

Abstract
High-quality evidence indicates that regular use of aspirin is effective in reducing the risk for precancerous colorectal neoplasia and colorectal cancer (CRC). This has led to US and international guidelines recommending aspirin for the primary prevention of CRC in specific populations. In this review, we summarize key questions that require addressing prior to broader adoption of aspirin-based chemoprevention, review recent evidence related to the benefits and harms of aspirin use among specific populations, and offer a rationale for precision prevention approaches. We specifically consider the mechanistic implications of evidence showing differences in aspirin’s effects according to age, the potential role of modifiable mechanistic biomarkers for personalizing prevention, and emerging evidence that the gut microbiota may offer novel aspirin-associated preventive targets to reduce high-risk neoplasia.
Keywords: chemoprevention, precision chemoprevention, personalized medicine, colorectal cancer, nonsteroidal anti-inflammatory drugs
INTRODUCTION
Aspirin (acetylsalicylic acid) has emerged as the agent with the most consistently observed chemopreventive effect on colorectal neoplasia (1–3). The preponderance of randomized controlled trials (RCTs) of aspirin in prevention of adenomatous polyps (adenoma), the precursor lesion for the vast majority of colorectal cancers (CRCs), and CRC have demonstrated consistent benefits (4). This evidence led the US Preventive Services Task Force (USPSTF) to recommend aspirin for joint prevention of CRC and cardiovascular disease (CVD) for many adults in 2016 (5). Based on a comprehensive risk/benefit analysis, the USPSTF provided a B rating for individuals aged 50–59 years with a 10% ten-year risk for a CVD event. For individuals aged 60–69 years, the recommendation was lowered to a C rating, supporting use according to an individualized assessment of potential benefits versus harms. At the time, the data for other age groups was found to be insufficient, including individuals under age 50 or over age 70, to offer any recommendation. Furthermore, the USPSTF emphasized that preventive benefits are observed primarily after consistent long-term use for approximately a decade. Largely due to continued uncertainty surrounding aspirin’s anticancer mode of action, the harms associated with regular use, and recent conflicting data from an RCT of aspirin initiation in older adults (6), additional investigation is required before broader implementation of chemopreventive strategies can be adopted.
THE EVIDENCE FOR ASPIRIN AS A CHEMOPREVENTIVE
The evidence supporting aspirin’s anticancer effect has been previously reviewed by us and others (7, 8). Meta-analyses and other systematic reviews of large observational cohort studies have estimated that aspirin reduces risk for colorectal neoplasia by approximately 20–30%. Estimates from adenoma prevention trials typically enrolling higher-risk individuals with a personal history of colorectal adenoma have demonstrated that risk for recurrent neoplasia is reduced by 13–18% (9, 10). The decision analysis and data synthesis provided by the USPSTF comprise a comprehensive resource of data on benefits and harms through late 2015 (11). As a result, several concepts have largely been accepted by the field.
First, epidemiologic data support that regular (at least two times per week), long-term aspirin use is associated with prevention of CRC in the general population if used for at least a decade (12–14). Additionally, this benefit may not be evident for 10 years after initiation, although some analyses support a significant effect within 5 years (15–18). These findings have largely been confirmed through long-term follow-up of CVD prevention trials. It is unclear whether this lag represents a “legacy” effect of aspirin, whereby a period of aspirin use in the past confers a benefit that is borne out in the future, or a “delayed” effect of aspirin, meaning that a reduced risk of early-stage neoplasia may not be manifest as a reduction in CRC risk for several years since the time necessary for an early neoplastic lesion to develop into CRC is estimated as 5–10 years. Nonetheless, the duration of exposure and potential lag/legacy effects are among the reasons that led the USPSTF to caution that aspirin use in primary prevention of CVD and CRC should only be considered for those with a reasonable life expectancy that exceeds a decade.
Second, daily use of low-dose aspirin (81–100 mg) appears sufficient for cancer prevention, but emerging evidence indicates that height, weight, or body mass index may impact aspirin bioavailability such that higher doses may be required in some individuals (19, 20). Other individual factors including cigarette smoking may counteract aspirin chemoprevention (21, 22). Moreover, while many studies have not provided precise estimates of an association according to dose, chemopreventive benefit has been observed across a range of doses. In some studies, standard or higher doses (325 mg/day or greater) were shown to prevent neoplasia and presumed to be potentially required (23–27), whereas other studies demonstrated that alternate-day use of low-dose aspirin was sufficient (15, 16, 18). In fact, in the Aspirin/Folate Polyp Prevention Study (AFPPS), prevention of adenoma was observed only for those receiving low-dose aspirin but not standard-dose aspirin (28). Most recently, the Systematic Evaluation of Aspirin and Fish Oil (seAFOod) Polyp Prevention Trial found that neither aspirin nor eicosapentaenoic acid nor both together achieved their primary endpoint of a reduction in the adenoma detection rate in high-risk individuals (those with three or more adenomas, at least one of which is 1 cm in diameter or greater, or five adenomas of <1 cm) at one-year surveillance endoscopy compared to placebo (29). However, aspirin did result in the significant reduction of total numbers of adenomas, especially proximal adenomas and serrated adenomas, and conventional adenomas throughout the colon, supporting a direct anticancer benefit on early neoplasia. Although adenoma detection rate has been a traditional outcome in prior polyp prevention trials, these results highlight a clear chemopreventive benefit. Owing to advances in screening endoscopy and the emphasis on quality assurance in colonoscopy, in which differences in adenoma detection rate are difficult to achieve, adenoma number is a better measure of chemopreventive efficacy (30).
Third, increasing data from large, well-designed clinical trials with CRC as the primary endpoint support that among individuals with Lynch syndrome, the most common hereditary CRC syndrome, aspirin may have significant preventive benefit. Most recently, long-term follow-up of the Cancer Prevention Program 2 (CaPP2) RCT demonstrated that use of aspirin (600 mg/day) for 2 years resulted in significant reduction of CRC risk among individuals with Lynch syndrome that became apparent 5 years after first use (31). These findings, along with the results of a large retrospective cohort study of carriers of mismatch repair gene mutations, led the National Institute for Health and Care Excellence (NICE) in the United Kingdom to update their recommendations for CRC prevention to specifically advocate daily aspirin for at least 2 years to prevent CRC among individuals with Lynch syndrome (32). However, NICE, like the USPSTF in its 2016 recommendation, cited balancing the harms with the perceived benefit and a need to clarify the optimal dose for preventive benefit.
Current evidence to support use of aspirin for primary prevention of CRC in many individuals is compelling. Nonetheless, in 2020, several significant questions remain for the field to address.
Question 1: What Is the Impact of Age on Aspirin Chemoprevention?
After the 2016 USPSTF guidelines were released, many looked to the ASPirin in Reducing Events in the Elderly (ASPREE) RCT to provide more clarity regarding aspirin chemoprevention among older populations. ASPREE enrolled 19,114 adults (16,703 in Australia and 2,411 in the United States) who were otherwise healthy to determine if daily low-dose (100 mg) aspirin had an effect on reducing “events,” a composite of death, dementia, and physical disability, among older individuals (70 years or older for white individuals; 65 years or older for minorities). Surprisingly, ASPREE reported that low-dose aspirin trended toward increased all-cause mortality [hazard ratio (HR) = 1.14, 95% confidence interval (CI) 1.01–1.29] that was driven by cancer deaths (HR = 1.31, 95% CI 1.10–1.56), including death from CRC specifically, after just 4.7 years of follow-up. Similarly, an analysis of RCTs of aspirin in primary prevention with long-term follow-up found that aspirin was associated with an increase in cancer incidence within the first 3 years of follow-up in participants at least 70 years of age (HR = 1.20, 95% CI 1.03–1.47), and highest among those with smaller body size (20). However, a more detailed analysis of the effect of aspirin on cancer incidence in ASPREE did not demonstrate an increase in overall cancer incidence (HR = 1.04, 95% CI 0.95–1.14) and CRC incidence (HR = 1.02, 95% CI 0.81–1.30) (33). There are several possible explanations for the lack of benefit, and perhaps harm, of initiating aspirin in older adults. First, the duration of treatment and/or follow-up may have been insufficient. This may be clarified with longer-term follow-up of this cohort. Second, aspirin may have potential biological effects that vary according to the timing of exposure relative to the life course. Importantly, the vast majority of ASPREE participants (89%) had never used aspirin regularly prior to enrollment at an age of at least 65 years. Thus, aspirin may not be effective in reducing cancer risk when initiated late in life. Third, older age may result in distinct colorectal carcinogenesis pathways that are less sensitive to aspirin. For example, aging is associated with alterations in DNA methylation which may affect susceptibility to cancer (34–36). CRCs in older individuals arise more commonly in the right side of the colon (37, 38), and tumors have a higher prevalence of specific molecular changes, including positive or high CpG island methylator phenotype and BRAF mutations, compared to CRCs in younger individuals (39, 40). In addition, there is a greater incidence of KRAS A146, K117, and Q61 oncogenic mutations in older patients, whereas in younger patients (<40 years) G12 mutations are more frequent (41). With respect to aspirin efficacy, it has been previously reported that regular aspirin use is associated with a lower risk of CRCs that have wild-type BRAF and KRAS genotypes but not CRCs carrying these oncogenic mutations, which may partially explain these age-related differences (42, 43). These results underscore the need to advance our understanding of individual characteristics, especially age, as modifiers of aspirin’s chemopreventive effects. Future studies will need to specifically consider the impact of age on both efficacy and harms of aspirin use.
On the other side of the aging spectrum, the data supporting aspirin chemoprevention among younger (<50 years) adults were also considered by the USPSTF to be insufficient in 2016. Concerningly, the incidence of early-onset CRCs is rising, with 2.5 more cases per year per 100,000 individuals since 2000 despite improved adoption of screening endoscopy over that time period (44). The rise was enough to prompt the American Cancer Society to recommend screening endoscopy to begin at age 45 for most individuals rather than age 50 (45). In a case-control study among US Veterans (44), aspirin users had a significantly reduced risk of early-onset CRC [odds ratio (OR) = 0.66, 95% CI 0.52–0.84] compared to nonusers. The results appeared to be restricted to individuals in the 40–49-year-old age stratum, leading the authors to conclude that longitudinal exposure to aspirin may be required.
Question 2: What Is the Optimal Age to Commence a Prevention Regimen?
The early-onset CRC findings raise an important consideration for patients and providers alike. When is the best time to start an aspirin chemoprevention regimen? For what duration does a person need to use aspirin to have long-lasting benefit? Studies in ApcMin/+ mice demonstrate that the effects of aspirin on tumorigenesis depend on age of initiation: Continuous exposure from conception suppresses intestinal tumorigenesis whereas limited exposure in adulthood does not (46). The Cancer Prevention Program 3 (CaPP3) trial is specifically designed to test the optimal dose for prevention in individuals with Lynch syndrome. However, given the broad age range of the study, there may be some insight gained into optimal age of initiation. As previously described, use over several years appears to result in risk reduction that becomes evident 5–10 years after commencing an aspirin regimen. Taken together with the USPSTF evidence synthesis, these findings suggest that regular use of low-dose aspirin should be considered starting at age 50 for individuals likely to experience CVD and CRC prevention benefits. From an implementation standpoint, initiating an aspirin regimen could be considered alongside initiating one’s first screening colonoscopy. Although limited, the data supporting a possible benefit for early-onset CRC may warrant discussion of an aspirin regimen at earlier ages for individuals who initiate screening between ages 45 and 50.
Question 3: What Is the Overall Risk/Benefit Ratio of an Aspirin Regimen?
The concern with widespread implementation of aspirin use, despite established potential benefits, stems from the increased risk of bleeding. While the USPSTF decision concluded that for some individuals a clear benefit for CVD and CRC prevention outweighed the potential harms, bleeding remains a significant concern, especially in older populations. The ASPREE findings highlighted the risks of aspirin intervention among older adults. Among the 19,114 individuals in ASPREE, the risk of hemorrhage was significantly elevated among those receiving aspirin (HR = 1.38, 95% CI 1.18–1.62) compared to those receiving placebo. This estimate is consistent with the broader risks for bleeding reported among average-risk individuals. A meta-analysis of 35 aspirin RCTs estimated the risks for a major gastrointestinal (GI) bleeding event at 1–2 events per 1,000 person-years (HR = 1.31, 95% CI 1.21–1.42) (47). Aspirin is also associated with intracranial bleeding (OR = 1.27, 95% CI 0.98–1.66), but the absolute incidence of intracranial bleeding is significantly lower. However, in ASPREE, aspirin appeared to increase overall GI bleeding risk by 60% (HR = 1.61, 95% CI 1.26–2.08) compared to placebo. This translated into a modest absolute 5-year risk of 5.03% risk for bleed for an 80-year-old taking aspirin who has additional risk factors, including smoking, hypertension, chronic kidney disease, and obesity versus 0.25% for a healthy 70-year-old not taking aspirin (48). Most studies have similarly demonstrated increasing risk with higher dose (49). A recent meta-analysis concluded that use of <300 mg/day of aspirin was associated with increased bleeding risk within 3 years of use, but not after more than 3 years, and that overall risk increased with age (50). The estimated adjusted incidence rate ratio for major bleeding events with each year of advancing age is 1.05 (95% CI: 1.05–1.05) (51). These findings suggest the possibility that if aspirin is well tolerated in the short term, long-term use may not pose any additional risk, and underscore the need for informed decision making that considers life expectancy and other risk factors for bleeding among older patients.
Although the data are not as definitive as for CRC, additional potential benefits of aspirin for other cancers should also be considered. The Cancer Prevention Study II demonstrated that regular use of standard-dose aspirin for at least 5 years was associated with lower overall cancer incidence among men [relative risk (RR) = 0.84, 95% CI 0.76–0.93] over 11 years of follow-up (52). Relative risk reductions were similar among women, but not statistically significant. After 32 years of follow-up within the Nurses’ Health Studies and Health Professionals Follow-up Study, we have shown that regular use of aspirin was associated with a 3% reduction in risk of cancer overall (RR = 0.97, 95% CI 0.94–0.99), which was driven primarily by prevention of GI cancers and further by CRC (53). In addition, we recently reported that long-term aspirin use was associated with reduced hepatocellular carcinoma risk (HR = 0.51, 95% CI 0.28–0.96) in the same populations (54). A meta-analysis of six trials of low-dose aspirin in primary prevention also determined that the risk of overall cancer incidence after 3 years was reduced by approximately 25% (in women, OR = 0.75, 95% CI 0.59–0.94; in men, OR = 0.76, 95% CI 0.66–0.88) (55).
Future risk/benefit analyses may also consider the effect on overall cancer death. An analysis using data from eight RCTs found that allocation to aspirin was associated with a decreased OR of 0.79 (95% CI 0.68–0.92) for cancer death, and the 20-year risk of cancer death remained lower among those receiving aspirin for all solid cancers (HR = 0.80, 95% CI 0.72–0.88), with the benefit increased with longer duration of exposure (56). This was extended in a larger meta-analysis of 51 RCTs, which demonstrated that aspirin reduced cancer deaths by approximately 15% (OR = 0.85, 95% CI 0.76–0.96) (55). Similar prevention of cancer death and all-cause mortality was observed within the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial (57). Our recent findings using Swedish national registries have established a potent signal for low-dose aspirin in prevention of hepatocellular carcinoma and lower liver-related mortality in individuals with chronic viral hepatitis, with no significant increase in bleeding risk (58). Based on this evidence, it may be reasonable to consider potential broader anticancer effects of aspirin when weighing the potential benefits in light of harms. However, tailoring individual recommendations to improve the therapeutic index by widening the harms gap may require a better understanding of aspirin’s chemopreventive mode of action. As discussed below, the differential preventive effects of aspirin according to age may offer clues.
Question 4: What Is Aspirin’s Mechanism of Action?
Because we still lack clarity on the mechanism of action for aspirin chemoprevention, it is difficult to know for whom—or alternatively, for what tumorigenesis pathways—aspirin will be most beneficial. Broadly, aspirin’s anti-inflammatory and antiplatelet properties have remained central to its proposed preventive mechanism. We have previously described an integrated, multimodal framework for aspirin’s chemoprevention stemming from preclinical, molecular epidemiology, and clinical trial studies (59). The framework centers on aspirin’s effects to inhibit prostaglandin synthase (PTGS) 1 and 2, also known as cyclooxygenase (COX) 1 and 2, which synthesize downstream prostaglandins and eicosanoids from arachidonic acid. In platelets, aspirin exerts antiplatelet effects by irreversibly inhibiting PTGS-1, which may indirectly influence PTGS-2 signaling in epithelial cells. Aspirin may also directly inhibit PTGS-2, resulting in downstream inhibition of prostanoids, including prostaglandin E2 (PGE2), that play key roles in inflammatory signaling and cellular proliferation, survival, and growth and are upregulated in many colorectal tumors. Through a combination of these factors, aspirin may further influence WNT, cAMP-PKA (cyclic AMP–protein kinase A), and β-catenin signaling and inhibit tumorigenesis. Molecular pathologic epidemiology studies have demonstrated that aspirin may more potently inhibit tumors that form via prostaglandin-mediated pathways. Aspirin reduces incidence of tumors that overexpress PTGS-2 (60) and reduces incidence of tumors in individuals who have intact HPGD expression in normal mucosa (61). HPGD (hydroxyprostaglandin dehydrogenase), the metabolic antagonist of PTGS-2, catabolizes PGE2 into downstream major prostaglandin metabolites (PGE-M; 11α-hydroxy-9,15-dioxo-2,3,4,5-tetranor-prostane-1,20-dioic acid). In total these results, combined with evidence that elevated urinary PGE-M levels may predict risk for recurrent adenoma among individuals at risk for CRC (62, 63), support a central role for modulation of prostaglandin balance as aspirin’s primary preventive mode of action.
Differential preventive effects according to age may further reveal aspirin’s mechanism. Aging is associated with increased chronic inflammation, termed inflammaging, such that even healthy older adults have elevated levels of multiple circulating inflammatory cytokines, including PGE2 and growth differentiation factor-15 (GDF-15)/macrophage inhibitory cytokine−1 (MIC-1), compared to younger adults (64, 65). Moreover, while inflammation and PGE2 dysregulation are associated with colorectal tumorigenesis (59, 66), they also influence intestinal stem cell (ISC) differentiation through Hippo/Yes-associated protein 1 (YAP) signaling pathways (67–70). Recent data indicate that aspirin-like nonsteroidal anti-inflammatory drugs (NSAIDs) preferentially eliminate premalignant Lgr5+ ISCs (71, 72), largely hypothesized to be the primary cell of origin for the majority of GI cancers. However, in preclinical experiments, aged mice compared to young adult mice have fewer, less regenerative Lgr5+ ISCs (73, 74), which are also less tumorigenic in an Apc tumor suppressor model (73). Consequently, multistep models suggest that sequential mutations transform normal cells, and exposure time to carcinogens associated with advanced age elevates cancer risk (75). Further, stem cell mutational burden increases with age in the colon (76), and other evidence suggests that cell- and noncell autonomous changes associated with aging may elevate cellular susceptibility to transformation. These changes include attenuated DNA repair, accumulation of oxidative damage, impaired proteostasis, defective immune surveillance, increased numbers of stem cell divisions (77, 78), and promotion of tumor growth and metastasis (75, 79). Although the mechanisms of age-related inflammation are complex [i.e., myeloid skewing (80)], PGE2 is a particularly relevant driver of inflammaging in the intestine. It is one of the major proinflammatory factors that is produced constitutively during homeostasis and is elevated during injury/repair, which may further contribute to tumorigenesis (81, 82). Similarly, signaling through Ptger4 (EP4 receptor for which PGE2 is a ligand) in the intestinal epithelium promotes tumorigenesis and affects wound healing processes during repair (69, 83–85). Thus, as a clear direct target of aspirin, PGE2 signaling via the Hippo/YAP pathways in intestinal epithelial cells may play a critical role in directing aspirin effects on ISC homeostasis and preventing alterations associated with inflammaging. The detrimental effects of inflammaging in older adults may therefore compromise the healthy ISC pool, leading to activation of atypical ISCs that may be less sensitive to aspirin chemoprevention treatment if initiated later in life (e.g., after inflammaging effects have taken hold).
Beyond age-related changes to prostaglandin pathways as a result of inflammaging, different molecular alterations appear to differentiate early-onset CRC from late-onset CRC. It has been found that frequency of microsatellite instability (MSI) status due to mismatch repair (MMR) deficiency is higher among younger adults without the increased frequency of CpG island methylator phenotype observed among older individuals in case series (86), likely driven by potential Lynch syndrome or inherited MMR defect cases. In preclinical in vitro experiments, aspirin and other NSAIDs have been shown to reduce MSI in MMR-deficient cells by promoting apoptosis in MSI+ CRC cells (87). However, perhaps epigenetically MSI phenotypes in late-onset CRC and higher rates of BRAF mutations (88–90) may render these neoplasias less sensitive to aspirin than are MMR mutation–driven MSI phenotypes. These molecular differences may at least partially explain age-related differences for aspirin prevention. In aggregate, these results suggest that aspirin may be more effective at preventing sporadic tumorigenesis that arises either through prostaglandin-mediated pathways (no apparent age association) or through non–epigenetically driven MMR deficiency.
With clarification of the preventive mode of action, aspirin chemoprevention strategies which leverage aspirin’s mechanism and mechanistically linked biomarkers may empower a precision approach by which individuals can be identified who are most likely to benefit.
THE NEED FOR PRECISION CHEMOPREVENTION
Over the last few decades, cancer prevention has largely focused on early detection (screening) or chemoprevention to inhibit, delay, or intercept and reverse carcinogenesis. Although we have enjoyed many notable successes with screening, there are barriers to implementation, associated harms, and inherent limitations to its efficacy (91). For example, colonoscopy reduces the risk of CRC but is not optimal in preventing proximal cancers, particularly tumors with specific molecular subtypes (2). For chemoprevention, the field has largely rigorously tested putative agents through expensive phase III RCTs of generally unselected individuals. However, with the exception of aspirin, this one-size-fits-all approach has yielded few successes. For example, for CRC, RCTs of colon polyp prevention using antioxidants, fiber supplements, folate, calcium/vitamin D, and ω-3 fatty acids have yielded null or inconsistent results (92, 93). These disappointing findings are likely due to incomplete information about the optimal formulation, dose, and duration of treatment as well as lack of selection of participants more likely to benefit. To date, the paradigm of precision medicine, or use of genomic or molecular biomarkers to optimize treatment regimens and predict therapeutic response, which has revolutionized the care of cancer patients, has not been translated to cancer prevention. This is largely due to knowledge gaps regarding the biological mode of action of most preventive agents. As noted by National Institutes of Health Director Francis Collins, understanding the molecular mechanisms underpinning cancer prevention is critical to achieve the promise of “precision prevention” (94).
Precision medicine approaches will be the key to advancing aspirin chemoprevention strategies more broadly. We have previously detailed the basis for a mechanistically informed precision chemoprevention strategy (59); however, additional findings have supported the need to update this paradigm to include additional factors that may contribute to the heterogeneous benefit observed across individuals and that mediate individual risk for colorectal neoplasia.
Modifiable Biomarkers
Identification of biomarkers that will empower individualized approaches to prevention is key to a precision chemoprevention strategy. We recently summarized many of the mechanistically informed biomarkers that need further study to determine their suitability for tailoring personal recommendations (59). These biomarkers were identified through molecular epidemiology methods demonstrating that biological characteristics, including expression of circulating plasma inflammatory markers, transcriptional activity in colorectal mucosa, and levels of urinary metabolites, not only stratify individual risk for CRC but also may predict who is most likely to benefit from aspirin chemoprevention. Ideally, precision prevention approaches may leverage biomarkers that are modifiable, through which surrogate measures of response or efficacy may be made.
One such promising biomarker is urinary PGE-M. Data have strongly supported the use of PGE-M as a marker for increased risk of colorectal neoplasia (95–97) and other types of cancer, including pancreas (98, 99), stomach (100, 101), lung (102), and breast (103, 104), where elevated PGE-M is associated with increased risk. Moreover, there have been suggested signals that PGE-M level may distinguish individuals more likely to benefit from an aspirin chemoprevention regimen (95). The ASPirin Intervention for the REDuction of colorectal cancer risk (ASPIRED) trial, a double-blind placebo-controlled trial, specifically aimed to determine if urinary PGE-M was modifiable by short-term aspirin intervention (105, 106). The trial’s primary outcome, change in urinary PGE-M levels, demonstrated that aspirin at 81 or 325 mg/day taken for 8 weeks significantly reduced high PGE-M levels in individuals with a recent history of colorectal adenoma compared to those taking placebo (106).
Similar results have recently been observed in a naproxen trial among Lynch syndrome patients, who demonstrated significant decreases in PGE-M and mucosal levels of PGE2, with resultant activation of resident immune cell types and changed tissue expression patterns toward epithelial cell differentiation and stem cell regulation associated with naproxen intervention (107). This “immune interception” may be a key mechanism for mounting a preventive response, and additional endpoints from ASPIRED may determine whether this effect extends to aspirin. Similarly, aspirin appears to more potently inhibit CRCs in individuals with higher levels of circulating inflammatory biomarkers MIC-1 (especially for those with PTGS-2-positive tumors) and soluble tumor necrosis factor receptor 2 (sTNFR2). These or other yet-to-be-identified markers of aspirin modulation of immunity may also provide key insights into individual response, but evidence demonstrating that these markers are modifiable by aspirin in the target population at risk has not yet been established. Combined, these studies highlight that inhibition of protumorigenic aberrant prostaglandin synthesis and catabolism and resultant changes to inflammation are likely central to the potential chemopreventive mechanism of aspirin and other NSAIDs, and that flux through this pathway may serve as a reliable marker of biological response and subsequent protection.
In secondary analyses, ASPIRED determined that reduction in urinary PGE-M was significant enough to predict a reduction of 10% of future recurrent advanced adenomas in approximately half of the individuals randomized to aspirin based on previously reported risk thresholds (106). Conversely, the results also highlighted that while aspirin beneficially lowered PGE-M in most individuals, for some individuals higher doses or longer durations of treatment may be required to meet potential target risk reduction thresholds, and that, for a smaller fraction of individuals, aspirin may have very little effect on elevated urinary PGE-M. Nonetheless, this heterogeneous response across the population underscores the potential utility for a modifiable biomarker to guide personal recommendations and provide key information for clinical decision making (Figure 1).
Figure 1.
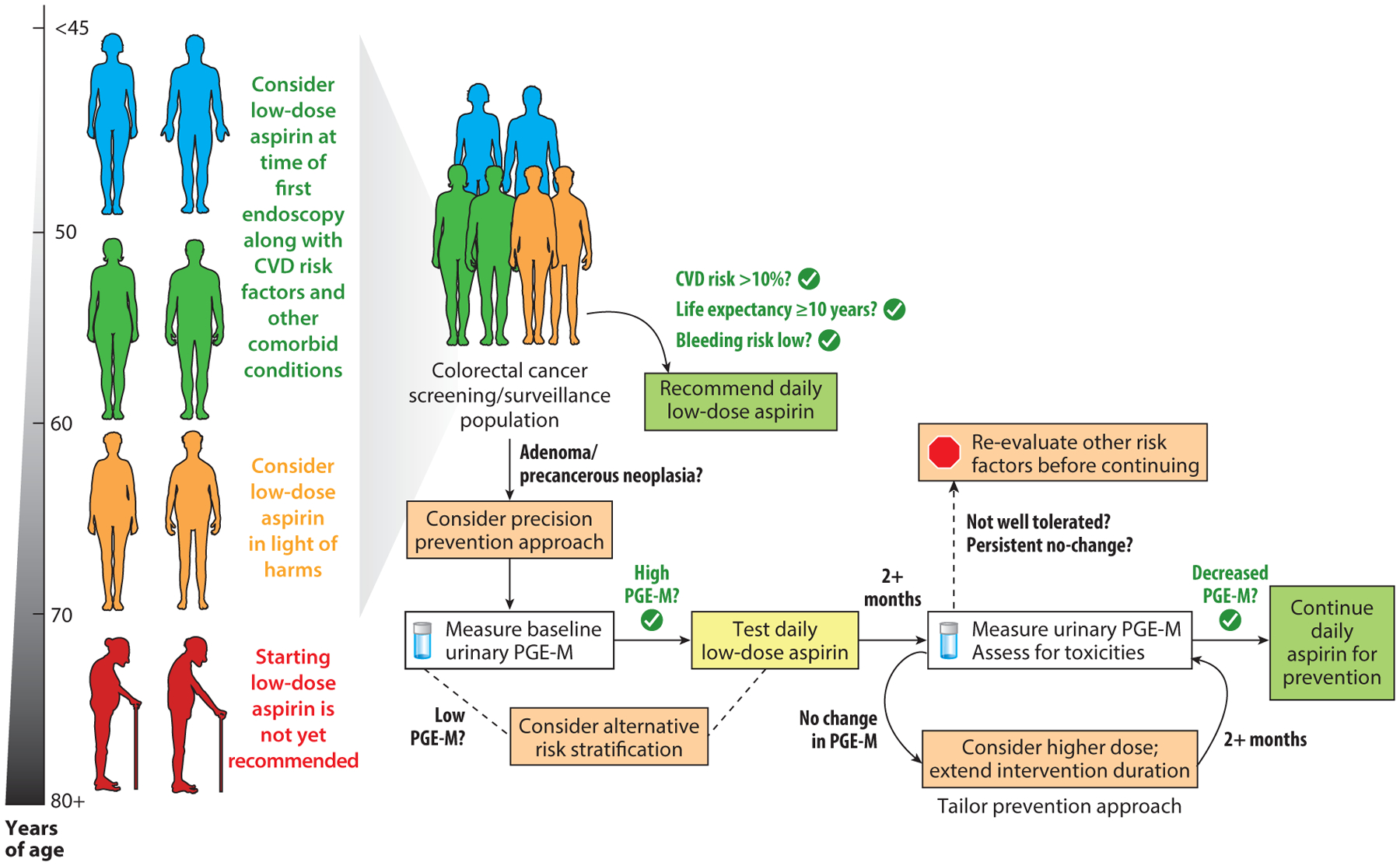
A model for precision chemoprevention in clinical decision making. Physicians may consider timing a discussion of use of aspirin for primary prevention to coincide with an individual’s first screening colonoscopy (recommended between ages 45 and 50). For individuals under age 60, after consideration of cardiovascular disease (CVD) risk factors, life expectancy, and risk for bleeding events, providers may recommend aspirin directly. For individuals over age 60, providers may want to consider colorectal cancer (CRC) risk factors, including the presence of adenomas and baseline levels of potentially modifiable biomarkers for CRC risk. In this example, we use urinary prostaglandin metabolites (PGE-M) as a risk biomarker that has recently been shown to be modifiable as a potentially useful precision tool to monitor a possible aspirin chemoprotective benefit while also monitoring for potential harms; PGE-M can help tailor individual strategies according to aspirin dose, duration, and continuation of use. Precision prevention strategies may be extended to those of advanced age (70 years or older); however, significant considerations of harms is warranted before starting an aspirin prevention regimen.
Emerging Role of the Microbiome
While data support a central role for prostaglandin modulation by aspirin or other NSAIDs in exerting chemoprotection, it is unclear whether aspirin’s influence is based primarily on direct inhibition of PTGS-1/2 in colonic epithelial cells or is exerted via indirect effects (59). Prostaglandins and, specifically, PGE2 have been shown to have a master regulator–like role during periods of inflammation (108, 109), mucosal injury repair (83, 84), and neoplastic development (66, 110–112). Similarly, the gut microbiome, as a key determinant for gut homeostasis, host immune activity (113), and intestinal stem cell proliferation/regeneration (114, 115), has increasingly been implicated in the development of colorectal neoplasia (116, 117). An emerging concept linking these areas is that the gut microbiome influences bile acid pools, which have recently been shown to inhibit farnesoid X receptor and downstream cytosolic phospholipase A2 (cPLA2) function and, thus, impact PGE2-mediated ISC renewal pathways (83, 115), thereby contributing to neoplastic initiation and CRC progression (113, 114).
There are significant interactions between the microbiome and nonantibiotic drugs (118), including nonaspirin NSAIDs (e.g., ibuprofen/naproxen) (119), proton pump inhibitors (120, 121), drugs treating diabetes (122), and atypical antipsychotics (123). Further, preclinical studies support that certain microbes promote a proinflammatory environment, leading to PTGS-2 induction (124–126). Elucidating the connectivity of the host, gut microbiome, cancer, and its chemoprevention is particularly important given the growing evidence linking changes in gut microbial communities with GI disease states (116, 117). Cross-sectional human studies have implicated a number of taxa in GI disease, including CRC, showing an increased abundance of immunomodulatory and tumor-permissive genera and a depletion of microbes that may protect against tumorigenesis (116, 127, 128). Perhaps the most convincing data to date are preclinical data demonstrating that chemoprevention of colon tumors in murine CRC models with intact gut microbiomes was less potent than in those with microbiomes depleted by antibiotics (129). This effect was attributed to the fact that certain aerobic gut microbes appear to degrade aspirin and limit circulating bioavailability of aspirin. However, beyond the effect gut microbes may have on aspirin absorption or metabolism, there was evidence that aspirin could lead to enrichment of putative beneficial commensals and reduction of pathobionts implicated in tumorigenesis. In humans, a small clinical study (n = 50) where healthy volunteers were randomized to aspirin (325 mg) or placebo for 6 weeks confirmed that aspirin intake can influence several microbial taxa as measured by 16S rRNA (130). Together, these data demonstrated that we must consider potential mediating functions of the gut microbiome in aspirin’s efficacy and bioavailability and points to possible sources of observed interindividual efficacy.
The diversity of microbes present in the gut, and the ability to modulate it either through broad-spectrum (i.e., pre-, pro-, or antibiotics) or specifically engineered approaches (131, 132), makes the gut microbiome a rich target for innovation in chemoprevention. Additional data from humans will be required to understand the intersection of aspirin chemoprevention of CRC and precancers. Strategies that target the microbiome as a way to promote capability of or bioavailability of anticancer agents or synergistically inhibit precancerous processes offer novel approaches to prevent tumorigenesis and may play a key role in the next chapter of understanding aspirin’s mode of action.
THE FUTURE FOR ASPIRIN CHEMOPREVENTION
The future for aspirin chemoprevention is bright, with additional studies building upon the existing evidence base. Based on this strong foundation and acceptance of aspirin as a protective agent, research can truly pivot from focusing on establishing a preventive benefit for all individuals to identification of personal characteristics, including biomarkers, to tailor prevention strategies. Further clarification of aspirin’s chemopreventive mechanisms may result in additional modifiable risk biomarkers that may guide patient and provider decisions or reveal novel targets for cancer prevention or interception. In addition, this research, especially efforts focused on disentangling the potentially harmful interaction with age-related effects, will be critical to promote wider adoption of aspirin chemoprevention strategies.
DISCLOSURE STATEMENT
A.T.C. has previously received grant support and consulting fees from Bayer Pharma AG. D.A.D. is supported by grants from the National Institutes of Health (NIH), including the National Institute of Diabetes and Digestive and Kidney Disorders (K01 DK120742) and National Cancer Institute (L30 CA209764). A.T.C. is supported by grants from the NIH/NCI (R35 CA253185; R01 CA137178) and is the Stuart and Suzanne Steele Massachusetts General Hospital Research Scholar. Both authors are also supported by the Aspirin for Cancer Prevention Collaboration (AsCaP) Cancer Research UK Catalyst Award.
LITERATURE CITED
- 1.Chan AT, Arber N, Burn J, et al. 2012. Aspirin in the chemoprevention of colorectal neoplasia: an overview. Cancer Prev. Res 5:164–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishihara R, Lochhead P, Kuchiba A, et al. 2013. Aspirin use and risk of colorectal cancer according to BRAF mutation status. JAMA 309:2563–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sutcliffe P, Connock M, Gurung T, et al. 2013. Aspirin for prophylactic use in the primary prevention of cardiovascular disease and cancer: a systematic review and overview of reviews. Health Technol. Assess 17:1–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan AT, McNeil J. 2018. Aspirin and cancer prevention in the elderly: Where do we go from here? Gastroenterology 156:534–38 [DOI] [PubMed] [Google Scholar]
- 5.Bibbins-Domingo K 2016. Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med 164:836–45 [DOI] [PubMed] [Google Scholar]
- 6.McNeil JJ, Nelson MR, Woods RL, et al. 2018. Effect of aspirin on all-cause mortality in the healthy elderly. N. Engl. J. Med 379:1519–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katona BW, Weiss JM. 2020. Chemoprevention of colorectal cancer. Gastroenterology 158:368–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thun MJ, Jacobs EJ, Patrono C. 2012. The role of aspirin in cancer prevention. Nat. Rev. Clin. Oncol 9:259–67 [DOI] [PubMed] [Google Scholar]
- 9.Dube C, Rostom A, Lewin G, et al. 2007. The use of aspirin for primary prevention of colorectal cancer: a systematic review prepared for the U.S. Preventive Services Task Force. Ann. Intern. Med 146:365–75 [DOI] [PubMed] [Google Scholar]
- 10.Cole BF, Logan RF, Halabi S, et al. 2009. Aspirin for the chemoprevention of colorectal adenomas: meta-analysis of the randomized trials. J. Natl. Cancer Inst 101:256–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chubak J, Kamineni A, Buist DSM, et al. 2015. Aspirin Use for the Prevention of Colorectal Cancer: An Updated Systematic Evidence Review for the U.S. Preventive Services Task Force Evidence Synthesis No. 133, Rep. No. 15-05228-EF-1. Rockville, MD: Agency Healthc. Res. Quality; [PubMed] [Google Scholar]
- 12.Giovannucci E, Rimm EB, Stampfer MJ, et al. 1994. Aspirin use and the risk for colorectal cancer and adenoma in male health professionals. Ann. Intern. Med 121:241–46 [DOI] [PubMed] [Google Scholar]
- 13.Chan AT, Giovannucci EL, Meyerhardt JA, et al. 2005. Long-term use of aspirin and nonsteroidal anti-inflammatory drugs and risk of colorectal cancer. JAMA 294:914–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giovannucci E, Egan KM, Hunter DJ, et al. 1995. Aspirin and the risk of colorectal cancer in women. N. Engl. J. Med 333:609–14 [DOI] [PubMed] [Google Scholar]
- 15.Cook NR, Lee IM, Gaziano JM, et al. 2005. Low-dose aspirin in the primary prevention of cancer: the Women’s Health Study: a randomized controlled trial. JAMA 294:47–55 [DOI] [PubMed] [Google Scholar]
- 16.Cook NR, Lee IM, Zhang SM, et al. 2013. Alternate-day, low-dose aspirin and cancer risk: long-term observational follow-up of a randomized trial. Ann. Intern. Med 159:77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothwell PM, Price JF, Fowkes FGR, et al. 2012. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 379:1602–12 [DOI] [PubMed] [Google Scholar]
- 18.Rothwell PM, Wilson M, Elwin CE, et al. 2010. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 376:1741–50 [DOI] [PubMed] [Google Scholar]
- 19.Petrucci G, Zaccardi F, Giaretta A, et al. 2019. Obesity is associated with impaired responsiveness to once-daily low-dose aspirin and in vivo platelet activation. J. Thromb. Haemost 17:885–95 [DOI] [PubMed] [Google Scholar]
- 20.Rothwell PM, Cook NR, Gaziano JM, et al. 2018. Effects of aspirin on risks of vascular events and cancer according to bodyweight and dose: analysis of individual patient data from randomised trials. Lancet 392:387–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drew DA, Goh G, Mo A, et al. 2016. Colorectal polyp prevention by daily aspirin use is abrogated among active smokers. Cancer Causes Control. 27:93–103 [DOI] [PubMed] [Google Scholar]
- 22.Ishikawa H, Mutoh M, Suzuki S, et al. 2014. The preventive effects of low-dose enteric-coated aspirin tablets on the development of colorectal tumours in Asian patients: a randomised trial. Gut 63:1755–59 [DOI] [PubMed] [Google Scholar]
- 23.Flossmann E, Rothwell PM. 2007. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet 369:1603–13 [DOI] [PubMed] [Google Scholar]
- 24.Burn J, Bishop DT, Mecklin JP, et al. 2008. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N. Engl. J. Med 359:2567–78 [DOI] [PubMed] [Google Scholar]
- 25.Burn J, Gerdes AM, Macrae F, et al. 2011. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 378:2081–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandler RS, Halabi S, Baron JA, et al. 2003. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N. Engl. J. Med 348:883–90 [DOI] [PubMed] [Google Scholar]
- 27.Logan RF, Grainge MJ, Shepherd VC, et al. 2008. Aspirin and folic acid for the prevention of recurrent colorectal adenomas. Gastroenterology 134:29–38 [DOI] [PubMed] [Google Scholar]
- 28.Baron JA, Cole BF, Sandler RS, et al. 2003. A randomized trial of aspirin to prevent colorectal adenomas. N. Engl. J. Med 348:891–99 [DOI] [PubMed] [Google Scholar]
- 29.Hull MA, Sprange K, Hepburn T, et al. 2018. Eicosapentaenoic acid and aspirin, alone and in combination, for the prevention of colorectal adenomas (seAFOod Polyp Prevention Trial): a multicentre, randomised, double-blind, placebo-controlled, 2 × 2 factorial trial. Lancet 392:2583–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeCensi A, Gescher A. 2019. An abstract provides “seAFOod” for thought. Cancer Prev. Res 12:123–24 [DOI] [PubMed] [Google Scholar]
- 31.Burn J, Sheth H, Elliott F, et al. 2020. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet 395:1855–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.NICE (Natl. Inst. Health Care Excellence). 2020. Colorectal cancer. NICE Guideline, January 29 https://www.nice.org.uk/guidance/ng151/resources/colorectal-cancer-pdf-66141835244485 [Google Scholar]
- 33.McNeil JJ, Gibbs P, Orchard SG, et al. 2020. Effect of aspirin on cancer incidence and mortality in older adults. J. Natl. Cancer Inst In press 10.1093/jnci/djaa114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez RF, Tejedor JR, Bayon GF, et al. 2018. Distinct chromatin signatures of DNA hypomethylation in aging and cancer. Aging Cell 17:e12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christensen BC, Houseman EA, Marsit CJ, et al. 2009. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLOS Genet. 5:e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hannum G, Guinney J, Zhao L, et al. 2013. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49:359–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nawa T, Kato J, Kawamoto H, et al. 2008. Differences between right- and left-sided colon cancer in patient characteristics, cancer morphology and histology. J. Gastroenterol. Hepatol 23:418–23 [DOI] [PubMed] [Google Scholar]
- 38.Jess P, Hansen IO, Gamborg M, et al. 2013. A nationwide Danish cohort study challenging the categorisation into right-sided and left-sided colon cancer. BMJ Open. 3:e002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonsalves WI, Mahoney MR, Sargent DJ, et al. 2014. Patient and tumor characteristics and BRAF and KRAS mutations in colon cancer, NCCTG/Alliance N0147. J. Natl. Cancer Inst 106:dju106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ang PW, Loh M, Liem N, et al. 2010. Comprehensive profiling of DNA methylation in colorectal cancer reveals subgroups with distinct clinicopathological and molecular features. BMC Cancer 10:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Serebriiskii IG, Connelly C, Frampton G, et al. 2019. Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat. Commun 10:3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amitay EL, Carr PR, Jansen L, et al. 2019. Association of aspirin and nonsteroidal anti-inflammatory drugs with colorectal cancer risk by molecular subtypes. J. Natl. Cancer Inst 111:475–83 [DOI] [PubMed] [Google Scholar]
- 43.Nishihara R, Lochhead P, Kuchiba A, et al. 2013. Aspirin use and risk of colorectal cancer according to BRAF mutation status. JAMA 309:2563–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Low EE, Demb J, Liu L, et al. 2020. Risk factors for early-onset colorectal cancer. Gastroenterology 159:492–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolf AMD, Fontham ETH, Church TR, et al. 2018. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J. Clin 68:250–81 [DOI] [PubMed] [Google Scholar]
- 46.Sansom OJ, Stark LA, Dunlop MG, Clarke AR. 2001. Suppression of intestinal and mammary neoplasia by lifetime administration of aspirin in ApcMin/+ and ApcMin/+, Msh2−/− mice. Cancer Res. 61:7060–64 [PubMed] [Google Scholar]
- 47.Lanas A, Wu P, Medin J, Mills EJ. 2011. Low doses of acetylsalicylic acid increase risk of gastrointestinal bleeding in a meta-analysis. Clin. Gastroenterol. Hepatol 9:762–68.e6 [DOI] [PubMed] [Google Scholar]
- 48.Mahady SE, Margolis KL, Chan A, et al. 2020. Major GI bleeding in older persons using aspirin: incidence and risk factors in the ASPREE randomised controlled trial. Gut. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Serebruany VL, Steinhubl SR, Berger PB, et al. 2005. Analysis of risk of bleeding complications after different doses of aspirin in 192,036 patients enrolled in 31 randomized controlled trials. Am. J. Cardiol 95:1218–22 [DOI] [PubMed] [Google Scholar]
- 50.Rothwell PM, Wilson M, Price JF, et al. 2012. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 379:1591–601 [DOI] [PubMed] [Google Scholar]
- 51.Whitlock EP WS, Burda BU, Feightner A, Beil T. 2015. Aspirin Use in Adults: Cancer, All-Cause Mortality, and Harms: A Systematic Evidence Review for the U.S. Preventive Services Task Force Evidence Synthesis No. 132, Rep. No. 13-05193-EF-1. Rockville, MD: Agency Healthc. Res. Quality; [PubMed] [Google Scholar]
- 52.Jacobs EJ, Thun MJ, Bain EB, et al. 2007. A large cohort study of long-term daily use of adult-strength aspirin and cancer incidence. J. Natl. Cancer Inst 99:608–15 [DOI] [PubMed] [Google Scholar]
- 53.Cao Y, Nishihara R, Wu K, et al. 2016. Population-wide impact of long-term use of aspirin and the risk for cancer. JAMA Oncol. 2:762–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simon TG, Ma Y, Ludvigsson JF, et al. 2018. Association between aspirin use and risk of hepatocellular carcinoma. JAMA Oncol. 4:1683–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rothwell PM, Price JF, Fowkes FG, et al. 2012. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 379:1602–12 [DOI] [PubMed] [Google Scholar]
- 56.Rothwell PM, Fowkes FG, Belch JF, et al. 2011. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 377:31–41 [DOI] [PubMed] [Google Scholar]
- 57.Loomans-Kropp HA, Pinsky P, Cao Y, Chan AT, Umar A. 2019. Association of aspirin use with mortality risk among older adult participants in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. In JAMA Netw. Open 2:e1916729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simon TG, Duberg AS, Aleman S, et al. 2020. Association of aspirin with hepatocellular carcinoma and liver-related mortality. N. Engl. J. Med 382:1018–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Drew DA, Cao Y, Chan AT. 2016. Aspirin and colorectal cancer: the promise of precision chemoprevention. Nat. Rev. Cancer 16:173–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan AT, Ogino S, Fuchs CS. 2007. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N. Engl. J. Med 356:2131–42 [DOI] [PubMed] [Google Scholar]
- 61.Fink SP, Yamauchi M, Nishihara R, et al. 2014. Aspirin and the risk of colorectal cancer in relation to the expression of 15-hydroxyprostaglandin dehydrogenase (HPGD). Sci. Transl. Med 6:233re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bezawada N, Song M, Wu K, et al. 2014. Urinary PGE-M levels are associated with risk of colorectal adenomas and chemopreventive response to anti-inflammatory drugs. Cancer Prev. Res 7:758–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fedirko V, Bradshaw PT, Figueiredo JC, et al. 2015. Urinary metabolites of prostanoids and risk of recurrent colorectal adenomas in the Aspirin/Folate Polyp Prevention Study (AFPPS). Cancer Prev. Res 8:1061–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franceschi C, Campisi J. 2014. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci 69(Suppl. 1):S4–S9 [DOI] [PubMed] [Google Scholar]
- 65.Franceschi C, Zaikin A, Gordleeva S, et al. 2018. Inflammaging 2018: an update and a model. Semin. Immunol 40:1–5 [DOI] [PubMed] [Google Scholar]
- 66.Wang D, DuBois RN. 2018. Role of prostanoids in gastrointestinal cancer. J. Clin. Investig 128:2732–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nusse YM, Savage AK, Marangoni P, et al. 2018. Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature 559:109–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yui S, Azzolin L, Maimets M, et al. 2018. YAP/TAZ-dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem. Cell 22:35–49.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roulis M, Kaklamanos A, Schernthanner M, et al. 2020. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature 580:524–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Y, Chiang IL, Ohara TE, et al. 2019. Long-term culture captures injury-repair cycles of colonic stem cells. Cell 179:1144–59.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qiu W, Wang X, Leibowitz B, et al. 2010. Chemoprevention by nonsteroidal anti-inflammatory drugs eliminates oncogenic intestinal stem cells via SMAC-dependent apoptosis. PNAS 107:20027–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moon CM, Kwon JH, Kim JS, et al. 2014. Nonsteroidal anti-inflammatory drugs suppress cancer stem cells via inhibiting PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in colorectal cancer. Int. J. Cancer 134:519–29 [DOI] [PubMed] [Google Scholar]
- 73.Mihaylova MM, Cheng CW, Cao AQ, et al. 2018. Fasting activates fatty acid oxidation to enhance intestinal stem cell function during homeostasis and aging. Cell Stem. Cell 22:769–78.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pentinmikko N, Iqbal S, Mana M, et al. 2019. Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nature 571:398–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Balducci L, Ershler WB. 2005. Cancer and ageing: a nexus at several levels. Nat. Rev. Cancer 5:655–62 [DOI] [PubMed] [Google Scholar]
- 76.Blokzijl F, de Ligt J, Jager M, et al. 2016. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538:260–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tomasetti C, Vogelstein B. 2015. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347:78–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tomasetti C, Li L, Vogelstein B. 2017. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355:1330–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ershler WB, Stewart JA, Hacker MP, et al. 1984. B16 murine melanoma and aging: slower growth and longer survival in old mice. J. Natl. Cancer Inst 72:161–64 [DOI] [PubMed] [Google Scholar]
- 80.Signer RA, Montecino-Rodriguez E, Witte ON, et al. 2007. Age-related defects in B lymphopoiesis underlie the myeloid dominance of adult leukemia. Blood 110:1831–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ishikawa TO, Herschman HR. 2006. Conditional knockout mouse for tissue-specific disruption of the cyclooxygenase-2 (Cox-2) gene. Genesis 44:143–49 [DOI] [PubMed] [Google Scholar]
- 82.Manieri NA, Mack MR, Himmelrich MD, et al. 2015. Mucosally transplanted mesenchymal stem cells stimulate intestinal healing by promoting angiogenesis. J. Clin. Invest 125:3606–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jain U, Lai CW, Xiong S, et al. 2018. Temporal regulation of the bacterial metabolite deoxycholate during colonic repair is critical for crypt regeneration. Cell Host Microbe 24:353–63.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miyoshi H, VanDussen KL, Malvin NP, et al. 2017. Prostaglandin E2 promotes intestinal repair through an adaptive cellular response of the epithelium. EMBO J. 36:5–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Montrose DC, Nakanishi M, Murphy RC, et al. 2015. The role of PGE2 in intestinal inflammation and tumorigenesis. Prostaglandins Other Lipid Mediat. 116–117:26–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Silla IO, Rueda D, Rodriguez Y, et al. 2014. Early-onset colorectal cancer: a separate subset of colorectal cancer. World J. Gastroenterol 20:17288–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ruschoff J, Wallinger S, Dietmaier W, et al. 1998. Aspirin suppresses the mutator phenotype associated with hereditary nonpolyposis colorectal cancer by genetic selection. PNAS 95:11301–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Perea J, Rueda D, Canal A, et al. 2014. Age at onset should be a major criterion for subclassification of colorectal cancer. J. Mol. Diagn 16:116–26 [DOI] [PubMed] [Google Scholar]
- 89.Giraldez MD, Lopez-Doriga A, Bujanda L, et al. 2012. Susceptibility genetic variants associated with early-onset colorectal cancer. Carcinogenesis 33:613–19 [DOI] [PubMed] [Google Scholar]
- 90.Goel A, Nagasaka T, Spiegel J, et al. 2010. Low frequency of Lynch syndrome among young patients with non-familial colorectal cancer. Clin. Gastroenterol. Hepatol 8:966–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Loomans-Kropp HA, Umar A. 2019. Cancer prevention and screening: the next step in the era of precision medicine. NPJ Precis. Oncol 3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chan AT, Giovannucci EL. 2010. Primary prevention of colorectal cancer. Gastroenterology 138:2029–43.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Song M, Garrett WS, Chan AT. 2015. Nutrients, foods, and colorectal cancer prevention. Gastroenterology 148:1244–60.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Collins F 2015. Precision medicine: who benefits from aspirin to prevent colorectal cancer. NIH Director’s Blog, March 24 https://directorsblog.nih.gov/2015/03/24/precision-medicine-who-benefits-from-aspirin-to-prevent-colorectal-cancer/ [Google Scholar]
- 95.Mehta RS, Song M, Bezawada N, et al. 2014. A prospective study of macrophage inhibitory cytokine-1 (MIC-1/GDF15) and risk of colorectal cancer. J. Natl. Cancer Inst 106:dju016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cai Q, Gao YT, Chow WH, et al. 2006. Prospective study of urinary prostaglandin E2 metabolite and-colorectal cancer risk. J. Clin. Oncol 24:5010–16 [DOI] [PubMed] [Google Scholar]
- 97.Johnson JC, Schmidt CR, Shrubsole MJ, et al. 2006. Urine PGE-M: a metabolite of prostaglandin E2 as a potential biomarker of advanced colorectal neoplasia. Clin. Gastroenterol. Hepatol 4:1358–65 [DOI] [PubMed] [Google Scholar]
- 98.Cui Y, Shu XO, Li HL, et al. 2017. Prospective study of urinary prostaglandin E2 metabolite and pancreatic cancer risk. Int. J. Cancer 141:2423–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao J, Wang J, Du J, et al. 2015. Urinary prostaglandin E2 metabolite and pancreatic cancer risk: case-control study in urban Shanghai. PLOS ONE 10:e0118004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dong LM, Shu XO, Gao YT, et al. 2009. Urinary prostaglandin E2 metabolite and gastric cancer risk in the Shanghai women’s health study. Cancer Epidemiol. Biomarkers. Prev 18:3075–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang T, Cai H, Zheng W, et al. 2017. A prospective study of urinary prostaglandin E2 metabolite, Helicobacter pylori antibodies, and gastric cancer risk. Clin. Infect. Dis 64:1380–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Murphey LJ, Williams MK, Sanchez SC, et al. 2004. Quantification of the major urinary metabolite of PGE2 by a liquid chromatographic/mass spectrometric assay: determination of cyclooxygenase-specific PGE2 synthesis in healthy humans and those with lung cancer. Anal. Biochem 334:266–75 [DOI] [PubMed] [Google Scholar]
- 103.Cui Y, Shu XO, Gao YT, et al. 2014. Urinary prostaglandin E2 metabolite and breast cancer risk. Cancer Epidemiol. Biomarkers. Prev 23:2866–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim S, Taylor JA, Milne GL, Sandler DP. 2013. Association between urinary prostaglandin E2 metabolite and breast cancer risk: a prospective, case-cohort study of postmenopausal women. Cancer Prev. Res 6:511–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Drew DA, Chin SM, Gilpin KK, et al. 2017. ASPirin Intervention for the REDuction of colorectal cancer risk (ASPIRED): a study protocol for a randomized controlled trial. Trials 18:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Drew DA, Schuck MM, Magicheva-Gupta MV, et al. 2020. Effect of low-dose and standard-dose aspirin on PGE2 biosynthesis among individuals with colorectal adenomas: a randomized clinical trial. Cancer Prev. Res In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reyes-Uribe L, Wu W, Gelincik O, et al. 2020. Naproxen chemoprevention promotes immune activation in Lynch syndrome colorectal mucosa. Gut. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ricciotti E, FitzGerald GA. 2011. Prostaglandins and inflammation. Arterioscler Thromb. Vasc. Biol 31:986–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kalinski P 2012. Regulation of immune responses by prostaglandin E2. J. Immunol 188:21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Roberts HR, Smartt HJ, Greenhough A, et al. 2011. Colon tumour cells increase PGE2 by regulating COX-2 and 15-PGDH to promote survival during the microenvironmental stress of glucose deprivation. Carcinogenesis 32:1741–47 [DOI] [PubMed] [Google Scholar]
- 111.Wang D, Fu L, Sun H, et al. 2015. Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology 149:1884–95.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bellamkonda K, Chandrashekar NK, Osman J, et al. 2016. The eicosanoids leukotriene D4 and prostaglandin E2 promote the tumorigenicity of colon cancer-initiating cells in a xenograft mouse model. BMC Cancer 16:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jia W, Xie G, Jia W. 2018. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol 15:111–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fu T, Coulter S, Yoshihara E, et al. 2019. FXR regulates intestinal cancer stem cell proliferation. Cell 176:1098–112.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kaiko GE, Ryu SH, Koues OI, et al. 2016. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell 165:1708–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ahn J, Sinha R, Pei Z, et al. 2013. Human gut microbiome and risk for colorectal cancer. J. Natl. Cancer Inst 105:1907–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brennan CA, Garrett WS. 2016. Gut microbiota, inflammation, and colorectal cancer. Annu. Rev. Microbiol 70:395–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maier L, Pruteanu M, Kuhn M, et al. 2018. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555:623–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rogers MAM, Aronoff DM. 2016. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin. Microbiol. Infect 22:178.e1–e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Imhann F, Bonder MJ, Vich Vila A, et al. 2016. Proton pump inhibitors affect the gut microbiome. Gut 65:740–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jackson MA, Goodrich JK, Maxan ME, et al. 2016. Proton pump inhibitors alter the composition of the gut microbiota. Gut 65:749–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Forslund K, Hildebrand F, Nielsen T, et al. 2015. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528:262–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Flowers SA, Evans SJ, Ward KM, et al. 2017. Interaction between atypical antipsychotics and the gut microbiome in a bipolar disease cohort. Pharmacotherapy 37:261–67 [DOI] [PubMed] [Google Scholar]
- 124.Biarc J, Nguyen IS, Pini A, et al. 2004. Carcinogenic properties of proteins with pro-inflammatory activity from Streptococcus infantarius (formerly S. bovis). Carcinogenesis 25:1477–84 [DOI] [PubMed] [Google Scholar]
- 125.Ellmerich S, Scholler M, Duranton B, et al. 2000. Promotion of intestinal carcinogenesis by Streptococcus bovis. Carcinogenesis 21:753–56 [DOI] [PubMed] [Google Scholar]
- 126.Wang X, Huycke MM. 2007. Extracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology 132:551–61 [DOI] [PubMed] [Google Scholar]
- 127.Kostic AD, Gevers D, Pedamallu CS, et al. 2012. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22:292–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhu Q, Gao R, Wu W, Qin H. 2013. The role of gut microbiota in the pathogenesis of colorectal cancer. Tumour Biol. 34:1285–300 [DOI] [PubMed] [Google Scholar]
- 129.Zhao R, Coker OO, Wu J, et al. 2020. Aspirin reduces colorectal tumor development in mice and gut microbes reduce its bioavailability and chemopreventive effects. Gastroenterology 159(3):969–83 [DOI] [PubMed] [Google Scholar]
- 130.Prizment AE, Staley C, Onyeaghala GC, et al. 2020. Randomised clinical study: oral aspirin 325 mg daily vs placebo alters gut microbial composition and bacterial taxa associated with colorectal cancer risk. Aliment. Pharmacol. Ther In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Drew DA, Chan AT. 2018. Towards a cancer-chemopreventive diet. Nat. Biomed. Eng 2:6–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ho CL, Tan HQ, Chua KJ, et al. 2018. Engineered commensal microbes for diet-mediated colorectal-cancer chemoprevention. Nat. Biomed. Eng 2:27–37 [DOI] [PubMed] [Google Scholar]