

Summary
Mitochondrial Ca2+ uptake has long been considered crucial for meeting the fluctuating energy demands of cells in the heart and other tissues. Increases in mitochondrial matrix [Ca2+] drive mitochondrial ATP production via stimulation of Ca2+-sensitive dehydrogenases. Mitochondria-targeted sensors have revealed mitochondrial matrix [Ca2+] rises that closely follow the cytoplasmic [Ca2+] signals in many paradigms. Mitochondrial Ca2+ uptake is mediated by the Ca2+ uniporter (mtCU). Pharmacological manipulation of the mtCU is potentially key to understanding its physiological significance, but no specific, cell-permeable inhibitors were identified. In the past decade, as the molecular identity of the mtCU was brought to light, efforts have focused on genetic targeting. However, in the cells/animals that are able to survive impaired mtCU function, robust compensatory changes were found in the mtCU as well as other mechanisms. Thus, the discovery, through chemical library screens on normal and mtCU-deficient cells, of new small-molecule inhibitors with improved cell permeability and specificity might offer a better chance to test the relevance of mitochondrial Ca2+ uptake. Success with the development of small molecule mtCU inhibitors is also expected to have clinical impact, considering the growing evidence for the role of mitochondrial Ca2+ uptake in a variety of diseases, including heart attack, stroke and various neurodegenerative disorders. Here, we review the progress in pharmacological targeting of mtCU and illustrate the challenges in this field using data obtained with MCU-i11, a new small molecule inhibitor.
Keywords: calcium, MCU, MICU1, permeability transition, mPTP, Ruthenium Red, Ru360, Ru265, mitoxantrone, DS16570511
Why is it important to target mtCU?
In unstimulated cells, the mitochondrial matrix [Ca2+] ([Ca2+]m) is similar to the cytoplasmic [Ca2+] ([Ca2+]c), and upon exposure of mitochondria to an uncoupler that releases their entire Ca2+ content, little or no increase in [Ca2+]c is detected [1]. These results suggest that mitochondria do not serve as intracellular Ca2+ stores. However, during the response to physiological Ca2+-mobilizing agonists, [Ca2+]m shows transient increases, leading to the stimulation of energy metabolism through the Ca2+-sensitive matrix dehydrogenases (CSMDH)/Krebs cycle and to activity changes in other mitochondrial Ca2+ sensing mechanisms [2, 3]. Furthermore, during prolonged elevations of [Ca2+]c, the matrix Ca2+ content rises progressively, providing a trigger to stress signaling or cell death via opening of the mitochondrial permeability transition pore (mPTP) [4, 5]. [Ca2+]m signals elicited by physiological [Ca2+]c oscillations can also trigger mPTP opening if they coincide with factors that enhance the sensitivity of the mPTP to Ca2+ [6]. Most relevant in this regard is the interplay between [Ca2+]m and reactive oxygen species (ROS), which includes [Ca2+]m-induced stimulation of ROS production and ROS-mediated sensitization of the mPTP to [Ca2+]m [7, 8]. Thus, via mtCU-mediated Ca2+ uptake, mitochondria sense and respond to the [Ca2+]c fluctuations. The fraction of the mobilized Ca2+ taken up by mitochondria during [Ca2+]c signals is tissue specific but even if the amount is small, the mitochondria can affect the activity of cytoplasmic Ca2+ sources and targets because their strategic localization allows them to effectively control local [Ca2+]c at the relevant sites [9, 10]. This is particularly relevant at the interface with the sarcoplasmic reticulum, where mitochondrial Ca2+ uptake can sense and affect Ca2+ mobilization [11, 12]. By modulating these pathways, mtCU might have a broad role in both physiology and pathogenesis (Figure 1).
Figure 1. Ca2+ transport mediated by the mtCU: pharmacological inhibitors and human disorders that might benefit from mtCU targeting.
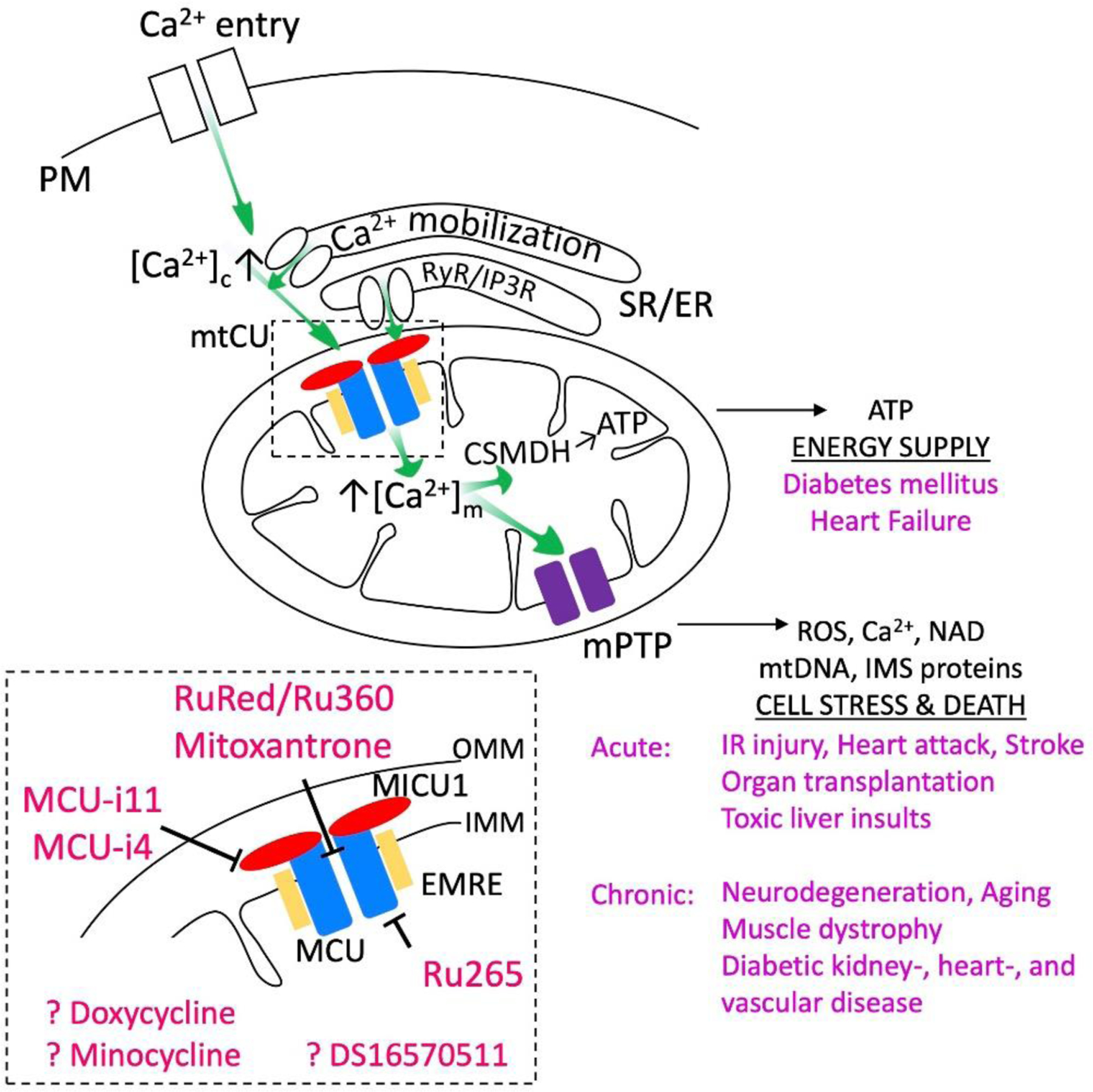
Mechanism of Ca2+ transfer to the mitochondria (green arrows) with an emphasis on the mtCU (rectangle with dashed lines) and the inhibitors of Ca2+ uptake (red characters) are depicted. The targets of Ca2+ in the mitochondrial matrix (CSMDH, mPTP) and the outcome of their activation (export of ATP, release of ROS, Ca2+, NAD, mtDNA and IMS proteins, respectively) is shown (on the right). The medical conditions linked to altered activity of these targets are listed in purple. Note that components of the Ca2+ transport and sensing without direct involvement in these pathways are omitted from the scheme.
Demonstrating the loss of functions associated with the perturbation of mtCU is essential to understand the relevance of mitochondrial Ca2+ uptake. This is particularly important in cells and tissues where a direct measurement of [Ca2+]m has not been feasible or where results obtained in different studies are divergent. Cardiomyocytes, in particular, remain controversial in terms of [Ca2+]c signal propagation to the mitochondria. Studies support both beat-to-beat regulation of [Ca2+]m and integration of the [Ca2+]c spikes in a frequency-dependent manner, with each [Ca2+]m phenotype expected to result in a different metabolic response [13–17]. The high frequency beating in mice favors integration of the [Ca2+]c spikes, whereas the lower heart rate in larger mammals, including human might be compatible with beat-to-beat rises of [Ca2+]m (10–50ms rise time and ≤1s decay time) [18]. Both in the frequency modulation and beat-to-beat paradigms, specific interference with mtCU is central to establishing the physiological role of mtCU activity.
Targeting of mtCU has also been envisioned as a potential therapeutic approach. In conditions of energy deficit including acute heart failure, stimulation of mtCU to enhance the activity of the Ca2+-dependent matrix enzymes and mitochondrial ATP production might be beneficial [19, 20] (Figure 1). Conversely, in mitochondrial Ca2+ overload-favoring conditions such as the reperfusion phase of ischemia-reperfusion injury, organ transplant or toxic liver injury, impaired control of the mtCU by MICU1 aggravates the injury [21–23], and acute suppression of mtCU activity helps to prevent mPTP opening and might offer therapeutic value [24–26]. In addition to these acute conditions, mtCU abundance and activity changes have been linked to other cardiac impairments including arrhythmias (e.g. [27]) and cardiomyopathies (e.g. [28]). Mitochondrial Ca2+ dysregulation and Ca2+ overload have also been implicated in the progression of various neurodegenerative diseases [29, 30] (Figure 1). Blocking mitochondrial Ca2+ uptake for a prolonged period likely has negative consequences in cardiomyocytes and neurons, but pharmacological “tuning” of mtCU activity might be considered as a therapeutic strategy. Lastly, altered expression and/or function of mtCU components has been linked to cancer progressions [31, 32], which might also become candidate for mtCU-directed therapy.
Molecular composition of the mtCU
Cytoplasmic Ca2+ has to cross both the outer and inner mitochondrial membranes (OMM, IMM) to enter the matrix (Figure 1). The predominant pathways for Ca2+ diffusion across the OMM are the Voltage Dependent Anion-selective Channels (VDACs)[33, 34]. Because of their high density and high conductance for cations in their “closed” state, VDACs allow rapid Ca2+ diffusion. The primary driving force for Ca2+ entry into the matrix is the inside-negative membrane potential (ΔΨm) across the IMM maintained by the proton-extruding activity of the electron transport chain. The predominant mechanism by which Ca2+ crosses the IMM inward is the mtCU [35] (Figure 1). In most cells, the mtCU-mediated Ca2+ uptake is negligible at submicromolar [Ca2+]c and the transport velocity increases by a supralinear manner above ≥1μM [Ca2+]c. This is because the mtCU has both a transport site and a separate activation site for Ca2+ [36]. By patch-clamping mitoplasts, the mtCU has been characterized as a ruthenium red-sensitive and highly selective inward-rectifying Ca2+-gated Ca2+ channel [37].
mtCU has now been established as a hetero-oligomeric channel complex. The major mtCU-forming proteins have been identified, including the pore, MCU [38, 39], a paralog that can produce a dominant-negative phenotype, MCUb [40], a scaffold/regulator, EMRE [41], and Ca2+-sensitive regulators, MICU1 [42], MICU2 [43] and MICU3 [44]. To date, a MICU complex (a hetero/homo-dimer of MICU1 and MICU2 or MICU1 and MICU3) appears to determine both the threshold and cooperative activation of the mtCU by Ca2+, thus providing a mechanism for the supralinear [Ca2+]c activation of the mtCU [45, 46]. Indeed, the MICU proteins possess EF-hand Ca2+-binding domains [42, 43] localized to the intermembrane space [45, 47], making them suitable to sense [Ca2+]c signals for the control of mitochondrial Ca2+ uptake. MICU1 and MICU2 are broadly expressed [43, 48], whereas MICU3 is mostly expressed in the brain [44]. Recent evidence indicates that MICU1 also interacts with the MICOS complex and this interaction might be relevant for both the control of Ca2+ uptake and cristae structure [49, 50].
The cryo-EM structure of the metazoan MCU-EMRE complex has shown a tetrameric complex stabilized by N-terminal domain (NTD) interactions in the matrix among the MCU subunits and EMRE-mediated gating through the interaction between the extended tail of EMRE and the juxtamembrane loop of MCU [51]. Notably, modification of a 10 amino acid long region in the TM2 domain of the human MCU allowed the formation of an EMRE independent channel [52]. Based on mutational analysis, MICU1 can be anchored to the MCU-EMRE complex via the interaction of its polybasic region with the IMS localized poly-aspartate tail of EMRE [41, 53]. However, recent evidence by us and others supports a direct interaction of MICU1 with the selectivity filter (-D261-x-x-E264- motif or D-ring formed by D261s in the tetrameric pore) of the MCU, which is central to minimizing the Ca2+ flux through the MCU at low [Ca2+]c [54, 55]. Very recently the structure has become available for the MCU-EMRE-MICU1-MICU2 complex from 3 cryo-EM studies [56–58]. These studies confirm the interaction of MICU1 with the D-ring and emphasize the role of several residues of MICU1 (Y114, Y121, K126, R129, R259, R261, and R263) in direct interaction with MCU.
Genetic targeting of the mtCU
The first MCU-deficient mouse was created on a mixed genetic background. The heterozygous breeding of these mice yielded KO/KO mice below the Mendelian ratio, but the few KO/KO offspring showed normal development and had only minor functional problems like the adaptation of striated muscle to work. Mitochondria harvested from the tissues failed to take up added Ca2+ [59]. By contrast, homozygous MCU-deficient mice created on the C57BL/6 background failed to give any viable pups [60, 61]. Heart-specific inducible deletion of MCU in adult C57BL/6 mice resulted in an impaired “fight-or-flight” response and protection against acute cardiac ischemia-reperfusion injury [24, 25]. Thus, in the absence of mitochondrial Ca2+ uptake post-uterine life is tenable, at least in mice, but some steps of early development seem to be impaired and the ability to meet increased energy needs is compromised in adult mice. Importantly, in MCU-deficient mice, several compensatory changes in Ca2+ handling and metabolism were found, including upregulation of fatty acid oxidation [62] and post-translational modification of cyclophilin D (CyPD), which mediates the effect of [Ca2+]m on mPTP activation [63], while in mouse hearts expressing a dominant negative form of MCU, cytoplasmic Ca2+ homeostasis was altered [64]. Mitochondria isolated from MCU-deficient cells were shown to have releasable Ca2+, fueling speculation as to a compensatory mechanism that can deliver Ca2+ to the mitochondrial matrix in the absence of the MCU [25].
Adaptive rearrangements of the Ca2+ homeostasis are even more striking in MICU1-deficient cells, mice and patients. MICU1-deficient mice show normal embryonic development, but all the KO/KO pups (on C57BL/6 background), or most of them (mixed background) die at birth without starting regular breathing [23, 65]. Neuron loss in the brainstem where the respiration is controlled likely results from mitochondrial Ca2+ overload, to which MICU1-deficient cells are prone [23]. Liver- or skeletal muscle-specific MICU1 deletion is tolerated at rest but the response to increased functional demand or injury is severely impaired [23, 66]. In the surviving MICU1 KO/KO pups and in the affected tissues of organ specific MICU1 knockout mice, EMRE shows a compensatory decrease [65, 66]. Furthermore, EMRE deletion restores the viability of the MICU1-deficient pups [65]. EMRE loss likely causes a decrease in the number of the functional MCU pores and so contributes to attenuation of the Ca2+ overload and cell injury. The Ca2+ chelation capacity in the mitochondrial matrix is also enhanced in some MICU1-deficient models [45].
Genetic deficiency in MCU in humans has not been documented to date. By contrast, many MICU1-deficient patients have been identified who all show motor dysfunction and commonly have learning impairments [67–69]. The patients’ cells lack MICU1 protein and also show compensatory changes in other components of the mtCU, for example EMRE down-regulation has been observed [66], though some patients seem to have an EMRE increase [70]. The mitochondrial Ca2+ transport phenotype of the patient cells replicates that of the MICU1-deficient mouse cells or cell lines, including an excessive Ca2+ accumulation at resting [Ca2+]c, and an attenuated Ca2+ uptake during [Ca2+]c signals and supraphysiological [Ca2+]c exposures [66, 67].
Based on studies of cell lines, some adaptive changes seen in response to genetic targeting of mtCU components are likely transcriptional, but others closely follow the targeting of MCU or MICU1, indicating control at the level of the protein turnover. For example the mAAA proteases, AFG3L2 and SPG7, mediate the rapid degradation of EMRE [71, 72]. Thus, even in short-term silencing experiments, the observed Ca2+ phenotype can reflect the combination of the effects on the primary target and adaptive changes.
In summary, genetic targeting approaches are effective for the different mtCU components but also induce robust compensatory mechanisms both at the mtCU level, and in other mitochondrial and cellular pathways. Some of the prior studies were clearly aware of the potential compensation and thus selected the approaches to eliminate the gene relatively acute fashion both in cells system and in vivo. However, the compensations often develop quickly and likely play a role in the survival of the initial insults. Complete understanding of the adaptive changes will require further studies. Thus, at this point, we cannot rely only on genetic targeting to understand the physiological and pathophysiological role of the mtCU.
Pharmacological targeting
Many pharmacological inhibitors of the mtCU are divalent/trivalent cations or polycations. Divalents/trivalents can either traverse mtCU and act as competitive inhibitors of Ca2+ like Sr2+ or bind to the channel without permeation like Mg2+ or La3+ [35]. These inhibitors have been useful to explore the biophysical properties of the mtCU but offer little help in intact cell or tissue paradigms. The most frequently used is Ruthenium Red (RuRed), an oxo-bridged triruthenium polycation stain that was first used to visualize extracellular acidic mucopolysaccharides and glycoproteins. RuRed effectively inhibits the mtCU (Ki ~30nM in rat liver mitochondria) [73] but also blocks a range of other channels including the ryanodine receptor [74]. It was recognized early, that the commercial RuRed preparations contained contamination that was more potent than RuRed itself. First, an oxo-bridged diruthenium compound contaminant, Ru360 was identified (IC50 ~0.18nM for Ru360 vs ~6.8nM for RuRed in cardiac mitochondria). Ru360 does not affect the ryanodine receptor, the L-type Ca2+ channels or the Na+/Ca2+ exchanger [75], making it particularly useful in the studies of Ca2+ transport in striated muscle that contain both mtCU and all the above Ca2+ channels/transporters. However, Ru360 is poorly membrane permeable [76] and is unstable in aqueous solution and loses its activity in days. Most recently, a new diruthenium compound, Ru265 was synthesized that seems to be more effective in the inhibition of the mtCU than Ru360 in permeabilized cells (IC50 ~2.5 nM in HeLa cells) [77].
mtCU inhibition by RuRed or Ru360 is dramatically decreased by S259A mutation of MCU adjacent to the -D261-x-x-E264- motif in the IMS loop [39]. Two NMR studies indicated that RuRed/Ru360 binds to and plug the D-ring in the selectivity filter of pentameric MCU [78, 79] (Figure 1). Furthermore, when MICU1 is deleted or its -D-x-x-E-interacting motif is mutated, the mtCU is sensitized towards inhibition by RuRed/Ru360 [54]. Thus, RuRed/Ru360 seems to exert mtCU inhibition by binding directly to MCU’s selectivity filter in the IMS, where the control of MCU by MICU1 also takes place. Unexpectedly, the inhibitory effect of Ru265 was found to be unaltered by the S259A mutation, but suppressed by mutating a cysteine in the N-terminal matrix motif of MCU (C97A) [77] (Figure 1). A matrix site of action would require Ru265 to transit 3 membranes.
The ruthenium compounds are extremely useful in blocking the mtCU in isolated mitochondria and other subcellular preparations. However, a major limitation in their use is that they bind to many targets and that their validated or not excluded off-target effects on various channels, including the two-pore-domain K+ channels (TASK-3, TREK-2, TRAAK), TRPV1–4, Piezo, and CalHM [80–82], prevent their use in validating mtCU physiological relevance or in therapy. Another concern is about their cell permeability. Ru360 has been used in intact cells but, in most studies, its effect on the intended target was not validated, while other studies with intact cells failed to document an inhibition of the [Ca2+]m signal [76]. Ru265 was also described as a cell permeable inhibitor of the mtCU, and after incubation of HeLa cells with 50μM Ru265 for 30min, inhibition of the agonist-induced [Ca2+]m rise was reported [77] but at such high concentrations of the drug, many off-target effects might come into play. Also, using the same Ru265 and treatment protocol, we found no significant change in the [Ca2+]m rise in intact histamine-stimulated HeLa cells (Figure 2). Thus, there is ambiguity about the future of these ruthenium compounds as selective mtCU inhibitors in intact cells or tissues.
Figure 2. Ru265 fails to inhibit mtCU in intact HeLa cells.
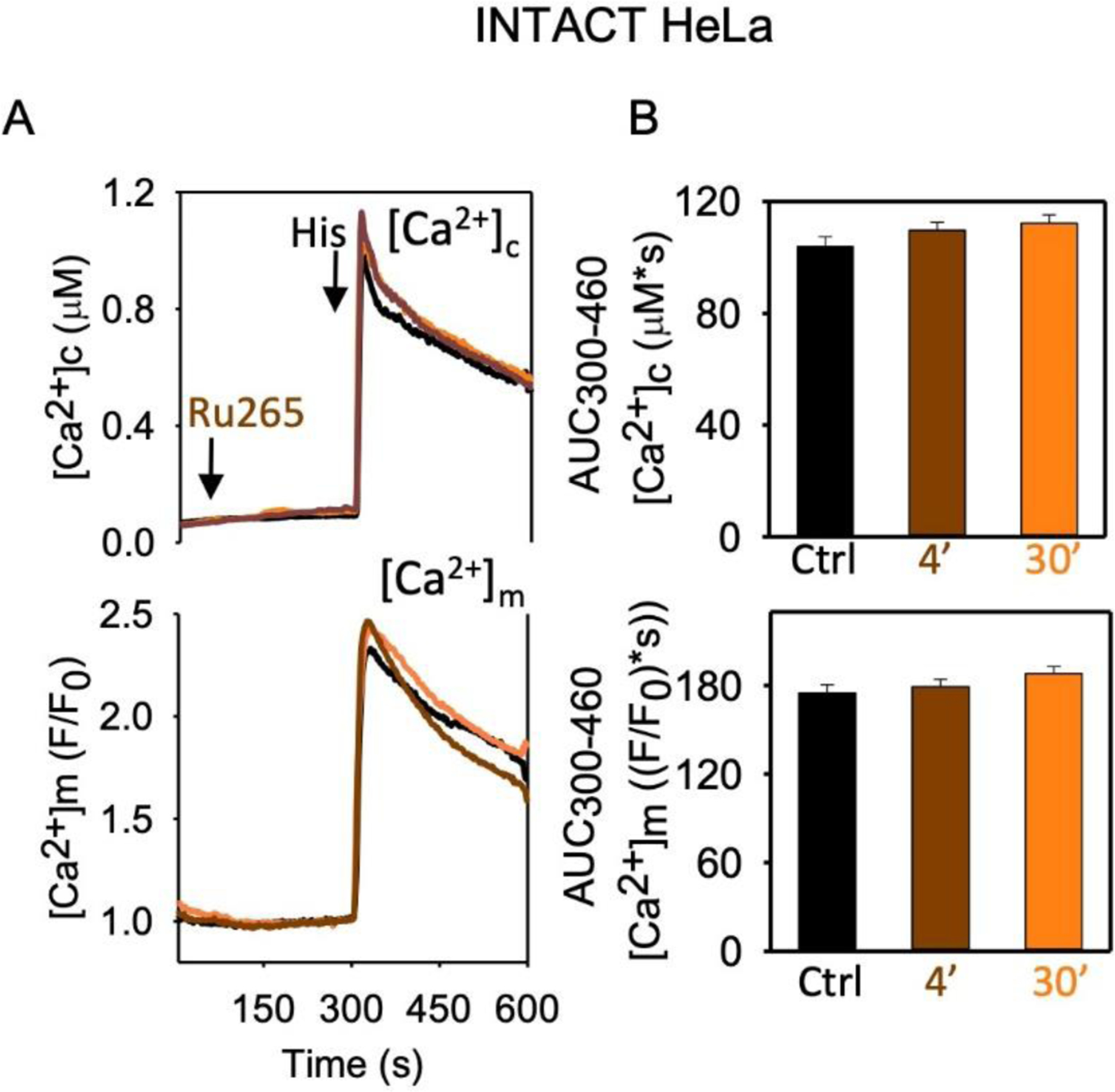
A [Ca2+]c (fura2) and [Ca2+]m (mtCepia) were measured in intact HeLa cells treated with 50μM Ru265/DMSO for 4 or 30min, before 100μM histamine addition [45]. Mean traces are shown for 3 independent cell cultures (each tested in quadruplicate measurements, 110–180 cells in total/condition). B Area under the curve (AUC, mean ± SEM) for the 300–460s time period.
Early studies of mitochondrial Ca2+ uptake also showed that some medically relevant drugs like doxycycline and minocycline exert an inhibitory effect on the mtCU in isolated mitochondria and permeabilized cells [83–85]. These drugs have been shown to be protective in numerous models of ischemia/hypoxia and other toxicities in 4–50 μM concentration [85–87]. However, in intact cells, minocycline up to 80 μM failed to inhibit the [Ca2+]m rise [84] and was shown to affect other mitochondrial targets including respiratory complexes, VDAC channel [88], and mPTP [89].
Recently, high throughput screening of small-molecule libraries have identified new candidates for cell permeable mtCU inhibitors: Mitoxantrone [78], DS16570511 [90], and most recently, MCU-i4 and MCU-i11 [91]. DS16570511 and MCU-i4 have been shown to cause depolarization of the mitochondria limiting their application as specific mtCU inhibitors [92][91]. Mitoxantrone has an IC50 ~13 μM in HeLa cells, and some of its applications are limited by its blue color. Also, mitoxantrone was actually developed as a cytotoxic topoisomerase inhibitor for use in cancer - this would obviously limit its application as an MCU inhibitor [93, 94]. At this point, MCU-i11 appears promising in several respects, therefore, we discuss the action of this small-molecule in more details and provide some data illustrating MCU-i11 strengths and limitations in inhibition of mtCU in cell lines and primary cells/tissues.
Lessons from MCU-i11
MCU-i11 and MCU-i4 are two small-molecule compounds, identified as mtCU inhibitors effective in intact HeLa cells, mouse embryonic fibroblasts and skeletal muscle fibers in a MICU1-dependent manner [91] (Figure 1). MCU-i4 causes mitochondrial depolarization, making the interpretation of the phenotypes in terms of mtCU-dependence difficult so our further analysis here is focused on MCU-i11 (Figures 3–4). The experiments described below also provide some guidance for testing future candidates for inhibition of mtCU.
Figure 3. mtCU inhibition by MCU-i11 in WT, MICU1KO and MCUKO cell lines.
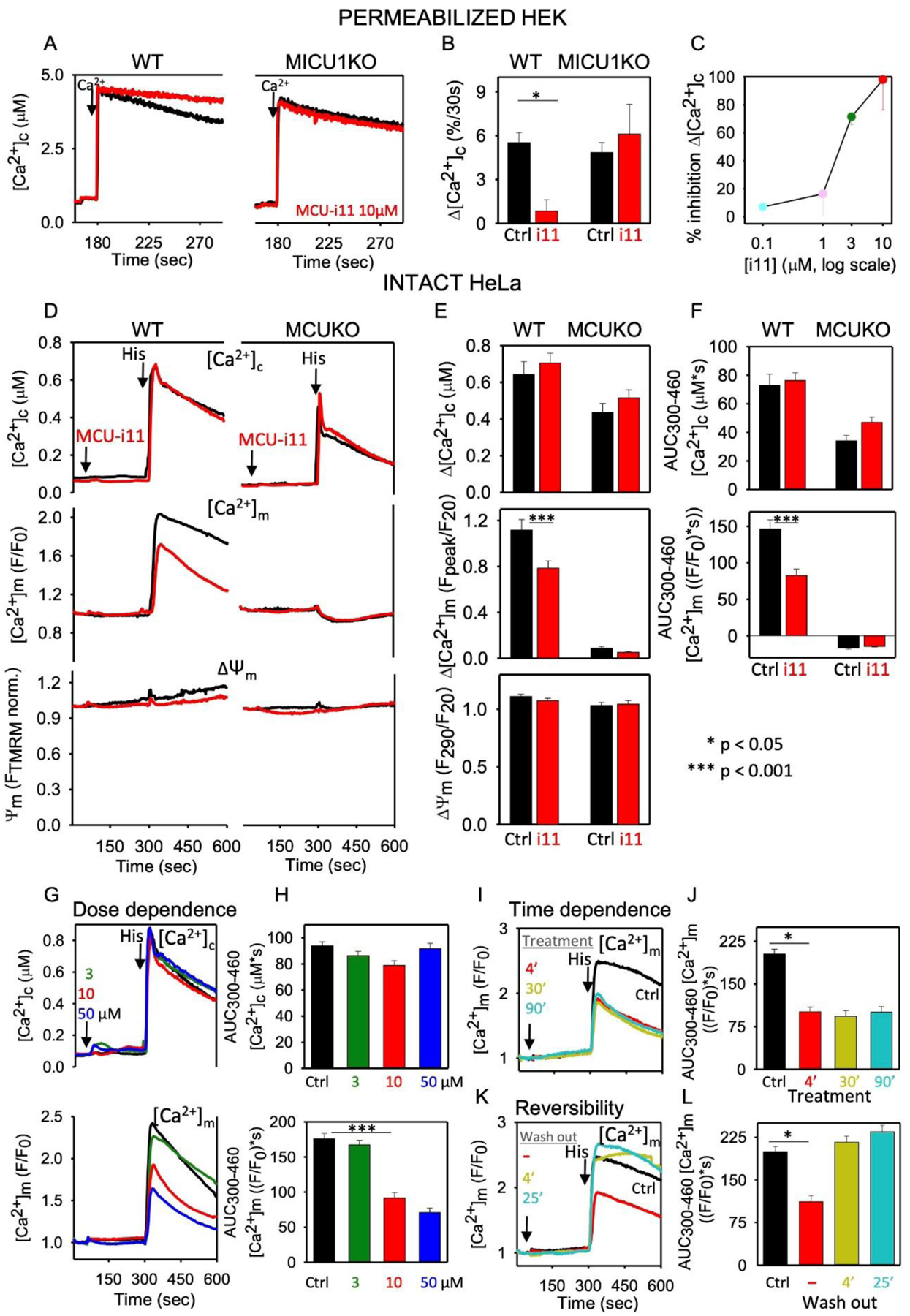
A Clearance of a Ca2+ bolus (10μM CaCl2) was monitored in saponin-permeabilized WT and MICU1KO HEK cells in the presence of 2μM thapsigargin and 10μM CGP37157 to inhibit ER Ca2+ uptake and NCLX, respectively as described in [54]. Clearance of Ca2+ was completely prevented by RuRed (100%, n=3), confirming that clearance was due to mitochondrial Ca2+ uptake via the mtCU. Representative traces show the inhibition of mitochondrial Ca2+ uptake by 10μM MCU-i11 in WT, and the loss of inhibition in MICU1KO. B Percentage of Ca2+ uptake (30s) inhibition by MCU-i11 (i11). C Dose response curve of MCU-i11’s inhibition of mitochondrial Ca2+ uptake (n=6). D [Ca2+]c (fura2), [Ca2+]m (mtCepia) and ΔΨm (TMRM) were simultaneously monitored in intact wild type (WT) and MCUKO HeLa cells treated sequentially with 10 μM MCU-i11/DMSO and 100μM histamine [45]. Traces represent the means of cells from 6–8 measurements of 2 experimental days. E Baseline subtracted peak amplitude of [Ca2+]c and [Ca2+]m, and the ΔΨm measured at the time of the peak normalized to the prestimulation value. F AUC for [Ca2+]c and [Ca2+]m. GH MCU-i11 (3, 10, or 50 μM) was added to WT HeLa cells before stimulation with histamine. Mean [Ca2+]c and [Ca2+]m traces in G and AUC in H. IJ WT HeLa were treated with 10μM MCU-i11 for 4, 3, or 90min, before histamine addition. Mean [Ca2+]m traces in I and AUC in J for the results of quadruplicate measurements for each condition. KL Intact WT HeLa cells were pretreated with 10μM MCU-i11 for different intervals (4, 30, 90min) and then it was washed out, or not, 4 or 25min before stimulation with histamine. Mean [Ca2+]m traces in K and AUC in L. Every single cell fluorescence imaging experiment was reproduced with ≥3 different cell cultures in quadruplicate measurements each day. Results are expressed as mean ± SEM (*p<0.05, ***p<0.001).
Figure 4. mtCU inhibition by MCU-i11 in liver and heart.
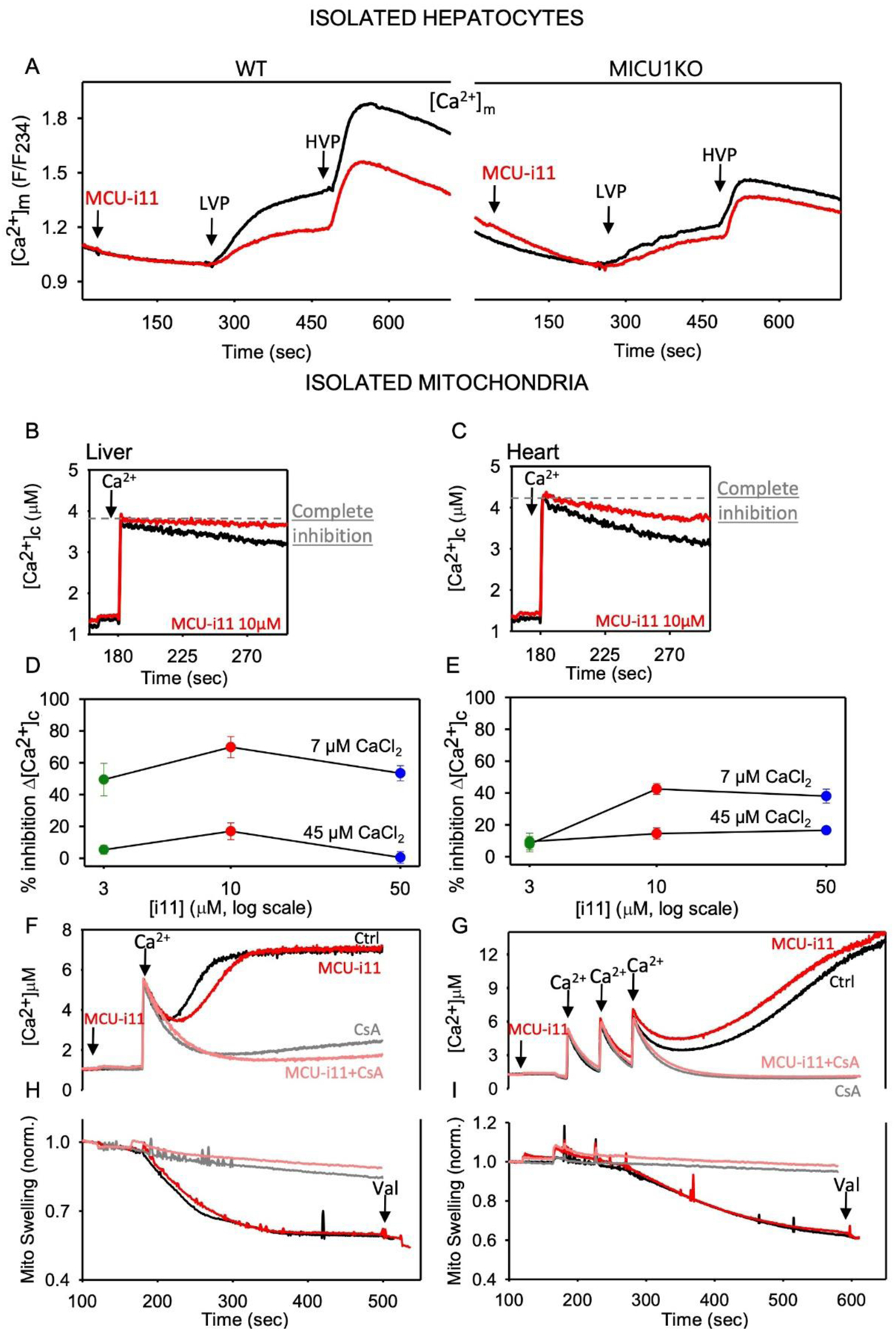
A Mean [Ca2+]m (mtRCaMP) is shown for mouse hepatocytes treated sequentially by a submaximal dose of vasopressin (LVP, 1–1.5nM), and a maximal dose (HVP, 100nM). Measurements were done as described previously [45] n=8 for WT and n=4 for MICU1KO. Baseline subtracted highest value of [Ca2+]m was calculated for each condition and compared to each other by Two-way ANOVA test (Mean±SEM). MCU-i11 significantly decreased the [Ca2+]m peak in WT (0.99±0.035 vs. 0.65±0.032, p<0.001) and failed to alter in MICU1KO (0.53±0.038 vs. 0.43±0.022, p=0.132). BC Clearance of a Ca2+ bolus (7μM CaCl2) was measured in isolated liver (B) and heart (C) mitochondria fluorometrically as described in [23]. DE MCU-i11 dose-response for the inhibition of the initial Ca2+ clearance in liver (D) and heart (E) isolated mitochondria upon 7 or 45μM CaCl2 addition (n=3 for liver, 6 for heart). F mPTP opening in isolated liver mitochondria was visualized as a delayed [Ca2+] increase upon 7μM CaCl2 addition in the absence of Mg-ATP. Traces were recorded without and with MCU-i11, and in the presence of 5μM CsA (inhibitor of mPTP). G In heart mitochondria, mPTP opening elicited by three pulses of 7μM CaCl2 was recorded in the absence and presence of MCU-i11. H&I mPTP opening manifests as mitochondrial swelling as measured by light scattering (side-scatter at 520nm). The Ca2+-induced light scattering decrease was recorded without and with MCU-i11 in liver (H) and heart (I) mitochondria. CsA was used as a positive control both in liver and heart (Mean traces for ≥4 measurements in each condition).
Ten μM MCU-i11 almost completely inhibited mitochondrial Ca2+ uptake in permeabilized HEK cells when extramitochondrial [Ca2+] was elevated to ~4μM, but exerted no inhibition in MICU1KO HEK cells (Figure 3AB). No MCU-i11-dependent change in ΔΨm was detected (TMRM uptake: 25.4±0.4 and 25.7±0.7% of total fluorescence in WT and MICU1KO, respectively, n=3). The IC50 in this model was between 1 and 3μM (Figure 3C). Thus, MCU-i11 is a potent inhibitor of the mtCU, though the IC50 is higher than that for RuRed/Ru360/Ru265. A striking difference between MCU-i11 and RuRed/Ru360 is that MICU1 is required for the inhibition by MCU-i11 ([91], current data), whereas MICU1 decreased the affinity to inhibition by RuRed/Ru360 [54].
In intact HeLa cells stimulated with histamine, which mobilizes Ca2+ from the endoplasmic reticulum via the IP3 receptor, 10μM MCU-i11 attenuated the [Ca2+]m signal without altering ΔΨm or the [Ca2+]c transient that activates the [Ca2+]m rise (Figure 3DE). The lack of off-target effects on cytoplasmic Ca2+ handling is further illustrated by the same measurement performed in HeLa cells devoid of MCU (Figure 3DF). The attenuation of the [Ca2+]m increase by MCU-i11 was partial, and the IC50 was between 3 and 10μM (Figure 3GH). Impressively, 4min preincubation with MCU-i11 was sufficient to attain a full effect (Figure 3I, as in [91]), and it was completely reversed by washout of MCU-i11 (Figure 3KL). In primary cell models, MCU-i11 was shown to decrease the RyR-mediated [Ca2+]m rise in mouse skeletal muscle fibers [91]. Furthermore, the IP3 receptor-mediated [Ca2+]m rise was partially attenuated by 10μM MCU-i11 in wild type (WT) but not in MICU1-deficient mouse hepatocytes (Figure 4A). In summary, MCU-i11 seems to be a useful tool to specifically and acutely suppress Ca2+ delivery to the mitochondrial matrix in a variety of cell types, without interfering with the extramitochondrial Ca2+ transport or the ΔΨm.
MCU-i11’s membrane permeation might contribute to the lesser inhibition in intact HeLa cells and hepatocytes than that in permeabilized HEK cells. However, another difference between the intact and permeabilized experiments is that in the former, mitochondria are exposed locally to 10–30μM [Ca2+] nanodomains [95, 96], whereas in latter, the extramitochondrial [Ca2+] was raised to 4μM or less. Thus, lesser potency of MCU-i11 at high [Ca2+] might be the cause of the partial mtCU inhibition in the intact cells. Notably, mtCU [Ca2+] sensing is provided by MICU1, which is needed for inhibition by MCU-i11, and MICU1 is more abundant relative to MCU in the liver than that in the heart [97].
To test the [Ca2+]c and MICU1/MCU dependence of the inhibition by MCU-i11, isolated mitochondria from heart and liver were compared side by side using 4μM and 16μM [Ca2+] exposures by the addition of 7μM and 45μM CaCl2, respectively (Figure 4B–E). In liver mitochondria, upon 4μM [Ca2+] exposure the maximal inhibition by MCU-i11 was ~70%, whereas at 16μM [Ca2+], it was only 17% (Figure 4BC). In heart mitochondria, the maximal inhibition by MCU-i11 was ~40%, and 16% during 4μM and 16μM [Ca2+] exposures, respectively (Figure 4DE). Thus, the effectiveness of MCU-i11’s inhibition seems to be reduced at high [Ca2+], which might explain the relatively small effect of MCU-i11 during local, IP3 receptor- or RyR-mediated Ca2+ transfer from the endo/sarcoplasmic reticulum (e.g. Figure 3B vs 3E). Furthermore, in tissues with higher MICU1 abundance in the mtCU complexes, like the liver, MCU-i11 might be more effective than in tissues where MICU1 is less abundant like in cardiac muscle [43, 48]. Mechanistically, MCU-i11 appears to support the gatekeeping action of MICU1 on the mtCU but is unable to prevent the Ca2+ binding and activation of MICU1 during high [Ca2+] exposure. Therefore, MCU-i11 might be more useful to inhibit mtCU in paradigms with abundant MICU1 and moderate [Ca2+]c elevations. An important question, then, is whether MCU-i11 might be of any use during mitochondrial Ca2+ overload conditions, which commonly occur during ischemia-reperfusion injury.
To model this condition, liver and heart mitochondria were exposed to Ca2+ loads in the absence of ATP, a condition that promotes mPTP opening. Ca2+ overload and mPTP opening manifests as a precipitous delayed [Ca2+]c rise that is suppressed by cyclosporin A (CsA), an inhibitor of the mPTP. Based on the [Ca2+]c traces, MCU-i11 slightly delayed mPTP opening in liver mitochondria and seemed to have a small effect in the opposite direction in the heart mitochondria (Figure 4FG). Because the [Ca2+]c traces show the combination of the Ca2+ uptake inhibition and the mPTP-mediated Ca2+ release, a more straightforward assay for the permeability transition is the monitoring of the light scattering by the mitochondria, which reflects the mPTP opening-induced swelling as a decrease in light scattering. Based on this assay, MCU-i11 did not prevent the Ca2+-induced permeability transition in either liver mitochondria or heart mitochondria (Figure 4HI). To directly assess the potential protective effect of MCU-i11 against ischemia-reperfusion, we evaluated cell death after hypoxia (45 min)-reoxygenation (3hrs) in isolated mouse cardiomyocytes. MCU-i11 did not decrease cardiomyocyte death induced by hypoxia-reoxygenation (without MCU-i11: 70.1±10.9%; preconditioning with MCU-i11: 67.3±11.4%; postconditioning with MCU-i11: 70.0±10.4% propidium-iodide positive cardiomyocytes, Mean±SEM, n=3). Thus, unfortunately, MCU-i11 is predicted to have little or no use in ischemia-reperfusion diseases and other disorders that progress through acute mitochondrial Ca2+ overload but could be of interest in chronic mild Ca2+ overload conditions, such as certain inherited cardiomyopathies.
Future directions
The demand has been growing for means to target the mtCU for research and, potentially, for clinical applications (see Figure 1). Genetic targeting has succeeded for each component of the mtCU in cells and mice. The phenotype seems to be more severe for deletion of the main Ca2+ sensing regulator, MICU1, than for the pore-forming unit, MCU, though outcomes depend on the genetic background. The loss of gatekeeping sensitizes cells to mitochondrial Ca2+ overload and cell death/tissue injury. Furthermore, loss or attenuation of the dynamic mitochondrial Ca2+ uptake during [Ca2+]c fluctuations seems to cause some problems in handling increased energy needs. However, both MICU1 and MCU deletion are followed by compensatory changes in cellular Ca2+ handling and mtCU composition, which are, per se, evidence of the physiological relevance of mtCU [98]. But the compensation makes it difficult to understand how the mtCU serves as an effector of physiology. Although the compensation for mtCU components seems to be rapid, silencing or conditional deletion of MCU or MICU1 may still offer a time window to test the specific roles of the primary target. Because of the growing evidence of altered abundance of various mtCU components, and corresponding changes in mitochondrial Ca2+ transport in various diseases, genetic manipulations of the mtCU are expected to be considered for human diseases, too.
In terms of pharmacological targeting, the need seems to be more urgent for mtCU antagonists than agonists, though positive regulators are speculated to be helpful in certain conditions. For decades the biggest obstacles were the membrane permeability and specificity of compounds. Recently, screening large libraries identified several small molecule inhibitors which rapidly cross the plasma membrane. Some of these molecules inhibit the uniporter without a direct effect on cytoplasmic Ca2+ homeostasis and mitochondrial energetics but future studies will be needed to test their specificity. However, none of the currently available inhibitors are sufficiently potent in intact cells, so either the current molecules will need further modification or new candidates identified.
It was already established that compounds can exert mtCU inhibition through either MCU or MICU1. The inhibition by RuRed/Ru360 targets the small IMS loop of MCU, which serves as the pore’s selectivity filter [78]. Further developments targeting this locus should recognize that MICU1’s presence affects the accessibility to this region of MCU [54]. MCU-i11, by contrast, targets MICU1 to stabilize the closed state of the mtCU, but this drug cannot prevent activation of the mtCU by high [Ca2+]c [91], and present data). Deciphering the mechanism of the interaction between MCU-i11 and Ca2+ on MICU1 will require further studies. It will also be useful to identify further compounds that create resistance to the Ca2+ effect on MICU1. Because the inhibitory efficacy of both RuRed/Ru360 and MCU-i11 depends on the relative abundance of MICU1 and MCU, the sensitivity to all these and related compounds is expected to be different in tissues with varying MICU1/MCU, and the sensitivity can also be altered by conditions with selective regulation of each mtCU component. In addition to targeting the mtCU components, potential future strategies might also target the factors that have been implicated in their regulation at the pre-or post-translational level (for a recent review see [99]).
Finding molecules that can potently and selectively inhibit the mtCU in intact cells and tissues is just the first step towards developing drugs for human applications. However, unlike RuRed, some of the new inhibitors can be modified in many ways to improve their pharmacodynamics. In any case, pharmacological inhibition of mtCU has progressed impressively in just a few years, and, building on the molecular definition and structural characterization of the mtCU constituents, and with the feasibility of screening extensive libraries of small molecules, the conditions are favorable for drug development.
Supplementary Material
Highlights.
Pharmacological approach is needed to test mtCU’s physiological and medical relevance
Ruthenium compounds fail to inhibit the mtCU in intact cells
MCU-i11 inhibits the mtCU in intact cells dependent on MICU1 expression
Inhibition is inversely proportional to [Ca2+] and fails to decrease overload injury
Plan for testing the efficacy, specificity and reversibility of mtCU drug candidates
ACKNOWLEDGEMENTS
The authors thank Justin Wilson and Joshua Woods for providing Ru265, Suresh K Joseph for providing MCUKO HeLa cells and comments on the manuscript, the entire Foundation Leducq “Mitocardia” team (Tish Murphy, Paolo Bernardi, Fabio di Lisa, Jeff Molkentin, Michel Cohen, Michael Ovize, and Mike Forte) and Diego De Stefani and Cristina Mammucari for the stimulating discussions on the subject of this review and David Weaver, György Csordás and Erin L. Seifert for comments on the manuscript. The work was funded by the Foundation Leducq and an NIH grant (RO1 HL142271) to G.H. K.M acknowledges support by the Rosztoczy Foundation.
Abbreviations
- [Ca2+]c
cytoplasmic [Ca2+]
- [Ca2+]m
mitochondrial matrix [Ca2+]
- CyPD
cyclophilin D
- IMM
inner mitochondrial membrane
- IMS
intermembrane space
- KO
knocked out
- MCU-i4
Mitochondrial calcium uniporter- inhibitor 4
- MCU-i11
Mitochondrial calcium uniporter- inhibitor 11
- mPTP
mitochondrial permeability transition pore
- mtCU
Ca2+ uniporter
- NTD
N-terminal domain
- OMM
outer mitochondrial membrane
- ROS
Reactive Oxygen Species
- RuRed
Ruthenium Red
- VDAC
Voltage Dependent Anion-selective Channel
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DECLARATION OF ETHICAL APPROVAL
Some experiments involve the use of animal subjects, here we state that all procedures were performed in compliance with the relevant laws and institutional guidelines - Institutional Animal Care & Use Committee (IACUC) - and that the appropriate institutional committee(s) have approved them (Protocol No. 01338).
Biography
- 1.Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+ homeostasis in intact cells. J Cell Biol. 1994;126(5):1183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82(3):415–24. [DOI] [PubMed] [Google Scholar]
- 3.Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A. 1999;96(24):13807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ichas F, Mazat JP. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochimica et biophysica acta. 1998;1366(1–2):33–50. [DOI] [PubMed] [Google Scholar]
- 5.Bernardi P, Petronilli V, Di Lisa F, Forte M. A mitochondrial perspective on cell death. Trends in biochemical sciences. 2001;26(2):112–7. [DOI] [PubMed] [Google Scholar]
- 6.Szalai G, Krishnamurthy R, Hajnoczky G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. Embo J. 1999;18(22):6349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287(4):C817–33. [DOI] [PubMed] [Google Scholar]
- 8.Csordas G, Hajnoczky G. SR/ER-mitochondrial local communication: calcium and ROS. Biochimica et biophysica acta. 2009;1787(11):1352–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89(7):1145–53. [DOI] [PubMed] [Google Scholar]
- 10.Hajnoczky G, Hager R, Thomas AP. Mitochondria suppress local feedback activation of inositol 1,4, 5-trisphosphate receptors by Ca2+. J Biol Chem. 1999;274(20):14157–62. [DOI] [PubMed] [Google Scholar]
- 11.Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr. 2000;32(1):97–104. [DOI] [PubMed] [Google Scholar]
- 12.Eisner V, Csordas G, Hajnoczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca(2)(+) and reactive oxygen species signaling. Journal of cell science. 2013;126(Pt 14):2965–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huser J, Blatter LA, Sheu SS. Mitochondrial calcium in heart cells: beat-to-beat oscillations or slow integration of cytosolic transients? J Bioenerg Biomembr. 2000;32(1):27–33. [DOI] [PubMed] [Google Scholar]
- 14.Griffiths EJ. Mitochondrial calcium transport in the heart: physiological and pathological roles. Journal of molecular and cellular cardiology. 2009;46(6):789–803. [DOI] [PubMed] [Google Scholar]
- 15.Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99(2):172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chacon E, Ohata H, Harper IS, Trollinger DR, Herman B, Lemasters JJ. Mitochondrial free calcium transients during excitation-contraction coupling in rabbit cardiac myocytes. FEBS Lett. 1996;382(1–2):31–6. [DOI] [PubMed] [Google Scholar]
- 17.Trollinger DR, Cascio WE, Lemasters JJ. Mitochondrial calcium transients in adult rabbit cardiac myocytes: inhibition by ruthenium red and artifacts caused by lysosomal loading of Ca(2+)-indicating fluorophores. Biophys J. 2000;79(1):39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wust RC, Helmes M, Martin JL, van der Wardt TJ, Musters RJ, van der Velden J, et al. Rapid frequency-dependent changes in free mitochondrial calcium concentration in rat cardiac myocytes. J Physiol. 2017;595(6):2001–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dietl A, Maack C. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Current heart failure reports. 2017;14(4):338–49. [DOI] [PubMed] [Google Scholar]
- 20.Diaz-Juarez J, Suarez J, Cividini F, Scott BT, Diemer T, Dai A, et al. Expression of the mitochondrial calcium uniporter in cardiac myocytes improves impaired mitochondrial calcium handling and metabolism in simulated hyperglycemia. Am J Physiol Cell Physiol. 2016;311(6):C1005–C13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banavath HN, Roman B, Mackowski N, Biswas D, Afzal J, Nomura Y, et al. miR-181c Activates Mitochondrial Calcium Uptake by Regulating MICU1 in the Heart. J Am Heart Assoc. 2019;8(24):e012919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xue Q, Pei H, Liu Q, Zhao M, Sun J, Gao E, et al. MICU1 protects against myocardial ischemia/reperfusion injury and its control by the importer receptor Tom70. Cell Death Dis. 2017;8(7):e2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, Bolon B, et al. MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nature communications. 2016;7:10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell reports. 2015;12(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, et al. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell reports. 2015;12(1):15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemasters JJ, Nieminen AL, Qian T, Trost LC, Herman B. The mitochondrial permeability transition in toxic, hypoxic and reperfusion injury. Molecular and cellular biochemistry. 1997;174(1–2):159–65. [PubMed] [Google Scholar]
- 27.Yu Z, Gong X, Yu Y, Li M, Liang Y, Qin S, et al. The mechanical effects of CRT promoting autophagy via mitochondrial calcium uniporter down-regulation and mitochondrial dynamics alteration. J Cell Mol Med. 2019;23(6):3833–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sommakia S, Houlihan PR, Deane SS, Simcox JA, Torres NS, Jeong MY, et al. Mitochondrial cardiomyopathies feature increased uptake and diminished efflux of mitochondrial calcium. Journal of molecular and cellular cardiology. 2017;113:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krieger C, Duchen MR. Mitochondria, Ca2+ and neurodegenerative disease. European journal of pharmacology. 2002;447(2–3):177–88. [DOI] [PubMed] [Google Scholar]
- 30.Rossi A, Pizzo P, Filadi R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochimica et biophysica acta Molecular cell research. 2019;1866(7):1068–78. [DOI] [PubMed] [Google Scholar]
- 31.Marchi S, Vitto VAM, Danese A, Wieckowski MR, Giorgi C, Pinton P. Mitochondrial calcium uniporter complex modulation in cancerogenesis. Cell Cycle. 2019;18(10):1068–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui C, Yang J, Fu L, Wang M, Wang X. Progress in understanding mitochondrial calcium uniporter complex-mediated calcium signalling: A potential target for cancer treatment. British journal of pharmacology. 2019;176(9):1190–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. The Journal of cell biology. 2002;159(4):613–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bathori G, Csordas G, Garcia-Perez C, Davies E, Hajnoczky G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). The Journal of biological chemistry. 2006;281(25):17347–58. [DOI] [PubMed] [Google Scholar]
- 35.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258(5 Pt 1):C755–86. [DOI] [PubMed] [Google Scholar]
- 36.Vinogradov A, Scarpa A. The initial velocities of calcium uptake by rat liver mitochondria. J Biol Chem. 1973;248(15):5527–31. [PubMed] [Google Scholar]
- 37.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427(6972):360–4. [DOI] [PubMed] [Google Scholar]
- 38.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011. [DOI] [PMC free article] [PubMed]
- 40.Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32(17):2362–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342(6164):1379–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467(7313):291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PloS one. 2013;8(2):e55785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patron M, Granatiero V, Espino J, Rizzuto R, De Stefani D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell death and differentiation. 2019;26(1):179–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell metabolism. 2013;17(6):976–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Molecular cell. 2014;53(5):726–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hung V, Zou P, Rhee HW, Udeshi ND, Cracan V, Svinkina T, et al. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Molecular cell. 2014;55(2):332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paillard M, Csordas G, Szanda G, Golenar T, Debattisti V, Bartok A, et al. Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca2+ Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell reports. 2017;18(10):2291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gottschalk B, Klec C, Leitinger G, Bernhart E, Rost R, Bischof H, et al. MICU1 controls cristae junction and spatially anchors mitochondrial Ca(2+) uniporter complex. Nature communications. 2019;10(1):3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tomar D, Thomas M, Garbincius JF, Kolmetzky DW, Salik O, Jadiya P, et al. MICU1 regulates mitochondrial cristae structure and function independent of the mitochondrial calcium uniporter channel. bioRxiv. 2019:803213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Nguyen NX, She J, Zeng W, Yang Y, Bai XC, et al. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell. 2019;177(5):1252–61 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacEwen MJ, Markhard AL, Bozbeyoglu M, Bradford F, Goldberger O, Mootha VK, et al. Evolutionary divergence reveals the molecular basis of EMRE dependence of the human MCU. Life Sci Alliance. 2020;3(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsai MF, Phillips CB, Ranaghan M, Tsai CW, Wu Y, Willliams C, et al. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. eLife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paillard M, Csordas G, Huang KT, Varnai P, Joseph SK, Hajnoczky G. MICU1 Interacts with the D-Ring of the MCU Pore to Control Its Ca(2+) Flux and Sensitivity to Ru360. Molecular cell. 2018;72(4):778–85 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Phillips CB, Tsai CW, Tsai MF. The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex. eLife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fan M, Zhang J, Tsai CW, Orlando BJ, Rodriguez M, Xu Y, et al. Structure and mechanism of the mitochondrial Ca(2+) uniporter holocomplex. Nature. 2020;582(7810):129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang C, Jacewicz A, Delgado BD, Baradaran R, Long SB. Structures reveal gatekeeping of the mitochondrial Ca(2+) uniporter by MICU1-MICU2. eLife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, Han Y, She J, Nguyen NX, Mootha VK, Bai XC, et al. Structural insights into the Ca(2+)-dependent gating of the human mitochondrial calcium uniporter. eLife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nature cell biology. 2013;15(12):1464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murphy E, Pan X, Nguyen T, Liu J, Holmstrom KM, Finkel T. Unresolved questions from the analysis of mice lacking MCU expression. Biochemical and biophysical research communications. 2014;449(4):384–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pendin D, Greotti E, Pozzan T. The elusive importance of being a mitochondrial Ca(2+) uniporter. Cell calcium. 2014;55(3):139–45. [DOI] [PubMed] [Google Scholar]
- 62.Altamimi TR, Karwi QG, Uddin GM, Fukushima A, Kwong JQ, Molkentin JD, et al. Cardiac-specific deficiency of the mitochondrial calcium uniporter augments fatty acid oxidation and functional reserve. Journal of molecular and cellular cardiology. 2019;127:223–31. [DOI] [PubMed] [Google Scholar]
- 63.Parks RJ, Menazza S, Holmstrom KM, Amanakis G, Fergusson M, Ma H, et al. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovascular research. 2019;115(2):385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci U S A. 2015;112(29):9129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, Fergusson MM, et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell reports. 2016;16(6):1561–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Debattisti V, Horn A, Singh R, Seifert EL, Hogarth MW, Mazala DA, et al. Dysregulation of Mitochondrial Ca(2+) Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1. Cell reports. 2019;29(5):1274–86 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet. 2014;46(2):188–93. [DOI] [PubMed] [Google Scholar]
- 68.Lewis-Smith D, Kamer KJ, Griffin H, Childs AM, Pysden K, Titov D, et al. Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurology Genetics. 2016;2(2):e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Musa S, Eyaid W, Kamer K, Ali R, Al-Mureikhi M, Shahbeck N, et al. A Middle Eastern Founder Mutation Expands the Genotypic and Phenotypic Spectrum of Mitochondrial MICU1 Deficiency: A Report of 13 Patients. JIMD reports. 2019;43:79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bhosale G, Sharpe JA, Koh A, Kouli A, Szabadkai G, Duchen MR. Pathological consequences of MICU1 mutations on mitochondrial calcium signalling and bioenergetics. Biochimica et biophysica acta. 2017;1864(6):1009–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsai CW, Wu Y, Pao PC, Phillips CB, Williams C, Miller C, et al. Proteolytic control of the mitochondrial calcium uniporter complex. Proc Natl Acad Sci U S A. 2017;114(17):4388–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.König T, Tröder SE, Bakka K, Korwitz A, Richter-Dennerlein R, Lampe PA, et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Molecular cell. 2016;64(1):148–62. [DOI] [PubMed] [Google Scholar]
- 73.Reed KC, Bygrave FL. The inhibition of mitochondrial calcium transport by lanthanides and ruthenium red. The Biochemical journal. 1974;140(2):143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma J Block by ruthenium red of the ryanodine-activated calcium release channel of skeletal muscle. The Journal of general physiology. 1993;102(6):1031–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, et al. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem. 1998;273(17):10223–31. [DOI] [PubMed] [Google Scholar]
- 76.Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, et al. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell calcium. 2006;40(5–6):553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Woods JJ, Nemani N, Shanmughapriya S, Kumar A, Zhang M, Nathan SR, et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS central science. 2019;5(1):153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arduino DM, Wettmarshausen J, Vais H, Navas-Navarro P, Cheng Y, Leimpek A, et al. Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening. Molecular cell. 2017;67(4):711–23 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cao C, Wang S, Cui T, Su XC, Chou JJ. Ion and inhibitor binding of the double-ring ion selectivity filter of the mitochondrial calcium uniporter. Proc Natl Acad Sci U S A. 2017;114(14):E2846–E51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mathie A, Veale EL. Therapeutic potential of neuronal two-pore domain potassium-channel modulators. Curr Opin Investig Drugs. 2007;8(7):555–62. [PubMed] [Google Scholar]
- 81.St Pierre M, Reeh PW, Zimmermann K. Differential effects of TRPV channel block on polymodal activation of rat cutaneous nociceptors in vitro. Experimental brain research. 2009;196(1):31–44. [DOI] [PubMed] [Google Scholar]
- 82.Gnanasambandam R, Gottlieb PA, Sachs F. The Kinetics and the Permeation Properties of Piezo Channels. Current topics in membranes. 2017;79:275–307. [DOI] [PubMed] [Google Scholar]
- 83.Theruvath TP, Zhong Z, Pediaditakis P, Ramshesh VK, Currin RT, Tikunov A, et al. Minocycline and N-methyl-4-isoleucine cyclosporin (NIM811) mitigate storage/reperfusion injury after rat liver transplantation through suppression of the mitochondrial permeability transition. Hepatology. 2008;47(1):236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Csordas G, Varnai P, Golenar T, Sheu SS, Hajnoczky G. Calcium transport across the inner mitochondrial membrane: Molecular mechanisms and pharmacology. Molecular and cellular endocrinology. 2012;353(1–2):109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schwartz J, Holmuhamedov E, Zhang X, Lovelace GL, Smith CD, Lemasters JJ. Minocycline and doxycycline, but not other tetracycline-derived compounds, protect liver cells from chemical hypoxia and ischemia/reperfusion injury by inhibition of the mitochondrial calcium uniporter. Toxicol Appl Pharmacol. 2013;273(1):172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav Brain Res. 2009;196(2):168–79. [DOI] [PubMed] [Google Scholar]
- 87.Hu J, Kholmukhamedov A, Lindsey CC, Beeson CC, Jaeschke H, Lemasters JJ. Translocation of iron from lysosomes to mitochondria during acetaminophen-induced hepatocellular injury: Protection by starch-desferal and minocycline. Free Radic Biol Med. 2016;97:418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Garcia-Martinez EM, Sanz-Blasco S, Karachitos A, Bandez MJ, Fernandez-Gomez FJ, Perez-Alvarez S, et al. Mitochondria and calcium flux as targets of neuroprotection caused by minocycline in cerebellar granule cells. Biochem Pharmacol. 2010;79(2):239–50. [DOI] [PubMed] [Google Scholar]
- 89.Mansson R, Morota S, Hansson MJ, Sonoda I, Yasuda Y, Shimazu M, et al. Minocycline sensitizes rodent and human liver mitochondria to the permeability transition: implications for toxicity in liver transplantation. Hepatology. 2010;51(1):347–8; author reply 9–50. [DOI] [PubMed] [Google Scholar]
- 90.Kon N, Murakoshi M, Isobe A, Kagechika K, Miyoshi N, Nagayama T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017;3:17045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Di Marco G, Vallese F, Jourde B, Bergsdorf C, Sturlese M, De Mario A, et al. A High-Throughput Screening Identifies MICU1 Targeting Compounds. Cell reports. 2020;30(7):2321–31 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Payne R, Li C, Fernandez-Garcia E, Vais H, Foskett K The MCU Inhibitor Ds16570511 has Off-Target Effects on Mitochondrial Membrane Potential. Biophysical Journal. 2019; Volume 116(Issue 3):270A.30612713 [Google Scholar]
- 93.Durr FE, Wallace RE, Citarella RV. Molecular and biochemical pharmacology of mitoxantrone. Cancer Treat Rev. 1983;10 Suppl B:3–11. [DOI] [PubMed] [Google Scholar]
- 94.Koeller J, Eble M. Mitoxantrone: a novel anthracycline derivative. Clin Pharm. 1988;7(8):574–81. [PubMed] [Google Scholar]
- 95.Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, et al. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Molecular cell. 2010;39(1):121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, et al. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Molecular cell. 2010;38(2):280–90. [DOI] [PubMed] [Google Scholar]
- 97.Paillard M, Csordas G, Szanda G, Golenar T, Debattisti V, Bartok A, et al. Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca(2+) Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell reports. 2017;18(10):2291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu JC, Parks RJ, Liu J, Stares J, Rovira II, Murphy E, et al. The In Vivo Biology of the Mitochondrial Calcium Uniporter. Advances in experimental medicine and biology. 2017;982:49–63. [DOI] [PubMed] [Google Scholar]
- 99.Mishra J, Jhun BS, Hurst S, J OU, Csordas G, Sheu SS. The Mitochondrial Ca(2+) Uniporter: Structure, Function, and Pharmacology. Handb Exp Pharmacol. 2017;240:129–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.