

Abstract
The response to Poly (ADP-ribose) polymerase inhibitors (PARPi) is dictated by homologous recombination (HR) DNA repair and the abundance of lesions that trap PARP enzymes. It remains unclear, however, if the established role of PARP in promoting chromatin accessibility impacts viability in these settings. Using a CRISPR-based screen, we identify the PAR-binding chromatin remodeler, ALC1/CHD1L, as a key determinant of PARPi toxicity in HR-deficient cells. ALC1 loss reduced viability of BRCA-mutant cells and enhanced sensitivity to PARPi by up to 250-fold, while overcoming several resistance mechanisms. ALC1 deficiency reduced chromatin accessibility concomitant with a decrease in the association of base damage repair factors. This resulted in an accumulation of replication associated DNA damage, increased PARP trapping, and a reliance on HR. These findings establish PAR-dependent chromatin remodeling as a mechanistically distinct aspect of PARPi responses and therapeutic target in HR-deficient cancers.
Introduction
The complex chromatin environment of eukaryotic genomes necessitates rapid nucleosome remodeling events in response to specific cues. Poly (ADP-ribose) polymerases, PARP1 and PARP2, are ideally suited to sense and transduce DNA damage signals through their high affinity interactions with DNA lesions, which allosterically activate PARP enzymatic activity1,2. PARP dependent histone PARylation promotes the rapid recruitment of PAR-binding effector proteins and mediates chromatin decompaction3–6.While PAR-recognition is critical to a myriad of damage responses7–15, the extent to which PARylation-directed chromatin remodeling impacts these pathways is less understood.
PARP inhibitors (PARPi) are selectively toxic in homologous recombination (HR)-deficient cells16,17. PARPi increases requirements for BRCA-dependent HR 18 in part by trapping the PARP enzymes on chromatin19. Acquired resistance due to HR restoration, reduced PARP1 trapping, and drug efflux are major limitations to PARPi clinical efficacy20. Rational design of orthogonal approaches to overcome resistance are therefore needed.
Here we reveal the PAR-dependent nucleosome sliding enzyme, ALC1/CHD1L (Amplified in Liver Cancer 1), as a key determinant of PARPi toxicity in BRCA-mutant cells. ALC1 deficiency conferred up to 250-fold increases in PARPi sensitivity in HR-deficient cells and overcame several resistance mechanisms as a consequence of this expanded therapeutic window. ALC1 function in the damage response was reliant on its ability to alter chromatin structure in a cooperative manner with PARP activity. These features uncover PARP-dependent chromatin accessibility as a vulnerability in HR-deficient cancers.
Results
Loss of ALC1 confers PARPi hypersensitivity in BRCA-mutant cells
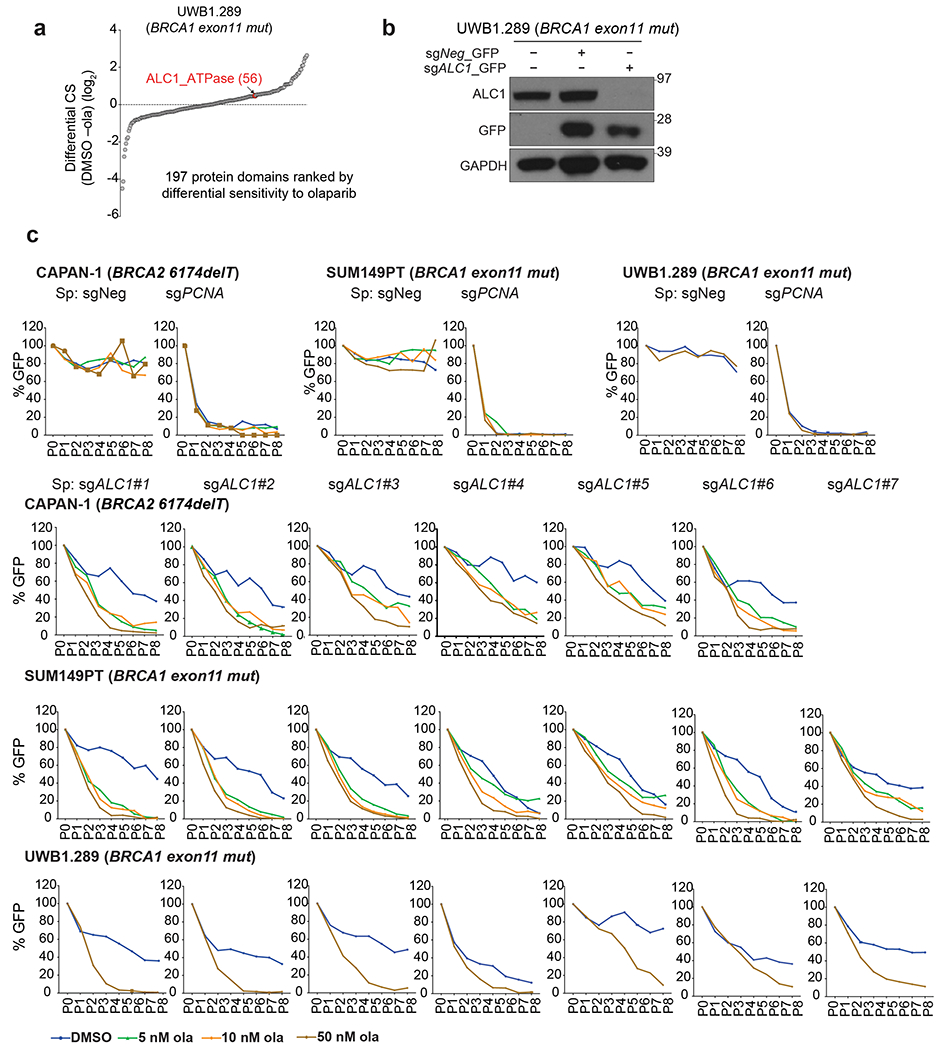
We performed a CRISPR-Cas9 genetic screen in BRCA-mutant cells to identify loss-of-function mutations in chromatin regulators that generate PARPi hypersensitivity. The single-guide RNAs (sgRNAs) targeted functional domains, an approach that imparts higher editing efficiency21. A sgRNA library targeting 197 functional domains of 179 chromatin regulators was transduced into Streptococcus pyogenes (SpCas9) expressing BRCA-mutant cells. These included the BRCA1 exon 11 mutant ovarian and breast cancer cell lines UWB1.289 and SUM149PT, respectively, and CAPAN-1, a pancreatic cancer line that harbors the 6174delT BRCA2 mutation. The screen was performed at 10 nM olaparib, which approximates the lethal dose 20 for these BRCA-mutant lines in a two-week clonogenic assay (Fig.1a, Supplementary Table1).
Fig. 1. Loss of ALC1 reduces proliferation and confers olaparib hypersensitivity in BRCA-mutant cells.
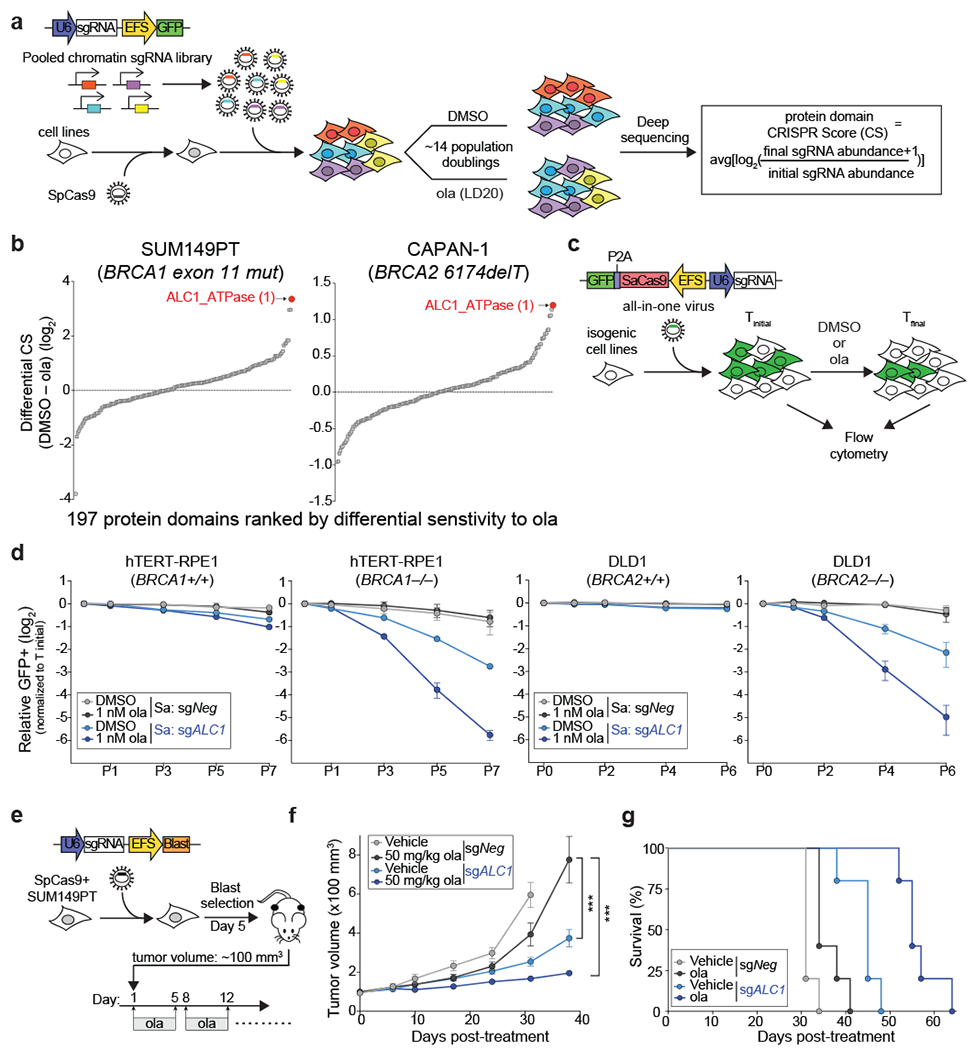
a, Schematic of the CRISPR screen to identify regulators of olaparib (ola) sensitivity. b, Protein domains ranked on the basis of CRISPR score (CS) for ola sensitivity in BRCA1-mutant SUM149PT cells (left) and BRCA2-mutant CAPAN-1 cells (right). c, Schematic of the GFP competition experiments. For a given cell line, Tinitial indicates the day when maximum GFP expression is achieved for a sgRNA targeting an essential gene. Tfinal indicates the final day of the data collection. d, GFP competition assay in isogenic BRCA-mutant lines upon transduction of sgNeg or sgALC1 (n=3-6 independent transductions). Data are mean ± s.e.m., normalized to Tinitial. After every two population doublings, cells were passaged (P) and percent GFP was recorded. e, Schematic of the xenograft experiment. f, Measurement of tumor volume after ola treatment was initiated (n=10 tumors for sgNeg + ola, sgALC1 + vehicle, sgALC1 + ola and n=8 tumors for sgNeg + vehicle). Data are mean ± s.e.m., p-values are derived from a 2-way ANOVA. g, Kaplan-Meier survival analysis (n=5 mice per group). A significantly improved survival was observed in vehicle (light blue) and ola (dark blue) treated sgALC1 xenografts compared to the ola treated sgNeg counterpart (black), p=0.04 and p=0.0015 respectively using Log-rank (Mantel-Cox) test. ***P ≤ 0.001. Cas9 from S. aureus (Sa) and S. pyogenes (Sp) was used as indicated. Source data are provided.
ALC1 loss conferred olaparib hypersensitivity across all cell lines and ranked as the top hit in both SUM149PT and CAPAN-1 cells (Fig.1b, Extended Data Fig.1a). Olaparib hypersensitivity was validated in a growth-based competition assay that monitors changes in the percent of GFP co-expressed from the sgRNA vector (Fig.1c and Extended Data Fig.1b, c). Loss of ALC1 also conferred a growth defect in all lines tested, albeit to a lesser degree in UWB1.289 cells, suggesting a synthetic sick relationship between ALC1 loss and BRCA deficiency.
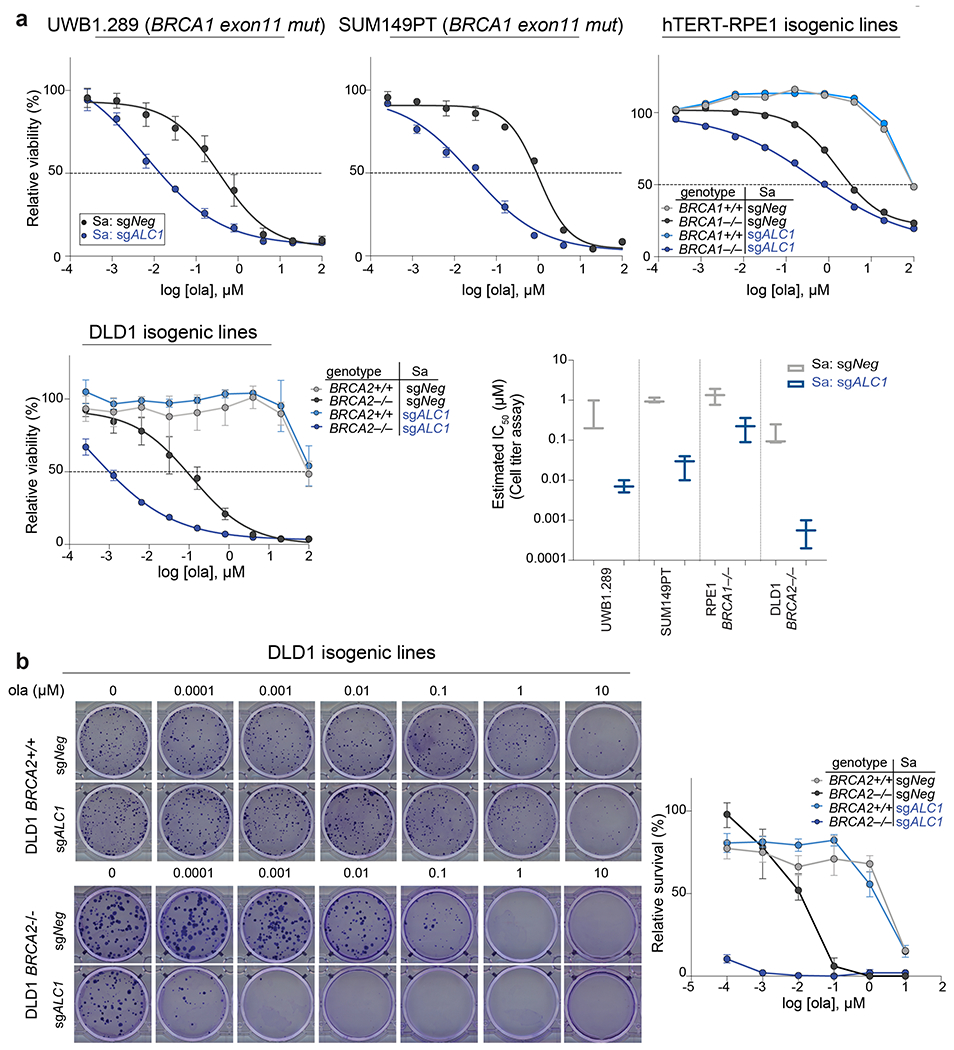
We next employed paired isogenic cell lines to examine if ALC1 loss selectively impacts PARPi sensitivity of BRCA-knockout (KO) cells in comparison to their wild-type (WT) counterparts. We utilized the dual Staphylococcus aureus SaCas9 and SpCas9 approach to achieve efficient simultaneous knockdown of two proteins22. Loss of ALC1 selectively impaired proliferation in both BRCA1 KO hTERT- retinal pigmented epithelial cells (RPE1) and BRCA2 KO colon cancer DLD1 cells, revealing a synthetic sick relationship with BRCA loss. Additionally, ALC1 depletion sensitized BRCA-mutant cells to low doses of olaparib (1 nM), while having minimal effects on their WT counterparts (Fig.1d). Given the half-maximal inhibitory concentration (IC50) of olaparib for purified PARP1 is 5 nM23, ALC1 loss appears to sensitize HR-deficient cells to PARPi concentrations that do not inhibit the majority of cellular PARP activity. Moreover, clonogenic survival and cellular ATP content-based viability assays revealed up to ~250-fold decreases in IC50 for PARPi upon ALC1 loss in several BRCA1- and BRCA2-mutant cell lines (Extended Data Fig.2, 3a).
To examine the impact of ALC1 in a BRCA-mutant tumor model, SUM149PT Cas9 cells were transduced with either sgNegative (sgNeg) or sgALC1 and the heterogeneous pool of cells was subcutaneously implanted in NSG (NOD-scid gamma) mice on day 5 after transduction, when maximum sgRNA expression occurs. The gap between sgRNA expression and protein depletion with CRISPR editing allowed the tumors to reach ~100 mm3, when olaparib treatment was initiated (Fig.1e). Tumor growth in sgALC1 xenografts was significantly slower compared to the olaparib treated sgNeg group, consistent with the synthetic sick interaction between ALC1 loss and BRCA mutation. The volume of PARPi treated sgNeg tumors increased by ~7.8-fold compared to a ~2-fold increase in sgALC1 counterparts by 38 days post olaparib administration (Fig. 1f). Kaplan-Meier analyses indicated that the median overall survival was significantly longer for untreated sgALC1 group compared to the olaparib administered sgNeg counterparts, and further extended in the PARPi-treated sgALC1 cohort (Fig.1g). Immuno-blot of late stage sgALC1 tumors revealed residual ALC1 protein, potentially resulting from cells that escaped genetic editing. In contrast, ALC1 was not detected 15 days post-transduction in sgALC1 cells that were used to inoculate the mice (Extended Data Fig.3b). These findings suggest that selective pressure exists to maintain ALC1 expression in BRCA-mutant tumors.
PARPi sensitivity in ALC1 deficient cells is not epistatic with other DNA repair pathways
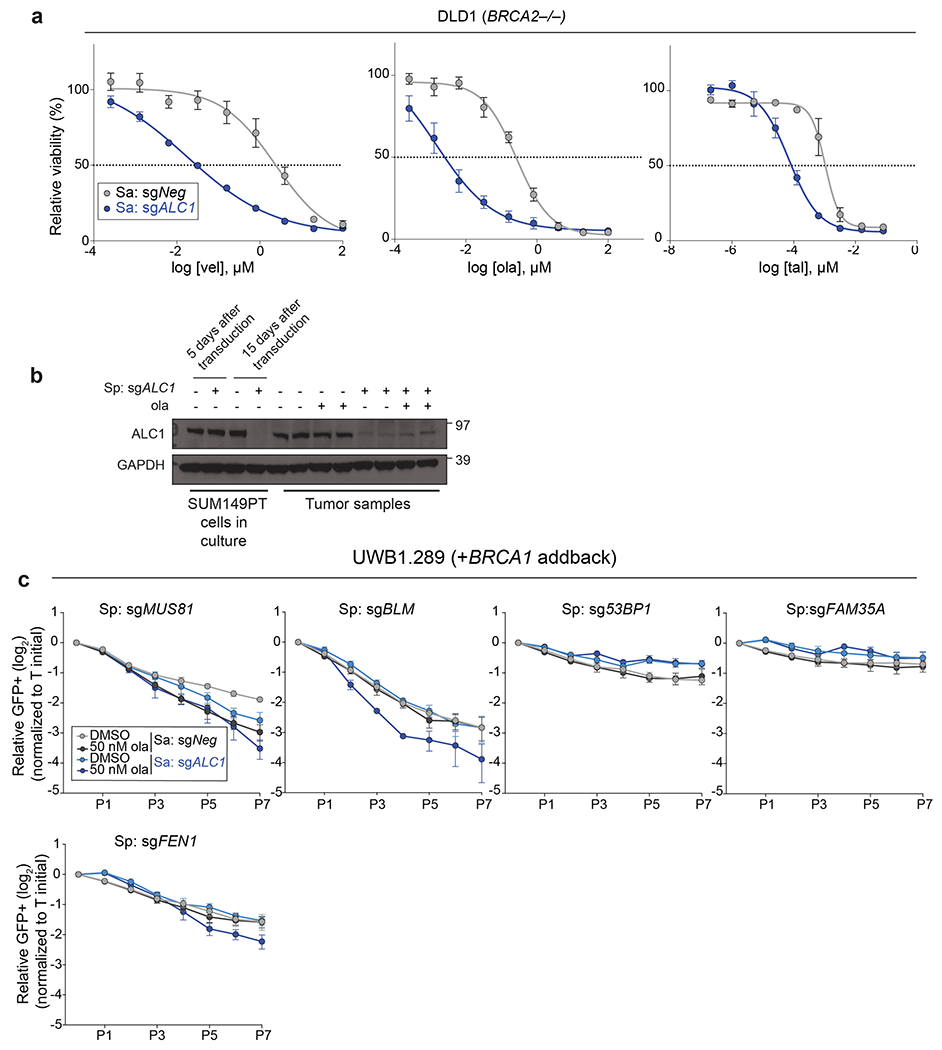
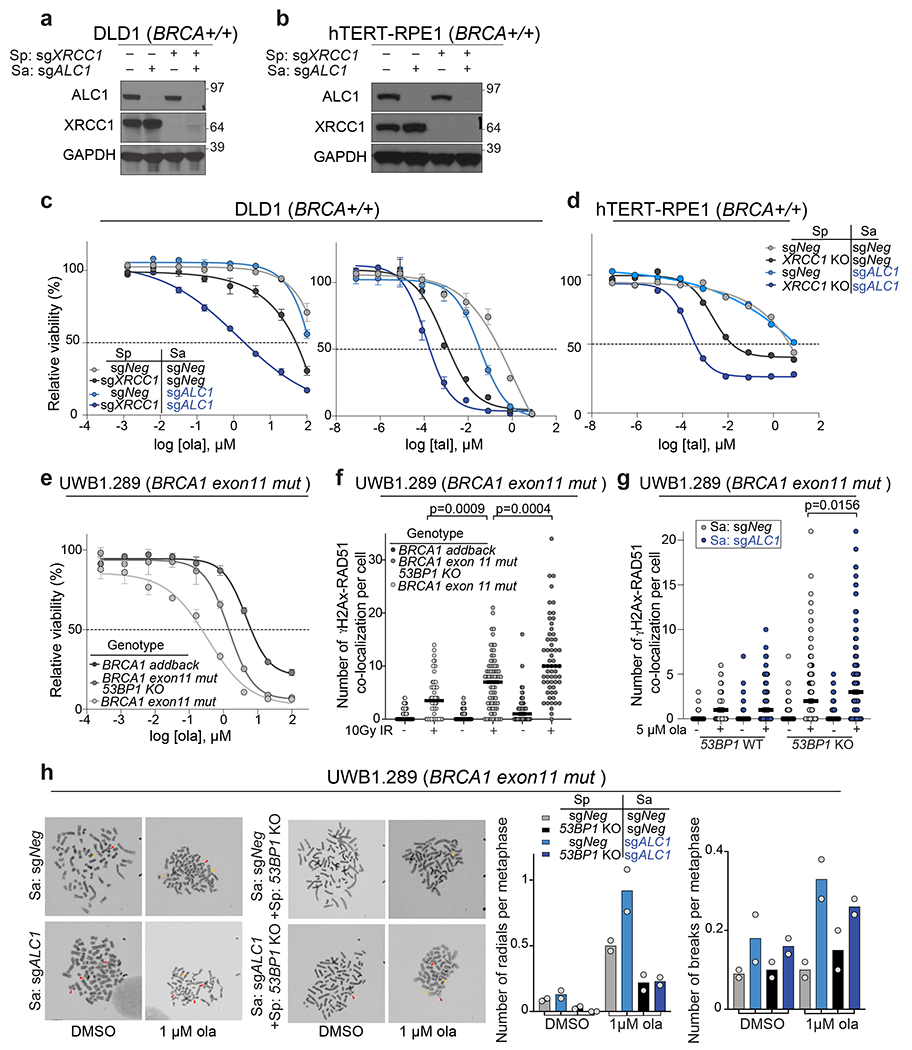
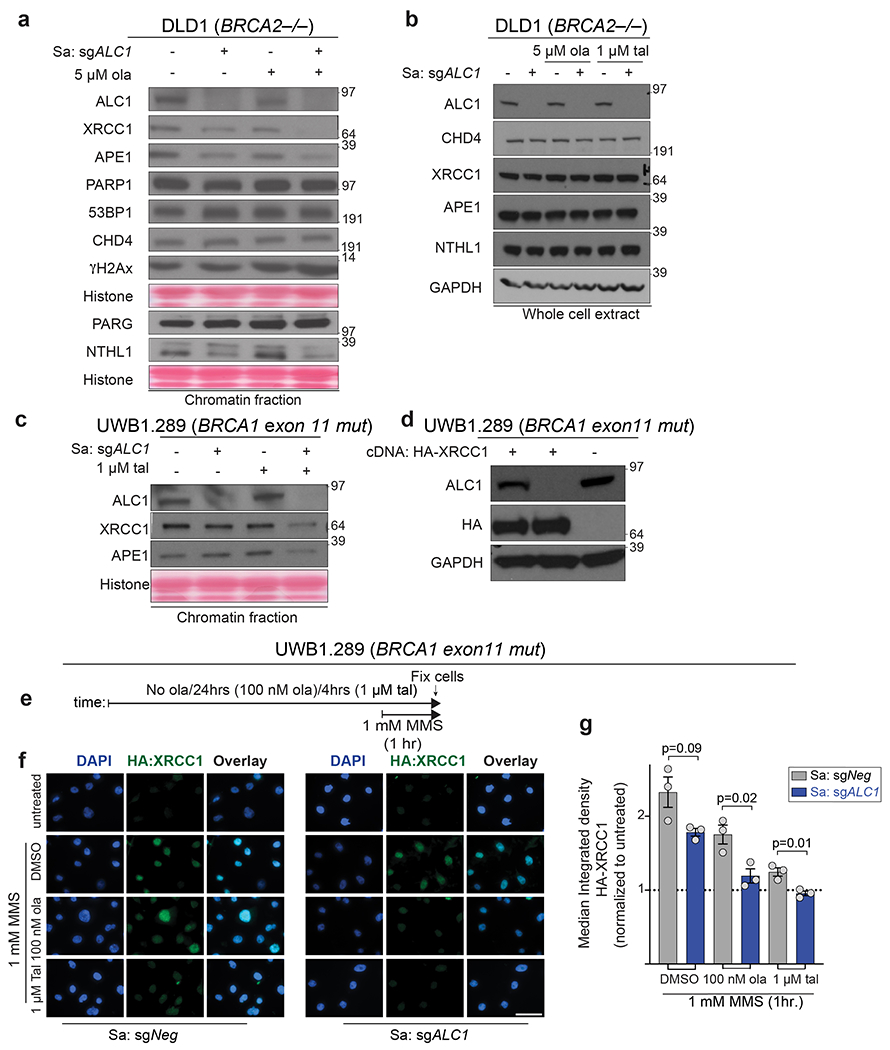
We systematically examined epistasis between ALC1 and a selected group of DNA repair pathways for PARPi response. An arrayed sgRNA library was designed that targeted genes in the HR, non-homologous end joining (NHEJ), single-strand break repair (SSBR), nucleotide excision repair (NER) and microhomology-mediated end joining (MMEJ) pathways. The proliferation of BRCA1-proficient UWB1.289 ALC1 KO cells was monitored following the loss of targeted genes in the absence or presence of 50 nM olaparib (Fig.2a). Targeting ATR, BLM, SLX4, Mus81 and Fen1 selectively sensitized ALC1 KO cells to 50 nM olaparib (Fig.2b, Extended Data Fig.3c). ALC1 deficiency was also synthetically sick with XRCC1 loss and further increased PARPi sensitivity, suggesting that ALC1 loss may generate lesions that increase reliance on SSBR in addition to HR. Reciprocal experiments with ALC1 depletion in both sgXRCC1 DLD1 cells and XRCC1 KO hTERT-RPE1 cells also enhanced PARPi sensitivity. Notably, loss of ALC1 alone conferred a modest increase in talazoparib sensitivity of BRCA-proficient DLD1 cancer cells, while having no effects in hTERT-RPE1 counterparts (Extended Data Fig.4a–d). ALC1 inactivation did not affect PARPi response following the loss of c-NHEJ or NER proteins (Fig.2b, Extended Data Fig.3c). Collectively, these findings reveal that ALC1 loss is not epistatic to either of the primary determinants of PARPi response, HR or SSBR24,25.
Fig. 2. ALC1 loss causes PARPi hypersensitivity in HR-deficient cells.
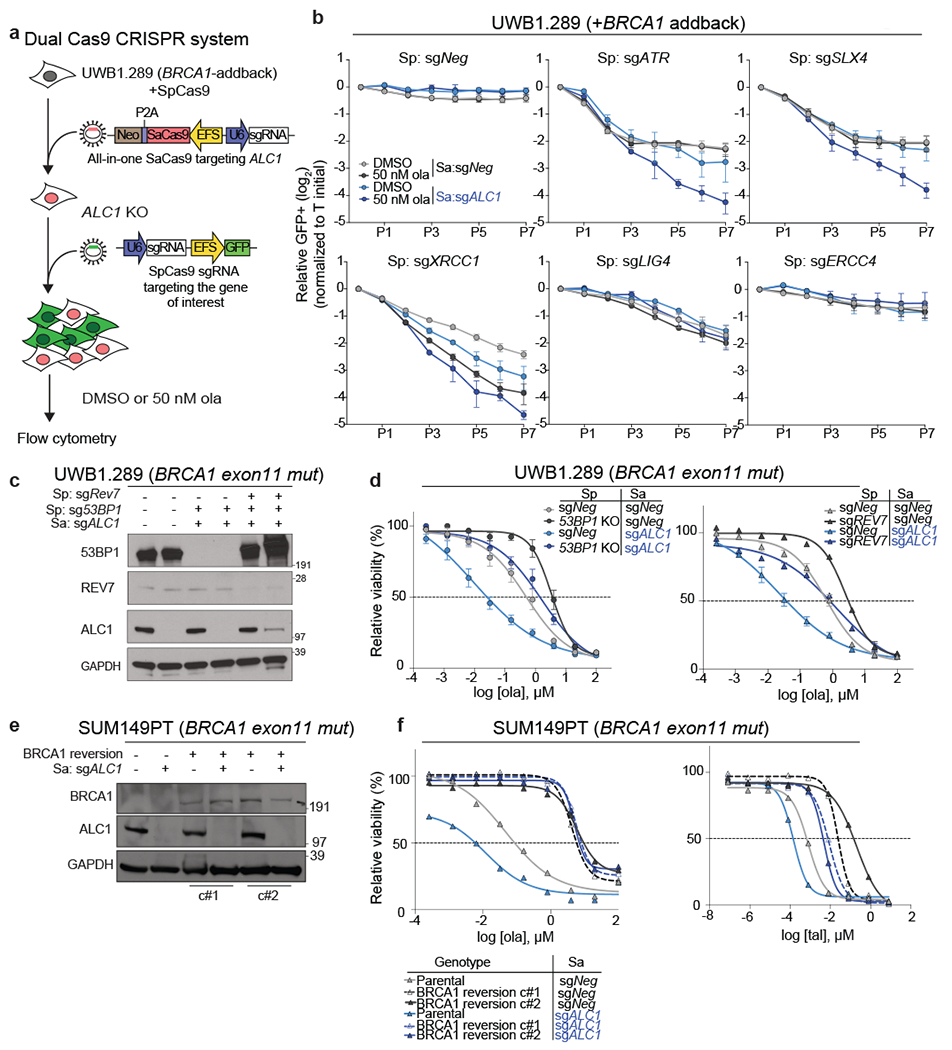
a, Schematic of the dual Cas9 CRISPR system for the GFP competition experiments. b, GFP competition experiment in the UWB1.289+BRCA1 addback line to examine cell proliferation and ola sensitivity following the combined loss of ALC1 and the indicated DNA repair protein. Data are mean ± s.e.m., normalized to Tinitial. After every two population doublings, cells were passaged (P) and percent GFP was recorded (n=4 independent transductions, except for sgXRCC1 and sgERCC4, where n = 6 independent transductions were made). c, Immunoblot showing ALC1, 53BP1 and Rev7 depletion in UWB1.289 cells. Consistent results were obtained across two independent blots. d, Sensitivities of the indicated UWB1.289 cell lines to ola using the CellTiter-Glo assay; n=3 biologically independent experiments. Data are mean ± s.e.m. e, Immunoblot showing BRCA1 and ALC1 levels in the indicated SUM149PT cells. Consistent results were obtained across two independent blots. f, Sensitivities of the indicated SUM149PT cell lines to ola (left) and talazoparib (tal) (right) using the CellTiter-Glo assay. c#1 and c#2 indicate two different clones with restored BRCA1 reading frames. Data are mean from 2 biologically independent experiments. Source data are provided.
ALC1 deficiency restores PARPi sensitivity in cells with engineered resistance
The profound PARPi response in ALC1-deficient BRCA-mutant cells led us to examine if this relationship could be leveraged to overcome therapeutic resistance. Loss of 53BP1 or Rev7 permits partial restoration of HR in BRCA1-mutant cells coincident with both reduced radial chromosome formation and PARPi resistance26–28. In agreement, PARPi sensitivity and Rad51 foci formation in 53BP1 KO UWB1.289 cells were intermediate between the parental and BRCA1-addback counterparts (Extended Data Fig.4e–f). ALC1 loss in either 53BP1- or Rev7-deficient BRCA1-mutant cells restored PARPi sensitivity, albeit at levels comparable to the parental control (Fig.2c, d). ALC1-deficient UWB1.289 cells formed increased radial chromosomes upon PARPi treatment that were eliminated by concomitant 53BP1 depletion (Extended Data Fig.4g–h). Increased chromosome breaks and Rad51 foci were still observed in PARPi treated ALC1-deficient 53BP1 KO BRCA1-mutant cells, potentially accounting for the continued drug sensitivity.
We extended this analysis to reversion mutations that produced functional BRCA1 protein. ALC1 loss failed to increase olaparib sensitivity in SUM149PT cells that have become resistant owing to the restoration of the BRCA1 reading frame29. In contrast, some degree of sensitivity was observed with the more potent PARPi, talazoparib (Fig.2e, f).
Reduced PARP1 expression renders PARPi resistance, suggesting that PARP1 trapping is the major contributor to PARPi cytotoxicity19,30. Remarkably, cells depleted of both PARP1 and ALC1 remained PARPi hypersensitive compared to the parental controls (Fig.3a–c). PARPi also traps PARP219,31 potentially accounting for the continued sensitivities in ALC1 and PARP1 depleted cells. Using a quantitative immunofluorescence assay, we observed increased PARP1 and PARP2 trapping in ALC1-deficient cells compared to the parental control (Extended Data Fig.5). Combined loss of PARP1 and PARP2 rendered ALC1-deficient BRCA1-mutant cells fully resistant to PARPi, whereas PARP1 loss alone caused resistance when ALC1 was present (Fig.3a–c). We propose that ALC1 loss increases the abundance of genomic lesions that trap both PARP1 and PARP2.
Fig. 3. ALC1 loss mitigates PARPi resistance in BRCA-mutant cells that are deficient in PARP1 or PARG.
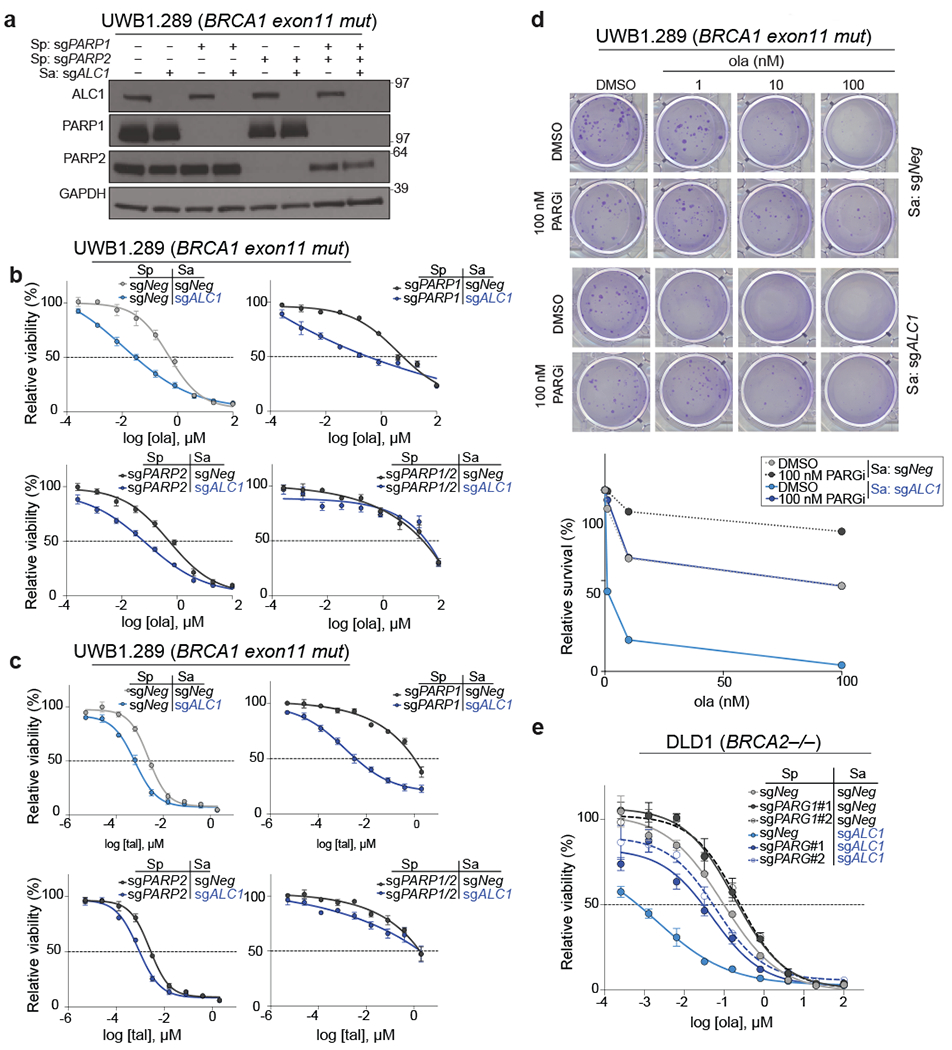
a, Immunoblot showing ALC1, PARP1 and PARP2 depletion in UWB1.289 cells. Consistent results were obtained across two independent blots. b-c, Sensitivities of the indicated UWB1.289 cell lines to ola (b) and tal (c) using the CellTiter-Glo assay. Data are mean ± s.e.m. from n = three biologically independent experiments. d, Representative images (top) and quantification (bottom) of the clonogenic survival assay of UWB1.289 cells treated with the indicated doses of ola and PARG inhibitor (PARGi). Colonies with more than 50 cells were included in the quantification. Data are mean from two biologically independent experiments. e, Sensitivities of the indicated DLD1 BRCA2−/− cell lines to ola using the CellTiter-Glo assay. sgPARG#1 and sgPARG#2 indicate two sgRNAs targeting PARG. Data are mean ± s.e.m. from n = three biologically independent experiments. Source data are provided.
Loss of poly (ADP-ribose) glycohydrolase (PARG) results in PAR accumulation concomitant with less PARP trapping and resistance32. ALC1 depletion restored olaparib sensitivity at levels comparable to parental controls in cells treated with either an inhibitor or sgRNA targeting PARG (Fig.3d–e). These observations (Figs.2 and 3) demonstrate that ALC1 loss restores PARPi efficacy across a broad range of resistance mechanisms.
ALC1 deficiency increases genomic instability and reliance on BRCA-dependent HR
We next characterized the genomic lesions arising from ALC1 loss in BRCA-mutant cells. Depletion of ALC1 in DLD1 BRCA2-deficient cells increased breaks and radial chromosomes, which were exacerbated by olaparib treatment (Fig.4a,b). Loss of ALC1 increased Rad51 foci formation upon olaparib treatment in BRCA-proficient U-2 OS and UWB1.289 cells, suggesting the genesis of lesions that require HR (Fig.4c and Extended Data Fig.6a). ALC1 loss did not enhance sensitivity to cisplatin in BRCA-proficient or deficient settings, emphasizing that it does not contribute to homology-directed repair as is observed for factors that affect PARPi sensitivity through HR or MMEJ (Fig.4d)33.
Fig. 4. Loss of ALC1 increases genomic instability.
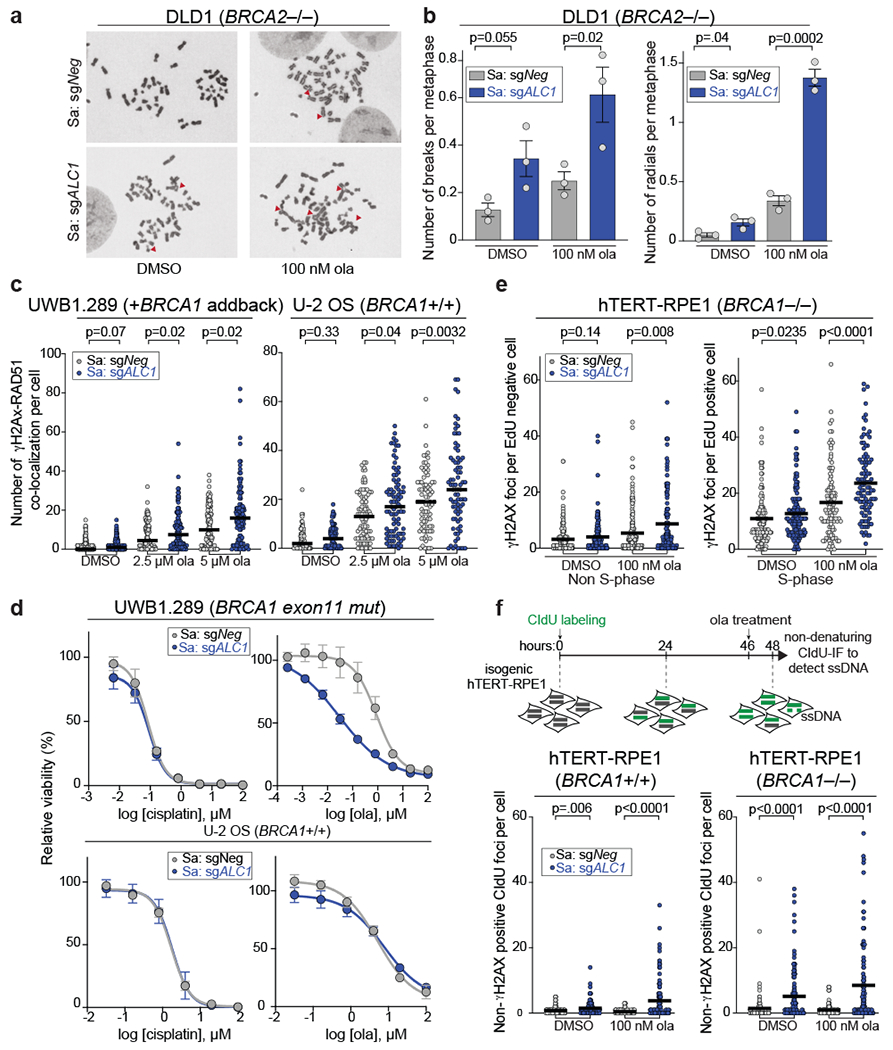
a-b, Representative images (a) of the chromosomal aberrations (indicated by red arrow heads) and quantification (b) of breaks and radials per metaphase upon ALC1 depletion in DLD1 BRCA2−/− cells. Data are mean ± s.e.m. from n = three biologically independent experiments, p-value, unpaired t-test. For each experiment, at least 50 spreads were analyzed per sample. c, Quantification of γH2AX-RAD51 co-localization in indicated UWB1.289 + BRCA1 add back and U-2 OS cells. Median is indicated. p-value determined by Mann-Whitney was derived from n≥67 cells examined over two biologically independent experiments. d, Sensitivities of the indicated cell lines to cisplatin and ola as determined by CellTiter-Glo assay. Data are mean ± s.e.m. from n = three biologically independent experiments. e, Quantification of γH2AX foci in non-S-phase (left) and S-phase (right) cells. Median is indicated. p-value determined by Mann-Whitney was derived from n≥99 cells examined over two biologically independent experiments. f, Schematic and quantification of non-γH2AX positive CIdU foci in the indicated cell lines to specifically detect single strand (ss)-DNA that was not generated by end-resection at DSBs. Median is indicated. p-value determined by Mann-Whitney was derived from n≥105 cells sampled over two biologically independent experiments. Cells were incubated with the indicated concentrations of ola for either 2 hrs (f) or 24 hrs (b, c, e). Source data are provided.
ALC1 depletion in BRCA-mutant cells increased γH2AX foci specifically in S-phase, consistent with the generation of replication-coupled DSBs (Fig.4e and Extended Data Fig.6b). We next quantified single strand-DNA (ss-DNA) using non-denaturing CIdU immunofluorescence. ALC1 loss resulted in a significant increase in non-γH2AX positive CIdU foci, an outcome that was more pronounced in BRCA-mutant settings and was further elevated upon olaparib treatment (Fig.4f).
ALC1 deficiency confers sensitivity to agents that induce single-strand breaks (SSBs)34–36. We could recapitulate the reported MMS sensitivity across several ALC1 depleted lines, with a notable exception in hTERT-RPE1 cells. BRCA loss exacerbated MMS sensitivity in ALC1 depleted cells consistent with channeling of unrepaired damage into HR10,37. In contrast, ALC1 depletion did not confer sensitivity to camptothecin (CPT) (Fig.5a, Extended Data Fig.6c). Repair of MMS damage necessitates base excision and nick generation. In contrast, CPT traps the TOP1 enzyme on already nicked DNA38. While repair of MMS induced lesions would require nucleosome sliding to provide accessibility to damaged bases, this may not be required for CPT induced lesions (Fig.5b). In agreement, ALC1 has been shown to function in the pre-incision step of NER by recognizing damaged nucleotides in a PARP1 dependent manner13. Moreover, ALC1 loss enhanced MMS but not CPT sensitivity in XRCC1 depleted cells (Fig. 5c and Extended Data Fig.6d).
Fig. 5. ALC1 function in base damage repair is not epistatic with HR and single-strand break repair (SSBR).
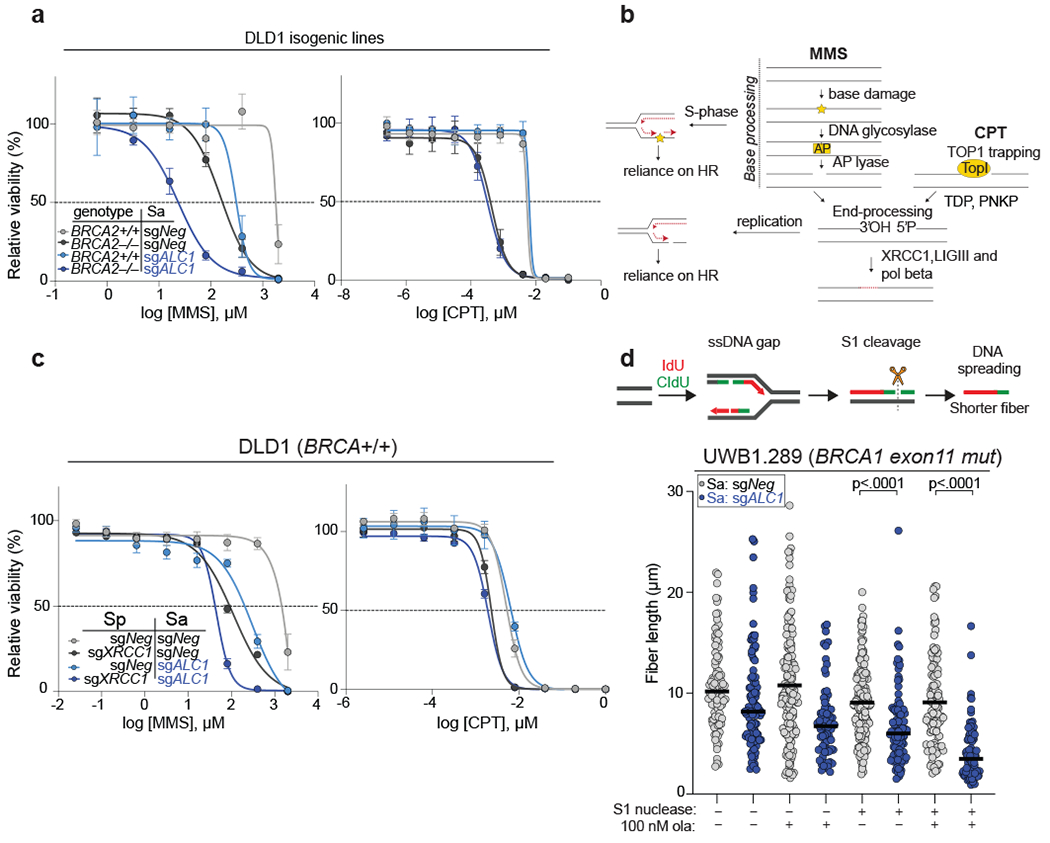
a, Sensitivities of the indicated DLD1 isogenic lines to methyl methanesulfonate (MMS) and camptothecin (CPT) in the CellTiter-Glo assay. Data are mean ± s.e.m. from n = three biologically independent experiments. b, Schematic of SSBR pathway. While base damage requires processing events at the chromatin, CPT traps TOP1 on already nicked substrates. Replication of damaged bases results in gaps and SSBs, that increase reliance on HR for repair. c, Sensitivities of the indicated DLD1 cell lines to MMS and CPT in the CellTiter-Glo assay. Data are mean ± s.e.m. from n = three biologically independent experiments. d, Schematic and quantification of fiber length in the absence and presence of S1 nuclease in the indicated cell lines. Median is indicated. p-value determined by Mann-Whitney was derived from n≥75 fibers sampled over two independent experiments. Cells were incubated with 100 nM ola for 24 hrs. Source data are provided.
The presence of persistent base lesions can lead to the generation of replication-coupled ss-gaps39. Indeed, loss of ALC1 increased the S1 nuclease sensitivity of nascent replication tracts and this was exacerbated by PARPi treatment (Fig.5d and Extended Data Fig.6e). Together, these results demonstrate that ALC1 prevents the accumulation of toxic genomic lesions that feed into SSB and HR repair pathways.
ALC1 localizes to chromatin in a PARP1 and PARP2 dependent manner
ALC1 recruitment to damaged chromatin requires PARylation36,40–42. We reasoned that owing to its nanomolar binding affinity for PAR chains42, ALC1 localization to damaged chromatin would not be diminished by low dose PARPi treatments. Treatment of cells with PARGi revealed accumulation of ALC1 specifically on S-phase chromatin. This suggests that ALC1 primarily associates with replicating DNA, consistent with PAR levels being highest in S-phase43. ALC1 chromatin association was evident in both replicating and non-S-phase cells following MMS treatment and did not require PARGi to elicit detection (Fig.6a,b). ALC1 chromatin localization was retained in cells pre-incubated with 100 nM olaparib for 24 hrs or after 4 hrs treatment with 5 μM olaparib or 1 μM talazoparib. Conversely, ALC1 localization was eliminated at 20 μM PARPi, consistent with the dose of olaparib required to nullify chromatin recruitment of other PAR-binding proteins (Extended Data Fig.7)44. In accordance with PAR-dependent recruitment, ALC1 chromatin association was abolished in cells lacking both PARP1 and PARP2, and ALC1 loss did not increase MMS sensitivity in these lines (Fig.6c–g).
Fig. 6. ALC1 function in the DNA damage response requires PARP1 and PARP2.
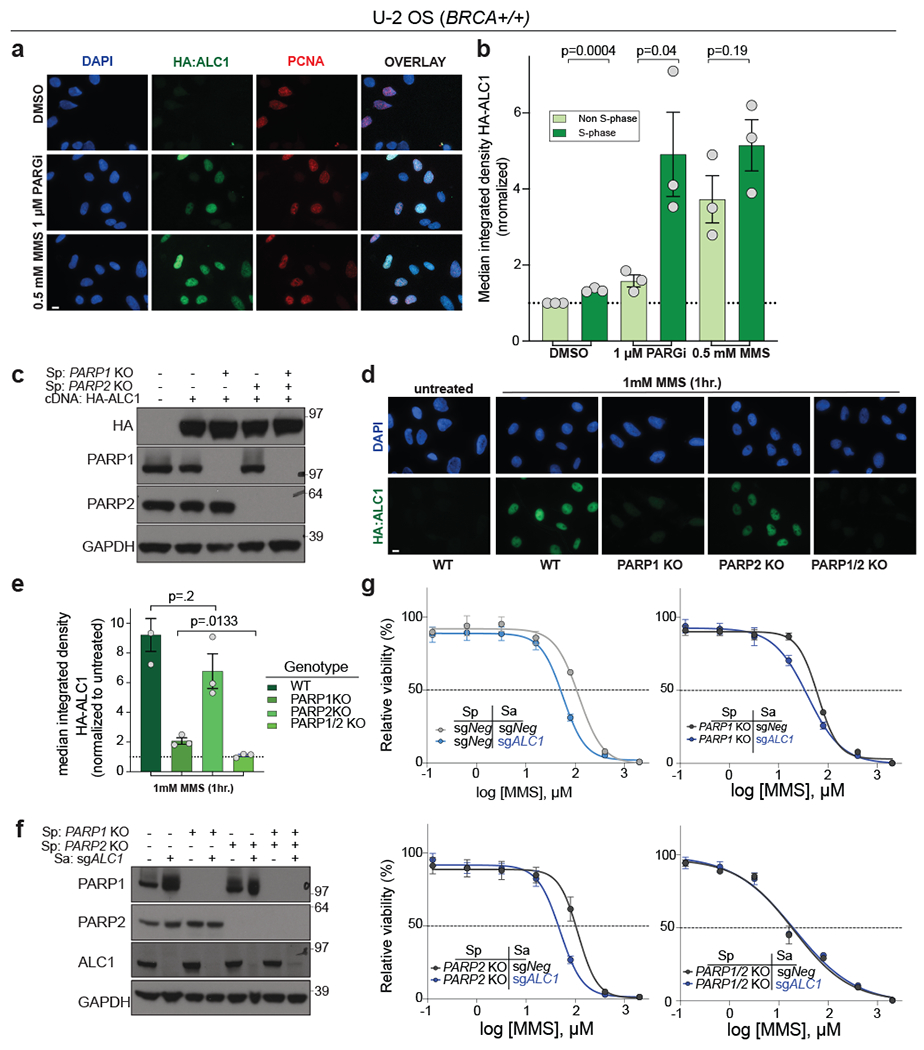
a-b, Representative images (a) and quantification (b) of HA-ALC1 localization to chromatin upon the indicated treatments. Scale bar, 10 microns. The median value was normalized to non-S-phase untreated control. Data are mean ± s.e.m. from n= three biologically independent experiments. p-value, unpaired Student’s t-test. For each experiment, at least 50 cells were analyzed per sample. c, Immunoblot showing stable expression of HA-ALC1 in indicated cell lines. Samples were analyzed once to examine the level of overexpression prior to localization experiments. d-e, Representative images (d) and quantification (e) of HA-ALC1 localization to chromatin, scale bar 10 microns. For each cell line, the median value upon MMS treatment was normalized to its respective untreated control. Data are mean ± s.e.m. from n=three biologically independent experiments, p-value, unpaired Student’s t-test. For each experiment, at least 50 cells were analyzed per sample. f, Immunoblot showing depletion of ALC1 in the indicated cell lines. Consistent results were obtained across two independent blots. g, Sensitivities of the indicated U-2 OS lines to MMS in the CellTiter-Glo assay. Data are mean ± s.e.m. from n=three biologically independent experiments. Source data are provided.
ALC1 responses to PARPi and MMS require its chromatin remodeling and PAR binding activities
We investigated the biochemical properties underlying ALC1 function in PARPi responses. The K77R mutation in the Walker A motif of the ALC1 ATPase domain abrogates its nucleosome sliding activity36. Overexpression of the K77R ALC1 mutant led to a pronounced loss of viability of BRCA-mutant cells, suggesting a dominant negative phenotype. Depletion of the endogenous ALC1 protein in the surviving population of K77R expressing cells resulted in PARPi hypersensitivity comparable to ALC1-deficient cells (Fig.7a,b and Extended Data Fig. 8a,b).
Fig. 7. ALC1 PAR-recognition and chromatin-remodeling activities are essential for responses to PARPi and MMS.
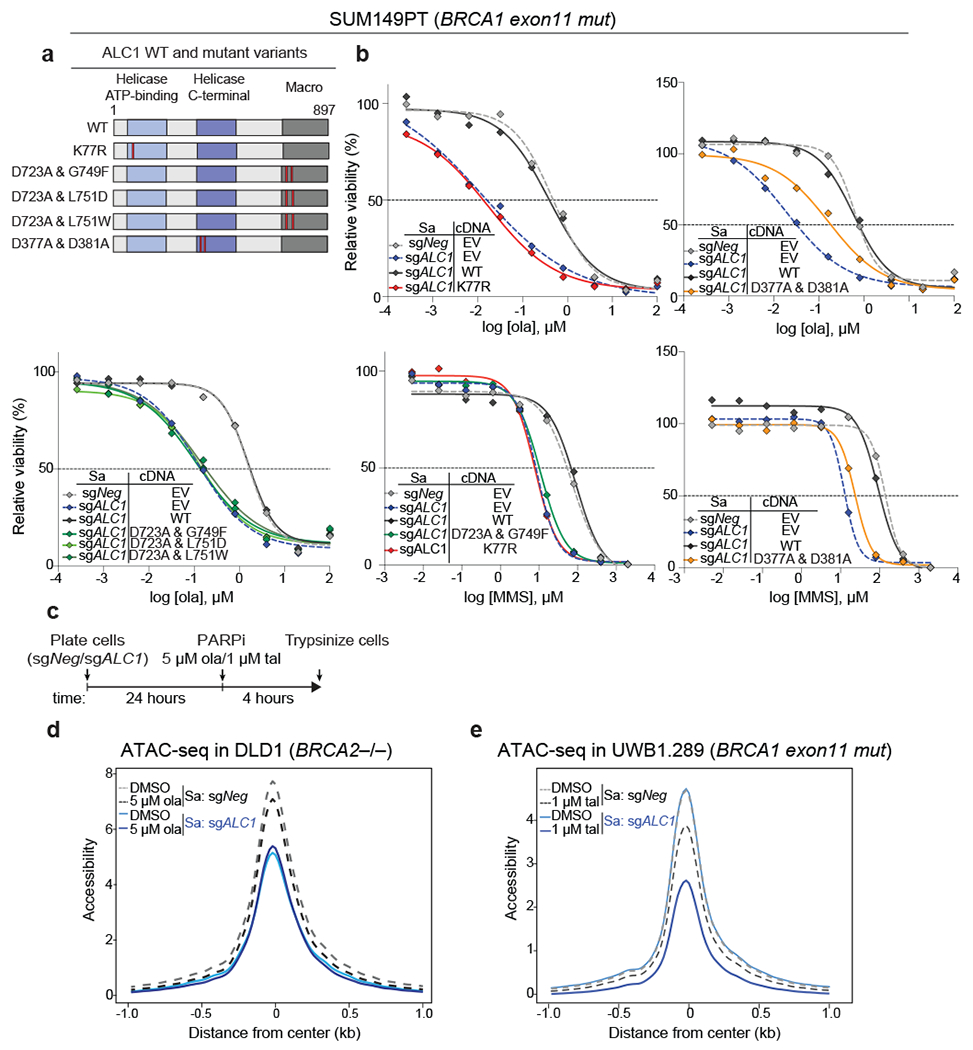
a, Domain organization and mutants of ALC1 used in this study. The helicase ATP-binding (light blue), helicase C-terminal (dark blue), and macro domains (grey) are indicated. Red bars show the position of the residues that were mutated. b, Sensitivities of SUM149PT cells expressing various ALC1 mutants to ola and MMS using the CellTiter-Glo assay (EV: empty vector and WT: wild type). Data are mean from two biologically independent experiments. c, Schematic of the experiment used to examine the effects of ALC1 loss and PARPi treatment on chromatin accessibility d, ATAC-seq analysis in DLD1 BRCA2−/− cells to assess global accessibility of chromatin. Cells were treated with ola for 4 hours. Data is from three biologically independent replicates for each condition. e, ATAC-seq analysis in UWB1.289 cells to assess global accessibility of chromatin. Cells were treated with tal for 4 hrs. Data is from three biologically independent replicates of untreated sgNeg and sgALC1+tal and two biologically independent replicates of sgNeg+tal and untreated sgALC1 cells. For plotting of both the ATAC-seq graphs (d,e) the 2000 base-pairs flanking the center of the accessible sites (i.e. from −1 to 1 on x-axis) were divided into equally sized 20 bp regions. The differential accessibility was calculated at each of these 20 bp regions. Source data are provided.
Like other chromatin remodelers, interaction of ALC1 with the basic histone H4 tail is essential for its nucleosome sliding activity36. Based on the cryo-EM structure of the ATPase domain of an ISWI remodeler (PDB:6PWF)45, we mapped the conserved acidic residues in ALC1 (D377,D381) that would interact with the histone H4 tail (Extended Data Fig.8c). D377A+D381A mutations in ALC1 reduced the association of ALC1 with histone H4 and failed to rescue PARPi responses (Fig. 7a,b and Extended Data Fig.8d).
To inactivate the macrodomain in ALC1, a previously reported D723A substitution that reduces affinity for PAR was combined with other rationally designed point mutations based on the crystal structure of the PAR-binding module of AF1521 (PDB:2BFQ)46. Amino acids G749 and L751 in ALC1 structurally correspond to AF1521 G41 and V43 residues, which form hydrogen bonding interactions with ADP-ribose in the macrodomain. While complementation with the single mutants D723A, G749F, L751D and L751W showed complete or partial rescue of PARPi sensitivity, no protection was observed with any of the double mutants (D723A+G749F, D723A+L751D, D723A+L751W) (Fig.7a,b and Extended Fig.8a,e). The ATPase activity, H4 interaction and macro domain of ALC1 were also essential to rescue MMS sensitivity (Fig.7a,b).
ALC1 and PARP activity cooperate to promote chromatin accessibility
ALC1 affects chromatin decondensation at laser-induced damage sites 47. To examine if ALC1 loss altered genome-wide chromatin accessibility, we performed ATAC-seq (Assay for Transpose-Accessible Chromatin) analysis in BRCA2-mutant DLD1 cells. Chromatin accessibility was assessed in ALC1-deficient cells treated with 5 μM olaparib for four hours (Fig.7c). A longer treatment was avoided to negate chromatin changes owing to differences in cell cycle. Pairwise comparisons of accessible sites using DESeq2 analysis did not identify large locus-specific changes (log2 fold change ≥0.5 or ≤-0.5, FDR <0.05), suggesting that ALC1 does not play a major role in regulating gene expression (Supplementary Table2). In agreement, RNA-seq analysis in ALC1 depleted delp53 BRCA1−/− hTERT-RPE1 and BRCA2−/− DLD1 cells revealed no consistent gene expression changes (Supplementary Table3).
We next examined if ALC1 loss had an effect on overall chromatin accessibility. Metagene analysis revealed a significant 1.6 fold overall reduction in chromatin accessibility upon ALC1 depletion alone. A four-hour olaparib treatment resulted in 1.3 and 1.84 fold changes in overall accessibility in sgNeg and sgALC1 cell respectively, compared to the control (paired t-test p-value <1E-15) (Fig.7d). We reasoned that for a short duration treatment, a more potent PARPi may manifest in larger accessibility changes. We performed ATAC-seq analysis in UWB1.289 cells, which are less sensitive to ALC1 loss in the absence of PARPi treatment and thus avoid confounding issues of toxicity from genetic interactions. ALC1 loss alone revealed no significant reduction in overall chromatin accessibility in UWB1.289 cells. In contrast, ALC1 depletion had a profound impact on chromatin accessibility when combined with the potent PARPi, talazoparib. A four hour administration of talazoparib (1 μM) alone resulted in a 1.3 fold overall reduction in chromatin accessibility while combining talazoparib with ALC1 loss yielded a 3.5 fold reduction (paired t-test p-value < 1E-15) (Fig.7e). These data reveal that the concerted activities of ALC1 and PARP regulate chromatin accessibility.
PARP-dependent chromatin relaxation has been implicated in the recruitment of repair proteins to the sites of laser damage48,49. Strikingly, loss of ALC1 alone reduced the association of several proteins dedicated to processing base damage in DLD1 BRCA2-mutant cells (Extended Data Fig.9a,b). This included the DNA glycosylase NTHL1, the apurinic/apyrimidinic endonuclease APE1, and XRCC1. These reductions were more pronounced upon combining ALC1 loss with olaparib, suggesting that ALC1 and PARP act together to ensure chromatin accessibility to certain repair factors. Similar changes in chromatin association of repair factors after PARPi treatment were also observed in UWB1.289 cells (Extended Data Fig.9c). XRCC1 chromatin localization was also significantly lower in PARPi treated ALC1-deficient UWB1.289 cells compared to its WT counterpart, even in the presence of MMS that induce PARylation (Extended Data Fig.9d–g).
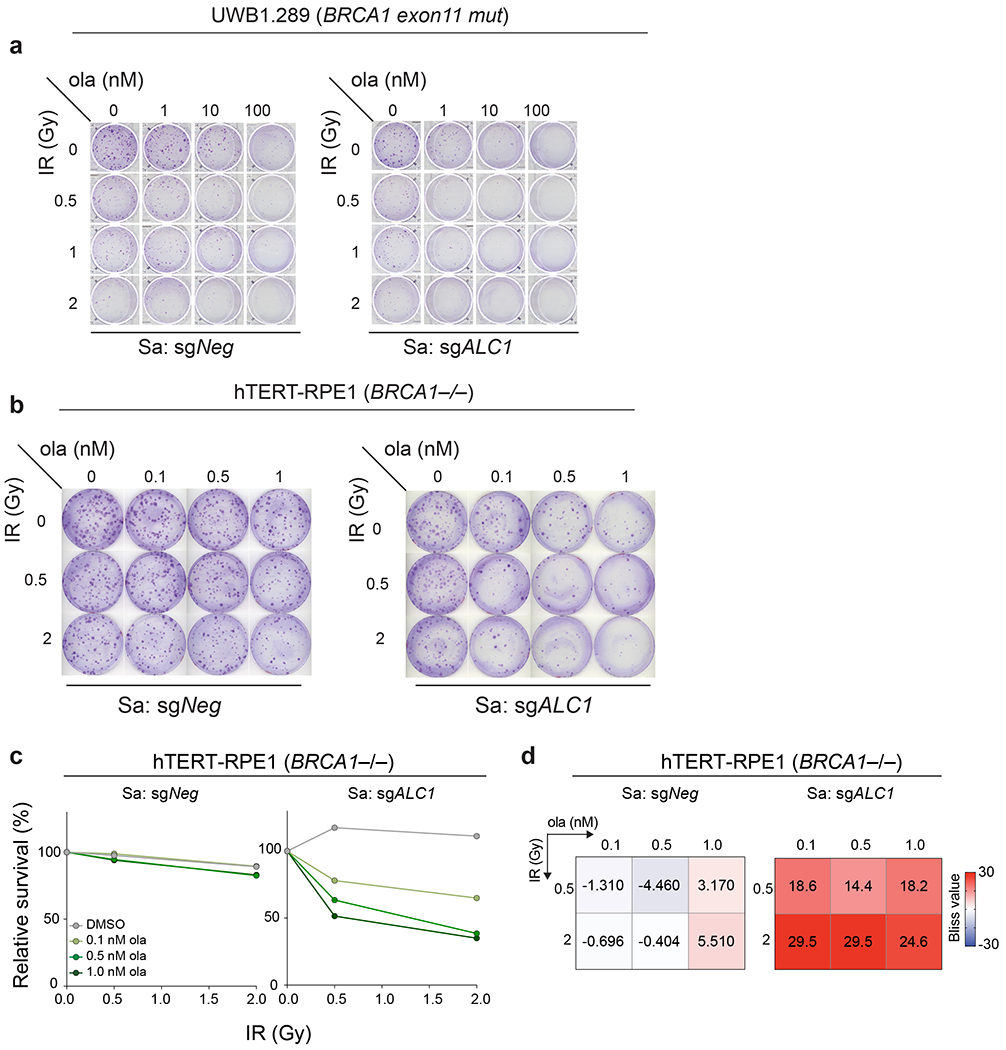
To determine if this coordination affects response to other genotoxic insults, we combined PARPi with ALC1 loss and examined responses to ionizing radiation (IR). ALC1 loss by itself did not increase sensitivity to IR in either BRCA1-mutant hTERT-RPE1 or UWB1.289 cells. In contrast, treatment with nanomolar concentrations of PARPi showed synergy with ALC1 loss to achieve cell killing at low doses of IR. Only modest or no synergy with IR was observed at these PARPi concentrations in ALC1-proficient cells (Fig.8a–c, Extended Data Fig.10). These results support a role for ALC1 in promoting diverse PARP-dependent damage responses.
Fig. 8. Co-operation between ALC1 with PARP activity facilitates chromatin-directed DNA repair.
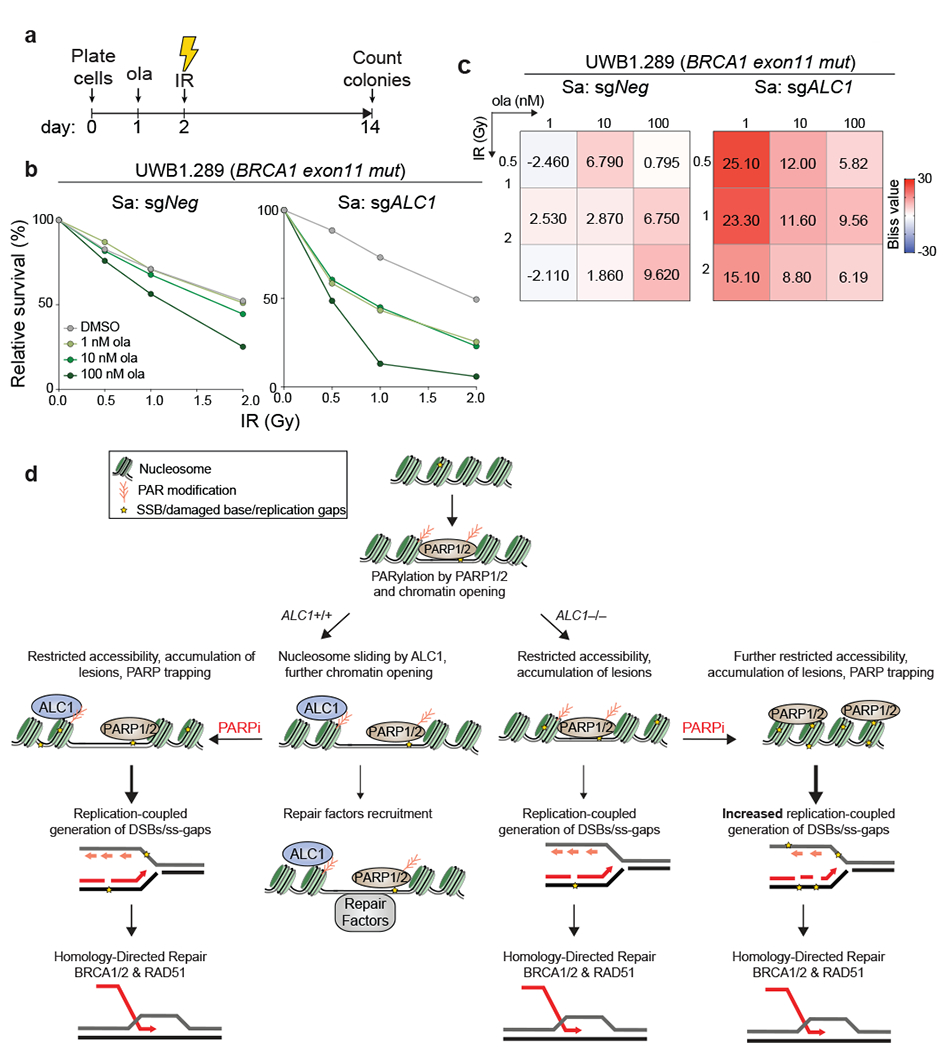
a, Schematic of the experiment to examine the effects of combining low doses of PARPi with ionizing radiation (IR) in ALC1 depleted cells. b-c, Quantification of clonogenic survival (b) and heat map of bliss scores (c) obtained from UWB1.289 cells treated with the indicated doses of ola and IR. Data are mean from two biologically independent experiments. Bliss score >0, synergistic; Bliss score <0, antagonistic; Bliss score = 0, additive. Number of colonies in IR-treated conditions were normalized to their respective un-irradiated counterparts. Colonies with more than 50 cells were included in the analysis. d, Model depicting cooperation between ALC1 and PARylation in response to DNA damage. PARylation results in ALC1 recruitment and chromatin decondensation. Nucleosome sliding by ALC1 further enhances chromatin accessibility. Combined loss of ALC1 and PARP activities reduces the access of base damage repair factors to the chromatin. Unrepaired base and SSBs result in the generation of replication-coupled ss-gaps and DSBs during S-phase that necessitate repair by BRCA-dependent HR. Source data are provided.
Discussion
PAR-dependent changes in chromatin accessibility were reported nearly four decades ago4, yet its relationship to the selective toxicity of PARPi in HR-deficient cells has remained unexplored. Our study identifies PARP-dependent chromatin remodeling by ALC1 as a vulnerability in HR-deficient cancers. ALC1 deficiency reduced the proliferation of BRCA-mutant cells and enhanced the efficacy of PARPi in a distinct manner from perturbations in canonical SSBR or HR pathways24,25,50. ALC1 modulates chromatin accessibility to enable processing of damaged bases. We propose that PARPi treatment in ALC1-deficient cells results in an accumulation of persistent lesions that trap PARP1 and PARP2, necessitating a transition to BRCA-dependent HR repair during S-phase (Fig.8d). The requirements for both PARP1 and PARP2 loss for resistance to PARPi or MMS provides evidence that PARP2 becomes important upon ALC1 loss. This differentiates PARPi response determinants from BRCA-deficiency alone, where PARP1 trapping is largely responsible for therapeutic outcome19.
Persistent base lesions in ALC1 deficient cells may increase the frequency of replication-coupled gaps that trap PARPi. Several recent studies reported the accumulation of ss-DNA enriched post-replicative territories upon treatments with alkylating agents or PARPi51,52. Replication restart by re-priming events downstream of DNA lesion can leave ss-DNA gaps to be filled after replication53,54 by translesion polymerases or repaired by replication restart or template switching with the sister chromatid55. The role of HR proteins in replication restart and template switching may account for the lower frequency of gaps in BRCA-proficient settings upon ALC1 loss (Fig.4f)56,57. ALC1 has been reported as a component of active replisomes, raising possibilities for its role in replication-associated repair58.
A recent genome-wide CRISPR screen reported that loss of enzymes involved in processing base damage confers PARPi sensitivity59. Together with our findings, these observations suggest that increased accumulation of base damage and resultant replication-coupled gaps may provide an avenue for enhancing PARPi cytotoxicity. ALC1 loss has also been reported to produce PARPi sensitivity in genome-wide screens in BRCA-proficient cell lines passaged over several weeks at olaparib concentrations of 0.5-2 μM (50-200 fold higher doses than used in our studies) or 5 μM veliparib, albeit investigation of underlying mechanisms was not described 59–61. These findings, together with our data from isogenic cell lines (Extended data Fig.2) indicate that a large therapeutic window can be obtained by targeting ALC1 in HR-deficient tumors.
The ATPase and macrodomain of ALC1 were essential for mediating PARPi response, presenting two moieties for small molecule inhibitor development. The relative dispensability of ALC1 in cellular settings with intact DNA repair presents potential advantages over combinatorial approaches that include cytotoxic agents with PARPi. Targeting PAR-dependent chromatin accessibility may offer possibilities that extend beyond inactivation of a singular repair pathway.
Methods
Cell culture:
UWB1.289 and UWB1.289+BRCA1 cells were purchased from ATCC and were maintained in 1:1 MEBM (Lonza):RPMI 1640 with GlutaMAX (Thermo Fisher) and supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals) and 1x penicillin and streptomycin (P/S, 100 U/ml, Gibco). DLD1 WT and DLD1 BRCA2−/− were purchased from Horizon Discovery and cultured in RPMI 1640 with GlutaMAX media and supplemented with 10% FBS and 1x P/S. 293T and U-2 OS cells were purchased from ATCC and were maintained in DMEM media (Thermo Fisher) with 10% Bovine Calf Serum (BCS, GE HyClone) and 1x P/S. CAPAN-1, hTERT-RPE1 p53−/− Cas9 and hTERT-RPE1 p53−/−BRCA−/− Cas9 cells were grown in DMEM with 10% FBS and 1x P/S. SUM149PT were cultured in Ham’s F-12 (Thermo Fisher) media supplemented with 5% FBS, 1x P/S, hydrocortisone (1 mg/ml, Sigma) and insulin (5 mg/ml, Sigma). hTERT-RPE1 and DLD1 cells were maintained at 3% oxygen, while other lines were maintained at atmospheric oxygen.
Chemicals used in this study include:
Olaparib (LC laboratories, 0-9201), talazoparib (Selleck Chemicals, S7048), camptothecin (Cayman Chemicals, 11694), H2O2 (Sigma, H1009), cisplatin (Tocris, 2251), MMS (Sigma, 129925), and PARGi (PDD 00017273, Fisher 5952). Most solid compounds were reconstituted in DMSO at 1000X of the required concentration, such that the final percentage of DMSO was 0.1%. To all untreated controls, a final concentration of 0.1% DMSO was added. Cisplatin was freshly dissolved in water to yield a stock concentration of 5 mM.
Vector construction and sgRNA cloning:
Single knockouts were generated either using a two-vector Streptococcus pyogenes (Sp) Cas9 system: LentiV_SpCas9_puro (Addgene, 108100) and LRG2.1 backbone (Addgene, 108098) or an all-in-one (expressing both SaCas9 and sgRNA) single vector Staphylococcus aureus (Sa) Cas9 system. The SaCas9 all-in-one vector, henceforth referred to as SaLgC, was derived by cloning the SaCas9 coding sequence (dCAS-VP64_Blast, Addgene: 61425) and its associated sgRNA expression cassette into a lentiviral vector. For double knockout experiments, ALC1 was knocked out using the single vector SaCas9 system and the two-vector SpCas9 system was employed for the depletion of the second protein of interest. Plasmids generated in this study, which will available through Addgene: (1) SaLgC (U6-sgRNA-EFS-SaCas9-P2A-GFP), (2) SaLgCp (U6-sgRNA-EFS-SaCas9-P2A-Puro) and (3) SaLgCn (U6-sgRNA-EFS-SaCas9-P2A-Neo). sgRNAs were designed to target the functional domain of protein and were cloned by annealing the two complementary DNA oligos into a BsmB1-digested vector using T4 DNA ligase. To improve U6 promoter transcription efficiency, an additional 5’ G nucleotide was added to all sgRNA oligos that did not already start with a 5’ G. List of sgRNAs used in the study are listed in Supplementary Table 4.
Lentivirus generation and transduction:
Lentivirus production and stable cell line generation was performed as described before62. Puromycin (2 mg/ml), Blasticidin (5 mg/ml), Hygromycin (100 μg/ml) and Neomycin (400 mg/ml) were used for selecting all cell lines except hTERT-RPE1, which were selected using puromycin (10 mg/ml) and neomycin (800 mg/ml). For all biological replicates, independent lentivirus infection and antibiotic selection was performed each time.
Competition-based cellular proliferation assays:
Competition-based cellular proliferation assays were performed as previously described63. For the purposes of these growth competition experiments and CRISPR screen, Tinitial was determined for each cell line. For a given cell line, Tinital denotes the day when the highest GFP expression was achieved for a sgRNA targeting an essential gene, for example, PCNA or RPA3. To determine the Tinitial, cells were transduced with sgRNA targeting an essential gene and GFP positivity was recorded for six consecutive days. Peak GFP was reached at Day 3 for hTERT-RPE1 (p53−/−, BRCA1−/−), Day 4 for UWB1.289 and UWB1.289+BRCA1, Day 5 for SUM149PT, DLD1 BRCA2−/− and CAPAN-1 and Day 6 for DLD1 WT. sgRNAs were cloned into a vector that also expressed GFP as a marker of transduction. Lentivirus were generated in an array format. Cells were transduced at an MOI of <0.5 to allow competition between the GFP-positive cells (that carried sgRNA) and GFP-negative cells. On the day of Tinitial, the percentage of GFP positive cells was recorded and cells were split into media containing either DMSO or olaparib. After every two population doublings, the percentage of GFP was measured using Guava InCyte (version 3.3) software on Guava Easycyte 10HT instrument and cells were passaged 1:4. GFP readings were taken for 6-7 passages (12-14 population doublings). Two population doubling time of cell lines used in the study was determined to be two days for hTERT-RPE1p53−/−, DLD1 WT, three days for hTERT-RPE1 BRCA1−/−, four days for UWB1.1289+BRCA1 and DLD1 BRCA2−/−, and five days for UWB1.289, SUM149PT and CAPAN-1.
Retrovirus plasmids and transduction:
ALC1 cDNA was cloned into a retroviral pOZ-N vector as well as in a pMSCV-puromycin vector using Gibson Assembly (NEB, E5510S) with an N-terminus FLAG-HA tag. FLAG-HA XRCC1 was cloned into the pMSCV-puromycin vector using Gibson Assembly. Site-directed mutagenesis was performed using Q5 Site-Directed Mutagenesis Kit (NEB, E0552S). Retrovirus was generated by transfection of Phoenix cells with 2:1 ratio of target plasmid: pCL ampho DNA using LipoD293 (Fisher, SL100668). Virus were collected at 48 and 72 hrs after transduction and target cells were spin infected at 500g for 30 min. Selection for pOZ constructs was done using IL-2-conjugated magnetic beads and pMSCV-puromycin expressing cells were selected using 1 mg/ml of puromycin.
Construction of domain-focused sgRNA pooled library:
A gene list of chromatin regulators in the human genome was manually curated based on the presence of chromatin regulatory protein domains32. The chromatin regulatory protein domain sequence information was retrieved from NCBI Conserved Domains Database. Approximately 6 sgRNAs were designed against individual protein domains (Supplementary Table 4). The design principle of sgRNA was based on previous reports and the sgRNAs with the predicted high off-target effect were excluded68. All of the sgRNAs oligos including positive and negative control sgRNAs were synthesized in a pooled format (Twist Bioscience) and then amplified by PCR. PCR amplified products were cloned into BsmB1-digested LRG2.1 (a lentiviral sgRNA expression vector, U6-sgRNA-EFS-GFP, Addgene: 108098) using the Gibson Assembly kit (NEB#E2611). To verify the identity and relative representation of sgRNAs in the pooled plasmids, a deep-sequencing analysis was performed on a MiSeq instrument (Illumina) and confirmed that 100% of the designed sgRNAs were cloned into the LRG2.1 vector and the abundance of >95% of individual sgRNA constructs was within 5-fold of the mean. This chromatin regulatory domain CRISPR sgRNA pooled library will be available through Addgene.
CRISPR-based pooled library screening:
CAPAN-1, SUM149PT and UWB1.289 were engineered to stably express SpCas9 (a lentiviral expression vector, EFS-Cas9-P2A-Puro vector, Addgene: 108100). Lentivirus of pooled sgRNA library targeting functional domains of chromatin factors was generated as described above. To ensure a single copy sgRNA transduction per cell, the multiplicity of infection (MOI) was set to 0.3-0.4. At Tinitial a fraction of cells were collected to prepare the reference representation and the rest were propagated either in the presence of DMSO or 10 nM olaparib, which approximately corresponds to the lethal dose 20 (in a two-week clonogenic assay) of the cells used for screening. Cells were passaged for 14 population doublings and were harvested at the final time point. To maintain the representation of sgRNAs during the screen, the number of sgRNA positive cells was kept at least 1000 times the sgRNA number in the library. CRISPR library was generated as described before62. A protein domain CRISPR Score (CS) was calculated by averaging the log2 fold-change of all CRISPR RNA targeting a given protein domain. Log2 fold-change= (final CRISPR RNA abundance + 1)/(initial CRISPR RNA abundance). The log2 fold-change values for each protein domain are provided in Supplementary Table 1.
Clonogenic assay:
For clonogenic experiments with SUM149PT, DLD1, UWB1.289 and UWB1.289+BRCA1, 500 cells were plated in technical triplicates and analyzed after 10-14 days. For hTERT-RPE-1, 250 cells were plated in a 10 cm dish. hTERT-RPE1 p53−/− type and p53−/− BRCA1−/− cells were grown at 3% oxygen for 8 and 11 days, respectively. For cellular complementation analysis with mutants, cells were directly plated in media containing olaparib (0.5 or 1 nM). For clonogenic experiments with PARGi, cells were plated on Day 0, PARGi was added on Day 1 and olaparib was added after a 24 hr incubation with PARGi. For IR+PARPi experiments, cells were plated on Day 0, treated with PARPi on Day 1 and IR treatment was performed 24 hrs after PARPi addition. After 14 days cells were washed with PBS and stained with 0.4% crystal violet in 20% methanol for 30 min at room temperature. Plates were then washed with deionized water and air-dried. Colonies were manually counted. Bliss scores for analyzing combinatorial responses were generated using Combenefit64.
CellTiter-Glo assay:
1000 cells in a volume of 100 μl were plated into each well of 96-well clear bottom black plates (Corning, Neta Scientific 3904) on Day 0. On the next day, 2X drug dilutions were prepared and 100 μl of the drug dilution was added to cells in technical triplicates. While other drugs were retained, MMS-treated cells were released into fresh media after 24 hrs. The viability was measured using CellTiter-Glo Luminescent Cell Viability Assay (Fisher, G7572) either after 5 days of drug addition (for RPE-1 and U-2 OS) or after 7 days (for DLD1, UWB1.289, UWB1.289+BRCA1 and SUM149PT) using the Gen5 software (v2.01.14) on Biotek Synergy microplate reader. When plotting the survival curves, the luminescence of the drug-treated population was normalized to solvent-exposed cells. The data was fitted in GraphPad Prism using the following equation: y = min+ (max-min)/1+10logEC50-x.
Xenograft experiment:
Xenograft studies were carried out under protocol number 803170 approved by the Institutional Animal Care & Use Committee at the University of Pennsylvania. 5-week-old female NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) mice were procured from Jackson Mice were kept at 72°F with average humidity of 60% and 12 hr day and 12 hr night cycle. Laboratory and tumor implantation was performed once these mice were 7-weeks old. SUM149PT-Cas9 cells were engineered to stably express either sgNeg or sgALC1. 5 days after transduction (Tinitial for SUM149PT), 2 x 106 cells mixed 1:1 with Matrigel (Fisher Scientific, 356237) and were subcutaneously implanted into both flanks of the mice. Once the tumor volume reached between 80-130 mm3, mice were randomized into vehicle and drug treated groups, such that the average tumor volume was approximately 100 mm3. Olaparib (LC laboratory) was dissolved in DMSO to a concentration of 50 mg/ml and was further diluted 1:10 in 15% hydroxypropyl b-cyclodextrin (Sigma) (w/v in PBS 7.4). 50 mg/kg of the drug was administered for five consecutive days of a week using oral gavage. 1:10 dilution of DMSO in 15% hydroxypropyl b-cyclodextrin (w/v in PBS 7.4) was used as vehicle control. Tumor dimensions were measured twice a week using a digital caliper and the volume was calculated using the formula: (length × width2)/2. Mice were euthanized once the tumor size reached >10.5 mm in any direction. The harvested tumors were snap frozen in liquid nitrogen for western studies.
Metaphase spreads:
Metaphase spreads were prepared as described before63. Slides were stained with Giemsa for 3-6 minutes, washed, and mounted using Permount.
Immunofluorescence:
PARP1 and PARP2 trapping was performed as described previously31,65. Images from two biologically independent experiments were captured at the same time using Zeiss Axio Widefield (20x/0.8) microscope. Immunostaining results were analyzed using CellProfiler 3.0 software, whereby a specialized pipeline was implemented to identify and measure PARP1, PARP2 and DAPI signal intensity (CellProfiler Module:IdentifyPrimaryObjects/Measure ObjectIntensity). Illumination correction was applied to each image prior to intensity measurements to account for non-uniformities in illumination. In addition, PARP2 signal was masked with DAPI-stained nuclei (CellProfiler Module:MaskImage – nuclei object) to ensure that only nuclear PARP2 signal intensity was measured. From this analysis, an output measure of IntegratedIntensity/per object/per field-of-view was obtained for each experimental condition.
To examine γH2AX foci during S-phase, cells were grown on Poly-K coated coverslips and were treated with 10 mM EdU for 20 min before harvesting. Cells were pre-extracted on ice for 5 min using Triton X-100 (0.5%) in CSK buffer (10 mM PIPES pH6.8, 100 mM NaCl, 300 mM Sucrose, 3 mM MgCl2 and 1 mM EGTA) and fixed using PFA (4%) in PBS for 15 min at RT. Following 3-4 washes with PBS, cells were permeabilized using Triton X-100 (0.5%) in PBS (0.5% PBSTx) for 5 min on ice and blocked using BSA (3%) in PBS for 30 min at room temperature. Alexa Fluor dye was conjugated to EdU by click chemistry using Click-iT Cell Reaction Buffer kit (C10269, Thermo Fisher). After washes with PBST (0.2% Tween in PBS), cells were fixed again with PFA (4%) for 10 min. Following PBS washes, cells were incubated with a 1:2000 dilution of anti-phospho-Histone H2A.X (Ser139) clone JBW301 antibody (Millipore, 05-636-1) for an hour at room temperature. Cells were washed with PBST and were next incubated with the Alexa Fluor conjugated mouse secondary (1:1000, Life Technologies) for an hour at RT. Coverslips were mounted using the Vectashield with DAPI. Images were acquired using a CoolSnap Myo camera connected to the Nikon NIS-Elements software. Images were processed using Fiji2.0, numbers of γH2AX foci were determined using Find Maxima function and statistical analysis was performed using GraphPad Prism.
For quantifying RAD51-γH2AX co-localization, cells grown on Poly-K coverslips were pre-extracted on ice for 3 min using Triton X-100 (0.5%) in CSK buffer and fixed using PFA (4%) in PBS for 15 min at room temperature. Following washes with PBS and permeabilization with 0.5% PBSTx, cells were blocked using blocking buffer (5% normal donkey serum, 0.1% fish skin gelatin, 0.1% Triton X-100, 0.05% Tween-20, and 0.05% sodium azide in PBS) for 1 hour at RT. Cells were incubated overnight with 1:200 dilution of RAD51 antibody (H-92, sc-8349, Santacruz) and 1:2000 dilution of anti-phospho-Histone H2A.X (Ser139) clone JBW301 antibody made in blocking buffer. Following washes with PBST, cells were incubated with 1:200 dilution of Alexa Fluor 488 conjugated anti-rabbit and 1:1000 dilution of Alexa Fluor 568 conjugated anti-mouse secondary for an hour at RT, mounted and imaged as described above. Co-localization events were manually counted keeping brightness and contrast throughout the analysis.
For quantifying ss-DNA, cells growing on Poly-K coverslips were labeled with CIdU (10 mM, C6891, Sigma) for 48 hours. After pre-extraction on ice for 5 min using Triton X-100 (0.5 %) in CSK buffer, cells were fixed in PBS containing PFA (4%) for 15 min at room temperature, washed with PBS, and blocked using the blocking buffer for an hour at room temperature. Cells were incubated with 1:100 dilution of CIdU antibody (Abcam, ab6326) and 1:2000 dilution of Anti-Histone H2A.X (phosphor S139) [EP854(2)Y] (Abcam, ab81299) for an hour at 37°C. This was followed by washes with PBST and incubation with Alexa Fluor 488 conjugated anti-mouse (1:200 dilution) and Alexa Fluor 568 conjugated anti-rabbit secondary antibodies (1:1000 dilution) for an hour at RT. Coverslips were mounted and imaged as described above. CIdU foci that did not co-localize with γH2AX were manually counted, keeping the values for image brightness and contrast constant throughout the analysis.
To quantify ALC1 and XRCC1 on chromatin, cell lines stably expressing HA-ALC1 or HA-XRCC1 were grown on Poly-K coverslips. After indicated treatments, cells were pre-extracted on ice for 2 min using 0.2% PBSTx (for U-2 OS and UWB1.289) or for 1 min using 0.5% CSK buffer (for SUM149PT) and fixed using PFA (4%) in PBS for 15 min at room temperature. Coverslips were then washed with PBS and permeabilized using ice cold Methanol: Acetic Acid (1:1) on ice. Cells were blocked using DMEM/FBS media for 1 hour at RT followed by incubation with 1:1000 dilution mouse anti-HA (Biolegend, 901514) and rabbit anti-PCNA (Cell Signaling, 13110S) for 1 hour at RT. Secondary antibody staining and mounting was performed as described above. To quantify the chromatin bound signal, the DAPI channel was used to define the ROI using the magic wand tool in Fiji2.0. Integrated density for the defined ROI was then computed using the command M function.
S1 nuclease fiber labeling:
S1 nuclease fiber labeling was performed as described before66. CIdU and IdU were detected using mouse anti-IdU (BD-347580) and rat anti-CIdU (Serotec-OBT0030G) antibodies. The line-scan function of ImageJ was used to calculate the length of the fibers.
Chromatin fractionation:
Chromatin fractionation was performed using the Thermo Scientific Subcellular Protein Fractionation Kit for cultured cells (Fisher, 78840) following the manufacturer’s instruction. Modifications in the protocol included scraping cells in cold PBS supplemented with PARPi used for the respective experiment and MNase digestion was performed at 37°C for 15 min. An extra wash step with the nuclear extraction buffer was introduced before MNase digestion.
Immunoprecipitation:
Exponentially growing HEK293T cells were transfected in a 10 cm dish with 3x FLAG-tagged ALC1 constructs using 3:1 ratio of DNA:PEI. 48 hours after transfection, cells were scrapped in cold PBS and the cell pellet was collected after spinning down at 500g for 5 minutes at 4°C. Cell pellets were suspended in lysis buffer (1 mL, 50 mM Tris, pH8, 137 mM NaCl, 1% Triton X-100, 1 mM MgCl2, 1 mM EDTA) supplemented with 1x protease inhibitor cocktail (Roche), PMSF (1 mM, Sigma), DTT (1 mM, Sigma) and benzonase (50 units/mL, 70746-3, Sigma) and incubated on an end-to-end rotor for 1 hour at 4°C. Cell lysates were then collected after centrifugation at 20,000 g for 15 min at 4°C. 15ul of lysis buffer equilibrated FLAG-M2 beads (A2220, Sigma) was incubated with 2mg of lysates for another hour at 4°C on an end-to-end rotor. The beads were then washed 4x times using the protease inhibitor and DDT supplemented lysis buffer. Bound proteins were eluted by adding 2X NativePAGE™ sample buffer (BN2003) and heating at 95°C for 10 min. before analyzing by immunoblotting.
Antibodies used for immunoblotting:
ALC1 (Santacruz Biotechnology, sc-81065, 1:200 dilution), XRCC1 (Abcam, ab1838, 1:200 dilution), PARG (Millipore,MABS61, 1:200 dilution), CHD4 (Proteintech, ab264417), APE-1 (Santacruz Biotechnology, sc-17774, 1:200 dilution), NTHL1 (Santacruz Biotechnology, sc-2660C1a, 1:200 dilution), 53BP1 (Novus Biological NB100-94, 1:1000 dilution), PARP1 (Cell Signaling mAB#9532, 1:2000 dilution), PARP2 antibody (Active Motif, 39743, 1:1000 dilution), HA (Biolegend, 901514, 1:1000 dilution), GFP (Cell Signaling mAb#2956, 1:1000 dilution), Rev7 (Invitrogen PA5-49352, 1:1000 dilution), GAPDH (Cell Signaling 2118S, 1:2000 dilution), mouse Anti-FLAG (F1804, Sigma), anti-Histone H4 (Millipore, 05-858).
Assay for ATAC-seq:
ATAC-seq was performed as previously described67. Reads from ATAC-seq experiments were trimmed with Trim Galore (version 0.4.1) with parameters -q 15 --phred33 --gzip --stringency 5 -e 0.1 --length 20. Trimmed reads were aligned to the Ensembl GRCh37.75 primary assembly including chromosome 1-22, chrX, chrY, chrM and contigs using BWA (version 0.7.13) with parameters bwa aln -q 5 -l 32 -k 2 -t 12 and paired-end reads were group with bwa sample -P -o 100000068. Reads mapped to contigs, ENCODE blacklist and marked as duplicates by Picard (version 2.1.0) were discarded and the remaining reads were used in downstream analyses. Peaks in each sample were identified using MACS (version 2.0.9) with parameters -p 1E-5 --nomodel --nolambda --format=BAM -g hs --bw=300 --keep-dup=1. A union of all peaks in untreated wildtype, PARPi treated wildtype, untreated ALC1 KO and PARPi treated ALC1 KO samples were generated using bedtools (version 2.27.1) ‘merge’ function.
To determine the change in accessibility, aligned reads of each ATAC-seq sample were quantified on the union of peaks and normalized to FPKM. Log2 fold change of accessibility was calculated as log2 averaged FPKM on replicates of other conditions versus untreated wildtype. Significance of change was determined using unnormalized quantification as input to ‘DESeq’ function from DESeq2 package in R (version 3.6.1) with parameters test = “Wald”, betaPrior = F, fitType = “parametric”. P-values were adjusted for multiple hypothesis testing using FDR. The average ATAC-seq signal +/− 1Kb round the center of the union of peaks were calculated using the ‘metagene’ R package and the metagene plot was generated using ‘ggplot2’ R package. One-tailed paired t-test of the average profiles was performed with other conditions versus untreated wildtype using ‘t.test’ function in R with parameters paired = ‘TRUE’ and alternative = ‘less’.
RNA-seq:
BRCA1-mutant hTERT-RPE1 cells and BRCA2-mutant DLD1 cells were infected with lentivirus expressing sgRNAs targeting ALC1 and selected using appropriate selection marker. 1-5 million cells were harvested at the 4th population doubling time after their respective Tinital. RNA seq library was prepared and analyzed as described before62. Software used for analysis included Lexogen Quantseq 2.3.1 FWD platform, STAR Aligner with modified ENCODE settings,HTSeq-count, DESeq2 (1.14.1).
Statistics and Reproducibility:
All statistical analyses were performed using Microsoft Excel 2011 and GraphPad PRISM 8 software. Significance was calculated either by the two-tailed student’s t-test or Mann-Whitney test unless otherwise specified. All experiments were biologically repeated and confirmed.
Extended Data
Extended Data Fig. 1. ALC1 loss renders olaparib hypersensitivity and proliferation defects in various BRCA-mutant lines.
a, Protein domains ranked on the basis of the CRISPR score (CS) for ola sensitivity in BRCA1-mutant UWB1.289 cells. b, Immunoblot showing depletion of ALC1 using a sgRNA vector with GFP selection marker. GFP positive cells were sorted and analyzed. The blot is a representative image of two biologically independent experiments. c, GFP competition assay to examine the effects of ALC1 depletion on ola sensitivity in CAPAN-1, SUM149PT and UWB1.289 cells. Ola sensitivity in CAPAN-1 and SUM149PT was confirmed using six and seven independent guides respectively and data for each guide are from one experiment performed at three different ola concentrations. Ola sensitivity in UWB1.289 was confirmed using seven independent guides and data for each guide is mean of two biologically independent experiments in the absence and presence of 50 nM ola concentration. Source data are provided.
Extended Data Fig. 2. ALC1 loss enhances the therapeutic window of olaparib sensitivity in BRCA-mutant cells.
a, Sensitivities of the indicated cell lines to ola using CellTiter-Glo. Data are mean of 2 (hTERT-RPE1) or mean ± s.e.m. of 3 (UWB1.289, SUM149PT and DLD1) biologically independent experiments. b, Representative images (left) and quantification (right) of the clonogenic survival of ALC1-depleted DLD1 WT and BRCA2-mutant cells grown in the presence of increasing concentrations of ola. Data are mean ± s.e.m. from 3 biologically independent experiments. Source data are provided.
Extended Data Fig. 3. Extended analysis of PARPi sensitivity upon ALC1 loss.
a, Sensitivities of the indicated DLD1 BRCA2−/− cells to vel (veliparib), ola and tal in CellTiter-Glo assay. Data are mean ± s.e.m. from n = 3 biologically independent experiments. b, Immunoblot showing ALC1 levels in cells used for xenograft studies (first four left lanes) and in tumors that reached >10.5 mm in any dimension, which is when the mice were euthanized. Data from two biologically independent tumors per condition are shown. c, GFP competition experiment in UWB1.289+BRCA1 addback line to examine the effects of the combined loss of ALC1 and the indicated DNA repair proteins on cell proliferation and ola sensitivity. Data are normalized to Tinitial and indicate mean ± s.e.m. After every two population doublings, cells were passaged (P) and GFP percent was recorded (n=4 independent transductions except for sgFEN1, where n = 6 independent transductions were performed). Source data are provided.
Extended Data Fig. 4. Genomic lesions in PARPi treated ALC1 deficient cells are repaired by SSBR and NHEJ.
a-b, Immunoblot showing levels of ALC1 and XRCC1 in indicated DLD1 (a) and hTERT-RPE1 (b) cells. The western samples were analyzed once to check the efficiency of the sgRNAs for protein depletion before drug sensitivity assays. c, Sensitivities of the indicated DLD1 cells lines to ola and tal using the CellTiter-Glo assay. Data are mean ± s.e.m. from n = 3 biologically independent experiments. d, Sensitivities of the indicated hTERT-RPE1 cells lines to tal using the CellTiter-Glo assay. Data are mean from n=2 biologically independent experiments. e, Sensitivities of the indicated UWB1.289 cell lines to ola using the CellTiter-Glo assay. Data are mean ± s.e.m. from n = 3 biologically independent experiments. f, Quantification of γH2AX-Rad51 foci in indicated cell lines. Cells were fixed 16 hrs. after treatment with 10 Gy ionizing radiation (IR). Median is indicated. p-value determined by Mann-Whitney was derived from n≥54 cells examined over two biologically independent experiments. g, Quantification of γH2AX-Rad51 foci in indicated cell lines. Cells were treated with 5 μM ola for 24 hrs. before fixation. Median is indicated. p-value determined by Mann-Whitney was derived from n≥114 cells examined over three biologically independent experiments. h, Representative images and quantification of radials (indicated by red arrow heads) and breaks (indicated by yellow arrowheads) in the indicated UWB1.289 cell lines, post treatment with 1 μM ola for 24 hrs. For each experiment, at least 50 spreads were analyzed per sample. Data are mean from two biologically independent experiments. Source data are provided.
Extended Data Fig. 5. ALC1 deficiency results in increased trapping of PARP1 and PARP2 by PARPi upon DNA damage.
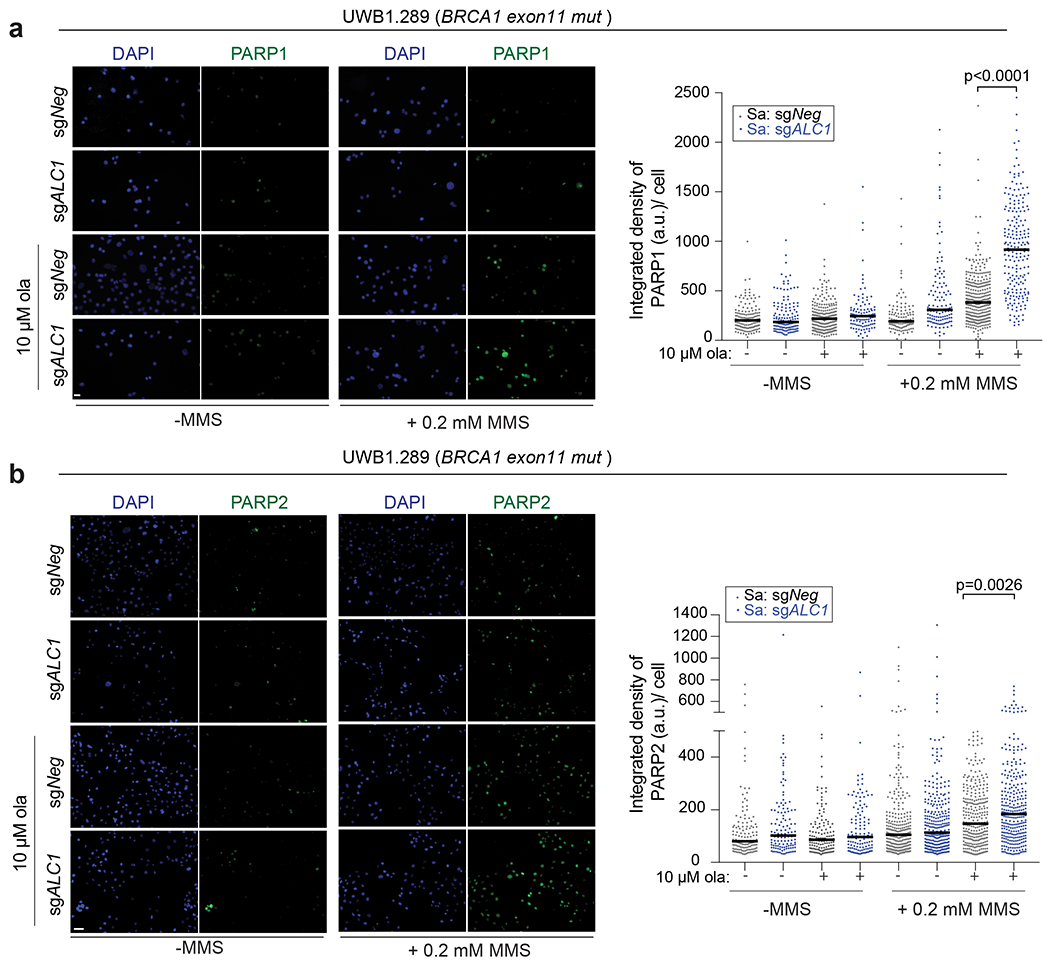
a-b Representative images (left) and quantification (right) of PARP1(a) and PARP2 (b) trapping in UWB1.289 cells. Indicated treatments were performed for 4 hours. Median is indicated. p-value determined by Mann-Whitney was derived from n≥107 cells sampled over two biologically independent experiments.
Extended Data Fig. 6. ALC1 loss confers MMS sensitivity and results in replication-coupled gaps.
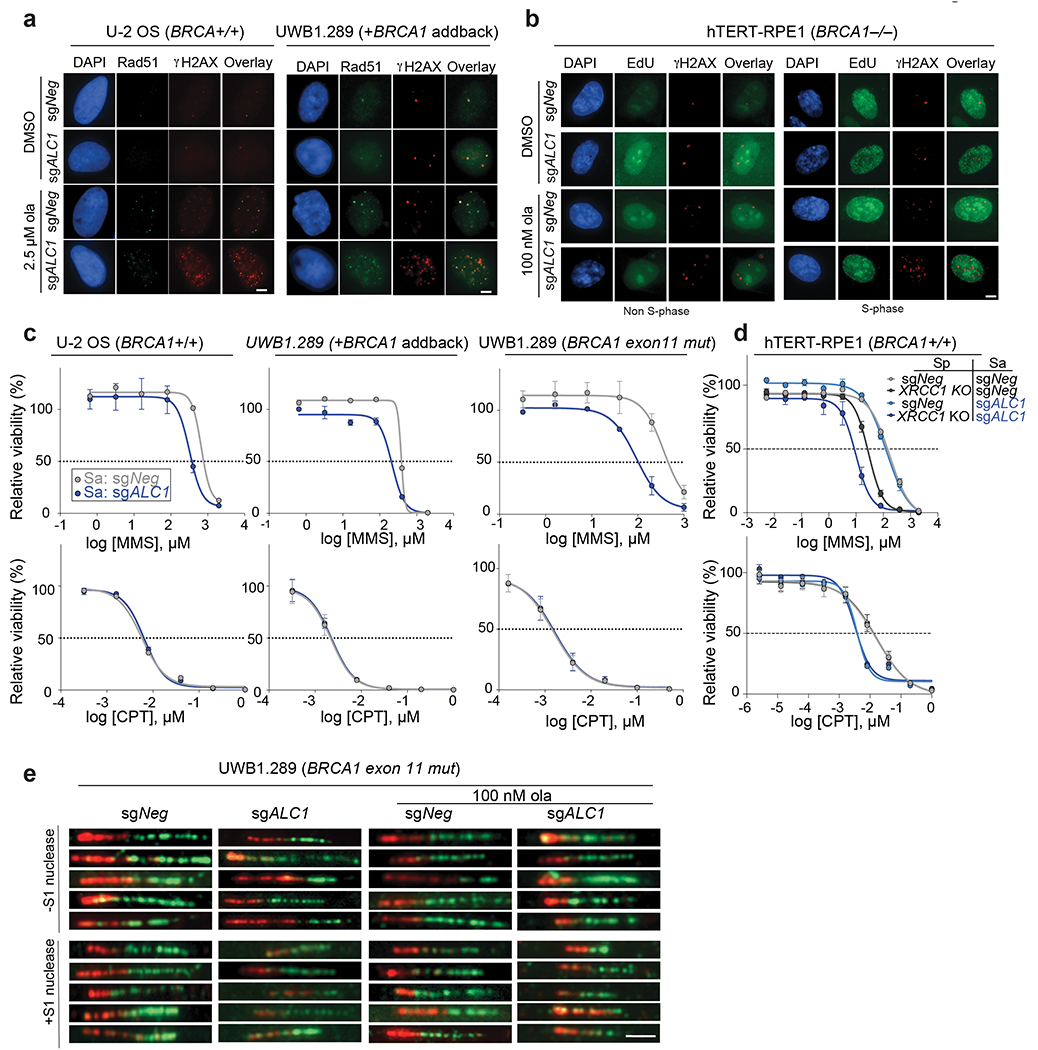
a, Representative images of Rad51-γH2AX foci in U-2 OS (left) and UWB1.289+BRCA1 (right) cell lines Scale bar: 10 microns. Images represent n≥67 cells examined over two biologically independent experiments. b, Representative images of γH2AX signal in EdU negative (left) and positive (right) hTERT-RPE1 BRCA1−/− cells. Scale bar: 10 microns. Images represent n≥99 cells examined over two biologically independent experiments. c, Sensitivities of the indicated cells lines to MMS and CPT using the CellTiter-Glo assay. Data are mean ± s.e.m. from n=3 biologically independent experiments. d, Sensitivities of the indicated hTERT-RPE1 cells lines to MMS and CPT using the CellTiter-Glo assay. Data are mean ± s.e.m. from n = three biologically independent experiments. e, Representative images of fibers from the S1 nuclease experiment. Scale bar: 2 microns. Images represent n≥75 fibers examined over two biologically independent experiments. Source data are provided.
Extended Data Fig. 7. ALC1 is recruited to the damaged chromatin under conditions of reduced PARylation.
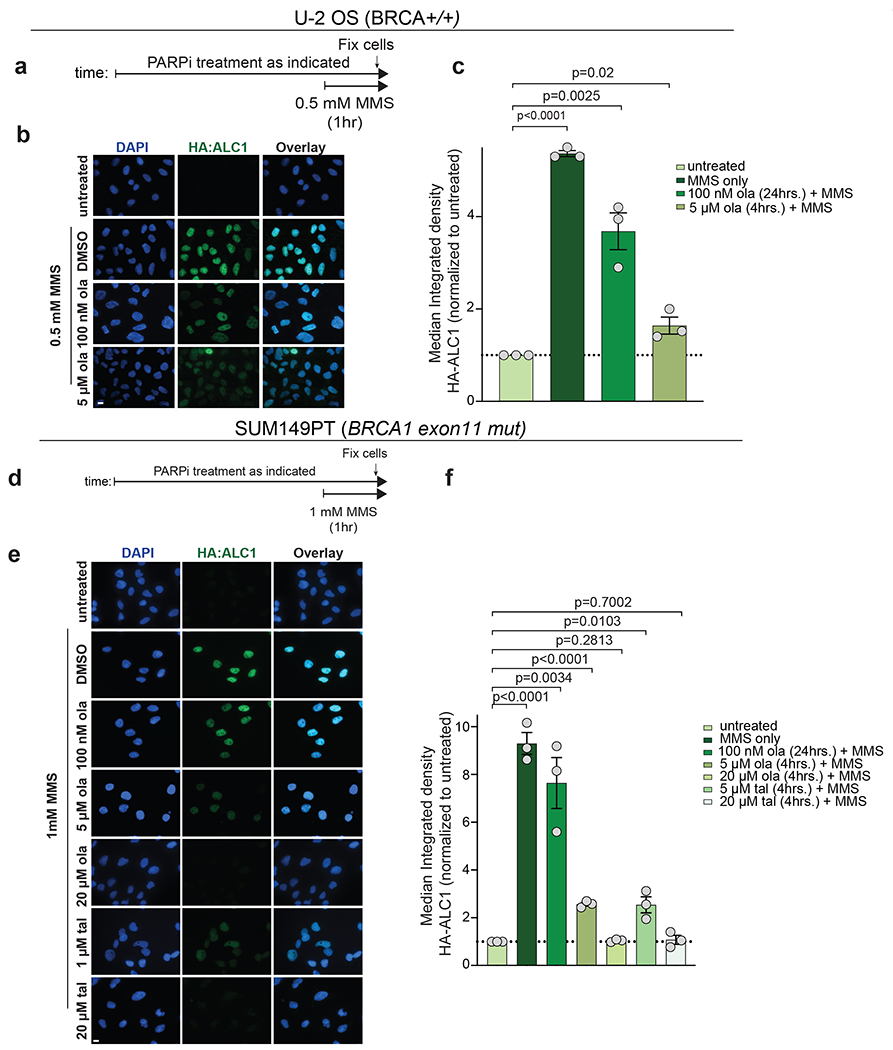
a, Schematic of the experiment. b-c, Representative images (b) and quantification (c) of HA-ALC1 localization to chromatin upon indicated treatments in U-2 OS cells. d, Schematic of the experiment. e-f, Representative images (e) and quantification (f) of HA-ALC1 localization to chromatin upon indicated treatments in SUM149PT cells. Scale bar, 10 microns. The median value was normalized to untreated control. Data are mean ± s.e.m. from n = three biologically independent experiments, p-value, unpaired Student’s t-test. For each experiment, at least 50 cells were analyzed per sample. Source data are provided.
Extended Data Fig. 8. ATPase activity, H4 interaction and macrodomains of ALC1 are essential for protecting BRCA-mutant cells from ola hypersensitivity.
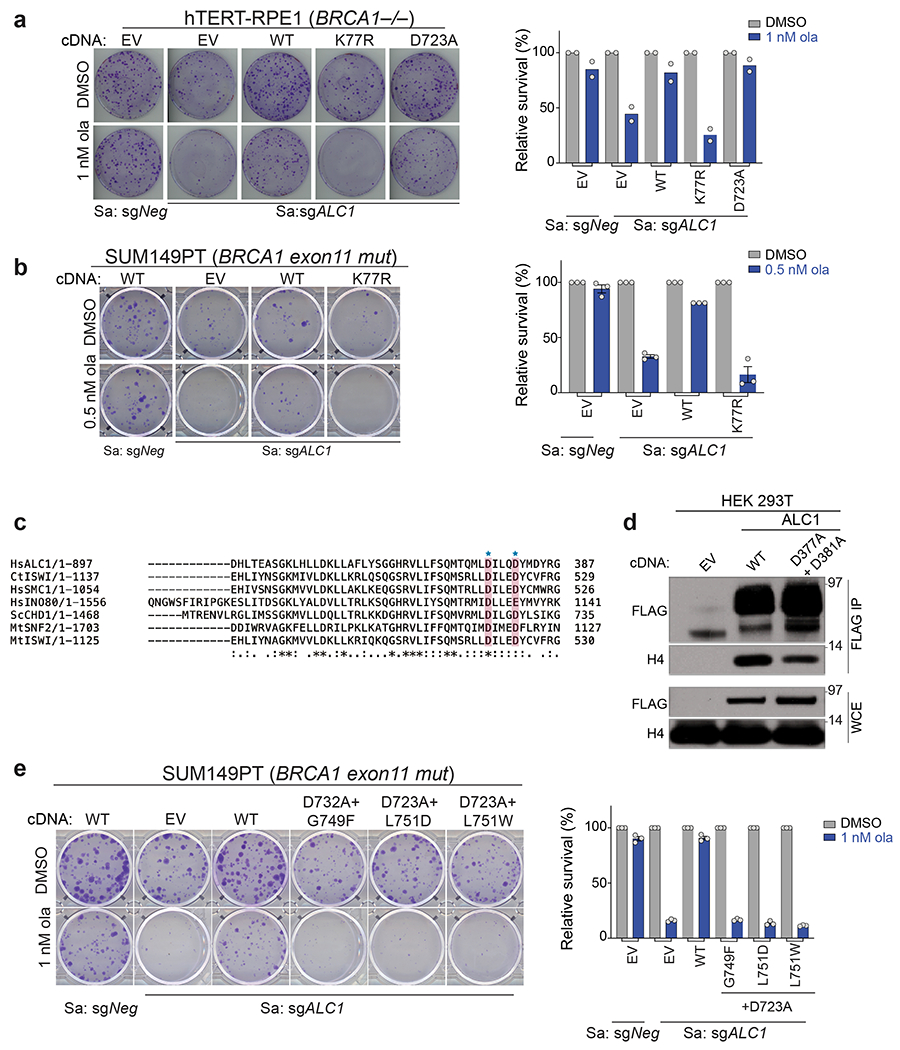
a, Representative images (left) and quantification (right) of the clonogenic survival assay using hTERT-RPE1 BRCA1−/− cells expressing sgALC1 and the indicated ALC1 mutants treated with ola (1 nM). Data are mean from two biologically independent experiments. b, Representative images (left) and quantification (right) of the clonogenic survival assay (left) using SUM149PT cells expressing sgALC1 and ALC1 K77R mutant treated with ola (0.5 nM). Data are mean ± s.e.m from n = three independent experiments. Number of colonies in the ola treated condition were normalized to its respective untreated counterpart. c, Sequence alignment of various chromatin remodelers using Clustal Omega. Histone H4 interacting residues as predicted by PDB:6PWF are highlighted and marked by a blue star. d, Immunoblots showing interactions of FLAG ALC1 WT and FLAG ALC1 D377A+ D381A with histone H4. Experiment was repeated twice with similar outcomes. e, Representative images of the clonogenic survival assay (left) and quantification (right) of SUM149PT cells expressing sgALC1 and indicated ALC1 macrodomain mutants treated with ola (1 nM). Data are mean ± s.e.m. from n = three independent experiments. Number of colonies in the ola treated condition were normalized to its respective untreated counterpart. Source data are provided.
Extended Data Fig. 9. ALC1 co-operates with PARP activity to permit association of repair proteins with chromatin.
a, DLD1 BRCA2−/− cells were fractionated and the chromatin-bound proteins were immunoblotted. Cells were treated with 5 μM ola for 4 hrs. Data for DLD1 BRCA2−/− cells are from the same sample, from two different western blots and the histone levels for each blot are shown by the ponceau staining. The data for XRCC1 is representative of 5 biologically independent experiments and the data for NTHL1 and APE1 is representative of 3 biologically independent experiments. b, Immunoblot of whole cell lysates of DLD1 BRCA2−/− cells showing total proteins levels upon ALC1 depletion and PARPi treatment. Cells were treated with indicated PARPi for 4 hrs. Data is representative of two biologically independent experiments. c, UWB1.289 cells were fractionated and the chromatin-bound proteins were immunoblotted. Cells were treated with 1 μM tal for 4 hrs. Data for XRCC1 is representative of 4 biologically independent experiments and the data for APE1 is representative of 3 biologically independent experiments. d, Immunoblot showing expression levels of HA-XRCC1. The blot was performed once to access the expression level of the tagged protein. e, Schematic of the IF experiment. f-g, Representative images (f) and quantification (g) of HA-XRCC1 localization to chromatin upon indicated treatments. Scale bar, 50 microns. Data are mean ± s.e.m. from n = three biologically independent experiments, p-value, unpaired Student’s t-test. For each cell line, the median value upon MMS treatment was normalized to its respective untreated control. Source data are provided.
Extended Data Fig. 10. ALC1 loss synergistically enhances IR sensitivity at low olaparib doses.
a-b Representative images of two independent clonogenic survival assay to monitor the effect of combining low doses of ola and IR upon ALC1 depletion in UWB1.289 (a) and hTERT-RPE1 BRCA1−/− cells (b). c-d, Quantification of clonogenic survival (c) and heat map of bliss scores (d) obtained from BRCA1−/− hTERT-RPE1 cells treated with the indicated doses of ola and IR. Data are mean from two biologically independent experiments. Bliss score >0, synergistic; Bliss score <0, antagonistic; Bliss score = 0, additive. Number of colonies in IR-treated conditions were normalized to their respective un-irradiated counterparts. Colonies with more than 50 cells were included in the analysis. Source data are provided.
Supplementary Material
Acknowledgements
We thank D. Durocher (Univ. Toronto, Lunenfeld) for sharing hTERT-RPE1 p53−/−BRCA1−/− cells and controls, J. and R. Connaway (Stowers) for ALC1 plasmids, Keith Caldecott (Univ. of Sussex) for sharing GFP-XRCC1 plasmid and hTERT-RPE1 XRCC1−/− and parental control cells, Nick Lakin (Univ. of Oxford) for sharing U-2 OS PARP1−/−, PARP2−/− and PARP1/2−/− cells, N. Johnson (Fox Chase) for SUM149PT BRCA1 reversion mutant cell lines and helpful discussions on mouse xenograft experiments, M. Altmeyer (Univ. of Zurich) for helpful suggestions on PARP1 trapping experiments. This work was supported by NIH grants GM101149 and CA17494 (to RAG), who is also supported by funds from the Penn Center for Genome Integrity, the Basser Center for BRCA, a V Foundation Team Convergence Award, and a Gray Foundation Team Science Award. PV was supported by the Ann and Sol Schreiber Mentored Investigator Award (Ovarian Cancer Research Fund Alliance) and Pilot funds from the Ovarian Cancer Translational Center for Excellence (UPenn). JS was supported by the Michele and Kevah Konner Award through the Basser Center for BRCA.
Footnotes
Competing interests
R.A.G. is a founder and scientific advisory board member of RADD Pharmaceuticals. No other authors declare any competing interests.
Data availability statement: Sequencing data generated in this study has be deposited in the Gene Expression Omnibus with accession code # GSE149104 (for RNA-seq) and # GSE150955 (for ATAC-seq). Data from the CRISPR screen has been provided as mapped reads in Supplementary Table 1. Functionally conserved domains were identified using either NCBI Conserved Domain Search or Uniprot. Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding author upon reasonable request.
Code availability statement: All the analyses were based on standard algorithms described in the methods and referenced accordingly. There are no custom algorithms to make available.
References
- 1.Langelier M-F, Planck JL, Roy S & Pascal JM Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 336, 728–732 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langelier M-F, Riccio AA & Pascal JM PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 42, 7762–7775 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupte R, Liu Z & Kraus WL PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 31, 101–126 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poirier GG, de Murcia G, Jongstra-Bilen J, Niedergang C & Mandel P Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. U. S. A. 79, 3423–3427 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krishnakumar R et al. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 319, 819–821 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Hakmé A, Wong H-K, Dantzer F & Schreiber V The expanding field of poly(ADP-ribosyl)ation reactions. ‘Protein Modifications: Beyond the Usual Suspects’ Review Series. EMBO Rep. 9, 1094–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pascal JM The comings and goings of PARP-1 in response to DNA damage. DNA Repair 71, 177–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luijsterburg MS et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell 61, 547–562 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mateos-Gomez PA et al. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ronson GE et al. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 9, 746 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ray Chaudhuri A et al. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 19, 417–423 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Fisher AEO, Hochegger H, Takeda S & Caldecott KW Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol. Cell. Biol. 27, 5597–5605 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pines A et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. The Journal of Cell Biology vol. 199 235–249 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bryant HE et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Golia B, Singh HR & Timinszky G Poly-ADP-ribosylation signaling during DNA damage repair. Front. Biosci. 20, 440–457 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Bryant HE et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Farmer H et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Helleday T The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Molecular Oncology vol. 5 387–393 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murai J et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 72, 5588–5599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gogola E, Rottenberg S & Jonkers J Resistance to PARP Inhibitors: Lessons from Preclinical Models of BRCA-Associated Cancer. Annual Review of Cancer Biology vol. 3 235–254 (2019). [Google Scholar]
- 21.Shi J et al. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 33, 661–667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Najm FJ et al. Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 36, 179–189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menear KA et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem. 51, 6581–6591 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Horton JK et al. XRCC1 and DNA polymerase beta in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 18, 48–63 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lord CJ & Ashworth A BRCAness revisited. Nat. Rev. Cancer 16, 110–120 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Xu G et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bunting SF et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell vol. 141 243–254 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaspers JE et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 3, 68–81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y et al. The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 76, 2778–2790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pettitt SJ et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 9, 1849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michelena J et al. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat. Commun. 9, 2678 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gogola E et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 35, 950–952 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ceccaldi R et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 518, 258–262 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsuda M et al. ALC1/CHD1L, a chromatin-remodeling enzyme, is required for efficient base excision repair. PLoS One 12, e0188320 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ooka M et al. Chromatin remodeler ALC1 prevents replication-fork collapse by slowing fork progression. PLoS One 13, e0192421 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahel D et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325, 1240–1243 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ensminger M et al. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 206, 29–43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caldecott KW Single-strand break repair and genetic disease. Nature Reviews Genetics vol. 9 619–631 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Lehmann AR & Fuchs RP Gaps and forks in DNA replication: Rediscovering old models. DNA Repair 5, 1495–1498 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Gottschalk AJ et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. U. S. A. 106, 13770–13774 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lehmann LC et al. Mechanistic Insights into Autoinhibition of the Oncogenic Chromatin Remodeler ALC1. Mol. Cell 68, 847–859.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh HR et al. A Poly-ADP-Ribose Trigger Releases the Auto-Inhibition of a Chromatin Remodeling Oncogene. Mol. Cell 68, 860–871.e7 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Hanzlikova H et al. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 71, 319–331.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hanzlikova H, Gittens W, Krejcikova K, Zeng Z & Caldecott KW Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 45, 2546–2557 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chittori S, Hong J, Bai Y & Subramaniam S Structure of the primed state of the ATPase domain of chromatin remodeling factor ISWI bound to the nucleosome. Nucleic Acids Res. 47, 9400–9409 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karras GI et al. The macro domain is an ADP-ribose binding module. EMBO J. 24, 1911–1920 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sellou H et al. The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage. Mol. Biol. Cell 27, 3791–3799 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith R, Sellou H, Chapuis C, Huet S & Timinszky G CHD3 and CHD4 recruitment and chromatin remodeling activity at DNA breaks is promoted by early poly(ADP-ribose)-dependent chromatin relaxation. Nucleic Acids Res. 46, 6087–6098 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith R et al. Poly (ADP-ribose)-dependent chromatin unfolding facilitates the association of DNA-binding proteins with DNA at sites of damage. Nucleic Acids Res. 47, 11250–11267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tarsounas M & Sung P The antitumorigenic roles of BRCA1–BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 21, 284–299 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong RP, García-Rodríguez N, Zilio N, Hanulová M & Ulrich HD Processing of DNA Polymerase-Blocking Lesions during Genome Replication Is Spatially and Temporally Segregated from Replication Forks. Mol. Cell 77, 3–16.e4 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Cong K et al. PARPi synthetic lethality derives from replication-associated single-stranded DNA gaps. doi: 10.1101/781989. [DOI] [Google Scholar]
- 53.Lopes M, Foiani M & Sogo JM Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 21, 15–27 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Quinet A et al. PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Molecular Cell vol. 77 461–474.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Branzei D & Szakal B Building up and breaking down: mechanisms controlling recombination during replication. Crit. Rev. Biochem. Mol. Biol. 52, 381–394 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Marians KJ Lesion Bypass and the Reactivation of Stalled Replication Forks. Annu. Rev. Biochem. 87, 217–238 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagaraju G & Scully R Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair 6, 1018–1031 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dungrawala H et al. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell 59, 998–1010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X et al. ERCC6L2 promotes DNA orientation-specific recombination in mammalian cells. Cell Res. 30, 732–744 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zimmermann M et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeWeirdt PC et al. Genetic screens in isogenic mammalian cell lines without single cell cloning. Nat. Commun. 11, 752 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gier RA et al. High-performance CRISPR-Cas12a genome editing for combinatorial genetic screening. Nat. Commun. 11, 3455 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Verma P et al. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes Dev. 33, 221–235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Di Veroli GY et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics 32, 2866–2868 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gatti M, Imhof R, Huang Q, Baudis M & Altmeyer M The Ubiquitin Ligase TRIP12 Limits PARP1 Trapping and Constrains PARP Inhibitor Efficiency. Cell Rep. 32, 107985 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quinet A, Carvajal-Maldonado D, Lemacon D & Vindigni A DNA Fiber Analysis: Mind the Gap! Methods Enzymol. 591, 55–82 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Petrovic J et al. Oncogenic Notch Promotes Long-Range Regulatory Interactions within Hyperconnected 3D Cliques. Mol. Cell 73, 1174–1190.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.