

Abstract
Objectives:
Pancreatic ductal adenocarcinoma is the fourth-leading cause of cancer death in the United States and there is an urgent need for effective therapies. Stearoyl-CoA desaturase (SCD) is an enzyme localized in the endoplasmic reticulum and generates monounsaturated fatty acid from saturated fatty acid. In this study, we examined the role of SCD in pancreatic cancer.
Methods:
We isolated EPCAM+ pancreatic tumors from the Pdx1Cre;LSL-KrasG12D mouse and formed organoids in matrigel. Using a SCD inhibitor, A939572, we tested its effects on growth and cell death in tumor organoids, tumors developed in the Pdx1Cre;LSL-KrasG12D mouse, and a human pancreatic ductal adenocarcinoma cell line, PANC-1.
Results:
A939572 treatment rapidly induced degeneration of mouse tumor organoids and activated the unfolded protein response (UPR). Co-treatment of oleic acid, but not stearic acid, reduced the UPR in the organoids and rescued the inhibitory effect of the SCD inhibitor on their growth. Administration of A939572 to Pdx1Cre;LSL-KrasG12D mice caused cell death in early pancreatic tumors, but not in acini or islets. The SCD inhibitor induced the UPR in PANC-1 and suppressed their growth, but did not induce cell death.
Conclusions:
The inhibition of the SCD enzyme causes an UPR and cell death in early pancreatic tumors.
Keywords: cell death, ER stress, organoid, pancreatic ductal adenocarcinoma, pancreatic intraepithelial neoplasia
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a relatively rare cancer type, but is the fourth-leading cause of cancer death in the United States.1,2 Due to its aggressive nature and lack of sensitive diagnostic markers, 80% of patients with PDAC are not eligible for surgical resection.3 Although chemotherapy and immunotherapy have been used for patients with PDAC, the overall five-year survival rate is 8%. Pancreatic ductal adenocarcinoma is expected to become the second-leading cause of cancer death by 2030 and there is an urgent need for effective therapies.4,5
The pancreas has both exocrine and endocrine functions. Acinar cells secrete digestive enzymes and bicarbonate through the pancreatic duct to the duodenum and facilitate digestion. Islets secrete hormones and regulate blood glucose levels. Cell lineage tracing studies indicate acinar cells are the major source of PDAC in mice.6 Acinar cells undergo acinar-ductal metaplasia and give rise to early pancreatic lesions named pancreatic intraepithelial neoplasia (PanIN).7 Pancreatic intraepithelial neoplasia express duct cell markers, such as epithelial cell adhesion molecule (EPCAM), keratin 19 (KRT19), and prominin 1 (PROM1/CD133).8–10 They gradually loses their polarity and become invasive PDAC.11
Over 90% of patients with PDAC show oncogenic mutation of KRAS GTPase.12,13 Overexpression of oncogenic KrasG12D in acinar cells is known to induce PanIN in pancreas/duodenum homeobox protein 1 (Pdx1)Cre and LSL-KrasG12D-flox mice.14 By introducing additional gene mutations, such as p53, KrasG12D-driven PanIN further develops to metastatic PDAC in mice.15 In addition to the genetic factors, environmental factors including smoking, obesity, chronic pancreatitis, and alcohol have been shown to induce PDAC.9,16–18
Lipid metabolism is linked to cancer growth and metastasis.19,20 Stearoyl-CoA desaturase (SCD) catalyzes the Δ−9 fatty acid desaturation in the endoplasmic reticulum (ER) and converts saturated fatty acyl-CoA into unsaturated fatty acyl-CoA.21 Stearoyl-CoA desaturase typically generates palmitoleic acid (16:1) and oleic acid (18:1) from palmitic acid (16:0) and stearic acid (18:0), respectively.21,22 There are two SCD genes (SCD1, SCD5) in humans, and four Scd genes (Scd1–4) in mice are orthologues to the human SCD1 gene.21,23 De novo biogenesis of unsaturated fatty acid is essential for maintaining the fluidity and integrity of lipid membranes and signaling in cells. Accumulation of long-chain saturated fatty acid in cells often shows cytotoxicity.19 The lipid-mediated lipotoxicity may cause stress to the ER and provoke the unfolded protein response (UPR) in cells. The UPR consists of three branches of a cascade, including protein kinase R-like ER kinase (PERK/EIF2AK3), inositol requiring enzyme 1 α (IRE1α), and activating transcription factor (ATF) 6.24,25 PERK induces phosphorylation of eukaryotic initiation factor 2α (elF2α) and reduces global protein translation. Phosphorylated elF2α also induces translation of ATF4, which induces expression of C/EBP homologous protein (CHOP/DDIT3), a proapoptotic transcription factor. CHOP negatively regulates elF2α by dephosphorylation through induction of growth arrest and DNA damage-inducible protein (GADD34/PPP1R15A). Activation of IRE1α initiates splicing of X-box binding protein 1 (Xbp1) mRNA and generates sXBP1, which induces the expression of genes involved in protein folding and lipid synthesis. Activation of ATF6 induces the transcription of genes involved in protein folding and ER-associated degradation. The UPR is a process for cells suffering from ER stress and is necessary for cell survival.24,25 However, if the UPR fails to restore ER homeostasis, cells undergo apoptosis.
It is known that SCD is involved in the growth and survival of cancers in the breast, lung, liver, and colon.22 Patients with PDAC show an increase of serum monounsaturated fatty acid generated by SCD1.26 However, the role of SCD in pancreatic cancer largely remains to be clarified. In the present study, we found that inhibition of the SCD enzyme strongly induces the UPR and results in degeneration of tumor organoids formed from the Pdx1Cre;LSL-KrasG12D mouse pancreas. We showed that unsaturated fatty acid, but not saturated fatty acid, rescues the negative effect of the inhibition of SCD enzyme on the organoids. The SCD inhibitor also selectively causes apoptosis in pancreatic tumors developed in the Pdx1Cre;LSL-KrasG12D mouse pancreas. Our data suggest that the SCD enzyme is required for early pancreatic cancer growth and will be a therapeutic target for suppression of pancreatic cancer.
MATERIALS AND METHODS
Mice
Pdx1Cre and LSL-KrasG12D mice were purchased from Jackson Laboratory (Bar Harbor, Maine).14 Pancreas tissue from Pdx1Cre/+;LSL-KrasG12D/+ mice (5–6 month of age) were used for fluorescence immunostaining and organoid formation. All animal experiments were performed in accordance with the National Institutes of Health guidelines under the protocol approved by the Institutional Animal Care and Use Committee at the University of Southern California.
Tumor Organoid Culture
Pancreas tissue from Pdx1Cre;LSL-KrasG12D mice were digested with 125 μg/ml collagenase type XI (Sigma, St. Louis, Mo) and 125 μg/ml Dispase II (ThermoFisher Scientific, Waltham, Mass) at 37°C for 1 hour.27 Digested cells were collected by centrifugation and be incubated with rat monoclonal EPCAM antibody (G8.8, DSHB, Iowa City, Iowa) at 4°C for 30 min. After washing, cells were incubated with anti-rat IgG microbeads (Miltenyi Biotech, Auburn, Calif) and EPCAM+ tumor cells were separated by MACS Pro separator (Miltenyi Biotech). EPCAM+ cells (2 × 104 cells) were suspended in 20 μl of matrigel (Corning, Tewksbury, Mass), plated on a round bottom 96-well plate, and incubated in a CO2 incubator at 37°C for 30 min. After polymerization of the matrigel, the cells were cultured in 100 μl of advanced DMEM/F-12 glutamax (ThermoFisher Scientific) supplemented with 1X penicillin/streptomycin, 1x B27 supplement, 10 mM HEPES (Sigma), 1.25 mM N-acetylcystein, 10 mM nicotinamide, 10 nM Gastrin I, 0.5 μM A83–01 (R&D Systems, Minneapolis, Minn), 0.1 ng/ml FGF-10, 100 ng/ml Noggin, 50 ng/ml EGF, 1 μg/ml R-Spondin-1 (PeproTech, Rocky Hill, NJ), and 10 nM Y-27632 (Tocris, Minneapolis, Minn).28 Tumor organoids formed in matrigel were disrupted by pipetting and were passaged from 1 well to 3 wells.
Treatment of Tumor Organoids
A SCD1 inhibitor (A939572), 4-(2-chlorophenoxy)-N-[3-[(methylamino)carbonyl] phenyl]-1-piperidinecarboxamide (Tocris) was dissolved in dimethylsulfoxide (DMSO) and added to the culture medium (20 μM at a final concentration). Oleic acid (18:1, O1008), stearic acid (18:0, S4751), and fatty acid free-BSA (A8806) were purchased from Sigma. BSA-conjugated fatty acids were prepared by diluting 50 mM stock fatty acids in DMSO with 100 mg/ml BSA in 1x PBS to produce a 3 mM working solution.29 Cells were then treated with a final concentration of 30 μM BSA-conjugated fatty acid (a final concentration of 0.1% BSA). Cells were treated with or without A939572 or 30 μM of fatty acid for 16 hours.
Quantitative Polymerase Chain Reaction
RNAs were isolated using a RNA miniprep kit (Zymo Research, Irvine, Calif). cDNA was synthesized using the Maxima first strand cDNA synthesis kit (ThermoFisher Scientific). Quantitative polymerase chain reaction (QPCR) was performed with the SYBR FAST qPCR kit (KAPA Biosystems, Wilmington, Mass) in the ViiA7 Real-Time PCR System (Applied Biosystems, Carlsbad, Calif). Primer sequences for mouse genes are Atf4 (5’-GAA ACC TCA TGG GTT CTC CA, 5’-GCC AAT TGG GTT CAC TGT CT), Atf6 (5’-TTT CGA AGG GAT CAT CTG CT, 5’-GGG TCG TCT CTG TGG TTG TT), Chop (5’-CGG AAC CTG AGG AGA GAG TG, 5’-GGA CGC AGG GTC AAG AGT AG), Epcam (5’-CGG CTC AGA GAG ACT GTG TC, 5’-GAT CCA GTA GGT CCT CAC GC), Gadd34 (5’-GAG GGA CGC CCA CAA CTT C, 5’-TTA CCA GAG ACA GGG GTA GGT), 78-kDa glucose-regulated protein (Grp78/Hspa5: 5’-TAC TCG GGC CAA ATT TGA AG, 5’-GGG GAC AAA CAT CAA GCA GT), glyceraldehyde-3-phosphate dehydrogenase (Gapdh: 5’-CGT CCC GTA GAC AAA ATG GT, 5’-GAA TTT GCC GTG AGT GGA GT), Krt19 (5’-CTC GGA TTG AGG AGC TGA AC, 5’-TCA CGC TCT GGA TCT GTG), Perk (5’-ATT CAG CAC CCA GAT GGA AC, 5’-CTG GGC TGT CTG GAA TGT TT), Prom1 (5’-GAA AGT TGC TCT GCG AAC C, 5’-TCT CAA GCT GAA AAG CAG CA), Scd1 (5’-TGC GAT ACA CTC TGG TGC TC, 5’-TAG TCG AAG GGG AAG GTG TG), Scd2 (5’-GCT CTC GGG AGA ACA TCT TG, 5’-GCT TCT GGA ACA GGA ACT GC), and sXbp1 (5’-GAG TCC GCA GCA GGT G, 5’-GTG TCA GAG TCC ATG GGA). Primer sequences for human genes are ATF4 (5’-TCA AAC CTC ATG GGT TCT CC, 5’-GTG TCA TCC AAC GTG GTC AG), CHOP (5’-CAG AGC TGG AAC CTG AGG AG, 5’-CGA AGG AGA AAG GCA ATG AC), GADD34 (5’-GGA CCT GTG ATC GCT TCT G, 5’-CAG GCC AGC TTC TAC CAG AG), GAPDH (5’-GAG TCA ACG GAT TTG GTC GT, 5’-TTG ATT TTG GAG GGA TCT CG), and sXBP1 (5’-GCC AGT GGC CGG GTC T, 5’-GGT CCA AGT TGT CCA GAA TGC). The samples were run in triplicate. The relative mRNA levels per sample were calculated by subtracting the detection limit (40 Ct) from the cycle threshold value (Ct) of each gene in the same sample to obtain the ΔCt value. Taking the log2 of -ΔCt resulted in the relative expression value of each gene for each sample, expressed in arbitrary units. Each value was normalized against Gapdh.
Fluorescence Immunostaining of Pancreatic Tumor Tissue and Organoids
Pancreas tissue were fixed with 4% paraformaldehyde at 4°C overnight. Fixed tissue were incubated with 30% sucrose in PBS overnight and then embedded in freezing medium. Tumor organoids cultured in matrigel in a 96-well plate were fixed with 4% paraformaldehyde for 10 min and the matrigel containing organoids was embedded in freezing medium. We made cryosections (7 μm) with a Cryostat (CM1900, Leica, Buffalo Grove, Ill). Cryosections were blocked with 5% donkey serum and 0.2% bovine serum albumin for 30 min and incubated with primary antibodies at 4°C overnight. The primary antibodies used were CDH1 conjugated with AlexaFluor 488 (100-fold dilution, 560061, BD Biosciences, San Jose, Calif), doublecortin like kinase 1 (DCLK1, 100-fold dilution, PA5–20908, ThermoFisher Scientific), PROM1 (50-fold dilution, 14–1331), EPCAM (100-fold dilution, G88, DSHB), and KRT19 (50-fold dilution, TROMA-III). The unconjugated primary antibodies were detected with secondary antibodies conjugated with AlexaFluor 488 and 568 dyes (ThermoFisher Scientific). The sections were counterstained with DAPI. Images were captured with a 90i microscope (Nikon, Melville, NY).
Detection of Apoptosis in Pancreatic Tumors
The SCD inhibitor was given orally to Pdx1Cre;LSL-KrasG12D mice (5 mg/kg body weight) 2 days before collecting the pancreas (n = 4 for A939572, n = 2 for DMSO). Pancreas tissue were fixed with 4% paraformaldehyde in PBS overnight at 4°C. Cryosections (7 μm) were used for detection of apoptosis using In Situ Apoptosis Detection Kit (Trevigen, Caithersburg, Md). DNA fragments were visualized with antibodies conjugated with horseradish peroxidase and diaminobenzidine according to the manufacturer’s instructions.
Culture of Human PDAC Cell Line
A human PDAC cell line, PANC-1, was purchased from American Type Culture Collection (Manassas, Va). PANC-1 was cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) with penicillin and streptomycin. After plating in 96-well plates (3 × 103 cells), PANC-1 was treated with A939572 (0.5–50 μM) for 48 hours. Cell proliferation was measured by Cell Counting Kit-8 (Dojindo, Rockville, Md) using a PowerWave 200 spectrophotometer (BioTech, Winooski, Vt). Cytotoxicity of A939572 on PANC-1 was assessed by measuring lactate dehydrogenase (LDH) release using a Cytotoxicity LDH assay kit (Dojindo).
Western Blot
PANC-1 cultured on 100 mm dishes were washed with PBS and lysed with 100 μl of a RIPA buffer containing 1 mM phenylmethanesulfonyl fluoride, 1 mM sodium orthovanadate, and a protease inhibitor cocktail (Cell Signaling, Danvers, Mass). After centrifugation, the supernatant was collected and the protein concentration was measured by the Bradford dye-binding method (Bio-Rad, Hercules, Calif). Cell lysates (40 μg protein) mixed with an SDS sample buffer were run in a 10% polyacrylamide gel and were transferred to a nitrocellulose membrane (Bio-Rad). Membranes were blocked with TBS buffer (20 mM Tris HCl pH 7.6, 150 mM NaCl) containing 0.1% Tween-20 (Sigma) and 5% BSA for 1 hour and were incubated with the first antibodies for 16 hours at 4°C. Primary antibodies used were Phospho-eIF2α Ser51 (500-fold dilution, #9721, Cell Signaling), eIF2α (1000-fold dilution, #9722), cleaved Caspase-3 Asp175 (500-fold dilution, #9661), poly (ADP-ribose) polymerase (PARP, 1000-fold dilution, #9542), and GAPDH (1000-fold dilution, sc-32233, Santa Cruz, Dallas, Texas). After washing with TBS containing 0.1% Tween-20, the membranes were incubated with anti-mouse or anti-rabbit secondary antibodies conjugated with horseradish peroxidase (5000-fold dilution, Jackson ImmunoResearch, West Grove, Pa) for 1 hour. After washing, the secondary antibodies were detected using an ECL Western blotting substrate (ThermoFisher Scientific) and the Chemidoc Plus imaging system (Bio-Rad).
Statistical Analysis
Statistical tests for the significance of differences were assessed by one-way ANOVA followed by a Tukey HSD post-hoc test. A P value of less than 0.05 was considered statistically significant.
RESULTS
Organoid Culture of Pancreatic Cancer Generated in the Pdx1Cre;LSL-KrasG12D Mouse Pancreas
Organoid culture in Matrigel has been used for studying epithelial cells in the intestine, liver, and pancreas.28 To examine lipid metabolism in pancreatic cancer, we used organoid culture prepared from Pdx1Cre;LSL-KrasG12D mice that develop PanIN in their pancreas. After digestion of the Pdx1Cre;LSL-KrasG12D pancreas, we isolated EPCAM+ tumor cells by MACS and confirmed high expression of pancreatic cancer markers, including Epcam, Krt19, and Prom1, in EPCAM+ cells compared to those in EPCAM- cells (Fig. 1A). EPCAM+ tumor cells embedded in matrigel formed organoids in culture medium containing R-Spondin1 within a few days (Fig. 1B). In contrast, EPCAM- cells did not form organoids. After passaging mechanically disrupted EPCAM+ tumor cell-derived organoids into new wells, the fragmented organoids re-formed whole organoids (Fig. 1C). To characterize tumor organoids, we compared the marker expression by fluorescence immunostaining in cryosections from the Pdx1Cre;LSL-KrasG12D mouse pancreas and tumor organoids. We confirmed PanIN developed in the pancreas is entirely positive for E-cadherin (CDH1), EPCAM, and KRT19 (Fig. 1D). PROM1 is expressed at the lumen of the PanIN and some of them are positive for DCLK1 as previously described.30 Tumor organoids showed sphere morphology formed by CDH1+ epithelial cell layers (Fig. 1D). These epithelial tumor cells are positive for EPCAM and KRT19 and express PROM1 at the lumen side, similar to tumors in vivo. Our data indicate that organoids maintain the apical-basal polarity of PanIN in culture. Tumor cells near the lumen side are positive for DCLK1 in organoids. This staining pattern is distinct from that in vivo and suggests that the culture condition drives tumor cells near the lumen side toward DCLK1+ cells.
FIGURE 1.
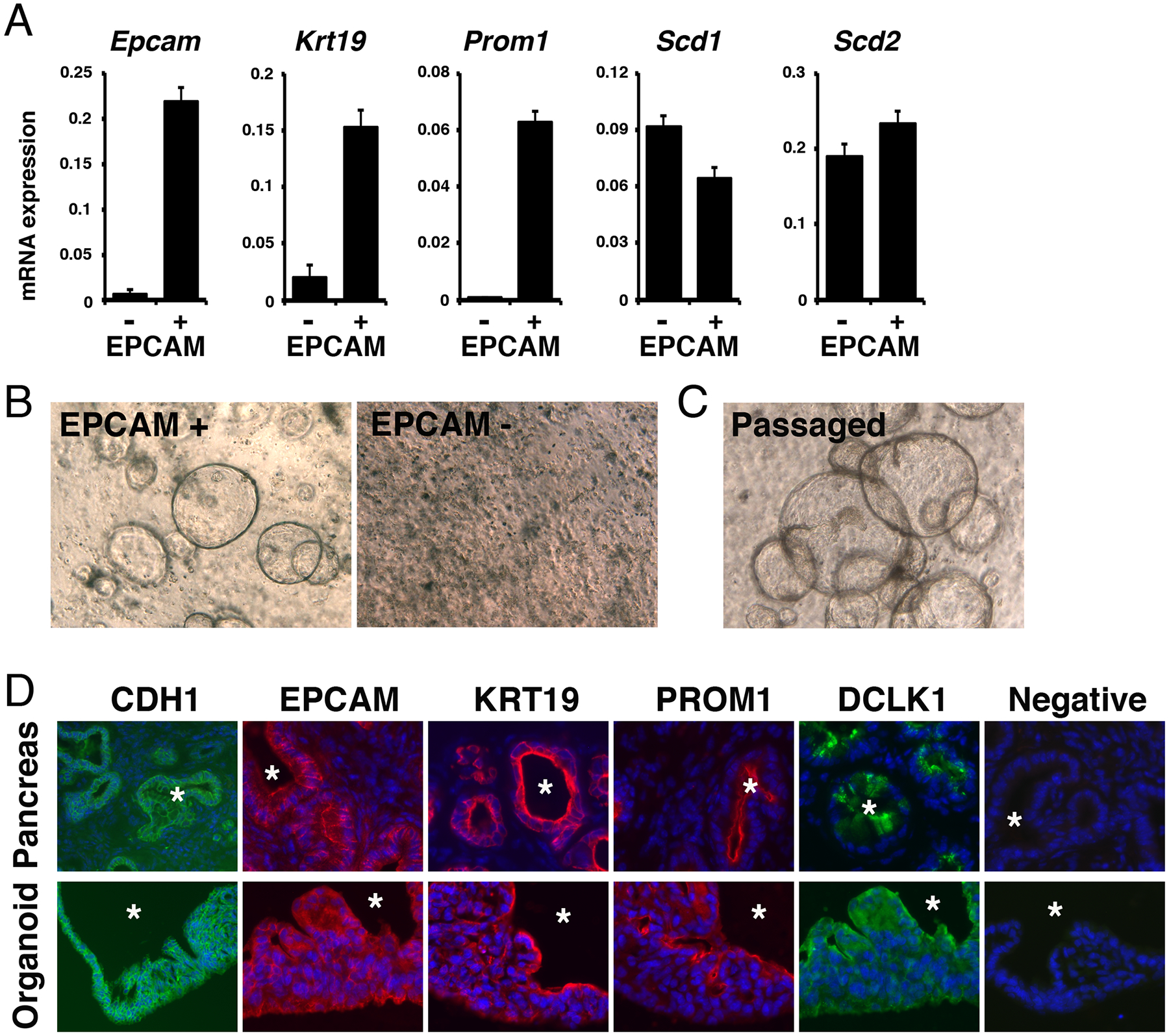
Organoid formation of EPCAM+ pancreatic tumor cells isolated from Pdx1Cre;LSL-KrasG12D mice. A, EPCAM+ and EPCAM- cells were isolated from the Pdx1Cre;LSL-KrasG12D mouse pancreas by MACS and were subjected to QPCR for measuring mRNAs. Expression values were normalized against Gapdh. B, Organoid formation from EPCAM+ cells, but not from EPCAM- cells. C, Organoid formation after passaging EPCAM+ organoids. D, Fluorescence immunostaining of CDH1, EPCAM, KRT19, PROM1, and DCLK1 in pancreatic tumors developed in the Pdx1Cre;LSL-KrasG12D mouse pancreas (first row) and organoids formed from EPCAM+ tumor cells (second row). Asterisks indicate lumens of the pancreatic tumors and tumor organoids. Immunostaining without primary antibodies were used as negative controls. Nuclei were counterstained with DAPI. Bars; 20 μm (B, C) and 50 μm (D).
Degeneration of Tumor Organoid by Inhibition of SCD
Stearoyl-CoA desaturase is an enzyme that functions in the ER by converting monounsaturated fatty acid from saturated fatty acid by introducing a single double bond.21 Our previous RNA-seq analysis showed expression of Scd1 and Scd2 mRNAs in the pancreas bearing tumors in Pdx1Cre;LSL-KrasG12D mouse (GSE139357).9 The QPCR showed that both EPCAM+ and EPCAM- cells isolated from the Pdx1Cre;LSL-KrasG12D mouse pancreas express Scd1 and Scd2 mRNAs (Fig. 1A). Having observed their expression in EPCAM+ pancreatic tumors, we examined the role of lipid metabolism regulated by the SCD enzyme in PDAC growth and survival using organoid culture. A939572 is a specific inhibitor for the SCD1 enzyme.31 To test whether pancreatic tumors requires SCD activity in their growth, we treated tumor organoids with 20 μM of A939572 dissolved in DMSO. In the control organoid treated with 0.2% of DMSO, they increased their size in culture over 16 hours (Fig. 2A). Interestingly, the SCD inhibitor not only suppressed their growth, but also induced their degeneration (Fig. 2D).
FIGURE 2.
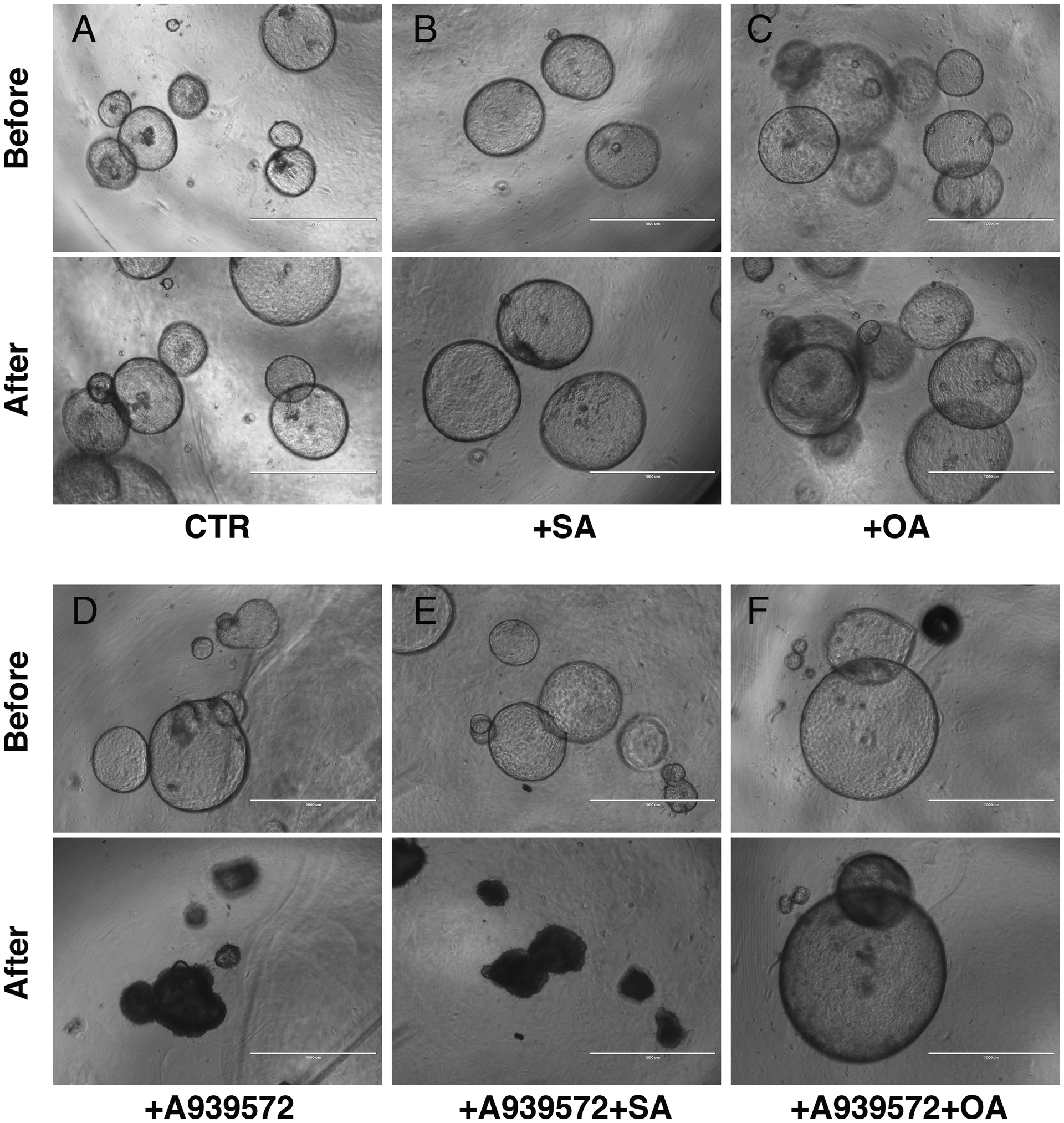
Inhibition of SCD causes degeneration of tumor spheroids in vitro. Tumor organoids formed from the Pdx1Cre;LSL-KrasG12D mouse pancreas were treated with DMSO (A-C) or the SCD inhibitor, A939572 (D-F) for 16 hours. They were also co-treated with stearic acid (SA: B, E) or oleic acid (OA: C, F). Addition of oleic acid (F), but not stearic acid (E), rescues the inhibitory effect of A939572. Bar: 1000 μm.
Saturated fatty acid is known to induce lipotoxicity.19 However, the tumor organoids treated with 30 μM of saturated fatty acid (stearic acid, 18:0) or unsaturated fatty acid (oleic acid, 18:1) equally grew in culture (Figs. 2B, C), suggesting that addition of saturated fatty acid does not provoke lipotoxicity, at least in this culture condition. To test whether the SCD inhibitor induces degeneration of the organoids by disrupting fatty acid metabolism, we treated them with A939572 with stearic acid or oleic acid. We expected that if a change of balance by accumulation of saturated fatty acid and decrease of unsaturated fatty acid is responsible for degeneration of the organoids, addition of oleic acid would rescue the negative effect of A939572. In fact, the addition of oleic acid, but not stearic acid, reverses the negative effect of A939572 (Figs. 2E, F). This experiment indicates that accumulation of saturated fatty acids and reduction of de novo biogenesis of unsaturated fatty acid inhibit the growth of pancreatic tumor organoids.
Induction of Unfolded Protein Response by the Inhibition of SCD in Tumor Organoids
Imbalance of lipogenesis might induce the UPR in cells. To test whether the SCD inhibitor induced the UPR in tumor organoids, we analyzed gene expression of UPR markers in the organoids by QPCR. Treatment with A939572 for 16 hours induced expression of Perk and its downstream effectors including Atf4, Chop, and Gadd34 mRNAs (Fig. 3A). The spliced form of Xbp1 mediated by IRE1 was similarly up-regulated by A939572 (Fig. 3B). mRNAs for Atf6 and Grp78, a chaperone for the misfolded proteins, were also increased by A939572 (Figs. 3C, D). The gene expression analysis indicates induction of the UPR in tumor organoids by A939572. Addition of stearic acid or oleic acid alone did not induce the expression of these markers in organoids, suggesting no induction of the UPR by these fatty acids. In the presence of A939572, co-treatment with stearic acid did not further increase the expression of these markers. On the other hand, addition of oleic acid reduced the expression of these markers induced by A939572 (Figs. 3A–D, lanes 6). Our data suggest that reduction of de novo biogenesis of unsaturated fatty acid and accumulation of saturated fatty acid suppress the growth of the tumor organoids through the induction of the UPR.
FIGURE 3.
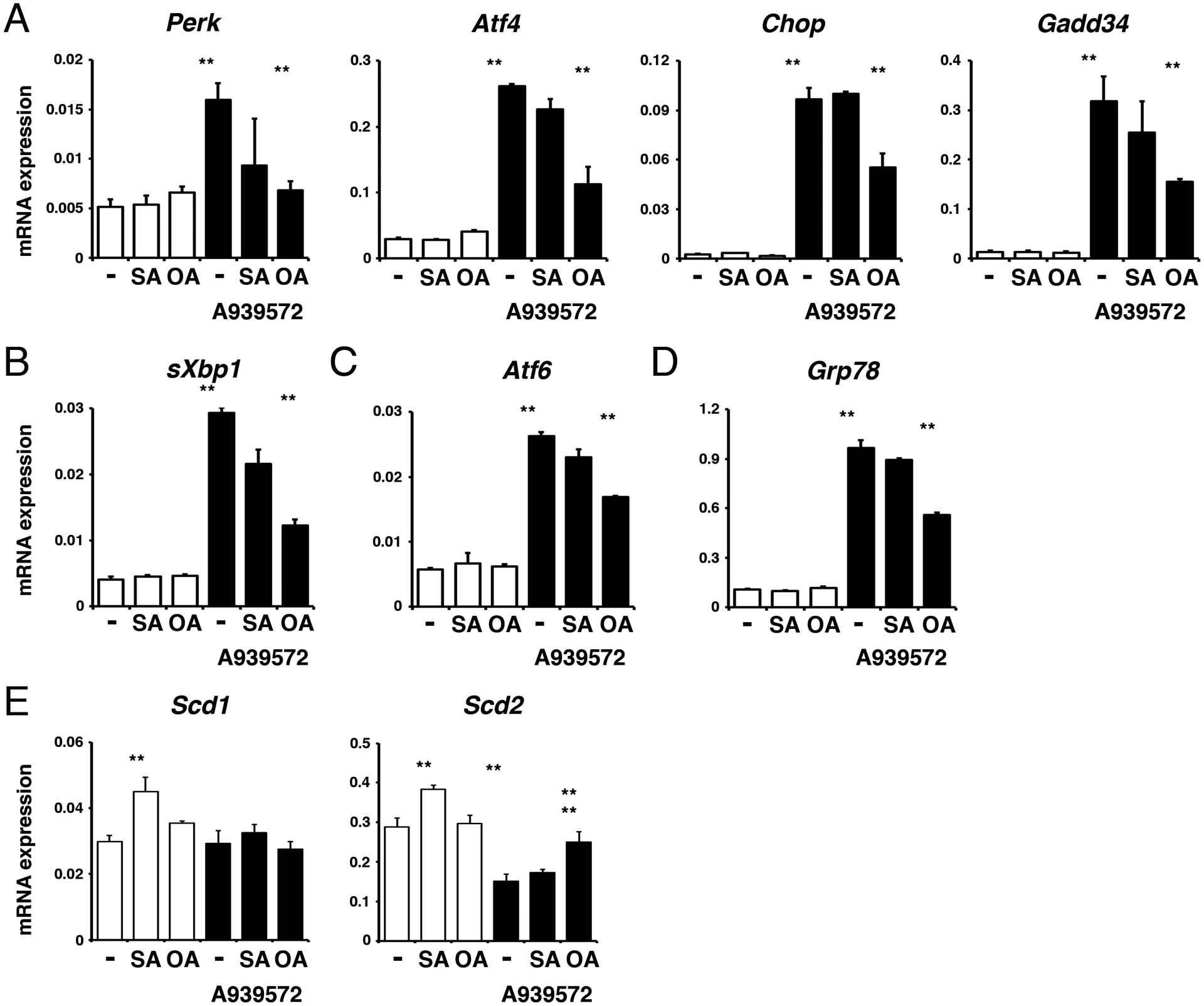
Suppression of the SCD enzyme induces the UPR in pancreatic tumor organoids.
Pancreatic tumor spheroids were treated with or without the SCD inhibitor, A939572, for 16 hours in the presence or absence of stearic acid (SA, 18:0) or oleic acid (OA, 18:1). A, mRNAs regulated by PERK-mediated UPR. B-D, Induction of sXbp1 (B), Atf6 (C), and Grp78 mRNAs (D) by A939572. Note that oleic acid, but not stearic acid, suppresses the induction of these genes. (E) Expression of Scd1 and Scd2 mRNAs. Expression values were normalized against Gapdh. **P < 0.01.
We confirmed the organoids express both Scd1 and Scd2 genes (Fig. 3E). Addition of stearic acid increased expression of these genes, suggesting a positive regulation of these genes by saturated fatty acid. Treatment with A939572 reduced the expression of Scd2, but not Scd1 mRNAs in organoids. Oleic acid weakly increased expression of Scd2 suppressed by A939572.
The SCD Inhibitor Induces Cell Death in Pancreatic Tumor In Vivo
To test whether the SCD inhibitor induces cell death in pancreatic tumors in vivo, we treated Pdx1Cre;LSL-KrasG12D mice bearing PanIN with A939572 (5 mg/kg body weight). Two days after treatment, we assessed cell death in the pancreas by TUNEL assay. The control mice treated with DMSO showed no TUNEL+ cells in the tumor (Fig. 4A). The inhibitor-treated pancreas showed TUNEL+ apoptotic cells in PanIN, but not in islets or acini (Figs. 4B–D), indicating that inhibition of SCD selectively induces apoptosis in pancreatic cancer in vivo.
FIGURE 4.
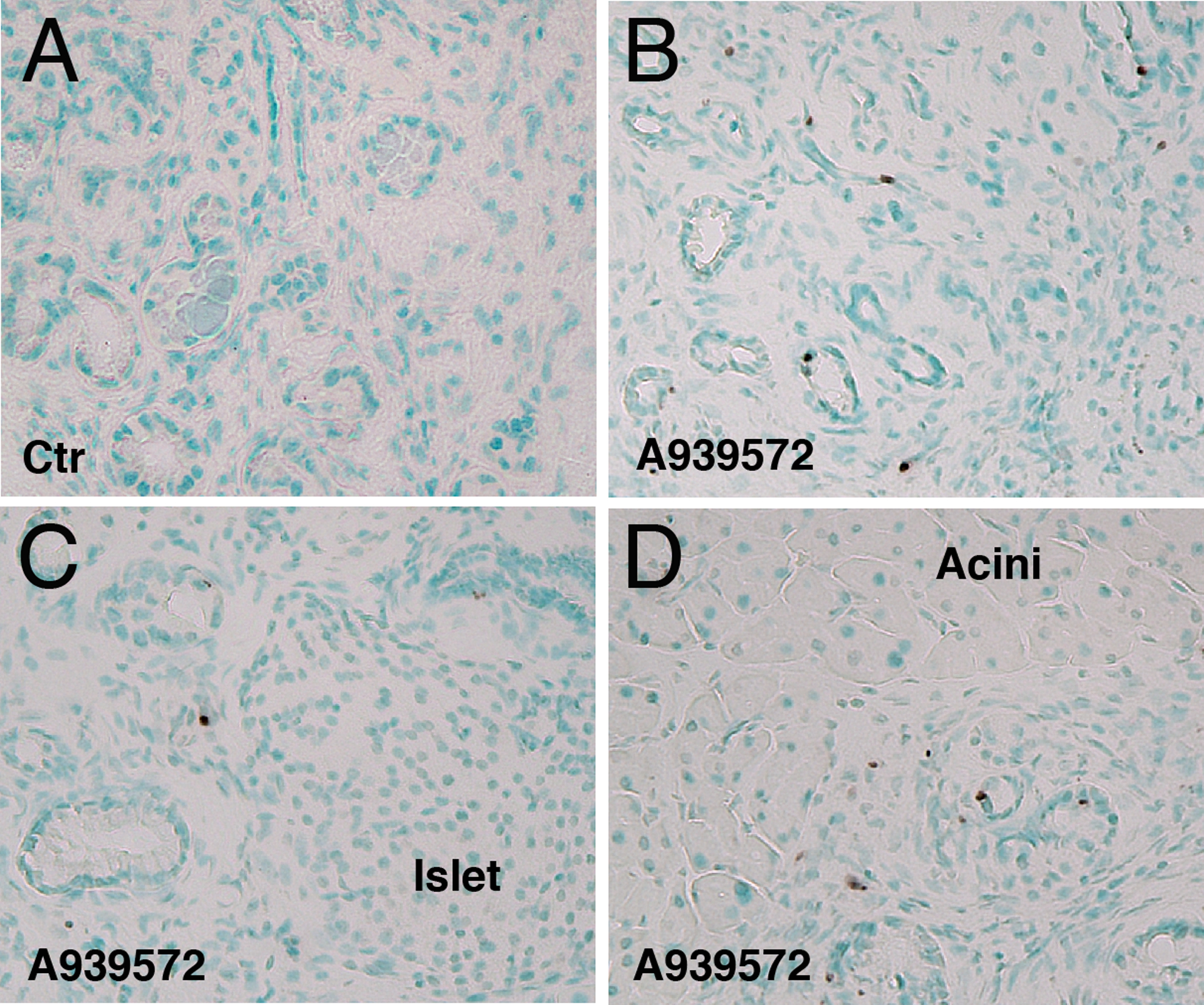
Inhibition of the SCD enzyme selectively induces apoptosis in pancreatic tumor in vivo. The Pdx1Cre;KrasG12D mice bearing PanIN were treated with A939572 or control DMSO. Pancreas tissues were analyzed by TUNEL assay. A, Tumors from control mice treated with DMSO. No TUNEL+ cells in the tumor. B-D, Tumors from mice treated with A939572. Arrows indicate TUNEL+ tumors in the pancreas. No TUNEL+ cells are observed in the islet or acini (C, D). Sections were counterstained with methyl green. Bar, 50 μm.
Inhibition of SCD Enzyme Suppresses Proliferation and Induces the UPR in PANC-1
To test whether SCD activity is necessary for PDAC, we analyzed the role of SCD1 in human PDAC cell line, PANC-1. We confirmed expression of SCD1 mRNA in PANC-1 by QPCR (data not shown). PANC-1 reduced its proliferation by treatment with A939572 (10–100 μM) for 2 days (Fig. 5A). However, we did not observe induction of cytotoxicity by A939572 (Fig. 5B). Western blot analysis showed induction of phosphorylation and expression of eIF2α by 5 and 20 μM of A939572 in PANC-1 (Fig. 5C). However, A939572 treatment did not induce the cleavage of Caspase-3 (Fig. 5C) or PARP (data not shown), indicating that the suppression of SCD1 induces the UPR, but does not induce cell death in PANC-1. Next, we measured UPR gene expression in PANC-1. Addition of stearic acid or oleic acid did not change expression of ATF4, CHOP, GADD34, and sXBP1 in PANC-1 (Fig. 5D). Similar to mouse tumor organoids, PANC-1 increased the expression of these genes by A939572 treatment (Fig. 5D). Co-treatment with oleic acid significantly suppressed the expression of these genes induced by A939572. Our data indicate that the suppression of SCD1 induces the UPR but does not induce cell death in PANC-1.
FIGURE 5.
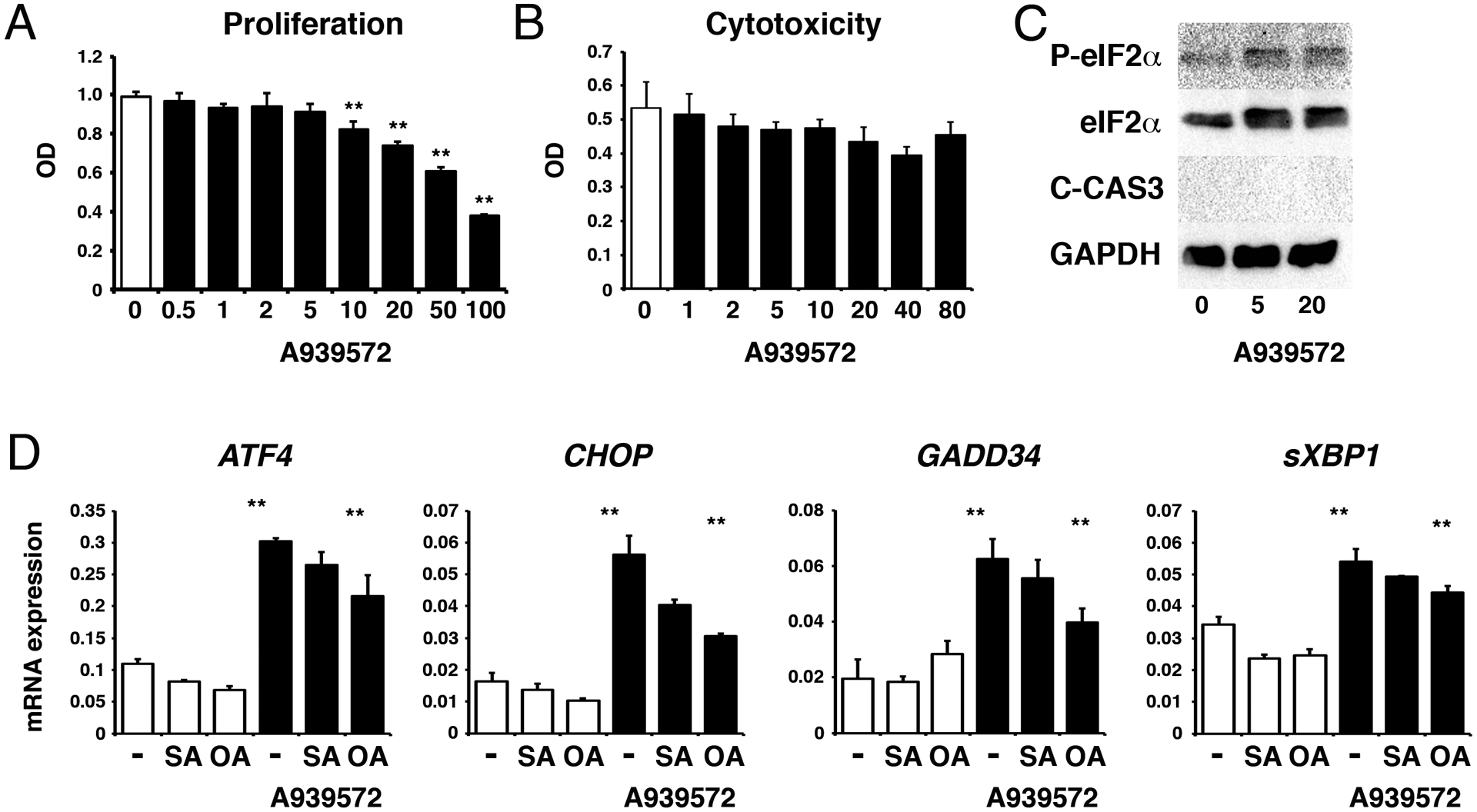
Inhibition of SCD1 induces the UPR, but not cell death in PANC-1. PANC-1 was treated with A939572 for 2 days. A, Proliferation of PANC-1 was suppressed by A939572 (10 μM to 100 μM). B, Release of LDH. No induction of cytotoxicity in PANC-1 by A939572. C, Western blot of phosphorylated eIF2α, eIF2α, cleaved Caspase-3 (C-CAS3), and GAPDH. A939572 induces expression and phosphorylation of eIF2α, but does not induce C-CAS3, in PANC-1. D, QPCR of PANC-1 treated with or without A939572 in the presence or absence of stearic acid (SA) and oleic acid (OA). **P < 0.01.
DISCUSSION
Due to its aggressive nature and lack of sensitive diagnostic markers, 80% of patients with PDAC are not eligible for surgical resection. Although chemotherapy and immunotherapy have been used for patients with PDAC, their effects have been limited and there is an urgent need for effective therapies. In the present study, we found that the inhibition of SCD causes the UPR and cell death in early pancreatic tumors. Our data suggest that the SCD enzyme might be a new therapeutic target for suppression of PDAC.
The inhibition of SCD by A939572 has been shown to increase saturated fatty acid and decrease unsaturated fatty acid in cells.31,32 We found that treatment with A939572 strongly induces degeneration of tumor organoids and the UPR in 16 hours. Long-chain saturated fatty acid, such as stearic acid, is known to cause lipotoxicity in cells.19 Accumulation of saturated fatty acid may disrupt ER homeostasis and induce the UPR via ER stress.24,25 Excessive UPR caused by ER lipotoxic stress may result in cell death. We found that A939572 treatment induces the UPR markers involved in the three UPR branches in mouse pancreatic tumor organoids. A939572 treatment also caused selective cell death in PanIN in the Pdx1Cre;LSL-KrasG12D mouse pancreas. Our data suggest that accumulation of saturated fatty acid in pancreatic tumor by A939572 causes the UPR and cell death in mice.
Exogenously added stearic acid did not induce the UPR in organoids and their degeneration. In addition, we did not observe further induction of the UPR by addition of stearic acid in the presence of A939572. It remains unclear whether the UPR was fully activated by endogenously accumulated stearic acid caused by A939572 or exogenous stearic acid is incorporated in cellular compartments different from the ER in pancreatic cancer. Interestingly, exogenously added oleic acid could rescue the negative effect caused by A939572 in organoids. Decreased level of unsaturated fatty acid might cause the UPR by disrupting the integrity of lipid membranes in pancreatic cancer.22,23 Another possibility is that modification by unsaturated fatty acid may be necessary for activity of proteins in pancreatic cancer. For example, palmitoylation is essential in Wnt ligands for their biological activity.33 However, oleic acid used in the present study is not a lipid species used for biogenesis of Wnt ligand.34 Thus, the rescue of the inhibitor-induced degeneration of the organoids by oleic acid is not attributable to biogenesis of active Wnt ligands.
Organoids from the intestine grow with budding carrying a crypt villus axis.35 On the other hand, organoids formed from EPCAM+ pancreatic tumor cells from the Pdx1Cre;LSL-KrasG12D mouse show a sphere morphology without budding and are comprised of epithelial cell layers. Similar to PanIN in vivo, the pancreatic tumor organoids show a duct cell phenotype with an apical-basal polarity. During the progression of PanIN to PDAC, cancer cells lose an epithelial polarity.9 Our data showed that tumor organoids in matrigel maintain an early pancreatic cancer phenotype of PanIN having epithelial cell polarity. We found that A939572 rapidly induces degeneration of these organoids. In addition, A939572 selectively induced apoptosis in PanIN in the Pdx1Cre;LSL-KrasG12D mouse pancreas. Our data indicate that the SCD inhibitor is effective for induction of apoptosis in early pancreatic cancer. In mouse pancreatic cancer, both EPCAM+ and EPCAM- cells express Scd1 and Scd2 mRNAs. Although we observed the strong effect of A939572 on tumor organoids, A939572 might cause apoptosis in PanIN indirectly by changing phenotypes of EPCAM- cells, including tumor-associated stromal cells and infiltrating leukocytes in the pancreas.
Similar to the mouse tumor organoid, we observed that A939572 induces the UPR in PANC-1. Chemical inhibitors for SCD1 have been shown to suppress the growth of lung cancer, hepatocellular carcinoma, colorectal cancer, and renal carcinoma.29,32,36,37 However, we did not observe induction of cell death in PANC-1 by the SCD inhibitor. PANC-1 is a poorly differentiated human PDAC cell line and has metastasizing capacity.38 Daemen et al classified heterogeneous pancreatic cancers into lipogenic and glycolytic subtypes.39 The glycolytic pancreatic cancer was shown to be insensitive for inhibition of SCD. We assume that the UPR induced by A939572 may result in apoptosis in early pancreatic cancer, but may not be sufficient in PDAC. We found that treatment with A939572 increased the expression of Grp78 mRNA in pancreatic tumor organoids. GRP78 is known to play an important role in pancreatic tumor progression and increase chemoresistance in PDAC.40,41 Induction of the UPR with reduction of GRP78 by triptolide was shown to cause cell death in PDAC.42 In hepatocellular carcinoma, a SCD inhibitor negates their resistance to sorafenib by inducing ER stress.43 Further studies are necessary to assess whether the UPR caused by the SCD inhibitor has synergistic effects on pancreatic cancer if combined with anti-cancer drugs.
ACKNOWLEDGMENT
We thank Dr. Hide Tsukamoto for discussion of this study.
Financial support: This work was supported by the pilot project from the Southern California Research Center for ALPD & Cirrhosis (National Institutes of Health P50AA11999) and USC Dean’s pilot project program.
Footnotes
Disclosure: The authors declare no conflict of interest.
REFERENCES
- 1.Chiaravalli M, Reni M, O’Reilly EM. Pancreatic ductal adenocarcinoma: State-of-the-art 2017 and new therapeutic strategies. Cancer Treat Rev. 2017;60:32–43. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 3.Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 4.Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. [DOI] [PubMed] [Google Scholar]
- 5.Saluja AK, Dudeja V, Banerjee S. Evolution of novel therapeutic options for pancreatic cancer. Curr Opin Gastroenterol. 2016;32:401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kopp JL, von Figura G, Mayes E, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liou GY, Doppler H, Necela B, et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J Cell Biol. 2013;202:563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hezel AF, Kimmelman AC, Stanger BZ, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. [DOI] [PubMed] [Google Scholar]
- 9.Asahina K, Balog S, Hwang E, et al. Moderate alcohol intake promotes pancreatic ductal adenocarcinoma development in mice expressing oncogenic Kras. Am J Physiol Gastrointest Liver Physiol. 2020;318:G265–G276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nomura A, Gupta VK, Dauer P, et al. NFkappaB-mediated invasiveness in CD133(+) pancreatic TICs Is regulated by autocrine and paracrine activation of IL1 signaling. Mol Cancer Res. 2018;16:162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morris JPt Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144:1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitcomb DC, Shelton CA, Brand RE. Genetics and genetic testing in pancreatic cancer. Gastroenterology. 2015;149:1252–1264. [DOI] [PubMed] [Google Scholar]
- 14.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. [DOI] [PubMed] [Google Scholar]
- 15.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. [DOI] [PubMed] [Google Scholar]
- 16.Chang HH, Moro A, Takakura K, et al. Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS One. 2017;12:e0184455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu S, Chheda C, Ouhaddi Y, et al. Characterization of mouse models of early pancreatic lesions induced by alcohol and chronic pancreatitis. Pancreas. 2015;44:882–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barone E, Corrado A, Gemignani F, et al. Environmental risk factors for pancreatic cancer: an update. Arch Toxicol. 2016;90:2617–2642. [DOI] [PubMed] [Google Scholar]
- 19.Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016;5:e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mancini R, Noto A, Pisanu ME, et al. Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene. 2018;37:2367–2378. [DOI] [PubMed] [Google Scholar]
- 21.Paton CM, Ntambi JM. Biochemical and physiological function of stearoyl-CoA desaturase. Am J Physiol Endocrinol Metab. 2009;297:E28–E37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Igal RA. Stearoyl CoA desaturase-1: New insights into a central regulator of cancer metabolism. Biochim Biophys Acta. 2016;1861:1865–1880. [DOI] [PubMed] [Google Scholar]
- 23.Koeberle A, Loser K, Thurmer M. Stearoyl-CoA desaturase-1 and adaptive stress signaling. Biochim Biophys Acta. 2016;1861:1719–1726. [DOI] [PubMed] [Google Scholar]
- 24.Corazzari M, Gagliardi M, Fimia GM, et al. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front Oncol. 2017;7:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clarke HJ, Chambers JE, Liniker E, et al. Endoplasmic reticulum stress in malignancy. Cancer Cell. 2014;25:563–573. [DOI] [PubMed] [Google Scholar]
- 26.Macasek J, Vecka M, Zak A, et al. Plasma fatty acid composition in patients with pancreatic cancer: correlations to clinical parameters. Nutr Cancer. 2012;64:946–955. [DOI] [PubMed] [Google Scholar]
- 27.Boj SF, Hwang CI, Baker LA, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huch M, Bonfanti P, Boj SF, et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013;32:2708–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai KKY, Kweon SM, Chi F, et al. Stearoyl-CoA desaturase promotes liver fibrosis and tumor development in mice via a Wnt positive-signaling loop by stabilization of low-density lipoprotein-receptor-related proteins 5 and 6. Gastroenterology. 2017;152:1477–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westphalen CB, Takemoto Y, Tanaka T, et al. Dclk1 defines quiescent pancreatic progenitors that promote injury-induced regeneration and tumorigenesis. Cell Stem Cell. 2016;18:441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xin Z, Zhao H, Serby MD, et al. Discovery of piperidine-aryl urea-based stearoyl-CoA desaturase 1 inhibitors. Bioorg Med Chem Lett. 2008;18:4298–4302. [DOI] [PubMed] [Google Scholar]
- 32.Chen L, Ren J, Yang L, et al. Stearoyl-CoA desaturase-1 mediated cell apoptosis in colorectal cancer by promoting ceramide synthesis. Sci Rep. 2016;6:19665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El Helou R, Pinna G, Cabaud O, et al. miR-600 acts as a bimodal switch that regulates breast cancer stem cell fate through WNT signaling. Cell Rep. 2017;18:2256–2268. [DOI] [PubMed] [Google Scholar]
- 34.Rios-Esteves J, Resh MD. Stearoyl CoA desaturase is required to produce active, lipid-modified Wnt proteins. Cell Rep. 2013;4:1072–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eriguchi Y, Nakamura K, Yokoi Y, et al. Essential role of IFN-γ in T cell-associated intestinal inflammation. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noto A, Raffa S, De Vitis C, et al. Stearoyl-CoA desaturase-1 is a key factor for lung cancer-initiating cells. Cell Death Dis. 2013;4:e947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.von Roemeling CA, Marlow LA, Wei JJ, et al. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin Cancer Res. 2013;19:2368–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deer EL, Gonzalez-Hernandez J, Coursen JD, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daemen A, Peterson D, Sahu N, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci U S A. 2015;112:E4410–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen J, Ha DP, Zhu G, et al. GRP78 haploinsufficiency suppresses acinar-to-ductal metaplasia, signaling, and mutant Kras-driven pancreatic tumorigenesis in mice. Proc Natl Acad Sci U S A. 2017;114:E4020–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gifford JB, Huang W, Zeleniak AE, et al. Expression of GRP78, master regulator of the unfolded protein response, increases chemoresistance in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2016;15:1043–1052. [DOI] [PubMed] [Google Scholar]
- 42.Mujumdar N, Banerjee S, Chen Z, et al. Triptolide activates unfolded protein response leading to chronic ER stress in pancreatic cancer cells. Am J Physiol Gastrointest Liver Physiol. 2014;306:G1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma MKF, Lau EYT, Leung DHW, et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J Hepatol. 2017;67:979–990. [DOI] [PubMed] [Google Scholar]