

Abstract
Prostate cancer is diagnosed mostly in men over the age of 50 years, and has favorable 5-year survival rates due to early cancer detection and availability of curative surgical management. However, progression to metastasis and emergence of therapeutic resistance are responsible for the majority of prostate cancer mortalities. Recent advancement in sequencing technologies and computational capabilities have improved the ability to organize and analyze large data, thus enabling the identification of novel biomarkers for survival, metastatic progression and patient prognosis. Large-scale sequencing studies have also uncovered genetic and epigenetic signatures associated with prostate cancer molecular subtypes, supporting the development of personalized targeted-therapies. However, the current state of mainstream prostate cancer management does not take full advantage of the personalized diagnostic and treatment modalities available. This review focuses on interrogating biomarkers of prostate cancer progression, including gene signatures that correspond to the acquisition of tumor lethality and those of predictive and prognostic value in progression to advanced disease, and suggest how we can use our knowledge of biomarkers and molecular subtypes to improve patient treatment and survival outcomes.
Introduction
The introduction of prostate specific antigen (PSA) in 1987 has led to prostate cancer (PCa) becoming the most commonly diagnosed malignancy in men in the United States, and it is predicted to have the second highest cancer-related mortality in men after lung cancer [1–4]. Although advancements in diagnostic and imaging technology have allowed PCa to be discovered in earlier stages, it remains a disease that afflicts primarily older men, with only 9.2% of all cases between 2012 and 2016 diagnosed in men younger than 55 years of age [2, 3]. The probability of developing invasive PCa also increases with age, with men over 70 years old having an 8.2% chance of developing invasive PCa, whereas men in younger age group (50–59 years-old) have only a 1.8% chance [4]. Most patients diagnosed with PCa have favorable prognoses, with an estimated mean 5-year survival rate of 98% based on data from the most recent Surveillance, Epidemiology, and End Results registry [3]. This high survival rate is attributed to the high incidence of localized disease at diagnosis, for which curative treatment with surgery or radiation achieves a 5-year survival rate of 100% [3]. In contrast, patients diagnosed with distant metastatic disease had a 5-year survival rate of 30.5% [3].
Tumor recurrence after primary treatment is a major cause of mortality in PCa, significantly contributing to the halted decline in PCa mortality rates in recent years [4]. Although androgen deprivation therapy (ADT) in the form of gonadotropin-releasing hormone (GnRH) and androgen receptor (AR) antagonists can improve patient survival and clinical outcomes, therapeutic resistance and lethal disease eventually develop in most cases as they progress to castration resistant prostate cancer (CRPC) and metastatic CRPC (mCRPC). The overall survival of patients with advanced mCRPC is variable, ranging from a few months to a few years, and particularly diverse in racial minorities with a significantly higher incidence and lethal disease among African American patients, compared to their white counterparts [5]. Therefore, genomic profiling using plasma DNA or circulating tumor cells (CTCs) can be critically important in guiding clinical decision making by improving our understanding of treatment resistance profiles, metastatic potential, personalized therapeutics, and patient prognosis with noninvasive issue biopsy [6, 7]. In the last decade, bioinformatics and system biology approaches have allowed for significant advancements in PCa risk stratification, treatment strategies, and disease monitoring toward personalized therapy. In this review, we examine PCa specific gene signatures associated with treatment resistance and tumor recurrence as predictive markers of clinical outcomes. We will also discuss advantages of big data analysis and tumor subtyping toward advancing personalized treatment strategies to overcome lethal PCa.
Contributors to tumor journey to metastasis
Growth kinetics: cell proliferation and apoptosis
Increased tumor burden by way of increased proliferation and decreased tumor cell death is a key driver of PCa aggressiveness. Although tumor migration via epithelial to mesenchymal transition (EMT) is generally associated with decreased proliferation, tumor growth and proliferation is necessary for the establishment of new tumor colonies at distant sites and the development of metastases [8]. A highly aggressive PCa Pb-Cre4 mouse model, harboring Pten and Smad4 (Ptenpc−/−Smad4pc−/−) conditional knockout alleles developed prostatic intraepithelial neoplasia (PIN) lesions at 7 weeks of age, focally invasive PCa at 11- weeks, and ultimately highly aggressive PCa with invasive characteristics by 15-weeks, with death resulting at 32 weeks of age. Ptenpc−/−Smad4pc−/− mice also demonstrated greater metastatic potential compared to Ptenpc−/− prostate tumor-bearing mice, developing metastases to draining lumbar nodes in all cases and lung metastases in a subset of cases [9]. Interestingly, cyclin D1 (CCND1) and osteopontin (SPP1) levels are significantly increased in the metastatic Ptenpc−/−Smad4pc−/− model compared to the Ptenpc−/− mice, and enforced expression of CCND1 and SPP1 significantly enhanced the in vivo prostate tumor xenograft proliferation and invasion, respectively [9]. Other cell growth and proliferation genes in addition to CCND1 are also highly enriched in the metastatic Ptenpc−/−Smad4pc−/− PCa transcriptome, leading to neutralization of oncogene-induced senescence, as a consequence of increased tumor proliferation. Combined with the Gleason score, this four-gene signature consisting of PTEN, SMAD4, CCND1, and SPP1 improves prognostic accuracy for biochemical recurrence (BCR) and development of metastasis, and highlights the “functional intimacy” and interactive rigor in the dynamic interplay between the process of tumor growth and invasion.
In addition to cellular growth, resistance of primary tumor epithelial cells to anoikis is integral to the initiation of metastatic spread. Defined as apoptosis induced by cellular detachment from the surrounding extracellular matrix (ECM), anoikis is a major barrier to cancer metastasis as tumor cells must survive in circulation after extravasation from the primary site. Aberrant activation of receptor tyrosine kinase signaling, MAPK signaling, and Ras family of small GTPases have been linked to anoikis-resistance across a variety of cancers [10]. Though the underlying mechanism for PCa anoikis is not yet fully understood, the heart of PCa anoikis resistance is dependent on aberrant anti-apoptotic signaling in the ECM-integrin cell survival pathway, the mitochondrial-mediated survival pathway, as well as an increase in EMT activity that activate similar downstream signaling as seen in other tumor types [11–15]. Basal cells that reside between the basement membrane and the luminal epithelial cells are prime examples of cells that escape detachment-mediated apoptosis by expressing cell surface adhesion molecules and ECM proteins that allow for cell-autonomous interactions. Moreover, prostate basal cells exhibit an inherently mesenchymal phenotype, a pro-EMT gene signature, and high levels of MAPK and AKT activity that also protect them against anoikis [16]. In contrast, luminal epithelial cells make up a terminally differentiated cell population without the ability to regenerate, and is therefore highly susceptible to anoikis-related cell death. Interestingly, dissociated murine luminal epithelial cells overexpressing Notch intracellular domain (NICD) can become anoikis-resistant, gaining the ability to form luminal cell-predominant spheres that appear morphologically similar to stable basal cell spheres. Furthermore, a small percentage of NICD-expressing luminal cells demonstrate enhanced survival and proliferation, likely due to increased PI3K-AKT activity caused by Notch-mediated Pten repression [16]. Interestingly, increased Notch activity could be a driver for the development of mCRPC, as Notch targets Hey1/2 and HeyL can act as AR corepressors.
Nuclear factor (NF)-κB activity has also been implicated in mediating anoikis resistance, both as a downstream target of Notch activity via a Hes1-dependent mechanism and also through a pro-inflammatory mechanism involving overexpression of leukotriene B4 receptor-2 (BLT2) [17]. Sphere-forming luminal cells that overexpress NICD showed approximately a twofold increase in NF-κB activity, as well as increased expression of anti-apoptotic, NF-κB targets Bcl-2, Bcl-xL, and cIAP1 [16]. In anoikis-resistant PC3 prostate tumor cells, cell detachment resulted in a time-dependent increase in BLT2 expression and subsequent generation of NADPH oxidase (NOX)-mediated reactive oxygen species (ROS) [17]. This BLT2-ROS cascade results in redox-initiated NF-κB activation in detached PC3 cells and subsequent upregulation of anti-apoptotic signals in the absence of cell attachment. Though Lee et al. demonstrated upregulated NF-κB activity in luminal epithelial cells that overexpress NICD, NF-κB-mediated anoikis-resistance is generally more commonly associated with cells with progenitor characteristics with inherently increased NF-κB signaling [18].
Phenotypic interconversions of EMT to MET
Human PCa invasion and metastatic progression is causally driven by EMT and phenotypic interconversion to mesenchymal to epithelial transition (MET). Originally identified as a critical phenomenon during embryonic development, EMT is phenotypically defined by the loss of epithelial traits and the gain of mesenchymal characteristics orchestrated by a complex series of genetic, epigenetic, and post-translational events within the tumor and its microenvironment [19]. The EMT phenotype is characteristically activated in invasive carcinoma, and the emerging journey to metastasis subsequently engages phenotypic configurations with dynamic EMT interconversions to MET. In epithelial cancers, EMT induction confers a cancer stem-cell phenotype, enabling direct colonization of foreign tissue sites and consequential metastasis formation, acquisition of therapeutic resistance, and self-renewal capabilities. Loss of tumor E-cadherin expression is a well-known hallmark of EMT initiation contributing to tumor invasive and metastatic properties [20]. In PCa, E-cadherin functions to preserve cellular organization by recruiting polarity-determining proteins to the lateral cell membrane, including Scribble cell polarity protein and spindle positioning determinant leucine-glycine-asparagine repeat protein [21]. Loss of E-cadherin due to disruption of cell-cell adherence interactions (Fig. 1) results in detachment-induced cellular division and can directly lead to the formation of PIN lesions in mice with subsequent carcinogenesis [21, 22]. E-cadherin loss of protein expression in PCa cells is commonly the result of transcriptional repression. For example, Wilms tumor 1 (WT1) transcription factor and trefoil factor 1 (TFF1) are important promoters of EMT and have been shown to decrease E-cadherin expression via direct transcriptional modulation of the E-cadherin promoter PCa cell lines [23]. Enhancer of Zeste homolog 2 (EZH2), a methyltransferase catalytic component of Polycomb repressive complex 2 and a known marker of aggressive PCa phenotypes, can also directly silence E-cadherin expression via promoter methylation [11, 24, 25]. This is initiated by eHsp90-dependent activation of MEK/ERK, which facilitates the recruitment of EZH2 to the E-cadherin promoter [11].
Fig. 1. Molecular contributors to prostate cancer aggressiveness.
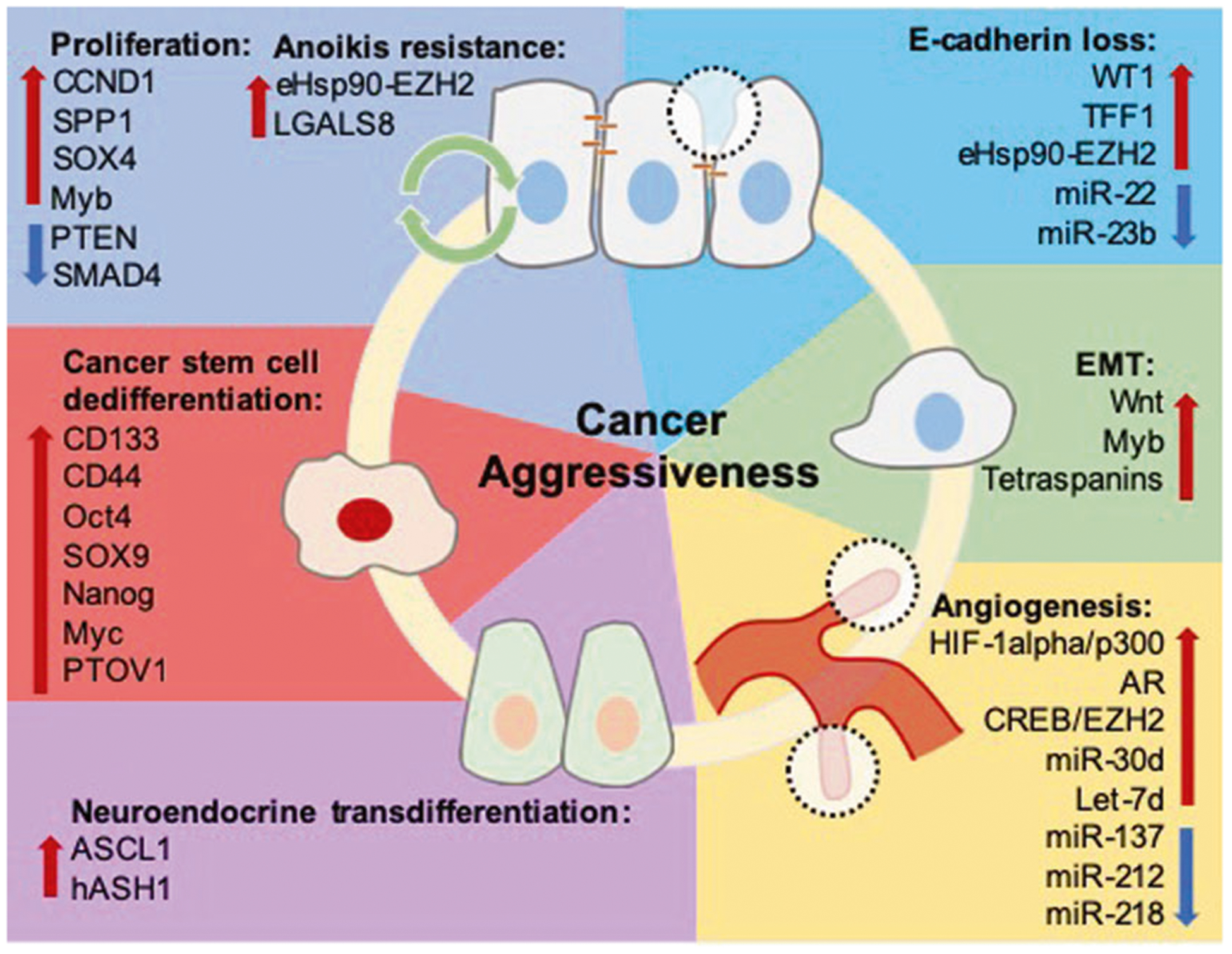
Tumor proliferation, apoptosis/anoikis evasion, and EMT are functional contributing process to tumor recurrence, invasion, and metastasis. Genes including CCND1, SPP1, SOX4, and Myb promote tumor proliferation and survival, with Myb also demonstrating a role in enhancing survival in the context of ADT-resistance. SMAD4 and PTEN are known tumor suppressors, and decreased expression of these genes is known to increase prostate cancer growth and invasion. Resistance to anoikis-related cell death is crucial for tumor survival in the circulation during metastasis. Overactivation of the eHsp90-EZH2 pathway and overexpression of LGALS8 have been shown to promote tumor cell survival and invasion. WT1 and TFF1 promote EMT by decreasing E-cadherin expression via direct transcriptional modulation, and eHsp90 upregulates expression of EZH2, which decreases E-cadherin expression by direct interaction with the E-cadherin promoter to enhance prostate cancer invasion. miR-22 and miR-23b have tumor suppressor roles and their loss is associated with decreased E-cadherin expression. Consequential to E-cadherin loss, β-catenin is released from the plasma membrane and canonical Wnt signaling is activated, leading to acquisition of invasive properties. Myb is associated with promotion of filopodia formation, and tetraspanins such as TSPAN1 and CD151 are also associated with increased expression of classical EMT machinery, promoting cellular motility and dissolution of cell polarity. VEGFA is a direct target of HIF-1α and is transcriptionally regulated by AR, and the HIF-1α/p300 pathway and AR-signaling promotes VEGFA production. In the context of ADT-resistance, the CREB/EZH2 axis promotes vascularization by allowing tumor cells to bypass androgen dependent angiogenesis. miR-30d and let-7d are pro-angiogenic and associated with increased tumor vascularity, while miR-137, miR-212, and miR-218 have inhibitory roles in angiogenesis and are frequently downregulated in prostate cancer. Finally, transdifferentiation and acquisition of a NE phenotype is a critical means of achieving ADT-resistance. ASCL1 and hASH1 are cell fate regulators that respond to decreased androgen signaling by downstream formation of NE characteristics. CSCs exhibit resistance to chemotherapy, and CD133+ cells demonstrating DTX-resistance often have increased CD44, Oct4, SOX9, and Nanog expression. Expression of renewal and stemness genes such as Nanog and Myc are also induced by PTOV1 during CSC formation.
In addition to E-cadherins, the tretraspanin protein family has been recently recognized as valuable biomarkers in PCa progression [26, 27]. Tetraspanins are cell-surface proteins with important roles in maintaining cellular organization and structure, including strengthening integrin-mediated cellular adhesion, cell morphology, motility, and tumor progression. Consequential to the complexity of interacting molecular networks navigating the tumor cell surface, integrin-associated tetraspanin CD151 remains controversial regarding its clinical significance and functional impact in human PCa progression to lethal disease. Induced by androgens, Tetraspanin 1 (TSPAN1) is significantly upregulated in PCa compared to normal prostate and benign prostatic hyperplasia. Increased TSPAN1 expression causes downstream upregulation of EMT-related genes such as SLUG and ARF6, leading to cell invasion and metastatic progression [26]. Specifically, tetraspanin CD151 has been functionally implicated in initiating the metastatic journey of PCa and studies in pre-clinical models (CD151-KO) demonstrated that loss of CD151 leads to decreased prostate tumor metastases to the lung [27]. However, recent studies integrating the dynamics of CD151 protein localization in clinical PCa cohorts with bioinformatic interrogation of the TCGA database, and functional evaluation at the cellular level, revealed that CD151 serves as a tumor suppressor in PCa progression. Of major translational significance was the observation that in primary prostate tumors from patients with metastatic disease, CD151 largely localized at cell-cell junctions was inversely associated with Gleason grade and tumor stage [28].
Angiogenesis: sustaining tumor vascularity
Angiogenesis is critically important for PCa survival, proliferation, and invasion. In tumor angiogenesis, poor vessel integrity impairs delivery of oxygen, nutrients, and drugs, while also facilitating tumor intravasation [29]. Impaired oxygen levels stimulate a hypoxic response, causing further tumor secretion of vascular endothelial growth factor A (VEGFA) and the initiation of a vicious pro-angiogenic cycle [29]. Earlier studies have established that in PCa that increased levels of androgens and activated AR signaling play critical roles in upregulating vascular endothelial growth factor (VEGF) expression. Thus, VEGFA is transcriptionally regulated by AR, and overexpression of AR coregulator KDM1A promotes VEGFA expression to promote PCa recurrence [30]. This dependency on AR signaling also accounts for the high vascularity observed in CRPC, as most CRPC tumors exhibit aberrant AR activation in the setting of androgen deprivation [31]. Interestingly, angiogenesis remains active in CRPC tumors exhibiting a neuroendocrine (NE) phenotype despite low AR signaling, as NE CRPCs are known to be highly vascular. This is attributed to an ADT-mediated increase in CREB (cAMP response element-binding protein) activity, leading to NE marker expression, notably EZH2, and subsequent transition to a NE phenotype [32, 33]. This series of events allows CRPCs to bypass androgen-dependent angiogenesis by direct epigenetic silencing of anti-angiogenic factor TSP1 via a CREB/EZH2/TSP1 axis. It is also useful to note that TSP1 is highly expressed in mCRPC compared to localized PCa, making it a potential biomarker for identifying aggressive CRPC subtypes [33].
Molecular determinants of tumor progression and therapeutic resistance
An array of molecular markers with high prognostic significance in PCa are typically genes associated with the development of aggressive metastatic disease, such as those functionally contributing to the regulation of phenotypic EMT, proliferation, invasion, resistance to apoptosis, and anoikis evasion (as schematically shown on Fig. 1) [34–37]. A classic example, is evidence by Cuzick et al. who created a proliferation-based prognostic score derived from mRNA expression levels of 31 genes involved in cell cycle progression (CCP), including TOP2A, RRM2, and BIRC5 that were previously associated with PCa prognosis [36]. These genes were selected based on their significant “signature expression profile” correlation, resulting in a get set that is robust in measuring tumor proliferation, but containing little information related to other causes for tumor aggressiveness. Nonetheless, this signature significantly predicted time to BCR and mortality in a retrospective study of a prostatectomy cohort and a conservatively managed transurethral resection (TURP) cohort, respectively. However, focusing solely on tumor proliferative ability does not provide a complete picture, as PSA still demonstrated significant prognostic ability independently of the CCP score in the two retrospective cohorts. This suggests that PSA might be capturing information that is unrelated to tumor proliferation, and the combination of PSA with markers that measure tumor proliferative ability can greatly improve the accuracy in predicting patient prognosis [36].
As previously discussed, the initiation of EMT inevitably leads to a more aggressive tumor type and poor patient outcome, and genes regulating in EMT and tumor invasion are thus valuable markers for patient prognosis as well [38]. Univariate cox regression analysis of a 230-case cohort showed that overexpression of SOX4, a member of the SOX family of transcription factors associated with increased cell proliferation and EMT, can significantly predict PCa mortality [39]. In addition, upregulation of SPOCK1 (Sparc/osteonectin, cwcv, and kazal-like domains proteoglycan 1), an oncogene encoding a protein member of the SPARC (secreted protein, acidic, cysteine-rich) family, has been associated with shorter disease free survival time in PCa likely due to SPOCK1-mediated upregulation of EMT [40]. Hypoxia within the tumor microenvironment seems to play a major role as an inducer of EMT, and has been associated with selecting for aggressive tumor cell survival [41]. Estrogen receptor β1 (ERB1) has been shown to negatively regulate hypoxia-induced EMT by repressing VEGFA transcription via regulating key response elements in the VEGFA promoter as well as destabilizing hypoxia inducible factor 1α (HIF-1α). In addition, one must consider that expression of ERB1 in the prostate glandular epithelium, can prevent EMT induction by preserving an epithelial phenotype, via its ability to sustain E-cadherin transcription, and sequester the transcriptional regulator Snail1 in the cytoplasm [41]. Interestingly, the β2 variant of the estrogen receptor (ERB2) stabilizes HIF-1α protein expression to promote a hypoxia gene signature in a normoxia state, assigning layers of functional complexity to the role of the ERB protein family members in PCa progression [42].
Finally, noncoding genes can also significantly influence patient outcome due to their regulatory roles in tumor invasion, proliferation, angiogenesis, and apoptosis. Though small nucleolar RNAs and long noncoding RNAs have been shown to play prognostic roles, perhaps the most well-studied subset of noncoding RNAs in PCa are microRNAs (miR) [43–46]. There exists dramatic downregulation of microRNA expression in PCa as a result of global promoter hypermethylation, resulting in the upregulation of many major cancer promoting pathways [47]. miR-200b, a member of the miR-200 family EMT regulators, is one such microRNA silenced via CpG island methylation in PCa [48]. miR-200b upregulation in PCa is associated with decreased tumor aggressiveness and reduced metastasis, likely mediated by the acquisition of epithelial markers such as E-cadherin, leading to EMT reversal. Interestingly, miR-200b is a downstream target of the AR, and its expression is increased in response to increased AR signaling [48]. It would thus be noteworthy to understand the role of miR-200b in mCRPC, especially in those tumors whose resistance is due to paradoxical downregulation of AR signaling. In addition to miR-200b, miR-23b and miR-34b are microRNA regulators of EMT also silenced by hypermethylation. Majid et al. demonstrated that both miR-23b and miR-34b are associated with favorable overall survival and recurrence free survival in PCa as a result of their roles in Akt-mediated inhibition of tumor cell proliferation, migration, invasion, as well as inducing G0/G1 cell cycle arrest, making them valuable prognostic markers with potential for tracking PCa disease progression and predicting patient outcome [37, 49]. A number of angiogenesis regulators are downregulated as well, such as the tumor suppressor miR-137, miR-212, and miR-218 [30, 50, 51]. In contrast, overexpression of pro-angiogenic miR-30d and let-7d is associated with increased tumor vascularity and poor clinical outcomes [52, 53]. Last, one must not forget that resistance to anoikis-mediated cell death is a critical characteristic of cells undergoing EMT. miR-132 methylation in PCa is correlated with high Gleason scores and advanced tumor stage, likely due to its role in maintaining tumor cell-to-ECM integrity, as re-expression of miR-132 in PC3 cells resulted in tumor cell detachment and eventual anoikis-mediated cell death [54].
Resistance to hormone therapy (targeting androgen axis)
Prostate cancer recurrence is an unfortunate clinical outcome after primary surgical and/or radiotherapy, and is monitored by tracking time to PSA doubling, radiographic progression, and nomograms to assess recurrence risk [1]. To minimize the chance of recurrence, ADT is typically administered after primary therapy, and first-line ADT consists of GnRH agonist-induced chemical castration to lower testosterone levels. To enhance the androgen deprivation effects of a GnRH-agonists, combination therapy with a competitive AR antagonist, i.e., an antiandrogen, is frequently administered to minimize acute systemic testosterone surges and improve treatment outcome [31]. Although most patients are initially responsive to ADT, the majority of patients will experience treatment resistance and eventual progression to CRPC [31]. Next generation AR inhibitors such as abiraterone, an inhibitor of the enzyme 17α-hydroxylase required for androgen production, and enzalutamide, an AR antagonist, are currently used in the treatment of CRPC and directly abrogate AR signaling. However, mCRPC development and lethal therapeutic resistance are still major concerns [31].
A major driver of the development of CRPC is the high intratumoral levels of androgens that activate AR signaling even under ADT-depleted conditions. The adrenal glands have been shown to provide circulating androgens in LuCaP35CR and LuCaP96CR mouse CRPC xenograft models, including androstenedione (AED), dehydroepiandrosterone (DHEA), and testosterone, that are found in high levels intratumorally [55]. Castrating these mice did not affect their levels of adrenal androgens or their downstream metabolites, and mice that underwent only castration demonstrated poorer median survival compared those that also underwent adrenalectomy in addition to castration [55]. Although adrenalectomy is usually not a feasible therapeutic option in PCa, this study nonetheless demonstrated the significance of extra-gonadal sources of androgens and their impact on PCa therapeutic resistance.
Perhaps what would be more actionable targets in CRPC are steroidogenic enzymes that contribute to high levels of intratumoral androgens. 17β-hydroxysteroid dehydrogenase type 5 (HSD17B5), a key steroidogenic enzyme encoded by the AKR1C3 gene, facilitates the conversion of weak androgens such as AED and 5α-androstanedione to more active androgens such as testosterone and DHT [56]. HSD17B5 can further promote tumor aggressiveness in CRPC by acting as a coactivator of AR as well as increasing AR-V7 protein expression through enhancing protein stability [57, 58]. Interestingly, AKR1C3 expression is not detected in the normal prostate, but is present at high levels in metastatic PCa and is associated with both abirarterone and enzalutamide resistance [59, 60].
Another important steroidogenic enzyme is 3β-hydroxysteroid dehydrogenase type 1 (3βHSD1), which is critical in the conversion of adrenal DHEA to DHT and catalyzes the rate limiting step in this reaction. A subset of CRPCs express N367T (1245A > C) gain-of-stability mutation, which protects the enzyme from ubiquitin-dependent degradation, leading to significantly increased levels of DHT and subsequent AR activation and ADT resistance [61]. Patients that inherit one or two copies of the HSD3B1 (1245A > C) allele of 3βHSD1 demonstrated significantly decreased progression free survival, metastasis-free survival, and overall survival compared to patients with two wild type copies of this gene [62]. This makes the HSD3B1 (1245C) allele a potential biomarker for predicting ADT resistance and rapid development of CRPC.
Resistance to ADT can also be attributed to reactivation of AR signaling as a result of gain of function mutations, AR gene amplification, structural alterations, or, paradoxically, even loss of AR expression and/or downstream AR signaling [63]. For example, patients with prior abiraterone exposure demonstrated increased AR expression and AR nuclear localization that is associated with upregulation of tumor Ki-67 expression and ADT resistance [64]. The presence of the AR splice variant 7 (AR-V7), which contains a truncated version of the ligand-binding domain, also predicts lack of clinical benefit to AR axis-targeted therapies (AATT) as well as antiandrogen cross-resistance in patients with mCRPC [65–67].
In addition to AR pathway alterations, the development of neuroendocrine prostate cancer (NEPC) is another mechanism of ADT-resistance and is highly correlated with exposure to androgen deprivation [68]. Similar to CRPCs and primary PCa, a large portion of NEPCs exhibit ERG rearrangements and PTEN deletions. However, NEPCs have little to no AR expression, and consequently absent PSA as well [68, 69]. Instead, NEPCs exhibit distinct NE lineage markers, including neuronal-specific enolase, chromsogranin A, and synaptophysin, as well as loss of tumor suppressors such as RB1 and TP53 [32, 70, 71]. Overexpression of N-Myc (MYCN) is also a defining characteristic of NEPCs and is a key driver of NEPC aggressiveness. N-Myc repression of AR signaling not only leads to prostate tumor desensitization to androgen antagonists and castration therapy, it also regulates AR-AKT crosstalk by downregulating AR target FKBP5, a scaffolding protein for AKT and PHLPP that promotes PHLPP dephosphorylation of AKT to negatively regulate AKT activity [72, 73]. Consequently, increased N-Myc expression in mouse PCa organoids results in increased phosphorylated-AKT levels and increased AKT activity, making PI3K/AKT inhibitors a potentially highly effective treatment strategy in NEPC [74]. Finally, recent studies have demonstrated the close relationship between N-Myc and Aurora kinase A (AURKA) coexpression. Independent from its catalytic activity, Aurora-A stabilizes N-Myc, and treatment with Aurora-A inhibitor MLN8237 resulted in rapid N-Myc degradation in LNCaP-N-Myc PCa cells [68, 74].
In addition, cell fate regulators ASCL1 and hASH1 are also closely involved in the development of NE-like features in CRPC after androgen deprivation. Decreased androgen signaling induces ASCL1 transcript and protein expression as well as hASH1 nuclear localization in LNCaP PCa cells, leading to the development of NE characteristics such as neuronal projections [75]. Moreover, ASCL1 expression was persistently elevated post-ADT even after the reintroduction of androgen, and further rounds of ADT mimicking cycles of intermittent ADT (iADT) caused incremental increases in hASH1 expression [75]. This cellular response supports that iADT may actually enhance tumor aggressiveness by rapidly inducing NE transdifferentiation and clinical emergence of CRPC with each round; thus the ability to trace ASCL1/hASH1 expression during iADT could be of relevant value in predicting prognosis in advanced disease [75].
Interestingly, MMP9 expression in androgen deprived (androgen-sensitive) LNCaP cells decreased with androgen supplementation, suggesting that presence of androgens may decrease the invasive potential of PCa epithelial cells [75]. This finding aligns well with recent evidence showing that enzalutamide promotes EMT by decreasing E-cadherin expression in mCRPC, in addition to inducing NE marker expression [76]. Not only EMT contributes to the emergence of mCRPC, but one must also consider that the EMT phenotype been causally linked to accelerated development of therapeutic resistance, treatment failure and ultimately and lethal disease [77].
Last but not least, there is compelling evidence to support that prostate tumors can acquire ADT-resistance by the process of dedifferentiation into a stem cell-like state [63, 78–81]. Indeed, a tumor subpopulation identified by ALDH-hi, CD44+, α2β1+ characteristics demonstrated cancer stem cell (CSC)-like qualities such as enhanced tumor-initiation and tumor-propagation in androgen-ablated male mice [82]. This triple marker positive population is enriched in a LAPC9 cell line-based CRPC model, as well as cells and in primary tumors under androgen-deprived culture conditions [82]. Activation of the Wnt signaling pathway has also been identified a contributor to post-ADT prostate tumor recurrence, and both canonical and non-canonical Wnt signaling are associated with treatment failure after ADT [83, 84]. Wnt activation and signaling opposes the antiproliferative effects of ADT, and data from single cell RNA sequencing of CTCs demonstrated both intra- and inter-patient heterogeneity of non-canonical (nc) Wnt signaling activation in recurrent disease [84]. Patients undergoing ADT had increased Wnt5a ligand expression and ncWnt signaling relative to controls and Wnt5a is induced by the second generation antiandrogen, enzalutamide [84]. In abiraterone-resistant mCRPC, components of the canonical Wnt/β-catenin pathway were frequently mutated, and negative regulators of Wnt/B-catenin were found to be downregulated or deleted [83].
Resistance to chemotherapy
Acquired resistance to 1st line chemotherapy docetaxel (DTX) is a major driver of patient mortality in CRPC. RNA sequencing of DTX-sensitive and DTX-resistant PCa cell lines demonstrated increased expression of CSC-associated genes such as NES, TSPAN8, DPP4, DNAJC12, and MYC in DTX-resistant cells [85]. CD133 (prominin-1), a biomarker of DTX-resistance, is also present in cells expressing markers for CSC (CD44, OCT4, SOX9, NANOG), EMT (c-myc, BMI1), and osteoblastic differentiation (Runx2) and skeletal morphogenesis (BMP2) [86]. DTX-resistant CD133+ cells have greater proliferative potential, as demonstrated by increased Ki-67 expression in CD133+ CTCs compared to CD133− CTCs [87]. In addition, Expression of renewal and stemness genes such as MYC, NANOG, ALDH1A1, and LIN28A in DTX-resistant cells can also be induced by PTOV1, a PCa oncogenic protein predictive of metastasis and poor prognosis. Consequently, PTOV1 expression is directly correlated with DTX-resistance, and the simultaneous presence of increased PTOV1, CCNG2, and ALDH1A1 expression in primary tumors is predictive of tumor invasion and metastasis [88]. Other mechanisms of DTX-resistance include loss of p53 function, increased β-tubulin isoform expression, decreased BRCA1 expression, and increased expression of the drug efflux pump ABCB1 (ATP Binding Cassette Subfamily B Member 1) [89].
In the face of DTX-resistance, cabazitaxel (CBZ), a second-line taxane chemotherapy is used to treat patients with mCRPC, though it does not alter disease course but merely prolongs patient survival in late stage disease [90]. Cabazitaxel treatment targets the microtubule depolymerizing kinesins toward inducing mitotic spindle collapse and multinucleation, and resistance is typically the result of mutations in CBZ targets such as tubulin subunits and participants of microtubule assembly, as well as mutations in AR [91]. However, the development of cross-resistance to CBZ in PCa patients previously treated with DTX, leads to PCa patient mortality consequential to taxane chemotherapy. Thus ABCB1 is not only a marker for DTX-resistance, but its overexpression also contributes to the development of CBZ cross resistance, and ABCB1 small molecule inhibitor elacridar can resensitize DTX-resistant cells to CBZ [89]. TUBB3 (Tubulin B 3 Class III) overexpression is also implicated in cross resistance between DTX and CBZ, as treatment with TUBB3 inhibitor LY294002 enhanced PTEN expression and re-sensitized cancer cells to both DTX and CBZ [92].
In contrast to DTX/CBZ cross-resistance, resistance to 2nd line taxane as a monotherapy (CBZ) in PCa patients is not a consequential to antiandrogen treatment [93–96]; rather, reduced AR activity is associated with enhanced sensitivity to CBZ and better patient outcomes [93–96]. Thus CBZ is highly effective in the targeting prostate of under reduced AR activity. Mechanistically 1st and 2nd generation antiandrogens bicalutamide and enzalutamide (respectively) increase therapeutic vulnerability of androgen-independent prostate tumors to CBZ via decreased ABCB1 ATPase activity inhibition [89, 97, 98]. The anti-tumor effects of 2nd line taxane chemotherapy (CBZ) are abolished under high AR activity, implicating an optimized therapeutic benefit with a sequential/combinatorial strategy of CBZ with antiandrogens/ADT, when given in patients with hormone-naïve early stage PCa, not lethal disease [96, 99].
The challenge of biomarker validation
Currently, the modified Gleason classification system is the primary method for clinical characterization of PCa aggressiveness, and serves as the main guide for downstream management [1]. However, due to variables such as insufficient sampling, fragility of biopsy tissue, tissue heterogeneity, and inconsistent biopsy practices, discrepancies are not uncommon between pre-operative and post-prostatectomy GS [1]. Although post-prostatectomy GS is one of the most accurate prognostic tools for disease risk stratification, prostatectomy is invasive and associated with significant risk for patient morbidity and mortality [100]. Blood PSA, though less invasive, does not always reflect the presence or severity of recurrence and is insufficient for purposes of risk stratification and disease monitoring [1, 101]. However, despite favorable PSA levels, tumors can still harbor aggressive characteristics [102]. The shortcomings of the Gleason classification system and serum PSA levels make it necessary to push for more accurate methods for diagnosis, risk stratification, prognosis prediction, and recurrence monitoring.
Identification of critical molecular players in tumor initiation and progression from large-scale data analysis has led to characterization of biomarkers for invasive and therapeutic resistant phenotypes in lethal disease [103–105]. SCNAs, gene fusions, and mutational burden in post-prostatectomy samples are strongly predictive of PCa relapse and poor prognosis [78, 88, 106–114]. Moreover, large-scale transcriptomic studies have identified several gene signatures that predict BCR, lymph node invasion, and metastases, including PCa specific genes BTG2, IGFBP2, SIRT1, MXI1, FDPS, SPINK1, as well as ERG fusion and noncoding RNAs [114–117]. Clinically, these expression-, fusion-, and mutation-based gene signatures can be adapted into a post-RP risk-stratification workflow to identify patients requiring intensive multi-modal therapy before showing radiological or clinical evidence of metastasis.
With advancements in the understanding of disease markers, combining current PSA-based screening protocols with noninvasive biomarker panels will allow a better understanding of the patient’s risk before biopsy or prostatectomy [118, 119]. For instance, CTCs with divergent expression of genes related to DNA repair, cancer stemness, EMT, therapeutic resistance, and tumor-microenvironment crosstalk with potential for prognostic value have been successfully identified [120]. Synaptophysin expression on CTC surface is useful in tracking ADT-resistance due to development of NE characteristics, and detection of AR-V7 expression in CTCs can help identify patients with poor outcomes after AATT treatment [65, 121]. Furthermore, stem and progenitor cell marker CD133, heat shock proteins, clusterin, interleukin-6, and macrophage inhibitory cytokine-1 can also serve as noninvasive, blood-based markers for monitoring taxane-resistance [64, 122]. Prognostic markers in mCRPC patient sera was also shown to perform significantly better than current nomograms in predicting 12- and 24-month survival, and markers in patient urine also show immense promise in PCa detection and risk stratification [7, 123–126]. Lastly, large extracellular vesicles shed by tumor cells can be isolated from plasma of patients with metastatic disease. These vesicles, termed large oncosomes, contain useful biomarkers of disease progression, including AKT1, α-V integrin, and microRNAs that can condition distant sites to create a tumor microenvironment favorable for cancer invasion and metastasis [127–131].
Genomic-based PCa subtyping toward the synthesis of a molecular atlas in the context of precision medicine is not without limitations. A major concern in cancer genomics studies is confidence in distinguishing normal versus tumor tissue, especially in cancers such as PCa where there is extensive tumor heterogeneity. Technologies such as whole genome sequencing and RNA sequencing (RNA-Seq) have provided new insights into point mutations, copy number alterations, and structural variations, as well as patterns of prostate tumor heterogeneity, multifocality, and multiclonality toward the development of treatment resistance [132]. But what neither technology addresses is how to navigate the tremendous clonal heterogeneity and significant inter- and intra-patient variability that impacts tumor progression to lethal disease [133–135]. Both divergent evolution of tumor subpopulations and tumor clones with independent tumor origins are key contributors to PCa heterogeneity [133, 135]. This heterogeneity results in the formation of distinct molecular subtypes, diverse treatment responses, resistance mechanisms, and ultimately, clinical outcomes [136–139]. The Cancer Genome Atlas (TCGA) Research Group identified seven PCa subpopulations with distinct genetic and molecular features [134]. The majority of these features consisted of gene fusion events involving members of the ETS family, with other cases defined by mutations in SPOP, FOXA1, or IDH1 [134]. Transmembrane protease, serine 2 (TMPRSS2) was found to be the most frequent fusion partner in all ETS fusions, and TMPRSS2-ERG fusion expression in the blood is associated with poor response to docetaxel therapy in mCRPC following enzalutamide treatment [76, 134]. Transcriptomic analysis also identified other subtypes including those with PTEN inactivation, SPOP inactivation, and Aurora A kinase, N-myc, or SPINK1 overexpression [140]. Most notably, subtype-based mutation profiles led to the identification of a mismatch repair gene defect and microsatellite instability subtype susceptible to poly(ADP-ribose) polymerase (PARP) inhibitor therapy [141, 142].
Large-scale, multi-omics studies have allowed for the identification of both non-progressive patient subtypes and highly lethal, treatment-resistant CRPCs [143–145]. For instance, the presence of the HSD3B1 (1245C) allele is a predictor of positive patient response to extragonadal androgen ablation with nonsteroidal CYP17A1 inhibitors such as ketoconazole, suggesting a way for personalized treatment stratification CRPC [146]. In addition, AKR1C3 overexpression leads to both abiraterone and enzalutamide resistance due to increased intracrine androgen synthesis, prompting a different therapeutic strategy for affected patients [147]. Finally, analysis of genome-wide expression profiles from prostatectomy or biopsy samples of 19,470 patients led to the discovery of an aggressive, AR-low subtype with shorter time to recurrence and metastasis [148]. The decreased DNA repair activity associated with this subtype led to increased sensitivity to PARP inhibitors, platinum chemotherapy, and radiotherapy [148].
Recognizing the confounding effects of extensive tumor heterogeneity in interpreting bulk sequencing data, single cell RNA-Seq can overcome this hurdle to enable researchers to identify individual cell subpopulations harboring driver mutations, tumor stem cells, or transdifferentiated cell types such as NEPC to a level of granularity not easily feasible with bulk sequencing techniques. Noninvasive, single cell-based analysis of CTCs in blood and urine samples have already demonstrated utilities in tumor diagnosis and disease tracking in PCa and other cancers [84, 149]. In a recent study, genomic profiling of single CTCs collected via apheresis liquid biopsies successfully identified patients with tremendous heterogeneity in copy number aberrations, suggesting the presence of distinct tumor cell lineages within each patient [150]. These cell subpopulations uniquely clustered with each patient’s tumor samples collected over the course of their disease, such as diagnostic prostatectomy, TURP, mCRPC bone biopsy, and lymph node biopsy [150]. Challenging as it might be, predicting the functional cooperation and temporal coordination of multiple drivers of diverse resistance mechanisms, noninvasive techniques such as this allows one to follow the clinical subtype closely and frequently to be able to trace tumor evolution, detect genomic alterations, and ultimately provide personalized intervention prior to the manifestation of clinical symptoms [151]. Methods of effective acquisition of clinical specimens, such as liquid biopsy or circulating tumor DNA, are constantly evolving and therefore deserving of further development and investigation [152].
Finally, the rapid increase of germline testing in PCa has brought several issues to the foreground, including standardization of testing indications, prioritizing gene panels, and the availability of result interpretation and genetic counseling services [153]. The 2019 Philadelphia Prostate Cancer Consensus Conference created a standardized approach to testing indications, which include men with metastatic PCa or family history of hereditary PCa, as well as defining genes such as BRCA2, BRCA1, and other DNA mismatch repair genes as part of a high priority panel for germline mutation testing. However, the role of germline testing for genes with lower aggression potential in PCa still remains unclear, especially considering our limited knowledge in the setting of cancer stage, grade, patient ancestry, or the presence of other inherited tumor syndromes [153]. Although recent works have both acknowledged the importance of patient ethnicity and made great strides in the understanding of ancestry in the context of genomic alteration profiles, disease progression, and patient outcomes, much greater numbers of PCa specimens from minority groups are still needed before we can truly appreciate the many complexities of this disease [154, 155].
Conclusions
Advanced PCa is a strikingly heterogeneous disease in (a) the clinical state that affects patients with varying metastatic burden and symptoms and (b) the mutational landscape of lethal tumors. Thus, incorporating validated biomarkers into workflows and tracing their role in PCa diagnosis, treatment, and therapeutic response, will be critical in improving clinical outcomes, understanding racial disparities, and overcoming lethal disease. The tremendous inter-patient and intra-patient heterogeneity that characterize PCa, calls for avoiding considering further the “one size fits all” concept in efforts to make relevant science-based recommendations in the clinical management of the disease (Fig. 2). Instead, empowered by advanced technology in NGS and big data analysis, personalized profiling methods of patient classification into candidates for surgery versus active surveillance would eliminate much of the current morbidity and mortality associated with both over- and under-treatment. Moreover, genomic sequencing of biopsy or prostatectomy tissue samples will allow for improved risk stratification and precisely guide the selection of subsequent therapeutic approaches tailored for individual patients. Our ongoing efforts focus on the development of a “molecular atlas” based on genomic profiling of CTCs, exosome secretion and plasma tumor transcriptome that would navigate noninvasive tracking of therapeutic response and tumor recurrence, as well as monitoring clinical tumor progression to metastasis and lethal disease.
Fig. 2. Tracing prostate cancer trajectory using a molecular atlas-based personalized strategy.
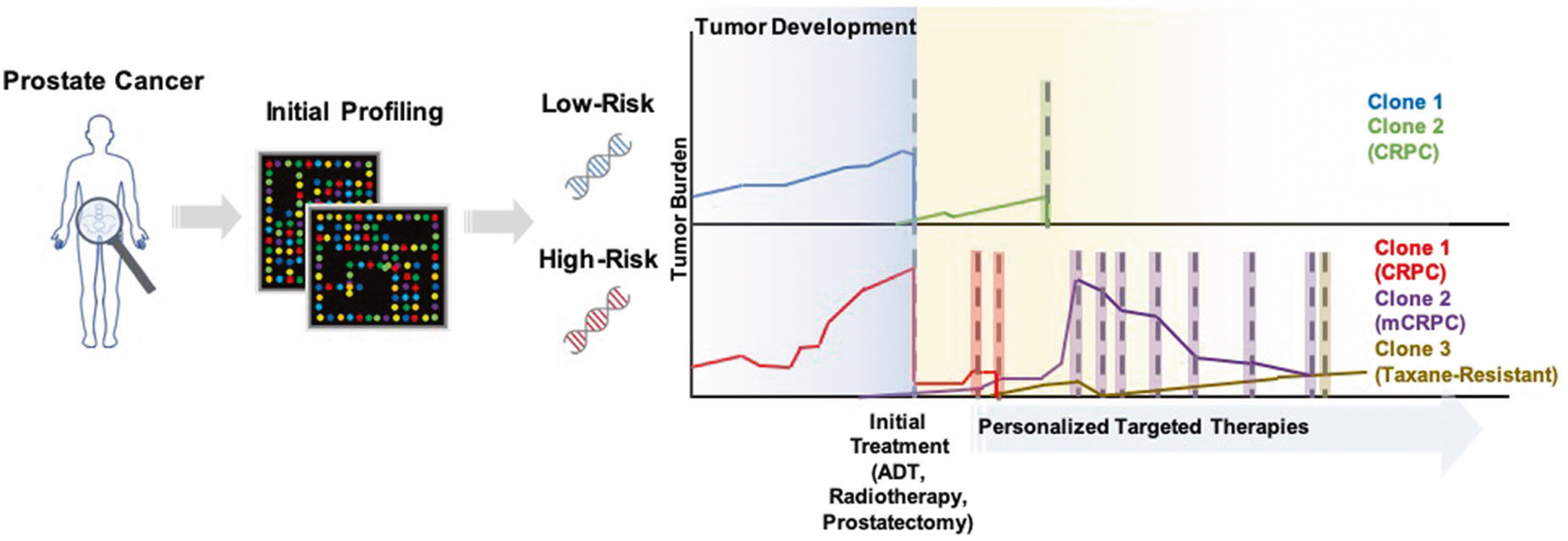
Personalized tumor profiling from the onset of cancer detection can facilitate stratification of patients by disease risk to avoid potential morbidities and mortalities associated with over- and under-treatment. While initial therapies may impart significant decreases in disease burden, continuous monitoring via serial molecular profiling of patient blood and urine for CTCs, exosomes, and/or tumor DNA is critical for timely identification of disease recurrence and new tumor populations that may arise. The continued compilation of a patient-specific molecular profile throughout the disease course will also encourage early identification of potential therapeutics and timely initiation of treatments to control disease progression.
Funding
NIH/NCI R01 CA232574.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Oon SF, Pennington SR, Fitzpatrick JM, Watson RW. Biomarker research in prostate cancer–towards utility, not futility. Nat Rev Urol. 2011;3:131–8. [DOI] [PubMed] [Google Scholar]
- 2.Salinas CA, Tsodikov A, Ishak-Howard M, Cooney KA. Prostate cancer in young men: an important clinical entity. Nat Rev Urol. 2014;6:317–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howlader NNA, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, et al. (eds). SEER Cancer Statistics Review (CSR), 1975–2016. National Cancer Institute, Bethesda, MD: [online]. 2019. [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;1:7–30. [DOI] [PubMed] [Google Scholar]
- 5.Merrill RM, Lyon JL. Explaining the difference in prostate cancer mortality rates between white and black men in the United States. Urology. 2000;5:730–5. [DOI] [PubMed] [Google Scholar]
- 6.Sun YF, Yang XR, Zhou J, Qiu SJ, Fan J, Xu Y. Circulating tumor cells: advances in detection methods, biological issues, and clinical relevance. J Cancer Res Clin Oncol. 2011;8:1151–73. [DOI] [PubMed] [Google Scholar]
- 7.Kalin M, Cima I, Schiess R, Fankhauser N, Powles T, Wild P, et al. Novel prognostic markers in the serum of patients with castration-resistant prostate cancer derived from quantitative analysis of the pten conditional knockout mouse proteome. Eur Urol. 2011;6:1235–43. [DOI] [PubMed] [Google Scholar]
- 8.Osorio LA, Farfan NM, Castellon EA, Contreras HR. SNAIL transcription factor increases the motility and invasive capacity of prostate cancer cells. Mol Med Rep. 2016;1:778–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;7333:269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer. 2014;9:632–41. [DOI] [PubMed] [Google Scholar]
- 11.Nolan KD, Franco OE, Hance MW, Hayward SW, Isaacs JS. Tumor-secreted Hsp90 subverts polycomb function to drive prostate tumor growth and invasion. J Biol Chem. 2015;13:8271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Broustas CG, Zhu A, Lieberman HB. Rad9 protein contributes to prostate tumor progression by promoting cell migration and anoikis resistance. J Biol Chem. 2012;49:41324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gajula RP, Chettiar ST, Williams RD, Thiyagarajan S, Kato Y, Aziz K, et al. The twist box domain is required for Twist1-induced prostate cancer metastasis. Mol Cancer Res. 2013;11:1387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee YC, Jin JK, Cheng CJ, Huang CF, Song JH, Huang M, et al. Targeting constitutively activated β1 integrins inhibits prostate cancer metastasis. Mol Cancer Res. 2013;4:405–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang K, Myllymäki SM, Gao P, Devarajan R, Kytölä V, Nykter M, et al. Oncogenic K-Ras upregulates ITGA6 expression via FOSL1 to induce anoikis resistance and synergizes with αV-Class integrins to promote EMT. Oncogene 2017;41:5681–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon OJ, Valdez JM, Zhang L, Zhang B, Wei X, Su Q, et al. Increased Notch signalling inhibits anoikis and stimulates proliferation of prostate luminal epithelial cells. Nat Commun. 2014;5:4416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JW, Kim JH. Activation of the leukotriene B4 receptor 2-reactive oxygen species (BLT2-ROS) cascade following detachment confers anoikis resistance in prostate cancer cells. J Biol Chem. 2013;42:30054–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat Commun. 2011;2:162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;2:69–84. [DOI] [PubMed] [Google Scholar]
- 20.Clarke NW, Hart CA, Brown MD. Molecular mechanisms of metastasis in prostate cancer. Asian J Androl. 2009;1:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Dong B, Zhang K, Ji Z, Cheng C, Zhao H, et al. E-cadherin bridges cell polarity and spindle orientation to ensure prostate epithelial integrity and prevent carcinogenesis in vivo. PLoS Genet. 2018;8:e1007609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olson A, Le V, Aldahl J, Yu EJ, Hooker E, He Y, et al. The comprehensive role of E-cadherin in maintaining prostatic epithelial integrity during oncogenic transformation and tumor progression. PLoS Genet. 2019;10:e1008451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bougen NM, Amiry N, Yuan Y, Kong XJ, Pandey V, Vidal LJ, et al. Trefoil factor 1 suppression of E-CADHERIN enhances prostate carcinoma cell invasiveness and metastasis. Cancer Lett. 2013;1:19–29. [DOI] [PubMed] [Google Scholar]
- 24.Borno ST, Fischer A, Kerick M, Falth M, Laible M, Brase JC, et al. Genome-wide DNA methylation events in TMPRSS2-ERG fusion-negative prostate cancers implicate an EZH2-dependent mechanism with miR-26a hypermethylation. Cancer Disco. 2012;11:1024–35. [DOI] [PubMed] [Google Scholar]
- 25.Karanikolas BD, Figueiredo ML, Wu L. Comprehensive evaluation of the role of EZH2 in the growth, invasion, and aggression of a panel of prostate cancer cell lines. Prostate 2010;6:675–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munkley J, McClurg UL, Livermore KE, Ehrmann I, Knight B, McCullagh P, et al. The cancer-associated cell migration protein TSPAN1 is under control of androgens and its upregulation increases prostate cancer cell migration. Sci Rep. 2017;1:5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Copeland BT, Bowman MJ, Ashman LK. Genetic ablation of the tetraspanin CD151 reduces spontaneous metastatic spread of prostate cancer in the TRAMP model. Mol Cancer Res. 2013;1:95–105. [DOI] [PubMed] [Google Scholar]
- 28.Han R, Hensley PJ, Li J, Zhang Y, Stark TW, Heller A, et al. Integrin-associated CD151 is a suppressor of prostate cancer progression. Am J Transl Res. 2020;4:1428–42. [PMC free article] [PubMed] [Google Scholar]
- 29.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;6:873–87. [DOI] [PubMed] [Google Scholar]
- 30.Nilsson EM, Laursen KB, Whitchurch J, McWilliam A, Odum N, Persson JL, et al. MiR137 is an androgen regulated repressor of an extended network of transcriptional coregulators. Oncotarget. 2015;34:35710–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;12:701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;3:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;7406:239–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berney DM, Gopalan A, Kudahetti S, Fisher G, Ambroisine L, Foster CS, et al. Ki-67 and outcome in clinically localised prostate cancer: analysis of conservatively treated prostate cancer patients from the Trans-Atlantic Prostate Group study. Br J Cancer. 2009;6:888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fisher G, Yang ZH, Kudahetti S, Moller H, Scardino P, Cuzick J, et al. Prognostic value of Ki-67 for prostate cancer death in a conservatively managed cohort. Br J Cancer. 2013;2:271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cuzick J, Swanson GP, Fisher G, Brothman AR, Berney DM, Reid JE, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol. 2011;3:245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Majid S, Dar AA, Saini S, Arora S, Shahryari V, Zaman MS, et al. miR-23b represses proto-oncogene Src kinase and functions as methylation-silenced tumor suppressor with diagnostic and prognostic significance in prostate cancer. Cancer Res. 2012;24:6435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Figiel S, Vasseur C, Bruyere F, Rozet F, Maheo K, Fromont G. Clinical significance of epithelial-mesenchymal transition markers in prostate cancer. Hum Pathol. 2017;61:26–32. [DOI] [PubMed] [Google Scholar]
- 39.Bilir B, Osunkoya AO, Wiles WGT, Sannigrahi S, Lefebvre V, Metzger D, et al. SOX4 is essential for prostate tumorigenesis initiated by PTEN ablation. Cancer Res. 2016;5:1112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chien MH, Lin YW, Wen YC, Yang YC, Hsiao M, Chang JL, et al. Targeting the SPOCK1-snail/slug axis-mediated epithelial-to-mesenchymal transition by apigenin contributes to repression of prostate cancer metastasis. J Exp Clin Cancer Res. 2019;1:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, et al. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer Cell. 2010;4:319–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dey P, Velazquez-Villegas LA, Faria M, Turner A, Jonsson P, Webb P, et al. Estrogen receptor β2 induces hypoxia signature of gene expression by stabilizing HIF-1α in prostate cancer. PLoS ONE. 2015;5:e0128239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yi C, Wan X, Zhang Y, Fu F, Zhao C, Qin R, et al. SNORA42 enhances prostate cancer cell viability, migration and EMT and is correlated with prostate cancer poor prognosis. Int J Biochem Cell Biol. 2018;102:138–50. [DOI] [PubMed] [Google Scholar]
- 44.Zheng J, Zhao S, He X, Zheng Z, Bai W, Duan Y, et al. The upregulation of long non-coding RNA CCAT2 indicates a poor prognosis for prostate cancer and promotes metastasis by affecting epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2016;4:508–14. [DOI] [PubMed] [Google Scholar]
- 45.Ramalho-Carvalho J, Martins JB, Cekaite L, Sveen A, Torres-Ferreira J, Graca I, et al. Epigenetic disruption of miR-130a promotes prostate cancer by targeting SEC23B and DEPDC1. Cancer Lett. 2017;385:150–9. [DOI] [PubMed] [Google Scholar]
- 46.Xu S, Yi XM, Tang CP, Ge JP, Zhang ZY, Zhou WQ. Long noncoding RNA ATB promotes growth and epithelial-mesenchymal transition and predicts poor prognosis in human prostate carcinoma. Oncol Rep. 2016;1:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skvortsova K, Masle-Farquhar E, Luu PL, Song JZ, Qu W, Zotenko E, et al. DNA hypermethylation encroachment at CpG island borders in cancer is predisposed by H3K4 mono-methylation patterns. Cancer Cell. 2019;2:297–314. e8. [DOI] [PubMed] [Google Scholar]
- 48.Williams LV, Veliceasa D, Vinokour E, Volpert OV. miR-200b inhibits prostate cancer EMT, growth and metastasis. PLoS ONE. 2013;12:e83991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Majid S, Dar AA, Saini S, Shahryari V, Arora S, Zaman MS, et al. miRNA-34b inhibits prostate cancer through demethylation, active chromatin modifications, and AKT pathways. Clin Cancer Res. 2013;1:73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guan B, Wu K, Zeng J, Xu S, Mu L, Gao Y, et al. Tumor-suppressive microRNA-218 inhibits tumor angiogenesis via targeting the mTOR component RICTOR in prostate cancer. Oncotarget 2017;5:8162–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramalinga M, Roy A, Srivastava A, Bhattarai A, Harish V, Suy S, et al. MicroRNA-212 negatively regulates starvation induced autophagy in prostate cancer cells by inhibiting SIRT1 and is a modulator of angiogenesis and cellular senescence. Oncotarget 2015;33:34446–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin ZY, Chen G, Zhang YQ, He HC, Liang YX, Ye JH, et al. MicroRNA-30d promotes angiogenesis and tumor growth via MYPT1/c-JUN/VEGFA pathway and predicts aggressive outcome in prostate cancer. Mol Cancer. 2017;1:48. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Wang Z, Xu L, Hu Y, Huang Y, Zhang Y, Zheng X, et al. miRNA let-7b modulates macrophage polarization and enhances tumor-associated macrophages to promote angiogenesis and mobility in prostate cancer. Sci Rep. 2016;6:25602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Formosa A, Lena AM, Markert EK, Cortelli S, Miano R, Mauriello A, et al. DNA methylation silences miR-132 in prostate cancer. Oncogene. 2013;1:127–34. [DOI] [PubMed] [Google Scholar]
- 55.Mostaghel EA, Zhang A, Hernandez S, Marck BT, Zhang X, Tamae D, et al. Contribution of adrenal glands to intratumor androgens and growth of castration-resistant prostate cancer. Clin Cancer Res. 2019;1:426–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the major oxidative 3alpha-hydroxysteroid dehydrogenase in human prostate that converts 5alpha-androstane-3alpha,17beta-diol to 5alpha-dihydrotestosterone: a potential therapeutic target for androgen-dependent disease. Mol Endocrinol. 2006;2:444–58. [DOI] [PubMed] [Google Scholar]
- 57.Liu C, Yang JC, Armstrong CM, Lou W, Liu L, Qiu X, et al. AKR1C3 promotes AR-V7 protein stabilization and confers resistance to AR-targeted therapies in advanced prostate cancer. Mol Cancer Ther. 2019;10:1875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yepuru M, Wu Z, Kulkarni A, Yin F, Barrett CM, Kim J, et al. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin Cancer Res. 2013;20:5613–25. [DOI] [PubMed] [Google Scholar]
- 59.Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol Cancer Ther. 2017;1:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, et al. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015;7:1413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;5:1074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hearn JWD, AbuAli G, Reichard CA, Reddy CA, Magi-Galluzzi C, Chang KH, et al. HSD3B1 and resistance to androgen-deprivation therapy in prostate cancer: a retrospective, multi-cohort study. Lancet Oncol. 2016;10:1435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beltran H, Hruszkewycz A, Scher HI, Hildesheim J, Isaacs J, Yu EY, et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin Cancer Res. 2019;23:6916–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reyes EE, VanderWeele DJ, Isikbay M, Duggan R, Campanile A, Stadler WM, et al. Quantitative characterization of androgen receptor protein expression and cellular localization in circulating tumor cells from patients with metastatic castration-resistant prostate cancer. J Transl Med. 2014;12:1313–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang T, Karsh LI, Nissenblatt MJ, Canfield SE. Androgen receptor splice variant, AR-V7, as a biomarker of resistance to androgen axis-targeted therapies in advanced prostate cancer. Clin Genitourin Cancer. 2020;1:1–10. [DOI] [PubMed] [Google Scholar]
- 66.Liu C, Armstrong C, Zhu Y, Lou W, Gao AC. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget. 2016;22:32210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao J, Ning S, Lou W, Yang JC, Armstrong CM, Lombard AP, et al. Cross-resistance among next-generation antiandrogen drugs through the AKR1C3/AR-V7 axis in advanced prostate cancer. Mol Cancer Ther. 2020;8:1708–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, Tagawa ST, et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia. 2013;1:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lotan TL, Gupta NS, Wang W, Toubaji A, Haffner MC, Chaux A, et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod Pathol. 2011;6:820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;4:890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akamatsu S, Wyatt AW, Lin D, Lysakowski S, Zhang F, Kim S, et al. The placental gene PEG10 promotes progression of neuroendocrine prostate cancer. Cell Rep. 2015;6:922–36. [DOI] [PubMed] [Google Scholar]
- 72.Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;6:917–31. [DOI] [PubMed] [Google Scholar]
- 73.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;5:575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell. 2016;4:563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fraser JA, Sutton JE, Tazayoni S, Bruce I, Poole AV. hASH1 nuclear localization persists in neuroendocrine transdifferentiated prostate cancer cells, even upon reintroduction of androgen. Sci Rep. 2019;1:19076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marin-Aguilera M, Reig O, Mila-Guasch M, Font A, Domenech M, Rodriguez-Vida A, et al. The influence of treatment sequence in the prognostic value of TMPRSS2-ERG as biomarker of taxane resistance in castration-resistant prostate cancer. Int J Cancer. 2019;7:1970–81. [DOI] [PubMed] [Google Scholar]
- 77.Yang YJ, Kong YY, Li GX, Wang Y, Ye DW, Dai B. Phenotypes of circulating tumour cells predict time to castration resistance in metastatic castration-sensitive prostate cancer. BJU Int. 2019;2:258–67. [DOI] [PubMed] [Google Scholar]
- 78.Beltran H, Wyatt AW, Chedgy EC, Donoghue A, Annala M, Warner EW, et al. Impact of therapy on genomics and transcriptomics in high-risk prostate cancer treated with neoadjuvant docetaxel and androgen deprivation therapy. Clin Cancer Res. 2017;22:6802–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harris KS, Kerr BA. Prostate cancer stem cell markers drive progression, therapeutic resistance, and bone metastasis. Stem Cells Int. 2017;2017:8629234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lassi K, Dawson NA. Update on castrate-resistant prostate cancer: 2010. Curr Opin Oncol. 2010;3:263–7. [DOI] [PubMed] [Google Scholar]
- 81.Labrecque MP, Takhar MK, Nason R, Santacruz S, Tam KJ, Massah S, et al. The retinoblastoma protein regulates hypoxia-inducible genetic programs, tumor cell invasiveness and neuroendocrine differentiation in prostate cancer cells. Oncotarget. 2016;17:24284–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen X, Li Q, Liu X, Liu C, Liu R, Rycaj K, et al. Defining a population of stem-like human prostate cancer cells that can generate and propagate castration-resistant prostate cancer. Clin Cancer Res. 2016;17:4505–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang L, Dehm SM, Hillman DW, Sicotte H, Tan W, Gormley M, et al. A prospective genome-wide study of prostate cancer metastases reveals association of wnt pathway activation and increased cell cycle proliferation with primary resistance to abiraterone acetate-prednisone. Ann Oncol. 2018;2:352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA-Seq of single prostate CTCs implicates non-canonical Wnt signaling in antiandrogen resistance. Science. 2015;6254:1351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cajigas-Du Ross CK, Martinez SR, Woods-Burnham L, Duran AM, Roy S, Basu A, et al. RNA sequencing reveals upregulation of a transcriptomic program associated with stemness in metastatic prostate cancer cells selected for taxane resistance. Oncotarget. 2018;54:30363–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kanwal R, Shukla S, Walker E, Gupta S. Acquisition of tumorigenic potential and therapeutic resistance in CD133+ subpopulation of prostate cancer cells exhibiting stem-cell like characteristics. Cancer Lett. 2018;430:25–33. [DOI] [PubMed] [Google Scholar]
- 87.Reyes EE, Gillard M, Duggan R, Wroblewski K, Kregel S, Isikbay M, et al. Molecular analysis of CD133-positive circulating tumor cells from patients with metastatic castration-resistant prostate cancer. J Transl Sci. 2015;1:1–6. [PMC free article] [PubMed] [Google Scholar]
- 88.Canovas V, Punal Y, Maggio V, Redondo E, Marin M, Mellado B, et al. Prostate tumor overexpressed-1 (PTOV1) promotes docetaxel-resistance and survival of castration resistant prostate cancer cells. Oncotarget. 2017;35:59165–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lombard AP, Liu C, Armstrong CM, Cucchiara V, Gu X, Lou W, et al. ABCB1 mediates cabazitaxel-docetaxel cross-resistance in advanced prostate cancer. Mol Cancer Ther. 2017;10:2257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ramachandran K, Speer C, Nathanson L, Claros M, Singal R. Role of DNA methylation in cabazitaxel resistance in prostate cancer. Anticancer Res. 2016;1:161–8. [PubMed] [Google Scholar]
- 91.Galletti G, Leach BI, Lam L, Tagawa ST. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat Rev. 2017;57:16–27. [DOI] [PubMed] [Google Scholar]
- 92.Sekino Y, Han X, Kawaguchi T, Babasaki T, Goto K, Inoue S, et al. TUBB3 Reverses resistance to docetaxel and cabazitaxel in prostate cancer. Int J Mol Sci. 2019;20:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Al Nakouzi N, Le Moulec S, Albiges L, Wang C, Beuzeboc P, Gross-Goupil M, et al. Cabazitaxel remains active in patients progressing after docetaxel followed by novel androgen receptor pathway targeted therapies. Eur Urol. 2015;2:228–35. [DOI] [PubMed] [Google Scholar]
- 94.van Soest RJ, de Morree ES, Kweldam CF, de Ridder CMA, Wiemer EAC, Mathijssen RHJ, et al. Targeting the androgen receptor confers in vivo cross-resistance between enzalutamide and docetaxel, but not cabazitaxel, in castration-resistant prostate cancer. Eur Urol. 2015;6:981–5. [DOI] [PubMed] [Google Scholar]
- 95.van Soest RJ, Nieuweboer AJ, de Morree ES, Chitu D, Bergman AM, Goey SH, et al. The influence of prior novel androgen receptor targeted therapy on the efficacy of cabazitaxel in men with metastatic castration-resistant prostate cancer. Eur J Cancer. 2015;17:2562–9. [DOI] [PubMed] [Google Scholar]
- 96.Pezaro CJ, Omlin AG, Altavilla A, Lorente D, Ferraldeschi R, Bianchini D, et al. Activity of cabazitaxel in castration-resistant prostate cancer progressing after docetaxel and next-generation endocrine agents. Eur Urol. 2014;3:459–65. [DOI] [PubMed] [Google Scholar]
- 97.Mout L, de Wit R, Stuurman D, Verhoef E, Mathijssen R, de Ridder C, et al. Testosterone diminishes cabazitaxel efficacy and intratumoral accumulation in a prostate cancer xenograft model. EBioMedicine. 2018;27:182–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Conteduca V, Castro E, Wetterskog D, Scarpi E, Jayaram A, Romero-Laorden N, et al. Plasma AR status and cabazitaxel in heavily treated metastatic castration-resistant prostate cancer. Eur J Cancer. 2019;116:158–68. [DOI] [PubMed] [Google Scholar]
- 99.Martin SK, Pu H, Penticuff JC, Cao Z, Horbinski C, Kyprianou N. Multinucleation and mesenchymal-to-epithelial transition alleviate resistance to combined cabazitaxel and antiandrogen therapy in advanced prostate cancer. Cancer Res. 2016;4:912–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Helpap B, Egevad L. Modified Gleason grading. An updated review. Histol Histopathol. 2009;5:661–6. [DOI] [PubMed] [Google Scholar]
- 101.Pound CR, Partin AW, Eisenberger MA, Chan DW, Pearson JD, Walsh PC. Natural history of progression after PSA elevation following radical prostatectomy. JAMA. 1999;17:1591–7. [DOI] [PubMed] [Google Scholar]
- 102.Tzelepi V, Efstathiou E, Wen S, Troncoso P, Karlou M, Pett-away CA, et al. Persistent, biologically meaningful prostate cancer after 1 year of androgen ablation and docetaxel treatment. J Clin Oncol. 2011;18:2574–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hudson BD, Kulp KS, Loots GG. Prostate cancer invasion and metastasis: insights from mining genomic data. Brief Funct Genomics. 2013;5:397–410. [DOI] [PubMed] [Google Scholar]
- 104.Luo Y, Jiang QW, Wu JY, Qiu JG, Zhang WJ, Mei XL, et al. Regulation of migration and invasion by Toll-like receptor-9 signaling network in prostate cancer. Oncotarget. 2015;26:22564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ok S, Kim SM, Kim C, Nam D, Shim BS, Kim SH, et al. Emodin inhibits invasion and migration of prostate and lung cancer cells by downregulating the expression of chemokine receptor CXCR4. Immunopharmacol Immunotoxicol. 2012;5:768–78. [DOI] [PubMed] [Google Scholar]
- 106.Agell L, Hernandez S, Nonell L, Lorenzo M, Puigdecanet E, de Muga S, et al. A 12-gene expression signature is associated with aggressive histological in prostate cancer: SEC14L1 and TCEB1 genes are potential markers of progression. Am J Pathol. 2012;5:1585–94. [DOI] [PubMed] [Google Scholar]
- 107.Glinsky GV, Glinskii AB, Stephenson AJ, Hoffman RM, Gerald WL. Gene expression profiling predicts clinical outcome of prostate cancer. J Clin Invest. 2004;6:913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Larkin SE, Holmes S, Cree IA, Walker T, Basketter V, Bickers B, et al. Identification of markers of prostate cancer progression using candidate gene expression. Br J Cancer. 2012;1:157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Penney KL, Sinnott JA, Fall K, Pawitan Y, Hoshida Y, Kraft P, et al. mRNA expression signature of Gleason grade predicts lethal prostate cancer. J Clin Oncol. 2011;17:2391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ross RW, Galsky MD, Scher HI, Magidson J, Wassmann K, Lee GS, et al. A whole-blood RNA transcript-based prognostic model in men with castration-resistant prostate cancer: a prospective study. Lancet Oncol. 2012;11:1105–13. [DOI] [PubMed] [Google Scholar]
- 111.Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci USA. 2007;18:7564–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stephenson AJ, Smith A, Kattan MW, Satagopan J, Reuter VE, Scardino PT, et al. Integration of gene expression profiling and clinical variables to predict prostate carcinoma recurrence after radical prostatectomy. Cancer. 2005;2:290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;5:393–406. [DOI] [PubMed] [Google Scholar]
- 114.Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS ONE. 2013;6:e66855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Long Q, Xu J, Osunkoya AO, Sannigrahi S, Johnson BA, Zhou W, et al. Global transcriptome analysis of formalin-fixed prostate cancer specimens identifies biomarkers of disease recurrence. Cancer Res. 2014;12:3228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tomlins SA, Alshalalfa M, Davicioni E, Erho N, Yousefi K, Zhao S, et al. Characterization of 1577 primary prostate cancers reveals novel biological and clinicopathologic insights into molecular subtypes. Eur Urol. 2015;4:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Oh JJ, Park S, Lee SE, Hong SK, Lee S, Lee HM, et al. A clinicogenetic model to predict lymph node invasion by use of genome-based biomarkers from exome arrays in prostate cancer patients. Korean J Urol. 2015;2:109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nordstrom T, Adolfsson J, Gronberg H, Eklund M. Effects of increasing the PSA cutoff to perform additional biomarker tests before prostate biopsy. BMC Urol. 2017;1:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Leon-Mateos L, Casas H, Abalo A, Vieito M, Abreu M, Anido U, et al. Improving circulating tumor cells enumeration and characterization to predict outcome in first line chemotherapy mCRPC patients. Oncotarget 2017;33:54708–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gorges TM, Kuske A, Rock K, Mauermann O, Muller V, Peine S, et al. Accession of tumor heterogeneity by multiplex transcriptome profiling of single circulating tumor cells. Clin Chem. 2016;11:1504–15. [DOI] [PubMed] [Google Scholar]
- 121.Pal SK, He M, Chen L, Yang L, Pillai R, Twardowski P, et al. Synaptophysin expression on circulating tumor cells in patients with castration resistant prostate cancer undergoing treatment with abiraterone acetate or enzalutamide. Urol Oncol. 2018;4:162 e1–162.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Magadoux L, Isambert N, Plenchette S, Jeannin JF, Laurens V. Emerging targets to monitor and overcome docetaxel resistance in castration resistant prostate cancer (review). Int J Oncol. 2014;3:919–28. [DOI] [PubMed] [Google Scholar]
- 123.Chen M, Wang K, Zhang L, Li C, Yang Y. The discovery of putative urine markers for the specific detection of prostate tumor by integrative mining of public genomic profiles. PLoS ONE. 2011;12:e28552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li M, Zhou D, Zhang W, Gao S, Zhou X. Urine PCA3 mRNA level in diagnostic of prostate cancer. J Cancer Res Ther. 2018;4:864–6. [DOI] [PubMed] [Google Scholar]
- 125.Bu H, Bormann S, Schafer G, Horninger W, Massoner P, Neeb A, et al. The anterior gradient 2 (AGR2) gene is overexpressed in prostate cancer and may be useful as a urine sediment marker for prostate cancer detection. Prostate. 2011;6:575–87. [DOI] [PubMed] [Google Scholar]
- 126.O’Reilly E, Tuzova AV, Walsh AL, Russell NM, O’Brien O, Kelly S, et al. epiCaPture: A urine DNA methylation test for early detection of aggressive prostate cancer. JCO Precis Oncol. 2019;2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ciardiello C, Leone A, Lanuti P, Roca MS, Moccia T, Minciacchi VR, et al. Large oncosomes overexpressing integrin alpha-V promote prostate cancer adhesion and invasion via AKT activation. J Exp Clin Cancer Res. 2019;1:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Minciacchi VR, You S, Spinelli C, Morley S, Zandian M, Aspuria PJ, et al. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget. 2015;13:11327–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009;13:5601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Di Vizio D, Morello M, Dudley AC, Schow PW, Adam RM, Morley S, et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am J Pathol. 2012;5:1573–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Minciacchi VR, Spinelli C, Reis-Sobreiro M, Cavallini L, You S, Zandian M, et al. MYC mediates large oncosome-induced fibroblast reprogramming in prostate cancer. Cancer Res. 2017;9:2306–17. [DOI] [PubMed] [Google Scholar]
- 132.Fraser M, Berlin A, Bristow RG, van der Kwast T. Genomic, pathological, and clinical heterogeneity as drivers of personalized medicine in prostate cancer. Urol Oncol. 2015;2:85–94. [DOI] [PubMed] [Google Scholar]
- 133.Lindberg J, Klevebring D, Liu W, Neiman M, Xu J, Wiklund P, et al. Exome sequencing of prostate cancer supports the hypothesis of independent tumour origins. Eur Urol. 2013;2:347–53. [DOI] [PubMed] [Google Scholar]
- 134.Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015;4:1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Boutros PC, Fraser M, Harding NJ, de Borja R, Trudel D, Lalonde E, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet. 2015;7:736–45. [DOI] [PubMed] [Google Scholar]
- 136.Pettersson A, Lis RT, Meisner A, Flavin R, Stack EC, Fiorentino M, et al. Modification of the association between obesity and lethal prostate cancer by TMPRSS2:ERG. J Natl Cancer Inst. 2013;24:1881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhao L, Yu N, Guo T, Hou Y, Zeng Z, Yang X, et al. Tissue biomarkers for prognosis of prostate cancer: a systematic review and meta-analysis. Cancer Epidemiol Biomark Prev. 2014;6:1047–54. [DOI] [PubMed] [Google Scholar]
- 138.You S, Knudsen BS, Erho N, Alshalalfa M, Takhar M, Al-Deen Ashab H, et al. Integrated classification of prostate cancer reveals a novel luminal subtype with poor outcome. Cancer Res. 2016;17:4948–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;5:920–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Santoni M, Scarpelli M, Mazzucchelli R, Lopez-Beltran A, Cheng L, Epstein JI, et al. Current histopathologic and molecular characterisations of prostate cancer: towards individualised prognosis and therapies. Eur Urol. 2016;2:186–90. [DOI] [PubMed] [Google Scholar]
- 141.Yang L, Wang S, Zhou M, Chen X, Jiang W, Zuo Y, et al. Molecular classification of prostate adenocarcinoma by the integrated somatic mutation profiles and molecular network. Sci Rep. 2017;1:738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Modena A, Iacovelli R, Scarpa A, Brunelli M, Ciccarese C, Fantinel E, et al. Investigating BRCA mutations: a breakthrough in precision medicine of castration-resistant prostate cancer. Target Oncol. 2016;5:569–77. [DOI] [PubMed] [Google Scholar]
- 143.Kamoun A, Cancel-Tassin G, Fromont G, Elarouci N, Armenoult L, Ayadi M, et al. Comprehensive molecular classification of localized prostate adenocarcinoma reveals a tumour subtype predictive of non-aggressive disease. Ann Oncol. 2018;8:1814–21. [DOI] [PubMed] [Google Scholar]
- 144.Ketola K, Munuganti RSN, Davies A, Nip KM, Bishop JL, Zoubeidi A. Targeting prostate cancer subtype 1 by forkhead Box M1 pathway inhibition. Clin Cancer Res. 2017;22:6923–33. [DOI] [PubMed] [Google Scholar]
- 145.Wu YM, Cieslik M, Lonigro RJ, Vats P, Reimers MA, Cao X, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 2018;7:1770 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Almassi N, Reichard C, Li J, Russell C, Perry J, Ryan CJ, et al. HSD3B1 and response to a nonsteroidal CYP17A1 inhibitor in castration-resistant prostate cancer. JAMA Oncol. 2018;4:554–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kim W, Zhang L, Wilton JH, Fetterly G, Mohler JL, Weinberg V, et al. Sequential use of the androgen synthesis inhibitors ketoconazole and abiraterone acetate in castration-resistant prostate cancer and the predictive value of circulating androgens. Clin Cancer Res. 2014;24:6269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Spratt DE, Alshalalfa M, Fishbane N, Weiner AB, Mehra R, Mahal BA, et al. Transcriptomic heterogeneity of androgen receptor activity defines a de novo low AR-active subclass in treatment naive primary prostate cancer. Clin Cancer Res. 2019;22:6721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;136:136ra68. [DOI] [PubMed] [Google Scholar]
- 150.Lambros MB, Seed G, Sumanasuriya S, Gil V, Crespo M, Fontes M, et al. Single-cell analyses of prostate cancer liquid biopsies acquired by apheresis. Clin Cancer Res. 2018;22: 5635–44. [DOI] [PubMed] [Google Scholar]
- 151.Oliveira KCS, Ramos IB, Silva JMC, Barra WF, Riggins GJ, Palande V, et al. Current perspectives on circulating tumor dna, precision medicine, and personalized clinical management of cancer. Mol Cancer Res. 2020;4:517–28. [DOI] [PubMed] [Google Scholar]
- 152.Ku SY, Gleave ME, Beltran H. Towards precision oncology in advanced prostate cancer. Nat Rev Urol. 2019;11:645–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Giri VN, Knudsen KE, Kelly WK, Cheng HH, Cooney KA, Cookson MS, et al. Implementation of Germline Testing for Prostate Cancer: Philadelphia Prostate Cancer Consensus Conference 2019. J Clin Oncol. 2020:Jco2000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Murphy AB, Carbunaru S, Nettey OS, Gornbein C, Macias V, Sharifi R, et al. A 17-gene panel genomic prostate score has similar predictive accuracy for adverse pathology at radical prostatectomy in African American and European American Men. Urology. 2020;142:166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tan SH, Petrovics G, Srivastava S. Prostate cancer genomics: recent advances and the prevailing underrepresentation from racial and ethnic minorities. Int J Mol Sci. 2018;19:4. [DOI] [PMC free article] [PubMed] [Google Scholar]