

Abstract
Background/Aim: We investigated the hypothesis that dichloroacetate (DCA), a pyruvate dehydrogenase kinase inhibitor, and metformin (MET), an antidiabetic agent and complex I inhibitor, have synergistic cytotoxic effects in glioblastoma cells in vitro and in vivo. Materials and Methods: We performed dose response experiments and combination index calculation. Apoptotic and necrotic cells were estimated by flow cytometry. Cell metabolism was evaluated by Seahorse analysis and lactate export. Overall survival and tumor volume growth experiments were performed in C57BL/6 mice GL-261 allograft model. Results: DCA and MET showed dose-dependent cytotoxicity and synergistic effects. DCA alleviated the increase in lactate production induced by MET. Seahorse analysis showed that DCA treatment results in increased oxygen consumption rate, which is decreased by MET. DCA and MET significantly inhibited tumor growth and increased overall survival in mice. Conclusion: Compounds targeting tumor cell metabolism could become potential treatment options for glioblastoma multiforme
Keywords: Dichloroacetate, metformin, glioblastoma, Warburg effect
Glioblastoma multiforme (GBM) is an aggressive primary brain tumor that affects a huge population worldwide (1). Existing standard therapies, such as surgery, radiotherapy, chemotherapy and tumor targeting fields manifest extremely poor responses (2). The average overall survival since the diagnosis rarely exceeds 15 months (1,3) and, upon recurrence, it is approximately 6 months (4). Thus, new therapeutic modalities increasing the lifespan of GBM patients are in high demand.
Altered energy metabolism is hallmarked as one of the features of cancer (5). Many tumors, including glioblastomas, undergo the so-called Warburg effect, when energy metabolism is geared towards aerobic glycolysis, resulting in fewer ATP molecules, but increased extracellular lactate (6). In addition, malignant gliomas depend on glucose and, like most cancers, GBMs have dysregulated mitochondria, disrupting an oxidative phosphorylation cycle (7,8).
Dichloroacetate (DCA), a small molecule, used to treat lactic acidosis (9) has been reported to reverse the Warburg effect by inhibiting pyruvate dehydrogenase kinase (PDK) and indirectly activating the gate keeping enzyme pyruvate dehydrogenase (PDH) (10). Generally, DCA shifts aerobic glycolysis towards glucose oxidation in cancer, without affecting normal cells. Numerous studies have evaluated DCA effects in tumors, including case studies and several small human trials (11,12). In initial research, a significant 70% shrinkage of tumors was observed after just 3 weeks of DCA treatments in rodents (13). This study encouraged further studies mostly in brain, breast, colorectal, lung, prostate and gynecologic cancers. It was soon realized that DCA is not universally effective in all types of tumor cells. This was attributed to hormesis, a biphasic dose-response phenomenon in which a low concentration of DCA may stimulate tumor cell proliferation or viability, while high doses would inhibit it (14,15). For example, DCA at concentrations of 20 mM or lower stimulated cellular viability while at concentrations 40 mM and higher reduced cell viability under normoxic conditions in vitro (15).
Metformin (MET), a biguanide, is a widely used oral antidiabetic agent that has the potential to decrease hyperglycemia in patients with type 2 diabetes. MET targets electron transport chain complex I, thus preventing oxidative mitochondrial metabolism (16). MET can activate AMP activated protein kinase (AMPK) (17,18), a powerful mediator in cellular energy metabolism (19). AMPK inhibits protein synthesis and induces cell cycle arrest (20), affects cell growth by inhibiting the mTOR pathway, which in turn plays an important role in metabolism, growth and proliferation of cancer cells (21). It was suggested by Evans and her team in 2005 that the use of MET may be related to reduced incidence of cancer (22). It has been shown, that patients who suffered type 2 diabetes and breast cancer and administered MET, responded to chemotherapy in a sufficiently better way than patients who did not receive MET (23). In vivo studies have shown the MET’s ability to prevent lung tumorigenesis (24) and eliminate tumor initiating cells in liver cancer (25,26). These findings complement in vitro studies where MET inhibited cell proliferation and reduced colony formation of breast cancer cells (27).
It has been observed that existing glycolysis inhibitors, such as DCA, 3-BrPA (3-bromopyruvate) or 2-DG (2-deoxyglucose), do not maintain effective shrinking of tumor volume when used as monotherapy and are much more efficient when used in combination with other cytotoxic agents (28-30). Numerous studies indicated that coupling DCA with other potential anticancer substances, such as curcumin (31), omeprazole and tamoxifen (28), 5-fluorouracil (32), sulindac (33) enhances therapeutic potential. Moreover, previously, it has been shown that a combination of DCA and MET synergistically suppressed the growth of ovarian, breast, and prostate cancer (34-36). Based on these recent findings, we hypothesized that the two metabolic modulators DCA and MET could potentially act as cytotoxic compounds in glioblastoma multiforme.
Materials and Methods
Cell lines and reagents. Human U-87 MG (U-87) and murine GL-261 glioblastoma cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). U-87 and GL-261 were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Waltham, MA, USA), supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, M, USA) and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin) (Gibco) and regularly passaged after reaching confluence. During all experiments, cells were maintained at 37˚C in a 5% CO2 humidified incubator. Sodium DCA (Sigma Aldrich, St. Louis, MO, USA) and MET (1,1-dimethylbiguanide hydrochloride) (Sigma Aldrich) stocks were stored in accordance with manufacturer’s recommendations. Drug solutions in medium were prepared fresh on the day of each experiment.
Animal study. All experiments were carried out in compliance with the ARRIVE guidelines (37) and following the approval of the State Food and Veterinary Service, Lithuania. For the animal experiments, 8 to 12-week-old female C57BL/6 mice (18-22 g) were bought from the State Research Institute Centre for Innovative Medicine (Vilnius, Lithuania). The mice were housed under normal daylight conditions with ad libitum access to water and food. Subcutaneous implantation of GL-261 cells was used for the glioblastoma model. 5×105 GL-261 (38) cells were subcutaneously implanted into the right flank of each mouse. When tumors became palpable, mice were randomly allocated into treatment and control groups (n=7 per group). On day 10, after tumor implantation, the treatment was started. DCA (150 mg/kg/day), MET (75 mg/kg/day) or both were injected intraperitoneally, and phosphate-buffered saline (PBS) injection was used in controls. The tumor size was monitored every other day with a sliding caliper. Tumor volume was estimated using the following formula: volume=width2 × length × 1/2. All animals were euthanized by cervical dislocation at a critical endpoint for individual mice, which was set subjectively by the technical staff.
Cell viability assay. U-87 cells were seeded in 96-well plates at a density of 5,000 cells/well in 200 μl of culture media and left overnight to attach. After 24 h, cells were treated with different concentrations of DCA or MET (1, 5, 10, 15, 20 mM) and their combination. After incubation for 48 h, the medium was exchanged and cells were treated with MTT reagent (Sigma Aldrich), to measure cell viability. Plates were incubated for 1.5 h at 37˚C, then formazan crystals were dissolved in DMSO and the absorbance was measured at 490 nm.
Apoptosis and necrosis assessment by flow cytometry. For apoptotic and necrotic cell examination, Alexa Fluor 488 annexin V/Dead Cell Apoptosis Kit (Invitrogen Ltd., Paisley, UK) was used. U-87 cells were seeded in a 6-well plate at a density of 3×105 cells per well. After 24 h, cells were exposed to either DCA, MET or their combination. After 48 h, cells were harvested, washed in cold PBS, stained and incubated in the dark according to the manufacturer’s protocol. Following incubation with Annexin V and PI, cells were acquired on flow cytometer BD LSR II (BD Biosciences, San Jose, CA, USA) and analysed by FlowJo v10 (BD Biosciences).
Lactate export. U-87 cells were seeded in 6-well plates and left overnight. The next day, the culture media were replaced and treatment was applied. After 24 h, the medium was collected and the concentrations of L-lactate were determined using L-lactate Assay Kit (Sigma Aldrich).
Cellular bioenergetics analysis. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were determined using the Seahorse XFp Analyzer (Agilent Technologies, Santa Clara, CA, USA). U-87 cells were seeded in XFp 8-well plates at a density of 3,000 cells/well. The next day, adherent cells were observed and treatment with DCA, MET or both drugs was applied for 24 h. Twenty-four h later, on the day of analysis, the cell culture medium was changed into Seahorse XF base medium (minimal Dulbecco’s modified Eagle’s medium) supplemented with 10 mM glucose, 2 mM glutamine, and 1 mM sodium pyruvate (pH 7.4) followed by incubation at 37˚C in a non-CO2 incubator for 1 h. For evaluation of the mitochondrial function, 3 metabolic inhibitors were sequentially dispensed as follows: oligomycin (inhibitor of ATP synthase, 500 nM), followed by carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, uncoupler of mitochondrial oxidative phosphorylation, 1 μM) and a combination of rotenone (mitochondrial complex I inhibitor, 100 nM) and antimycin A (complex III inhibitor, 100 nM). Basal OCR, ECAR, and changes induced by the above inhibitors were measured. Experiments were performed in triplicate. The key parameters of mitochondrial respiration were measured using the Seahorse XF Cell Mito Stress Test Kit and analyzed as described (39). OCR and ECAR were automatically recorded and calculated by the Seahorse XFe96 software (Wave 2.3.0). All the measurements were normalized to cell numbers and intracellular protein concentration. Protein concentration was assessed by Bradford assay.
Statistical analysis. All statistics were performed using GraphPad Prism 8 software (San Diego, CA, USA). Kaplan–Meier survival curves were plotted and compared using the Mantel–Cox log-rank test. One-way or two-way ANOVA was used for groupwise comparisons. Results represent three independent experiments and are expressed as mean±standard error mean (SEM). The synergistic effect was analyzed according to the Chou-Talalay method (40) using CompuSyn software and a combination index (CI) was evaluated. When CI <1 - synergistic, CI=1 – additive, CI>1 – antagonistic effect. p-values are follows: *p<0.05; **p<0.001; ***p<0.0001.
Results
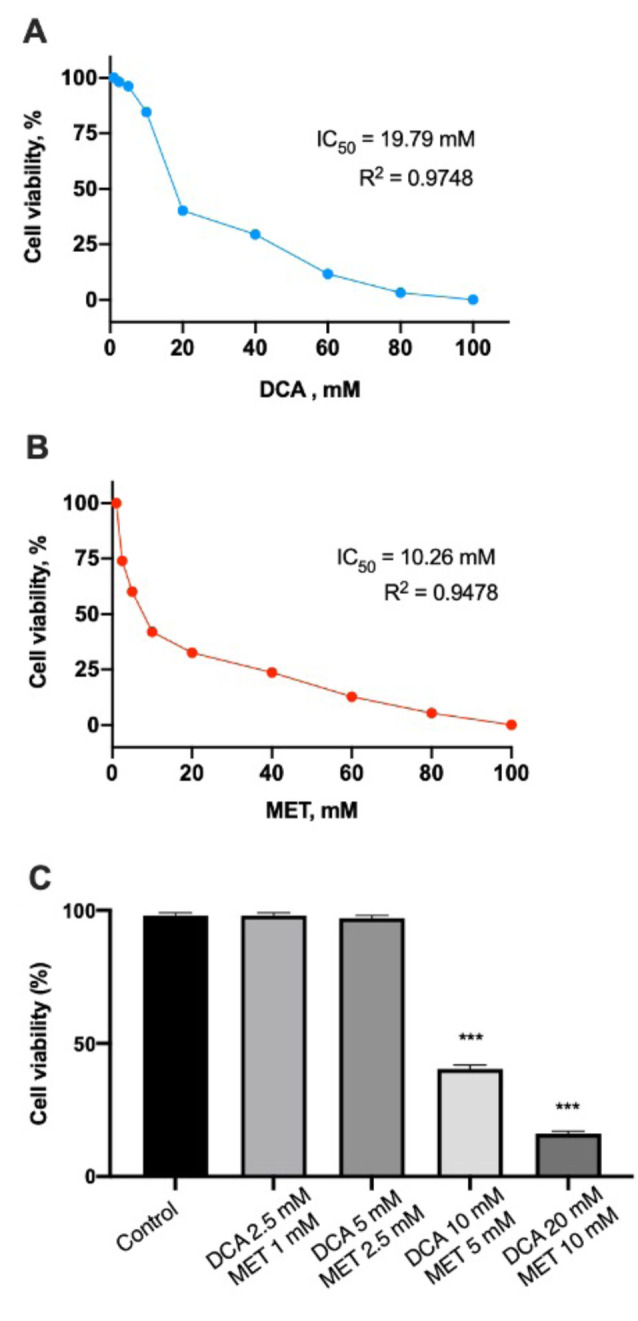
MET and DCA show dose dependent cytotoxicity. First, we investigated the effects of DCA, MET or their combination on U-87 glioblastoma cells. To assess cytotoxicity of the drugs, the MTT assay was used. We conducted drug concentration response experiments and evaluated the half maximal inhibitory concentrations (IC50) for each drug, which were 19.79 mM for DCA and 10.26 mM for MET (Figure 1A and B). Then, we examined whether these drugs could exhibit a synergistic effect when combined. Taking this into consideration, DCA concentrations used in the study were 20, 10, 5 and 2.5 mM, while MET concentrations were 10, 5, 2.5, 1 mM. Data were analyzed using the Calcusyn Software, by Chou-Talalay (40), generating a combination index (CI) for each single combination point. CI values below 1 suggest synergy, whereas CI values above 1 indicate antagonism. MET enhanced the potency of DCA to inhibit cancer cell proliferation compared with the effect of these drugs individually (Figure 1C). After a series of experiments, we observed that combination treatment with DCA and MET was dose dependent and only high concentrations, such as 10 mM DCA and 5 mM MET or 20 mM DCA and 10 mM MET, showed a synergistic effect, inhibiting cell viability by 60%±1.5%, CI 0.95, and 85%±0.87%, CI 0.94, respectively (Figure 1C). At lower doses, when 1 and 5 mM DCA were combined with 1 and 2.5 mM MET, only antagonistic effects were observed. Ten mM of DCA alone reduced the percentage of cell number to 85% and 5 mM of MET to 60%, while their combination reduced the cell number to 40%, compared with the control. For this combination the CI value was equal to 0.94, which is interpreted as strong drug synergy.
Figure 1. Dichloroacetate (DCA) in combination with metformin (MET) leads to reduced cell growth. A. Concentration response curve of DCA, B. concentration response curve of MET in U-87 cell line. U-87 cells were treated with DCA (1-100 mM) and MET (1-100 mM) and their IC50 values were determined. To evaluate the efficacy of their combinations, data was analyzed using the Calcusyn Software, and combination (CI) values were generated. C. Synergistic cytotoxic effect of DCA and MET, (***p<0.001).
DCA and MET synergistically induce apoptosis in glioblastoma cancer cells. DCA has been reported to induce apoptosis in cancer cells; however, some authors have reported that this is cancer cell type specific (41). Therefore, we investigated whether the combination of DCA and MET increased apoptosis in the human glioblastoma U-87 cell line. Cells were exposed to 10 mM DCA, 5 mM MET or their combination for 48 h, and the apoptotic rate was evaluated by flow cytometry. Each drug alone did not have a statistically significant effect compared with control; however, similar to what was observed at dose response experiments, once the drugs were applied in combination, the apoptotic rate of cancer cells was increased significantly (Figure 2A). The percentages of healthy (Q4), early- (Q3) and late-apoptotic (Q2), as well as necrotic (Q1) cells, are presented in appropriate quadrants. As shown in representative dot plots in Figure 2, the combination of MET and DCA produced a synergistic effect dramatically increasing the early (30.9%) cell populations after 48-h exposure, compared with control (0.9%) and to the single agents - DCA (10.8) and Met (15.1) (Figure 2, Q3). Synergistic effect of DCA and Met also reflected in elevated late apoptotic cell percentage (13.6%), compared to control (2.56%) and also to the single agents – DCA (4.42%) and Met (2.35%) (Figure 2, Q2). These results complement cell viability experiments, indicating the potentiating effect of combinational therapy of DCA and Met.
Figure 2. Dichloroacetate (DCA) and metformin (MET) synergistically induce apoptosis in glioblastoma cancer cells. A. Average level of apoptosis calculated from three independent flow cytometry experiments. B. Effect of 10 mM DCA, 5 mM MET or both on early, late apoptosis or necrosis of the U-87 cell line, after incubation for 48 h. C. Cell fractions of early, late apoptotic and necrotic cells after 10 mM DCA and 5 mM MET treatment, compared to control (PBS). *p<0.05; **p<0.01.

DCA alleviated MET induced lactate production and restored OXPHOS in glioblastoma cells. DCA, as a PDK inhibitor, is able to increase OXPHOS rate in cancer cell lines; on the contrary, MET, as a complex I inhibitor, has the ability to increase lactate production, which is associated with reduced oxidative phosphorylation. In these series of experiments, we examined whether cancer cell treatment with DCA and MET would influence lactate levels (Figure 3D). Not only did we show that DCA reduced the levels of lactate in cancer culture media, but it also increased the mitochondrial respiration rate. The cellular oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR) were measured. Figure 3 represents the OCR ratio of U-87 cells after DCA and MET exposure for 24 h. MET decreased the ratio of OCR, which was dramatically reversed by DCA, suggesting that DCA can restore oxidative phosphorylation and recover mitochondrial respiration.
Figure 3. Dichloroacetate (DCA) alleviates metformin (MET)-induced lactate production and restores mitochondrial respiration. U-87 cells were cotreated with 10 mM DCA and 5 mM MET, or each alone for 24 h. PBS was used as control. A. and C. Real-time metabolism analysis by Seahorse mitochondrial stress test kit that was used to measure the ECAR (extracellular acidification rate) representing the amount of glycolysis, and OCR (oxygen consumption rate), defining the amount of mitochondrial respiration. B. Histograms of glycolytic function parameters. D. Lactate release into media after drug exposure. Values represent the mean±SEM of three independent experiments. *p<0.05; **p<0.01; ***p<0.001.

DCA and MET synergistically suppresses the growth of glioblastoma cells in vivo. After investigating the synergistic effects of DCA and MET in vitro, we evaluated glioblastoma cell growth dynamics in vivo, together with overall survival of mice. C57BL/6 mice were chosen for this study. Treatment began when palpable tumors were formed, on day 10 after inoculation of GL-261 cells. Mice were injected with PBS in the control group, and in the treatment groups with 150 mg/kg/day DCA, 75 mg/kg/day MET, or both. Tumor volume was evaluated every other day for 22 days. On day 18, no significant reduction in tumor growth was observed; however, on day 22, all treatments showed a significant reduction in tumor volume compared with the control group, suggesting that each drug alone and their combination can reduce tumor growth (Figure 4A). However, only the combination therapy achieved a statistically significant clinical result, with that treatment group having longer overall survival compared with the control or monotherapy groups (Figure 4B).
Figure 4. Dichloroacetate (DCA) in combination with metformin (MET) leads to reduced tumor growth and increased overall survival in mice. A. Tumor volume growth curve, statistically significant reduction after 22 days of treatment with DCA, MET or their combination, compared with control (***p<0.001). B. Kaplan-Meier survival curves for mice receiving DCA, MET or both. Mice treated with a combination therapy showed higher median overall survival mOS=29.71 days, p=0.0073; compared with DCA mOS=24.29 days; MET mOS=22.71 days, control mOS=20 days. Only combination treatment with DCA and MET showed a statistically different effect compared with the control and each drug alone.

Discussion
In our work, we analyzed the synergistic anti-cancer effect of DCA and MET, both in vitro and in vivo. Similar experiments with the same compounds were performed in 2016 (35), but instead of ovarian carcinoma, this study focuses on glioblastoma, as this disease currently has less viable treatment options and is reported as the most common and deadly brain tumor. Therefore, it attracts the attention of scientists trying to improve clinical outcomes by conducting in vivo experiments in a variety of murine models (42). To tackle this tumor, we exploited what is commonly known as the Warburg effect that describes the phenomenon of aerobic glycolysis in cancer cells (6). Since this process is observed in glioblastoma multiforme (43), we hypothesized that targeting mitochondria might be a viable option for reducing GBM growth. In the present study, we showed that drugs targeting cell bioenergetics could significantly reduce human U-87 cell growth in vitro, as well as inhibit tumor growth and, importantly, increase overall survival of C57BL/6 mice in an in vivo GL-261 allograft model.
Firstly, we demonstrate that DCA and MET cytotoxicity is dose dependent, which confirmed previous studies performed in other tumor models (44). Secondly, we showed that DCA alleviated the increase in lactate production induced by MET. Furthermore, OCR for DCA and MET cotreatment was drastically higher compared with MET alone, demonstrating how DCA restores mitochondrial respiration. These results are in line with the findings in an ovarian cancer model reported previously (35), indicating that the synergistic mechanism of DCA and MET can be universal across different cancer types.
Both agents could induce apoptosis as monotreatments; however, once combined, the apoptotic rate increased and flow cytometric analysis showed a higher proportion of early/late apoptotic and necrotic cells than in monotherapy or control samples. These in vitro results in U-87 cells are similar to those reported previously in breast, colorectal, endometrial, and ovarian cancer cells (34,45-47).
Our study showed that tumor growth in the GL-261 in vivo allograft model was inhibited by DCA or MET individually, and their combination. There were no statistically significant differences in the tumor growth rate between the 3 treatment groups; however, only the combination of DCA and MET resulted into overall survival benefit (mOS=29.71 days vs. 20 days control mice), demonstrating a synergistic effect. In a similar tumor growth experiment in a xenograft ovarian model in NUDE mice (35), DCA and MET suppressed tumor more efficiently than DCA or MET alone, resembling our findings. That study did not present survival data; therefore, no comparison can be made in that aspect.
When combined, DCA and MET potentiated anticancer activity against GBM. We hypothesized that when the drugs are applied as a combination therapy, lower concentration of each drug is needed in order to obtain a similar cytotoxic effect compared to monotherapy. Micromolar concentrations of drugs are highly desirable in clinical practice, especially in oncology. However, only at high doses, DCA and MET synergistically suppressed cancer cell proliferation. These results supplement other authors’ findings, where only high concentrations could inhibit cancer (34,35,48). Although these findings seem to be promising, the dose causes concerns and possibly limits their application. In our study, effective DCA and MET doses were at millimolar concentration range, which is quite high for any drug that is given daily in chemotherapy regimen. MET is usually administered in doses between 500-2,500 mg daily and mild gastrointestinal side-effects are observed (49). On the other hand, DCA can induce peripheral neuropathy in patients receiving high doses, such as 50 mg/kg/day. One study observed that 50-500 mg/kg/day could induce tactile allodynia in both juvenile and adult rats (50). In our study, we used 150 mg/kg of DCA, which would be equivalent to ~5,000 mg/day for an average 70 kg human (51). Since we have not monitored the potential damage to rodent nerves or any other side effects that DCA could cause, this should be addressed in future studies.
Conclusion
In times of highly advanced cancer therapies, such as immunotherapy, targeted therapies, monoclonal antibodies, etc., some tumors still cannot be repressed, due to underlying complex mechanisms supporting their survival. The most important factor for GBM patients is the extended overall survival and increased quality of life. An ideal therapy should attain GBM cells apoptosis, cope with cellular heterogeneity, inhibit angiogenesis of the tumor and cross the blood-brain barrier. Based on this, we conclude that treatment with the combination of DCA and MET produces synergistic antiproliferative, proapoptotic, metabolism-altering effects, together with inhibition of glioblastoma growth in vivo. However, we embrace the limitations of this study, and further investigations on the potential mechanisms should be performed.
Conflicts of Interest
The Authors declare no conflicts of interest in relation to this study.
Authors’ Contribution
Conceptualization, L.K., J.A.K. and E.S.; Data curation, L.K. and J.A.K.; Investigation, L.K. and J.A.K.; Methodology, L.K.; Resources, E.S. Supervision E.S.; Writing – original draft, L.K. and J.A.K.; Writing - review & editing, E.S.
References
- 1.Marenco-Hillembrand L, Wijesekera O, Suarez-Meade P, Mampre D, Jackson C, Peterson J, Trifiletti D, Hammack J, Ortiz K, Lesser E, Spiegel M, Prevatt C, Hawayek M, Quinones-Hinojosa A, Chaichana KL. Trends in glioblastoma: outcomes over time and type of intervention: a systematic evidence based analysis. J Neuro-oncol. 2020;147:297–307. doi: 10.1007/s11060-020-03451-6. [DOI] [PubMed] [Google Scholar]
- 2.Weller M, Bent M van den, Hopkins K, Tonn JC, Stupp R, Falini A, Cohen-Jonathan-Moyal E, Frappaz D, Henriksson R, Balana C, Chinot O, Ram Z, Reifenberger G, Soffietti R, Wick W, European Association for Neuro-Oncology (EANO) Task Force on Malignant Glioma EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014;15:e395–e403. doi: 10.1016/s1470-2045(14)70011-7. [DOI] [PubMed] [Google Scholar]
- 3.Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: From molecular pathology to targeted treatment. Annu Rev Pathology Mech Dis. 2014;9:1–25. doi: 10.1146/annurev-pathol-011110-130324. [DOI] [PubMed] [Google Scholar]
- 4.Linde Me, Brahm C, Hamer Pc, Reijneveld J, Bruynzeel A, Vandertop W, Ven Pm, Wagemakers M, Weide Hl, Enting R, Walenkamp A, Verheul H. Treatment outcome of patients with recurrent glioblastoma multiforme: a retrospective multicenter analysis. J Neuro-oncol. 2017;135:183–192. doi: 10.1007/s11060-017-2564-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Warburg O. On the origin of cancer cells. Science (New York, NY) 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 7.Seyfried T, Flores R, Poff A, D’Agostino D. Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis. 2014;35:515–527. doi: 10.1093/carcin/bgt480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seyfried T, Flores R, Poff A, D’Agostino D, Mukherjee P. Metabolic therapy: A new paradigm for managing malignant brain cancer. Cancer Lett. 2015;356:289–300. doi: 10.1016/j.canlet.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 9.Stacpoole PW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol. 2003;43:683–691. [PubMed] [Google Scholar]
- 10.Stacpoole P. Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J Natl Cancer Inst. 2017;109:djx071. doi: 10.1093/jnci/djx071. [DOI] [PubMed] [Google Scholar]
- 11.Stacpoole P, Kerr D, Barnes C, Bunch S, Carney P, Fennell E, Felitsyn N, Gilmore R, Greer M, Henderson G, Hutson A, Neiberger R, O’Brien R, Perkins L, Quisling R, Shroads A, Shuster J, Silverstein J, Theriaque D, Valenstein E. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519–1531. doi: 10.1542/peds.2005-1226. [DOI] [PubMed] [Google Scholar]
- 12.Inc K. A clinical trial to evaluate dichloroacetate (DCA) as a treatment for endometriosis-associated pain. Case Medical Res. 2019 doi: 10.31525/ct1-nct04046081. [DOI] [Google Scholar]
- 13.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 14.Calabrese E. Cancer biology and hormesis: human tumor cell lines commonly display hormetic (Biphasic) dose responses. Crit Rev Toxicol. 2008;35:463–582. doi: 10.1080/10408440591034502. [DOI] [PubMed] [Google Scholar]
- 15.Robey IF, Martin NK. Bicarbonate and dichloroacetate: evaluating pH altering therapies in a mouse model for metastatic breast cancer. BMC Cancer. 2011;11:235. doi: 10.1186/1471-2407-11-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348 Pt 3:607–614. [PMC free article] [PubMed] [Google Scholar]
- 17.Rocha GZ, Dias MMM, Ropelle ER, Osório-Costa F, Rossato FA, Vercesi AE, Saad MJ, Carvalheira JBB. Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clin Cancer Res. 2011;17:3993–4005. doi: 10.1158/1078-0432.CCR-10-2243. [DOI] [PubMed] [Google Scholar]
- 18.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Wang L, Zhou X, Ke J, Waal Pw, Gu X, Tan M, Wang D, Wu D, Xu H, Melcher K. Erratum: Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015;25:398–398. doi: 10.1038/cr.2015.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahra I, Marchand-Brustel Y, Tanti J, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug. Mol Cancer Ther. 2010;9:1092–1099. doi: 10.1158/1535-7163.mct-09-1186. [DOI] [PubMed] [Google Scholar]
- 21.Kasznicki J, Drzewoski J. A case of autoimmune urticaria accompanying autoimmune polyglandular syndrome type III associated with Hashimoto’s disease, type 1 diabetes mellitus, and vitiligo. Endokrynol Pol. 2014;65:320–323. doi: 10.5603/ep.2014.0044. [DOI] [PubMed] [Google Scholar]
- 22.Evans J, Donnelly L, Emslie-Smith A, Alessi D, Morris A. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304. doi: 10.1136/bmj.38415.708634.f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiralerspong S, Palla S, Giordano S, Meric-Bernstam F, Liedtke C, Barnett C, Hsu L, Hung M, Hortobagyi G, Gonzalez-Angulo A. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27:3297–3302. doi: 10.1200/jco.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Memmott R, Mercado J, Maier C, Kawabata S, Fox S, Dennis P. Metformin prevents tobacco carcinogen–induced lung tumorigenesis. Cancer Prev Res. 2010;3:1066–1076. doi: 10.1158/1940-6207.capr-10-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saito M, Yano K, Kamigaki T, Goto S. A patient with scirrhous stomach cancer treated with combination of hyperthermotherapy and 5-aminolevulinic acid (ALA) Anticancer Res. 2013;33:2957–2963. [PubMed] [Google Scholar]
- 26.Saito T, Chiba T, Yuki K, Zen Y, Oshima M, Koide S, Motoyama T, Ogasawara S, Suzuki E, Ooka Y, Tawada A, Tada M, Kanai F, Takiguchi Y, Iwama A, Yokosuka O. Metformin, a diabetes drug, eliminates tumor-initiating hepatocellular carcinoma cells. PLoS One. 2013;8:e70010. doi: 10.1371/journal.pone.0070010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alimova I, Liu B, Fan Z, Edgerton S, Dillon T, Lind S, Thor A. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8:909–915. doi: 10.4161/cc.8.6.7933. [DOI] [PubMed] [Google Scholar]
- 28.Ishiguro T, Ishiguro R, Ishiguro M, Iwai S. Co-treatment of dichloroacetate, omeprazole and tamoxifen exhibited synergistically antiproliferative effect on malignant tumors: in vivo experiments and a case report. Hepatogastroenterology. 2012;59:994–996. doi: 10.5754/hge10507. [DOI] [PubMed] [Google Scholar]
- 29.Ishiguro T, Ishiguro M, Ishiguro R, Iwai S. Cotreatment with dichloroacetate and omeprazole exhibits a synergistic antiproli-ferative effect on malignant tumors. Oncol Lett. 2012;3:726–728. doi: 10.3892/ol.2012.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong F, Peng X, Sang Y, Qiu M, Luo C, He Z, Zhao X, Tong A. Dichloroacetate induces protective autophagy in LoVo cells: involvement of cathepsin D/thioredoxin-like protein 1 and Akt-mTOR-mediated signaling. Cell Death Dis. 2013;4:e913. doi: 10.1038/cddis.2013.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kan PC, Chang YJ, Chien CS, Su CY, Fang HW. Coupling dichloroacetate treatment with curcumin significantly enhances anticancer potential. Anticancer Res. 2018;38:6253–6261. doi: 10.21873/anticanres.12981. [DOI] [PubMed] [Google Scholar]
- 32.Tong J, Xie G, He J, Li J, Pan F, Liang H. Synergistic antitumor effect of dichloroacetate in combination with 5-fluorouracil in colorectal cancer. J Biomed Biotechnol. 2011;2011:740564. doi: 10.1155/2011/740564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayyanathan K, Kesaraju S, Dawson-Scully K, Weissbach H. Combination of sulindac and dichloroacetate kills cancer cells via oxidative damage. PLoS One. 2012;7:e39949. doi: 10.1371/journal.pone.0039949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haugrud AB, Zhuang Y, Coppock JD, Miskimins WK. Dichloroacetate enhances apoptotic cell death via oxidative damage and attenuates lactate production in metformin-treated breast cancer cells. Breast Cancer Res Treat. 2014;147:539–550. doi: 10.1007/s10549-014-3128-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li B, Li X, Ni Z, Zhang Y, Zeng Y, Yan X, Huang Y, He J, Lyu X, Wu Y, Wang Y, Zheng Y, He F. Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells. Oncotarget. 2016;7:59458–59470. doi: 10.18632/oncotarget.10694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kiliccioglu I, Konac E, Albayrak G, Dönmez C, Bilen CY. Combination of metformin and dichloroacetate inhibits proliferation and induce intrinsic pathway of apoptosis in PC-3 human prostate cancer cells. GMJ. 2015;26 doi: 10.12996/gmj.2015.65. [DOI] [Google Scholar]
- 37.Kilkenny C, Browne W, Cuthill I, Emerson M, Altman DG, National Centre for the Replacement, Refinement and Reduction of Amimals in Research Animal research: reporting in vivo experiments—the ARRIVE guidelines. J Cereb Blood Flow Metab. 2011;31:991–993. doi: 10.1038/jcbfm.2010.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szatmári T, Lumniczky K, Désaknai S, Trajcevski S, Hídvégi E, Hamada H, Sáfrány G. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci. 2006;97:546–553. doi: 10.1111/j.1349-7006.2006.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Y, Al-Saaidi R, Fernandez-Guerra P, Freude K, Olsen R, Jensen U, Gregersen N, Hyttel P, Bolund L, Aagaard L, Bross P, Luo Y. Mitochondrial spare respiratory capacity is negatively correlated with nuclear reprogramming efficiency. Stem Cells Dev. 2017;26:166–176. doi: 10.1089/scd.2016.0162. [DOI] [PubMed] [Google Scholar]
- 40.Chou T. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 41.Kankotia S, Stacpoole PW. Dichloroacetate and cancer: new home for an orphan drug. Biochim Biophys Acta. 2014;1846:617–629. doi: 10.1016/j.bbcan.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 42.Miyai M, Tomita H, Soeda A, Yano H, Iwama T, Hara A. Current trends in mouse models of glioblastoma. J Neurooncol. 2017;135:423–432. doi: 10.1007/s11060-017-2626-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poteet E, Choudhury G, Winters A, Li W, Ryou M, Liu R, Tang L, Ghorpade A, Wen Y, Yuan F, Keir S, Yan H, Bigner D, Simpkins J, Yang S. Correction: Reversing the Warburg effect as a treatment for glioblastoma. J Biol Chem. 2018;293:14973–14973. doi: 10.1074/jbc.aac118.005625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Florio R, Lellis LD, Veschi S, Verginelli F, Giacomo di V, Gallorini M, Perconti S, Sanna M, Mariani-Costantini R, Natale A, Arduini A, Amoroso R, Cataldi A, Cama A. Effects of dichloroacetate as single agent or in combination with GW6471 and metformin in paraganglioma cells. Sci Rep. 2018;8:13610. doi: 10.1038/s41598-018-31797-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer. 2010;102:1746–1752. doi: 10.1038/sj.bjc.6605701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong JYY, Huggins GS, Debidda M, Munshi NC, Vivo ID. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 2008;109:394–402. doi: 10.1016/j.ygyno.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saed GM, Fletcher NM, Jiang ZL, Abu-Soud HM, Diamond MP. Dichloroacetate induces apoptosis of epithelial ovarian cancer cells through a mechanism involving modulation of oxidative stress. Reprod Sci. 2011;18:1253–1261. doi: 10.1177/1933719111411731. [DOI] [PubMed] [Google Scholar]
- 48.Ward NP, Poff AM, Koutnik AP, D’Agostino DP. Complex I inhibition augments dichloroacetate cytotoxicity through enhancing oxidative stress in VM-M3 glioblastoma cells. PLoS One. 2017;12:e0180061. doi: 10.1371/journal.pone.0180061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanto K, Ito H, Noso S, Babaya N, Hiromine Y, Taketomo Y, Toma J, Niwano F, Yasutake S, Kawabata Y, Ikegami H. Effects of dosage and dosing frequency on the efficacy and safety of high-dose metformin in Japanese patients with type 2 diabetes mellitus. J Diabetes Invest. 2017;9:587–593. doi: 10.1111/jdi.12755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calcutt NA, Lopez VL, Bautista AD, Mizisin LM, Torres BR, Shroads AL, Mizisin AP, Stacpoole PW. Peripheral neuropathy in rats exposed to dichloroacetate. J Neuropathol Exp Neurol. 2009;68:985–993. doi: 10.1097/NEN.0b013e3181b40217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nair A, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27–31. doi: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]