

Abstract
Regulation of gene expression plays a fundamental role in cardiac stress-responses. Modification of coding transcripts by adenosine methylation (m6A) has recently emerged as a critical post-transcriptional mechanism underlying heart disease. Thousands of mammalian mRNAs are known to be m6A-modified, suggesting that remodeling of the m6A landscape may play an important role in cardiac pathophysiology. Here we found an increase in m6A content in human heart failure samples. We then adopted genome-wide analysis to define all m6A-regulated sites in human failing compared to non-failing hearts and identified targeted transcripts involved in histone modification as enriched in heart failure. Further, we compared all m6A sites regulated in human hearts with the ones occurring in isolated rat hypertrophic cardiomyocytes to define cardiomyocyte-specific m6A events conserved across species. Our results identified 38 shared transcripts targeted by m6A during stress conditions, and 11 events that are unique to unstressed cardiomyocytes. Of these, further evaluation of select mRNA and protein abundances demonstrates the potential impact of m6A on post-transcriptional regulation of gene expression in the heart.
Keywords: heart failure, cardiomyocyte hypertrophy, m6A, gene expression, epigenetics
Introduction
Gene expression regulation underlies the ability of the heart to cope with stress factors that perturb homeostasis [1, 2]. A classic phenotypic feature of the stressed heart is represented by hypertrophic growth of the cardiomyocytes. Cardiac hypertrophy is a risk factor for heart failure, highlighting the importance of understanding the molecular events leading to stress-induced remodeling of the proteome in cardiomyocytes.
Changes in the transcriptional output of cardiomyocytes, through chromatin remodeling and regulation of transcription factor activity, indisputably contribute to cardiac injury responses [3–6]. Nevertheless, mechanisms that act post-transcriptionally to regulate mRNA translation have been shown to heavily contribute to the response to cardiac injury [7–11]. Post-transcriptional chemical modification of RNA represents a major mechanism used by cells to determine the fate of specific transcripts.
Despite the chemical diversity of existing RNA modifications, methylation on position N6 of adenosines (m6A) is particularly important for the life of mRNAs as it is directly and abundantly deposited on internal sites of coding transcripts [12, 13]. Additionally, the proven dynamic nature of the m6A landscape allows cells to regulate mRNA translation to ultimately respond to environmental challenges and accordingly modify cell morphology and/or behavior [12, 13].
Our group and others have recently demonstrated the importance of m6A for cardiac pathophysiology [14–19]. Changes in the overall amount of this modification have indeed been proven to not only mark disease states but can also act as a sufficient determinant of cardiomyocyte remodeling [14–17]. Sequencing of m6A-modified mRNAs have further revealed the identity of thousands of transcripts that are subjected to modification by this pathway in the heart, pointing to the overwhelming complexity of this regulatory mechanism [14, 15, 17, 18].
A consistent observation from studies on m6A is the insult- and cell-dependent presence of this mark on specific mRNA sites. Despite the growing body of information that sequencing methods have generated, defining key evolutionarily conserved m6A events that could be targetable for heart disease has proven challenging, as independent groups have found various subsets of m6A-modified transcripts unique to different forms of cardiac injury and the cell specificity of such changes has often remained elusive.
Here, we defined how the m6A landscape remodels in the human failing heart and cross-analyzed these results with data collected by genome-wide m6A analysis on isolated rat cardiomyocytes subjected to hypertrophic stimuli. Our study reveals key post-transcriptional events that underlie cardiomyocyte hypertrophy and are relevant across species.
Results
m6A level increases in heart failure, but shows preserved distribution
Global m6A content can be modulated during different pathophysiological states. Using mass spectrometry we found higher m6A levels in mRNA from human non-ischemic failing hearts as compared to non-failing control samples (Figure 1A). Analysis of protein expression for key enzymes responsible for m6A deposition (Methyltransferase like 3, RNA methylase METTL3) and removal (AlkB Homolog 5, RNA demethylase ALKBH5; Fat mass and obesity-associated protein, FTO) revealed increased METTL3 levels with concomitant reduction in FTO expression in human failing hearts and no change in ALKBH5 levels (Figure 1B–E). These results suggest a balance in favor of m6A methylation during heart failure in humans.
Figure 1. m6A and associated catalyzing enzyme are enriched in failing human heart tissue.
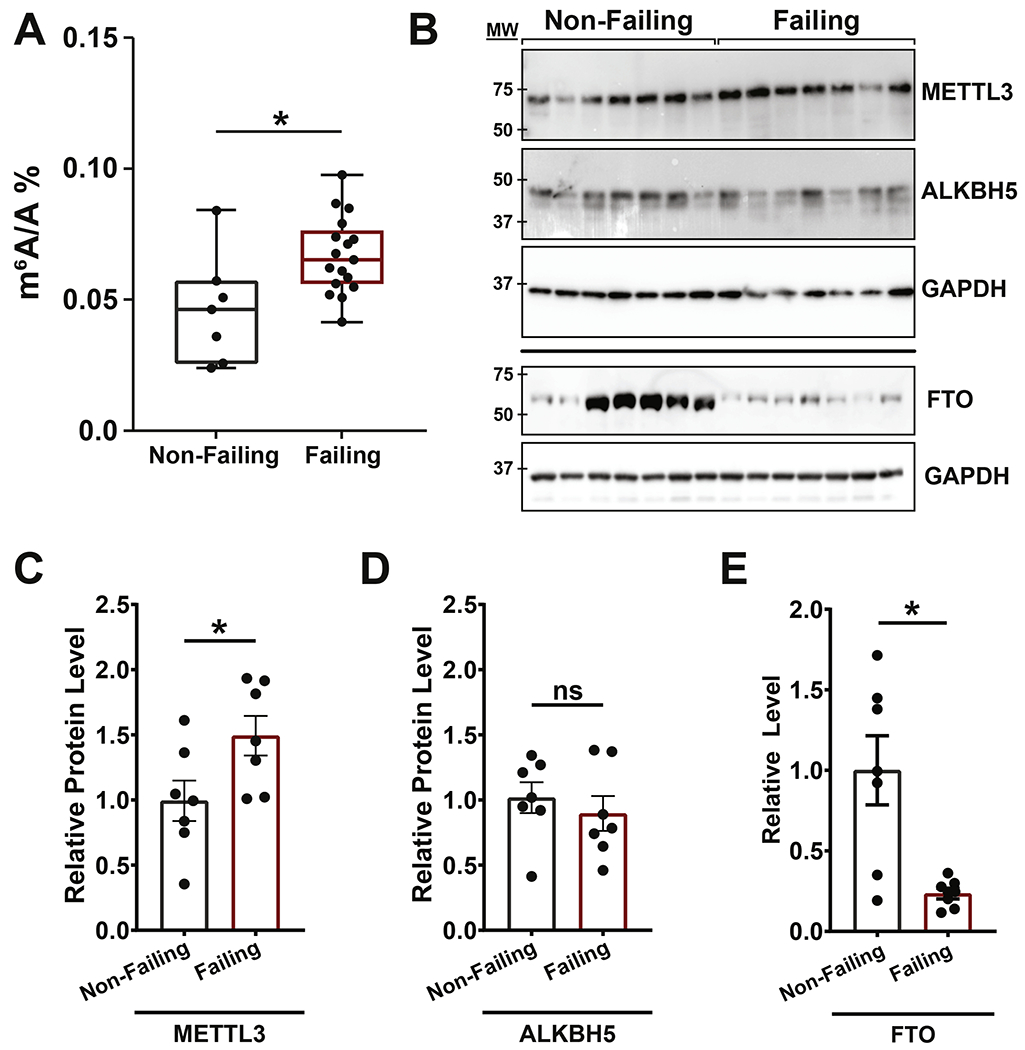
(A) RNA isolated from non-failing and failing human heart samples analyzed by UHPLC-MS/MS for global levels of methyl-6-adenosine (m6A) in relation to unmodified adenosine (A) showed a significant increase in total m6A in failing hearts (non-failing: n = 7; failing: n =17). (B) Western blots on non-failing and failing heart protein lysates measuring expression of METTL3 (m6A methylase), ALKBH5 and FTO (m6A demethylases). GAPDH and ponceau staining are shown as loading controls. (C-D) Quantification of METTL3 (C), ALKBH5 (D) and FTO (E) protein amount using GAPDH as control showing increased levels of METTL3 and decreased levels of FTO in failing hearts while ALKBH5 levels were stable (non-failing and failing: n = 7). Dots on graphs indicate individual data point from biological replicates, ns = nonsignificant, * = p < 0.05. Statistics carried out using two tailed t-test.
Regardless of global m6A content, dynamic regulation of the m6A landscape occurs to specifically affect stress-responsive mRNAs. To define the genome-wide m6A targets underlying non-ischemic heart failure in humans in an unbiased manner, we performed high throughput sequencing of mRNA fragments pulled down using antibodies against m6A-modified RNA (meRIP) (Figure 2A and Supplemental Figure 1A). Analysis of total m6A sites revealed that, despite the majority of peaks being shared between diseased and control hearts, a substantial number of modification sites were uniquely occurring in each condition (Figure 2B). Most m6A modifications were found in protein coding transcripts and were not significantly impacted by disease state (Supplemental Figure 1B and 1B’). Regardless from the assessed condition, enrichment in m6A site density occurred near the start and stop codons of cardiac transcripts (Figure 2C), while modification frequency was overall higher in coding sequences (Figure 2D). These results suggest that, while m6A increases globally in heart failure, the overall methylation distribution on transcripts is not impacted by disease state.
Figure 2. meRIP-seq analysis of total detected peaks from human non-failing and failing hearts.
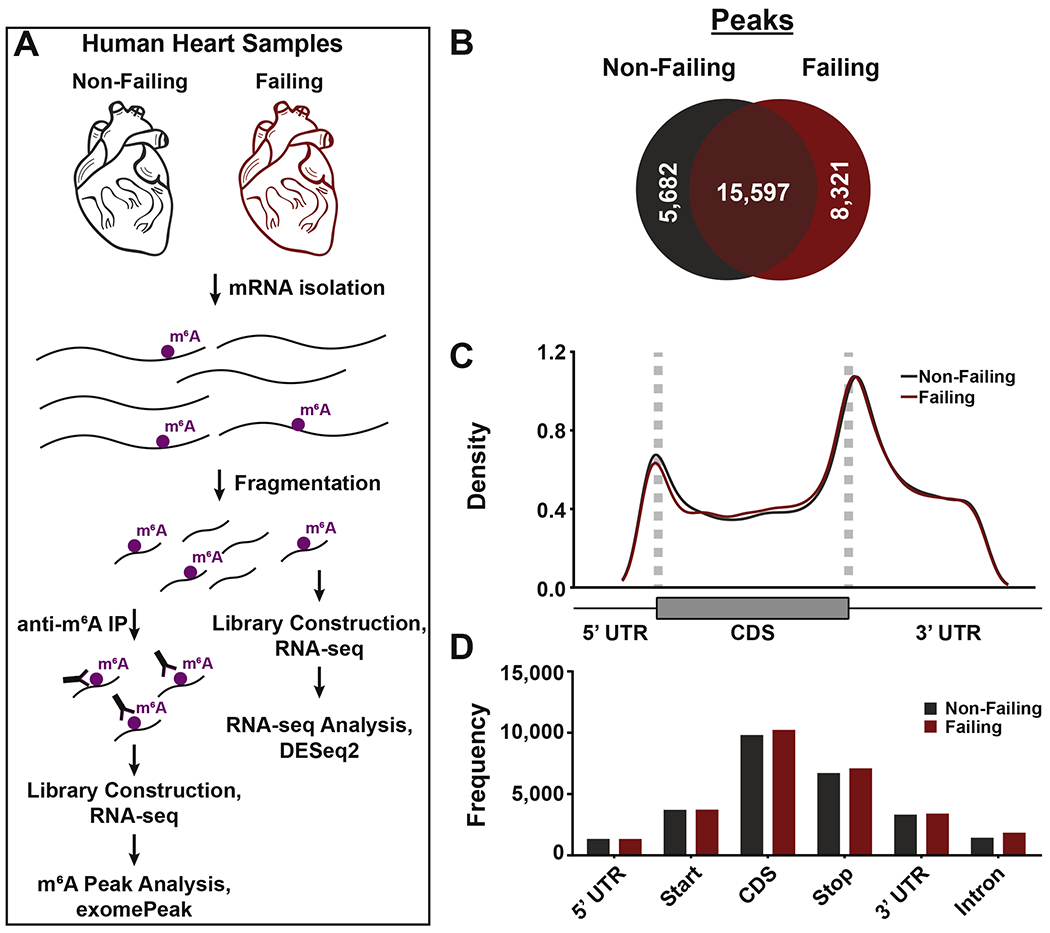
(A) General schematic for meRIP-seq pipeline. (B) Analysis of total number of m6A peaks in non-failing (grey) and failing (red) hearts. (C) Peaks detected in non-failing (grey) and failing (red) hearts were plotted via density across mRNA regions (including 5’ UTR, start codon, coding sequence (CDS), stop codon, and 3’ UTR). UTR = untranslated region. (D) Peaks detected in non-failing (grey) and failing (red) samples were plotted via frequency across the indicated mRNA regions.
m6A differentially targets subsets of transcripts in the failing heart
It is critical to discover the identity of m6A-targeted mRNAs that are responsive to disease state to define how m6A might differentially affect the transcriptome when cardiac homeostasis is perturbed. Analysis of significantly enriched sites using a logaritmic 1.5 fold change cutoff revealed that 864 peaks on 780 transcripts were characteristic of failing hearts (Figure 3A and 3B). Interestingly, we also observed a number of methylation events differentially occurring in non-failing hearts (734 peaks on 698 mRNAs), suggesting that remodeling of m6A in the cardiac transcriptome is characterized by specific enrichments in both homeostatic and pathological conditions (Figure 3A and 3B). Analysis of density and frequencies for m6A sites specifically enriched in either non-failing or failing hearts (excluding common peaks) revealed a comparatively higher amount of modifications occurring in coding sequences with disease (Figure 3C and 3D). Specifically, non-failing enriched peaks have higher density around start codons, while failing enriched peaks have higher density across coding sequences (Figure 3C). Failing samples also have more enriched peaks in total, with greater frequency observed in the coding sequence, stop codon, 3’ untranslated region (UTR), and intronic regions (Figure 3D). In-depth assessment of consensus sequences in both failing and non-failing enriched sites highlighted the expected METTL3 target site (GGAC), but found a loss of cytosine dominance in position −3 in heart failure (Figure 3E and 3E’). Gene ontology analysis for differentially m6A-regulated transcripts in heart failure further revealed that this modification targets specific subsets of mRNAs in response to stress with histone modification being the most enriched category of m6A-modified mRNAs in heart failure (Figure 3F and 3F’; Supplemental Table 1). Importantly, these enrichment categories were not overlapping with the ones obtained from the total transcript analysis (DESeq2; Supplemental Table 2). These results reinforce the idea that post-transcriptional events constitute an additional layer of regulation of gene expression that can uniquely affect specific subsets of transcripts.
Figure 3. Non-failing and failing human hearts are enriched for distinct m6A peaks.
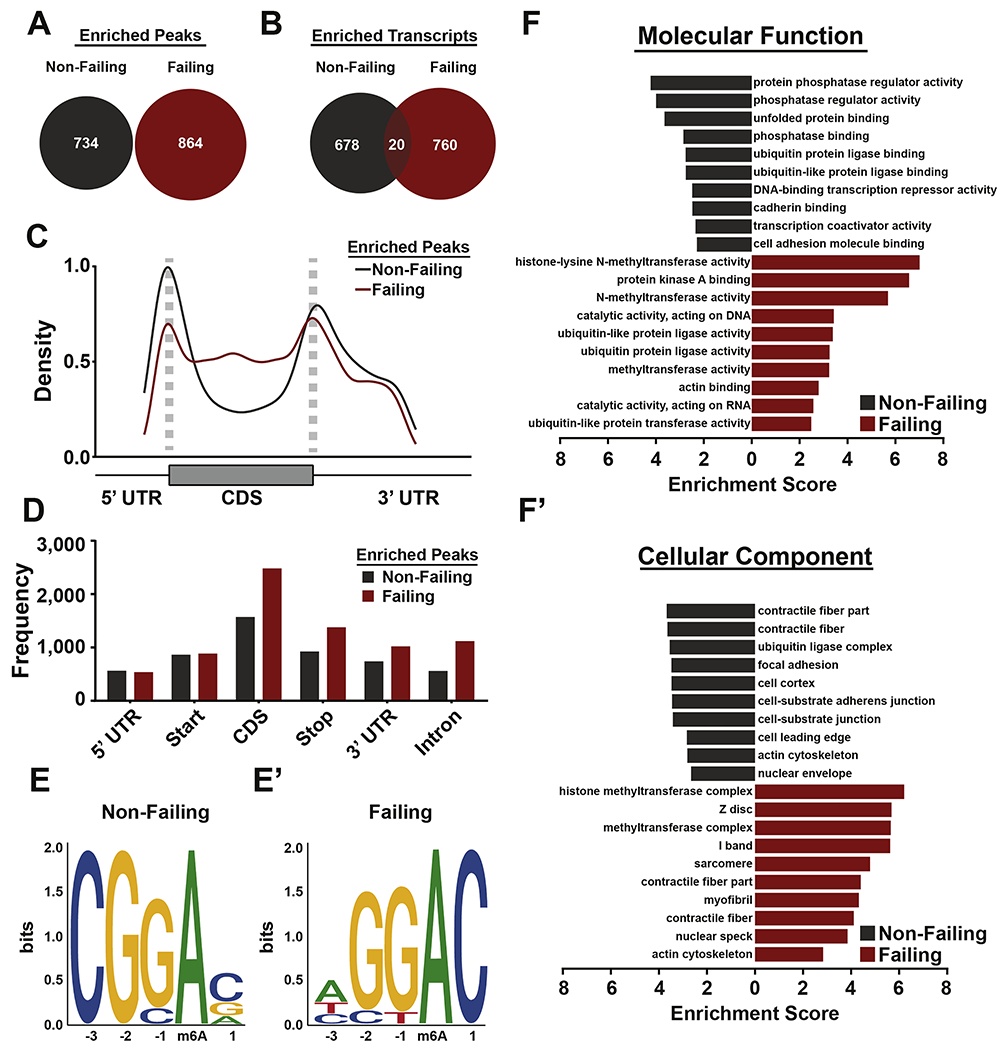
(A) Venn diagram of peaks enriched (FC > 1.5) in non-failing (gray) and failing (red) heart samples. (B) Venn diagram of transcripts with m6A peaks that were found to be m6A-targeted in nonfailing (gray), failing (red), or common between both (maroon). (C) Peaks enriched in nonfailing (grey) and failing (red) hearts were plotted via density across the indicated mRNA regions. Non-failing enriched peaks have higher density around start codons, while failing enriched peaks have higher density across coding sequences. (D) Peaks enriched in non-failing (grey) and failing (red) hearts were plotted via frequency across the indicated RNA regions. Failing samples have more enriched peaks in total, with an enrichment in frequency in coding sequence, stop codon, 3’ UTR, and intronic regions. (E, E’) Motif-based sequence analysis of peaks enriched in either non-failing (E) or failing (E’) samples through the software MEME. The motifs shown were found to be significant (non-failing: p = 1x10−38, failing p = 1x10−63). (F, F’) Gene ontology (GO) analysis of transcripts with enriched m6A peaks in either failing (grey) and non-failing (red) human samples. Enrichment score is based on a reference database of protein coding genes, and all GO categories plotted were found to have a false discovery rate (FDR) < 0.05.
Overall, these data reveal how the cardiac disease state is accompanied by changes in the m6A landscape that appear to be highly regulated in specific gene locations and functional categories of transcripts.
Conservation of stress-responsive m6A-transcripts between rat and human reveals key modification sites underlying cardiomyocyte hypertrophy
Whole-tissue analyses highlight changes underlying global organ remodeling, but reconstitution of cell-specific events is often challenging. As cardiomyocytes represent the key contractile working cell in the heart, we decided to compare our genome-wide m6A sequencing analysis performed on total human heart samples with results obtained when the same analysis was performed on isolated neonatal rat cardiomyocytes undergoing hypertrophic remodeling (Figure 4A). This experiment allowed us to narrow down our focus to 38 shared transcripts that undergo greater m6A-modification in both rat hypertrophic cardiomyocytes and human failing hearts (Figure 4B and Supplemental Table 3). Remarkably, 37% of the transcripts shared between hypertrophic cardiomyocytes and failing hearts were implicated in gene expression regulation (Figure 4C). These data reinforce our result presented in Figure 3F where we discovered histone modification as an enriched m6A-targeted pathway in heart failure and suggests an intriguing cross-talk between transcriptional and post-transcriptional gene regulation in the heart. Indeed, gene ontology analysis of methylated transcript in rat cardiomyocytes also revealed enrichment for gene expression-related pathways (Supplemental Table 4). More in-depth analysis of location conservation for the identified sites revealed 5 total transcripts with conserved sequence location containing the METTL3 consensus sequence, 4 of which fell into the gene expression category (Figure 4C - bolded genes; and Supplemental Figure 2). Genome view of two representative transcripts with conserved m6A peak location, REST and SF3B4, are presented in Figure 4D and 4E, respectively.
Figure 4. m6A-modified transcripts enriched in failing human heart samples are conserved in neonatal rat cardiomyocytes cultured in hypertrophic conditions.
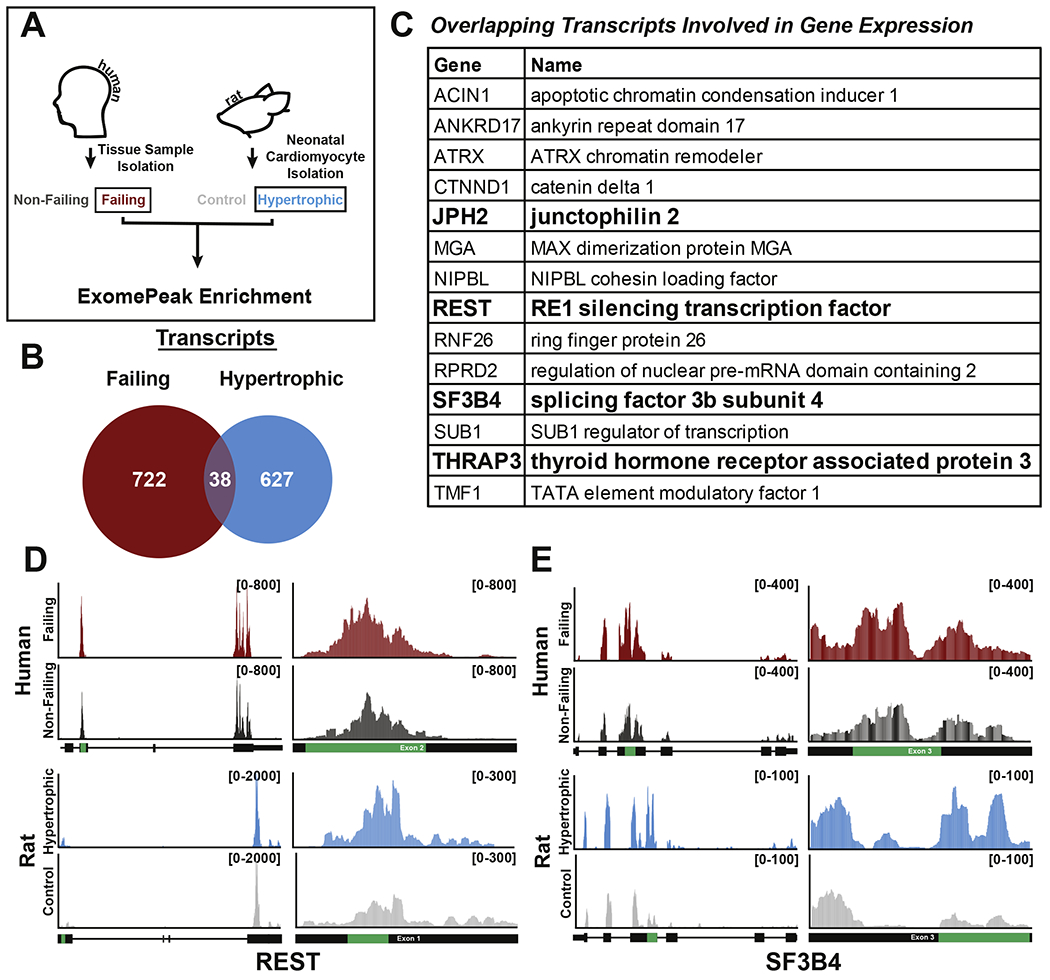
(A) General schematic for m6A peak comparison between human failing and neonatal rat cardiomyocyte hypertrophic samples. (B) Venn diagram of transcripts containing enriched m6A peaks in failing (red) and rat hypertrophic cardiomyocytes (blue) (FC >1.5 by exomePeak). Comparison found 38 total transcripts to be co-enriched. (C) Table outlining 14 transcripts co-enriched in failing and hypertrophic samples that play roles in gene expression. Genes in bold have conserved m6A peaks at the nucleotide level. (D,E) Gene view of either REST (D) or SF3B4 (E) found to contain a conserved m6A peak enriched in both human failing (top, red) and rat hypertrophic (bottom, blue) as compared to respective controls. The enriched peak calculated by exomePeak (green) is shown in relation to the total gene (left) and in relation to the specific exon (right). Read counts are found bracketed (top right, black).
Considering that enrichment of m6A-peaks were also found in the non-failing heart (indicating decreased modification on specific transcripts in heart failure), we further assessed if this phenomenon was conserved when comparing transcripts enriched in non-failing hearts and transcripts enriched in isolated rat control cardiomyocytes (Figure 5A). This analysis revealed 11 shared transcripts of which only one, CORO6, presented m6A peak location conservation across species (Figure 5B–D, Supplemental Figure 2 and Supplemental Table 5). Importantly, m6A-modification on CORO6 mRNA occurred within the 3’UTR, a known critical location for the regulation of transcript stability and translation. We therefore decided to test CORO6 mRNA and protein levels in rat cardiomyocytes and human hearts from the different conditions used in our study. Aditionally, we investigated the impact of m6A on two genes, REST and SF3B4, which methylation was enriched in failing heart and hypertorphic cardiomyocytes and found on coding regions. Interestingly, levels of either mature of precursor mRNAs were overall not significantly affected in any condition with the exception of an increase in mature REST mRNA in rat hyperotrophic cardiomyoctes (Figure 6A and 6B). However, protein content analysis showed reduced levels of CORO6 and increased level of REST with stress in both human hearts (Figure 6C and 6D) and rat cardiomyocytes (Figure 6E and 6F), although higher variability was seen with human samples. Interestingly, despite no differences were overall seen for SF3B4 in any of the tested conditions, overexpression of METTL3 in neonatal rat cardiomyocytes was sufficient to recapitulate the m6A-dependent regulation of REST and CORO6 (Figure 6G and 6H). Importantly, m6A content on REST was higher in failing hearts and hypertrophic cardiomyocytes (Figure 4D), while in the case of CORO6 we observed greater m6A content in non-failing heart and control cardiomyocytes (Figure 5D). In both of these cases protein expression was higher in condition of greater m6A content, and potentiating the m6A pathway by overexpression of METTL3 was sufficient to positively affect the translation of REST and CORO6. This suggests a direct role for this modification in post-transcriptional control of gene expression in cardiomyocytes.
Figure 5. CORO6 mRNA contains a conserved m6A peak in non-failing human and control rat samples.
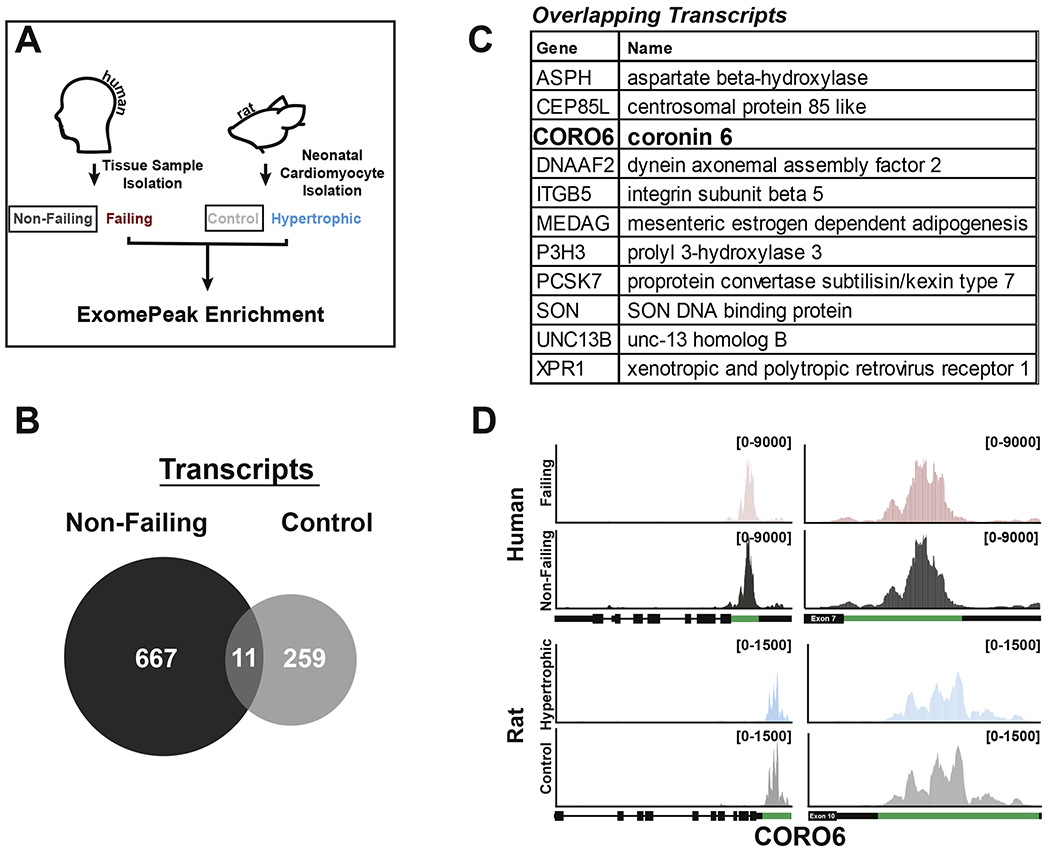
(A) General schematic for m6A peak comparison between human non-failing and neonatal rat control cardiomyocytes samples. (B) Venn diagram of transcripts containing enriched m6A peaks in non-failing (dark gray) and rat cardiomyocytes control (light gray) (FC >1.5 by exomePeak). Comparison found 11 total transcripts to be co-enriched. (C) Table outlining 11 transcripts co-enriched in non-failing and isolated rat cardiomyocytes control samples. Genes in bold have conserved m6A peaks at the nucleotide level. (D) Gene view of CORO6, which contains a conserved m6A peak enriched in both human non-failing (top, dark gray) and rat control (bottom, light gray) samples when compared to failing (red) or hypertrophic (blue) samples, respectively. The enriched peak calculated by exomePeak (green) is shown in relation to the total gene (left) and in relation to the specific exon (right). Read counts are found bracketed (top right, black).
Figure 6. m6A enrichment on REST and CORO6 mRNA impacts protein expression.
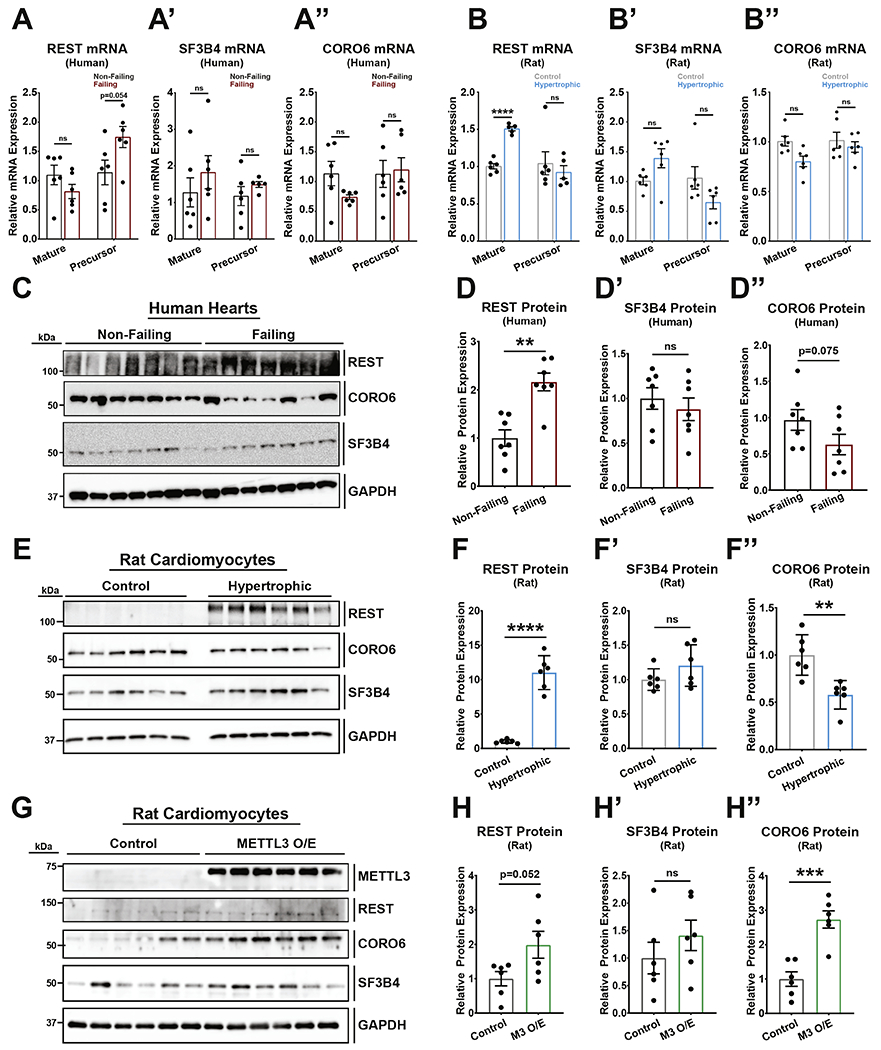
(A) qPCR against human (A) REST, (A’) SF3B4, and (A”) CORO6 show no differences in mature or precursor mRNA levels between non-failing and failing RNA samples (non-failing and failing: n=6). (B) qPCR against rat (B) REST, (B’) SF3B4, and (B”) CORO6 show an increase in REST mature mRNA but no differences in mature SF3B4, mature CORO6, and all precursor mRNAs between control and hyertrophic samples (control and hypertrophic: n=6). (C) Western blot of human non-failing and failing heart samples with antibodies against REST, CORO6, SF3B4, and control GAPDH (n=7, each). (D) Protein quantification of the human western blot (C) for REST (D), SF3B4 (D’), and CORO6 (D”). (E) Western blot of rat neonatal cardiomyocyte control and hypertrophic samples with antibodies against REST, CORO6, SF3B4, and control GAPDH (n=6, each). (D) Protein quantification of the rat western blot (E) for REST (F), SF3B4 (F’), and CORO6 (F”). (G) Western blot of rat neonatal cardiomyocyte control or METTL3 overexpression samples (M3 O/E) with antibodies against METTL3, REST, CORO6, SF3B4, and control GAPDH (n=6, each). (D) Protein quantification of the rat western blot (G) for REST (H), SF3B4 (H’), and CORO6 (H”). Dots on graphs indicate individual data point from biological replicates. Statistics carried out using two tailed t-test. ns = nonsignificant;** = p < 0.01. *** = p < 0.001. **** = p < 0.0001.
Overall, our data reveal key transcripts subjected to conserved m6A-modification during cardiomyocyte remodeling in rats and humans, therefore defining novel hypertrophy-dependent targets for post-transcriptional gene regulation in the heart.
Materials and Methods
Human Heart Samples
All human heart tissue research was approved by The Ohio State University Institutional Review Board in compliance with all relevant ethical regulations. Informed consent for tissue collection was obtained from transplant patients and families of donors. Human heart tissues used in this study were de-identified and labeled with 6-digit random reference codes. Failing samples are obtained from patients with left ventricular hypertrophy and non-ischemic heart failure, and nonfailing samples are from healthy donors without history of heart failure. Samples were obtained from The Ohio State University Cardiac Research Tissue Program or LifeLine of Ohio Organ Procurement Organization.
Cardiomyocyte isolation and treatments
Neonatal rat ventricular cardiomyocytes were isolated as previously published [11, 20]. Briefly, hearts were incubated with trypsin at 4°C overnight, followed by trypsin inhibitor and collagenase incubation at 37°C for lhr (Worthington Biochemical). Hearts were mechanically dissociated and incubated for an additional 15min, followed by re-suspension in media supplemented with fetal bovine serum and pre-plating to remove non-cardiomyocytes. Cardiomyocytes were plated on 0.1% gelatin coated dishes in the presence of 10% serum. The following day cells were washed twice and cultured in M199 media without serum (Corning). Hypertrophy was induced by culturing cardiomyocytes for 48 hours in the presence of 2% serum.
Quantitative analysis of m6A level on mRNA via UHPLC-MS/MS
Total RNA was purified with TRIzol reagents from human heart samples. Polyadenylated RNA was then isolated from total RNA with two rounds of polyA tail purification using Dynabeads® mRNA DIRECT™ kit (Thermo Scientific). rRNA was further removed from polyadenylated RNA using RiboMinus Eukaryote kit (Thermo Scientific). 50 ng polyadenylated RNA after rRNA depletion was digested by nuclease P1 (Sigma, N8630), respectively, in 20 μl of buffer containing 25 mM NaCl and 2.5 mM ZnCl2 for 1 h at 42 °C. Subsequently, 1 unit of FastAP (Thermo Scientific) in 10× FastAP buffer was added and the samples were incubated for 4h at 37 °C. After digestion, the samples were filtered (0.22 μm, Millipore) and injected into a C18 reverse-phase column coupled online to Agilent 6460 LC-MS/MS spectrometer in positive electrospray ionization mode. The nucleosides were identified by using retention time and the nucleoside to base ion mass transitions (268 to 136 for A; 282 to 150 for m6A). Quantification was performed by comparing with the standard curve obtained from pure nucleoside standards running with the same batch of samples. The m6A level was calculated as the ratio of m6A to A.
mRNA m6A MeRIP-seq
Polyadenylated RNA was purified as aforementioned. 1 ug polyadenylated RNA was adjusted to 10 ng/μl in 100 μl and fragmented using a BioRuptor ultrasonicator (Diagenode) with 30 s on/off for 30 cycles. After fragmentation, 5% of the fragmented RNA was saved as input. m6A marked fragments were enriched using EpiMark N6-Methyladenosine Enrichment Kit (NEB) following the manufacturer’s protocols and collected as IP. Both input and IP samples were subjected to library preparation using TruSeq Stranded mRNA Library Prep Kit (Illumina) following the manufacturer’s protocols. Sequencing was carried out at the University of Chicago Genomics Facility on an Illumina HiSeq 2000 machine in single-end read mode with 50 bp per read.
Data preparation for sequencing analysis
Fastq files were checked for quality using FASTQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and then processed using CutAdapt [21] to remove 10bp from the 5’ side of each read fragment. Processed fastq files were aligned using Hisat2 [22] against the Ensembl index for humans (Genome Reference Consortium Human Build 38, CRCh38.p13) or rat (INSDC Assembly, Rnor_6.0) [23] and sorted using Samtools [24].
Analysis using DESeq2
Sorted BAM files from Hisat2 were used as input for calculating transcript counts using Htseq [25]. GTF files matched to their respective Ensembl alignment indexes were used [23]. Htseq count tables were then used as input for DESeq2 [26]. Significantly enriched transcripts were calculated with a fold change greater than 1.5 and a Benjamini and Hochberg adjusted p-value less than 0.05.
Analysis of m6A peaks and motifs
Sorted BAM files from Hisat2 were used as input for exomePeak analysis [27, 28]. GTF files matched to their respective Ensembl alignment indexes were used. Peak prediction used general default settings for exomePeak, but in short, minimal peak length was set to 25bp, peak cutoff FDR was set to 0.05, and fold enrichment was set to 4. When looking for enriched peaks between sample groups (Non-failing vs Failing, Control vs Hypertrophic), fold enrichment was set to 1.5.
Output bed files from exomePeak analysis were used as input for global peak distribution analysis (RNA features, biotypes, density plots, etc.) using RNAmod, an integrated system for the annotation of mRNA modifications [29]. Analysis was done with general default settings. Motif analysis on enriched peaks were calculated using HOMER motif analysis [30] with default parameters looking for motifs ranging from 4-6bp. Sequncing data are publicly available under the GEO submission code GSE159243.
Gene Ontology analysis using Webgestalt
Gene ontology analysis on detected and enriched peaks from exomePeak data was carried out using the WEB-based Gene set analysis toolkit (Webgestalt) [31]. Over-representation analysis (ORA) in cellular component and molecular mechanism categories were carried out using the ‘genome protein coding’ reference set as background control as well as an heart-specific background.
Western blotting
Western blotting was carried out using 7.5% SDS PAGE gels for both human and neonatal rat cardiomyocytes protein extracts. Primary antibody incubation occurred at 4°C overnight and secondary incubation occurred at room temperature for 1 hour. Antibodies used are: anti CORO6 (Proteintech, 17243-1-AP, 1:500), anti GAPDH (Fitzgerald Industries, 1:20,000), anti METTL3 (Abcam, ab195352, 1:2,000), ALKBH5 (proteintech, 16837-1-AP, 1:1,000), FTO (Santa Cruz, sc98768, 1:1,000), REST (Proteintech, 22242-1-AP, 1:500) and SF3B4 (Proteintech, 10482-1-AP, 1:500). Secondary antibodies (Jackson Immunoresearch) were diluted to 1:20,000. SuperSignal™ ECL substrate (Thermo Scientific) was incubated with membranes for 2 minutes at room temperature and then exposed using the BioRad Chemidoc® imager.
mRNA expression analysis by RT-qPCR
RNA was extracted from neonatal rat cardiomyocytes or human heart samples using Trizol (Invitrogen), and reverse transcription was performed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). COR06 mRNA were quantified using SYBR green (Applied Biosystems) and the species specific primers (listed below). Quantified mRNA expression was normalized to Rpl7 (ribosomal protein L7) and expressed relative to controls. Primers used include: rat Coro6 mature mRNA (5′- TCACTGGACACACTGGCCCT-3’, 5′- GGGTGTAGTCTGGAATCTGCC-3’), rat Coro6 precursor mRNA (5’- TCACTGGACACACTGGCCCT, 5’- GCCTGGCAGCATCCCCTT-3’), rat Rest mature mRNA (5′- GTTCAACACGTTCGAACTCACAC-3’, 5′- ACGCATGTGTCGAGTTAGATGA-3’), rat Rest precursor mRNA (5’- GTTCAACACGTTCGAACTCACAC, 5’- ACGCATGTGTCGAGTTAGATGA-3’), rat Sf3b4 mature mRNA (5′- GATAGAGTCACTGGCCAGCAC-3’, 5′- TGTTGTGAGCCGAGGCTTTG-3’), rat Sfib4 precursor mRNA (5’- GATAGAGTCACTGGCCAGCAC, 5’-AGTTGTGACTGGAGTTGTGACTC-3’), rat Rpl7 (5’-AAAAGAAGGTTGCCGCTG-3’, 5′- TAGAAGTTGCCAGCTTTCC-3’), human Coro6 mature mRNA (5′- AGGCGGGGGTGCCTTCATC-3’, 5′- CCAGTGACCAGTGGGTAGTTC-3’), human Coro6 precursor mRNA (5′- AGGCGGGGGTGCCTTCATC-3’, 5′- GATGCTTCTCCTGACTATGG- 3’), human Rest mature mRNA (5′- GGAAAATGCAACTATTTTTCAGACAG-3’, 5′- AAGGACAAAGTTCACATTTATATGG-3’), human Rest precursor mRNA (5′- GGAAAATGCAACTATTTTTCAGACAG-3’, 5′- AAGCACTACTTAACTAACCACTC-3’), human Sf3b4 mature mRNA (5′- TTCTCCAGGCTGGACCAGTA-3’, 5′- CATAGTCAGCATCTTCCTCA-3’), human Sf3b4 precursor mRNA (5′- TTCTCCAGGCTGGACCAGTA-3’, 5′- GTGAATACTGCTGGGACCCT-3’), and human Rpl7 (5’-ATGCGCCAATTCCTCTTT-3’, 5’-CAGCTCTGCGAAATTCCTTC-3’).
Statistics
Results were presented as mean ± SEM. Statistical analysis between two groups was performed using an unpaired 2-tailed t test assuming equal variance. P-values < 0.05 were considered significant for RT-qPCR and western blot quantification. Bioinformatic statistics used default settings for each individual pipeline. In short, DESeq2 used a Benjamini and Hochberg adjusted p-value. exomePeak used both FDR calculation and rescaled hypergeometric test to calculate predicted and differentially enriched peaks.
Discussion
N6-methyladenosine, or m6A, is conserved in all domains of life and is the most abundant internal modification on mRNA. Increasing evidence for the biological importance of m6A in all tested mammalian systems has emerged, and the cardiovascular system is no exception. Our group has previously regulated the level of the m6A-depositing enzyme, Methyltransferase-like 3 or METTL3, to prove the critical role of this pathway for cardiomyocyte hypertrophy and cardiac homeostasis [14]. Similarly, other groups had proven the relevance of m6A in the heart in the context of cardiomyocyte ischemic and hypertrophic remodeling, and the RNA demethylase Fat mass and obesity-associated protein (FTO) has been manipulated, in addition to METTL3, to affect global m6A content [15, 17, 18]. Despite the different strategies and model systems used, all previously published work suggests an importance of m6A in the heart.
In this study we found increased levels of m6A and METTL3 in human non-ischemic failing hearts, mimicking the previously reported increase in m6A observed in ischemic and dilated cardiomyopathy [15, 17]. These results are important, as they suggest m6A addition can be a common mechanism to regulate cardiac remodeling independent of disease origin. Increased level of METTL3, the catalytically active component of the m6A methyltransferase complex, was accompanied by decreased expression of the RNA demethylase FTO. This is in agreement with a previous study showing that the reduction of this demethylase could account for increased m6A levels in the heart [15]. Altogether, our results suggest that in heart failure the balance between RNA methylation and demethylation favors m6A accumulation.
How specific transcripts are preferentially regulated by m6A following pathologic stimuli has been less clear, likely due to each study revealing hundreds of modified mRNAs and focusing on different subsets of select transcripts for follow up studies. An interesting result of our current work relates to our finding that during non-ischemic heart failure m6A-methylation occurs preferentially on a METTL3 consensus sequence which loses dominance of cytosine in position −3 from the modified adenosine. Although more work will be required to understand the mechanism behind this change, it is exciting to speculate how remodeling of the m6A methyltransferase complex in the stressed heart could lead to changes in METTL3 target preference.
In line with the previously described concept, we have observed differential enrichment of m6A on specific subsets of mRNA in heart failure. In the tested non-ischemic human failing samples, regulation of histone modification appeared as the most significant category of affected transcripts by gene ontology analysis. When cross-analyzing our failing human heart data with the m6A landscape occurring in hypertrophic rat cardiomyocytes, we find that enrichment in gene regulation remained a key conserved pathway. This is particularly interesting as it would suggest coordination between several steps of gene expression with potential feedback from post-transcriptional modifications to regulated transcription and gene expression. Importantly, by comparing such divergent sample groups, human hearts and rat cardiomyocytes, our study allowed for identification of the few critical post-trancriptional events that could be of primary importance for hypertrophic cardiomyocyte remodeling. Indeed, when analyzing total heart tissue multiple cell types are contributing to the results and the complex environement that the end-stage failing heart is subjected to can also complicate data interpretation. In addition, m6A is an evolutionarily conserved modification and knowledge on key methylated sites that are preserved across species can greatly inform on the most essential m6A-depednent regulatory processes.
Although the final outcome of m6A deposition on mRNA transcripts may vary based on mechanisms not fully understood, location of the modification on the mRNA may impact which step of transcript processing m6A is most likely to affect [32]. The 3’ untranslated sequence (UTR) is a typical regulatory region used for post-transcriptional decisions impacting translation [33–37]. Our work identified Coronin-6 (or CORO6) as a conserved transcript undergoing m6A regulation at the 3’UTR in both rat cardiomyocytes and human hearts. Interestingly, CORO6 mRNA abundance was unchanged in hypertrophic conditions but protein content decreased with hypertrophy, indicating that, indeed, this transcript is regulated at the post-transcriptional level and that m6A could play a direct role in this process. CORO6 belongs to the Coronin family of actin filament binding proteins [38]. Although the functional role of CORO6 in the heart is currently unknown, our study revealed this gene as potential novel regulatory node underlying hypertrophic cardiomyocyte remodeling. On the other hand, our analysis of two transcripts containing m6A in their coding regions, RE1-silencing transcription factor (REST) and Splicing factor 3b subunit 4 (SF3B4), showed clear impact on protein expression on REST only, suggesting a more complex and perhaps isoform-specific role of this modification on SF3B4. Importantly, REST has been previously described as a critical regulator of cardiomyocyte remodeling [39], validating the importance of our results. Altogether, our study highlighted conserved m6A-regulated events that mark stress-responses in the heart and shed light on new targets of post-transcriptional gene expression regulation during cardiac disease.
Supplementary Material
Highlights.
The m6A landscape remodels in human failing hearts
Stress responsive m6A modifications target transcripts regulating gene expression
Few critical m6A sites mark stress response in an evolutionarily conserved way
mRNA methylation affects transcript translation potential
Acknowledgments
Sources of funding:
This work was supported by NIH grant R01HL 136951 to F.A.
Non-standard abbreviations:
- (m6A)
methyl-6-adenosine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
None.
References
- [1].Backs J, Olson EN, Control of cardiac growth by histone acetylation/deacetylation, Circ Res 98(1) (2006) 15–24. [DOI] [PubMed] [Google Scholar]
- [2].McKinsey TO, E., Toward transcriptional therapies for the failing heart: chemical screens to modulate genes, Journal of Clinical Investigation (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang CM, A T; S Chang; Antos CL; Hill JA; Olson EN, Class II Histone Deacetylases Act as Signal-Responsive Repressors of Cardiac Hypertrophy, Cell (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liang Q, De Windt LJ, Witt SA, Kimball TR, Markham BE, Molkentin JD, The transcription factors GATA4 and GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo, J Biol Chem 276(32) (2001) 30245–53. [DOI] [PubMed] [Google Scholar]
- [5].Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, Rindt H, Gorczynski RJ, Olson EN, Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors, J Biol Chem 278(31) (2003) 28930–7. [DOI] [PubMed] [Google Scholar]
- [6].Wallner M, Eaton DM, Berretta RM, Liesinger L, Schittmayer M, Gindlhuber J, Wu J, Jeong MY, Lin YH, Borghetti G, Baker ST, Zhao H, Pfleger J, Blass S, Rainer PP, von Lewinski D, Bugger H, Mohsin S, Graier WF, Zirlik A, McKinsey TA, Birner-Gruenberger R, Wolfson MR, Houser SR, HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction, Sci Transl Med 12(525) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Seimetz J, Arif W, Bangru S, Hernaez M, Kalsotra A, Cell-type specific polysome profiling from mammalian tissues, Methods 155 (2019) 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].van Heesch S, Witte F, Schneider-Lunitz V, Schulz JF, Adami E, Faber AB, Kirchner M, Maatz H, Blachut S, Sandmann CL, Kanda M, Worth CL, Schafer S, Calviello L, Merriott R, Patone G, Hummel O, Wyler E, Obermayer B, Mucke MB, Lindberg EL, Trnka F, Memczak S, Schilling M, Felkin LE, Barton PJR, Quaife NM, Vanezis K, Diecke S, Mukai M, Mah N, Oh SJ, Kurtz A, Schramm C, Schwinge D, Sebode M, Harakalova M, Asselbergs FW, Vink A, de Weger RA, Viswanathan S, Widjaja AA, Gartner-Rommel A, Milting H, Dos Remedios C, Knosalla C, Mertins P, Landthaler M, Vingron M, Linke WA, Seidman JG, Seidman CE, Rajewsky N, Ohler U, Cook SA, Hubner N, The Translational Landscape of the Human Heart, Cell 178(1) (2019) 242–260 e29. [DOI] [PubMed] [Google Scholar]
- [9].Doroudgar S, Hofmann C, Boileau E, Malone B, Riechert E, Gorska AA, Jakobi T, Sandmann C, Jurgensen L, Kmietczyk V, Malovrh E, Burghaus J, Rettel M, Stein F, Younesi F, Friedrich UA, Mauz V, Backs J, Kramer G, Katus HA, Dieterich C, Volkers M, Monitoring Cell-Type-Specific Gene Expression Using Ribosome Profiling In Vivo During Cardiac Hemodynamic Stress, Circ Res 125(4) (2019) 431–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guo Q, Zhang Y, Zhang S, Jin J, Pang S, Wu X, Zhang W, Bi X, Zhang Y, Zhang Q, Jiang F, Genome-wide translational reprogramming of genes important for myocyte functions in overload-induced heart failure, Biochim Biophys Acta Mol Basis Dis 1866(3) (2020) 165649. [DOI] [PubMed] [Google Scholar]
- [11].Accornero F, Schips TG, Petrosino JM, Gu SQ, Kanisicak O, van Berlo JH, Molkentin JD, BEX1 is an RNA-dependent mediator of cardiomyopathy, Nat Commun 8(1) (2017) 1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G, Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq, Nature 485(7397) (2012) 201–6. [DOI] [PubMed] [Google Scholar]
- [13].Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR, Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons, Cell 149(7) (2012) 1635–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M, Hanna JH, van Berlo JH, Accornero F, The N(6)-Methyladenosine mRNA Methylase METTL3 Controls Cardiac Homeostasis and Hypertrophy, Circulation 139(4) (2019) 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mathiyalagan P, Adamiak M, Mayourian J, Sassi Y, Liang Y, Agarwal N, Jha D, Zhang S, Kohlbrenner E, Chepurko E, Chen J, Trivieri MG, Singh R, Bouchareb R, Fish K, Ishikawa K, Lebeche D, Hajjar RJ, Sahoo S, FTO-Dependent N(6)-Methyladenosine Regulates Cardiac Function During Remodeling and Repair, Circulation 139(4) (2019) 518–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Song H, Feng X, Zhang H, Luo Y, Huang J, Lin M, Jin J, Ding X, Wu S, Huang H, Yu T, Zhang M, Hong H, Yao S, Zhao Y, Zhang Z, METTL3 and ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes, Autophagy 15(8) (2019) 1419–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kmietczyk V, Riechert E, Kalinski L, Boileau E, Malovrh E, Malone B, Gorska A, Hofmann C, Varma E, Jurgensen L, Kamuf-Schenk V, Altmuller J, Tappu R, Busch M, Most P, Katus HA, Dieterich C, Volkers M, m(6)A-mRNA methylation regulates cardiac gene expression and cellular growth, Life Sci Alliance 2(2) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Berulava T, Buchholz E, Elerdashvili V, Pena T, Islam MR, Lbik D, Mohamed BA, Renner A, von Lewinski D, Sacherer M, Bohnsack KE, Bohnsack MT, Jain G, Capece V, Cleve N, Burkhardt S, Hasenfuss G, Fischer A, Toischer K, Changes in m6A RNA methylation contribute to heart failure progression by modulating translation, Eur J Heart Fail 22(1) (2020) 54–66. [DOI] [PubMed] [Google Scholar]
- [19].Dorn LE, Tual-Chalot S, Stellos K, Accornero F, RNA epigenetics and cardiovascular diseases, J Mol Cell Cardiol 129 (2019) 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dorn LE, Petrosino JM, Wright P, Accornero F, CTGF/CCN2 is an autocrine regulator of cardiac fibrosis, J Mol Cell Cardiol 121 (2018) 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kehat I, Davis J, Tiburcy M, Accornero F, Saba-El-Leil MK, Maillet M, York AJ, Lorenz JN, Zimmermann WH, Meloche S, Molkentin JD, Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth, Circ Res 108(2) (2011) 176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim D, Paggi JM, Park C, Bennett C, Salzberg SL, Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype, Nat Biotechnol 37(8) (2019) 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM, Bennett R, Bhai J, Billis K, Boddu S, Cummins C, Davidson C, Dodiya KJ, Gall A, Giron CG, Gil L, Grego T, Haggerty L, Haskell E, Hourlier T, Izuogu OG, Janacek SH, Juettemann T, Kay M, Laird MR, Lavidas I, Liu Z, Loveland JE, Marugan JC, Maurel T, McMahon AC, Moore B, Morales J, Mudge JM, Nuhn M, Ogeh D, Parker A, Parton A, Patricio M, Abdul Salam AI, Schmitt BM, Schuilenburg H, Sheppard D, Sparrow H, Stapleton E, Szuba M, Taylor K, Threadgold G, Thormann A, Vullo A, Walts B, Winterbottom A, Zadissa A, Chakiachvili M, Frankish A, Hunt SE, Kostadima M, Langridge N, Martin FJ, Muffato M, Perry E, Ruffier M, Staines DM, Trevanion SJ, Aken BL, Yates AD, Zerbino DR, Flicek P, Ensembl 2019, Nucleic Acids Res 47(D1) (2019) D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome S Project Data Processing, The Sequence Alignment/Map format and SAMtools, Bioinformatics 25(16) (2009) 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Anders S, Pyl PT, Huber W, HTSeq--a Python framework to work with high-throughput sequencing data, Bioinformatics 31(2) (2015) 166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol 15(12) (2014) 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Meng J, Cui X, Rao MK, Chen Y, Huang Y, Exome-based analysis for RNA epigenome sequencing data, Bioinformatics 29(12) (2013) 1565–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Meng J, Lu Z, Liu H, Zhang L, Zhang S, Chen Y, Rao MK, Huang Y, A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package, Methods 69(3) (2014) 274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu Q, Gregory RI, RNAmod: an integrated system for the annotation of mRNA modifications, Nucleic Acids Res 47(W1) (2019) W548–W555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK, Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities, Mol Cell 38(4) (2010) 576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B, WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs, Nucleic Acids Res 47(W1) (2019) W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, Haripal B, Zucker-Scharff I, Moore MJ, Park CY, Vagbo CB, Kussnierczyk A, Klungland A, Darnell JE Jr., Darnell RB, A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation, Genes Dev 29(19) (2015) 2037–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Muhlrad DP, R., Aberrant mRNAs with extended 3′ UTRs are substrates for rapid degradation by mRNA surveillance, RNA (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lai EC, Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation, Nat Genet 30(4) (2002) 363–4. [DOI] [PubMed] [Google Scholar]
- [35].Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J, Involvement of microRNA in AU-rich element-mediated mRNA instability, Cell 120(5) (2005) 623–34. [DOI] [PubMed] [Google Scholar]
- [36].Chen CS, AB, AU-rich elements: characterization and importance in mRNA degradation, Trends in Biochemical Sciences (1995). [DOI] [PubMed] [Google Scholar]
- [37].N.S.A. Chen C.; Xu, mRNA Decay Mediated by Two Distinct AU-Rich Elemeents from c-fos and Granulocyte-Macrophage Colony-Stimulating Factor Transcripts: Different Deadenylation Kinetics and Uncoupling from Translation, Molecular and Cellular Biology (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].de Hostos EL, The coronin family of actin-associated proteins, Trends in Cell Biology (1999). [DOI] [PubMed] [Google Scholar]
- [39].Kuwahara K, Saito Y, Takano M, Arai Y, Yasuno S, Nakagawa Y, Takahashi N, Adachi Y, Takemura G, Horie M, Miyamoto Y, Morisaki T, Kuratomi S, Noma A, Fujiwara H, Yoshimasa Y, Kinoshita H, Kawakami R, Kishimoto I, Nakanishi M, Usami S, Saito Y, Harada M, Nakao K, NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function, EMBO J 22(23) (2003) 6310–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.