

Abstract
The leading cause of mutation due to oxidative damage is 8-oxo-2’-deoxyguanosine (8-oxoG, see Glossary) mispairing with adenine (Ade), which can occur in two ways. First, guanine of a G:C DNA base pair can be oxidized. If not repaired in time, DNA polymerases can mispair Ade with 8-oxoG in the template. This 8-oxoG:A can be repaired by enzymes that remove Ade opposite to template 8-oxoG, or 8-oxoG opposite to Cyt. Second, free 8-oxo-dGTP can be misincorporated by DNA polymerases into DNA opposite template Ade. However, there is no known repair activity that removes 8-oxoG opposite to template Ade. We suggest that a major role of N6-methyladenine in mammalian DNA is minimizing incorporation of 8-oxoG opposite to Ade by DNA polymerases following adduct formation.
Keywords: 8-oxoguanine, DNA adenine methylation, single-stranded DNA, MettL3-MettL14, YTHDC1
Generation of ssDNA segment during repair
Genomic DNA is constantly subject to damage, induced by incidents from UV irradiation and oxidative stress to environmental mutagens and cancer chemotherapeutic drugs [1]. Nucleotide excision repair (NER, see Glossary) is the main pathway used by mammals to remove bulky DNA lesions [2]. Mutations in NER proteins are associated with (at least) three inherited human diseases: Xeroderma pigmentosum with its tremendously increased risk of skin cancer, Cockayne syndrome with its developmental and neurological abnormalities, and Trichothiodystrophy, with its brittle hair, hematological disorders, and intellectual impairment [3, 4]. The NER pathway involves a set of coordinated enzyme activities that recognize a wide range of DNA substrates, including UV-radiation induced photoproducts (cyclopyrimidine dimers or 6,4-photoproducts). NER proteins unwind the DNA and excise a segment (~25–30 nucleotides) of the damaged strand, spanning the site of damage.
After removal of a stretch of single-strand (ss)DNA containing the lesion, repair-associated DNA synthesis requires DNA replication proteins for gap-filling synthesis by DNA polymerase(s), and ligation. DNA polymerase beta (β) is required for base excision repair [5], but other polymerases including Y family members delta (δ), epsilon (ε), and kappa (κ) are important for NER repair synthesis in human cells [6]. Significantly, some DNA polymerases have a higher tendency to misincorporate 8-oxo-dGTP into DNA opposite template adenosines, using Hoogsteen pairing [7–10] (Figure 1; compare panels A and C). This 8-oxoG:A pairing requires the exocyclic N6 amino group of adenosine, which has implications we will discuss below. First, we will summarize what is known about sources of 8-oxoG.
Figure 1. Examples of 8-oxoG:A mispairs by DNA polymerases.
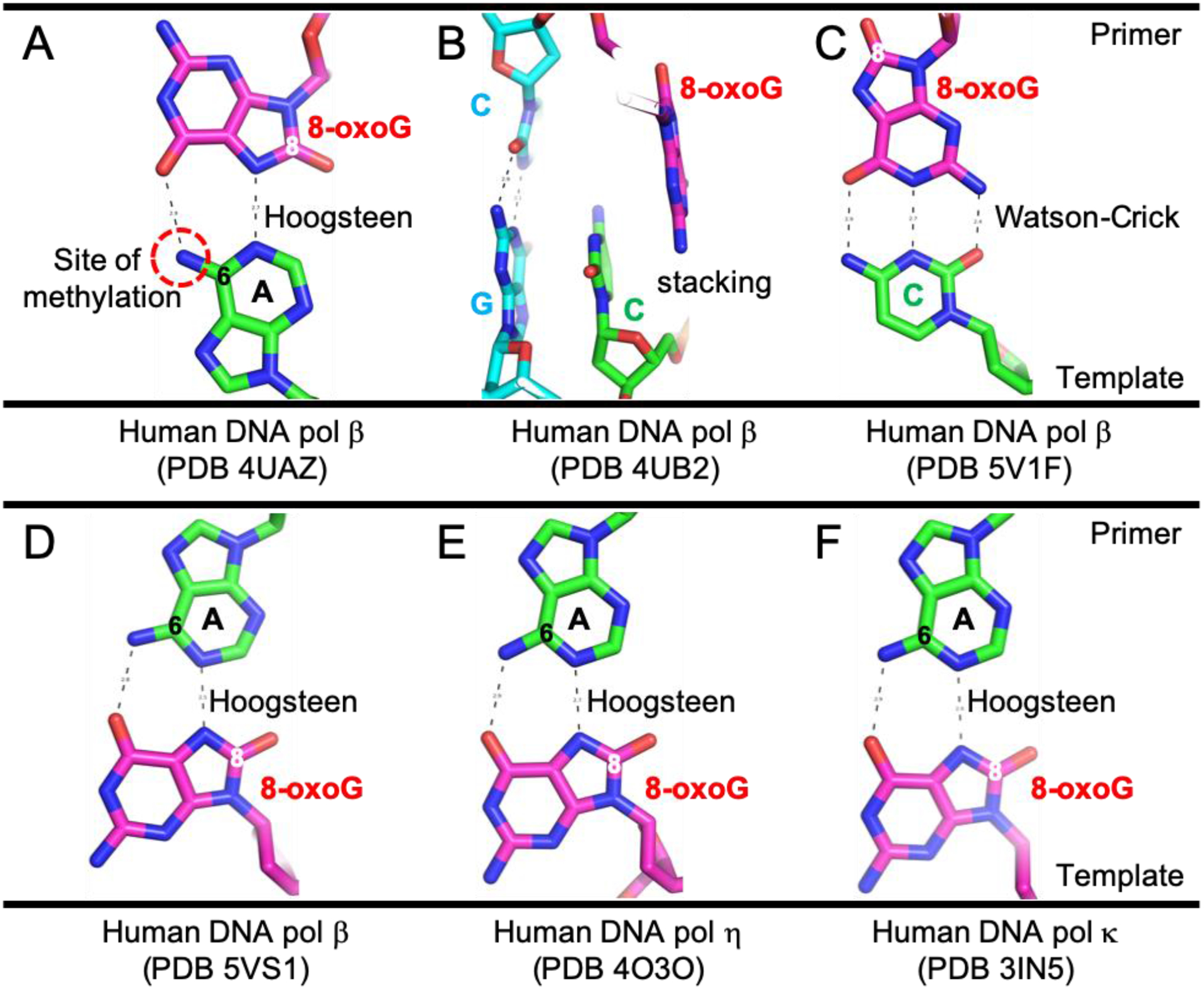
(A) 8-oxoG incorporation opposite to template Ade. (B-C) For comparison, 8-oxoG incorporation opposite to Cyt involves steps of stacking (panel B) and Watson-Crick pairing (panel C). 8-oxoG in the syn-conformation pairs with Ade (panel A), whereas 8-oxoG in the anti-conformation pairs with Cyt (panel C). The two conformations are rotated ~180° relative to each other, along the glycosidic bond that joins base and sugar. (D-F) Ade incorporation opposite to template 8-oxoG in the context of three different DNA polymerases.
Origins and repair of 8-oxoG in DNA
Free 8-oxo-dGTP in the deoxynucleotide triphosphate pool is believed to result from (age-related) oxidative stress [11–14]. There are cellular quality control activities such as MTH1 8-oxo-dGTPase (a MutT ortholog also called oxidized purine nucleoside triphosphatase). Mutant studies reveal that MTH1 reduces 8-oxoG nucleotide pool levels by about a third – from a range of 3–6% down to 2–4% of all guanine nucleotides [15] – but the remainder still poses a significant risk for incorporation into DNA. Surprisingly, the efficiency of β polymerase 8-oxo-dGTP incorporation opposite a template Ade is higher, by a factor of 40, than that opposite the normal (for Gua) Watson-Crick template Cyt [10, 16]. This 8-oxoG:A misincorporation, if it cannot be prevented or repaired appropriately, results in a transversion mutation from T:A to G:C (or T→G in which the mutation is represented by the pyrimidine of the base pair) (Figure 2A). Earlier studies revealed that MutT-deficient Escherichia coli show 1000-fold higher frequency of T→G transversion than wild type cells [17]; mutations arising in MutT-deficient cells are strictly T→G transversions [18]. Mouse Mth1 protein substantially reduces T→G mutations when expressed in MutT-deficient bacterial cells [19]. Mice lacking Mth1 activity were shown to be hypersusceptible to lung, liver and stomach cancers [20]. In humans, age-associated T→G somatic mutations are more abundant in liver than in colon and small intestine [21], and are significantly elevated, for example, in esophageal adenocarcinoma and B-cell non-Hodgkin lymphoma [22].
Figure 2. A biochemically-derived model for a DNA N6mA role in nucleotide excision repair.
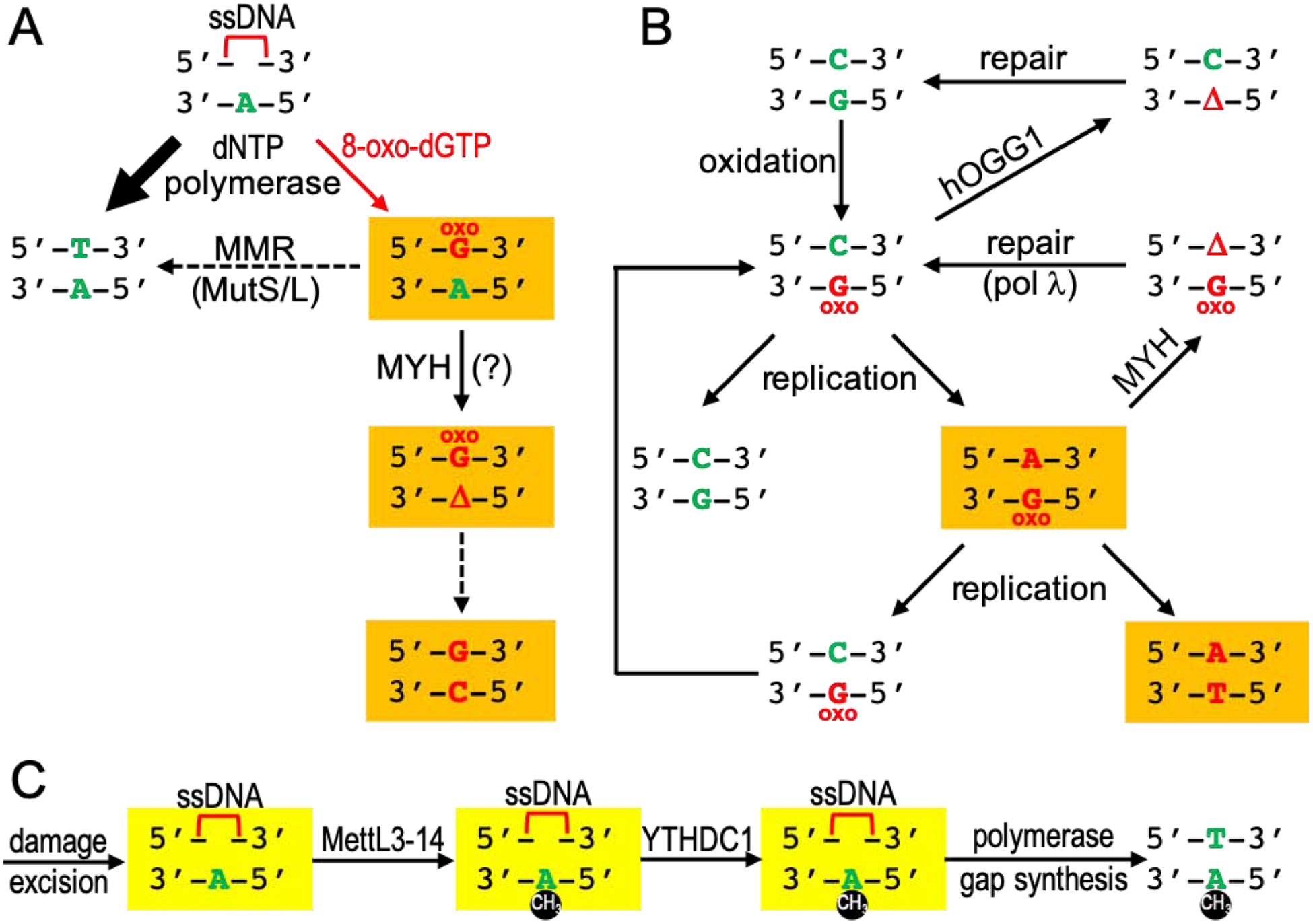
(A) The red bracket represents an ssDNA gap (~25–30 nt) after nucleotide excision (top). DNA synthesis would be largely accurate because T is preferentially inserted opposite A (left bold arrow). However, reactive oxygen species can lead to the formation of 8-oxo-dGTP in the dNTP pool, and in the event that 8-oxo-dGTP is incorporated into DNA during synthesis, it may be either correctly base-paired opposite C (see Figure 1C) or mispaired opposite Ade (right red arrow; see Figure 1A). In the latter event, MMR most likely excises incorporated 8-oxoG from newly synthesized daughter DNA (dashed line) or base excision of the (correct) Ade by the MYH DNA glycosylase can result in a T→G transversion mutation (orange boxes). (B) Oxidation can occur directly to a G:C base pair already in the DNA, resulting in an 8-oxoG:C pair. The adduct can be excised from DNA by hOGG1 glycosylase, and subsequently repaired via base excision repair (BER) to restore the original G:C base pair (top right). If the 8-oxoG adduct is not removed prior to DNA replication, DNA synthesis may retain the 8-oxoG and form an 8-oxoG:A mispair (middle right, orange box; see also Figure 1D–F). The misincorporated Ade can be removed by the MYH DNA glycosylase and replaced by C in the single-nucleotide gap via polymerase λ, yielding a further chance for removal of the 8-oxoG lesion by hOGG1. If not repaired in time, a second round of replication can yield a C→A transversion mutation (orange box, bottom right). (C) A model of N6mA reducing misincorporation of 8-oxoG opposite to Ade. Between the nucleotide excision and DNA synthesis, a transient ssDNA gap (~25–30 nt; the red bracket) can become methylated by MettL3–14 and protected by YTHDC1.
This opinion piece is focused on a means of preventing 8-oxoG incorporation during repair synthesis but, for completeness, we note that oxidation can occur directly to a G:C base pair already in the DNA, resulting in an 8-oxoG:C pair (Figure 2B, top). This is particularly likely to occur when the paired Cyt is methylated (5-methylcytosine), which may have epigenetic consequences [23]. 8-oxoG is the main product of oxidative damage to both DNA and nucleotide pools (reviewed in [24]). In fact, there is evidence that 8-oxoG presence in DNA is used as a gene-regulatory signal of oxidative damage [25].
Base excision repair (BER) enzymes such as the DNA glycosylase OGG1 from eukaryotes or formamidopyrimidine-DNA-glycosylase (Fpg) from Escherichia coli remove 8-oxoG from DNA when it is paired with cytosine (8-oxoG:C) [26] (Figure 2B). However, if not repaired in time, the replication DNA polymerases can misread 8-oxoG in the template and incorporate Ade instead of Cyt [27], and a second round of replication would generate a G:C to T:A transversion (C→A mutation) (Figure 2B). Importantly, after removing the misincorporated Ade by the MutY glycosylase homolog (MYH), DNA polymerase λ - together with proliferating cell nuclear antigen (PCNA) and replication protein A (RP-A) - correctly incorporates dCTP (instead of dATP) opposite 8-oxoG, leading to the formation of C:8-oxoG (Figure 2B) [28] (reviewed in [29] and references therein).
Misincorporation of 8-oxo-dGTP (Figure 2A), rather than direct oxidation of an existing G:C base pair, also yields an 8-oxoG:A mismatch, but the consequences of 8-oxoG:A mismatches are not equal in the two situations. OGG1 is unable to remove 8-oxoG from 8-oxoG:A pairs [30], but they are subject to repair by MYH, which removes the Ade, allowing its eventual replacement with Cyt (Figure 2A). Similarly, in Saccharomyces cerevisiae, the MSH2-MSH6 mismatch repair heterodimer removes Ade paired with 8-oxoG [31]. This is a double-edged sword, however: MYH or MSH2-MSH6 do correctly repair 8-oxoG:A resulting from misincorporation of Ade across from 8-oxoG (Figure 2B, middle right); but in the case of misincorporation of 8-oxoG across from Ade this “locks in” a T→G transversion (Figure 2A). Mismatch repair (MMR) systems [32, 33] may also provide an independent supplementary layer of protection by excising 8-oxoG from DNA following incorporation from the dNTP pool [30]; though the extent to which this occurs is not yet clear. The steady-state level of DNA 8-oxoG is significantly elevated in MMR-deficient mouse [34] and human cells [30], and the oxidized purine dNTP pool contributes significantly to the genetic instability of MMR-defective cells [35]. To our knowledge, there is no proven activity that removes 8-oxoG from 8-oxoG:A base pairs. Clearly, the mispairing of 8-oxoG with Ade poses an unacceptable mutation risk, despite the OGG1, MYH, and MMR-associated repair capabilities.
DNA adenine methylation may reduce misincorporation of 8-oxoG
It would be counterproductive to start repairing one type of DNA damage, and end up with another (due to 8-oxoG:A mispairing). A means of preventing 8-oxoG incorporation in the first place would presumably have strong selective value. In fact, there exists a mechanism by which 8-oxoG misincorporation across from Ade can be greatly reduced. Specifically, methylation of the template adenosine at the amino group - required for Hoogsteen pairing - generates N6-methyladenine (N6mA). N6mA:8-oxoG is much less stable than A:8-oxoG, resulting in less efficient misincorporation of 8-oxo-dGTP opposite N6mA by polymerases such as human pol β [36]. Examined polymerases exhibit a discrimination factor between 7–19 fold, meaning that there is on average about an order of magnitude lower incorporation of 8-oxo-dGTP opposite N6mA than across from unmethylated Ade [36]. We propose that a major role of N6mA in mammalian DNA is minimizing incorporation of 8-oxoG opposite to A by DNA polymerases.
DNA methylation, including generation of N6mA, is common and has long been known to occur in bacteria and archaea [37], where it plays roles in various cellular functions including adenine methylation-directed mismatch repair in E. coli [38]. In contrast, N6mA was detected in mammalian DNA only in 2016 [39], and its further study has grown in parallel with development and application of ultrasensitive mass spectrometry [40–44]. There are three currently-debated issues regarding N6mA in mammalian DNA: (A) whether low-level N6mA can be accurately detected [45–48]; (B) how N6mA in DNA is generated [42, 43, 49–53]; and (C) what the potential functions of N6mA in DNA might be. Here we suggest one functional, highly significant role for N6mA in DNA.
The following considerations prompt us to suggest the potential involvement of DNA adenine methylation in DNA repair. First, the initial observation in embryonic stem cells identified N6mA enrichment in H2A.X deposition regions [39], where H2A.X is a histone variant typically associated with DNA double-strand breaks (DSBs). N6mA DNA modification is also elevated in glioblastoma (a disease partially associated with the cumulative effects of high-dose exposure to ionizing radiation, or to chemical carcinogens), but not in normal adult tissues or mammalian cells [42]. Second, upon ultraviolet irradiation of human sarcoma U2OS cells pretreated with bromodeoxyuridine, the MettL3–14 methyltransferase complex – which generates N6mA in RNA or DNA – is recruited within 2 min to the damaged sites, and MettL3 catalytic activity is required for the DNA repair [54]. Further, DNA polymerase κ requires the catalytic activity of MettL3 for its immediate localization to sites of DNA damage [54]. Third, again in human sarcoma U2OS cells, irradiation with X-rays or treatment with chemicals that induce DSBs leads to colocalization of MettL3 and γH2A.X foci [55]. In addition, YTHDC1 – which binds to N6mA in RNA or DNA – is recruited to DSBs [55]. Fourth, MettL3–14 and YTHDC1 have been characterized extensively as, respectively, generating and binding N6mA in RNA ([56, 57] and references therein). The data on repair of UV-induced and DSB DNA damage suggested a plausible, if complex, RNA methylation-mediated mechanism [54, 55].
Here we offer a simpler model of DNA adenine methylation in preventing or reducing DNA polymerase incorporation of 8-oxoG opposite to A, based on MettL3–14 and YTHDC1 having the respective abilities to methylate and bind N6mA in ssDNA [58, 59] – an intermediate substrate, after excision and before the gap-filling synthesis (Figure 2C). We note that, while Occam’s Razor and evolutionary constraints tend to favor simplicity, the RNA-associated and DNA-only mechanisms are not mutually exclusive, and further experiments are needed to clearly distinguish between them.
MettL3–14 methyltransferase complex and YTHDC1 are active on ssDNA, including unpaired DNA
MettL3 and MettL14 are members of a family of class beta methyltransferases [60]. The beta methyltransferases act on the amino groups of adenine or cytosine in DNA or RNA, and have their conserved motifs in a particular order in their amino acid sequences [61]. The beta methyltransferases also have a unique requirement for being multimeric, forming either homo- or hetero-dimers (such as MettL3-MettL14). MettL3–14 has been studied widely for its role in generating N6mA in RNA [56, 57], at the degenerate consensus sequence RRACH (R=purine and H=not G) [62]. Prompted by the observations noted above, we investigated whether the MettL3–14 heterodimer, known for RNA methylation, also possesses methyl transfer activity onto DNA adenines in vitro. In fact, on synthetic substrate oligonucleotides, MettL3–14 shows >10-fold greater catalytic efficiency for methylation of ssDNA than for ssRNA under the same conditions [58]. In addition, MettL3–14 is active on unpaired DNA, but is inactive on double-stranded DNA [58], suggesting a means of targeting methylation to damaged regions of DNA.
For the same reason, we investigated the YTH domain of mammalian YTHDC1 as a N6mA DNA-binding domain in vitro. YTH domain-containing proteins have been studied extensively for their ability to bind N6mA-containing RNA in mammalian cells [63–65]. There are five YTH domain-containing proteins in humans (DC1, DC2, DF1, DF2, and DF3), and among them only YTHDC1 is known to localize in the nucleus [66]. We showed that the YTH domain of YTHDC1 binds to ssDNA containing N6mA. Under the same conditions, this YTH binding affinity for N6mA in a DNA context is stronger by a factor of 5 than such binding in an RNA context [59]. For comparison, YTHDF1 is predominantly cytoplasmic, while YTHDF2 migrates to mitotic chromatin in human induced pluripotent stem cells [67], and the YTH domains of these two proteins exhibited the opposite effect from that of YTHDC1, with ~1.5–2X stronger binding to N6mA in ssRNA than in the corresponding DNA. Importantly, no measurable binding was observed to double-stranded (ds)DNA, or to RNA/DNA hybrid oligos containing N6mA on one strand.
A biochemically-derived model for a DNA N6mA role in nucleotide excision repair
Here, we build on the established NER pathway, recruitment of MettL3–14 and YTHDC1 to the UV-induced or DSB damaged site [54, 55], and our in vitro characterization of MettL3–14 and YTHDC1 activity on ssDNA – an intermediate substrate between excision and the gap-filling synthesis. We hypothesize that damage-induced generation of N6mA in DNA reduces misincorporation of 8-oxoG opposite to Ade (Figure 2C). UV damage results in bulky DNA adducts – these adducts are mostly cyclothymine dimers and 6,4-photoproducts opposite to the AA dinucleotides. Recognition of this damage by XPC - one of the XP protein factors named from their association (when defective) with Xeroderma pigmentosa - leads to subsequent excision and removal of a segment of ssDNA that contains the lesion, resulting in single-stranded regions in the genome (Figure 2C). Recruitment of MettL3–14 to the damaged sites yields N6mA in the transiently-generated ssDNA, followed by the recruitment of YTHDC1 to N6mA-containing ssDNA. The YTHDC1 could protect against, for example, demethylation by Alkbh1 which also acts on damaged or unpaired DNA [68]. Finally, DNA polymerases use the undamaged, adenine-methylated ssDNA to synthesize a short complementary sequence with little or no 8-oxoG opposite to N6mA. Thus, in this model, the N6mA is not just a signal to recruit proteins, but is an intrinsically-protective modification to potential mutation hotspots.
We note that our simpler DNA-only model is not readily applicable to all steps along the DSB repair pathways, which are complex ([69] and references therein). Choice among the available DSB repair pathways (non-homologous end joining and homologous recombination) is affected by the initiating DNA lesion itself (whether the DNA ends contain single-stranded tails). Because our DNA-N6mA model is based on transiently ssDNA substrates for MettL3–14 and YTHDC1, the emergence of ssDNA regions (as well as non-B DNA structures of D-loops or resolution of the four-way Holliday junction) during repair – traditionally considered the error-prone step of single-strand annealing [70] – might be where N6mA methylation could have its greatest impact.
It is not yet clear which proteins control mammalian DNA N6mA or its effects
Ever since the initial report of N6mA existence in the DNA of mouse embryonic stem cells [39], the identification of potential adenine DNA methyltransferase(s) has been an unsettled issue. Two recent developments might point to where and when the enzyme or enzyme complex might function. First, the same laboratory that made the initial claim has reported that N6mA in DNA increases during the development of mouse trophoblast stem cells (which eventually give rise to the placenta), specifically at regions of stress-induced DNA double helix destabilization (SIDD) [44]. Single-stranded DNA that persists in that state is one of the possible SIDD aftereffects [71]. Second, the characterized candidates for DNA N6mA writer (MettL3–14), reader (YTHDC1), and eraser (ALKBH1), all prefer locally unpaired DNA substrates in vitro [58, 59, 68]. Of these three proteins, the activities of YTHDC1 and ALKBH1 are independent of sequence context aside from the methylated adenine itself. In contrast, MettL3–14 is an adenine methylase complex long studied for its activity on RNA, where it targets the sequence RRACH, with particular specificity for the ApC motif. Interestingly, many nucleic acid-modifying enzymes are able to modify both DNA and RNA, with examples including human MettL4 [51, 72] - another candidate adenine methylase. As noted above, MettL3–14 and YTHDC1 have the respective abilities to methylate and bind N6mA in ssDNA [58, 59]
We suggest that a proper biochemical investigation of proteins that copurify with DNA adenine methylase activity will be needed to properly identify all necessary components of the catalytically active methylase complex. A functional homolog DNA methyltransferase does exist in Escherichia coli (M.EcoGII), that meets the requirement of sequence independence aside from the target adenine, ability to act on both nucleic acid types (DNA and RNA), and impartiality regarding strandedness (single or double) [60]. Such a gene might only be expressed at specific time of development, and might be induced only under stress.
Concluding remarks
Our model predicts that the DNA N6mA level will be elevated in cells immediately after repair of DNA damage, since there is no immediate need to remove it from the template strand following repair synthesis. We do note that mammalian Alkbh1 and Alkbh4 have been implicated in DNA N6mA demethylation [39, 42, 43] so, in theory, N6mA accumulation could result from either targeted methylation or targeted depression of demethylating activity. In either case, as damage could occur anywhere in the genome, the appearance of N6mA would appear to be random. The role of N6mA we propose is not to attract repair enzymes, but to prevent 8-oxoG incorporation during repair, so there is in theory no need to remove the N6mA following repair. In the absence of MettL3–14 and/or YTHDC1, increased incorporation of 8-oxoG should be observed in the genome. We note that the recent study on damage-induced recruitment of MettL3–14 focused on its RNA methylation activity [54, 55]. A genome-wide study on the DNA N6mA induced by UV irradiation, particularly in skin cancer-prone cells, should be carefully examined with considerations of potential experimental caveats [48].
Because the methyltransferase complex MettL3–14 is recruited to damaged sites within 2 min of ultraviolet irradiation, it is possible that methylation occurs before excision. If so, the enzymatic activity of MettL3–14 should be tested on dsDNA containing a photoproduct or other lesion. We note that MettL3–14 is active on dsDNA-containing unpaired, mismatched bases centered on the target adenine, but is inactive on the fully-paired duplex [58]. The effects of template strand N6mA on the activities of NER enzymes should also be examined.
How repair-associated polymerases (δ, ε, and κ) discriminate between damaged and undamaged nucleotides is not well understood. The replicative fidelities of the Y-family enzymes (such as η and κ) are relatively low [73] – they induce mutations at a frequency of ~10−3 / generation or higher when acting on undamaged templates [74, 75], and this may be one reason for which polymerase κ is kept outside the nucleus under nonstress conditions [76]. Aside from cases in which an elevated mutation rate may yield beneficial adaptations [77–79], it seems counterproductive for cells to use an error-prone polymerase to carry out repair synthesis even for a short patch size of 30 nucleotides, which would result in about one error every 30 repair patches. Perhaps an error prevention mechanism such as proposed here serves to adjustably reduce the mutational price paid for the post-polymerase repair. In light of our model, it is critical to determine the effects of DNA damage on the rate of incorporation of 8-oxoG opposite to A vs. N6mA.
Finally, the effects of N6mA on the post-polymerase repair enzymes (such as MYH and MMS in Figure 2A) acting on mispaired DNA substrates (8-oxoG:N6mA vs. 8-oxoG:A) needs to be examined. Perhaps the adenine methylation provides a sign for the repair enzymes that, when 8-oxoG is nevertheless incorporated opposite Ade, the adenine should not be cleaved from the template (presumptively correct) strand – a strategy used in E. coli for strand discrimination during mismatch repair [80]. We have raised some outstanding questions (see Outstanding Questions), but much remains to be learned about the writers, erasers, and readers of N6mA in mammalian DNA, as well as its roles that may transcend control of gene expression.
Outstanding Questions Box:
Are other RNA adenine methyltransferases active on DNA?
Does RNaseH-resistant DNA N6mA increase following DNA damage that results in ssDNA?
Does DNA N6mA increase following DNA damage that results in ssDNA?
Are the ssDNA regions identified by permanganate enriched for N6mA?
Do MettL3–14 and/or YTHDC1 mutants show increased 8-oxoG levels in DNA or elevated T →G transversions (particularly in an MTH1 background following oxidative stress)?
What are the step-by-step mechanisms in recruitment of MettL3–14 and YTHDC1 to the damaged sites?
Are there direct interactions between MettL3–14 (and YTHDC1) and known DNA repair factors?
Highlights.
DNA polymerases can misincorporate 8-oxoguanine into DNA opposite template adenine.
DNA damage induced by ultraviolet and double-strand breaks recruit the adenine methyltransferase MettL3-MettL14, and N6-methyladenine reader YTHDC1, to the damaged sites.
MettL3-MettL14 and YTHDC1 are active on single-strand DNA, as well as RNA.
DNA polymerases exhibit reduced misincorporation of 8-oxoguanine opposite N6-methyladenine, compared to opposite unmethylated adenine.
DNA adenine methylation may thus provide a significant error prevention mechanism during repair.
Acknowledgements
We thank Drs. Clayton Woodcock and John Horton for their work on CcrM, MettL3–14 and YTHDC1, Drs. Tao Wu of Baylor College of Medicine and Yun Huang of Texas A&M University for discussion. We thank Drs. Richard Wood, Sankar Mitra, and John Tainer for comments. The thoughts discussed here were accumulated over the course of work on CcrM, HemK2/KMT9/N6AMT1, MettL3–14 and YTHDC1 (supported by U.S. National Institutes of Health [R35GM134744] and Cancer Prevention and Research Institute of Texas [RR160029]). R.M.B. and X.C. prepared the manuscript. X.C. is a CPRIT Scholar in Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Olivieri M et al. (2020) A Genetic Map of the Response to DNA Damage in Human Cells. Cell 182, 481–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scharer OD (2013) Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol 5, a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehmann AR (2001) The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev. 15, 15–23 [DOI] [PubMed] [Google Scholar]
- 4.Kraemer KH et al. (2007) Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience 145, 1388–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cabelof DC et al. (2002) Induction of DNA polymerase beta-dependent base excision repair in response to oxidative stress in vivo. Carcinogenesis 23, 1419–1425 [DOI] [PubMed] [Google Scholar]
- 6.Ogi T et al. (2010) Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 37, 714–727 [DOI] [PubMed] [Google Scholar]
- 7.Kamath-Loeb AS et al. (1997) Incorporation of the guanosine triphosphate analogs 8-oxo dGTP and 8-NH2-dGTP by reverse transcriptases and mammalian DNA polymerases. J. Biol. Chem 272, 5892–5898 [DOI] [PubMed] [Google Scholar]
- 8.Hanes JW et al. (2006) Incorporation and replication of 8-oxo-deoxyguanosine by the human mitochondrial DNA polymerase. J. Biol. Chem 281, 36241–36248 [DOI] [PubMed] [Google Scholar]
- 9.Pursell ZF et al. (2008) Trace amounts of 8-oxo-dGTP in mitochondrial dNTP pools reduce DNA polymerase gamma replication fidelity. Nucleic Acids Res. 36, 2174–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freudenthal BD et al. (2015) Uncovering the polymerase-induced cytotoxicity of an oxidized nucleotide. Nature 517, 635–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fraga CG et al. (1990) Oxidative damage to DNA during aging: 8-hydroxy-2’-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. U S A 87, 4533–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haghdoost S et al. (2006) The nucleotide pool is a significant target for oxidative stress. Free Radic. Biol. Med 41, 620–626 [DOI] [PubMed] [Google Scholar]
- 13.Nakabeppu Y (2014) Cellular levels of 8-oxoguanine in either DNA or the nucleotide pool play pivotal roles in carcinogenesis and survival of cancer cells. Int. J. Mol. Sci 15, 12543–12557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding Y et al. (2017) Sequencing the Mouse Genome for the Oxidatively Modified Base 8-Oxo-7,8-dihydroguanine by OG-Seq. J. Am. Chem. Soc 139, 2569–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kapoor I and Varshney U (2020) Diverse roles of nucleoside diphosphate kinase in genome stability and growth fitness. Curr. Genet 66, 671–682 [DOI] [PubMed] [Google Scholar]
- 16.Kim T et al. (2016) Insertion of oxidized nucleotide triggers rapid DNA polymerase opening. Nucleic Acids Res. 44, 4409–4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yanofsky C et al. (1966) The unusual mutagenic specificity of an E. Coli mutator gene. Proc. Natl. Acad. Sci. U S A 55, 274–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tajiri T et al. (1995) Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat. Res 336, 257–267 [DOI] [PubMed] [Google Scholar]
- 19.Kakuma T et al. (1995) Mouse MTH1 protein with 8-oxo-7,8-dihydro-2’-deoxyguanosine 5’-triphosphatase activity that prevents transversion mutation. cDNA cloning and tissue distribution. J. Biol. Chem 270, 25942–25948 [DOI] [PubMed] [Google Scholar]
- 20.Tsuzuki T et al. (2001) Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc. Natl. Acad. Sci. U S A 98, 11456–11461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blokzijl F et al. (2016) Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexandrov LB et al. (2020) The repertoire of mutational signatures in human cancer. Nature 578, 94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ming X et al. (2014) Mapping structurally defined guanine oxidation products along DNA duplexes: influence of local sequence context and endogenous cytosine methylation. J. Am. Chem. Soc 136, 4223–4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ba X and Boldogh I (2018) 8-Oxoguanine DNA glycosylase 1: Beyond repair of the oxidatively modified base lesions. Redox Biol. 14, 669–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sampath H and Lloyd RS (2019) Roles of OGG1 in transcriptional regulation and maintenance of metabolic homeostasis. DNA Repair (Amst) 81, 102667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boiteux S et al. (2017) Repair of 8-oxo-7,8-dihydroguanine in prokaryotic and eukaryotic cells: Properties and biological roles of the Fpg and OGG1 DNA N-glycosylases. Free Radic. Biol. Med 107, 179–201 [DOI] [PubMed] [Google Scholar]
- 27.Shibutani S et al. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349, 431–434 [DOI] [PubMed] [Google Scholar]
- 28.van Loon B and Hubscher U (2009) An 8-oxo-guanine repair pathway coordinated by MUTYH glycosylase and DNA polymerase lambda. Proc. Natl. Acad. Sci. U S A 106, 18201–18206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mazzei F et al. (2013) Role of MUTYH in human cancer. Mutat. Res 743–744, 33–43 [DOI] [PubMed] [Google Scholar]
- 30.Colussi C et al. (2002) The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr. Biol 12, 912–918 [DOI] [PubMed] [Google Scholar]
- 31.Ni TT et al. (1999) MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol. Cell 4, 439–444 [DOI] [PubMed] [Google Scholar]
- 32.Pena-Diaz J and Jiricny J (2012) Mammalian mismatch repair: error-free or error-prone? Trends Biochem. Sci 37, 206–214 [DOI] [PubMed] [Google Scholar]
- 33.Jiricny J (2013) Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol 5, a012633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeWeese TL et al. (1998) Mouse embryonic stem cells carrying one or two defective Msh2 alleles respond abnormally to oxidative stress inflicted by low-level radiation. Proc. Natl. Acad. Sci. U S A 95, 11915–11920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russo MT et al. (2004) The oxidized deoxynucleoside triphosphate pool is a significant contributor to genetic instability in mismatch repair-deficient cells. Mol. Cell. Biol 24, 465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S et al. (2017) The m(6)A methylation perturbs the Hoogsteen pairing-guided incorporation of an oxidized nucleotide. Chem. Sci 8, 6380–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanchez-Romero MA and Casadesus J (2020) The bacterial epigenome. Nat. Rev. Microbiol 18, 7–20 [DOI] [PubMed] [Google Scholar]
- 38.Wyrzykowski J and Volkert MR (2003) The Escherichia coli methyl-directed mismatch repair system repairs base pairs containing oxidative lesions. J. Bacteriol 185, 1701–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu TP et al. (2016) DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 532, 329–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao CL et al. (2018) N(6)-Methyladenine DNA Modification in the Human Genome. Mol Cell 71, 306–318 [DOI] [PubMed] [Google Scholar]
- 41.Koh CWQ et al. (2018) Single-nucleotide-resolution sequencing of human N6-methyldeoxyadenosine reveals strand-asymmetric clusters associated with SSBP1 on the mitochondrial genome. Nucleic Acids Res. 46, 11659–11670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie Q et al. (2018) N(6)-methyladenine DNA Modification in Glioblastoma. Cell 175, 1228–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kweon SM et al. (2019) An Adversarial DNA N(6)-Methyladenine-Sensor Network Preserves Polycomb Silencing. Mol. Cell 74, 1138–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Z et al. (2020) N6-methyladenine in DNA antagonizes SATB1 in early development. Nature 583, 625–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ratel D et al. (2006) Undetectable levels of N6-methyl adenine in mouse DNA: Cloning and analysis of PRED28, a gene coding for a putative mammalian DNA adenine methyltransferase. FEBS Lett. 580, 3179–3184 [DOI] [PubMed] [Google Scholar]
- 46.Schiffers S et al. (2017) Quantitative LC-MS Provides No Evidence for m(6) dA or m(4) dC in the Genome of Mouse Embryonic Stem Cells and Tissues. Angew Chem. Int. Ed. Engl 56, 11268–11271 [DOI] [PubMed] [Google Scholar]
- 47.Liu B et al. (2017) Metabolically Generated Stable Isotope-Labeled Deoxynucleoside Code for Tracing DNA N(6)-Methyladenine in Human Cells. Anal. Chem 89, 6202–6209 [DOI] [PubMed] [Google Scholar]
- 48.Douvlataniotis K et al. (2020) No evidence for DNA N (6)-methyladenine in mammals. Sci. Adv 6, eaay3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woodcock CB et al. (2019) Human HemK2/KMT9/N6AMT1 is an active protein methyltransferase, but does not act on DNA in vitro, in the presence of Trm112. Cell Discov. 5, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W et al. (2019) Structural insight into human N6amt1-Trm112 complex functioning as a protein methyltransferase. Cell Discov. 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hao Z et al. (2020) N(6)-Deoxyadenosine Methylation in Mammalian Mitochondrial DNA. Mol. Cell 78, 382–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Musheev MU et al. (2020) The origin of genomic N(6)-methyl-deoxyadenosine in mammalian cells. Nat. Chem. Biol 16, 630–634 [DOI] [PubMed] [Google Scholar]
- 53.Liu X et al. (2020) N(6)-methyladenine is incorporated into mammalian genome by DNA polymerase. Cell Res. 10.1038/s41422-020-0317-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiang Y et al. (2017) RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang C et al. (2020) METTL3 and N6-Methyladenosine Promote Homologous Recombination-Mediated Repair of DSBs by Modulating DNA-RNA Hybrid Accumulation. Mol. Cell 79, 1–18 [DOI] [PubMed] [Google Scholar]
- 56.Frye M et al. (2018) RNA modifications modulate gene expression during development. Science 361, 1346–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J et al. (2020) N (6)-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Woodcock CB et al. (2019) Human MettL3-MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discov. 5, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woodcock CB et al. (2020) Biochemical and structural basis for YTH domain of human YTHDC1 binding to methylated adenine in DNA. Nucleic Acids Res. gkaa604, 10.1093/nar/gkaa604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Woodcock CB et al. (2020) Beta class amino methyltransferases from bacteria to humans: evolution and structural consequences. Nucleic Acids Res. gkaa446, 10.1093/nar/gkaa446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malone T et al. (1995) Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol 253, 618–632 [DOI] [PubMed] [Google Scholar]
- 62.Schibler U et al. (1977) Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J. Mol. Biol 115, 695–714 [DOI] [PubMed] [Google Scholar]
- 63.Patil DP et al. (2018) Reading m(6)A in the Transcriptome: m(6)A-Binding Proteins. Trends Cell Biol. 28, 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao S et al. (2018) YTH Domain: A Family of N(6)-methyladenosine (m(6)A) Readers. Genomics Proteomics Bioinformatics 16, 99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tong J et al. (2018) RNA m(6)A modification and its function in diseases. Front. Med 12, 481–489 [DOI] [PubMed] [Google Scholar]
- 66.Nayler O et al. (2000) The ER repeat protein YT521-B localizes to a novel subnuclear compartment. J. Cell Biol 150, 949–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abakir A et al. (2020) N(6)-methyladenosine regulates the stability of RNA:DNA hybrids in human cells. Nat. Genet 52, 48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang M et al. (2020) Mammalian ALKBH1 serves as an N(6)-mA demethylase of unpairing DNA. Cell Res. 30, 197–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scully R et al. (2019) DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol 20, 698–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benitez A et al. (2018) FANCA Promotes DNA Double-Strand Break Repair by Catalyzing Single-Strand Annealing and Strand Exchange. Mol. Cell 71, 621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kouzine F et al. (2017) Permanganate/S1 Nuclease Footprinting Reveals Non-B DNA Structures with Regulatory Potential across a Mammalian Genome. Cell Syst. 4, 344–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen H et al. (2020) METTL4 is an snRNA m(6)Am methyltransferase that regulates RNA splicing. Cell Res. 30, 544–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McCulloch SD and Kunkel TA (2008) The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 18, 148–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ohashi E et al. (2000) Fidelity and processivity of DNA synthesis by DNA polymerase kappa, the product of the human DINB1 gene. J. Biol. Chem 275, 39678–39684 [DOI] [PubMed] [Google Scholar]
- 75.Zhang Y et al. (2000) Human DNA polymerase kappa synthesizes DNA with extraordinarily low fidelity. Nucleic Acids Res. 28, 4147–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Temprine K et al. (2020) Regulation of the error-prone DNA polymerase Polkappa by oncogenic signaling and its contribution to drug resistance. Sci. Signal 13, eaau1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bavoux C et al. (2005) Up-regulation of the error-prone DNA polymerase {kappa} promotes pleiotropic genetic alterations and tumorigenesis. Cancer Res. 65, 325–330 [PubMed] [Google Scholar]
- 78.Peng C et al. (2016) The Error-Prone DNA Polymerase kappa Promotes Temozolomide Resistance in Glioblastoma through Rad17-Dependent Activation of ATR-Chk1 Signaling. Cancer Res. 76, 2340–2353 [DOI] [PubMed] [Google Scholar]
- 79.Pribis JP et al. (2019) Gamblers: An Antibiotic-Induced Evolvable Cell Subpopulation Differentiated by Reactive-Oxygen-Induced General Stress Response. Mol. Cell 74, 785–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pukkila PJ et al. (1983) Effects of high levels of DNA adenine methylation on methyl-directed mismatch repair in Escherichia coli. Genetics 104, 571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]