq assay for use with FFPE clinical adrenal specimens. Despite targeting only 194 genes and utilizing only up to 15 ng of FFPE-extracted RNA, we demonstrated the ability of this approach to generate biologically relevant transcriptomic information and highlighted its potential clinical utility for the classification of benign and malignant adrenal cortical tumors.
First, utilizing pairwise differential expression analysis, we identified distinct DEG subsets among the different tumor types: APA-high genes were associated with GPCR signaling pathways; CPA-high genes were associated with the IGF signaling pathway and sex hormone biosynthesis; and, ACC-high genes were associated with the IGF signaling pathway, nucleotide biosynthesis, DNA replication, and the p53 and ubiquitin proteasome pathways. We also showed that, as expected, ACC had significantly higher indicators of cellular proliferation (i.e., Proliferation Score) than APA and CPA. These results support previously published genome-wide gene expression data indicating that APA, CPA, and ACC have distinct transcriptomic profiles [6–8]. One recent study explored transcriptomic differences among APA with different aldosterone-driver mutations [19]; however, most of the differences were observed between tumors with and without CTNNB1 mutations – which are present in only a small subset of APA. In contrast, relatively few gene expression differences were detected among APA with other aldosterone-driver mutations (i.e., KCNJ5, ATP1A1, ATP2B3, and CACNA1D) – although the total number of tumors profiled was relatively small, and not all aldosterone-driver mutation subgroups were adequately represented. Ongoing work in our laboratory is focused on applying our targeted RNAseq approach to analyze potential transcriptomic differences across the APA genomic subtypes; if necessary to identify non-targeted DEGs among these tumors, we have also pioneered the application of amplicon-based RNAseq for whole-transcriptome profiling using archival FFPE tissue [20].
We also identified a subset of high-confident DEGs among APA, CPA, and ACC that was able to accurately group samples by tumor type using an unbiased approach (i.e., unsupervised hierarchical clustering). Similar to prior genome-wide microarray-based studies, the ACC transcriptome is distinct from that of adrenal cortical adenomas (ACA; i.e., APA and CPA) and, in particular, shows IGF2 upregulation with increased expression of cell cycle/proliferation transcripts [6–8, 21–23]. While clearly not the primary focus of this initial validation study, these data suggest that our targeted RNAseq assay may have clinical utility for the classification of adrenal cortical tumors. This is particularly intriguing given its low input RNA requirements (up to 15 ng) and ability to generate robust transcriptomic information from clinical FFPE specimens. While the distinction between ACA and ACC is frequently straightforward based on well-established clinicopathologic parameters (i.e., modified Weiss criteria, etc.), the diagnosis of ACC may be challenging in low-grade tumors with oncocytic features, as well as in minute core biopsy specimens. This potential issue is highlighted by the ACC1 sample in our dataset. Pathologically, ACC1 is a low-grade ACC with a low mitotic count (less than 20 per 50 high-power fields); while our targeted RNAseq data shows that its IGF2 expression and Proliferation Score are low relative to other ACC in the cohort, integrative analysis clearly supports its classification as ACC (see Figures 3A and 3C). Indeed, previous studies have introduced the notion that this kind of integrative analysis is a possible approach to improve classification and prognostication of low-grade adrenal cortical tumors [24,25]. While this possibility remains specifically untested in the current study, ACC1 is an example which supports the use of such an integrative approach in classification of low-grade adrenal cortical tumors, and this is an area of future translational research interest for our group.
Despite initial validation of our targeted RNAseq panel, the relatively small number of samples analyzed in this study is a clear limitation, and additional transcriptomic profiling of APA, CPA, and ACC samples is warranted to confirm the robustness of this approach. Similarly, the small number of genes targeted by the panel limits its potential utility as a discovery tool; however, as noted above, our group has pioneered amplicon-based whole-transcriptome RNAseq using archival FFPE tissue, which could be applied in future studies.
In conclusion, our data support the use of targeted amplicon-based RNAseq for comprehensive transcriptomic profiling of archival FFPE adrenal tumor material and indicate that this approach may facilitate important translational research opportunities for the study of these tumors.
Supplementary Material
Figure S1. RNAseq gene correlation plot. Unsupervised hierarchical clustering of amplicon-level Pearson correlation coefficients from targeted RNAseq data highlights expected patterns of gene expression correlation (i.e., cell cycle/proliferation transcripts, adrenal medulla transcripts, etc.). Range = −1 to 1 (blue to red).
{kind=link}
Figure S2. RNAseq principal component analysis (PCA) plot. Sample-level visualization of targeted RNAseq data using the first two PCA components (F1 and F2) highlights discrete clustering by tumor type. ACC = adrenal cortical carcinoma, APA = aldosterone-producing adenoma, and CPA = cortisol-producing adenoma.
{kind=link}
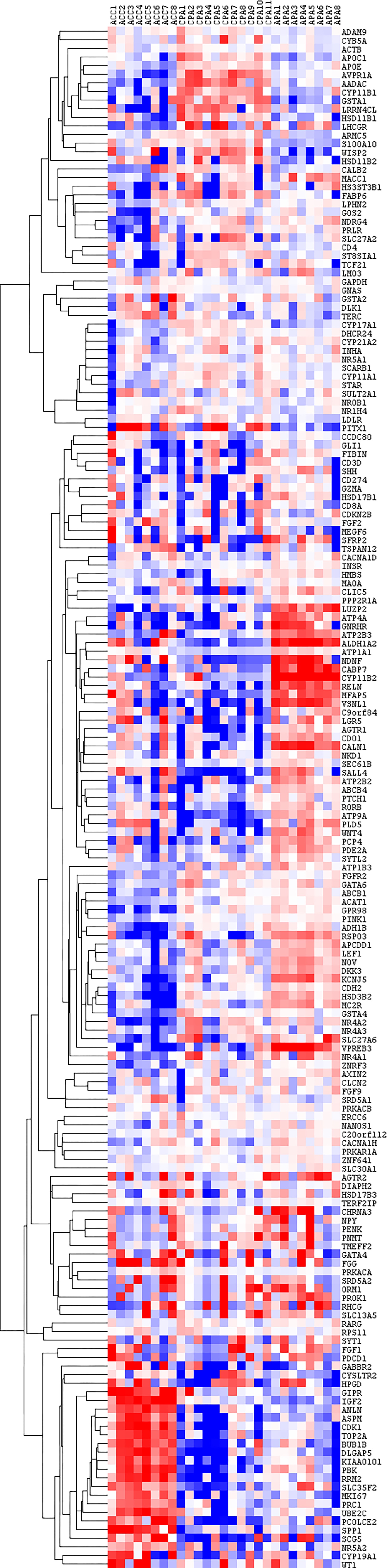
Figure S3. RNAseq gene expression heatmap. Amplicon-level unsupervised clustering of median-centered log2-normalized expression values from targeted RNAseq data reveals distinct transcriptional modules associated with specific tumor subtypes (i.e., high expression of cell cycle/proliferation genes in ACC, high expression of subsets of metabolic enzymes in APA and CPA, etc.). Range = −5 to 5 (blue to red). ACC = adrenal cortical carcinoma, APA = aldosterone-producing adenoma, and CPA = cortisol-producing adenoma.
{kind=link}
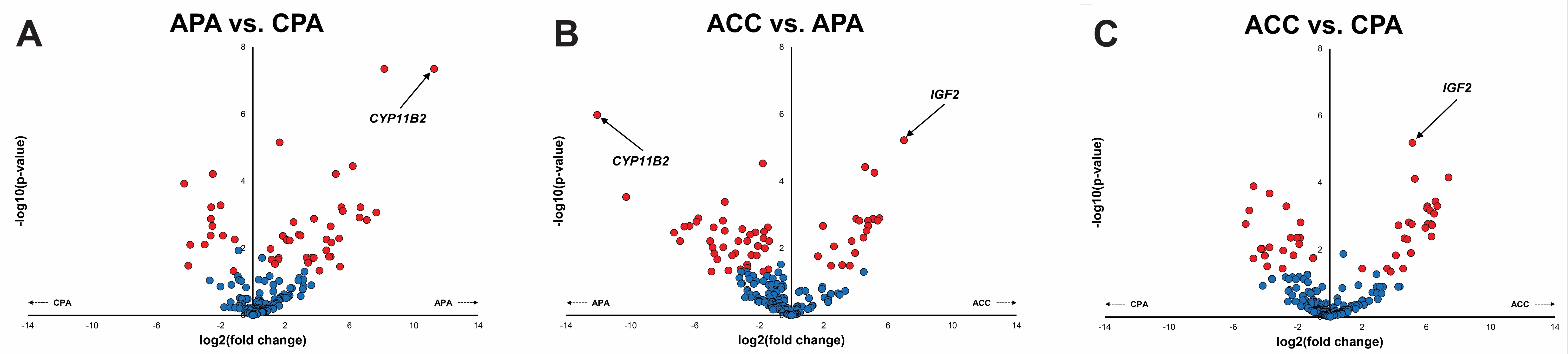
Figure S4. Differential expression volcano plots. Volcano plots depicting pairwise differentially expressed genes (DEG) – depicted in red – from targeted RNAseq data highlights known tumor type-specific DEGs, including CYP11B2 and IGF2.
{kind=link}
Table S1. Cohort clinical information
Table S2. qPCR assay information
Table S3. RNAseq panel information
Table S4. RNAseq sample QC
Table S5. RNAseq raw read counts
Table S6. RNAseq log2-normalized gene expression
Table S7. RNAseq pairwise DEG
Table S8. RNAseq pairwise DEG PANTHER pathway analysis
Table S9. qRT-PCR raw Ct values
Table S10. qRT-PCR DEG validation
Table S11. RNAseq multi-comparison DEG
Acknowledgements
We would like to thank Michelle Vinco and Farah Keyoumarsi at the University of Michigan for assistance in case identification and slide preparation, along with Amy Blinder and Jessica Baker at the University of Michigan for technical assistance.
Sources of Funding
This work was supported by grants from National Institute of Diabetes and Digestive and Kidney Diseases (DK106618 to W.E. Rainey). Drs. Hammer, Else and Lerario were supported by the US Department of Defense Grant W81XWH19-1-0530.
REFERENCES
- 1.Pignatti E, Leng S, Carlone DL et al. Regulation of zonation and homeostasis in the adrenal cortex. Mol Cell Endocrinol 2017; 441: 146–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conn JW. Presidential address. I. Painting background. II. Primary aldosteronism, a new clinical syndrome. J Lab Clin Med 1955; 45: 3–17 [PubMed] [Google Scholar]
- 3.Cushing H Further Notes on Pituitary Basophilism. JAMA 1932; 99: 281–284 [Google Scholar]
- 4.Dutta RK, Söderkvist P, Gimm O. Genetics of primary hyperaldosteronism. Endocr Relat Cancer 2016; 23: R437–454 [DOI] [PubMed] [Google Scholar]
- 5.Sato Y, Maekawa S, Ishii R et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 2014; 344: 917–920 [DOI] [PubMed] [Google Scholar]
- 6.Zheng S, Cherniack AD, Dewal N et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016; 30: 363. [DOI] [PubMed] [Google Scholar]
- 7.Slater EP, Diehl SM, Langer P et al. Analysis by cDNA microarrays of gene expression patterns of human adrenocortical tumors. Eur J Endocrinol 2006; 154: 587–598 [DOI] [PubMed] [Google Scholar]
- 8.Soon PSH, Gill AJ, Benn DE et al. Microarray gene expression and immunohistochemistry analyses of adrenocortical tumors identify IGF2 and Ki-67 as useful in differentiating carcinomas from adenomas. Endocr Relat Cancer 2009; 16: 573–583 [DOI] [PubMed] [Google Scholar]
- 9.Nanba K, Chen AX, Omata K et al. Molecular Heterogeneity in Aldosterone-Producing Adenomas. J Clin Endocrinol Metab 2016; 101: 999–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishimoto K, Tomlins SA, Kuick R et al. Aldosterone stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci U S A 2015; 112: E4591–E4599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hovelson DH, Udager AM, McDaniel AS et al. Targeted DNA and RNA Sequencing of Paired Urothelial and Squamous Bladder Cancers Reveals Discordant Genomic and Transcriptomic Events and Unique Therapeutic Implications. Eur Urol 2018; 74: 741–753 [DOI] [PubMed] [Google Scholar]
- 12.Salami SS, Hovelson DH, Kaplan JB et al. Transcriptomic heterogeneity in multifocal prostate cancer. JCI Insight 2018; 3: e123468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas PD, Kejariwal A, Guo N et al. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res 2006; 34: W645–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nanba K, Blinder AR, Rege J et al. Somatic CACNA1H Mutation As a Cause of Aldosterone Producing Adenoma. Hypertension 2020; 75: 645–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Omata K, Yamazaki Y, Nakamura Y et al. Genetic and Histopathologic Intertumor Heterogeneity in Primary Aldosteronism. J Clin Endocrinol Metab 2017; 102: 1792–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Omata K, Anand SK, Hovelson DH et al. Aldosterone-Producing Cell Clusters Frequently Harbor Somatic Mutations and Accumulate With Age in Normal Adrenals. J Endocr Soc 2017; 1: 787–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Omata K, Satoh F, Morimoto R et al. Cellular and Genetic Causes of Idiopathic Hyperaldosteronism. Hypertension 2018; 72: 874–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dutta RK, Arnesen T, Heie A et al. A somatic mutation in CLCN2 identified in a sporadic aldosterone-producing adenoma. Eur J Endocrinol 2019; 181: K37–K41 [DOI] [PubMed] [Google Scholar]
- 19.Backman S, Åkerström T, Maharjan R et al. RNA Sequencing Provides Novel Insights into the Transcriptome of Aldosterone Producing Adenomas. Sci Rep 2019; 9: 6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazo de la Vega L, Samaha MC, Hu K et al. Multiclonality and Marked Branched Evolution of Low Grade Endometrioid Endometrial Carcinoma. Mol Cancer Res 2019; 17: 731–740 [DOI] [PubMed] [Google Scholar]
- 21.Giordano TJ, Kuick R, Else T et al. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res 2009; 15: 668–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gara SK, Wang Y, Patel D et al. Integrated genome-wide analysis of genomic changes and gene regulation in human adrenocortical tissue samples. Nucleic Acids Res 2015; 43: 9327–9339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Assie G, Giordano TJ, Bertherat J. Gene expression profiling in adrenocortical neoplasia. Mol Cell Endocrinol 2012; 351: 111–117 [DOI] [PubMed] [Google Scholar]
- 24.Giordano TJ. Molecular pathology of adrenal cortical tumors: separating adenomas from carcinomas. Endocr Pathol 2006; 17: 355–363 [DOI] [PubMed] [Google Scholar]
- 25.Mete O, Gucer H, Kefeli M et al. Diagnostic and Prognostic Biomarkers of Adrenal Cortical Carcinoma. Am J Surg Pathol 2018; 42: 201–213 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. RNAseq gene correlation plot. Unsupervised hierarchical clustering of amplicon-level Pearson correlation coefficients from targeted RNAseq data highlights expected patterns of gene expression correlation (i.e., cell cycle/proliferation transcripts, adrenal medulla transcripts, etc.). Range = −1 to 1 (blue to red).
Figure S2. RNAseq principal component analysis (PCA) plot. Sample-level visualization of targeted RNAseq data using the first two PCA components (F1 and F2) highlights discrete clustering by tumor type. ACC = adrenal cortical carcinoma, APA = aldosterone-producing adenoma, and CPA = cortisol-producing adenoma.
Figure S3. RNAseq gene expression heatmap. Amplicon-level unsupervised clustering of median-centered log2-normalized expression values from targeted RNAseq data reveals distinct transcriptional modules associated with specific tumor subtypes (i.e., high expression of cell cycle/proliferation genes in ACC, high expression of subsets of metabolic enzymes in APA and CPA, etc.). Range = −5 to 5 (blue to red). ACC = adrenal cortical carcinoma, APA = aldosterone-producing adenoma, and CPA = cortisol-producing adenoma.
Figure S4. Differential expression volcano plots. Volcano plots depicting pairwise differentially expressed genes (DEG) – depicted in red – from targeted RNAseq data highlights known tumor type-specific DEGs, including CYP11B2 and IGF2.
Table S1. Cohort clinical information
Table S2. qPCR assay information
Table S3. RNAseq panel information
Table S4. RNAseq sample QC
Table S5. RNAseq raw read counts
Table S6. RNAseq log2-normalized gene expression
Table S7. RNAseq pairwise DEG
Table S8. RNAseq pairwise DEG PANTHER pathway analysis
Table S9. qRT-PCR raw Ct values
Table S10. qRT-PCR DEG validation
Table S11. RNAseq multi-comparison DEG