

Abstract
Background/Aim: Decreased mitochondrial DNA copy number (mtDNA-CN) has been associated with coronary artery disease (CAD). We aimed to clarify the difference between stable CAD (SCAD) and acute coronary syndrome (ACS) regarding mtDNA-CN and the DNA methylation ratio in regions influencing the regulation of mitochondrial biogenesis. Materials and Methods: Using quantitative real-time polymerase chain reaction, mtDNA-CN was measured in peripheral blood leukocytes sampled from 50 patients with SCAD and 50 with ACS. We then conducted bisulfite modification of DNA followed by methylation-specific polymerase chain reaction to quantify mtDNA methylation in the mitochondrial D-loop region (mtDLR) and nuclear DNA methylation in the promoter region of nuclear peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PPARGC1A) gene. Results: Compared to patients with SCAD, those with ACS had significantly lower relative mtDNA-CN (0.89±0.24 vs. 1.00±0.28, p=0.013) and higher DNA methylation ratio of the mtDLR (1.11±0.24 vs. 1.00±0.25, p=0.027) Conclusion: Our findings suggest that increased DNA methylation in the mtDLR, which translates into reduced mtDNA content, may affect the clinical phenotype of CAD
Keywords: Acute coronary syndrome ACS, mitochondrial dysfunction, mitochondrial DNA mtDNA copy number, peroxisome-proliferator-activated receptor γ co-activator-1α PPARGC1A, D-loop region, DNA methylation
Coronary artery disease (CAD) is the most common cause of death globally. The underlying mechanism of CAD involves reduced blood and oxygen flow to the heart muscle due to atherosclerosis of the coronary arteries. Atherosclerosis generally starts early in life and worsens with age. Almost all individuals aged above 65 years have some degree of atherosclerosis. Progression of atherosclerosis can lead to acute cardiovascular events such as acute myocardial infarction (AMI), unstable angina (UA) pectoris, and even sudden cardiac death. Atherosclerotic disease may also remain at a subclinical stage or manifest as chronic stable angina.
In patients with stable coronary artery disease (SCAD) such as subclinical coronary atherosclerosis or chronic stable angina, mortality and the incidence of myocardial infarction are relatively low (1,2). However, prognosis is poorer among patients with acute coronary syndrome (ACS), who develop obstructed coronary blood flow due to plaque rupture and thrombus formation, resulting in myocardial damage (3). Compared to patients with SCAD, those with ACS have traditionally been thought to be at higher risk of both short- and long-term mortality because of myocardial damage. Therefore, it is very important to predict whether patients with atheromatous coronary artery plaque will suffer from ACS. However, currently used imaging techniques and biomarkers do not provide sufficient predictive power for this.
Mitochondria play a critical role in energy homeostasis, apoptosis, and calcium signaling, also serving as a major cellular source of reactive oxygen species (ROS) generating oxidative stress. Mitochondrial DNA (mtDNA) is similar to nuclear DNA (nDNA) regarding the function of transcription and replication. However, unlike nDNA, mtDNA is believed to be vulnerable to ROS-induced damage because it does not incorporate histones and does not possess an efficient repair mechanism. Mitochondrial damage/dysfunction may play an important role in the initiation and progression of atherosclerosis, the main pathological mechanism underlying cardiovascular disease (4,5). Whereas each individual mitochondrion retains a relatively constant number of copies of mtDNA, the number of mitochondria per cell varies remarkably according to cell lineage and function (6). The mtDNA copy number (mtDNA-CN), while not a direct measure of mtDNA damage/dysfunction, is positively correlated with mitochondrial enzyme activity and ATP production (7) and can therefore be used as a biomarker of mitochondrial function. In their analysis of three large prospective studies, Ashar et al. reported that reduced mtDNA-CN is an independent risk factor for cardiovascular disease including CAD and may thus have clinical utility for improving cardiovascular risk classification (8). Others reported that mtDNA-CN is inversely related with CAD severity (9). Based on these results, we hypothesized that mtDNA-CN would be lower in patients with ACS than in those with SCAD.
Similarly to nDNA, mtDNA can be methylated by machinery within the mitochondria (10-12) and can serve to mediate the control of mitochondrial gene expression (13). Wallace et al. proposed the bioenergetic-epigenome hypothesis to explain the increasing number of epigenetic diseases being associated with mitochondrial dysfunction (14). When it occurs in a gene promoter, DNA methylation typically acts to repress gene transcription. A previous study suggested that platelet mtDNA methylation may be implicated in the etiology of cardiovascular disease (15). We thus postulated that in patients with CAD, changes in mitochondrial D-loop or methylation of nuclear peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PPARGC1A) gene may affect mtDNA-CN.
In the present study, we aimed to clarify the difference between ACS and SCAD in terms of mtDNA-CN, as well as in terms of the DNA methylation ratio in the mitochondrial D-loop region and in the promoter region of nuclear PPARGC1A, both known as key regulators of mtDNA replication and copy number.
Materials and Methods
Participant recruitment and DNA preparation. All study protocols were approved by the Institutional Review Board of Eulji Medical School Hospital, Korea (approval no. 2018-06-004) and were conducted in accordance with the Helsinki Declaration of 1975, as revised in 2000. Written informed consent was obtained from all participants.
Blood samples were collected from 50 patients with SCAD and 50 with ACS from the Biobank of Eulji Medi-Bio Research Institute. All patients underwent coronary angiography to investigate the cause of chest pain between January 2016 and December 2017, and all blood samples were obtained within 2 days after coronary angiography. The two groups of SCAD and ACS were frequency-matched according to age and sex. SCAD was defined as: (i) Subclinical coronary atherosclerosis with mild-to-moderate diameter stenosis on coronary angiography; or (ii) chronic stable angina diagnosed based on clinical symptoms and angiographic findings. ACS included UA and AMI showing substantial plaque burden or plaque rupture on coronary angiography. MI type I defined according to the third universal definition of myocardial infarction of the ESC guideline (16) was only eligible in this study.
The blood samples were collected in EDTA-containing tubes and stored at −70˚C. Genomic DNA was extracted from 500 μl of the blood sample using the G-spin Genomic DNA Extraction Kit (Intron, Daejeon, Korea) at Eulji University, Korea. The DNA quantity and purity were determined using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific Korea, Seoul, Korea), revealing optical density (OD 260/OD 280) values of 1.7-2.0 for all DNA samples. The samples of total DNA were also stored at −70˚C.
Measurement of mtDNA-CN. The mtDNA-CN was evaluated based on the ratio of mtDNA to nDNA, which were quantified based on the mitochondrial gene cytochrome B (CYTB) and based on the single-copy nuclear gene pyruvate kinase L/R (PKLR), respectively. The relative amounts of mtDNA and nDNA were measured using quantitative polymerase chain reaction (qPCR) with the primers described by Yoo et al. (17). The primers for the CYTB gene were forward 5’- CACGATTCTTTACCTTTCACTTCATC-3’ and reverse 5’-TGATCCCGTTTCGTGCAAG-3’ sequences, whereas the primers for the PKLR gene were forward 5’-AGCCCAAAT GGCCTTGAAG-3’ and reverse 5’-AGAGACAGAATGCCA GTGAGCTT-3’ sequences. Genomic DNA (20 ng) was used as template per 10 μl reaction with IQ SYBR Green Supermix (Bio-Rad Laboratories, Seoul, Korea) for qPCR, which was conducted under the following conditions: 95˚C for 10 min (pre-denaturation) and 40 cycles, each involving two steps (i.e. denaturation at 95˚C for 15 s, followed by annealing and extension at 60ºC for 1 min). Each measurement was performed in duplicate and the acceptable standard deviation (SD) of the duplicate threshold cycle (DCt) values was set at 0.7. The run was repeated if unacceptable SD values were obtained. The relative mtDNA-CN was calculated as 2−DDCt, where DCt=CtmtDNA CYTB-CtPK, according to a previous report (17).
Bisulfite modification of DNA and methylation-specific PCR. Genomic DNA (200-500 ng) was prepared for bisulfite conversion according to the manufacturer’s instructions (EpiJET Bisulfite conversion kit; Invitrogen, Carlsbad, CA, USA). During bisulfite treatment of genomic DNA, all unmethylated cytosines were converted to uracils, whereas methylated cytosines remained unaffected. The bisulfite-converted DNA was used as the template for methylation-specific PCR to determine the DNA methylation state of the promoter region of nuclear PPARGC1A and of the mitochondrial D-loop region. DNA methylation was quantified using suitable primers, as described previously (18,19): forward primer 5’-ATTTTTTATTGTTATGGGGGTAGTC-3’ and reverse primer 5’-AAAAATATTTAAAAACGCAAACGAA-3’ were used to quantify methylated DNA in the PPARGC1A promoter; forward primer 5’-TTTTATTGTTATGGGGGTAGTTGA-3’ and reverse primer 5’-AAAAAATATTTAAAAACACAAACAAA-3’ were used to quantify unmethylated DNA in the PPARGC1A promoter; forward primer 5’-TAGGAATTAAAGATAGATATTGCGA-3’ and reverse primer 5’-ACTCTCCATACATTTAATATTTTCGTC-3’ were used to quantify methylated DNA in the D-loop region; and forward primer 5’-GGTAGGAATTAAAGATAGATATTGTGA-3’ and reverse primer 5’-ACTCTCCATACATTTAATATTTTCATC-3’ were used to quantify unmethylated DNA in the D-loop region. The PCR conditions are summarized in Table I. The qPCR assay was performed using IQ SYBR Green Supermix (all from Bio-Rad Laboratories). Each determination was performed in duplicate using 20 ng of converted DNA per 10 μl reaction. The acceptable SD of the DCt values was set at 0.7. The DNA methylation ratio was expressed as the ratio of the estimated amount of methylated DNA to that of unmethylated DNA, calculated for each sample as 2−DDCt, where DCt=Ctmethyl-Ctunmethyl.
Table I. Conditions for methylation-specific polymerase chain reaction.

PPARGC1A: Peroxisome-proliferator-activated receptor γ co-activator-1α
Statistical analyses. The patients were stratified according to CAD status (SCAD versus ACS). Demographic and clinical characteristics were compared between the groups using the independent t-test for continuous variables and the Pearson chi-square test for categorical variables. The Mann-Whitney U-test was used to evaluate the difference in relative mtDNA-CN between subgroups of each group. Comparison of the methylation ratios of the PPARGC1A gene promoter was also performed using Mann-Whitney U-tests because the data did not follow a normal distribution. All statistical analyses were performed using SPSS version 22.0 (IBM, Armonk, NY, USA) and Prism 7 (GraphPad Software, Inc., La Jolla, CA, USA). Statistical significance was set at a probability level of p<0.05.
Results
Patient characteristics according to CAD status. The demographic and clinical characteristics of the 100 patients included in this study are summarized in Table II. The SCAD and ACS groups were well-matched in terms of age and sex (p=0.271 and p>0.99, respectively). Among the patients with SCAD, 38% (19/50) had mild stenosis (<40% diameter stenosis), whereas 62% (31/50) had subclinical CAD (>40% diameter stenosis) or chronic stable angina. Among the patients with ACS, 40% (20/50) had UA and 60% (30/50) had AMI. Compared to the SCAD group, the ACS group had significantly higher prevalence of current smokers (18.0% vs. 38%, p=0.026), higher white blood cell counts (5.90±1.59 vs. 7.97±2.44 ×109 cells/l, p<0.001), and higher peak creatine kinase levels (112±61 vs. 1341±1946 IU/l, p<0.001).
Table II. Demographic and clinical characteristics of study patients.

Data shown are mean±standard deviation or frequency (percentage). ACS: Acute coronary syndrome; BMI: body mass index; CK: creatine kinase; CRP: C-reactive protein; DBP: diastolic blood pressure; HbA1C: glycated hemoglobin; HDL-C: high-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol; SBP: systolic blood pressure; SCAD: stable coronary artery disease; TC: total cholesterol; TG: triglycerides; WBC: white blood cell. Statistically significant p-values are shown in bold
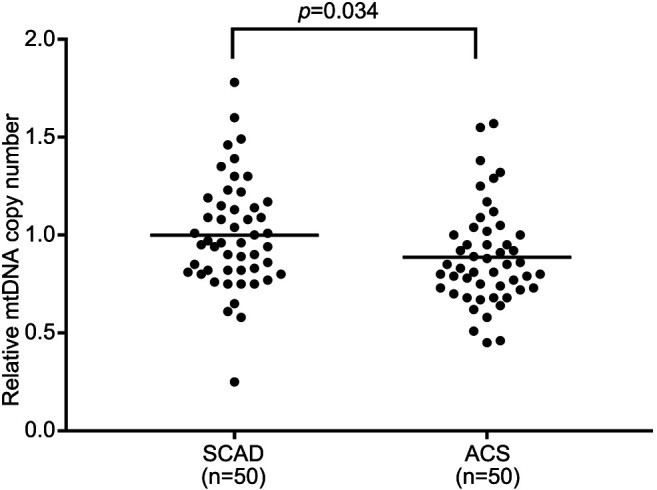
MtDNA-CN according to CAD status. The mtDNA-CN in the peripheral blood samples was significantly lower in the ACS group than in the SCAD group (1.0±0.28 vs. 0.89±0.24, p=0.034; Figure 1).
Figure 1. Relative mitochondrial DNA (mtDNA) copy number in peripheral blood. Relative mtDNA copy number was defined as the ratio of mtDNA to nuclear DNA. Data are shown for patients with stable coronary artery disease (SCAD) and for those with acute coronary syndrome (ACS). Bar: Mean value.
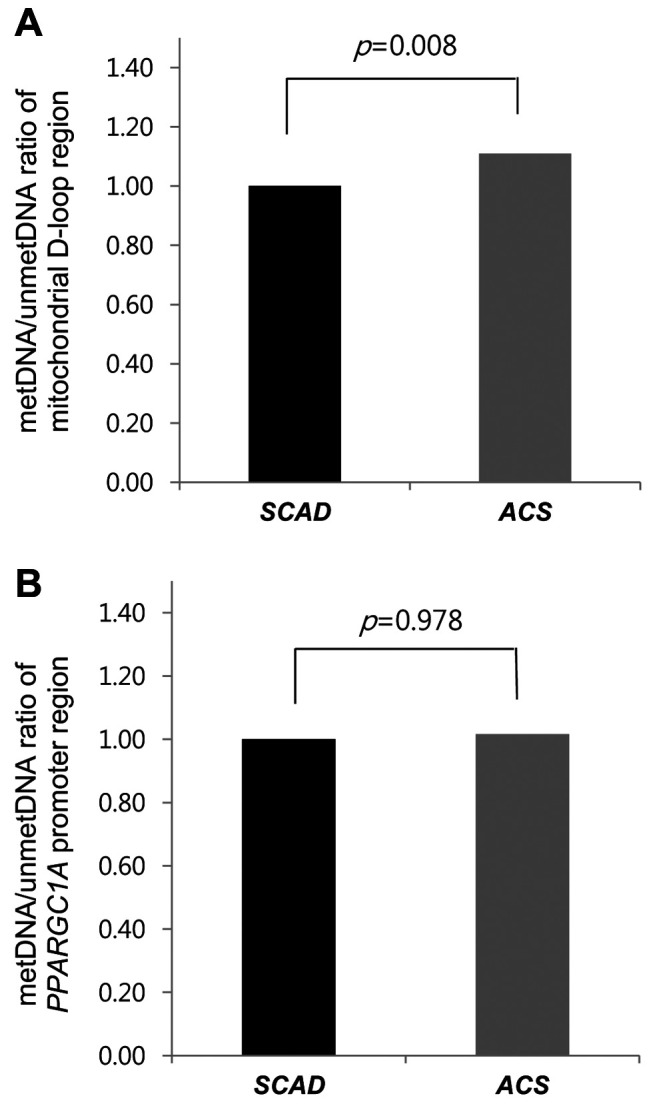
Methylation of the mitochondrial D-loop region and promoter region of nuclear PPARGC1A. Compared to the SCAD group, the ACS group had a higher DNA methylation ratio in the mitochondrial D-loop region (p=0.008; Figure 2A) but similar DNA methylation ratio in the promoter region of nuclear PPARGC1A (p=0.978, Figure 2B).
Figure 2. DNA methylation ratio in stable coronary artery disease (SCAD) and acute coronary syndrome (ACS). Data are shown for the mitochondrial D-loop region (A) and for the nuclear peroxisome-proliferator-activated receptor γ co-activator-1α (PPARGC1A) promoter region (B). SCAD was classified as mild-CAD and sub-CAD, whereas ACS was classified as unstable angina and acute myocardial infarction. metDNA: Methylated DNA; unmetDNA: unmethylated DNA.
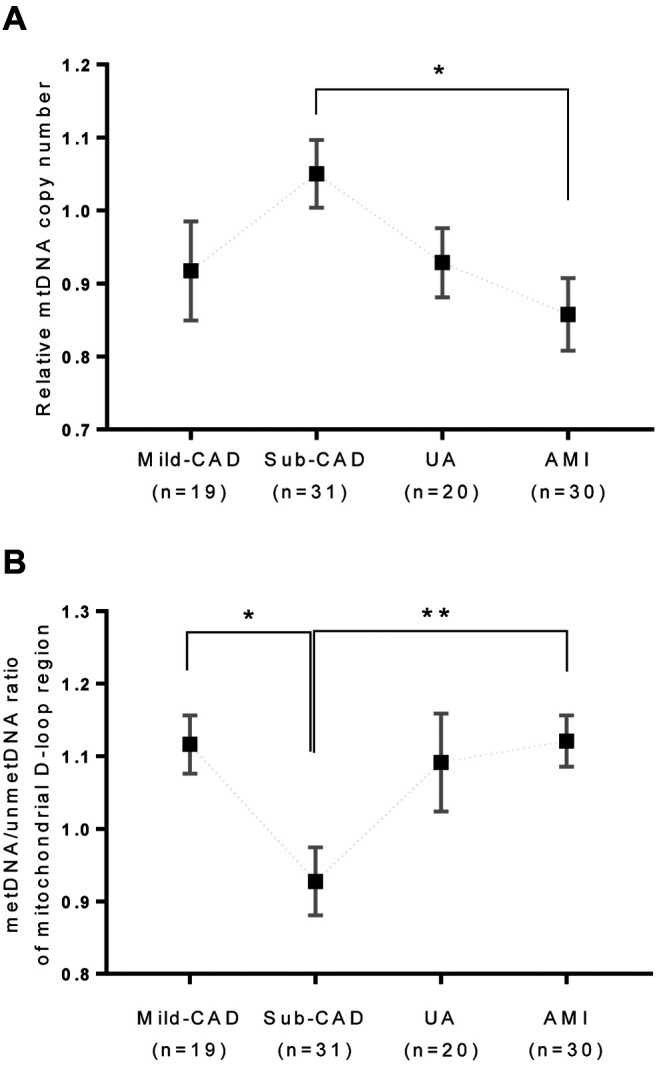
Relationship between mtDNA-CN and mitochondrial D-loop region methylation according to CAD status. We next aimed to determine whether mtDNA-CN is affected by the DNA methylation ratio in the mitochondrial D-loop region (Figure 3). In this analysis, SCAD was stratified into mild CAD (n=19; nearly normal or mild lesion with <40% diameter stenosis on coronary angiography) and substantial CAD (n=31; subclinical CAD or chronic stable angina, exhibiting lesions with substantial plaque burden characterized by >40% diameter stenosis on coronary angiography). Additionally, ACS was stratified into UA (n=20) and AMI (n=30). Relative mtDNA-CN differed among these four subgroups (mild CAD, substantial CAD, UA, and AMI), increasing up to substantial CAD but then decreasing; however, upon post-hoc Bonferroni correction, statistical significance was retained only for the difference between subclinical CAD and AMI (p=0.030). With increasing CAD severity, the pattern of DNA methylation ratio of the mitochondrial D-loop region was opposite to that of relative mtDNA-CN, with similar differences among the subgroups; however, upon post-hoc Bonferroni correction, statistical significance was retained for the difference between mild and subclinical CAD (p=0.045), as well as for the difference between subclinical CAD and AMI (p=0.012) (Figure 3).
Figure 3. Mitochondrial DNA methylation according to coronary artery disease (CAD) status and severity. The patients were stratified into two groups according to disease status: Stable coronary artery disease (SCAD) and acute coronary syndrome (ACS). SCAD was further classified as mild-CAD and sub-CAD, whereas ACS was classified as unstable angina (UA) and acute myocardial infarction (AMI). metDNA: Methylated DNA; mild-CAD: nearly normal or mild lesion with diameter stenosis <40% on coronary angiography; sub-CAD: subclinical CAD or chronic stable angina with lesions displaying substantial plaque burden (diameter stenosis >40% on coronary angiography); unmetDNA: unmethylated DNA. Data are displayed as mean values, with error bars indicating the standard error of the mean. Significantly different at *p<0.05, and **p=0.012.
Distribution of mtDNA-CN values according to patient characteristics. Upon stratifying the relative mtDNA-CN values according to patient characteristics, most subgroups of patients with SCAD and ACS continued to differ significantly in terms of relative mtDNA-CN (Table III), whereas no significant modulating effect of patient characteristics on relative mtDNA-CN was found within either group (Table III).
Table III. Distribution of relative mitochondrial DNA copy number (mtDNA-CN) values according to patient characteristics.

ACS: Acute coronary syndrome; BMI: body mass index; CRP: C-reactive protein; HbA1C: glycated hemoglobin; HDL-C: high-density lipoprotein cholesterol; LDL-C: low-density lipoprotein cholesterol; mtDNA: mitochondrial DNA; SCAD: stable coronary artery disease; SD: standard deviation; TC: total cholesterol; TG: triglycerides. *Between-subgroup differences in mtDNA content were evaluated using the Mann-Whitney U-test
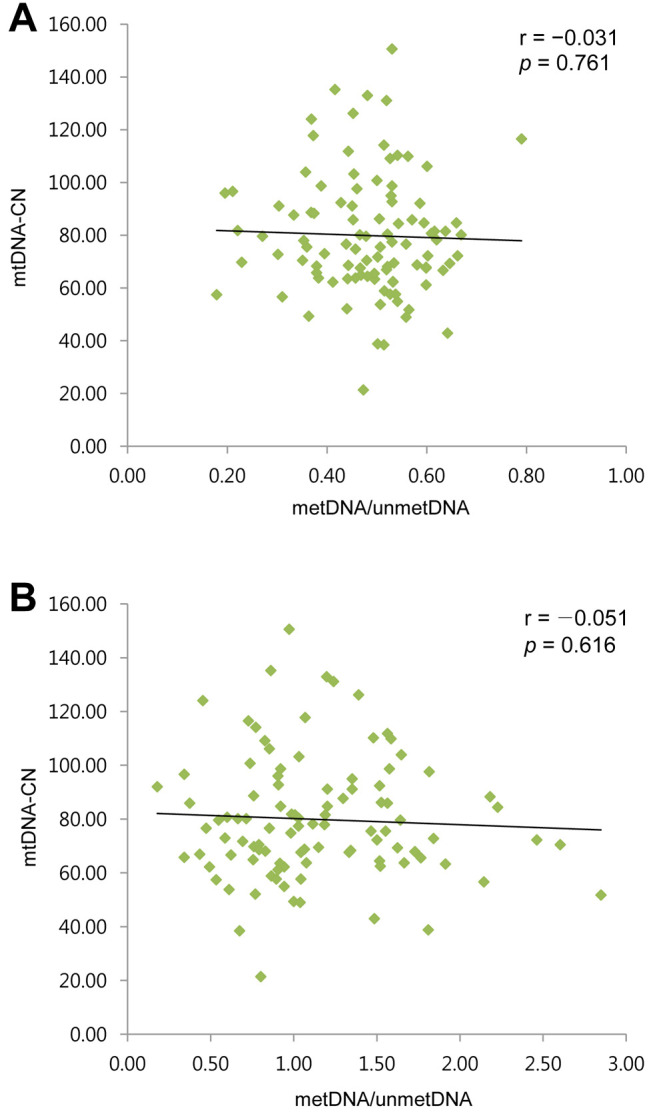
Correlation of mtDNA-CN with the methylation ratio of the mitochondrial D-loop region and of the promoter region of nuclear PPARGC1A. Across the 100 samples analyzed, low and statistically non-significant inverse correlation was noted for mtDNA-CN with the methylation status of the PPARGC1A promoter region and that of the mitochondrial D-loop region (Figure 4).
Figure 4. Correlation between mitochondrial DNA copy number (mtDNA-CN) and the ratio of methylated to unmethylated DNA (metDNA/unmetDNA) for the mitochondrial D-loop region (A) and for the promoter region of nuclear peroxisome-proliferator-activated receptor γ co-activator-1α (PPARGC1A) (B).
Discussion
We examined whether mtDNA-CN and DNA methylation ratios are affected by CAD status. We found that compared to patients with SCAD, those with ACS had significantly lower mtDNA-CN, which suggests that lower mtDNA-CN in the peripheral blood may be a risk factor for progression to ACS in patients with atheromatous coronary artery plaque. Furthermore, we were able to confirm previous findings that mtDNA-CN is correlated inversely with CAD severity (9). The concept that mtDNA mutation or content (i.e. copy number) alterations play a key role in the development of cardiovascular disease is not new. A previous study demonstrated that mtDNA damage not only promotes atherosclerosis and plaque vulnerability but also serves as a biomarker of higher risk in atherosclerosis (5). It was also reported that mitochondrial damage is more pronounced in patients with ACS than in those with with stable angina (5), which our present findings are relatively consistent with, assuming that reduced mtDNA-CN reflects increased mitochondrial damage. Previously, only a small case-control study investigated the difference in mtDNA content between patients with CAD and healthy controls (20). To our knowledge, the present study is the first to demonstrate the difference in mtDNA-CN between SCAD and ACS.
We also examined whether mtDNA-CN and CAD status are affected by the DNA methylation ratio in the mitochondrial D-loop region and in the gene region of nuclear PPARGC1A, both of which are involved in mitochondrial replication/dysfunction. We found that the methylation ratio of the D-loop region was higher in the ACS group than in the SCAD group, whereas no such difference was noted for the methylation ratio of the PPARGC1A promoter region, suggesting a potential association between epigenetic changes in the D-loop and mtDNA-CN changes.
We further evaluated mtDNA-CN according to CAD severity and found that patients with subclinical CAD or stable angina had higher relative mtDNA-CN than that of patients with minimal lesion on coronary angiography, although the difference was not statistically significant (Figure 3A). The increase in mtDNA-CN noted in patients with subclinical CAD or stable angina may reflect a compensatory response to mitochondrial dysfunction caused by reduced coronary blood flow or myocardial ischemia. A previous case-control study reported lower mtDNA content in patients with CAD than in healthy controls but did not perform stratified analysis according to CAD status or severity (20). On the other hand, we found that patients with UA or AMI, which are considered more advanced manifestations of CAD, had lower mtDNA-CN than that noted in patients with subclinical CAD or stable angina. These findings suggest that the disruption of mechanisms compensating for mitochondrial dysfunction may contribute to ACS. Further studies are needed to determine whether mtDNA-CN alteration contributes to the development of ACS.
Although an increasing number of studies report an association between mtDNA-CN and cardiovascular disease, few have focused on the biological cause of mtDNA-CN alteration in the cardiovascular system. Our findings suggest that increased methylation of the mitochondrial D-loop may affect mtDNA replication and result in reduced mtDNA-CN. The pattern of DNA methylation ratio in the mitochondrial D-loop was found to be opposite to the trend of mtDNA-CN according to CAD severity (Figure 3), further highlighting the potential role of D-loop methylation in mtDNA replication. Although whether or not mtDNA methylation occurs or plays any role has been debated, there is a growing body of evidence to support not only the occurrence of mtDNA methylation but also its function in various processes, such as mtDNA gene transcription (21).
PPARGC1A gene product is considered the main mediator of mtDNA replication. In addition, several factors such as mitochondrial transcription factor A (TFAM), nuclear respiratory factor-1 (NRF1), and -2 are also involved in mtDNA-CN regulation. TFAM is required for mtDNA transcription and replication initiation. Specifically, it regulates protein binding in the D-loop region, which is the cis-regulatory region of the mitochondrial genome (22). PPARGC1A induces TFAM expression, which promotes mtDNA replication. It can therefore be hypothesized that increased methylation of the PPARGC1A gene would suppress TFAM expression, resulting in reduced mtDNA replication. However, we found no significant difference between the SCAD and ACS groups regarding the methylation ratio in the PARGC1A promoter region. One possible explanation for this finding might be that mitochondrial D-loop methylation has a direct influence on mtDNA replication or copy number within the mitochondrion, whereas mtDNA-CN regulation by the remote nuclear PPARGC1A gene is indirect and thus subject to the influence of various mediators or signaling molecules.
A previous study reported a positive correlation of mtDNA-CN with mitochondrial mitochondrially encoded tRNA-Phe (UUU/C) (MT-TF) and mitochondrially encoded 12S rRNA (MT-RNR1) gene methylation in 40 patients exposed to airborne pollutants (10), concluding that the positive correlation reflected an mtDNA-based compensatory mechanism serving to maintain normal cellular function despite increased respiratory demands required for ROS clearance. Another human study reported inverse correlation between mtDNA-CN and the methylation ratio of the mitochondrial D-loop region in patients with insulin resistance (19). However, we found no significant correlation (positive or negative) between mtDNA-CN and the methylation ratio of the mitochondrial D-loop region or promoter region of nuclear PPARGC1A across the entire sample of 100 patients with CAD, nor separately in the SCAD group (n=50) or in the ACS group (n=50). The compensatory increase in mtDNA-CN among patients with subclinical CAD or chronic stable angina may have masked statistically significant linear correlations between mtDNA-CN and D-loop methylation. Furthermore, mtDNA-CN and mtDNA methylation can be influenced by various factors which remain to be clarified.
It is important to note that the methylation status of mtDNA is affected by environmental, lifestyle and pathological conditions such as colorectal cancer, nonalcoholic fatty liver disease, airborne pollutant exposure, and aging (10,13,23,24). Furthermore, it is suggested that mtDNA methylation changes involving both regulatory and coding regions may play a role in aging (25). Based on the findings of the study of mtDNA methylation in patients with insulin resistance (19), it may be hypothesized that the increase in DNA methylation of the mitochondrial D-loop region reduces the rate of mtDNA replication, resulting in reduced mtDNA-CN. In fact, recent findings suggest that mtDNA hypermethylation may represent the main contributor to mitochondrial malfunction (26).
Conventional epigenetic studies including evaluation of nDNA methylation or histone modification experiments have identified several nuclear genes that may serve as epigenetic candidates in cardiovascular disease. However, studies on mtDNA methylation in patients with cardiovascular disease are very rare. Only one study focused explicitly on mtDNA methylation (25) but included only 10 patients with CAD who were moreover not matched to healthy controls according to age, sex, or race. In the present study, we used age- and sex-matched groups of patients with SCAD and ACS, all Korean, with 50 patients per group.
Several limitations to the present study should be considered. Firstly, although we used age- and sex-matched groups, the overall sample size (n=100) may not have been sufficient to observe correlations between clinical manifestations and biological findings such as mtDNA-CN. Furthermore, our study did not target all genes affecting mtDNA-CN. Instead, we focused on candidate target regions such as nuclear PPARGC1A gene and the mitochondrial D-loop region. In addition, within the candidate gene, we did not distinguish among different methylation sites because we used methylation-specific PCR. Finally, our study enrolled only Korean patients, and thus the findings might not be generalizable to other ethnicities. In the light of these limitations, future studies should enroll larger samples, employ more research techniques, and examine multiple mitochondrial genes that may serve as epigenetics targets.
Conclusion
In this epigenetics study of the relationship between mtDNA methylation and the clinical phenotype of CAD, we found that the acute phenotype was associated with significantly lower mtDNA-CN and increased methylation ratio in the mitochondrial D-loop but not in a key nuclear gene regulating mtDNA replication. Our results confirm that mtDNA methylation is relevant in CAD and suggest that lower mtDNA-CN in the peripheral blood may be a risk factor for progression to ACS in patients with atheromatous coronary artery plaque. We hope that future studies on the role of mtDNA methylation in cardiovascular disease can clarify the importance of mtDNA-CN as a biomarker of cardiovascular risk.
Conflicts of Interest
The Authors declare that they have no competing interests
Authors’ Contributions
SAK provided the conception and design of the study and performed data analysis, interpretation and article writing. SHP was involved in data collection, analysis, interpretation and writing. SYL performed experiments.
Acknowledgements
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation of Korea, funded by the Korean Government (MSIP; grant No. 2016M3A9B694241).
References
- 1.Park GM, An H, Lee SW, Cho YR, Gil EH, Her SH, Park HW, Ahn JM, Park DW, Kang SJ, Kim YH, Lee CW, Yang DH, Kang JW, Lim TH, Kim HK, Choe J, Park SW, Park SJ. Impact of metabolic syndrome on subclinical atherosclerosis in asymptomatic individuals. Circ J. 2015;79(8):1799–1806. doi: 10.1253/circj.CJ-14-1197. [DOI] [PubMed] [Google Scholar]
- 2.Poole-Wilson PA, Lubsen J, Kirwan BA, van Dalen FJ, Wagener G, Danchin N, Just H, Fox KA, Pocock SJ, Clayton TC, Motro M, Parker JD, Bourassa MG, Dart AM, Hildebrandt P, Hjalmarson A, Kragten JA, Molhoek GP, Otterstad JE, Seabra-Gomes R, Soler-Soler J, Weber S, Coronary disease Trial Investigating Outcome with Nifedipine gastrointestinal therapeutic system I Effect of long-acting nifedipine on mortality and cardiovascular morbidity in patients with stable angina requiring treatment (action trial): Randomised controlled trial. Lancet. 2004;364(9437):849–857. doi: 10.1016/S0140-6736(04)16980-8. [DOI] [PubMed] [Google Scholar]
- 3.Hamm CW, Ravkilde J, Gerhardt W, Jorgensen P, Peheim E, Ljungdahl L, Goldmann B, Katus HA. The prognostic value of serum troponin t in unstable angina. N Engl J Med. 1992;327(3):146–150. doi: 10.1056/NEJM199207163270302. [DOI] [PubMed] [Google Scholar]
- 4.Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, Runge MS. Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106(5):544–549. doi: 10.1161/01.cir.0000023921.93743.89. [DOI] [PubMed] [Google Scholar]
- 5.Yu E, Calvert PA, Mercer JR, Harrison J, Baker L, Figg NL, Kumar S, Wang JC, Hurst LA, Obaid DR, Logan A, West NE, Clarke MC, Vidal-Puig A, Murphy MP, Bennett MR. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation. 2013;128(7):702–712. doi: 10.1161/circulationaha.113.002271. [DOI] [PubMed] [Google Scholar]
- 6.Robin ED, Wong R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J Cell Physiol. 1988;136(3):507–513. doi: 10.1002/jcp.1041360316. [DOI] [PubMed] [Google Scholar]
- 7.Jeng JY, Yeh TS, Lee JW, Lin SH, Fong TH, Hsieh RH. Maintenance of mitochondrial DNA copy number and expression are essential for preservation of mitochondrial function and cell growth. J Cell Biochem. 2008;103(2):347–357. doi: 10.1002/jcb.21625. [DOI] [PubMed] [Google Scholar]
- 8.Ashar FN, Zhang Y, Longchamps RJ, Lane J, Moes A, Grove ML, Mychaleckyj JC, Taylor KD, Coresh J, Rotter JI, Boerwinkle E, Pankratz N, Guallar E, Arking DE. Association of mitochondrial DNA copy number with cardiovascular disease. JAMA Cardiol. 2017;2(11):1247–1255. doi: 10.1001/jamacardio.2017.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu LP, Cheng K, Ning MA, Li HH, Wang HC, Li F, Chen SY, Qu FL, Guo WY. Association between peripheral blood cells mitochondrial DNA content and severity of coronary heart disease. Atherosclerosis. 2017;261:105–110. doi: 10.1016/j.atherosclerosis.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Byun HM, Panni T, Motta V, Hou L, Nordio F, Apostoli P, Bertazzi PA, Baccarelli AA. Effects of airborne pollutants on mitochondrial DNA methylation. Part Fibre Toxicol. 2013;10:18. doi: 10.1186/1743-8977-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byun HM, Baccarelli AA. Environmental exposure and mitochondrial epigenetics: Study design and analytical challenges. Hum Genet. 2014;133(3):247–257. doi: 10.1007/s00439-013-1417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Infantino V, Castegna A, Iacobazzi F, Spera I, Scala I, Andria G, Iacobazzi V. Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in down’s syndrome. Mol Genet Metab. 2011;102(3):378–382. doi: 10.1016/j.ymgme.2010.11.166. [DOI] [PubMed] [Google Scholar]
- 13.Feng S, Xiong L, Ji Z, Cheng W, Yang H. Correlation between increased nd2 expression and demethylated displacement loop of mtdna in colorectal cancer. Mol Med Rep. 2012;6(1):125–130. doi: 10.3892/mmr.2012.870. [DOI] [PubMed] [Google Scholar]
- 14.Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10(1):12–31. doi: 10.1016/j.mito.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baccarelli AA, Byun HM. Platelet mitochondrial DNA methylation: A potential new marker of cardiovascular disease. Clin Epigenetics. 2015;7:44. doi: 10.1186/s13148-015-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, Thygesen K, Alpert JS, White HD, Jaffe AS, Katus HA, Apple FS, Lindahl B, Morrow DA, Chaitman BA, Clemmensen PM, Johanson P, Hod H, Underwood R, Bax JJ, Bonow RO, Pinto F, Gibbons RJ, Fox KA, Atar D, Newby LK, Galvani M, Hamm CW, Uretsky BF, Steg PG, Wijns W, Bassand JP, Menasché P, Ravkilde J, Ohman EM, Antman EM, Wallentin LC, Armstrong PW, Simoons ML, Januzzi JL, Nieminen MS, Gheorghiade M, Filippatos G, Luepker RV, Fortmann SP, Rosamond WD, Levy D, Wood D, Smith SC, Hu D, Lopez-Sendon JL, Robertson RM, Weaver D, Tendera M, Bove AA, Parkhomenko AN, Vasilieva EJ, Mendis S. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551–2567. doi: 10.1093/eurheartj/ehs184. [DOI] [PubMed] [Google Scholar]
- 17.Yoo HJ, Park M, Kim SA. Difference in mitochondrial DNA copy number in peripheral blood cells between probands with autism spectrum disorders and their unaffected siblings. World J Biol Psychiatry. 2017;18(2):151–156. doi: 10.1080/15622975.2016.1234069. [DOI] [PubMed] [Google Scholar]
- 18.Sookoian S, Rosselli MS, Gemma C, Burgueno AL, Fernandez Gianotti T, Castano GO, Pirola CJ. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology. 2010;52(6):1992–2000. doi: 10.1002/hep.23927. [DOI] [PubMed] [Google Scholar]
- 19.Zheng LD, Linarelli LE, Liu L, Wall SS, Greenawald MH, Seidel RW, Estabrooks PA, Almeida FA, Cheng Z. Insulin resistance is associated with epigenetic and genetic regulation of mitochondrial DNA in obese humans. Clin Epigenetics. 2015;7:60. doi: 10.1186/s13148-015-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S, Xie X, Wang Y, Gao Y, Xie X, Yang J, Ye J. Association between leukocyte mitochondrial DNA content and risk of coronary heart disease: A case-control study. Atherosclerosis. 2014;237(1):220–226. doi: 10.1016/j.atherosclerosis.2014.08.051. [DOI] [PubMed] [Google Scholar]
- 21.Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci USA. 2011;108(9):3630–3635. doi: 10.1073/pnas.1012311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghivizzani SC, Madsen CS, Nelen MR, Ammini CV, Hauswirth WW. In organello footprint analysis of human mitochondrial DNA: Human mitochondrial transcription factor a interactions at the origin of replication. Mol Cell Biol. 1994;14(12):7717–7730. doi: 10.1128/mcb.14.12.7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pirola CJ, Gianotti TF, Burgueno AL, Rey-Funes M, Loidl CF, Mallardi P, Martino JS, Castano GO, Sookoian S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut. 2013;62(9):1356–1363. doi: 10.1136/gutjnl-2012-302962. [DOI] [PubMed] [Google Scholar]
- 24.Dzitoyeva S, Chen H, Manev H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol Aging. 2012;33(12):2881–2891. doi: 10.1016/j.neurobiolaging.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bacalini MG, D’Aquila P, Marasco E, Nardini C, Montesanto A, Franceschi C, Passarino G, Garagnani P, Bellizzi D. The methylation of nuclear and mitochondrial DNA in ageing phenotypes and longevity. Mech Ageing Dev. 2017;165(Pt B):156–161. doi: 10.1016/j.mad.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 26.Jia L, Li J, He B, Jia Y, Niu Y, Wang C, Zhao R. Abnormally activated one-carbon metabolic pathway is associated with mtdna hypermethylation and mitochondrial malfunction in the oocytes of polycystic gilt ovaries. Sci Rep. 2016;6:19436. doi: 10.1038/srep19436. [DOI] [PMC free article] [PubMed] [Google Scholar]