

Abstract
Background: Pulmonary cystadenoma is a very rare benign tumor of the lung with slow growth rate and most often, asymptomatic. Case Report: We present the case of a 58-year-old patient admitted in the hospital for coughing with hemoptoic sputum. Standard thoracic radiography revealed a 4/5 cm macronodular opacity in the right inferior lobe, paracardiac. Thoracic computed tomography (CT) with contrast discovered a 3.8/4.7 cm homogenous mass in the right inferior lobe. After intraoperative assessment of the lesion a lower right lobectomy with mediastinal and local lymphadenectomy was performed. Conclusion: Pulmonary mucinous cystadenoma is one of the primary pulmonary mucinous cystic neoplasia (PMCT) alongside PMCT of low malignancy and pulmonary mucinous cystadenocarcinoma (PMCAC). Because of this and because of the clinical and imagistic similarities between these main entities, establishing a preoperative diagnosis becomes very difficult. Therefore, histopathological and immunohistochemistry studies are mandatory in order to establish the correct diagnosis.
Keywords: Pulmonary mucinous cystadenoma, pulmonary cyst neoplasia, right inferior lobectomy, mediastinal lymphadenectomy
Pulmonary mucinous cystadenoma (PMC) is a benign tumor rarely found in the lung. It is more frequently found in women in the ovaries, breasts, liver, and urinary bladder or retroperitoneal (1-3). The first described case of a PMC belongs to Eck in 1969 (4), however, in 1978, Sam Brooke Goward described a separate pathological pulmonary entity – primary pulmonary mucinous cystadenoma neoplasia (PMCN). Several pathologies have been included in this category (PMCN) under different names: mucinous cyst tumor (MCT), mucinous cyst, mucinous cyst tumor of low malignancy, mucinous cystadenocarcinoma (MCAC). However, the most important entities are pulmonary mucinous cystadenoma (PMCA), PMCT of low malignancy and PMCAC. PMCN has clear histological criteria, which allows differentiation between other types of primary or secondary mucinous tumors. The 2015 WHO classification of pulmonary, pleural, thymus and heart tumors has not fully cleared the terminology used for mucinous cyst adenocarcinoma, this entity being included in the colloid adenocarcinoma category (5). In this study, we present a case of pulmonary mucinous cystadenoma discovered intraoperatively, in which we encountered difficulties in the preoperative diagnosis.
Case presentation
A 58-year-old patient with a history of smoking (23 AP) was admitted into the hospital for hemoptysis and physical weakness, which increased in the last 3 weeks prior to admission. Chest radiography revealed the presence of a 4/5 cm macronodular opacity of subcostal intensity, with undefined limits. Bloodwork discovered a mild anemia, with no other modified parameters in biochemistry and clotting tests.
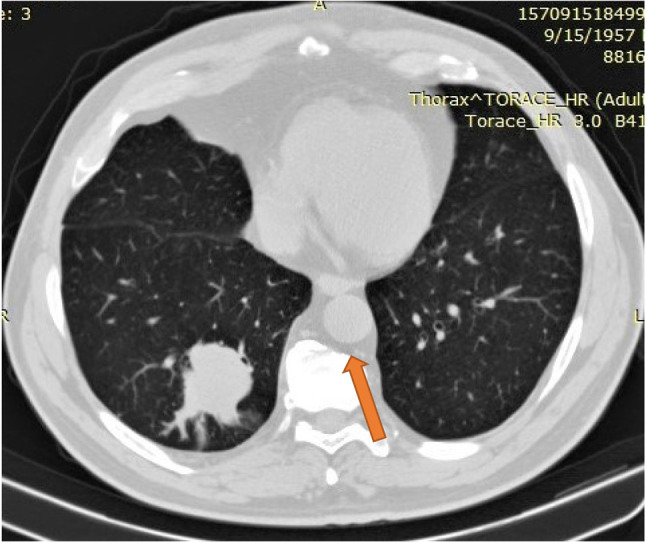
Thoracic and superior abdomen computed tomography (CT) scan with contrast (Figure 1) discovered a 38/47 mm dense, homogenous mass with irregular shape in the right lower lobe, as well as enlarged hilar and subcarinal lymph nodes. No secondary lesions were identified in the liver or adrenal glands. Both CT and chest radiography pointed the diagnosis to a primary pulmonary tumor of the right lower lobe, possibly malignant, given the presence of enlarged mediastinal lymph nodes and the smoking status of the patient.
Figure 1. Thoracic computed tomography view of the lesion.
Bronchoscopy did not identify any endobronchial modifications with the exception of erythematous mucosa of the bronchus, which is specific for chronic bronchiectasis. Lung volume tests revealed a slightly diminished forced expiratory volume (FEV) and forced vital capacity (FVC). Electrocardiogram and echocardiography were within normal limits, with no significant modifications.
Surgery was performed using an antero-lateral thoracotomy with access through the fifth intercostal space, discovering an elastic, renitent mass in the right lower lobe without affecting the visceral pleura. We performed a pneumotomy and attempted dissection of the mass (Figure 2). We discovered the presence of a 3/4 cm encapsulated, gelatinous pulmonary mass, which appeared more as a cystic mass. Mediastinal inspection also revealed the presence of enlarged paratracheal, hilar and paraesophageal lymph nodes. Based on these findings, after closing the pneumotomy, a right lower lobectomy with mediastinal lymphadenectomy was performed. Postoperative evolution was favorable, with no complications and with full reexpansion of the remaining lung; patient was discharged after seven days of hospital stay.
Figure 2. Intraoperative discovery of the lesion after pneumotomy.

Histopathological examinations of the surgical specimen identified a cystic mass with a diameter of 4 cm with mucous contents. The walls of the mass were thin, transparent, smooth, with a few dilated bronchus surrounding the tumor. Microscopic examination identified pulmonary tissue with modified architecture through benign proliferation, the tumor wall covered in cylindrical/cuboidal epithelium with high secretion of mucin along with focal giant cell reaction. The cyst wall was also covered with cylindrical/cuboidal epithelium with hyperchromatic nuclei and basophilic cytoplasm, with no cell atypia. Adjacent pulmonary parenchyma presented as an organizing pneumonia as well as a dilated bronchus with ulcerated epithelium and thickened walls. All examined lymph nodes were between 0.8 and 1.5 cm and presented follicular hyperplasia, anthracosis and sinus histiocytosis. Histopathological examination concluded that the lesion appeared as a pulmonary mucinous cystadenoma (PMCA) (Figure 3).
Figure 3. Histological view of the resected pulmonary lesion using hematoxylin-eosin staining at ×4 and ×40 magnification.

Immunohistochemistry performed on the resected specimen identified intense positive reactions for cytokeratin 7 and 20 (CK7 and CK20) as well as a slight positive reaction for AE1/AE3 (high molecular weight keratin/low molecular weight cytokeratin). Test was negative for cancer antigen 125 (CA125), carcino embryonic antigen (CEA) and thyroid transcription factor 1 (TTF-1). Immunohistochemistry findings pointed towards a benign pulmonary lesion.
Patient follow up through thoracic radiography and thoracic CT scan after 6 months and 1 year did not show any signs of recurrence.
Case presentation
A 58-year-old patient with a history of smoking (23 AP) was admitted into the hospital for hemoptysis and physical weakness, which increased in the last 3 weeks prior to admission. Chest radiography revealed the presence of a 4/5 cm macronodular opacity of subcostal intensity, with undefined limits. Bloodwork discovered a mild anemia, with no other modified parameters in biochemistry and clotting tests.
Thoracic and superior abdomen computed tomography (CT) scan with contrast (Figure 1) discovered a 38/47 mm dense, homogenous mass with irregular shape in the right lower lobe, as well as enlarged hilar and subcarinal lymph nodes. No secondary lesions were identified in the liver or adrenal glands. Both CT and chest radiography pointed the diagnosis to a primary pulmonary tumor of the right lower lobe, possibly malignant, given the presence of enlarged mediastinal lymph nodes and the smoking status of the patient.
Bronchoscopy did not identify any endobronchial modifications with the exception of erythematous mucosa of the bronchus, which is specific for chronic bronchiectasis. Lung volume tests revealed a slightly diminished forced expiratory volume (FEV) and forced vital capacity (FVC). Electrocardiogram and echocardiography were within normal limits, with no significant modifications.
Surgery was performed using an antero-lateral thoracotomy with access through the fifth intercostal space, discovering an elastic, renitent mass in the right lower lobe without affecting the visceral pleura. We performed a pneumotomy and attempted dissection of the mass (Figure 2). We discovered the presence of a 3/4 cm encapsulated, gelatinous pulmonary mass, which appeared more as a cystic mass. Mediastinal inspection also revealed the presence of enlarged paratracheal, hilar and paraesophageal lymph nodes. Based on these findings, after closing the pneumotomy, a right lower lobectomy with mediastinal lymphadenectomy was performed. Postoperative evolution was favorable, with no complications and with full reexpansion of the remaining lung; patient was discharged after seven days of hospital stay.
Histopathological examinations of the surgical specimen identified a cystic mass with a diameter of 4 cm with mucous contents. The walls of the mass were thin, transparent, smooth, with a few dilated bronchus surrounding the tumor. Microscopic examination identified pulmonary tissue with modified architecture through benign proliferation, the tumor wall covered in cylindrical/cuboidal epithelium with high secretion of mucin along with focal giant cell reaction. The cyst wall was also covered with cylindrical/cuboidal epithelium with hyperchromatic nuclei and basophilic cytoplasm, with no cell atypia. Adjacent pulmonary parenchyma presented as an organizing pneumonia as well as a dilated bronchus with ulcerated epithelium and thickened walls. All examined lymph nodes were between 0.8 and 1.5 cm and presented follicular hyperplasia, anthracosis and sinus histiocytosis. Histopathological examination concluded that the lesion appeared as a pulmonary mucinous cystadenoma (PMCA) (Figure 3).
Immunohistochemistry performed on the resected specimen identified intense positive reactions for cytokeratin 7 and 20 (CK7 and CK20) as well as a slight positive reaction for AE1/AE3 (high molecular weight keratin/low molecular weight cytokeratin). Test was negative for cancer antigen 125 (CA125), carcino embryonic antigen (CEA) and thyroid transcription factor 1 (TTF-1). Immunohistochemistry findings pointed towards a benign pulmonary lesion.
Patient follow up through thoracic radiography and thoracic CT scan after 6 months and 1 year did not show any signs of recurrence.
Discussion
Being a rare pathology, there are few cases of PMCA in the literature. Preoperative diagnosis is difficult to establish, especially since 90% of the tumor volume consists of mucin (6), as was the case for the patient presented, with the lesion being discovered intraoperatively. In most cases, PMCA is asymptomatic (68% of cases) and the most common symptoms are coughing (14% of cases), hemoptysis (5% of cases) and recurrent pneumonia (7). PMCA consists of unilocular, occasionally multilocular cysts filled with mucus and enclosed by a fibrous capsule (8). However, PMCA is a lesion with an uncertain natural history, which is why it must be taken seriously (9).
Some authors have established a series of histological criteria that define PMCN: cystic gross appearance with or without a fibrous wall; solitary well circumscribed lesions in the lung parenchyma with no evidence of carcinoma in situ in the bronchial mucosa to suggest endobronchial origin of the tumor; greater than 90% of the tumor bulk made of mucin, neoplastic epithelial cells are mucin-producing cells that can be floating in the mucin or lining the fibrous wall; no other primary mucinous malignant process after intensive clinical workup (7). During the differential diagnosis of PMCA, bronchial cystadenoma must be considered; while PMCA is a unilocular or multilocular cyst filled with mucus, surrounded by a fibrous capsule, bronchial cystadenoma is frequently endobronchial as a well-defined, solitary tumor, visible during bronchoscopy. Histological components consist of mucosecretant cells of the trachea-bronchial seromucinous glands (8). Although pulmonary cystadenomas are located in the lung parenchyma (8, 10), there has only been one described case of a peripheral bronchial cystadenoma, which brought up a very difficult differential diagnosis (11).
Differential diagnosis must also include other non-neoplastic lesions such as bronchogenic cyst, congenital adenomatoid malformation or postinfectious bronchogenic cyst; all of which present with areas of mucous epithelium covered with ciliated columnar epithelium (12, 13). In the case of bronchogenic cysts, cartilaginous areas and seromucinous glands can be found in the cyst wall (9). On the other hand, pulmonary cystic adenomatoid malformations appear during the juvenile stage and are most times cystic lesions; histology usually identifies smooth muscular fibers in the cyst wall with the epithelium formed of columnar or cuboidal cells (9).
Congenital cystic adenomatoid malformation, especially type 1, is capable of extracystic mucinous proliferation similar to that of bronchoalveolar mucinous carcinomas, with some authors considering it a precursor for this type of neoplastic lesion (14). However, mucinous cystadenoma must first be differentiated from pulmonary mucinous tumor of low malignancy (PMCT) and pulmonary mucinous cystadenocarcinoma (PMCAC). Further lesions that must be differentiated are mucoepidermoid carcinoma, mucinous bronchoalveolar carcinoma or other types of metastatic mucinous ovarian or pancreatic tumors (7, 10, 15, 16).
PMCT are extremely rare tumors that present as a gelatinous mass with areas of cellular atypia, which resemble a pulmonary mucinous adenocarcinoma, but with a much more favorable outcome, with no local recurrence or distant metastases (10, 17). Furthermore, this type of lesion is not associated with local invasion, with the exception areas with a malignant cytology in the cyst wall (9). Some authors consider pulmonary mucinous cysts of low malignancy to have a good prognosis and must be differentiated from pulmonary adenocarcinomas (18, 19). Even so, there has been one study reporting recurrence of low malignancy lesions when using limited surgical resections (20). Immunohistochemistry shows these types of lesions to have a strong expression for CK20 and a weaker one for CK7 (21).
Regarding pulmonary mucinous adenocarcinoma, there have been cases with bilateral multifocal cystic lesions (22-24). Some authors consider PMCAC to be a cystic presentation of a mucin productive pulmonary adenocarcinoma, with a much higher incidence compared to PMCA (25, 26).
Histological differentiation from other pulmonary metastatic cystic mucoid lesions is difficult to establish. In these situations, immunohistochemistry examinations are fundamental. Pulmonary mucinous cyst neoplasia is positive for CK7, CK20 and CDX-2 and negative for CA125, D2-40, and WT-1. Ovarian cyst neoplasia is positive for CA125 and negative for CK20. Histologically, PMCA is the equivalent of ovarian and pancreatic mucinous cystadenoma with the exception of the stroma (fibrous stroma in PMCA versus ovarian stroma) (3).
There have been forms of multifocal pulmonary mucinous cystic neoplasia described with ovarian type stroma that appears positive for AE1/AE3, EMA and CK7 and negative for CK20 and CK19 (1). However, pulmonary mucinous cyst adenocarcinoma appears positive for CK7, related homeobox gene 2 (CDX2), CA, TTF-1 and CK20 (2, 22, 27). In the case of primary mucinous cyst neoplasia, immunohistochemistry is positive for AE1/AE3 and CK7 and negative for CK20 (7). PMCA has a low expression of the proliferating cell nuclear antigen (PCNA) marker, MIB1 gene and CAE (28). Because a diagnosis of PMCN is difficult to establish, some authors tried using positron emission tomography with 2-deoxy-2-[fluorine-18]fluoro- D-glucose integrated with computed tomography (PET-CT 18F-FDG). This revealed low and diffuse capture in the pulmonary lesion, but much more intense in the hilar and paratracheal lymph nodes, especially in cystadenocarcinomas (29). In other cases, capture was intense in the solid portion of the lesion wall and significantly lower in the cystic part (26). There have also been cases of PMCA discovered using Pet-F-FDG/CT with high capture of 18F-FDG, mimicking lung cancer (30, 31). In order to establish a preoperative histological diagnosis, some authors resorted to a CT guided fine-needle aspiration biopsy (32). Few cases are like this due to the low number of neoplastic cells compared to the high quantity of mucin (33).
In the case presented, PET F-FDG/CT or CT guided biopsy was not used, considering that it would not assist the preoperative diagnosis. Intraoperatively, once the lesion was identified as a PMCT, right lower lobectomy with hilar and mediastinal lymphadenectomy was performed.
Many authors recommend early and complete resection of pulmonary mucinous cysts due to their risk of malignant transformation or masking a borderline lesion (9, 34). There have been cases of recurrent PMCA after 20 years, especially when anatomical resections were not performed (35). A series of authors suggest long-term monitoring of patients with PMCN, especially since the evolution of this type of pulmonary pathology is not yet completely understood (16).
Prognosis for these patients is usually good, with recurrence being rare and metastases from malignant lesions being exceptional. Depending on the local status, resection of these lesions can also be minimally invasive (6).
Discussion
Being a rare pathology, there are few cases of PMCA in the literature. Preoperative diagnosis is difficult to establish, especially since 90% of the tumor volume consists of mucin (6), as was the case for the patient presented, with the lesion being discovered intraoperatively. In most cases, PMCA is asymptomatic (68% of cases) and the most common symptoms are coughing (14% of cases), hemoptysis (5% of cases) and recurrent pneumonia (7). PMCA consists of unilocular, occasionally multilocular cysts filled with mucus and enclosed by a fibrous capsule (8). However, PMCA is a lesion with an uncertain natural history, which is why it must be taken seriously (9).
Some authors have established a series of histological criteria that define PMCN: cystic gross appearance with or without a fibrous wall; solitary well circumscribed lesions in the lung parenchyma with no evidence of carcinoma in situ in the bronchial mucosa to suggest endobronchial origin of the tumor; greater than 90% of the tumor bulk made of mucin, neoplastic epithelial cells are mucin-producing cells that can be floating in the mucin or lining the fibrous wall; no other primary mucinous malignant process after intensive clinical workup (7). During the differential diagnosis of PMCA, bronchial cystadenoma must be considered; while PMCA is a unilocular or multilocular cyst filled with mucus, surrounded by a fibrous capsule, bronchial cystadenoma is frequently endobronchial as a well-defined, solitary tumor, visible during bronchoscopy. Histological components consist of mucosecretant cells of the trachea-bronchial seromucinous glands (8). Although pulmonary cystadenomas are located in the lung parenchyma (8,10), there has only been one described case of a peripheral bronchial cystadenoma, which brought up a very difficult differential diagnosis (11).
Differential diagnosis must also include other non-neoplastic lesions such as bronchogenic cyst, congenital adenomatoid malformation or postinfectious bronchogenic cyst; all of which present with areas of mucous epithelium covered with ciliated columnar epithelium (12,13). In the case of bronchogenic cysts, cartilaginous areas and seromucinous glands can be found in the cyst wall (9). On the other hand, pulmonary cystic adenomatoid malformations appear during the juvenile stage and are most times cystic lesions; histology usually identifies smooth muscular fibers in the cyst wall with the epithelium formed of columnar or cuboidal cells (9).
Congenital cystic adenomatoid malformation, especially type 1, is capable of extracystic mucinous proliferation similar to that of bronchoalveolar mucinous carcinomas, with some authors considering it a precursor for this type of neoplastic lesion (14). However, mucinous cystadenoma must first be differentiated from pulmonary mucinous tumor of low malignancy (PMCT) and pulmonary mucinous cystadenocarcinoma (PMCAC). Further lesions that must be differentiated are mucoepidermoid carcinoma, mucinous bronchoalveolar carcinoma or other types of metastatic mucinous ovarian or pancreatic tumors (7,10,15,16).
PMCT are extremely rare tumors that present as a gelatinous mass with areas of cellular atypia, which resemble a pulmonary mucinous adenocarcinoma, but with a much more favorable outcome, with no local recurrence or distant metastases (10,17). Furthermore, this type of lesion is not associated with local invasion, with the exception areas with a malignant cytology in the cyst wall (9). Some authors consider pulmonary mucinous cysts of low malignancy to have a good prognosis and must be differentiated from pulmonary adenocarcinomas (18,19). Even so, there has been one study reporting recurrence of low malignancy lesions when using limited surgical resections (20). Immunohistochemistry shows these types of lesions to have a strong expression for CK20 and a weaker one for CK7 (21).
Regarding pulmonary mucinous adenocarcinoma, there have been cases with bilateral multifocal cystic lesions (22-24). Some authors consider PMCAC to be a cystic presentation of a mucin productive pulmonary adenocarcinoma, with a much higher incidence compared to PMCA (25,26).
Histological differentiation from other pulmonary metastatic cystic mucoid lesions is difficult to establish. In these situations, immunohistochemistry examinations are fundamental. Pulmonary mucinous cyst neoplasia is positive for CK7, CK20 and CDX-2 and negative for CA125, D2-40, and WT-1. Ovarian cyst neoplasia is positive for CA125 and negative for CK20. Histologically, PMCA is the equivalent of ovarian and pancreatic mucinous cystadenoma with the exception of the stroma (fibrous stroma in PMCA versus ovarian stroma) (3).
There have been forms of multifocal pulmonary mucinous cystic neoplasia described with ovarian type stroma that appears positive for AE1/AE3, EMA and CK7 and negative for CK20 and CK19 (1). However, pulmonary mucinous cyst adenocarcinoma appears positive for CK7, related homeobox gene 2 (CDX2), CA, TTF-1 and CK20 (2,22,27). In the case of primary mucinous cyst neoplasia, immunohistochemistry is positive for AE1/AE3 and CK7 and negative for CK20 (7). PMCA has a low expression of the proliferating cell nuclear antigen (PCNA) marker, MIB1 gene and CAE (28). Because a diagnosis of PMCN is difficult to establish, some authors tried using positron emission tomography with 2-deoxy-2-[fluorine-18]fluoro- D-glucose integrated with computed tomography (PET-CT 18F-FDG). This revealed low and diffuse capture in the pulmonary lesion, but much more intense in the hilar and paratracheal lymph nodes, especially in cystadenocarcinomas (29). In other cases, capture was intense in the solid portion of the lesion wall and significantly lower in the cystic part (26). There have also been cases of PMCA discovered using Pet-F-FDG/CT with high capture of 18F-FDG, mimicking lung cancer (30,31). In order to establish a preoperative histological diagnosis, some authors resorted to a CT guided fine-needle aspiration biopsy (32). Few cases are like this due to the low number of neoplastic cells compared to the high quantity of mucin (33).
In the case presented, PET F-FDG/CT or CT guided biopsy was not used, considering that it would not assist the preoperative diagnosis. Intraoperatively, once the lesion was identified as a PMCT, right lower lobectomy with hilar and mediastinal lymphadenectomy was performed.
Many authors recommend early and complete resection of pulmonary mucinous cysts due to their risk of malignant transformation or masking a borderline lesion (9,34). There have been cases of recurrent PMCA after 20 years, especially when anatomical resections were not performed (35). A series of authors suggest long-term monitoring of patients with PMCN, especially since the evolution of this type of pulmonary pathology is not yet completely understood (16).
Prognosis for these patients is usually good, with recurrence being rare and metastases from malignant lesions being exceptional. Depending on the local status, resection of these lesions can also be minimally invasive (6).
Conclusion
In conclusion, PMCA is a rare benign lesion with a yet unknown possibility of malignant transformation. Diagnosis of PMCA and implicitly of PMCN is difficult to establish preoperatively. When presented with such a case, complete resection with local lymphadenectomy is preferred, even if the lesion turns out to be benign.
Conflicts of Interest
There are no conflicts of interest to declare regarding this study.
Authors’ Contributions
CS, AM, AG performed the surgical procedure; NB, CS, IB, OS reviewed literature data; CD preoperative investigation the patient; IB, NB CS prepared the draft of the manuscript; CS was advisor of the surgical procedures; SV performed histopathological examinations; CS, NB reviewed the final version of the manuscript. The Authors read and approved the final version of the manuscript.
References
- 1.Geramizadeh B, Ziyaian B, Khaleghi S. Multifocal pulmonary mucinous cystic neoplasm with ovarian type stroma, a new finding in an extremely rare case report. Indian J Pathol Microbiol. 2014;57(1):92–93. doi: 10.4103/0377-4929.130910. [DOI] [PubMed] [Google Scholar]
- 2.Moneke I, Zeisel C, Elze M, Haager B, Passlick B, Kayser G. Mucinous cystadenocarcinoma arising from mucinous cystadenoma of the lung: case report and review of the literature. J Thorac Dis. 2018;10(4):E243–E249. doi: 10.21037/jtd.2018.04.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silverman JF, Zhu B, Liu Y, Lin X. Distinctive immunohistochemical profile of mucinous cystic neoplasms of pancreas, ovary and lung. Histol Histopathol. 2009;24(1):77–82. doi: 10.14670/HH-24.77. [DOI] [PubMed] [Google Scholar]
- 4.Eck H, Haupt R, Rothe G. Respiratory tract and lungs: the benign and malignant pulmonary tumors. Springer, Berlin, Heidelberg. 1969;Fourth part doi: 10.1007/978-3-642-47840-6_1. [DOI] [Google Scholar]
- 5.Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, Chirieac LR, Dacic S, Duhig E, Flieder DB, Geisinger K, Hirsch FR, Ishikawa Y, Kerr KM, Noguchi M, Pelosi G, Powell CA, Tsao MS, Wistuba I. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J Thorac Oncol. 2015;10(9):1243–1260. doi: 10.1097/JTO.0000000000000630. [DOI] [PubMed] [Google Scholar]
- 6.Haruki T, Nakamura H, Taniguchi Y, Miwa K, Adachi Y, Fujioka S. Pulmonary mucinous cystadenoma: a rare benign tumor of the lung. Gen Thorac Cardiovasc Surg. 2010;58(6):287–290. doi: 10.1007/s11748-009-0519-9. [DOI] [PubMed] [Google Scholar]
- 7.Gao ZH, Urbanski SJ. The spectrum of pulmonary mucinous cystic neoplasia: a clinicopathologic and immunohistochemical study of ten cases and review of literature. Am J Clin Pathol. 2005;124(1):62–70. doi: 10.1309/52XXR6E6U0J2JX0F. [DOI] [PubMed] [Google Scholar]
- 8.England DM, Hochholzer L. Truly benign "bronchial adenoma". Report of 10 cases of mucous gland adenoma with immunohistochemical and ultrastructural findings. Am J Surg Pathol. 1995;19(8):887–899. doi: 10.1097/00000478-199508000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Davison AM, Lowe JW, Da Costa P. Adenocarcinoma arising in a mucinous cystadenoma of the lung. Thorax. 1992;47(2):129–130. doi: 10.1136/thx.47.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickstein PJ, Amaral SM, Silva AM, Daltro PA, Ferreira AJ, Aiex A. Bronchial mucous gland adenoma presenting as bronchogenic cyst. Pediatr Pulmonol. 1993;16(6):370–374. doi: 10.1002/ppul.1950160609. [DOI] [PubMed] [Google Scholar]
- 11.Ulusan A, Bayramoglu Z, Sanli M, Bakir K. A rare case of peripherally located non-bronchial pulmonary mucous gland adenoma. Turk Gogus Kalp Damar Cerrahisi Derg. 2018;26(4):664–667. doi: 10.5606/tgkdc.dergisi.2018.15357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kragel PJ, Devaney KO, Meth BM, Linnoila I, Frierson HF Jr., Travis WD. Mucinous cystadenoma of the lung. A report of two cases with immunohistochemical and ultrastructural analysis. Arch Pathol Lab Med. 1990;114(10):1053–1056. [PubMed] [Google Scholar]
- 13.Horvat T, Savu C, Motas C, Tetu M. Pneumopericardium--complication of an unknown tuberculosis in a HIV positive patient. Eur J Cardiothorac Surg. 2004;26(5):1043. doi: 10.1016/j.ejcts.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Lantuejoul S, Nicholson AG, Sartori G, Piolat C, Danel C, Brabencova E, Goldstraw P, Brambilla E, Rossi G. Mucinous cells in type 1 pulmonary congenital cystic adenomatoid malformation as mucinous bronchioloalveolar carcinoma precursors. Am J Surg Pathol. 2007;31(6):961–969. doi: 10.1097/01.pas.0000249444.90594.27. [DOI] [PubMed] [Google Scholar]
- 15.Pirvu A, Angelescu D, Savu C. Localized fibrous tumor of the pleura an unusual cause of severe hypoglycaemia. Case report. Rev Med Chir Soc Med Nat Iasi. 2016;120(3):628–630. [PubMed] [Google Scholar]
- 16.Ishibashi H, Moriya T, Matsuda Y, Sado T, Hoshikawa Y, Chida M, Sato M, Sasano H, Kondo T. Pulmonary mucinous cystadenocarcinoma: report of a case and review of the literature. Ann Thorac Surg. 2003;76(5):1738–1740. doi: 10.1016/s0003-4975(03)00657-x. [DOI] [PubMed] [Google Scholar]
- 17.Savu C, Melinte A, Posea R, Galie N, Balescu I, Diaconu C, Cretoiu D, Dima S, Filipescu A, Balalau C, Bacalbasa N. Pleural solitary fibrous tumors-a retrospective study on 45 patients. Medicina (Kaunas) 2020;56(4) doi: 10.3390/medicina56040185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yaman B, Veral A, Cagirici U, Nart D. Pulmonary mucinous cystic tumour of borderline malignancy: A case report. Virchows Archiv. 2013;463:241. [Google Scholar]
- 19.Dediu M, Horvat T, Tarlea A, Anghel R, Cordos I, Miron G, Iorga P, Alexandru A, Nistor C, Grozavu C, Savu C. Adjuvant chemotherapy for radically resected non-small cell lung cancer: a retrospective analysis of 311 consecutively treated patients. Lung Cancer. 2005;47(1):93–101. doi: 10.1016/j.lungcan.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Mann GN, Wilczynski SP, Sager K, Grannis FW Jr. Recurrence of pulmonary mucinous cystic tumor of borderline malignancy. Ann Thorac Surg. 2001;71(2):696–697. doi: 10.1016/s0003-4975(00)02174-3. [DOI] [PubMed] [Google Scholar]
- 21.Monaghan H, Salter DM, Ferguson T. Pulmonary mucinous cystic tumour of borderline malignancy: a rare variant of adenocarcinoma. J Clin Pathol. 2002;55(2):156. doi: 10.1136/jcp.55.2.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shannon VR, Nanda AS, Middleton LP, Faiz SA. Pulmonary mucinous cystadenocarcinoma presenting as extensive multifocal cystic lesions. Am J Respir Crit Care Med. 2017;195(9):1267–1268. doi: 10.1164/rccm.201610-2106IM. [DOI] [PubMed] [Google Scholar]
- 23.Al Rashidi FM, Muqim AT, Mothafar FJ. Bilateral diffuse mucinous cystic adenocarcinoma of the lungs complicated by recurrent pneumothorax in a pregnant woman. Kuwait Med J. 2012;44(1):56–59. [Google Scholar]
- 24.Higashiyama M, Doi O, Kodama K, Yokouchi H, Tateishi R. Cystic mucinous adenocarcinoma of the lung. Two cases of cystic variant of mucus-producing lung adenocarcinoma. Chest. 1992;101(3):763–766. doi: 10.1378/chest.101.3.763. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez-Carpintero AM, Tamura Ezcurra MA, Torres Tajes JP. Primary pulmonary mucinous cystadenocarcinoma: presentation of a case and a review of the literature. Arch Bronconeumol. 2011;47(4):216–217. doi: 10.1016/j.arbres.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 26.Choi YA, Lee HY, Han J, Choi JY, Kim J, Kwon OJ, Lee KS. Pulmonary mucinous cystadenocarcinoma: report a case and review of CT findings. Korean J Radiol. 2013;14(2):384–388. doi: 10.3348/kjr.2013.14.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shawash S, Abdeljalil R, Haddad H. Multifocal pulmonary mucinous cystic neoplasm with ovarian-like stroma: Once in a blue moon-case report. Respir Med Case Rep. 2019;26:276–280. doi: 10.1016/j.rmcr.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roux FJ, Lantuejoul S, Brambilla E, Brambilla C. Mucinous cystadenoma of the lung. Cancer. 1995;76(9):1540–1544. doi: 10.1002/1097-0142(19951101)76:9<1540::aid-cncr2820760907>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 29.Kalkanis A, Palaiodimos L, Klinaki I, Karantanis D, Kalkanis D. (18)F-FDG PET/CT imaging of pulmonary mucinous cystadenocarcinoma with signet ring cells. Nucl Med Mol Imaging. 2017;51(3):277–280. doi: 10.1007/s13139-016-0402-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cafarotti S, Treglia G, Bongiovanni M, Ceriani L, Paone G, Giovanella L, Dutly A. A rare case of mucinous cystadenoma of the lung mimicking malignancy at 18 F-FDG PET/CT. Clin Nucl Med. 2014;39(6):e331–e333. doi: 10.1097/RLU.0b013e31829960bf. [DOI] [PubMed] [Google Scholar]
- 31.Cho J, Eo JS, In KH, Kim CH, Choe JG. 18F-FDG PET/CT of Bronchial Mucous Gland Adenoma. Clin Nucl Med. 2016;41(2):e118–e119. doi: 10.1097/RLU.0000000000000921. [DOI] [PubMed] [Google Scholar]
- 32.Chhieng DC. Fine-needle aspiration cytology of pulmonary mucinous cystadenocarcinoma. Diagn Cytopathol. 2008;36(8):581–585. doi: 10.1002/dc.20845. [DOI] [PubMed] [Google Scholar]
- 33.Butnor KJ, Sporn TA, Dodd LG. Fine needle aspiration cytology of mucinous cystadenocarcinoma of the lung: report of a case with radiographic and histologic correlation. Acta Cytol. 2001;45(5):779–783. doi: 10.1159/000328305. [DOI] [PubMed] [Google Scholar]
- 34.Cabibi D, Sciuto A, Geraci G, Lo NC, Modica G, Cajozzo M. Cystic mucinous adenocarcinoma of the lung: a case report. J Cardiothorac Surg. 2011;6:128. doi: 10.1186/1749-8090-6-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuo T, Yusuke KN, Takamori S, Shirouzu K. Recurrent pulmonary mucinous cystadenoma. Eur J Cardiothorac Surg. 2005;28(1):176–177. doi: 10.1016/j.ejcts.2005.04.006. [DOI] [PubMed] [Google Scholar]