

Abstract
Introduction:
Children with high-grade gliomas (pHGGs) represent a clinical population in substantial need of new therapeutic options given the inefficacy and toxicity of current standard-of-care modalities. Although immunotherapy has emerged as a promising modality, it has yet to elicit a significant survival benefit for pHGG patients. While preclinical studies address a variety of underlying challenges, translational clinical trial design and management also need to reflect the most updated progress and lessons from the field.
Areas Covered:
The authors will focus our discussion on the design of clinical trials, the management of potential toxicities, immune monitoring, and novel biomarkers. Clinical trial design should integrate appropriate patient populations, novel and preclinically optimized trial design, and logical treatment combinations, particularly those which synergize with standard of care modalities. However, there are caveats due to the nature of immunotherapy trials, such as patient selection bias, evidenced by the frequent exclusion of patients on high-dose corticosteroids. Robust immune-modulating effects of modern immunotherapy can have toxicities. As such, it is important to understand and manage these, especially in pHGG patients.
Expert Opinion:
Adequate integration of these considerations should allow us to effectively gain insights on biological activity, safety, and biomarkers associated with benefits for patients.
Keywords: biological correlates, clinical trial design, diffuse intrinsic pontine glioma, diffuse midline glioma, high-grade glioma, immunotherapy, toxicity
1. Introduction
Radiotherapy and chemotherapy for pediatric high-grade gliomas (pHGGs) are both associated with substantial toxicities and are generally unable to prevent tumor recurrence after tumor resection [1]. Unlike in the adult setting, pHGGs do not benefit from treatment with temozolomide (TMZ) plus radiotherapy, although radiotherapy followed by either the combination of TMZ and lomustine or TMZ, irinotecan, and bevacizumab [2] may elicit a modest increase in overall survival (OS) in some cases [3,4]. While pHGGs are far less common than low-grade gliomas (LGGs) and account for approximately 11% of primary pediatric central nervous system (CNS) tumors, the three year OS is just 22% [3,5].
Despite decades of innovative clinical studies, pHGG remains universally fatal. Recent successes in treating various hematological malignancies and solid tumors with immunotherapeutic strategies have driven a rapid increase in immunotherapy trials for patients with adult and pediatric CNS tumors [6,7]. Among many strategies, there are four immunotherapeutic strategies currently being explored for pHGG that we consider in this review. The first is checkpoint blockade, which uses targeted antibodies to inhibit the activity of molecular pathways that negatively regulate T cell activity (reviewed in [8]). Among the agents undergoing evaluation, pembrolizumab and nivolumab are both monoclonal antibodies targeting programmed cell death protein 1 (PD-1), while ipilimumab targets cytotoxic T-lymphocyte protein 4 (CTLA-4). Several studies have evaluated immune checkpoint inhibitors (ICI) alongside standard-of-care for adult and pediatric HGG but these therapies have not been FDA approved for usage in either setting [6,9–11]. The second strategy is vaccine-based therapies that deliver immunogenic glioma-associated peptides exogenously with immune adjuvant or loaded onto antigen-presenting cells (reviewed in [12–14]). Most recently, a peptide plus innate immune adjuvant strategy was exploited to generate an immune response against the histone variant 3.3 (H3.3) K27M mutation in a clinical trial for diffuse midline glioma (DMG) (NCT02960230) [15]. A third strategy is adoptive cell therapy (ACT) in which autologous T cells or NK cells are engineered ex vivo to target a tumor antigen before administration to the patient [reviewed in [16]. Chimeric antigen receptor T cells (CAR T) which target surface tumor antigens are an example of this strategy that is being evaluated for adult and pediatric HGG [17–22]. Finally, oncolytic virus strategies involve intra-tumoral administration of replication-competent viruses that have been engineered to selectively infect and kill glioma cells. This strategy has significant potential both because it can directly lyse tumor cells and because the active viral infection recruits antigen-presenting cells to the tumor site that can present tumor-associated antigens and provoke an adaptive immune response (reviewed in [23,24]). Currently, oncolytic virotherapy is being evaluated in clinical trials for various pediatric CNS malignancies, including diffuse intrinsic pontine glioma (DIPG) (reviewed in [25,26]).
Progress in molecular profiling of pHGG has revealed that the disease category includes a genetically and phenotypically heterogeneous collection of tumors [1]. Notably, this heterogeneity provides critical opportunities for the development of targeted immunotherapies to exploit pHGG sub-groups. Genomic and epigenomic analyses have identified specific driver mutations associated with age, tumor location, immune cell infiltration, and survival outcome [27–29]. These findings reaffirm the need for narrowly defined subgroups when evaluating prospective therapies and has been extensively reviewed [1,30–32]. With the ongoing characterization of a vast repository of genomic, epigenomic, and protein-level alterations characterizing pHGG subtypes and influencing prognosis, precisely targeted immunotherapies herald immense potential to improve upon standard-of-care treatment for pHGG.
While immunotherapies for HGG continue to garner excitement, no immunotherapy has managed to elicit a significant survival benefit for pHGG patients. The challenges facing development of immunotherapies for pHGG have been comprehensively reviewed and include disease heterogeneity [1], homing and retention of lymphocytes to the tumor microenvironment [33,34], paucity of targetable antigens due to lower mutational burden, and an immature peripheral immune system (reviewed in [35]). Ongoing and future clinical trials will continue to evaluate innovative strategies to overcome the unique challenges facing immunotherapies for pHGG. As these clinical trials are conducted, there is a critical need for consensus guidance on trial design and evaluation in order to limit variability based on different institutional protocols. As pHGG is a relatively rare disease, small numbers of enrollees and the narrow tumor subgrouping required in each trial compound the need for reliable comparison of patient outcomes between different institutions.
In addition to in-depth understanding of these scientific challenges and mechanisms, when it comes to clinical trial design and management, there are important considerations. In this review, we have established a multi-institutional team of experts in pHGG immunotherapy to provide consensus guidance on the design of clinical trials, management of potential toxicities associated with immunotherapy, and immune monitoring.
2. Clinical trial design
The ultimate development of safe and effective immunotherapy for children with CNS tumors requires thoughtful study design and managements. Typically, this is first done through clinical trials to assess real-world toxicity and efficacy with careful consideration of eligibility criteria, response endpoints, and dose escalation. When applied to immunotherapy for CNS tumors, each of these considerations must be specifically addressed through the lens of both immune cell infiltration of the CNS and systemic side effects, which is distinct from more traditional systemic treatments.
2.1. Novel trial design
The primary purpose of an early-phase clinical trial is to determine the appropriate dose of a new therapeutic agent. The ideal therapeutic index is usually obtained by administering a relatively high-dose that yields maximum tolerated doses (MTD) such that the probability of dose limiting toxicities (DLT) is no more than a pre-determined percentage. The MTD is often the recommended Phase II dose (RP2D) under the assumption that higher doses are likely to be more effective [36]. The endpoints for RP2D and DLT become less clear in immunotherapeutic trials as immunotherapy drug-related toxicities have been less well-defined than those for traditional cytotoxic therapies. As a result, it is conceivable that the traditional dose escalation approach may need to be altered or reconsidered for future trials.
The standard method of 3+3 dose escalation has been largely replaced by more innovative designs, such as the adaptive Bayesian Phase I design [37,38], which can lead to greater flexibility and efficiency by utilizing results accumulated during the trial. The continual reassessment method (CRM) starts at the dose level where toxicity rate is closest to the target toxicity level and restricts to no more than one dose level increase at a time. Alternatively, the modified CRM assigns more than one subject at a time to each dose level, reducing the duration, toxicity, and severity of toxicity in the trial [39]. Other designs, such as the extended CRM and the escalation with overdose control (EWOC), further attempt to reduce the numbers of patients required at each dose level while providing accurate estimates of toxicity.
As with standard cytotoxic agents, rapid and efficient escalation of immunotherapeutic approaches must leverage novel statistical designs, especially given the lack of traditional MTD seen in many pHGG-related immunotherapy trials.
2.2. Factors in patient selection criteria
The selection of patients with pHGG for enrollment on an immunotherapy clinical trial is complex, necessitating that a multitude of factors should be carefully considered, given the risk for pseudoprogression-related toxicity and the immunomodulatory effects afforded by many supportive therapies. Key variables to be considered include appropriate disease status (newly diagnosed vs. recurrent), location of disease, tumor phenotype and expression of targetable biomarkers, and use of steroids.
2.2.1. Diagnosis and phenotype
Traditionally, pediatric gliomas were grouped based on histopathology. Advances in genetic and epigenetic profiling of pHGG have enabled separation into two distinct groups: diffuse midline gliomas with H3 K27M mutations (e.g. brainstem, thalamus, and spine) and hemispheric pHGG [40]. DIPG can still be considered a clinical diagnosis as determined by consensus radiographic features including a T1-hypointense and T2-hyperintense tumor involving at least 50% of the pons by cross-sectional area[41]. Previously, the role for neurosurgery in the diagnosis of DIPG was to alleviate signs of intracranial pressure (ICP) and obtain a biopsy in cases of uncertain diagnosis. Recent clinical studies have demonstrated the safety and feasibility of pediatric brainstem biopsy, particularly stereotactic biopsy, enabling improved biological understanding and the development of new therapies[42]. Stereotactic biopsy requires immense skill and it may not be possible for every center to offer biopsy to patients. Therefore, not all trials will require a biopsy prior to enrollment for newly diagnosed DIPG patients. However, trials with biologically-targeted therapy, such as those targeting the histone 3 mutated tumors, should require a biopsy prior to enrollment.
Finally, these tumors are characterized by a high degree of tumor heterogeneity which complicates the selection of appropriate targets. The heterogeneous nature of antigen expression could theoretically alter the efficacy of therapies targeting a single antigen or molecule, as occurs with ACT. For example, CAR T cells may be ineffective if some pHGG target cells do not express the antigen, downregulate the antigen, or mutate in a form of antigen escape, as has been observed in CD19- relapsed B-ALL following CAR T therapy[43,44]. In addition, gliomas employ multiple mechanisms of immune suppression. Gliomas express inhibitory ligands that induce anergy and apoptosis of cytotoxic lymphocytes, along with immune checkpoints that impair the anti-tumoral immune response [45]. They also elicit tumoral infiltration by immunosuppressive cells, likely impacting pHGG immunogenicity. Some hypothesize that the emergence and selection of specific genetic alterations could be related to cells that evade the initial immune response [45]. For instance, pediatric glioblastoma (GBM) has up to 1.9 fold decreased PD-L1 expression compared to non-tumor controls and lower levels of expression of the T cell activation markers HLA-DR and CD64 [46]. Overall, understanding and including the appropriate patient phenotype is far more complex than previously understood, but may have substantive impacts on the efficacy of a candidate immunotherapeutic approach.
2.2.2. Newly diagnosed vs. recurrent
The current standard of care for the treatment of pHGG includes maximal tumor resection followed by focal radiation therapy. Newly-diagnosed patients are treated with resection and biopsy which allows for tissue phenotyping at the time of initial diagnosis. Immunotherapy approaches that require fresh tissue procurement are most easily considered at upfront resection following the initial diagnosis. Furthermore, in resectable HGG, active disease burden is it at its lowest following optimal resection and some trials have shown that survival outcomes were improved in patients with minimal residual disease after optimal tumor resection [47,48]. However, some patients are not offered immunotherapy until relapse given the investigational nature of such treatments. In most of these cases, a biopsy is not performed to determine changes in tumor phenotype. Mohm et al. demonstrated that tumor infiltrating lymphocytes (TILs) in recurrent GBM had an enlarged proportion of CD8+ and CD4+ effector memory T cells and lower CD8+ transitional memory TILs when compared with primary GBM [49]. In addition to a changing tumor phenotype at recurrence, patients are generally closer to tumor burden-related clinical deterioration and also may not have the time needed to generate personalized approaches. Unfortunately, enrollment on immunotherapeutic trials is logistically challenging at initial diagnosis and may consign patients to additional interventions compared with standard of care.
2.2.3. Location, accessibility of tumor and drug delivery
When designing an immunotherapy clinical trial, it is also important to consider the location of the tumor and how this will affect therapeutic response and toxicity. While the blood-brain-barrier (BBB) was historically understood to prevent immune cells from accessing tumors, it has been shown that immune responses against CNS tumors do occur. Both human studies and animal models of intracranial tumors show that strong antitumor immune responses are able to control tumor growth [50]. Further, soluble antigens are able to drain from the cerebrospinal fluid (CSF) space to the cervical lymph nodes where they are presented to T cells by dendritic cells (DC) [51]. An obstacle to effective treatment is the likely impedance to effector cell infiltration. To combat this, some immunotherapy trials utilize adjuvant treatment like poly-ICLC, a toll-like receptor 3 (TLR3) and melanoma differentiation-associated protein 5 (MDA5) agonist. TLR3 is the most abundant TLR expressed by astrocytes and microglial cells and its activation can enhance homing by inducing pro-inflammatory cytokines and chemokines [52,53]. Some studies, especially those investigating CAR T cells for pediatric gliomas, attempt to bypass the BBB via direct injection into the tumor bed or ventricular system (NCT03500991, 03638167, 04185038). Theoretically, this approach would enhance responses due to direct invasion of the tumor with effector cells. It is unknown whether direct intra-tumoral therapy results in sufficient recruitment of the innate and systemic immune system to develop a memory response. Furthermore, while indwelling catheters are well tolerated and widely used in pediatric patients, the added risks of infection and potential bleeding events should always be considered.
2.2.4. Corticosteroids
Many patients with pHGG will receive corticosteroids at some point during their treatment course for amelioration of tumor edema-related symptoms. However, this could have a deleterious effect on the efficacy of immunotherapy given that steroids decrease migration of leukocytes into inflamed tissues, reduce peripheral blood counts of all leukocyte subsets, especially T cells, and decrease IgG levels [54]. Adult data studying patient cohorts found that dexamethasone-induced anti-proliferative effects may confer protection from radiotherapy- and chemotherapy-induced genotoxic stress and therefore may decrease the effectiveness of treatment and shorten survival time for GBM patients.[55] Data from the PNOC-007 study, a pilot multi-center trial of H3.3K27M peptide vaccine in children with DMG (NCT02960230), demonstrated detrimental effects of steroid use on the induction of vaccine-specific T cell response, observing decreased expansion of vaccine-reactive CD8+ T cells in dexamethasone treated patients [15,56,57]. In vitro and animal models describe an antagonistic relationship between ICI and corticosteroids. This has been corroborated in human studies demonstrating decreased median OS in patients receiving ICI, although the contribution of corticosteroids to immunotherapy antagonism still needs to be well-defined [58–61].
2.2.5. Exclusion criteria
A final set of considerations for immunotherapy clinical trial design concerns exclusion criteria. Uncontrolled infections that may impair immunity or active autoimmune disease are frequently considered to exclude patients from immunotherapy trials. Examples of uncontrolled infections or conditions that impair immunity include hepatitis and HIV, where augmented immune responses may lead to undesirable off-target activation of infectious complications. Finally, some trials will also exclude patients with bulky tumors from consideration, such as those with pre-determined large tumors or those that have induced uncal herniation or midline shift, but this is not unanimously performed. These patients may require high-dose corticosteroid and have rapid deterioration in the context of potential immune cell infiltration that may become life-threatening. Moreover, these conditions may also introduce significant patient selection bias and caution should be applied when interpreting results by comparing with historical control data.
2.2.6. Neoadjuvant and adjuvant immunotherapy
Neoadjuvant design provides an opportunity to evaluate the effects of pre-surgically administered therapeutic agents in the resected tumor. Another advantage of neoadjuvant immunotherapy with standard-of care (SOC) is that it could provide an early start for a systemic immune response that can be sustained with adjuvant immunotherapy post-surgery and radiation therapy (RT). Therapy delivered in the adjuvant, post-surgery phase may benefit from the bulk of the tumor being resected, leaving a smaller tumor burden for T cells to eliminate and a denuded local immunosuppressive environment. In a recent clinical trial, patients who received neoadjuvant pembrolizumab with continued adjuvant therapy following surgery had significantly extended OS compared to patients that received adjuvant pembrolizumab post-surgery [62]. This study demonstrates that neoadjuvant PD-1 blockade induced infiltration of interferon-γ (IFN-γ) producing, PD-1-suppressed CD8+ T cells in the tumor microenvironment (TME) that was essential for generation of a systemic immune response [62]. Another phase II clinical trial with neoadjuvant nivolumab demonstrated similar immunological impacts [63]. Given that immune checkpoint blockade used primarily as an adjuvant therapy has not shown promising results in GBM, these findings highlight the importance of timing for the success of immunotherapy.
2.3. Combination therapy with radiation or chemotherapy
While some immunotherapeutic strategies have induced significant and remarkable responses [64–69], they have not yet dramatically improved the landscape of CNS tumor treatment. Thus, in addition to optimization, future strategies are likely to benefit from rational combinatorial approaches. Although a complete discussion of varying combinations with immunotherapy is beyond the scope of this review, utilization in the context of standard of care therapies, such as radiation and chemotherapy, will be discussed below.
Recent studies have expanded our knowledge of the complex effects of RT on the immune system. Radiation-induced cell death can facilitate antigen release and promote activation of the adaptive immune system through improved antigen-presenting cell (APC) cross-presentation, DC function, enhanced T cell priming, and increased infiltration of T cells into the TME [70,71]. In rare cases, local RT can also induce a systemic anti-tumor response that leads to regression of non-irradiated metastatic tumors at a distant site, known as the abscopal effect [71,72]. In contrast, localized RT can also induce the production of cytokines and chemokines that can recruit immunosuppressive cells such as myeloid-derived suppressor cells (MDSC) and regulatory T (Treg) cells into the TME [73]. In this section, we will discuss both positive and negative effects of these standard-of-care modalities. Figure 1 summarizes impacts of RT on the anti-tumor immune response.
Figure 1. Immunomodulatory effects of Radiation and Chemotherapy.
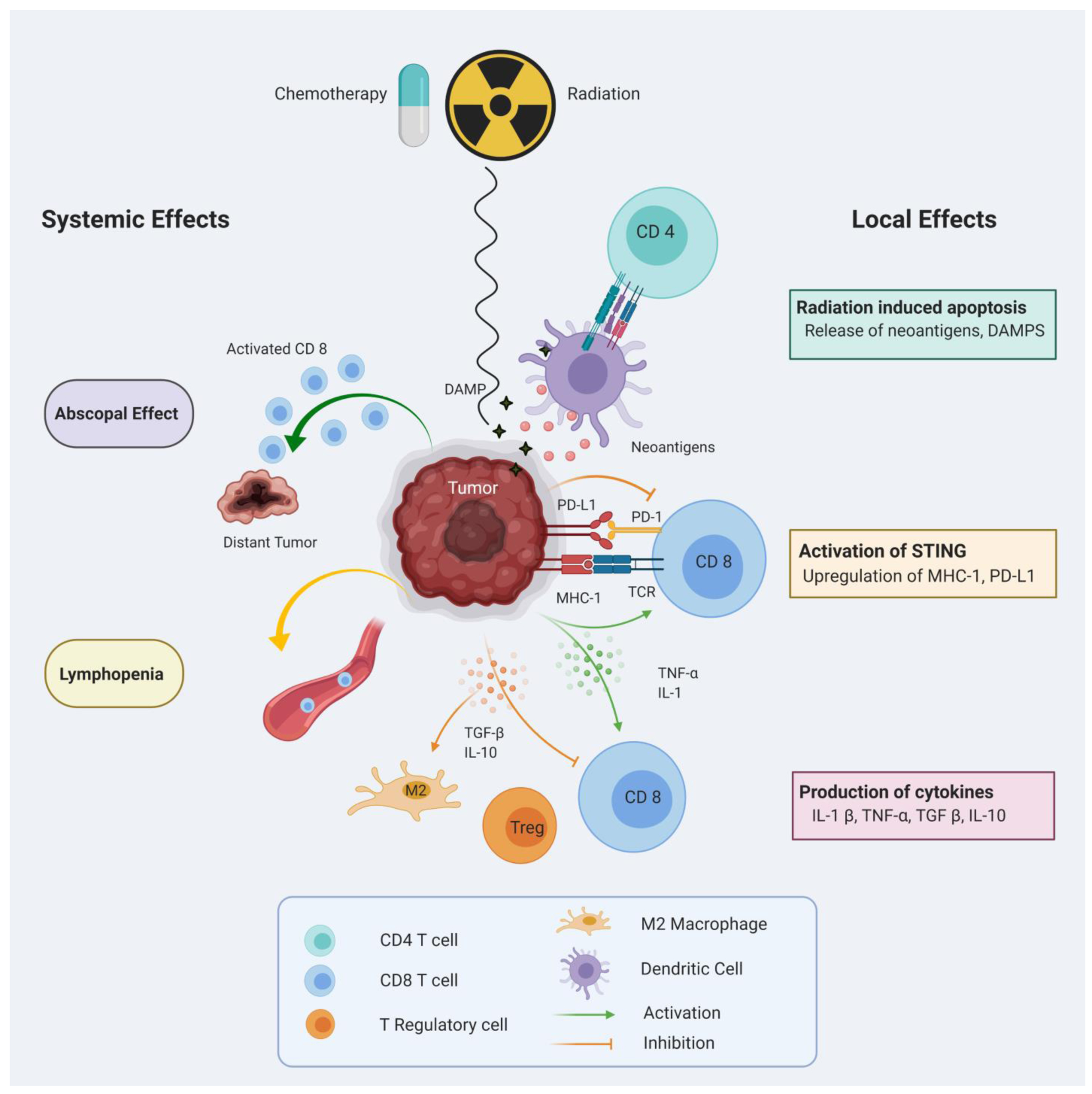
Cancer cell death induced by radiation or chemotherapy can release neoantigens and damage-associated molecular pattern molecules (DAMPS) into the tumor microenvironment (TME) which promote priming and activation of T cells. Up-regulation of surface molecules, such as MHC-I, facilitates T cell-mediated cytotoxicity. Activated T cells can mediate an abscopal effect on distant non-irradiated tumors. Potential immunosuppressive effects of radiation and chemotherapy include systemic lymphopenia, attraction of immunosuppressive cells and up-regulation of immuno-regulatory molecules in the TME.
2.3.1. Radiation and chemotherapy-induced immune activation
Radiation-induced apoptosis and necrosis release tumor neoantigens, rendering the tumor more accessible to the innate and adaptive immune systems. Localized radiation-induced cell death promotes release of damage-associated molecular pattern molecules (DAMP), such as calreticulin, high-mobility group box 1 (HMGB1), and adenosine triphosphate (ATP) into the extracellular environment, allowing recognition of tumor cells by DC [74,75]. Furthermore, RT induces the production of pro-inflammatory cytokines such as interleukin 1β (IL-1β), transforming growth factor β (TGF-β) and tumor necrosis factor (TNF). Studies have shown an up-regulation of major histocompatibility complex I (MHC-I) in human and murine tumor cells after irradiation, thus enhancing T cell mediated anti-tumor response when combined with vaccination. In a phase II clinical trial, adjuvant immunotherapy using DC pulsed with tumor lysate showed favorable survival rates with median OS of 39.1 months in the vaccinated group over 15 months in the non-vaccinated control group [76]. In another report, Lee et al showed that high-dose dramatically increases T cell priming and susceptibility to vaccine-mediated T cell killing [77].
There is ample proof suggesting interferon (IFN) levels are elevated following radiation by activation of stimulator of interferon genes (STING) in irradiated tumor cells [78,79]. Similar to its effect on MHC-I expression, IFN-γ is known to upregulate the expression of NKG2G ligands on tumor cells [80]. Similarly, radiation-induced IFN-γ production can lead to an increase in PD-L1 expression on tumor cells. Deng et al showed that RT can upregulate PD-L1 on cancer cells, supporting the use of ICI along with RT [71]. Additionally, RT has been observed to induce an immune-mediated abscopal effect when combined with checkpoint inhibition [72,81].
Many studies indicate that TMZ-induced lymphopenia can be a concern while combining with immunotherapy [82]. However, TMZ has been found to increase priming of tumor antigen-specific T cells [83] and improve efficacy of TAT-survivin pulsed DC vaccines in murine models [84]. Clinical trials with DC vaccination have also demonstrated immune stimulation and increased antigen-specific T cells following pre-treatment with RT-TMZ [85]. Furthermore, TMZ-induced hypermutation and increased mutational load is associated with better response to ICI in several cancers, including GBM [57,86].
2.3.2. Radiation and chemotherapy-induced immunosuppression
Conventional RT combined with TMZ is known to induce immunosuppressive effects, including lymphopenia. Given that lymphopenia is associated with poor survival in patients with HGG, altering the radiation dose and delivery techniques to reduce lymphopenia could potentially improve the therapeutic outcomes [87,88]. In an ongoing phase 1 clinical trial (NCT02313272), the efficacy of Hypofractionated Stereotactic Irradiation (HFSRT) in combination with pembrolizumab and bevacizumab is being evaluated in adult patients with recurrent HGG.
Most clinical trials for immunotherapy enroll patients that have already undergone RT and chemotherapy as a standard course of treatment. Few studies to date have addressed the question of whether patients have better outcomes when RT is used sequentially or concurrently with immunotherapy. In a murine GBM model, Beclaid et al concluded that concurrent, rather than sequential administration of radiation and immunotherapy is associated with better outcomes [89].
3. Toxicity assessment and management
The robust immune-modulating effects of novel immunotherapies can cause severe toxicities. It is critical to understand these adverse events and integrate proper assessment and management plans in clinical trials. In this section, we discuss these toxicity considerations in the context of CAR T cell therapy and ICI.
3.1. Toxicities related to immune checkpoint inhibition
Immune checkpoint inhibition has revolutionized the treatment landscape for many cancers, resulting in improved survival for some peripheral tumors [90]. In the case of two clinically relevant checkpoints, CTLA-4 and PD-1, blocking interactions with their ligands induces strong immunological responses in the host including increased T cell activation and cytotoxic T cell-mediated tumor clearance [62,91,92]. Indoleamine 2, 3-dioxygenases 1 (IDO1) is a crucial innate immunity regulator that modulates T cell and NK cell function, and its inhibition is a therapeutic opportunity that is currently being explored in translational research against brain cancers [93]. As recent studies have shown, altering the immune system often requires a fine balance between therapeutic efficacy and over activation on the verge of toxicity.
Although CNS malignancies are immunologically unique, innovative and novel uses of ICI against brain cancers are demonstrably promising. A recent study by Cloughsey et al demonstrate that neoadjuvant ICI using PD-1 inhibitor had extended overall survival in patients with recurrent GBM relative to a cohort receiving adjuvant PD-1 blockade [62]. In addition, anti-PD-1 is also frequently practiced as salvage therapy in patients with CNS malignancies [94].
The role of naturally expressed checkpoint molecules is to prevent uncontrollable hyperactivation of the host immune system by regulating activation. ICIs remove this regulatory mechanism, thus allowing increased effector T cell function. The lack of restraint of T cell function by ICIs can lead to a plethora of immune-related adverse events through undefined mechanisms. Research suggests that immune-related adverse events develop through multiple pathways including autoreactive T cells, autoantibodies, and cytokine release [95]. Although immune-related adverse events most commonly involve the gastrointestinal tract, endocrine glands, skin, and liver [96], the effects of anti-CLTA-4 and anti-PD-1 differ in humans. Those treated with anti-CTLA-1 experience higher frequency of colitis and hypophysistis, whereas those treated with anti-PD-1 experience more pneumonitis and thyroiditis [97,98]. Thyroid disorders are more likely to occur in patients that receive anti-PD-1 or anti-PD-L1 who have anti-thyroid antibodies. It is hypothesized that blocking the PD-1/PD-L1 axis may impact both T cell-mediated immunity as well as humoral immunity thus emphasizing the anti-thyroid antibodies [99].
Both autoantibodies and autoreactive T cells contribute to immune-related adverse events, but the mechanisms remain uncharacterized. For example, in two reported cases of myocarditis, the same T cell clones were observed in the tumor and in the myocardium, but no antibody deposits were found [100]. This suggests that the T cells were reactive to a normal antigen shared by both the tumor and the myocardium. Another frequently observed autoimmune symptom is vitiligo, an autoimmune reaction against melanocytes in the skin. Cytokines, specifically IL-17 secretion, may also play a role in immune-related adverse events. In both murine and human systems, elevated levels of IL-17 have been observed in hosts with ICI-induced colitis [101].
Despite the discussion above, ICI with PD-1 blockade is reported to be generally well tolerated with common treatment-related toxicities including muscle weakness, headache, and hyperglycemia [62]. The occurrence of immune-related adverse events may indicate strong anti-tumor responses. However, it is clear the toxicity of ICIs are similar across cancers, suggesting that the toxicities are independent of tumor and antigen, and more related to systemic immune responses. Table 1 summarizes toxicity assessments and managements in ICI therapy.
Table 1.
Toxicity assessments and managements in immune checkpoint inhibition therapy
| Target | Drug | Indications | Immune-related reactions | Management | |
|---|---|---|---|---|---|
| PD-1 | Nivolomab; pembrolizumab | Metastatic non-small cell lung cancer; melanoma; advanced renal cell carcinoma; small cell lung cancer; metastatic squamous cell carcinoma; hepatocellular carcinoma; metastatic urothelial carcinoma; metastatic colorectal cancer; classical Hodgkin lymphoma | Pneumonitis; colitis; hepatitis; neuropathies; endocrinopathies; nephritis and renal dysfunction; dermatitis; encephalitis; complications after receiving allogeneic bone marrow transplant; increased mortality with multiple myeloma; embryofetal toxicity | Hepatic adverse events GI adverse events Endocrine adverse events Pulmonary adverse events |
Methylprednisolone Methylprednisolone Immunosuppression Methylprednisolone Hormone therapy Methylprednisolone Immunosuppression |
| CTLA-4 | Ipilimumab | Melanoma; renal cell carcinoma; colorectal cancer; hepatocellular carcinoma; non-small cell lunch cancer; hepatocellular carcinoma; metastatic non-small cell lung cancer | Colitis; hepatitis; skin rashes; neuropathies; hormone gland problems; pneumonitis; kidney failure; encephalitis; vision problems; complications after receiving allogeneic bone marrow transplant | Hepatic adverse events GI adverse events Endocrine adverse events Pulmonary adverse events |
Methylprednisolone Methylprednisolone Immunosuppression Methylprednisolone Hormone therapy Methylprednisolone Immunosuppression |
3.2. Toxicities associated with CAR T cell therapy
The side effects of CAR T cells can be classified by anatomical compartment effected and specific etiology. Potential toxicities that are unrelated to the tumor itself can be systemic, such as cytokine release syndrome (CRS). They can also be neurotoxic, as in immune effector cell-associated neurotoxicity syndrome (ICANS), which is also known as CAR T cell-related encephalopathy syndrome (CRES). Additionally, changes in neurological symptoms that are associated with the tumor location can be described as peritumoral edema or pseudoprogression, based upon imaging and pathological findings. Table 2 summarizes toxicity assessments and managements in CAR T cell therapy.
Table 2.
Toxicity assessments and managements in CAR T cell therapy
| Toxicities | Etiology | Symptoms | Treatment |
|---|---|---|---|
| CRS | Systemic inflammation | Fever | Symptomatic |
| Hypotension | IL-6 antagonists (Tociluzumab) | ||
| Hypoxia | Corticosteroids (dexamethasone) | ||
| ICANS | Unclear, possible increased blood-brain barrier permeability | Headache | Corticosteriods (dexamethasone) |
| Tremor | |||
| Dysgraphia | |||
| Aphasia | |||
| Seizure | |||
| Encephalopathy | |||
| Cerebral Edema | |||
| Pseudoprogression | Peritumoral edema | Worsening of pre-existing neurological symptoms | Corticosteriods (dexamethasone) |
CRS and ICANS have primarily been described in CD19-specific CAR T for leukemia and lymphoma. CRS is a systemic inflammatory response caused by the release of cytokines by CAR T cells, including IL-6, IFN-γ, and IL-2, in particular [65]. Release of these cytokines leads to systemic symptoms that affect multiple organs but is hallmarked by fever as well as capillary leak leading to peripheral edema and hypotension [102,103]. While there are various grading systems available for CRS [104–107], the American Society for Transplantation and Cellular Therapy (ASTCT) recently developed consensus recommendations for the grading of CRS based on fever, hypotension, and hypoxia [102]. There have been a limited number of clinical trials testing CAR T cells in adult GBM patients. However, when considering available study data, most patients across various treatment regimens had mild to no CRS symptoms [17–19,108,109]. The most common side effects reported included fever, malaise, and myalgias (Grade 1–2), and in those who received lymphodepleting chemotherapy, lymphopenia could be observed as well [17–19,108,109]. One study assessing CAR T cells targeting epidermal growth factor receptor variant 3 (EGFRvIII) in adult GBM looked at serum cytokine levels and identified increases in IL-6, IL-5, and IL-10 following CAR T cell administration [18].
The neurotoxicity related to CAR T therapy (ICANS) is a distinct entity from CRS, characterized by various neurological symptoms ranging from mild to more severe [65,102,103]. Mild symptoms can include headache, tremor, and dysgraphia. Severe symptoms include aphasia, seizures, and encephalopathy, including fatal cerebral edema. Timing of ICANS can occur within days to weeks after infusion and can co-occur with CRS or present after CRS has resolved [65,102]. While further investigation into the mechanisms of the development of ICANS are needed, data have suggested increased BBB permeability as a potential mechanism [110,111]. In GBM patients, there has been concern about peritumoral inflammatory responses causing potentially severe or fatal neuroinflammation. However, in the existing clinical trials to date, patients have presented with mild to no neurotoxicity [17–19,109]. In the cases where mild symptoms were present, some were secondary to disease progression rather than CAR T related neurotoxicity [17–19,108,109]. The most common symptom secondary to CAR T was seizure but other symptoms included headache, weakness (including facial nerve weakness), and gait changes [17–19,108,109]. One study found increased levels of cytokines in the cerebrospinal fluid (CSF), such as IL-6, IFN-γ, IL-5, IL-10, when patients were experiencing ICANS symptoms without any corresponding increase in serum cytokines or presence of CAR T cells in the blood [17].
3.3. Peritumoral edema due to immunotherapy-induced local inflammation
In addition to systemic inflammatory responses resulting from CAR T such as CRS, neurological symptoms can present due to peritumoral edema. If neurological signs are consistent with dysfunction of the neuroanatomical compartment where the tumor is located, peritumoral edema is more likely to be responsible than ICANS. These signs and symptoms may present as worsening of pre-existing symptoms and may be associated with tumor pseudoprogression on MRI. The presence of local inflammation in the peritumoral region can make it difficult to distinguish pseudoprogression from true progression of disease [111]. Therefore, follow-up imaging as well as tissue biopsy with histopathological assessment for inflammatory infiltrate and tumor cell markers are the most appropriate manners in which to rule out pseudoprogression [111,112]. In the adult GBM setting, pseudoprogression following immunotherapy has made it challenging to assess treatment response in clinical trials. Therefore, the Immunotherapy Response Assessment in Neuro-Oncology (iRANO) working group has recommended comprehensive imaging guidelines for potential cases of pseudoprogression in the adult CNS tumor setting. iRANO suggests that pseudoprogression should be considered as a possible explanation for any radiographic progression that is observed during the first six months after administration of an immunotherapy [112]. As long as patients do not exhibit neurological decline associated with these findings, iRANO recommends repeated imaging follow-up to confirm progressive lesions before reclassifying patients as non-responsive. In the case of pHGG, where pseudoprogression may occur along the midline and brain stem, resultant hydrocephalus and other neurological symptoms may require anti-inflammatory agents for management.
3.4. Management of neurotoxicities
CRS treatment starts with addressing symptoms and if further treatment is required, CRS is treated with an IL-6 antagonist or corticosteroids in severe cases [104]. Available IL-6 antagonists include tocilizumab and siltuximab, which bind to the IL-6 receptor and IL-6, respectively. Systemic symptoms often respond to IL-6 antagonists and no subjects in GBM CAR T clinical trials to this point have required treatment for CRS symptoms. In the case of ICANS, the limited BBB penetration by tocilizumab and its reported capacity to transiently raise IL-6 levels risk inefficacy and increased neurotoxicity in some patients [104,113]. Therefore, tocilizumab is not a first line treatment unless symptoms of CRS are also present [103]. In one adult GBM trial, ICANS symptoms were treated with siltuximab but it was unclear if there was any clinical benefit [18]. Therefore, dexamethasone is the most appropriate agent for controlling acute neurological symptoms secondary to CAR T administration. However, while dexamethasone penetrates the brain parenchyma efficiently, it is highly immunosuppressive [114,115]. As discussed earlier in this review, clinical vaccine studies for malignant glioma, including DMG, suggest that dexamethasone diminishes activation of vaccine-specific T cell responses [15,116]. Nonetheless, for management of acute neuroinflammation, CRS, and ICANS, dexamethasone is the most appropriate treatment option. As an alternative to corticosteroids, bevacizumab can also be used to manage neurological symptoms and pseudoprogression in a manner that may be more appropriate for long-term use [117,118]. While these treatments are important considerations for toxicity management in pediatric clinical trials for high-grade glioma, no adult GBM patients in CAR T clinical trials have required intervention with corticosteroids for ICANS up to this point [17–19,108,109]. Finally, it should be noted that CRS, ICANS, and neurological symptoms associated with peritumoral edema or pseudoprogression can occur concurrently and may require distinct treatment consideration. The clinical team should assess imaging findings for evidence of tumor-related edema alongside assessments of elevated cytokines, neurotoxicity, and systemic symptoms.
4. Disease monitoring and biomarkers of immunotherapy response
4.1. Imaging
Assessing disease response following immunotherapy presents a unique challenge when applying traditional radiographic and clinical response criteria, particularly for CNS tumors [6,119]. Immunotherapeutic strategies, in achieving their intended purpose of activating or introducing immune activity, often result in imaging changes that can be difficult to distinguish from tumor progression or evolving effects of prior cancer-directed therapy. As experience with immunomodulators against CNS tumors has expanded, the field of neuroimaging has been forced to develop imaging criteria that address several complexities including immune-related pseudoprogression, heterogeneous enhancement patterns, abscopal effects, and delayed or mixed responses. As discussed earlier, iRANO criteria aim to preserve or improve the utility of response measures after immunotherapeutic agents using currently available modalities [112]. iRANO and other related guidelines of immunotherapy response monitoring (e.g. irRECIST, irRC) include specific recommendations on the assessment of new lesions, interpreting changes in the absence of clinical symptoms, and time intervals to determining response or progression [120,121]. For example, due to evidence that early radiographic progression and new lesions are often associated with subsequent therapeutic benefit [122,123], iRANO recommends repeat scans from initial radiographic progression prior to confirming progressive disease. This time window aims to decrease the likelihood of concluding drug inefficacy prematurely, while maintaining safeguards for patients who develop neurologic symptoms. Early experiences applying iRANO criteria to DMG patients treated with peptide-based vaccine immunotherapy identified 4/21 patients (19%) with documented pseudoprogression based on clinical and radiographic findings by conventional MRI with MR spectroscopy, diffusion, and perfusion techniques [124]. While further validation across disease entities and treatment regimens is warranted, this case series highlights feasibility of overlying novel imaging metrics on top of conventional imaging modalities to monitor disease and correlate with clinical outcomes. Ongoing efforts to implement advanced neuroimaging techniques such as immuno-PET imaging using novel molecular radiotracers (e.g. 18F-fluorothymidine, Granzyme B[125,126]) or labeled monoclonal antibodies (e.g. copper-64, zirconium-89 [127–129]) remain investigational, but hold promise for improved non-invasive imaging of tumor-specific responses and immune-related events.
4.2. Novel biomarkers
While clinical endpoints and imaging will remain critical to disease and toxicity assessment, correlative analyses of CSF, plasma, and even urine may offer accessible surrogate markers of anti-tumor immunity [130–132]. These biomarkers may include quantitative measures, such as circulating tumor DNA (ctDNA), and in the case of cellular therapies such as CAR T cells, circulating CAR T DNA (cartDNA). They also can include qualitative or functional assessments of immune response, such as immune cell phenotyping, effector-mediated activity panels measuring cytokines, chemokines, and growth factors (e.g. the secretome), or sequencing of extracellular vesicle (EV) cargo. Layering of appropriate correlative studies to maximize the power of minimally-invasive biospecimens will be a critical component of next-generation immunotherapy trials and, while no current standard exists for the timing of collections, efforts are ongoing to harmonize immune monitoring protocols among clinical trials and consortia [133]. Furthermore, some tumors that can be biopsied relatively safely but are unresectable, such as DIPG, may especially benefit from correlative disease monitoring. In these cases, serial collections will always be more ethical and feasible than repeat neurosurgical biopsies to assess biomarkers of efficacy and treatment response, particularly in children.
Common bioassays used to measure immune activation in plasma and CSF include multiplexed panels with the ability to analyze multiple cytokines in parallel on a single run, such as bead-based multiplex immunoassays (MIA; Luminex/Milliplex) and Meso Scale Discovery (MSD) electrochemiluminescence [134]. These immunoassays detect factors of innate and adaptive immunity, with molecules of interest often including granulocyte macrophage colony stimulating factor (GM-CSF), IFN-γ, soluble FAS and FAS ligand, soluble CD137, granzyme A and B, interleukin (IL)-2, IL-4, IL-5, IL-6, IL-10, IL-13, macrophage inflammatory protein (MIP)-1α MIP-1β, tumor necrosis factor (TNF-α), and perforin [135]. As we continue to gain knowledge from ongoing pediatric CNS immunotherapy trials, our ability to tailor monitoring parameters to disease-specific and immunomodulator-specific markers should not only improve but be employed systematically across working groups. For example, in ongoing CNS CAR T cell trials at Seattle Children’s [NCT03500991, NCT03618381, NCT04185038], patient’s serum and CSF are serially interrogated for CNS tumor-related and endothelial markers, including angiopoietin (Ang)-1, Ang-1, glial fibrillary acidic protein (GFAP), S100 calcium-binding protein (S100b), vascular endothelial growth factor (VEGF), and von Willebrand factor (VWF) using the Meso Scale Discovery platform [110,135]. Many of these markers are only available in specialized immunology labs, which combined with the relative instability of cytokines, chemokines, and growth factors, makes standardization of collection approaches difficult. However, the potential for gaining insight into predictive biomarkers of response and mechanisms of resistance provides a strong rationale for inclusion on current and future studies.
In addition to systemic immune activation, early signs of efficacy in patient responders versus non-responders to cellular therapies include direct measurements of the survival and activity of transferred product (e.g. CAR T cell, TCR, CTL). Techniques including flow cytometry, molecular analysis, and labeled radionucleotide imaging have all been utilized to detect and track cellular products following patient administration. Gene sequencing or flow cytometric analysis of the T cell receptor-β (TCR-β) are commonly applied to detect T cell-based therapies, with functional activity measured by ex vivo re-challenge or detection of exhaustion markers such as TIM-3, LAG-3, TIGIT, PD-1, CD39, and Nur77. The clonotypic repertoire of TCR Vβ can also be used to distinguish CAR T cells from endogenous tumor-infiltrating T cells, determining successful engraftment and survival advantage of transduced T cells over endogenous lymphocyte populations, or as in the case of an EGFRvIII CAR, an increase in polyclonal T cells with immunosuppressive or regulatory phenotypic predominance [18]. Early attempts at nuclear imaging approaches to detect T cells were restrained by poor BBB penetration of radioactive nucleotides and immunogenicity. However, newer techniques show promise in overcoming this barrier, although they are not yet widely clinically available [136,137]. Weist et al demonstrated that passive ex vivo CAR T cell labeling with 89Zr-oxine can be accurately tracked by PET imaging, providing a real-time assessment of T cell delivery into the brain parenchyma and CAR T cell tendency to persist in the tumor location or diffuse through the CNS following intratumoral (ICT) versus intraventricular (ICV) injections, respectively [138]. Furthermore, differential T cell tracking by route of therapeutic delivery holds clinical relevance. For instance, systemically-delivered CAR T cells may be directly evaluated by plasma sampling, while CNS tumor-targeting CAR T cells delivered locoregionally are expected to be absent from the periphery and may require CSF sampling for cell detection [139].
Delivery devices may assist with the feasibility of repeated locoregional (e.g. ICT or ICV) sampling and these minimally invasive procedures can be performed by a range of trained practitioners. Optimal frequency of sample collection and analysis is still under investigation, with recent trials demonstrating feasibility of frequent collection timepoints. Current guidelines from a Seattle Children’s active Phase 1 trial of repeatedly dosed locoregional B7-H3-specific CAR T cells for DIPG/DMG and recurrent/refractory pediatric CNS tumors [BrainChild-03, NCT04185038] includes routine correlative study collection including: (1) Monthly CSF collection by lumbar puncture for ICT-delivered CAR T cells; (2) Weekly pre- and post-infusion collection via Ommaya reservoir for ICV-delivered CAR T cells, with occasional mandated lumbar punctures; and (3) Peripheral blood obtained weekly.
Circulating immune cells may also serve as dynamic biomarkers of tumor-immune cell interactions. Griffiths et al. demonstrated that patient peripheral blood mononuclear cells (PBMCs) could be analyzed by single-cell RNA sequencing (scRNAseq) to detect abundance, activation, and differentiation states of T cell and monocyte populations following anti-PD-1 therapy [140]. Patient responsiveness was correlated to an early increase of T cell abundance and IFN signaling, providing a non-invasive and early predictor of response. More commonly-used techniques to detect T cell activity and potential for epitope spreading after immunotherapy include intracellular IFN-γ staining, tetramer analysis, or enzyme-linked immune absorbent spot (ELISPOT) assays. Cross-primed T cells recognizing cancer-testis antigens (CTA) or cancer-associated antigens (CAA) such as HER2, gp100, MAGE-A3, and others, may also signify early potential for anti-tumor activity. Notably, this has been hypothesized to be a contributor towards therapeutic efficacy observed following ICI and cytotoxic T lymphocyte therapy [141,142].
The challenge of antigen escape, whereby tumors downregulate expression of target epitopes after targeted therapy, is a well-documented mechanism of resistance to CNS-directed immunotherapy. This challenge highlights the importance of considering heterogeneity and adaptability of tumor surface expression throughout the course of disease and treatment [143–145]. Moreover, tumor antigens may serve as biomarkers of treatment response whether or not they are intentional targets [146,147]. Arguably one of the most clinically relevant of these antigen modifications is the induced expression of checkpoint proteins, such as PD-L1, which has been reported to increase in patients after even a single dose of intravenously delivered EGFRvIII CAR T cells [18]. However, recent studies assessing the predictive and prognostic value of PD-L1 expression prior to ICI, in the absence of mismatch repair deficiency, show mixed results [148–151]. While this data can potentially aid in the design of future combinatorial strategies, monitoring expression levels for patient-specific clinical benefit is limited and requires neurosurgical biopsy or resection. Tissue procurement is often complicated by tumor heterogeneity, repeat sampling error, and phenotypic variability among metastatic lesions with differential responses to immunomodulation. Incorporating serial biopsies into study design remains controversial due to concerns about reliability and feasibility of adequate tissue collection, along with ethical considerations in the pediatric population. Autopsy specimens from consenting families/patients circumvent a number of these concerns and can identify comparable intratumoral and TME changes following immunotherapy [152], but are inherently inadequate in assessing temporal and dynamic changes.
Advances in circulating tumor DNA (ctDNA) analysis have broadened the application of this technique beyond the detection of recurrent disease, with new sensitive approaches providing longitudinal analysis of treatment responses [153,154]. This was demonstrated in a subset of patients with DMG, wherein the driver histone 3 p.K27M (H3 K27M) mutation could be detected in 88% of plasma and CSF samples, with decreasing ctDNA levels over time corresponding to treatment response and radiographic tumor regression by MRI [155]. Additionally, EVs are cellular membrane components secreted by tumor cells harboring tumor-specific mRNAs, miRNAs, and proteins, that can be traced as another method of “liquid biopsy” in GBM patients [156–158]. Together, these novel molecular techniques for analysis of tumor-secreted factors in plasma and CSF may provide improved disease monitoring with the ability to detect early immunotherapeutic failures [159,160].
5. Expert opinion
We hope that our review and discussions above will assist future studies to accelerate progress and collaborations in the field of immunotherapy for pediatric patients with HGG. Because the field is relatively new and in its growing phase, there is not much data or established guidelines available specifically for immunotherapy in pediatric HGG. Nonetheless, careful comparison of anti-tumor immune response in successfully treated extracranial solid tumors would help the field gain more in-depth understanding on the critical factors that may need to be addressed in pediatric HGG patients. To this end, comparison of pre- and post-immunotherapy TME between HGG versus other tumors would be informative. However, while pre-immunotherapy biopsy has been established even in DMG patients [15], debulking surgery is often not feasible especially for DMG/DIPG. Regarding post-treatment tumor samples, to date, justification of post-treatment biopsy of patients with DMG remains a controversial topic in pediatric neuro-oncology due to multiple challenges, including the risk and costs [161,162]. Lesser and non-invasive biomarkers available through novel technologies, as discussed in this manuscript, may help us to overcome these challenges.
While the field still needs to develop a better understanding of the biological challenges facing pediatric HGG treatment, such as low mutation load, genetic and antigenic heterogeneity, and tumor- and therapy-induced immunosuppression. We must enhance our translational therapeutic efforts by collaborating with relevant experts to develop more effective therapeutics, such as T cell and antibody-based therapies. These efforts should be integrated with considerate trial designs that allow for therapeutic administration within the critical window of opportunity. Considering the relatively low frequency of pHGG, it is also essential to promote collaborative, multicenter clinical trials, such as those conducted through Pacific Pediatric Neuro-oncology Consortium (PNOC) and the COllaborative Network for NEuro-Oncology Clinical Trials (CONNECT). Collaborative endeavors and the sharing of primary data can help to speed up the understanding of how novel immunotherapeutic interventions improve anti-HGG immune mechanisms.
While brain malignancies represent the most common cancer-related mortality in children, it is often difficult to incite the interests of industry sponsors due to the small market size. We need to formulate and execute better strategies to advocate the importance of this area of research not only at an industry level but at the level of regulatory agencies, such as the FDA and NIH. Our continued and concerted efforts towards complete mechanistic understanding, rational treatment combinations and sophisticated clinical trial designs will be pivotal in achieving our shared goal of developing effective therapies for these children.
Article highlights:
The authors review and provide insights on key consideration points in immunotherapy of pHGGs
Clinical trial design should integrate appropriate patient populations, novel and preclinically optimized trial design, and logical treatment combinations, especially standard of care modalities. There are critical caveats, such as patient selection bias, due to the nature of immunotherapy trials, such as frequent exclusion of patients on high-dose corticosteroids.
Robust immune-activating/modulating effects of modern immunotherapy can have toxicities. It is important to understand and manage these, especially in patients with pHGG.
The rapid evolution of immune-biomarker detection should be integrated to provide intra- and inter-trial comparability.
Funding
This paper was supported by funding from grants from NIH/NINDS (1R35 NS105068), to H Okada, as well as from the V Foundation.
Footnotes
Declaration of interests
H Okada declares consultant fees from Bristol-Myers Squibb as well as royalties as inventor of: the H3.3K27M TCR, IL-13Ra2 (345–353:1A9V) peptide, and EGFRvIII-CAR for which an exclusive licensing agreement has been executed with Tmunity, Inc., Stemline, Inc. and Novartis Pharma, respectively. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or conflict with the subject matter or materials discussed in this manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- 1.Jones C, Karajannis MA, Jones DTW et al. Pediatric high-grade glioma: biologically and clinically in need of new thinking Neuro Oncol, 19, 153–61 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crotty EE, Leary SES, Geyer JR et al. Children with DIPG and high-grade glioma treated with temozolomide, irinotecan, and bevacizumab: the Seattle Children’s Hospital experience J Neurooncol, 148, 607–17 (2020) [DOI] [PubMed] [Google Scholar]
- 3.Cohen KJ, Pollack IF, Zhou T et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children’s Oncology Group Neuro Oncol, 13, 317–23 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jakacki RI, Cohen KJ, Buxton A et al. Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: a report of the Children’s Oncology Group ACNS0423 study Neuro Oncol, 18, 1442–50 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostrom QT, de Blank PM, Kruchko C et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011 Neuro Oncol, 16 Suppl 10, x1–x36 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lieberman NAP, Vitanza NA, Crane CA. Immunotherapy for brain tumors: understanding early successes and limitations Expert Rev Neurother, 18, 251–9 (2018) [DOI] [PubMed] [Google Scholar]
- 7.Mende AL, Schulte JD, Okada H et al. Current Advances in Immunotherapy for Glioblastoma Curr. Oncol. Rep, In Press (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy Cancer discovery, 8, 1069–86 (2018) [DOI] [PubMed] [Google Scholar]
- 9.Reardon DA, Brandes AA, Omuro A et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial JAMA Oncol, (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.AlHarbi M, Ali Mobark N, AlMubarak L et al. Durable Response to Nivolumab in a Pediatric Patient with Refractory Glioblastoma and Constitutional Biallelic Mismatch Repair Deficiency Oncologist, 23, 1401–6 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorsi HS, Malicki DM, Barsan V et al. Nivolumab in the Treatment of Recurrent or Refractory Pediatric Brain Tumors: A Single Institutional Experience J Pediatr Hematol Oncol, 41, e235–e41 (2019) [DOI] [PubMed] [Google Scholar]
- 12.Srinivasan VM, Ferguson SD, Lee S et al. Tumor Vaccines for Malignant Gliomas Neurotherapeutics, 14, 345–57 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nejo T, Yamamichi A, Almeida ND et al. Tumor antigens in glioma Semin Immunol, 47, 101385 (2020) [DOI] [PubMed] [Google Scholar]
- 14.Kwok D, Okada H. T-Cell based therapies for overcoming neuroanatomical and immunosuppressive challenges within the glioma microenvironment J Neurooncol, 147, 281–95 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller S, Taitt JM, Villanueva-Meyer JE et al. Mass cytometry detects H3.3K27M-specific vaccine responses in diffuse midline glioma Journal of Clinical Investigation, In Press (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Shen F, Yao Y et al. Adoptive Cell Therapy: A Novel and Potential Immunotherapy for Glioblastoma Frontiers in oncology, 10, 59 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown CE, Alizadeh D, Starr R et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy N Engl J Med, 375, 2561–9 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Rourke DM, Nasrallah MP, Desai A et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma Sci Transl Med, 9 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goff SL, Morgan RA, Yang JC et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma J Immunother, 42, 126–35 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majzner RG, Theruvath JL, Nellan A et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors Clinical Cancer Research, 25, 2560–74 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Theruvath J, Sotillo E, Mount CW et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors Nature Medicine, 26, 712–9 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravanpay AC, Gust J, Johnson AJ et al. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma Oncotarget, 10, 7080–95 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawler SE, Speranza MC, Cho CF et al. Oncolytic Viruses in Cancer Treatment: A Review JAMA Oncol, 3, 841–9 (2017) [DOI] [PubMed] [Google Scholar]
- 24.Lieberman NA, Moyes KW, Crane CA. Developing immunotherapeutic strategies to target brain tumors Expert review of anticancer therapy, 16, 775–88 (2016) [DOI] [PubMed] [Google Scholar]
- 25.Tejada S, Diez-Valle R, Dominguez PD et al. DNX-2401, an Oncolytic Virus, for the Treatment of Newly Diagnosed Diffuse Intrinsic Pontine Gliomas: A Case Report Frontiers in oncology, 8, 61 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varela-Guruceaga M, Tejada-Solis S, Garcia-Moure M et al. Oncolytic Viruses as Therapeutic Tools for Pediatric Brain Tumors Cancers (Basel), 10 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falsetti L, Schivardi MR, Prandini BD. A new low-dose estrogen oral contraceptive combination: effect on endocrine parameters and lipid status Contraception, 36, 489–97 (1987) [DOI] [PubMed] [Google Scholar]
- 28.Mackay A, Burford A, Molinari V et al. Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial Cancer Cell, 33, 829–42 e5 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu G, Diaz AK, Paugh BS et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma Nat Genet, 46, 444–50 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gajjar A, Pfister SM, Taylor MD et al. Molecular insights into pediatric brain tumors have the potential to transform therapy Clin Cancer Res, 20, 5630–40 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Northcott PA, Pfister SM, Jones DT. Next-generation (epi)genetic drivers of childhood brain tumours and the outlook for targeted therapies Lancet Oncol, 16, e293–302 (2015) [DOI] [PubMed] [Google Scholar]
- 32.Sturm D, Bender S, Jones DT et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge Nat Rev Cancer, 14, 92–107 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lieberman NAP, DeGolier K, Kovar HM et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy Neuro Oncol, 21, 83–94 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin GL, Nagaraja S, Filbin MG et al. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma Acta Neuropathol Commun, 6, 51 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hutzen B, Ghonime M, Lee J et al. Immunotherapeutic Challenges for Pediatric Cancers Mol Ther Oncolytics, 15, 38–48 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wages NA, Chiuzan C, Panageas KS. Design considerations for early-phase clinical trials of immune-oncology agents Journal for immunotherapy of cancer, 6, 81 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Quigley J. Sequential design and analysis of dose finding studies in patients with life threatening disease Fundam Clin Pharmacol, 4 Suppl 2, 81s–91s (1990) [DOI] [PubMed] [Google Scholar]
- 38.Storer BE. Design and analysis of phase I clinical trials Biometrics, 45, 925–37 (1989) [PubMed] [Google Scholar]
- 39.Goodman SN, Zahurak ML, Piantadosi S. Some practical improvements in the continual reassessment method for phase I studies Stat Med, 14, 1149–61 (1995) [DOI] [PubMed] [Google Scholar]
- 40.Juratli TA, Qin N, Cahill DP et al. Molecular pathogenesis and therapeutic implications in pediatric high-grade gliomas Pharmacol Ther, 182, 70–9 (2018) [DOI] [PubMed] [Google Scholar]
- 41.Chiang J, Diaz AK, Makepeace L et al. Clinical, imaging, and molecular analysis of pediatric pontine tumors lacking characteristic imaging features of DIPG Acta Neuropathol Commun, 8, 57 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams JR, Young CC, Vitanza NA et al. Progress in diffuse intrinsic pontine glioma: advocating for stereotactic biopsy in the standard of care Neurosurg Focus, 48, E4 (2020) [DOI] [PubMed] [Google Scholar]
- 43.Majzner RG, Mackall CL. Tumor Antigen Escape from CAR T-cell Therapy Cancer discovery, 8, 1219–26 (2018) [DOI] [PubMed] [Google Scholar]
- 44.Xu X, Sun Q, Liang X et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies Front Immunol, 10, 2664 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arrieta VA, Cacho-Diaz B, Zhao J et al. The possibility of cancer immune editing in gliomas. A critical review Oncoimmunology, 7, e1445458 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Griesinger AM, Birks DK, Donson AM et al. Characterization of distinct immunophenotypes across pediatric brain tumor types J Immunol, 191, 4880–8 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wheeler LA, Manzanera AG, Bell SD et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma Neuro Oncol, 18, 1137–45 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winograd EK, Ciesielski MJ, Fenstermaker RA. Novel vaccines for glioblastoma: clinical update and perspective Immunotherapy, 8, 1293–308 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mohme M, Schliffke S, Maire CL et al. Immunophenotyping of Newly Diagnosed and Recurrent Glioblastoma Defines Distinct Immune Exhaustion Profiles in Peripheral and Tumor-infiltrating Lymphocytes Clin Cancer Res, 24, 4187–200 (2018) [DOI] [PubMed] [Google Scholar]
- 50.Dutoit V, Migliorini D, Dietrich PY et al. Immunotherapy of Malignant Tumors in the Brain: How Different from Other Sites? Frontiers in oncology, 6, 256 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Louveau A, Smirnov I, Keyes TJ et al. Structural and functional features of central nervous system lymphatic vessels Nature, 523, 337–41 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu X, Fallert-Junecko BA, Fujita M et al. Poly-ICLC promotes the infiltration of effector T cells into intracranial gliomas via induction of CXCL10 in IFN-alpha and IFN-gamma dependent manners Cancer Immunol Immunother, 59, 1401–9 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu X, Nishimura F, Sasaki K et al. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models J.Transl.Med, 5, 10 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mitchell DA, Fecci PE, Sampson JH. Immunotherapy of malignant brain tumors Immunol Rev, 222, 70–100 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pitter KL, Tamagno I, Alikhanyan K et al. Corticosteroids compromise survival in glioblastoma Brain, 139, 1458–71 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petrelli F, Signorelli D, Ghidini M et al. Association of Steroids use with Survival in Patients Treated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis Cancers (Basel), 12 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Preusser M, Lim M, Hafler DA et al. Prospects of immune checkpoint modulators in the treatment of glioblastoma Nat Rev Neurol, 11, 504–14 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garant A, Guilbault C, Ekmekjian T et al. Concomitant use of corticosteroids and immune checkpoint inhibitors in patients with hematologic or solid neoplasms: A systematic review Crit Rev Oncol Hematol, 120, 86–92 (2017) [DOI] [PubMed] [Google Scholar]
- 59.Downey SG, Klapper JA, Smith FO et al. Prognostic factors related to clinical response in patients with metastatic melanoma treated by CTL-associated antigen-4 blockade Clin Cancer Res, 13, 6681–8 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Naidoo J, Page DB, Li BT et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies Annals of oncology : official journal of the European Society for Medical Oncology / ESMO, 26, 2375–91 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Min L, Hodi FS, Kaiser UB. Corticosteroids and immune checkpoint blockade Aging (Albany NY), 7, 521–2 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cloughesy TF, Mochizuki AY, Orpilla JR et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma Nat Med, 25, 477–86 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma Nat Med, 25, 470–6 (2019) [DOI] [PubMed] [Google Scholar]
- 64.Brentjens RJ, Davila ML, Riviere I et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia Sci Transl Med, 5, 177ra38 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee DW, Kochenderfer JN, Stetler-Stevenson M et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial Lancet, 385, 517–28 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singh N, Frey NV, Grupp SA et al. CAR T Cell Therapy in Acute Lymphoblastic Leukemia and Potential for Chronic Lymphocytic Leukemia Current treatment options in oncology, 17, 28 (2016) [DOI] [PubMed] [Google Scholar]
- 67.Tasian SK. Acute myeloid leukemia chimeric antigen receptor T-cell immunotherapy: how far up the road have we traveled? Ther Adv Hematol, 9, 135–48 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Queirolo P, Boutros A, Tanda E et al. Immune-checkpoint inhibitors for the treatment of metastatic melanoma: a model of cancer immunotherapy Semin Cancer Biol, 59, 290–7 (2019) [DOI] [PubMed] [Google Scholar]
- 69.Auslander N, Zhang G, Lee JS et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma Nat Med, 24, 1545–9 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Demaria S, Ng B, Devitt ML et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated Int J Radiat Oncol Biol Phys, 58, 862–70 (2004) [DOI] [PubMed] [Google Scholar]
- 71.Weichselbaum RR, Liang H, Deng L et al. Radiotherapy and immunotherapy: a beneficial liaison? Nat Rev Clin Oncol, 14, 365–79 (2017) [DOI] [PubMed] [Google Scholar]
- 72.Bramhall RJ, Mahady K, Peach AH. Spontaneous regression of metastatic melanoma - clinical evidence of the abscopal effect Eur J Surg Oncol, 40, 34–41 (2014) [DOI] [PubMed] [Google Scholar]
- 73.Schaue D, Kachikwu EL, McBride WH. Cytokines in radiobiological responses: a review Radiat Res, 178, 505–23 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bhalla N, Brooker R, Brada M. Combining immunotherapy and radiotherapy in lung cancer J Thorac Dis, 10, S1447–S60 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goto T. Radiation as an In Situ Auto-Vaccination: Current Perspectives and Challenges Vaccines (Basel), 7, 100 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cho DY, Yang WK, Lee HC et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: a phase II clinical trial World Neurosurg, 77, 736–44 (2012) [DOI] [PubMed] [Google Scholar]
- 77.Lee Y, Auh SL, Wang Y et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment Blood, 114, 589–95 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deng L, Liang H, Xu M et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors Immunity, 41, 843–52 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting Nat Rev Immunol, 6, 836–48 (2006) [DOI] [PubMed] [Google Scholar]
- 80.Kim JY, Son YO, Park SW et al. Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation Exp Mol Med, 38, 474–84 (2006) [DOI] [PubMed] [Google Scholar]
- 81.Dewan MZ, Galloway AE, Kawashima N et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody Clin Cancer Res, 15, 5379–88 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Litterman AJ, Zellmer DM, Grinnen KL et al. Profound impairment of adaptive immune responses by alkylating chemotherapy J Immunol, 190, 6259–68 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sanchez-Perez LA, Choi BD, Archer GE et al. Myeloablative temozolomide enhances CD8(+) T-cell responses to vaccine and is required for efficacy against brain tumors in mice PLoS One, 8, e59082 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim CH, Woo SJ, Park JS et al. Enhanced antitumour immunity by combined use of temozolomide and TAT-survivin pulsed dendritic cells in a murine glioma Immunology, 122, 615–22 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fadul CE, Fisher JL, Hampton TH et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy J Immunother, 34, 382–9 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rizvi NA, Hellmann MD, Snyder A et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer Science, 348, 124–8 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yovino S, Kleinberg L, Grossman SA et al. The etiology of treatment-related lymphopenia in patients with malignant gliomas: modeling radiation dose to circulating lymphocytes explains clinical observations and suggests methods of modifying the impact of radiation on immune cells Cancer Invest, 31, 140–4 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang X, Wang P, Zhao Z et al. A review of radiation-induced lymphopenia in patients with esophageal cancer: an immunological perspective for radiotherapy Therapeutic advances in medical oncology, 12, 1758835920926822 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Belcaid Z, Phallen JA, Zeng J et al. Focal radiation therapy combined with 4–1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model PLoS One, 9, e101764 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hodi FS, O’Day SJ, McDermott DF et al. Improved survival with ipilimumab in patients with metastatic melanoma N Engl J Med, 363, 711–23 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reardon DA, Gokhale PC, Klein SR et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model Cancer immunology research, 4, 124–35 (2016) [DOI] [PubMed] [Google Scholar]
- 92.Davidson TB, Lee A, Hsu M et al. Expression of PD-1 by T Cells in Malignant Glioma Patients Reflects Exhaustion and Activation Clin Cancer Res, 25, 1913–22 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu M, Wang X, Wang L et al. Targeting the IDO1 pathway in cancer: from bench to bedside J Hematol Oncol, 11, 100 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kurz SC, Cabrera LP, Hastie D et al. PD-1 inhibition has only limited clinical benefit in patients with recurrent high-grade glioma Neurology, 91, e1355–e9 (2018) [DOI] [PubMed] [Google Scholar]
- 95.Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade N Engl J Med, 378, 158–68 (2018) [DOI] [PubMed] [Google Scholar]
- 96.Weber JS, Hodi FS, Wolchok JD et al. Safety Profile of Nivolumab Monotherapy: A Pooled Analysis of Patients With Advanced Melanoma J Clin Oncol, 35, 785–92 (2017) [DOI] [PubMed] [Google Scholar]
- 97.Halsey T, Ologun G, Wargo J et al. Uncovering the role of the gut microbiota in immune checkpoint blockade therapy: A mini-review Semin Hematol, 57, 13–8 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kennedy LB, Salama AKS. A Review of Immune-Mediated Adverse Events in Melanoma Oncol Ther, 7, 101–20 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Osorio JC, Ni A, Chaft JE et al. Antibody-mediated thyroid dysfunction during T-cell checkpoint blockade in patients with non-small-cell lung cancer Annals of oncology : official journal of the European Society for Medical Oncology / ESMO, 28, 583–9 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Johnson DB, Balko JM, Compton ML et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade N Engl J Med, 375, 1749–55 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Harbour SN, Maynard CL, Zindl CL et al. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis Proc Natl Acad Sci U S A, 112, 7061–6 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee DW, Santomasso BD, Locke FL et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells Biol Blood Marrow Transplant, 25, 625–38 (2019) [DOI] [PubMed] [Google Scholar]
- 103.Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity CA Cancer J Clin, 70, 86–104 (2020) [DOI] [PubMed] [Google Scholar]
- 104.Lee DW, Gardner R, Porter DL et al. Current concepts in the diagnosis and management of cytokine release syndrome Blood, 124, 188–95 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Porter D, Frey N, Wood PA et al. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel J Hematol Oncol, 11, 35 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Park JH, Riviere I, Gonen M et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia N Engl J Med, 378, 449–59 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Neelapu SS, Tummala S, Kebriaei P et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities Nat Rev Clin Oncol, 15, 47–62 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brown CE, Badie B, Barish ME et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma Clin Cancer Res, 21, 4062–72 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ahmed N, Brawley V, Hegde M et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial JAMA Oncol, 3, 1094–101 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gust J, Hay KA, Hanafi LA et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells Cancer Discov, 7, 1404–19 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thust SC, van den Bent MJ, Smits M. Pseudoprogression of brain tumors J Magn Reson Imaging, (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Okada H, Weller M, Huang R et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group Lancet Oncol, 16, e534–e42 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nishimoto N, Terao K, Mima T et al. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease Blood, 112, 3959–64 (2008) [DOI] [PubMed] [Google Scholar]
- 114.Gustafson MP, Lin Y, New KC et al. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone Neuro Oncol, 12, 631–44 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vrabec M, Van Cauter S, Himmelreich U et al. MR perfusion and diffusion imaging in the follow-up of recurrent glioblastoma treated with dendritic cell immunotherapy: a pilot study Neuroradiology, 53, 721–31 (2011) [DOI] [PubMed] [Google Scholar]
- 116.Keskin DB, Anandappa AJ, Sun J et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial Nature, 565, 234–9 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nguyen TK, Perry J, Sundaram ANE et al. Rescue bevacizumab following symptomatic pseudoprogression of a tectal glioma post-radiotherapy: a case report and review of the literature J Neurooncol, 143, 475–81 (2019) [DOI] [PubMed] [Google Scholar]
- 118.Foster KA, Ares WJ, Pollack IF et al. Bevacizumab for symptomatic radiation-induced tumor enlargement in pediatric low grade gliomas Pediatric blood & cancer, 62, 240–5 (2015) [DOI] [PubMed] [Google Scholar]
- 119.Carter BW, Bhosale PR, Yang WT. Immunotherapy and the role of imaging Cancer, 124, 2906–22 (2018) [DOI] [PubMed] [Google Scholar]
- 120.Nishino M, Giobbie-Hurder A, Gargano M et al. Developing a Common Language for Tumor Response to Immunotherapy: Immune-Related Response Criteria Using Unidimensional Measurements Clinical Cancer Research, 19, 3936–43 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wolchok JD, Hoos A, O’Day S et al. Guidelines for the Evaluation of Immune Therapy Activity in Solid Tumors: Immune-Related Response Criteria Clinical Cancer Research, 15, 7412–20 (2009) [DOI] [PubMed] [Google Scholar]
- 122.Brandsma D, Stalpers L, Taal W et al. Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas The Lancet Oncology, 9, 453–61 (2008) [DOI] [PubMed] [Google Scholar]
- 123.Roth P, Valavanis A, Weller M. Long-term control and partial remission after initial pseudoprogression of glioblastoma by anti–PD-1 treatment with nivolumab Neuro-Oncology, now265 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Furtado AD, Ceschin R, Blüml S et al. Neuroimaging of Peptide-based Vaccine Therapy in Pediatric Brain Tumors Neuroimaging Clinics of North America, 27, 155–66 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bollineni VR, Kramer GM, Jansma EP et al. A systematic review on [18F]FLT-PET uptake as a measure of treatment response in cancer patients European journal of cancer, 55, 81–97 (2016) [DOI] [PubMed] [Google Scholar]
- 126.Lasalle T, Austin EE, Rigney G et al. Granzyme B PET imaging of immune-mediated tumor killing as a tool for understanding immunotherapy response Journal for immunotherapy of cancer, 8, e000291 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tavare R, Escuin-Ordinas H, Mok S et al. An Effective Immuno-PET Imaging Method to Monitor CD8-Dependent Responses to Immunotherapy Cancer Research, 76, 73–82 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Seo JW, Tavaré R, Mahakian LM et al. CD8+T-Cell Density Imaging with64Cu-Labeled Cys-Diabody Informs Immunotherapy Protocols Clinical Cancer Research, (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lesniak WG, Chatterjee S, Gabrielson M et al. PD-L1 Detection in Tumors Using [64Cu]Atezolizumab with PET Bioconjugate Chemistry, 27, 2103–10 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lynes JP, Nwankwo AK, Sur HP et al. Biomarkers for immunotherapy for treatment of glioblastoma Journal for immunotherapy of cancer, 8, e000348 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Saratsis AM, Yadavilli S, Magge S et al. Insights into pediatric diffuse intrinsic pontine glioma through proteomic analysis of cerebrospinal fluid Neuro-Oncology, 14, 547–60 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Siravegna G, Marsoni S, Siena S et al. Integrating liquid biopsies into the management of cancer Nature Reviews Clinical Oncology, 14, 531–48 (2017) [DOI] [PubMed] [Google Scholar]
- 133.Nierkens S, Lankester AC, Egeler RM et al. Challenges in the harmonization of immune monitoring studies and trial design for cell-based therapies in the context of hematopoietic cell transplantation for pediatric cancer patients Cytotherapy, 17, 1667–74 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Keustermans GCE, Hoeks SBE, Jenny et al. Cytokine assays: An assessment of the preparation and treatment of blood and tissue samples Methods, 61, 10–7 (2013) [DOI] [PubMed] [Google Scholar]
- 135.Gust J, Finney OC, Li D et al. Glial injury in neurotoxicity after pediatric CD19-directed chimeric antigen receptor T cell therapy Annals of neurology, 86, 42–54 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yaghoubi SS, Jensen MC, Satyamurthy N et al. Noninvasive detection of therapeutic cytolytic T cells with 18F–FHBG PET in a patient with glioma Nat Clin Pract Oncol, 6, 53–8 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Najjar AM, Manuri PR, Olivares S et al. Imaging of Sleeping Beauty-Modified CD19-Specific T Cells Expressing HSV1-Thymidine Kinase by Positron Emission Tomography, 18, 838–48 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Weist MR, Starr R, Aguilar B et al. PET of Adoptively Transferred Chimeric Antigen Receptor T Cells with 89Zr-Oxine Journal of Nuclear Medicine, 59, 1531–7 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Akhavan D, Alizadeh D, Wang D et al. CAR T cells for brain tumors: Lessons learned and road ahead Immunological Reviews, 290, 60–84 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Griffiths JI, Wallet P, Pflieger LT et al. Circulating immune cell phenotype dynamics reflect the strength of tumor–immune cell interactions in patients during immunotherapy Proceedings of the National Academy of Sciences, 201918937, 117, 16072–16082 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brossart P. The role of antigen-spreading in the efficacy of immunotherapies Clinical Cancer Research, clincanres.0305, 26, 4442–4447 (2020) [DOI] [PubMed] [Google Scholar]
- 142.Chapuis AG, Roberts IM, Thompson JA et al. T-Cell Therapy Using Interleukin-21–Primed Cytotoxic T-Cell Lymphocytes Combined With Cytotoxic T-Cell Lymphocyte Antigen-4 Blockade Results in Long-Term Cell Persistence and Durable Tumor Regression Journal of Clinical Oncology, 34, 3787–95 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kahlon KS, Brown C, Cooper LJN et al. Specific Recognition and Killing of Glioblastoma Multiforme by Interleukin 13-Zetakine Redirected Cytolytic T Cells Cancer Research, 64, 9160–6 (2004) [DOI] [PubMed] [Google Scholar]
- 144.Sampson JH, Heimberger AB, Archer GE et al. Immunologic Escape After Prolonged Progression-Free Survival With Epidermal Growth Factor Receptor Variant III Peptide Vaccination in Patients With Newly Diagnosed Glioblastoma Journal of Clinical Oncology, 28, 4722–9 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Weller M, Butowski N, Tran DD et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial The Lancet Oncology, 18, 1373–85 (2017) [DOI] [PubMed] [Google Scholar]
- 146.Phuphanich S, Wheeler CJ, Rudnick JD et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma Cancer Immunol Immunother, 62, 125–35 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lang FF, Conrad C, Gomez-Manzano C et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma Journal of Clinical Oncology, 36, 1419–27 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carbognin L, Pilotto S, Milella M et al. Differential Activity of Nivolumab, Pembrolizumab and MPDL3280A according to the Tumor Expression of Programmed Death-Ligand-1 (PD-L1): Sensitivity Analysis of Trials in Melanoma, Lung and Genitourinary Cancers PLOS ONE, 10, e0130142 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sharma P, Callahan MK, Bono P et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open-label, two-stage, multi-arm, phase 1/2 trial The Lancet Oncology, 17, 1590–8 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Berghoff AS, Kiesel B, Widhalm G et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma Neuro-Oncology, 17, 1064–75 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nduom EK, Wei J, Yaghi NK et al. PD-L1 expression and prognostic impact in glioblastoma Neuro-Oncology, 18, 195–205 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kambhampati M, Panditharatna E, Yadavilli S et al. Harmonization of postmortem donations for pediatric brain tumors and molecular characterization of diffuse midline gliomas Scientific Reports, 10 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.McDonald BR, Contente-Cuomo T, Sammut S-J et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer Science Translational Medicine, 11, eaax7392 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Markus H, Contente-Cuomo T, Farooq M et al. Evaluation of pre-analytical factors affecting plasma DNA analysis Scientific Reports, 8 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Panditharatna E, Kilburn LB, Aboian MS et al. Clinically Relevant and Minimally Invasive Tumor Surveillance of Pediatric Diffuse Midline Gliomas Using Patient-Derived Liquid Biopsy Clinical Cancer Research, 24, 5850–9 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Santiago-Dieppa DR, Steinberg J, Gonda D et al. Extracellular vesicles as a platform for ‘liquid biopsy’ in glioblastoma patients Expert Review of Molecular Diagnostics, 14, 819–25 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.García-Romero N, Carrión-Navarro J, Esteban-Rubio S et al. DNA sequences within glioma-derived extracellular vesicles can cross the intact Blood-Brain Barrier and be detected in peripheral blood of patients Oncotarget, 8, 1416–1428 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shi R, Wang P-Y, Li X-Y et al. Exosomal levels of miRNA-21 from cerebrospinal fluids associated with poor prognosis and tumor recurrence of glioma patients Oncotarget, 6, 26971–81 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Greytak SR, Engel KB, Parpart-Li S et al. Harmonizing Cell-Free DNA Collection and Processing Practices through Evidence-Based Guidance Clinical Cancer Research, clincanres.3015, 26, 3104–3109 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Murtaza M, Dawson S-J, Tsui DWY et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA Nature, 497, 108–12 (2013) [DOI] [PubMed] [Google Scholar]
- 161.Mueller S, Jain P, Liang WS et al. A pilot precision medicine trial for children with diffuse intrinsic pontine glioma-PNOC003: A report from the Pacific Pediatric Neuro-Oncology Consortium Int. J. Cancer, 145, 1889–901 (2019) [DOI] [PubMed] [Google Scholar]
- 162.Gupta N, Goumnerova LC, Manley P et al. Prospective feasibility and safety assessment of surgical biopsy for patients with newly diagnosed diffuse intrinsic pontine glioma Neuro-oncology, 20, 1547–55 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]