

Abstract
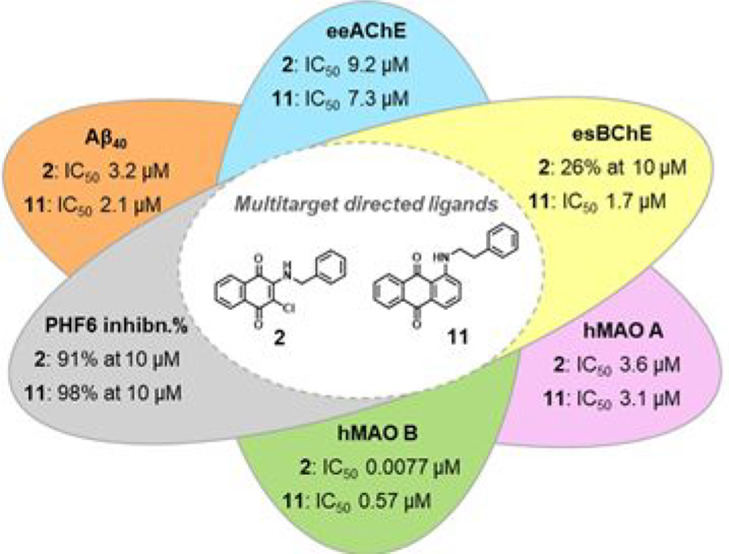
Two series of naphthoquinone and anthraquinone derivatives decorated with an aromatic/heteroaromatic chain have been synthesized and evaluated as potential promiscuous agents capable of targeting different factors playing a key role in Alzheimer’s disease (AD) pathogenesis. On the basis of the in vitro biological profiling, most of them exhibited a significant ability to inhibit amyloid aggregation, PHF6 tau sequence aggregation, acetylcholinesterase (AChE), and monoamine oxidase (MAO) B. In particular, naphthoquinone 2 resulted as one of the best performing multitarget-directed ligand (MTDL) experiencing a high potency profile in inhibiting β-amyloid (Aβ40) aggregation (IC50 = 3.2 μM), PHF6 tau fragment (91% at 10 μM), AChE enzyme (IC50 = 9.2 μM) jointly with a remarkable inhibitory activity against MAO B (IC50 = 7.7 nM). Molecular modeling studies explained the structure–activity relationship (SAR) around the binding modes of representative compound 2 in complex with hMAO B and hAChE enzymes, revealing inhibitor/protein key contacts and the likely molecular rationale for enzyme selectivity. Compound 2 was also demonstrated to be a strong inhibitor of Aβ42 aggregation, with potency comparable to quercetin. Accordingly, atomic force microscopy (AFM) revealed that the most promising naphthoquinones 2 and 5 and anthraquinones 11 and 12 were able to impair Aβ42 fibrillation, deconstructing the morphologies of its fibrillar aggregates. Moreover, the same compounds exerted a moderate neuroprotective effect against Aβ42 toxicity in primary cultures of cerebellar granule cells. Therefore, our findings demonstrate that these molecules may represent valuable chemotypes toward the development of promising candidates for AD therapy.
Keywords: Naphthoquinone and anthraquinone derivatives, multitarget-directed ligands (MTDLs), Aβ and Tau aggregation inhibition, AChE and BChE inhibition, MAO inhibition
1. Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder, mainly characterized by the loss of function and death of neurons in different areas of the brain. In spite of the high clinical and social impact owing to its global prevalence as the most common form of dementia, AD etiology still has to be clearly understood. Several factors concur to AD onset and progression, thus playing a pivotal role in the disease pathogenesis. The dysfunction of the basal acetylcholine (ACh) forebrain signaling,1 deposits of β-amyloid (Aβ)2 and tau-mediated neurofibrillary tangles (NFTs),3 and neuroinflammation are the major hallmarks of AD. Soluble Aβ oligomers in concert with hyperphosphorylated tau serve as the main pathogenic contributors of neurodegeneration in AD, acting in a synergistic fashion to cause cell death and neurotransmitter deficits.4,5 Also oxidative stress and free radical formation,6 metal dyshomeostasis,7 and neuroinflammatory processes8 are pathological components of the disease. The range of targets in AD is increasing, and for the most part, enzymes have been recognized as crucial contributors to AD onset and progression.9 Consequently, a number of molecules have entered clinical phase study with their targets, such as BACE1, phosphodiesterase, phospholipase A2, MAPK, and SIRT1, as examples (clinicaltrials.gov).
Bulk of evidence supports the AD multifactorial nature as a result of an intricate network of neurochemical factors that need to be simultaneously modulated to strive for a better disease outcome. In fact, the marketed anti-Alzheimer drugs, namely, the acetylcholinesterase (AChE) inhibitors and the NMDA receptor antagonist memantine, are regarded as merely symptomatic, since they work against a single target, respectively modulating the cholinergic or glutamatergic function.10 This scenario has drawn greater attention on the multitarget-directed ligands (MTDL) strategy as these molecules may exhibit significant potential on the road to therapeutics for AD.5,11−13
In this regard, the literature contains many examples of naphthoquinone and anthraquinone compounds from natural sources or synthetic as endowed with promising properties against diverse AD targets. Successfully, hybrid- and fragment-based drug design strategies allowed the yield of multifunctional molecules, also based on naphthoquinone14−16 and anthraquinone15,17,18 scaffolds which elicited antioxidant activity, AChE inhibition, BACE inhibition, inhibition of Aβ, and tau aggregation.
Previously we investigated a library of thioxanthen-9-one, xanthen-9-one, naphthoquinone (I–VIII) and anthraquinone derivatives (IX–XIV) decorated with a basic side chain (especially quinolizidinylalkyl chains), which displayed a multitarget behavior by inhibiting both AChE and BChE, and the spontaneous aggregation of β-amyloid with similar potencies16 (Figure 1). Naphthoquinones were dual AChE-preferring inhibitors (IC50 = 0.011–5.8 μM), while anthraquinones were equipotent toward both enzymes in the low micromolar range. On the contrary the tricyclic anthraquinone system was more suited to promoting the inhibition of Aβ aggregation (mean IC50 ∼ 8 μM) than smaller naphthoquinones, which were generally endowed with less efficacy.
Figure 1.
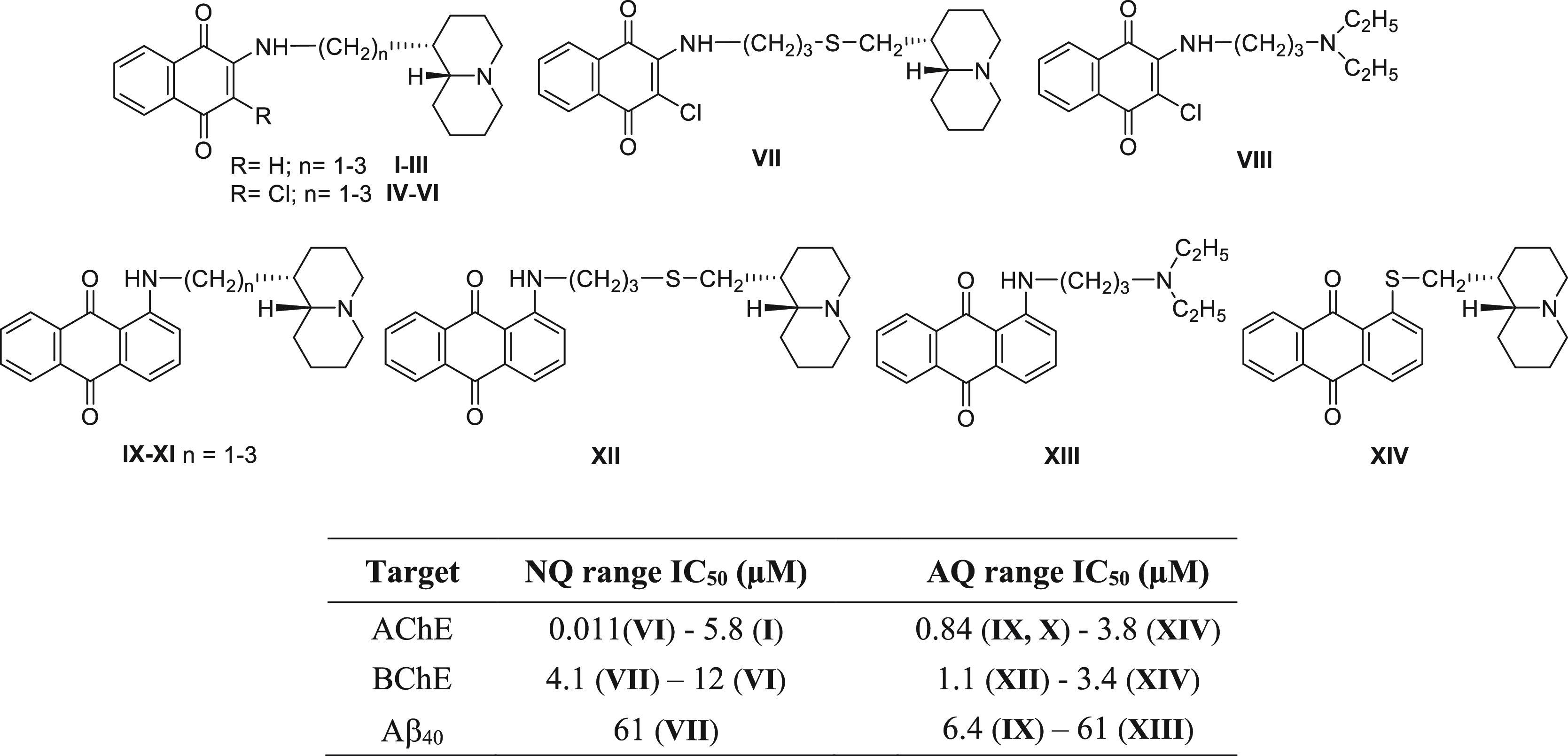
First series of the naphthoquinone and anthraquinone derivatives, bearing a dialkylaminoalkyl or a quinolizidinylalkyl chain, as multitarget agents inhibiting cholinesterases and β-amyloid aggregation: NQ = naphthoquinone; AQ = anthraquinone.
2. Results and Discussion
2.1. Design
This report details our investigations to probe new additional substitutions on the naphthoquinone and anthraquinone scaffolds that have been tethered to an aromatic or heteroaromatic ring through a polymethylene chain, with a view to gaining a better understanding of their potential multitarget (promiscuity) profiles for AD (Figure 2). These novel hydrophobic features have been included with the aim of favoring suited hydrophobic interactions with a sequence of aromatic amino acids (H14QKLVFF20) of Aβ, which plays an important role in the initial phases of molecular recognition and structural transition and leads to Aβ aggregation in soluble oligomers and fibrillary species.14,19,20 AChE continues to be a crucial target for AD therapy because of its noncholinergic functions as demonstrated by its chaperone role in β-amyloid toxicity,21 thus promoting the multitarget approach in order to hit at least Aβ aggregation and AChE activity.22 Besides AChE, BChE is another target of interest in the search for anti-Alzheimer drugs, as this enzyme exhibits a compensatory effect in response to a greatly decreased AChE activity in the central nervous system (CNS) during AD progression.23 Additionally, excessive monoamine oxidase (MAO) activity contributes to neurodegeneration in AD, inducing Aβ fibrillogenesis,24 imbalance of cholinergic, glutamatergic, and noradrenergic functions,25 and oxidative stress.26 Accordingly, the design of MAO inhibitors, or even better multipotent MAO and ChEs inhibitors, rapidly increased with a view to improving cognitive deficits and memory.27−29 Shikonin and acetylshikonin, characterized by the 5,8-dihydroxy-1,4-naphthoquinone scaffold (naphthazarin), were shown to be nonselective and reversible MAO inhibitors endowed with activity in the micromolar range.30,31 The unsaturated and lipophilic nature of their prenyl-like side chain resembled the hydrophobic features of substituents decorating the present quinone-based compounds (Figure 2), thus prompting us to also explore their potential as MAO inhibitors.
Figure 2.

Structures of the investigated naphthoquinone and anthraquinone derivatives 1–14 as MTDLs for AD.
On the whole, in terms of targets selection, we move from amyloid and ChEs as relevant players in AD pathology to other targets with more credentials for a disease-modifying effect, such as tau and MAO B.32
Herein, the biological profiling included the in vitro evaluation of (i) self-induced Aβ and tau aggregation inhibition, (ii) the inhibitory activities against AChE and BChE, and (iii) inhibition of MAO A and B isoforms. Atomic force microscopy (AFM) was then employed to assess the ability of the best performing Aβ40 inhibitors (2, 5, 11, and 12) to impair Aβ42 fibrillation and revealed clear differences between Aβ42 aggregate morphologies obtained in the presence or absence of the compounds. The same compounds have been assayed for their protective effect against Aβ42 toxicity in primary cultures of cerebellar granule cells from postnatal rats (P7).
2.2. Chemistry
Compounds 1, 3,332,345, 9,35 and 8(36) have been achieved according to the cited references. For compound 10(37) we applied a different synthetic route with respect to the literature; thus its experimental properties have been reported herein as follows. Naphthoquinones 4, 6, and 7 were obtained by reacting the 2,3-dichloro-1,4-naphthoquinone, previously suspended in ethanol or methanol, with 2 equiv of the proper amine (Schemes 1 and 2).
Scheme 1.

Reagents and conditions: (a) EtOH, Δ, 4 h.
Scheme 2.

Reagents and conditions: (a) LiAlH4 /anhydrous THF, Δ, 10 h; (b) MeOH, rt, 24 h.
The condensation at 160 °C of a mixture of 1-chloroanthraquinone with the proper aryl/heteroaryl alkylamine has given compounds 10–14 (Schemes 3 and 4).
Scheme 3.

Reagents and conditions: (a) 160 °C, 6 h.
Scheme 4.

Reagents and conditions: (a) LiAlH4/anhydrous THF, Δ, 8 h; (b) 160 °C, 6 h.
The intermediate (heteroaryl)ethylamines required for the synthesis of the indoles 5 and 13, benzimidazole 6, and benzotriazoles 7 and 14 were prepared by reduction with LiAlH4 of 2-(3-indolyl)acetamide, 1-benzimidazolyl, and 1-benzotriazolyl acetonitriles, respectively (Schemes 4 and 2). While 2-(3-indolyl)acetamide was commercially available, to synthesize 2-(benzimidazol-1-yl) and 2-(benzotriazol-1-yl) acetonitriles, benzimidazole and benzotriazole were condensed with 2-chloroacetonitrile in DMF in the presence of TEA according to the literature.38
The structures of the novel compounds have been confirmed using 1H and 13C NMR and elemental analysis. The purity of compounds (checked by elemental analysis) has been in all cases >95%.
2.3. In Silico and in Vitro Blood–Brain Barrier (BBB) Permeation
The use of naphthoquinone and anthraquinone-based derivatives as potential anti-AD agents requires their ability to enter the CNS. Accordingly, we tested in silico the propensity to cross the BBB by passive diffusion, calculating the LogPS values39 for the new compounds in comparison to the drugs donepezil, quercetin, safinamide, and the quinolizidine-containing naphthoquinone VI (Table 1, second column), whose BBB permeability profile was experimentally confirmed.16
Table 1. In Silico and in Vitro Evaluation of Naphthoquinone and Anthraquinone-Based Derivatives’ Propensity to Cross BBBc.

Rate of brain penetration, −3 (medium affinity) < table < −1 (high affinity). PS represents permeability–surface area product and is derived from the kinetic equation of capillary transport.
CNS permeation prediction based on the PAMPA-BBB classification range from Di et al.40
Measured as LogPS (ACD/Percepta Platform 2015 v14.0.0, https://www.acdlabs.com/). The last two columns report the experimental permeability results from the PAMPA-BBB assay (Pe, 10–6 cm/s) and the corresponding predictive penetration in the CNS for compounds 2, 5, 8, 11, 12 and for donepezil and quercetin as positive (CNS+) and negative (CNS−) controls, respectively.
The results, listed in Table 1, predicted that all the compounds are characterized by a brain-penetration ability comparable to the reference compounds and sufficient to obtain pharmacologically relevant concentrations within the CNS. As expected, the capability of amino acids naphthoquinones 8 and 9 to penetrate the BBB was poor, as LogPS values fell close to the lower limit of the recommended ranges (−3 < LogPS < −1), even if they could presumably reach the CNS by carrier-mediated active transport. The in vitro permeability (Pe) of compounds 2, 5, 8, 11, and 12 was also determined using the PAMPA-BBB assays40 (Table 1, third column). Successful assay validation was performed by comparing the experimental permeability (Pe,exp) with the corresponding reported values (Pe,rep) for 20 commercial drugs (Pe,exp = 0.9625Pe,rep + 0.2836, R2 = 0.9622, Figure S1 and Table S1). On the basis of the Pe values obtained, all analyzed compounds should be endowed with good-to-high BBB passive permeability, with Pe values ranging from 6.1 to 28.4 × 10–6 cm/s (Table 1, last two columns).
2.4. Inhibition of Self-Induced Aβ Aggregation and Cholinesterases
In vitro inhibition of Aβ40 aggregation was assessed following a previously reported thioflavin T (ThT) fluorescence-based method involving the use of hexafluoroisopropanol (HFIP) as aggregation enhancer. For the most active compounds (≥80% Aβ aggregation inhibition) IC50 values were determined under the same assay conditions as previously described.41 Inhibitory activities on AChE from electric eel (eeAChE) and BChE from equine serum (esBChE) were determined by the spectrophotometric method of Ellman42 and are reported in Table 2 as IC50 (μM) for the most active compounds or as percentage of inhibition at 10 μM for low active (i.e., <50%) compounds.
Table 2. Inhibitory Activitiesa (μM) of the Investigated Compounds 1–14 against Aβ Aggregation, ChEs, and MAOs.
| IC50 (μM) or (% inhibition)b |
|||||
|---|---|---|---|---|---|
| compd | Aβ40 aggr | eeAChE | esBChE | hMAO A | hMAO B |
| 1 | (63 ± 3)c | (39 ± 1) | (8 ± 3) | (<5) | (22 ± 4) |
| 2 | 3.2 ± 0.8 | 9.2 ± 0.6 | (26 ± 3) | 3.6 ± 0.3 | 0.0077 ± 0.0013 |
| 3 | 4.4 ± 0.3 | 7.9 ± 0.8 | (24 ± 1) | (41 ± 3) | 0.031 ± 0.001 |
| 4 | 8.2 ± 0.3 | 3.5 ± 0.3 | (35 ± 3) | 3.0 ± 0.2 | 0.054 ± 0.001 |
| 5 | 6.6 ± 0.1 | 8.7 ± 0.8 | (29 ± 4) | 5.0 ± 0.2 | 0.11 ± 0.01 |
| 6 | 19 ± 4 | 6.8 ± 0.7 | (24 ± 4) | (23 ± 4) | (24 ± 6) |
| 7 | 8.7 ± 0.4 | 1.7 ± 0.1 | (15 ± 3) | (21 ± 6) | 0.48 ± 0.08 |
| 8 | 17 ± 2 | 13 ± 2 | (14 ± 3) | (26 ± 6) | 2.7 ± 0.8 |
| 9 | 14 ± 2 | 11 ± 1 | (17 ± 1) | (22 ± 2) | (52 ± 3) |
| 10 | (62 ± 2)c | 8.7 ± 0.5 | (24 ± 3) | 1.1 ± 0.3 | (34 ± 3) |
| 11 | 2.1 ± 0.2 | 7.3 ± 0.7 | 1.7 ± 0.3 | 3.1 ± 0.3 | 0.57 ± 0.02 |
| 12 | 1.9 ± 0.3 | 7.84 ± 0.03 | 8.2 ± 0.4 | (50 ± 3) | 0.24 ± 0.05 |
| 13 | (57 ± 5)c | 1.85 ± 0.04 | (17 ± 1) | (25 ± 5) | (42 ± 1) |
| 14 | 11 ± 2 | 8.1 ± 0.5 | 3.4 ± 0.2 | (53 ± 3) | 0.98 ± 0.14 |
| quercetin | 0.82 ± 0.07 | ||||
| donepezil | 0.021 ± 0.002 | 2.3 ± 0.1 | |||
| safinamide | (18 ± 3) | 0.031 ± 0.001 | |||
Data are the mean ± SEM of n = 3 experiments.
Data in parentheses correspond to % of inhibition at 10 μM, or
100 μM for inhibition of Aβ40 aggregation.
At a concentration of 100 μM, all the compounds 1–14 were proved to inhibit Aβ aggregation, mostly showing IC50 values in the range of 1.9–19 μM. Among them, eight (2–9) and three (11, 12, and 14) were naphthoquinone and anthraquinone derivatives, respectively. The compounds 1, 10, and 13 exhibited an Aβ inhibition rate of about 60% at a concentration of 100 μM. The size of the aromatic bi- and tricyclic system seems to influence the biological effect, as the most potent compounds (12 and 11) sharing IC50 values equal to 1.9 and 2.1 μM, respectively, belong to the anthraquinone series. Previous studies have demonstrated the capability of (hetero)aromatic tricyclic systems to establish stronger interactions (hydrophobic and electrostatic) with the amino acid sequence H14QKLVFF20 of Aβ, which is more prone to aggregating.43 The length of spacer between the quinone scaffolds and the (hetero)aromatic rings influenced the biological activity with a different trend in the two series; that is, one methylene unit was found as the optimal distance for naphthoquinone-based derivatives (compare aryl derivatives 2–4), while for anthraquinones the increase of the carbon units from 1 to 3 (compare derivatives 10–12) resulted in a proportional increase of the inhibition potency against Aβ aggregation. Regarding the naphthoquinones linked to a heterocyclic skeleton, the activities of the indole (5, IC50 = 6.6 μM) and benzotriazole (7, IC50 = 8.7 μM) derivatives were comparable and about 2-fold higher than that of benzimidazole derivative (6). Consequently, the presence of a hydrogen bond donor group such as the NH group of indole ring rather than a hydrogen bond acceptor feature as experienced by the N(2) atom of the benzotriazole ring was permitted, allowing the yield of the same degree of activity. In silico studies previously highlighted the marginal role of NH indole in the tryptophan derivative 9, since its N-methylation did not affect the binding affinity to amyloid oligomers, while its NH group on naphthoquinone C(2) and its CO group of carboxylic function were reported as essential features for the inhibition of Aβ aggregation.44 Our experimental data, however, pointed out that compound 5, lacking in the COOH function, was 2-fold more effective than the amino acid analogue 9 such as Aβ aggregation inhibitor. The same potencies trend was observed for analogues 3 and 8, whereas the presence of the polar carboxylic group in the phenylalanine derivative 8 negatively impaired the activity. Our results corroborate the suitability of naphthoquinone and anthraquinone scaffolds in providing Aβ peptide inhibition, even more when properly substituted with hydrophobic moieties able to target the aromatic interactions occurring in the amyloid self-assembly process. Such behavior was confirmed by AFM studies, as shown below.
By comparison of compounds 1–14 with our first series of naphthoquinone and anthraquinones bearing a basic side chain (Figure 1), in the case of naphthoquinones the activity improved with 8 out of 9 compounds able to target Aβ peptide, while the anthraquinones exhibited only a slight increase of the potency but with a reduced number of active compounds. Therefore, the more apolar nature of the side chain (aromatic/heteroaromatic moiety), in place of polar basic groups, emerged as a relevant factor in enhancing the intrinsic capability of naphthoquinone and anthraquinone scaffolds to target Aβ fibrillation.
Apart from the phenylamino naphthoquinone 1, all the compounds have also been proven to inhibit AChE reaching a low micromolar potency range. Conversely, BChE has been found to be less sensitive to these series of compounds except for the anthraquinone derivatives 11, 12, and 14 whose IC50 ranged from 1.7 to 8.2 μM. The most potent and selective AChE inhibitors were the benzotriazole-based naphthoquinone 7 (IC50 = 1.7 μM) and the indole-based anthraquinone 13 (IC50 = 1.85 μM), while the anthraquinone derivatives 11 and 12 behaved as dual cholinesterase inhibitors; in particular, compound 11 exhibited a comparable potency profile toward the two enzymes, while 12 was 2.5-fold more selective for BChE. In general, the new substitution pattern explored in the present series of naphthoquinone and anthraquinone derivatives on one hand positively ameliorated the inhibitory potency against Aβ aggregation and on the other hand was responsible for a reduced efficacy against cholinesterases, especially BChE, in comparison to the previous series (Figure 1) which were shown to be more potent dual inhibitors of both enzymes.16
2.5. Inhibition of hMAOs
MAO inhibition was performed with a routine spectrophotometric assay, monitoring the fluorescence of 4-hydroxyquinoline produced in the MAO-catalyzed oxidation of kynuramine.45 With few exceptions, the title compounds acted as selective MAO B inhibitors, thus corroborating their potential as multitarget anti-AD agents, with IC50 values in the submicromolar range (Table 2). It is worthy to note the high potency of N-arylalkyl substituted naphthoquinones 2–4, whose IC50 values lay in the nanomolar range. Benzylamine derivative 2, with IC50 equal to 7.7 nM, resulted in a potency higher than reference drug safinamide, while 11 and 12, homologues of 3 and 4, were the most potent MAO B inhibitors within the anthraquinone series. Compared with phenethyl derivative 3, the substitution of phenyl with indole (5) retained good potency, while the introduction of different heteroaromatic systems was somehow detrimental.
The very high potency of naphthoquinone 2 deserved further biochemical investigation in order to clarify its inhibition mechanism. Preliminarily we confirmed that MAO B inhibition was not the consequence of a pan-assay interference of 2 in the fluorimetric assay,46 by performing the same kynuramine-based assay in spectrophotometric mode, following the increase of absorbance of 4-hydroxyquinoline at 316 nm.47 An IC50 equal to 18 nM was obtained, in good agreement with that reported in Table 2. Concerning the inhibition mechanism, we detected a competitive inhibition (inhibition constant Ki = 22 nM) when the coincubation with enzyme was limited to 5 min (Figure S2). For higher coincubation times (up to 2 h) the mechanism apparently turned noncompetitive, with the Ki increasing about 20-fold. This change could be ascribed to the formation of a covalent complex with the N5 of flavin,48 due to the presence of the chlorine leaving group. Indeed, a time course of absorption of 4-hydroxyquinoline at 316 nm revealed that the enzymatic activity was still present even after 3 h in the presence of 10 nM 2 (Figure 3). This activity profile was quite comparable to that of 10 nM safinamide, the reference drug for reversible and selective MAO B inhibition, when the assay was performed without preincubation with the enzyme (Figure 3, left). With a preincubation of 1 h, the time course of MAO B activity was still comparable for 2 and safinamide, although 2 suffered a strong decrease of potency (Figure 3, right). When incubated with 100 nM pargyline, a known irreversible MAO B inhibitor, the MAO B kinetic profiles were clearly different, since the enzymatic activity in the presence of pargyline came to saturation in about 1 h (Figure 3, left) and was completely inhibited with the preincubation (Figure 3, right). We speculate that the time-dependent change in potency of compound 2 may be due to its inactivation, either from chemical decomposition or from covalent reaction with nucleophilic residues of the enzyme, rather than to the formation of a covalent adduct with the flavin of catalytic site of the enzyme49 and by consequence that the inhibition of 2 may be considered as reversible.
Figure 3.

Time course of inhibition of MAO B by 2 (10 nM, blue line), safinamide (10 nM, red line), and pargyline (100 nM, green line): left, no preincubation; right, 1 h preincubation with enzyme. Data points represent the absorbance of 4-hydroxyquinoline at 316 nm (n = 3; mean ± SD).
The results listed in Table 2 so far highlighted the naphthoquinone 2 as the molecule endowed with the most effective inhibitory activity against MAO B (IC50 = 7.7 nM) and AChE (IC50 = 9.2 μM). In order to investigate the molecular determinants for the potency and selectivity of 2 toward MAO B and AChE, molecular modeling studies were next performed. Accordingly, the putative binding sites and modes for 2 were first identified on both human enzymes (Figure 4) and then molecular dynamics (MD) simulations of the resulting inhibitor/protein complexes were carried out to evaluate the corresponding free energy of binding (ΔGbind) following our consolidated approach.50−56 A per-residue binding free energy deconvolution (PRBFED) of the enthalpic (ΔHbind,res) terms50,51 was finally performed to define and describe the intermolecular interactions between compound 2 and the two proteins (Figure 5). The simulation results clearly show that 2 has a substantially higher affinity for MAO B (ΔGbind = −10.98 ± 0.16 kcal/mol) than for AChE (ΔGbind = −7.93 ± 0.13 kcal/mol), in agreement with the relevant experimental findings. The inspection of the relevant MD trajectories reveals that when in complex with MAO B (Figure 4A), the two carbonyl groups of 2 are engaged in two stable hydrogen bonds (HBs) with the monoamine oxidase side chains of Y326 (3.21 ± 0.13 Å) (ΔHbind,res = −1.77 kcal/mol, Figure 5A) and Y435 (2.87 ± 0.11 Å) (ΔHbind,res = −1.84 kcal/mol, Figure 5A), respectively. Moreover, one of the −C=O groups of 2 is also permanently H-bonded with the −-NH2 moiety of MAO B Q206 (3.23 ± 0.13 Å) (ΔHbind,res = −1.21 kcal/mol, Figure 5A), while the chlorine atom of 2 interacts via a halogen bond (HaB) with the thiol group of MAO B C172 at an optimal distance of 2.95 ± 0.11 Å (ΔHbind,res = −1.03 kcal/mol).57 In addition, the 2/MAO B complex is further stabilized in the putative binding site through an extended network of close van der Waals/hydrophobic contact interactions (CIs) between the ligand and the side chains of MAO B residues F103, P104, W119, L164, L167, L171, I198, I199, K296, F343, and Y398 (∑ΔHbind,res = −4.46 kcal/mol, Figure 5A), as highlighted in panel A of Figure 4.
Figure 4.

Details of compound 2 in the binding pocket of human MAO B (A) and human AChE (B). Compound 2 is shown as atom-colored sticks (C, gray; O, red; N, blue; Cl, green), while the side chains of the protein residues mainly interacting with the compound are highlighted as colored sticks and labeled. HBs/HaBs are shown as dark green broken lines, and their lengths are indicated (Å). Hydrogen atoms, water molecules, ions, and counterions are omitted for clarity.
Figure 5.

Per residue binding free energy deconvolution (PRBFED) of the enthalpic term (ΔHbind,res) for the MAO B (A) and AChE (B) residues involved in the complex with 2. Dark-colored bars highlights those protein residues involved in stronger interactions (e.g., HBs) with the compound in each complex.
On the other hand, within the putative binding site of AChE the naphthoquinone core of 2 is involved in a π–π interaction with AChE W286 (ΔHbind,res = −1.05 kcal/mol, Figure 5B) and is further stabilized by weak CIs with the side chains of the esterase residues V294, F338, and Y341 (∑ΔHbind,res = −1.36 kcal/mol, Figure 5B). One of the two −C=O groups of 2 directly engages the side chain of Y72 in a long but stable HB (3.48 ± 0.22 Å) (ΔHbind,res = −1.59 kcal/mol, Figure 5B), while the second carbonyl moiety interacts with the −OH group of AChE Y124 via a water-mediated HB (2.40 ± 0.33 (2···H2O), 2.38 ± 0.36 Å (Y124···H2O)) (ΔHbind,res = −1.84 kcal/mol, Figure 5A). Additionally, mildly favorable, unspecific contacts are detected between the phenyl moiety of 2 and AChE residues V73, T83, W86, N87, S125, and Y337 (∑ΔHbind,res = −1.69 kcal/mol, Figure 5B). Accordingly, the interactions between 2 and AChE are definitely less optimized than those detected in the alternative 2/MAO B complex and provide a molecular-based rationale for the weaker (μM) and stronger (nM) affinity of 2 for the esterase and the monoamino oxidase, respectively.
2.6. Inhibition of PHF6 Aggregation
New evidence has shifted our understanding about the role of tau in AD pathogenesis, acting as crucial partner of Aβ.58 The intracellular binding of soluble Aβ to nonphosphorylated tau was detected and possibly represents a precursor event to later self-aggregation of both molecules.59 Aβ has also been shown to affect tau pathology through the upregulation of kinases and proinflammatory cytokines that modulate tau phosphorylation.60 The high potency of compounds 1–14 in inhibiting Aβ aggregation prompted us to investigate the same feature in a smart in vitro model of tau aggregation, using as probe the highly repeated sequence (306)VQIVYK(311) (PHF6) responsible for aggregation of tau in paired helical fragments (PHF).61 To this aim we developed a fast assay method based on ThT fluorescence.45 Results in Figure 6 account for a high potency profile of the entire set compounds in inhibiting PHF6 aggregation, with inhibition values often >90% and comparable to reference drug quercetin. It is worthy of note that phenethyl derivatives 3 and 11 emerged as the most potent within their structural subsets (inhibition >95%), with IC50 values equal to 4.96 ± 0.75 μM and 1.78 ± 0.23 μM, respectively.
Figure 6.

Inhibition of aggregation of PHF6 tau sequence (50 μM) by 10 μM test molecules. Quercetin (QUR) was used as reference compound. Bars represent the mean ± SD of residual aggregation compared with control (CTR). Clear and dark filling patterns are for naphthoquinones and anthraquinones, respectively.
2.7. Inhibition of R3 Aggregation
The inhibition exerted by 3 and 11 toward PHF6 aggregation was confirmed in a kinetic experiment of dose-dependent aggregation of peptide R3. The 30-amino acid peptide (V306-Q336) R3 is a high-repeated domain containing the PHF6 sequence and present in all tau isoforms.62 Its high propensity to self-aggregate in buffered solutions, even in the absence of aggregation inducers such as heparin,63 makes R3 a reliable surrogate of full-length tau protein for in vitro experiments. In our assay protocol, the commercial R3 peptide was pretreated with 1,1,1-trifluoroethanol (TFE) overnight in order to dissolve any preformed aggregate and reestablish the random coil arrangement, then diluted in PBS at the test concentration of 25 μM. The R3 aggregation, detected by means of ThT fluorescence, resulted in a sigmoidal time course reaching the plateau after 4 h of incubation at 37 °C (Figure 7). The plateau values (as % of control) were used to derive the dose–response curve for the calculation of IC50 values, which were equal to 1.19 ± 0.44 μM for naphthoquinone 3 and 0.36 ± 0.02 μM for anthraquinone 11.
Figure 7.

Inhibition of aggregation of R3 tau sequence (25 μM) by 11: time course of aggregation in the presence of six concentrations of 11 (ranging from 30 to 0.1 μM).
2.8. Inhibition of Aβ42 Fibrillation by ThT Fluorescence Assay
Aβ42 is the main component of circulating amyloid peptides and majorly responsible for neurotoxicity in AD. Its high propensity to self-aggregate allows a fast execution of aggregation assays without the need of aggregation enhancers as for Aβ40. The antiamyloidogenic properties of the newly prepared compounds 1–14 were then tested also for Aβ42 in a coincubation assay at two concentrations (100 and 5 μM) at 37 °C for 48 h. Data presented in Figure 8 confirmed the activities already disclosed for Aβ40, with naphthoquinones 2–5 and anthraquinones 11, 12, and 14 displaying the best activity profile. Satisfactorily, compound 2 behaved as a strong inhibitor of Aβ42 aggregation even at the lower concentration used, being comparable for potency to quercetin.
Figure 8.

Inhibition of aggregation of Aβ42 (30 μM) by 100 μM (blue filling) and 5 μM (white filling) test molecules. Quercetin (QUR) was used as reference compound. Bars represent the mean ± SD of residual aggregation compared with control. Clear and dark filling patterns are for naphthoquinones and anthraquinones, respectively.
2.9. Inhibition of Aβ42 Fibrillation Studied by AFM
Among the investigated compounds, the naphthoquinone derivatives 2 and 5, and the anthraquinone derivatives 11 and 12 exhibited the highest efficiency against Aβ40 aggregation, as demonstrated by their low IC50 values (Table 2). Moreover, they also confirmed their antiamyloidogenic behavior against Aβ42 peptide (Figure 8). Tapping mode atomic force microscopy was then employed to test the ability of this selected subset of compounds to inhibit fibrillation of the highly amyloidogenic fragment Aβ42.
Figure 9 shows representative images of Aβ42 aggregated for 72 h in fibrillar morphology in the absence (Figure 9A) and in the presence (Figure 9B–E) of the compounds at a molar ratio 1:2 peptide/compound. The AFM inspection allowed a quantitative evaluation of the number of fibrils per unit area at a fixed aggregation time in the different conditions (Figure 9F). Compared with the control, in the presence of all the investigated compounds a significant decrease of the fibril surface density was observed. The inhibitory effect of the compounds was already observed after 24 h of aggregation. In fact, although at this stage of aggregation the sample was mainly oligomeric and the number of fibrils was low, in the presence of the compounds there was still a decrease in the fibrils surface density (Figure S3).
Figure 9.

Inhibition of Aβ42 fibrillation tested by AFM. Tapping mode AFM images of Aβ42 after 72 h of aggregation at room temperature in the absence (A) and in the presence of compounds 2 (B), 5 (C), 11 (D), and 12 (E). Scan size was 5.0 μM. Z range was 25 nm. (F) Quantitative evaluation of the fibril surface density (number of fibrils per unit area) in the absence and in the presence of the compounds. Mean values obtained on at least six different areas of 100 μm2 are reported. Errors were calculated using Student’s statistics, assuming a confidence level of 95%.
2.10. Protection against Aβ42-Induced Toxicity
The most promising MTDLs 2, 5, 11, and 12 were also tested for their ability to restore cell viability against the toxic effects exerted by Aβ42. Cerebellar granule cells (CGCs) were exposed for 48 h to 5 μM Aβ42 aggregated for 24 h with and without the compounds (at a molar ratio peptide/compound of 1:2). Cell viability after exposure was measured with the MTT test (Figure 10). Compared with the control, the viability of cells treated with Aβ42 alone decreased to 51%. In the presence of the compounds a partial recovery of the viability was generally observed, with compound 2 as the best neuroprotective agent.
Figure 10.

Protection against Aβ42 toxicity in cerebellar granule cells. The CGCs were treated with vehicle (control) or with Aβ42, with or without the tested compounds, for 48 h. Cell viability was measured by MTT reduction test and expressed as loss of viability in comparison with vehicle-treated controls. Values have been obtained from at least three experiments. Compound + Aβ vs Aβ: *p < 0.01, **p < 0.02, ***p < 0.05.
The incomplete recovery of cell viability is probably due to the fact that although the compounds are effective in inhibiting aggregation of the peptide, they display some degree of toxicity per se, as at 10 μM for any of the test compounds the cell viability remained as high as ≥80% compared to the untreated control cells (Table 3). However, on the whole, the biological results indicate for these molecules a rather narrow safety margin (as the ratio toxicity/activity), especially considering that most of their activities are in the micromolar range. For a more meaningful assessment of the value of the present compounds, the issue of their toxicity, in comparison to established drugs for AD, should deserve further investigation.
Table 3. In Vitro Cytotoxicity of Compounds 2, 5, 11, 12a.
| |
||||
|---|---|---|---|---|
| compound | 2 | 5 | 11 | 12 |
| cell viability vs control (%) | 89 ± 14 | 85 ± 14 | 85 ± 15 | 77 ± 12 |
The CGCs were treated with the compounds (10 μM) for 48 h. Cell viability was measured by MTT reduction test and expressed as loss of viability in comparison with vehicle-treated controls. Values have been obtained from at least three experiments. Data are expressed as the mean ± SEM.
3. Conclusions
The present work demonstrated the promiscuity profile of a novel set of naphthoquinone and anthraquinone-based derivatives able to tackle different factors of interest for the efficient management of AD. These molecules were purportedly designed to inhibit Aβ aggregation, being properly decorated with hydrophobic moieties able to target the aromatic interactions implicated in amyloid self-assembly. In agreement with the design rationale the compounds significantly inhibited Aβ40 aggregation and more interestingly the most active derivatives (2, 5, 11, and 12) were confirmed to disrupt fibrillation of the highly amyloidogenic fragment Aβ42. All the compounds exhibited a preferential inhibition of AChE than BChE and appeared to be efficient inhibitors of fast-aggregating tau peptides PHF6 and R3. Moreover, most of them turned out to be potent and selective MAO B inhibitors, being able to provide IC50 values in the nanomolar or submicromolar range with respect to the less sensitive MAO A isoform. Outstandingly, compound 2 exhibited a remarkable inhibitory activity against MAO B (IC50 = 7.7 nM) which was 473-fold higher than that versus MAO A. MAO enzymes significantly contribute to the selective degeneration of noradrenergic and cholinergic neurons in AD brains25 and through the formation of reactive aldehydes that showed facilitation of the conversion of β-amyloid to the hydrophobic β-sheet conformation and subsequent fibrillogenesis in vitro.64 It is worth noting that a comparable SAR trend was observed between the previously discussed inhibition of self-induced Aβ aggregation and MAO, thus suggesting a potential correlation between the two inhibition mechanisms. Finally, these MTDL molecules were also found to ameliorate the cytotoxicity caused by Aβ42 peptide. Collectively, these results indicate that our series of naphthoquinone and anthraquinone derivatives possess antiamyloidogenic properties and a multitarget profile, thus enabling them as valuable candidates for AD therapy.
4. Methods
4.1. Chemistry
4.1.1. General Methods
Chemicals and solvents were purchased from Sigma-Aldrich (Milan, Italy). Melting points were measured using a Büchi apparatus and were uncorrected. 1H NMR spectra and 13C NMR spectra were recorded on a Varian Gemini-200 instrument at 200 and 50 MHz, respectively. Chemical shifts are reported as δ (ppm) and are referenced to a solvent signal: CDCl3, singlet at 7.26 ppm (1H), triplet at 77.0 ppm (13C); DMSO-d6, quintet at 2.5 ppm (1H), septet at 39.5 ppm (13C). J is in Hz. Elemental analyses were performed on a Flash 2000 CHNS (Thermo Scientific) instrument in the Microanalysis Laboratory of the Department of Pharmacy, University of Genoa. NMR spectra of the novel compounds are shown in the Supporting Information. Results of elemental analyses indicated that the purity of all compounds was ≥95%.
4.1.2. General Method for the Synthesis of Naphthoquinone Derivatives (4–7)
A mixture of appropriate amine (5 mmol) and 2,3-dichloronaphthoquinone 1 (2.5 mmol) in absolute ethanol (20 mL) was stirred under reflux for 4 h with stirring. In the case of compounds 5–7 the reaction mixture was stirred at room temperature for 24 h. During the progress of the reaction, monitored by TLC, a change in the color from yellow to red was observed. After evaporation of the solvent, the residue was treated with a solution of 2 N NaOH and CHCl3. The layers were separated, and the chloroform solution was dried (Na2SO4), filtered, and evaporated, affording a residue that was purified by CC (SiO2, CHCl3 + 2%MeOH).
4.1.2.1. 2-Chloro-3-(phenylpropylamino)-1,4-naphthoquinone (4)
Yield: 75%. Mp 94–96 °C. 1H NMR (200 MHz, CDCl3) δ: 8.40–8.00 (m, 2H, arom); 7.84–7.58 (m, 2H, arom); 7.41–7.18 (m, 5H, arom); 6.15 (s, 1H, NH); 3.90 (q, J = 7 Hz, 2H, NHCH2CH2CH2Ar); 2.77 (t, J = 7.6 Hz, 2H, NHCH2CH2CH2Ar); 2.07 (quint, J = 6.8 Hz, 2H, NHCH2CH2CH2Ar). 13C NMR (50 MHz, CDCl3) δ: 179.4, 175.8, 143.0, 139.7, 133.9, 131.7, 131.4, 128.6, 127.5, 127.3, 125.8, 125.2, 43.2, 31.8, 31.5. Anal. Calcd for C19H16ClNO2: C 70.05; H 4.95; N 4.30. Found: C 70.30; H 5.17; N 4.17.
4.1.2.2. 2-[2-(1H-Benzimidazol-1-yl)ethylamino]-3-chloro-1,4-naphthoquinone (6)
Yield: 49%. Mp 205–207 °C. 1H NMR (200 MHz, CDCl3) δ: 8.22 (s, 1H, arom); 7.88 (s, 1H, arom); 7.58–7.18 (m, 4H, arom); 6.98–6.77 (m, 1H arom); 6.76–6.42 (m, 2H, arom); 6.24 (broad s, 1H, NH); 3.70–3.40 (m, 2H, NHCH2CH2-N(1)Benz); 3.34–2.98 (m, 2H, NHCH2CH2-(N1)Benz). 13C NMR (50 MHz, CDCl3) δ: 172.9, 153.6, 142.7, 140.0, 137.6, 131.4, 129.6, 128.5, 124.5, 124.0, 121.9, 118.5, 112.0, 105.6, 48.5, 44.9. Anal. Calcd for C19H14ClN3O2: C 64.87; H 4.01; N 11.94. Found: C 64.63; H 4.28; N 12.06.
4.1.2.3. 2-{[2-(1H-Benzotriazol-1-yl)ethy]amino}-3-chloro-1,4-naphthoquinone (7)
Yield: 39%. Mp 193–194 °C. 1H NMR (200 MHz, DMSO) δ: 8.05–7.65 (m, 5H, arom); 7.62–7.41 (m, 2H, arom); 7.39–7.23 (m, 1H, arom); 6.29 (s, 1H, NH); 4.98 (t, J = 5.8 Hz, 2H, NHCH2CH2-N(1)Bzt); 4.24 (q, J = 6.2 Hz, 2H, NHCH2CH2-N(1)Bzt). 13C NMR (50 MHz, DMSO) δ: 179.6, 174.9, 145.2, 144.7, 134.4, 134.2, 132.7, 132.3, 131.1, 129.6, 126.8, 126.0, 125.3, 123.5, 118.6, 110.1, 47.8, 43.0. Anal. Calcd for C18H13ClN4O2: C 61.28; H 3.71; N 15.88. Found: C 61.17; H 3.77; N 15.98.
4.1.3. General Method for the Synthesis of Anthraquinone Derivatives (10–14)
A mixture of 1-chloroanthraquinone (2.5 mmol) with the suitable amino compound (2.5 mmol) was heated at 160 °C in a sealed tube for 6 h. After cooling, the mixture was treated with 2 M NaOH until alkalinity and extracted with CHCl3. After removal of the solvent, the residue was purified by CC (SiO2/CHCl3 + 2% MeOH).
4.1.3.1. 1-(Benzylamino)-9,10-anthraquinone (10)
Yield: 90%. Mp 171–173 °C. 1H NMR (200 MHz, CDCl3) δ: 8.43–8.19 (m, 2H, arom); 7.96–7.61 (m, 4H, arom); 7.60–7.20 (m, 6H, arom); 7.17–6.98 (m, 1H, NH); 4.62 (s, 2H, NHCH2Ar). 13C NMR (50 MHz, CDCl3) δ: 184.2, 182.7, 150.4, 136.9, 136.7, 134.3, 133.9, 133.6, 133.5, 132.9, 132.6, 132.0, 127.8, 126.5, 126.4, 126.0, 125.7, 117.3, 115.1,112.4, 46.0. Anal. Calcd for C21H15NO2: C 80.49; H 4.82; N 4.47. Found: C 80.11; H 5.20; N 4.17.
4.1.3.2. 1-(Phenethylamino)-9,10-anthraquinone (11)
Yield: 71%. Mp 100–103 °C. 1H NMR (200 MHz, CDCl3) δ: 8.42–8.20 (m, 2H, arom); 7.90–7.70 (m, 2H, arom); 7.68–7.51 (m, 3H, arom); 7.48–7.22 (m, 5H, arom); 7.11 (dd, 1H, J = 1.6, 6.6 Hz, NH); 3.63 (t, J = 7.2 Hz, 2H, NHCH2CH2Ar); 3.10 (t, J = 7.8 Hz, 2H, NHCH2CH2Ar). 13C NMR (50 MHz, CDCl3) δ: 184.0, 182.8, 150.4, 137.7, 134.3, 134.0, 133.7, 131.9, 127.7, 126.5, 125.7, 116.7, 114.7, 112.1, 43.6, 34.6. Anal. Calcd for C22H17NO2: C 80.71; H 5.23; N 4.28. Found: C 80.62; H 5.04; N 3.98.
4.1.3.3. 1-[(3-Phenylpropryl)amino]-9,10-anthraquinone (12)
Yield: 76%. Mp 115–116 °C. 1H NMR (200 MHz, CDCl3) δ: 8.42–8.21 (m, 2H, arom); 7.90–7.70 (m, 2H, arom); 7.68–7.48 (m, 2H, arom); 7.42–7.18 (m, 6H, arom); 7.06 (dd, 1H, J = 1.6, 8.2 Hz, NH); 3.38 (t, J = 7.0 Hz, 2H, NHCH2CH2CH2Ar); 2.86 (t, J = 7.4 Hz, 2H, NHCH2CH2CH2Ar); 2.15 (quint, J = 7.2 Hz, 2H, NHCH2CH2CH2Ar). 13C NMR (50 MHz, CDCl3) δ: 183.9, 182.8, 150.6, 140.1, 134.2, 133.9, 133.5, 132.9, 131.9, 128.3, 127.5, 127.4, 125.7, 125.6, 125.1, 116.8, 114.6, 111.8, 41.0, 32.1, 29.5. Anal. Calcd for C23H19NO2: C 80.92; H 5.61; N 4.10. Found: C 80.73; H 5.98; N 3.93.
4.1.3.4. 1-{[2-(1H-Indol-3-yl)ethyl]amino}-9,10-anthraquinone (13)
Yield: 40%. Mp 240–241 °C. 1H NMR (200 MHz, CDCl3) δ: 10.84 (s, 1H, NH of indole); 8.18–7.41 (m, 7H, arom); 7.39–6.82 (m, 5H, arom); 6.60 (s, 1H, NH); 3.98 (pseudo s, 2H, NHCH2CH2Ind); 2.99 (pseudo s, 2H, NHCH2CH2Ind). 13C NMR (50 MHz, CDCl3) δ: 184.6, 182.6, 150.0, 135.8, 134.4, 134.2, 132.1, 131.5, 130.5, 129.5, 126.6, 126.0, 125.3, 122.7, 120.6, 117.9, 110.9, 110.3, 44.2, 33.0. Anal. Calcd for C24H18N2O2: C 78.67; H 4.95; N 7.65. Found: C 78.81; H 4.95; N 7.43.
4.1.3.5. 1-{[2-(1H-Benzotriazol-1-yl)ethyl]amino}-9,10-anthraquinone (14)
Yield: 42%. Mp 176–177 °C. 1H NMR (200 MHz, CDCl3) δ: 8.36–8.15 (m, 3H, arom); 7.86–7.57 (m, 5H, arom); 7.55–7.22 (m, 3H, arom); 6.82 (s, 1H, NH); 4.57 (t, J = 7.0 Hz, 2H, NHCH2CH2-N(1)Bzt); 3.03 (t, J = 7.0 Hz, 2H, NHCH2CH2-N(1)Bzt). 13C NMR (50 MHz, CDCl3) δ: 184.5, 183.7, 146.2, 144.0, 135.9, 133.9, 133.4, 132.9, 132.0, 127.8, 126.9, 125.8, 125.0, 121.9, 119.0, 48.5, 44.2. Anal. Calcd for C22H16N4O2: C 71.73; H 4.38; N 15.21. Found: C 71.59; H 4.31; N 15.20.
4.1.4. General Method for the Synthesis of Intermediates: Tryptamine, 2-(Benzimidazol-1-yl)ethylamine, and 2-(Benzotriazol-1-yl)ethylamine
To a suspension of LiAlH4 (40 mmol) in 20 mL of anhydrous THF cooled at 0–5 °C, a solution of 3-indolylacetamide or 2-(benzimidazol-1-yl)acetonitrile or 2-(benzotriazol-1-yl)acetonitrile (10 mmol) in anhydrous THF (5 mL) was added drop by drop in 20 min. Then the mixture was refluxed for 8–10 h. At rt 5 mL of H2O, 5 mL of 2N NaOH, and 5 mL of H2O were carefully added to the mixture to decompose the excess of LiAlH4, filtering and washing the inorganic residue with Et2O. The solution was dried with Na2SO4, filtered, and evaporated to dryness, yielding an orange oil corresponding to the title ethylamino derivative that was chromatographed on SiO2 eluting with Et2O.
These intermediates were already prepared through different procedures; thus they were characterized as follows.
4.1.4.1. Tryptamine
Yield: 81%. 1H NMR (200 MHz, CDCl3) δ: 10.11 (s, NH of indole), 7.71–7.52 (m, 2H, arom), 7.39–7.20 (m, 2H, arom), 7.16–7.03 (m, 1H, arom), 4.88 (broad s, NH2), 3.09–2.95 (m, 4H, CH2CH2-NH2). Anal. Calcd for C10H12N2: C 74.97; H 7.55; N 17.48. Found: C 74.81; H 7.95; N 17.23.
4.1.4.2. 2-(Benzimidazol-1-yl)ethylamine
Yield: 76%. 1H NMR (200 MHz, CDCl3) δ: 8.21 (s, 1H, arom), 7.65–7.49 (m, 2H, arom), 7.35–7.21 (m, 2H, arom), 4.97 (broad s, NH2), 4.19–4.01 (m, 2H, N-CH2), 3.16–3.07 (m, 2H, CH2-NH2). Anal. Calcd for C9H11N3: C 67.06; H 6.88; N 26.07. Found: C 66.91; H 6.79; N 26.31.
4.1.4.3. 2-(Benzotriazol-1-yl)ethylamine
Yield: 76%. 1H NMR (200 MHz, CDCl3) δ: 8.11–7.71 (m, 2H, arom), 7.52–7.31 (m, 2H, arom), 5.01 (broad s, NH2), 4.22 (pseudo s, 2H, N-CH2), 3.45 (pseudo s, 2H, CH2-NH2). Anal. Calcd for C8H10N4: C 59.24; H 6.21; N 34.54. Found: C 59.01; H 6.56; N 34.16.
4.2. Biological Tests
4.2.1. Inhibition of Self-Induced Aβ Aggregation
Aβ40 and Aβ42 peptides were purchased from EzBiolab, Carmel, IN, USA. In vitro inhibition assay of Aβ40 aggregation has been previously reported41 and adapted to a 96-well plate platform. Briefly, samples of Aβ40 (30 μM) were coincubated for 2 h at 25 °C with test molecules (100 μM for single point concentration assay, seven concentrations ranging from 100 to 0.1 μM for IC50 calculations) in PBS containing 2% v/v of HFIP.
Inhibition of Aβ42 aggregation was assayed by incubating the peptide (30 μM) alone (as the control) or with test compounds (5 or 100 μM) in PBS at 37 °C for 48 h.
After incubation in 96-well black, nonbinding microplates (Greiner Bio-One GmbH, Frickenhausen, Germany), thioflavin T (25 μM) was added and fluorimetric reads (ex 440, em 485 nm) were performed in a multiplate reader Infinite M1000 Pro (Tecan, Cernusco sul Naviglio, Italy). Experiments were run in triplicate. Results were expressed by statistical analysis while IC50 values were obtained by nonlinear regression using Prism software (GraphPad Prism version 5.00 for Windows, GraphPad Software, San Diego, CA, USA).
4.2.2. Inhibition of Cholinesterases
The classical Ellman’s method, modified to a 96-well plate platform, was used as already described.65 AChE from electric eel (463 U/mL), BChE from horse serum (13 U/mL), acetyl- and butyrylthiocholine, and dithiobis(2-nitrobenzoic acid) were purchased from Sigma-Aldrich, Milan, Italy. Experiments were run in triplicate, and inhibition values were obtained by nonlinear regression using Prism software.
4.2.3. Inhibition of Human Monoamine Oxidases
The fluorimetric assays of MAO A and B following the oxidation of kynuramine to 4-hydroxyquinoline (ex 310, em 400 nm in NaOH) were performed as described.45 All enzymes and reagents were purchased from Sigma-Aldrich. For inhibition kinetics, four concentrations of 2 (ranging from 0 to 20 nM) and five concentrations of kynuramine (from 2 to 30 μM) were used. Direct spectrophotometric measurement of 4-hydroxyquinoline absorption at 316 nm was performed as described.47 Inhibition values and kinetic parameters were calculated by means of Prism.
4.2.4. Inhibition of PHF6 Aggregation
The fluorimetric assay, using thioflavin T as the chromophoric reagent, has already been described.45 Briefly, samples of PHF6 (JPT Peptide Technologies GmbH, Berlin, Germany) (50 μM), inhibitor (10 μM), and ThT (10 μM) were prepared in triplicate in PBS containing 3.3% of 1,1,1-trifluoroethanol and read within 3 h at 30 °C with a Tecan Infinite M1000 Pro instrument. The plateau values (as % of residual activity compared to control) were used for the calculation of residual aggregation or in nonlinear regression by means of Prism to calculate the IC50 values as the mean of two independent experiments.
4.2.5. Inhibition of R3 Aggregation
A stock solution of 1.5 mM R3 (trifluoroacetate salt; Bachem, Bubendorf, Switzerland) was prepared in TFE and left overnight at room temperature. Incubation samples were set in nonbinding, flat-bottomed black microplates (Greiner Bio-One) in triplicate for each concentration and contained peptide (25 μM), inhibitor (seven concentrations ranging from 30 to 0.1 μM), and ThT (10 μM) in PBS containing 1.7% of 1,1,1-trifluoroethanol. Fluorescence was read within 4 h at 37 °C with a Tecan Infinite M1000 Pro instrument. IC50 values were calculated by means of Prism as the mean of two independent experiments.
4.2.6. Atomic Force Microscopy
Compounds 2, 5, 11, and 12 were dissolved in DMSO at concentrations ranging from 2.0 mg/mL to 3.3 mg/mL. Aβ42 (Bachem) was dissolved in TFA to obtain a 1 mg/mL stock solution. In each experiment, appropriate peptide aliquots were deposited in glass vials and dried under a gentle nitrogen stream. Aggregation was initiated by hydrating the peptide with PBS premixed with the compound under study. The final compound and peptide concentrations were 100 μM and 50 μM, respectively. In the control samples, PBS was premixed with DMSO volumes equivalent to those present in the compound aliquots employed for the experiments. Aggregation was performed at room temperature.
For AFM inspection, sample aliquots of 20 μL were deposited on a freshly cleaved mica surface and incubated for 7 min. Samples were then gently rinsed with Milli-Q water and dried overnight under mild vacuum. Tapping mode AFM images were acquired in air using a Dimension 3100 SPM (Bruker, Karlsruhe, Germany) equipped with “G” scanning head (maximum scan size 100 μm) and driven by a Nanoscope IIIa controller (Bruker). Single-beam uncoated silicon cantilevers (type TESPA_V2, Bruker) were used. The drive frequency was 300–320 kHz, and the scan rate was 0.5 Hz.
4.2.7. Primary Cultures
Cerebellar granule cells were prepared from 8-day-old Sprague Dawley rats as described previously.66 Cells were studied from the 6th to the 12th day in vitro. The experimental procedures and care of the animals were performed in compliance with the Directive of the EU Parliament and Council of September 22, 2010 (2010/63/EU) and were approved by the Italian Ministry of Health (COD. 75F11.N.6DX) in accordance with D.M. 116/1992. All efforts were made in order to minimize animal suffering and the number of animals necessary to obtain reliable results.
4.2.8. Cell Survival Assay
Cerebellar granule cells in 48-well plate were treated with Aβ42, aggregated in PBS for 24 h, in the absence and in the presence of each tested compound in the same conditions described in section 4.2.6. The final compound and Aβ42 concentrations after addition to the well plate were 10 μM and 5 μM, respectively. Cells were maintained in 5% CO2 at 37 °C. The cell viability was assessed by the MTT assay 48 h after the treatment. MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] (Sigma-Aldrich, Germany) was added to the medium at a concentration of 0.25 mg/mL (MTT) into each well, and the multiwell plates were incubated for 3 h at 37 °C. After the removal of the medium, formazan crystals were dissolved in DMSO and the values of optical density (OD) were measured spectrophotometrically at 570 nm using a BioTek ELx800 (Winooski, VT, USA) microplate reader. The survival rates of viable cells were calculated by comparing the optical absorbance of treated samples with that of the untreated controls. All experiments were repeated at least three times independently, and data are expressed as the mean ± SEM.
4.2.9. In Vitro Blood–Brain Barrier Permeation Assay (PAMPA-BBB)
Prediction of the brain permeation of compounds 2, 5, 8, 11, and 12 was evaluated using a parallel artificial membrane permeation assay (PAMPA-BBB), following a well-established procedure.40,50 Briefly, a semiautomated pipetting system (BenchSmart 96, Mettler Toledo) and a microplate spectrophotometer (SpectraMax Plus 384 microplate reader, Molecular Devices) were employed for pipetting and UV reading, respectively. All commercial drugs and reagents were purchased from Sigma-Aldrich. The porcine brain lipid (PBL) was acquired from Avanti Polar Lipids, while Millex filter units (PVDF membrane, pore size 0.45 μm) were obtained from Millipore. For the assay, the 96-well acceptor microplate (PTFE, Millipore) was filled with 300 μL of PBS:ethanol (70:30), whereas the artificial membrane of the donor microplate (PVDF membrane, pore size 0.45 μm, Millipore) was coated with 4 or 5 μL of PBL dissolved in dodecane (20 mg/L). All compounds were first dissolved in DMSO and then diluted with PBS/EtOH (70:30, pH 7.4) to reach the final concentration in the range 40–100 μM in the donor well, filtered through a Millex filter, and then added to the donor microplate wells (200 μL). The donor filter microplate was carefully placed onto the acceptor microplate so as to form a sandwich, which was left undisturbed for 16 h at 25 °C into a sealed container with wet paper towels to avoid evaporation. After incubation, the donor microplate was cautiously removed and the concentrations of the tested compounds in the acceptor and donor microplate wells were determined via UV–vis spectroscopy. Every sample was analyzed at five wavelengths in four wells and in three independent runs. Accordingly, the results given in Table 1 are reported as average values ± standard deviation. Pe was calculated by the following formula: Pe = {−VdVa/[(Vd + Va)At]} × ln(1 – drugacceptor/drugequilibrium), where Vd and Va are the volumes of the donor and acceptor wells, respectively, A is artificial membrane area, t is the permeation time, drugacceptor is the absorbance obtained in the acceptor well, and drugequilibrium is the theoretical equilibrium absorbance. Twenty known commercial drugs of known BBB permeability (Table S1, Figure S1) were used as quality control standards to validate and normalize the analysis set. Donepezil and quercetin were further tested as CNS+ and CNS– positive and negative controls, respectively (Table 1). Upon completion of the PAMPA-BBB assay for each present and standard compound, lipid membrane integrity was verified based on the transport of Lucifer Yellow (Sigma-Aldrich), a fluorescent molecule with very poor membrane permeability which, in the presence of a uniform and integral lipid membrane, should effectively be completely rejected.67 The Lucifer Yellow test was performed following Millipore protocol lit. no. PC1545EN00 (https://www.sigmaaldrich.com/technical-documents/protocols/biology/membrane-integrity-test-for-lipid-pampa-artificial-membranes.html). As the relevant fluorescence readings (SpectraMax Gemini XPS microplate reader, Molecular Devices) were comparable to background readings of buffer only (5% DMSO in PBS), the membrane integrity after all PAMPA-BBB assays was confirmed.
4.3. Molecular Modeling Studies
All simulations were carried our using Amber 2068 running on our own CPU/GPU cluster. Molecular graphics and analyses were performed with UCSF Chimera (v1.15),69 developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH Grant P41-GM103311. The starting molecular structure of MAO B in complex with E98 was obtained from the Protein Data Bank (PDB code 6FWC.pdb),70 while the AChE structure in complex with a flavonoid-based inhibitor was taken from our previous work50,51 (original PBD code 4EY7).71 The entire modeling and simulation procedure is also reported in detail in earlier papers.50,51 Briefly, the geometry- and energy-optimized structure of compound 2 was docked into each identified protein binding pocket using Autodock 4.2.6/Autodock Tools1.4.616172 on a win64 platform. The resulting complexes were further energy minimized to convergence. Each intermolecular complex was then solvated by a cubic box of TIP3P water molecules73 and energy minimized using a combination of molecular dynamics (MD) techniques.50,51 20 ns molecular dynamics (MD) simulations at 298 K were then employed for system equilibration, and further, 50 ns MD simulations were run for data production. The binding free energies of the 2/MAO B and 2/AChE complexes were calculated following the MM/PBSA methodology74 as previously described.50−56 The PRBFED analysis was carried out using the molecular mechanics/generalized Boltzmann surface area (MM/GBSA) approach,75 as already detailed,50,51 and was based on the same snapshots used in the binding free energy calculation.
Acknowledgments
M.T., A.R., and C.C. acknowledge the dedicated technical assistance of Dr. Mattia Siri to the AFM study. We thank Prof. Tullio Florio (University of Genoa, Department of Internal Medicine, Section of Pharmacology) for access to the microplate reader.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00624.
Permeability data (Pe, 10–6 cm/s) of 20 commercial drugs used in the PAMPA-BBB assay validation (Table S1) and linear correlation between experimental and reported Pe of the 20 commercial drugs using the PAMPA-BBB assay (Figure S1); Lineweaver–Burk plot of inhibition kinetics of MAO B by 2 (Figure S2); fibril surface density after 24 h of Aβ42 aggregation in the absence and in the presence of the compounds (2, 5, 11, and 12) (Figure S3); 1H and 13C NMR spectra of the newly synthesized compounds 4, 6, 7, and 10–14 (PDF)
Author Contributions
M.T. conceived the study. M.T. and M.Cam. synthesized the molecules. M.Cam. and B.T. provided their structural characterization. M.Cat. performed inhibition studies against Aβ and PHF6 aggregation, cholinesterases, and MAO enzymes. E.L. and S.P. performed PAMPA test and docking studies. C.C. and A.R. performed AFM studies. E.G. performed cytotoxicity and neuroprotection assays. M.T., M.Cat., A.R., and S.P. analyzed and discussed the results. M.T., M.Cat., A.R., and S.P. wrote and reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Contestabile A. (2011) The history of the cholinergic hypothesis. Behav. Brain Res. 221, 334–340. 10.1016/j.bbr.2009.12.044. [DOI] [PubMed] [Google Scholar]
- Masters C. L.; Selkoe D. J. (2012) Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer Disease. Cold Spring Harbor Perspect. Med. 2, a006262. 10.1101/cshperspect.a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M.; Shepardson N.; Yang T.; Chen G.; Walsh D.; Selkoe D. J. (2011) Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U. S. A. 108, 5819–5824. 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciarelli R.; Fedele E. (2017) The amyloid cascade hypothesis in Alzheimer’s disease: it’s time to change our mind. Curr. Neuropharmacol. 15, 926–935. 10.2174/1570159X15666170116143743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oset-Gasque M. J.; Marco-Contelles J. (2018) Alzheimer’s Disease, the “One-Molecule, One-Target” Paradigm, and the Multitarget Directed Ligand Approach. ACS Chem. Neurosci. 9, 401–403. 10.1021/acschemneuro.8b00069. [DOI] [PubMed] [Google Scholar]
- Butterfield D. A. (2018) Perspectives on Oxidative Stress in Alzheimer’s Disease and Predictions of Future Research Emphases. J. Alzheimer's Dis. 64, S469–S479. 10.3233/JAD-179912. [DOI] [PubMed] [Google Scholar]
- Li Y.; Jiao Q.; Xu H.; Du X.; Shi L.; Jia F.; Jiang H. (2017) Biometal Dyshomeostasis and Toxic Metal Accumulations in the Development of Alzheimer’s Disease. Front. Mol. Neurosci. 10, 339. 10.3389/fnmol.2017.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M. T.; O’Banion M. K.; Terwel D.; Kummer M. P. (2010) Neuroinflammatory processes in Alzheimer’s disease. J. Neural Transm. (Vienna) 117, 919–947. 10.1007/s00702-010-0438-z. [DOI] [PubMed] [Google Scholar]
- Alam J.; Sharma L. (2019) Potential enzymatic targets in Alzheimer’s: a comprehensive review. Curr. Drug Targets 20, 316–339. 10.2174/1389450119666180820104723. [DOI] [PubMed] [Google Scholar]
- Blaikie L.; Kay G.; Lin P. K. T. (2019) Current and emerging therapeutic targets of Alzheimer’s disease for the design of multi-target directed ligands. MedChemComm 10, 2052. 10.1039/C9MD00337A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosini M.; Simoni E.; Caporaso R.; Minarini A. (2016) Multitarget strategies in Alzheimer’s disease: benefits and challenges on the road to therapeutics. Future Med. Chem. 8, 697–771. 10.4155/fmc-2016-0003. [DOI] [PubMed] [Google Scholar]
- Prati F.; Cavalli A.; Bolognesi M. L. (2016) Navigating the chemical space of multitarget-directed ligands: from hybrids to fragments in Alzheimer’s disease. Molecules 21, 466. 10.3390/molecules21040466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesiti F.; Chavarria D.; Gaspar A.; Alcaro S.; Borges F. (2019) The chemistry toolbox of multitarget-directed ligands for Alzheimer’s disease. Eur. J. Med. Chem. 181, 111572. 10.1016/j.ejmech.2019.111572. [DOI] [PubMed] [Google Scholar]
- Paul A.; Viswanathan G. K.; Mahapatra S.; Balboni G.; Pacifico S.; Gazit E.; Segal D. (2019) Antagonistic activity of naphthoquinone-based hybrids toward amyloids associated with Alzheimer’s disease and type-2 diabetes. ACS Chem. Neurosci. 10, 3510–3520. 10.1021/acschemneuro.9b00123. [DOI] [PubMed] [Google Scholar]
- Campora M.; Francesconi V.; Schenone S.; Tasso B.; Tonelli M. (2021) Journey on Naphthoquinone and Anthraquinone Derivatives: New Insights in Alzheimer’s Disease. Pharmaceuticals 14, 33. 10.3390/ph14010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli M.; Catto M.; Tasso B.; Novelli F.; Canu C.; Iusco G.; Pisani L.; Stradis A. D.; Denora N.; Sparatore A.; Boido V.; Carotti A.; Sparatore F. (2015) Multitarget therapeutic leads for Alzheimer’s disease: quinolizidinyl derivatives of bi- and tricyclic systems as dual inhibitors of cholinesterases and β-Amyloid (Aβ) aggregation. ChemMedChem 10, 1040–1053. 10.1002/cmdc.201500104. [DOI] [PubMed] [Google Scholar]
- Zhang J. H.; Xin H. L.; Xu Y. M.; Shen Y.; He Y. Q.; Hsien-Yeh; Lin B.; Song H. T.; Juan-Liu; Yang H. Y.; Qin L. P.; Zhang Q. Y.; Du J. (2018) Morinda officinalis How. – A comprehensive review of traditional uses, phytochemistry and pharmacology. J. Ethnopharmacol. 213, 230–255. 10.1016/j.jep.2017.10.028. [DOI] [PubMed] [Google Scholar]
- Augustin N.; Nuthakki V. K.; Abdullaha M.; Hassan Q. P.; Gandhi S. G.; Bharate S. B. (2020) Discovery of helminthosporin, an anthraquinone isolated from Rumex abyssinicus Jacq as a dual cholinesterase inhibitor. ACS Omega 5, 1616–1624. 10.1021/acsomega.9b03693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit E. (2002) A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 16, 77–83. 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- Kirkitadze M. D.; Kowalska A. (2005) Molecular mechanisms initiating amyloid beta-fibril formation in Alzheimer’s disease. Acta Biochim. Polym. 52, 417–423. 10.18388/abp.2005_3454. [DOI] [PubMed] [Google Scholar]
- Chierrito T.; Pedersoli-Mantoani S.; Roca C.; Requena C.; Sebastian-Perez V.; Castillo W. O.; Moreira N.; Pérez C.; Sakamoto-Hojo E. T.; Takahashi C. S.; Jiménez-Barbero J.; Cañada F. J.; Campillo N. E.; Martinez A.; Carvalho I. (2017) From dual binding site acetylcholinesterase inhibitors to allosteric modulators: a new avenue for disease-modifying drugs in Alzheimer’s disease. Eur. J. Med. Chem. 139, 773–791. 10.1016/j.ejmech.2017.08.051. [DOI] [PubMed] [Google Scholar]
- Viayna E.; Sabate R.; Muñoz-Torrero D. (2013) Dual inhibitors of β-amyloid aggregation and acetylcholinesterase as multi-target anti-Alzheimer drug candidates. Curr. Top. Med. Chem. 13, 1820–1842. 10.2174/15680266113139990139. [DOI] [PubMed] [Google Scholar]
- Lane R. M.; Potkin S. G.; Enz A. (2006) Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 9, 101–124. 10.1017/S1461145705005833. [DOI] [PubMed] [Google Scholar]
- Caraci F.; Pappalardo G.; Basile L.; Giuffrida A.; Copani A.; Tosto R.; Sinopoli A.; Giuffrida M. L.; Pirrone E.; Drago F.; Pignatello R.; Guccione S. (2015) Neuroprotective effects of the monoamine oxidase inhibitor tranylcypromine and its amide derivatives against Aβ(1-42)-induced toxicity. Eur. J. Pharmacol. 764, 256–263. 10.1016/j.ejphar.2015.07.015. [DOI] [PubMed] [Google Scholar]
- Cai Z. (2014) Monoamine oxidase inhibitors: promising therapeutic agents for Alzheimer’s disease. Mol. Med. Rep. 9, 1533–1541. 10.3892/mmr.2014.2040. [DOI] [PubMed] [Google Scholar]
- Riederer P.; Danielczyk W.; Grünblatt E. (2004) Monoamine oxidase-B inhibition in Alzheimer’s disease. NeuroToxicology 25, 271–277. 10.1016/S0161-813X(03)00106-2. [DOI] [PubMed] [Google Scholar]
- Weinreb O.; Amit T.; Bar-Am O.; Youdim M. B. (2016) Neuroprotective effects of multifaceted hybrid agents targeting MAO, cholinesterase, iron and β-amyloid in ageing and Alzheimer’s disease. Br. J. Pharmacol. 173, 2080–2094. 10.1111/bph.13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew B.; Parambi D.; Mathew G. E.; Uddin M. S.; Inasu S. T.; Kim H.; Marathakam A.; Unnikrishnan M. K.; Carradori S. (2019) Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch. Pharm. (Weinheim, Ger.) 352, 1900177. 10.1002/ardp.201900177. [DOI] [PubMed] [Google Scholar]
- Ibrahim M. M.; Gabr M. T. (2019) Multitarget therapeutic strategies for Alzheimer’s disease. Neural Regener. Res. 14, 437–440. 10.4103/1673-5374.245463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W. H.; Hong S. S.; Lee S. A.; Han X. H.; Lee K. S.; Lee M. K.; Hwang B. Y.; Ro J. S. (2005) Monoamine oxidase inhibitory naphthoquinones from the roots of Lithospermum erythrorhizon. Arch. Pharmacal Res. 28, 400–404. 10.1007/BF02977668. [DOI] [PubMed] [Google Scholar]
- Mostert S.; Petzer A.; Petzer J. P. (2016) Evaluation of natural and synthetic 1,4-naphthoquinones as inhibitors of monoamine oxidase. Chem. Biol. Drug Des. 87, 737–746. 10.1111/cbdd.12708. [DOI] [PubMed] [Google Scholar]
- Banik A.; Brown R. E.; Bamburg J.; Lahiri D. K.; Khurana D.; Friedland R. P.; Chen W.; Ding Y.; Mudher A.; Padjen A. L.; Mukaetova-Ladinska E.; Ihara M.; Srivastava S.; Padma Srivastava M. V.; Masters C. L.; Kalaria R. N.; Anand A. (2015) Translation of pre-clinical studies into successful clinical trials for Alzheimer’s disease: what are the roadblocks and how can they be overcome?. J. Alzheimer's Dis. 47, 815–843. 10.3233/JAD-150136. [DOI] [PubMed] [Google Scholar]
- Prachayasittikul V.; Pingaew R.; Worachartcheewan A.; Nantasenamat C.; Prachayasittikul S.; Ruchirawat S.; Prachayasittikul V. (2014) Synthesis, anticancer activity and QSAR study of 1,4-naphthoquinone derivatives. Eur. J. Med. Chem. 84, 247–263. 10.1016/j.ejmech.2014.07.024. [DOI] [PubMed] [Google Scholar]
- Singh V. K.; Verma S. K.; Kadu R.; Mobin S. M. (2015) Identification of unusual C–Cl···π contacts in 2-(alkylamino)-3-chloro-1,4-naphthoquinones: effect of N-substituents on crystal packing, fluorescence, redox and anti-microbial properties. RSC Adv. 5, 43669–43686. 10.1039/C5RA02295A. [DOI] [Google Scholar]
- Shrestha-Dawadi P. B.; Bittner S.; Fridkin M.; Rahimipour S. (1996) On the synthesis of naphthoquinonyl heterocyclic amino acids. Synthesis 1996, 1468–1472. 10.1055/s-1996-4417. [DOI] [Google Scholar]
- Rivera-Ávalos E.; de Loera D.; Araujo-Huitrado J. G.; Escalante-García I. L.; Muñoz-Sánchez M. A.; Hernández H.; López J. A.; López L. (2019) Synthesis of amino acid-naphthoquinones and in vitro studies on cervical and breast cell lines. Molecules 24, 4285. 10.3390/molecules24234285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havlíčková L.; Arient J. (1970) Anthraquinone dyes. Part VI. Nuclear alkylation of 1-aminoanthraquinone leuco-derivative. J. Chem. Soc. C 570–572. 10.1039/J39700000570. [DOI] [Google Scholar]
- Carta A.; Palomba M.; Boatto G.; Busonera B.; Murreddu M.; Loddo R. (2004) Synthesis and antiproliferative activity of 3-aryl-2-[1H(2H)-benzotriazol-1(2)-yl]acrylonitriles variously substituted: Part 4. Farmaco 59, 637–644. 10.1016/j.farmac.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Carpenter T. S.; Kirshner D. A.; Lau E. Y.; Wong S. E.; Nilmeier J. P.; Lightstone F. C. (2014) A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys. J. 107, 630–641. 10.1016/j.bpj.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.; Fan K.; McConnell O. J.; Carter G. T. (2003) High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 38, 223–232. 10.1016/S0223-5234(03)00012-6. [DOI] [PubMed] [Google Scholar]
- Cellamare S.; Stefanachi A.; Stolfa D. A.; Basile T.; Catto M.; Campagna F.; Sotelo E.; Acquafredda P.; Carotti A. (2008) Design, synthesis, and biological evaluation of glycine-based molecular tongs as inhibitors of Abeta1-40 aggregation in vitro. Bioorg. Med. Chem. 16, 4810–4822. 10.1016/j.bmc.2008.03.052. [DOI] [PubMed] [Google Scholar]
- Ellman G. L.; Courtney D.; Andres V.; Feather-Stone R. M. (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95. 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Convertino M.; Pellarin R.; Catto M.; Carotti A.; Caflisch A. (2009) 9,10-Anthraquinone hinders beta-aggregation: how does a small molecule interfere with Abeta-peptide amyloid fibrillation?. Protein Sci. 18, 792–800. 10.1002/pro.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzer-Attali R.; Convertino M.; Pellarin R.; Gazit E.; Segal D.; Caflisch A. (2013) Methylations of tryptophan-modified naphthoquinone affect its inhibitory potential toward Aβ aggregation. J. Phys. Chem. B 117, 1780–1789. 10.1021/jp309066p. [DOI] [PubMed] [Google Scholar]
- Bolognino I.; Giangregorio N.; Pisani L.; de Candia M.; Purgatorio R.; Tonazzi A.; Altomare C. D.; Cellamare S.; Catto M. (2019) A prospective repurposing of dantrolene as multitarget agent for Alzheimer’s disease. Molecules 24, 4298. 10.3390/molecules24234298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasial S.; Hu Y.; Bajorath J. (2017) How frequently are pan-assay interference compounds active? Large-scale analysis of screening data reveals diverse activity profiles, low global hit frequency, and many consistently inactive compounds. J. Med. Chem. 60, 3879–3886. 10.1021/acs.jmedchem.7b00154. [DOI] [PubMed] [Google Scholar]
- Pisani L.; Iacobazzi R. M.; Catto M.; Rullo M.; Farina R.; Denora N.; Cellamare S.; Altomare C. D. (2019) Investigating alkyl nitrates as nitric oxide releasing precursors of multitarget acetylcholinesterase-monoamine oxidase B inhibitors. Eur. J. Med. Chem. 161, 292–309. 10.1016/j.ejmech.2018.10.016. [DOI] [PubMed] [Google Scholar]
- Chuang H. Y.; Patek D. R.; Hellerman L. (1974) Mitochondrial monoamine oxidase. Inactivation by pargyline. Adduct formation. J. Biol. Chem. 249, 2381–2384. [PubMed] [Google Scholar]
- Hubálek F.; Binda C.; Li M.; Herzig Y.; Sterling J.; Youdim M. B.; Mattevi A.; Edmondson D. E. (2004) Inactivation of purified human recombinant monoamine oxidases A and B by rasagiline and its analogues. J. Med. Chem. 47, 1760–1766. 10.1021/jm0310885. [DOI] [PubMed] [Google Scholar]
- Estrada-Valencia M.; Herrera-Arozamena C.; Pérez C.; Viña D.; Morales-García J. A.; Pérez-Castillo A.; Ramos E.; Romero A.; Laurini E.; Pricl S.; Rodríguez-Franco M. I. (2019) New flavonoid - N,N-dibenzyl(N-methyl)amine hybrids: Multi-target-directed agents for Alzheimer′s disease endowed with neurogenic properties. J. Enzyme Inhib. Med. Chem. 34, 712–727. 10.1080/14756366.2019.1581184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada Valencia M.; Herrera-Arozamena C.; de Andrés L.; Pérez C.; Morales-García J. A.; Pérez-Castillo A.; Ramos E.; Romero A.; Viña D.; Yáñez M.; Laurini E.; Pricl S.; Rodríguez-Franco M. I. (2018) Neurogenic and neuroprotective donepezil-flavonoid hybrids with sigma-1 affinity and inhibition of key enzymes in Alzheimer’s disease. Eur. J. Med. Chem. 156, 534–553. 10.1016/j.ejmech.2018.07.026. [DOI] [PubMed] [Google Scholar]
- Laurini E.; Marson D.; Aulic S.; Fermeglia M.; Pricl S. (2020) Computational Alanine Scanning and Structural Analysis of the SARS-CoV-2 Spike Protein/Angiotensin-Converting Enzyme 2 Complex. ACS Nano 14, 11821–11830. 10.1021/acsnano.0c04674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli M.; Sparatore A.; Basilico N.; Cavicchini L.; Parapini S.; Tasso B.; Laurini E.; Pricl S.; Boido V.; Sparatore F. (2020) Quinolizidine-Derived Lucanthone and Amitriptyline Analogues Endowed with Potent Antileishmanial Activity. Pharmaceuticals 13, 339. 10.3390/ph13110339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg E.; Weber F.; Brune S.; Schepmann D.; Almansa C.; Friedland K.; Laurini E.; Pricl S.; Wünsch B. (2019) Synthesis and Structure-Affinity Relationships of Spirocyclic Benzopyrans with Exocyclic Amino Moiety. J. Med. Chem. 62, 4204–4217. 10.1021/acs.jmedchem.9b00449. [DOI] [PubMed] [Google Scholar]
- Genini D.; Brambilla L.; Laurini E.; Merulla J.; Civenni G.; Pandit S.; D’Antuono R.; Perez L.; Levy D. E.; Pricl S.; Carbone G. M.; Catapano C. V. (2017) Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. U. S. A. 114, E4924–E4933. 10.1073/pnas.1615730114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons D. L.; Pricl S.; Posocco P.; Laurini E.; Fermeglia M.; Sun H.; Talpaz M.; Donato N.; Quintás-Cardama A. (2014) Molecular dynamics reveal BCR-ABL1 polymutants as a unique mechanism of resistance to PAN-BCR-ABL1 kinase inhibitor therapy. Proc. Natl. Acad. Sci. U. S. A. 111, 3550–3555. 10.1073/pnas.1321173111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Joerger A. C.; Boeckler F. M. (2013) Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 56, 1363–88. 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- Ittner L. M.; Götz J. (2011) Amyloid-β and tau--a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 12, 67–72. 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- Guo J. P.; Arai T.; Miklossy J.; McGeer P. L. (2006) Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 103, 1953–1958. 10.1073/pnas.0509386103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blurton-Jones M.; Laferla F. M. (2006) Pathways by which Abeta facilitates tau pathology. Curr. Alzheimer Res. 3, 437–448. 10.2174/156720506779025242. [DOI] [PubMed] [Google Scholar]
- Dammers C.; Yolcu D.; Kukuk L.; Willbold D.; Pickhardt M.; Mandelkow E.; Horn A. H.; Sticht H.; Malhis M. N.; Will N.; Schuster J.; Funke S. A. (2016) Selection and characterization of Tau binding D-enantiomeric peptides with potential for therapy of Alzheimer disease. PLoS One 11, e0167432. 10.1371/journal.pone.0167432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.; Cowan N.; Kirschner M. (1988) The primary structure and heterogeneity of tau protein from mouse brain. Science 239, 285–288. 10.1126/science.3122323. [DOI] [PubMed] [Google Scholar]
- Adamcik J.; Sánchez-Ferrer A.; Ait-Bouziad N.; Reynolds N. P.; Lashuel H. A.; Mezzenga R. (2016) Microtubule-binding R3 fragment from tau self-assembles into giant multistranded amyloid ribbons. Angew. Chem., Int. Ed. 55, 618–622. 10.1002/anie.201508968. [DOI] [PubMed] [Google Scholar]
- Chen K.; Maley J.; Yu P. H. (2006) Potential implications of endogenous aldehydes in beta-amyloid misfolding, oligomerization and fibrillogenesis. J. Neurochem. 99, 1413–1424. 10.1111/j.1471-4159.2006.04181.x. [DOI] [PubMed] [Google Scholar]
- Pisani L.; Catto M.; De Palma A.; Farina R.; Cellamare S.; Altomare C. D. (2017) Discovery of potent dual binding site acetylcholinesterase inhibitors via homo- and heterodimerization of coumarin-based moieties. ChemMedChem 12, 1349–1358. 10.1002/cmdc.201700282. [DOI] [PubMed] [Google Scholar]
- Robello M.; Amico C.; Cupello A. (1993) Regulation of GABAA receptor in cerebellar granule cells in culture: differential involvement of kinase activities. Neuroscience 53, 131–138. 10.1016/0306-4522(93)90291-M. [DOI] [PubMed] [Google Scholar]
- Berben P.; Bauer-Brandl A.; Brandl M.; Faller B.; Flaten G. E.; Jacobsen A. C.; Brouwers J.; Augustijns P. (2018) Drug permeability profiling using cell-free permeation tools: Overview and applications. Eur. J. Pharm. Sci. 119, 219–233. 10.1016/j.ejps.2018.04.016. [DOI] [PubMed] [Google Scholar]
- Case D. A., Belfon K., Ben-Shalom Y., Brozell S. R., Cerutti D. S., Cheatham T. E. III, Cruzeiro V. W. D., Darden T. A., Duke R. E., Giambasu G., Gilson M. K., Gohlke H., Goetz A. W., Harris R., Izadi S., Izmailov S. A., Kasavajhala K., Kovalenko A., Krasny R., Kurtzman T., Lee T. S., LeGrand S., Li P., Lin C., Liu J., Luchko T., Luo R., Man V., Merz K. M., Miao Y., Mikhailovskii O., Monard G., Nguyen H., Onufriev A., Pan F., Pantano S., Qi R., Roe D. R., Roitberg A., Sagui C., Schott-Verdugo S., Shen J., Simmerling C. L., Skrynnikov N. R., Smith J., Swails J., Walker R. C., Wang J., Wilson L., Wolf R. M., Wu X., Xiong Y., Xue Y., York D. M., and Kollman P. A. (2020) AMBER 2020, University of California, San Francisco, CA, U.S.
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Reis J.; Manzella N.; Cagide F.; Mialet-Perez J.; Uriarte E.; Parini A.; Borges F.; Binda C. (2018) Tight-Binding Inhibition of Human Monoamine Oxidase B by Chromone Analogs: A Kinetic, Crystallographic, and Biological Analysis. J. Med. Chem. 61, 4203–4212. 10.1021/acs.jmedchem.8b00357. [DOI] [PubMed] [Google Scholar]
- Cheung J.; Rudolph M. J.; Burshteyn F.; Cassidy M. S.; Gary E. N.; Love J.; Franklin M. C.; Height J. J. (2012) Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 55, 10282–10286. 10.1021/jm300871x. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Massova I.; Kollman P. A. (2000) Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discovery Des. 18, 113–135. 10.1023/A:1008763014207. [DOI] [Google Scholar]
- Tsui V.; Case D. A. (2000) Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 56, 275–291. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.