

Abstract
Rationale & Objective:
Characteristics of the transformation of primary to secondary calciprotein particles (CPP) in serum, including the size of secondary CPP (CPP2) aggregates and the time of transformation (T50), may be markers for arterial calcification in patients undergoing hemodialysis (HD). We examined the associations of CPP2 aggregate size and T50 with arterial calcification in incident HD patients.
Study Design:
Prospective cohort study
Setting & Participants:
Incident HD patients (n=402 with available CPP2 measures and n=388 with available T50 measures) from the Predictors of Arrhythmic and Cardiovascular Risk in ESRD (PACE) study
Predictors:
Serum CPP2 size and T50 at baseline
Outcomes:
Primary outcomes were baseline coronary artery and thoracic aorta calcifications. Exploratory outcomes included baseline arterial stiffness, measured by pulse wave velocity (PWV) and ankle brachial index, and longitudinally, repeated measures of PWV and all-cause mortality.
Analytical Approach:
Tobit regression, multiple linear regression, Poisson regression, linear mixed-effects regression, and Cox proportional hazards regression.
Results:
Mean age was 55±13 years; 41% were women; 71% were black and 57% had diabetes mellitus. Baseline CPP2 size and T50 were correlated with baseline fetuin-A (r=−0.59 for CPP2 and 0.44 for T50, p<0.001 for both), but neither was associated with baseline measures of arterial calcification or arterial stiffness. Baseline CPP2 size and T50 were not associated with repeated measures of PWV. During a median follow-up of 3.5 years (IQR 1.7–6.2), larger CPP2 was associated with higher risk of mortality [HR: 1.17 (95% CI 1.05, 1.31) per 100 nm larger CPP2 size] after adjusting for demographics and comorbidities, but there was no association between baseline T50 and risk of mortality.
Limitations:
Possible imprecision in assays, small sample size, limited generalizability to incident hemodialysis populations with different racial composition, and residual confounding.
Conclusions:
In incident HD patients, neither CPP2 size nor T50 was associated with prevalent arterial calcification and stiffness. Larger CPP2 was associated with risk of mortality, but this finding needs to be confirmed in future studies.
Keywords: calciprotein particle, arterial calcification, mortality, dialysis, vascular calcification
INTRODUCTION
Patients with end stage renal disease (ESRD) have a high prevalence of arterial calcification, which may contribute to their high cardiovascular morbidity and mortality.1,2 Disordered calcium and phosphate metabolism, deficiency in calcification inhibitors and phenotypic transformation of vascular smooth muscle cells (VSMCs) in the arterial media are among the leading proposed mechanisms for accelerated vascular calcification in chronic kidney disease (CKD).1,3–7,8 Calciprotein particles (CPPs), nanoparticles that are composed of calcium phosphate crystals and calcification inhibitors, are thought to mediate the transport and clearance of excess calcium and phosphate in circulation.9
There are 2 types of CPPs: primary and secondary. Primary CPPs (CPP1) are amorphous and soluble and are the predominant form of circulating CPP;10–12 in vitro, CPP1 spontaneously transform to secondary CPPs (CPP2), which are larger and more crystalline.13 CPP2 may mediate the effect of phosphate on arterial calcification,14 or directly induce oxidative stress in VSMCs leading to mineralization.15 The half-maximal time of transformation from CPP1 to CPP2 is referred to as T50.16 Lower T50 or a faster transformation of CPP1 to CPP2 has been shown to be associated with higher mortality in several CKD chorts.17–20 In predialysis CKD patients with prevalent coronary arterial calcification (CAC), lower T50 was associated with greater CAC.21 Several studies have measured T50 in the serum and used it as a surrogate marker of arterial calcification.22–24 However, whether circulating CPPs are pathogenic or directly involved in arterial calcification leading to increased mortality, especially in patients with ESRD, remains unknown.
We developed a high throughput, microplate-based assay to directly measure the hydrodynamic radius of CPP2 aggregates in addition to measuring T50.11 Using an existing cohort of ESRD patients—the Predictors of Arrhythmic and Cardiovascular Risk in ESRD (PACE) study, we hypothesized that larger CPP2 aggregates and lower T50 were associated with greater arterial calcification in patients on hemodialysis (HD). We also explored the associations of the CPP parameters with arterial stiffness and all-cause mortality.
METHODS
Study population
The PACE study is a prospective cohort designed to determine cardiovascular and dialysis-related risk factors associated with cardiac dysfunction in patients on HD. From 2008–2012, incident HD patients (on HD for <6 months) receiving regular outpatient HD thrice weekly were recruited from 27 outpatient units in Baltimore, Maryland and its surrounding area. The details of eligibility criteria and recruitment were described previously.25 The PACE study was approved by the Johns Hopkins School of Medicine and MedStar Institutional Review Boards. Of 568 participants who consented and enrolled at baseline, we measured CPP2 size in 402 individuals and T50 in 388 individuals. The flowchart of the study population is in Figure 1.
Figure 1. Participant flow sheet and analysis plan.
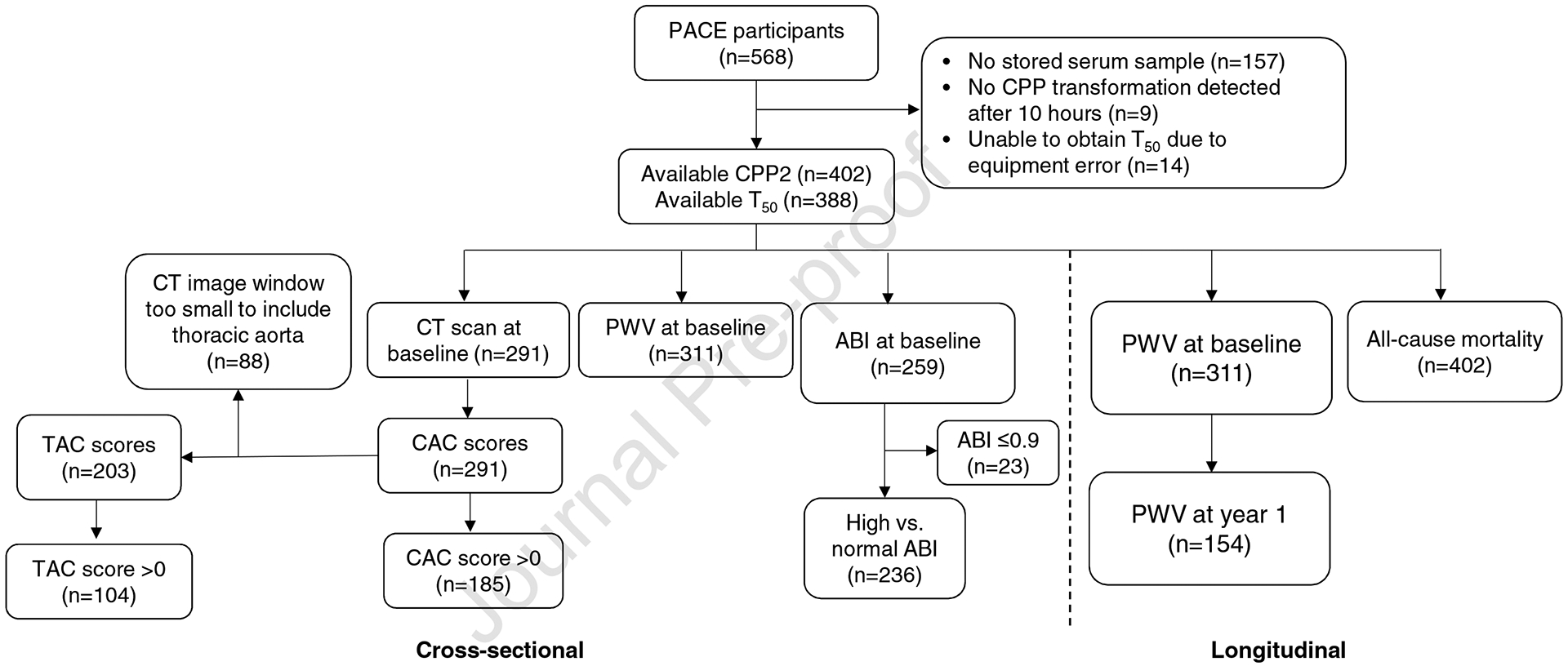
We measured characteristics of CPP transformation in incident HD patients from the PACE cohort with available stored serum samples. Measurements of CPP2 size were available in 402 participants and of T50 in 388 participants. We examined cross-sectional analyses of CPP2 and T50 with CAC, TAC, PWV, ABI as well as longitudinal analyses with PWV and all-cause mortality. Abbreviations: CPP2, secondary calciprotein particle; T50, half maximal transformation of primary to secondary calciprotein particle; CT, computed tomography; CAC, coronary arterial calcification; TAC, thoracic aortic calcification; PWV, pulse wave velocity; ABI, ankle brachial index.
Measurement of CPP transformation
At the baseline visit, serum was collected on a non-HD day after ~8 hours of fasting and stored at −80°C. CPP transformation of serum was measured using dynamic light scattering, as previously described.11 Briefly, after adding concentrated calcium and phosphate solutions (10 mM calcium and 6 mM phosphate) in the serum, we measured hydrodynamic radius of CPP1 and CPP2 at a constant temperature of 37°C for 10 hours using DynaPro Plate Reader II (Wyatt Technology, Santa Barbara, CA, USA, Supplementary Methods). T50 was the half-maximal time of transformation from CPP1 to CPP2.
Measurement of outcome variables
Our main objective was to examine the associations of CPP2 aggregate size and T50 with arterial calcification in incident HD patients. Therefore, our primary outcome variables were coronary arterial calcification (CAC) score and thoracic aortic calcification (TAC) score. Arterial calcification was measured in coronary arteries and the thoracic aorta using computed tomography (CT) at the baseline visit.26 CAC score was quantified based on CT using Agatston score.25 TAC score was calculated as the sum of calcium scores from the ascending and descending thoracic aorta. Of the 402 participants with available data on CPP2, 291 participants had CT examination and available CAC scores (Figure 1). TAC scores were available in 203 participants.
Exploratory outcomes included arterial stiffness, defined as high pulse wave velocity (PWV) or high ankle brachial index (ABI), and all-cause mortality. These outcomes were considered exploratory due to limited sample size. PWV was measured in 311 participants at baseline and also in 154 participants at year 1 (more details in Supplementary Methods). Participants with an ABI≤0.9 (n=23) were excluded from the analyses involving ABI because an ABI≤0.9 indicates the presence of peripheral arterial disease.27 High ABI was defined as an ABI>1.4 or having incompressible vessels, and normal ABI as >0.9 and ≤1.4.28 Mortality data were ascertained from the United States Renal Data System.
Measurement of covariates
Confounders were selected a priori and included self-reported demographic factors (age, sex, and race), educational level, smoking history, medical history, serum markers of mineral metabolism and inflammation, as well as circulating inhibitors of arterial calcification. Comorbidities such as diabetes mellitus (DM), coronary artery disease (CAD) and hypertension were adjudicated by a committee of physicians.25 Serum markers of mineral metabolism included serum calcium, phosphorous, intact parathyroid hormone (PTH), fibroblast growth factor-23 (FGF23), and soluble klotho. Circulating inhibitors of arterial calcification included serum albumin,13,29 dephosphorylated and uncarboxylated matrix glutamate (Gla) protein (dp-ucMGP),30,31 osteoprotegerin,32 and fetuin-A33. Please see Supplementary Methods for measurement of the covariates.
Statistical analyses
CPP2 size and T50 were examined as both continuous variables and categorical variables dichotomized at their median (small CPP2: < median, large CPP2: ≥ median; low T50:< median, high T50: ≥ median). Baseline participant characteristics were examined by CPP2 (small vs. large) and T50 (low vs. high) categories. Two-sample t-tests or Mann-Whitney U tests were used to examine continuous variables, and chi-squared tests were used to examine categorical variables. Because of skewed distributions of CPP2 size and T50, the relationship between CPP2 size and T50 as well as their correlations with covariates was examined using Pearson correlation with logarithmically transformed CPP2 size and T50.
At baseline, we examined cross-sectional associations between exposure variables of CPP2 size and T50 with outcomes variables of CAC, TAC, PWV, and high ABI. CAC was the primary outcome. CPP2 size and T50 were not transformed for these analyses. To simultaneously model the presence and severity of CAC and TAC, we transformed the calcification score [Ln(calcification score+1)] and used Tobit regression with left censoring at 0 and bootstrap techniques with 999 repetitions.34 Tobit regression of calcification score is more likely to identify associations with known risk factors of arterial calcification and provide more consistent results compared to other methods, such as linear regression.34 We performed natural logarithmic transformation of the calcification score +1 because of its right skewed distribution and the presence of many zero scores. For arterial stiffness, PWV was log-transformed to meet the normality assumption and multiple linear regression was used to examine the associations of CPP2 and T50 with PWV. The percentage changes in PWV were calculated by transforming the β coefficients [% change = 100 × (eβ−1)]. For ABI, we examined CPP2 and T50 with the outcome variable of high versus normal ABI, using Poisson regression with robust variance and estimated prevalence ratio. We performed longitudinal analyses of CPP2 and T50 with repeated measures of PWV and all-cause mortality. For repeated measures of PWV, we used linear mixed-effects regression with random intercept to account for correlation among individuals. For mortality, we used Cox proportional hazards regression. The proportional hazards assumption, tested using Schoenfeld residuals, was not violated. Censoring events included end of the study, transplantation, transfer to peritoneal dialysis, and loss to follow up.
For modeling building, we included demographics (age, sex and race) and comorbidities (DM and CAD) in our base model (Model 1). To investigate whether serum markers of mineral metabolism, circulating inhibitors of arterial calcification or inflammation markers confounded the associations of CPP2 size and T50 with the outcomes, we added covariates in each category to the Model 1. Only the covariates that were correlated with either CPP2 or T50 were included in the models. In addition to Model 1, Model 2 included serum calcium, phosphorous and FGF-23; Model 3, serum albumin and fetuin-A; Model 4, dp-ucMGP and osteoprotegerin; and Model 5, C-reactive protein. To examine whether DM and CAD were effect modifiers, we stratified by the status of DM and CAD after adjusting for demographics, then assessed effect modification qualitatively. For a secondary analysis, we grouped the participants into 4 categories: small CPP2 and high T50 (n=135, reference group), small CPP2 and low T50 (n=53), large CPP2 and high T50 (n=58) and large CPP2 and low T50 (n=141). We repeated the cross-sectional analyses for CAC, TAC and PWV using these categories. A two-sided p-value <0.05 was considered statistically significant for all analyses, including interaction terms. All analyses were conducted using STATA 14.1 (StataCorp, College Station, TX, USA).
RESULTS
Baseline participant characteristics
Mean age of the participants was 55 ± 13 years; 41% were women; 71% were African American; 57% had DM; 37% had CAD (Table 1). All participants had hypertension. Mean CPP1 size was 55 ± 9 nm (median: 55 nm, interquartile range (IQR): 49–61); mean size of CPP2 aggregates was 307 ± 146 nm (median: 291 nm, IQR: 201–379); mean T50 was 317 ± 122 min (median: 304 min, IQR: 229–387). Compared to those with small CPP2 aggregates, participants with large CPP2 aggregates were older and more likely to have DM and CAD. Compared to those with high T50, participants with low T50 were more likely to have smoked and less likely to have DM. Compared to those with a CAC=0, participants with a CAC score >0 were older, less likely to be African American, and had higher prevalence of CAD and higher osteoprotegerin levels (Table S1).
Table 1.
Baseline characteristics by CPP2 size and T50 dichotomized at their medians among incident HD participants from the PACE cohort
| Total (n=402) | Small CPP2 size (<median, n=202) | Large CPP2 size (≥median, n=200) | Low T50 (<median, n=194) | High T50 (≥median, n=194) | |
|---|---|---|---|---|---|
| Age, year | 55 ± 13 | 53 ± 12 | 57 ± 14 | 54 ± 14 | 56 ± 12 |
| Women, n (%) | 164 (41%) | 85 (42%) | 79 (40%) | 73 (38%) | 82 (42%) |
| African American, n (%) | 284 (71%) | 139 (69%) | 145 (73%) | 135 (70%) | 141 (73%) |
| Body Mass Index, kg/m2 | 30 ± 8 | 30 ± 8 | 29 ± 8 | 29 ± 8 | 30 ± 8 |
| History of smoking, n (%) | 229 (61%) | 118 (61%) | 111 (60%) | 116 (64%) | 102 (56%) |
| Education, less than grade 11, n (%) | 140 (37%) | 71 (37%) | 69 (38%) | 64 (36%) | 70 (39%) |
| Diabetes mellitus, n (%) | 228 (57%) | 107 (53%) | 121 (61%) | 104 (54%) | 120 (61%) |
| Coronary artery disease, n (%) | 148 (37%) | 70 (35%) | 78 (39%) | 73 (38%) | 71 (37%) |
| Serum calcium, mg/dL | 8.6 ± 0.6 | 8.7 ± 0.6 | 8.5 ± 0.6 | 8.5 ± 0.7 | 8.7 ± 0.6 |
| Serum phosphorous, mg/dL | 5.2 ± 1.1 | 5.1 ± 1.2 | 5.2 ± 1.1 | 5.4 ± 1.2 | 5.1 ± 1.1 |
| Serum magnesium, mEq/L | 1.8 ± 0.2 | 1.8 ± 0.2 | 1.8 ± 0.2 | 1.7 ± 0.2 | 1.8 ± 0.2 |
| Intact parathyroid hormone, pg/mL | 391 (262–578) | 390 (282–578) | 397 (239–583) | 374 (243–557) | 414 (275–590) |
| Serum albumin, g/dL | 3.6 ± 0.5 | 3.7 ± 0.5 | 3.6 ± 0.4 | 3.5 ± 0.5 | 3.7 ± 0.4 |
| Hemoglobin, g/dL | 10.8 ± 1.3 | 11.0 ± 1.3 | 10.6 ± 1.2 | 10.5 ± 1.3 | 10.9 ± 1.2 |
| LDL, mg/dL | 83 (60–108) | 99 (67–124) | 71 (55–91) | 81 (61–110) | 84 (58–107) |
| Fetuin-A, mg/L | 502 ± 163 | 581 ±168 | 420 ±108 | 439 ± 137 | 556 ± 167 |
| Dp-ucMGP, ppm | 1412 (962–2069) | 1400 (899–1986) | 1417 (972–2207) | 1434 (980–2378) | 1407 (896–1905) |
| Osteoprotegerin, pmol/L | 10.9 ± 4.8 | 10.4 ± 4.7 | 11.5 ± 4.9 | 10.7 ± 4.9 | 11.2 ± 4.8 |
| FGF23, RU/ml | 514 (241–1167) | 395 (218–1029) | 632 (265–1216) | 669 (256–1303) | 419 (242–924) |
| Klotho, pg/mL | 366 (273–500) | 372 (281–515) | 363 (265–483) | 359 (269–510) | 379 (274–490) |
| C-reactive protein, mg/mL | 5.9 (2.3–15.7) | 5.4 (2.3–15.8) | 6.1 (2.3–15) | 6.4 (2.6–16.7) | 5.1 (1.9–15.7) |
| CPP1, nm | 55 ± 9 | 54 ± 8 | 57 ± 9 | 55 ± 9 | 55 ± 8 |
| CPP2, nm | 291 (201–379) | 202 (147–243) | 379 (332–467) | 359 (281–445) | 227 (159–308) |
| T50, min | 304 (229–387) | 370 (291–449) | 254 (193–317) | 229 (179–269) | 387 (347–462) |
| Calcium-based phosphate binder, n (%) | 136 (37%) | 64 (34%) | 72 (40%) | 69 (39%) | 63 (35%) |
| Vitamin D therapy, n (%) | 290 (78%) | 146 (77%) | 144 (79%) | 138 (77%) | 140 (78%) |
| RAAS blockade, n (%) | 140 (41%) | 83 (48%) | 57 (35%) | 62 (39%) | 77 (47%) |
Abbreviations: LDL, low density lipoprotein; RAAS, renin-angiotensin-aldosterone system; dp-ucMGP, Dephosphorylated and uncarboxylated matrix Gla protein; FGF23, fibroblast growth factor-23; CPP1, primary calciprotein particle; CPP2, secondary calciprotein particle, T50, half-maximal time of transformation from CPP1 to CPP2
Note: If normally distributed, values for continuous variables with normal distribution are provided as mean ± standard deviation. Otherwise, they are provided as median (interquartile range). Categorical variables are presented as absolute number with percentage.
Correlations of laboratory covariates with CPP2 size and T50
The size of CPP2 aggregates was negatively correlated with T50 (r=−0.54, p<0.001; Figure S1). For serum markers of mineral metabolism, larger CPP2 was correlated with lower serum calcium (r=−0.15, p=0.006) and higher FGF23 levels (r=0.12, p=0.03; Figure S2). Higher T50 was correlated with higher serum calcium (r=0.19, p<0.001) and lower phosphorous levels (r=−0.15, p=0.006). For circulating inhibitors of arterial calcification, larger CPP2 was correlated with lower serum albumin (r=−0.16, p=0.003), lower fetuin-A (r=−0.59, p<0.001), and higher osteoprotegerin levels (r=0.13, p=0.01; Figure S3). Higher T50 was correlated with higher serum albumin (r=0.22, p<0.001), higher fetuin-A (r=0.44, p<0.001), and lower dp-ucMGP levels (p=−0.11, p=0.046). Lastly, higher level of C-reactive protein was associated with larger CPP2 (r=0.14, p=0.009), but not with T50.
Associations of CPP2 and T50 with arterial calcification and with arterial stiffness
There were 64% participants who had a CAC score >0; and among these participants, median CAC score was 310 (IQR 62–929). In both the unadjusted and adjusted Tobit regression models, there was no statistical evidence that CPP2 size or T50 was associated with the presence and severity of CAC (Table 2). There were 51% who had a TAC score>0, and among these participants, median TAC score was 424 (IQR 54–1345). CPP2 size and T50 were not associated with the presence and severity of TAC.
Table 2.
Cross-sectional associations of CPP2 size and T50 with arterial calcification (CAC and TAC) and with arterial stiffness (PWV)
| CPP2 size (per 100 nm) | T50 (per 100 min) | |||||
|---|---|---|---|---|---|---|
| CAC score | ||||||
| n | Coefficient (95% CI) | p | n | Coefficient (95% CI) | p | |
| Unadjusted | 291 | 0.02 (−0.36, 0.39) | 0.92 | 283 | 0.07 (−0.32, 0.46) | 0.73 |
| Model 1 | 291 | −0.09 (−0.37, 0.19) | 0.51 | 283 | −0.03 (−0.36, 0.29) | 0.83 |
| Model 2 | 286 | −0.23 (−0.55, 0.08) | 0.15 | 278 | 0.06 (−0.28, 0.41) | 0.71 |
| Model 3 | 286 | −0.19 (−0.53, 0.16) | 0.29 | 278 | 0.10 (−0.28, 0.47) | 0.62 |
| Model 4 | 272 | −0.15 (−0.44, 0.14) | 0.30 | 264 | −0.02 (−0.38, 0.33) | 0.90 |
| Model 5 | 289 | −0.14 (−0.43, 0.15) | 0.35 | 281 | −0.02 (−0.36, 0.32) | 0.93 |
| TAC score | ||||||
| n | Coefficient (95% CI) | p | n | Coefficient (95% CI) | p | |
| Unadjusted | 203 | 0.25 (−0.35, 0.84) | 0.42 | 197 | 0.06 (−0.71, 0.83) | 0.88 |
| Model 1 | 203 | 0.04 (−0.43, 0.51) | 0.86 | 197 | −0.12 (−0.74, 0.50) | 0.70 |
| Model 2 | 201 | −0.13 (−0.59, 0.32) | 0.56 | 195 | 0.06 (−0.59, 0.71) | 0.85 |
| Model 3 | 201 | −0.03 (−0.63, 0.58) | 0.93 | 195 | 0.11 (−0.65, 0.86) | 0.78 |
| Model 4 | 188 | −0.11 (−0.56. 0.34) | 0.62 | 182 | 0.003 (−0.65, 0.65) | >0.99 |
| Model 5 | 202 | 0.03 (−0.44, 0.49) | 0.91 | 196 | −0.10 (−0.74, 0.53) | 0.76 |
| PWV (m/s) | ||||||
| n | % Change (95% CI) | p | n | % Change (95% CI) | p | |
| Unadjusted | 311 | 1.64 (−0.93, 4.28) | 0.21 | 298 | 1.58 (−1.15, 4.38) | 0.26 |
| Model 1 | 306 | −0.02 (−2.19, 2.20) | 0.99 | 293 | 1.04 (−1.29. 3.44) | 0.38 |
| Model 2 | 302 | −0.32 (−2.60, 2.01) | 0.78 | 289 | 1.42 (−1.01, 3.92) | 0.25 |
| Model 3 | 302 | −0.61 (−3.19, 2.04) | 0.65 | 289 | 1.83 (−0.87, 4.60) | 0.19 |
| Model 4 | 289 | 0.33 (−2.59, 1.98) | 0.78 | 276 | 1.95 (−0.53, 4.49) | 0.12 |
| Model 5 | 305 | −0.17 (−2.38, 2.08) | 0.88 | 292 | 1.18 (−1.19, 3.60) | 0.33 |
For CAC and TAC scores, we transformed calcification score [Ln(calcification score+1)], then used Tobit regression with left censoring at 0 and bootstrap techniques with 999 repetitions. For PWV, we performed multiple linear regression after logarithmic transformation of PWV.
Abbreviations: CAC, coronary arterial calcification; TAC, thoracic aortic calcification; PWV, pulse wave velocity.
Model 1: adjusted for demographics (age, sex, race), comorbidities (diabetes, coronary artery disease)
Model 2: adjusted for demographics, comorbidities, serum markers of mineral metabolism (calcium, phosphorous, FGF-23)
Model 3: adjusted for demographics, comorbidities, serum albumin, fetuin-A
Model 4: adjusted for demographics, comorbidities, dp-MGP, osteoprotegerin
Model 5: adjusted for demographics, comorbidities, C-reactive protein
Median PWV was 10.3 m/s (IQR: 8.1–12.6) at baseline, and PWV did not change after 1 year [median PWV at Year 1: 10.1 m/s (IQR: 8.1–12.1), p=0.36]. There was no statistical evidence that CPP2 or T50 was associated with log-transformed PWV at baseline (Table 2) using linear regression and with repeated measures of log-transformed PWV (Table S2, S3) using linear mixed-effects regression. After adjusting for demographics and comorbidities, log-transformed PWV at baseline was 0.0002 lower per 100 nm larger CPP2, which was equivalent to 0.02% change in PWV; log-transformed PWV during 1-year follow-up was 0.002 higher per 100 nm increase in CPP2 size, which was equivalent to 0.19% change in PWV, but none of these were statistically significant (p=0.99 and 0.86, respectively). At baseline, 14% participants had high ABI. There was no statistical evidence that CPP2 or T50 was associated with high ABI (Table S4) using Poisson regression.
In stratified analyses, CPP2 and T50 were not associated with CAC score, TAC score or PWV cross-sectionally, regardless of the status of DM and CAD (Table S5). For secondary analyses, we repeated the analyses for CAC, TAC and PWV using 4 categories of CPP2 and T50 (Table S6). There was overall no association of CPP2 and T50 categories with CAC, except when comparing participants with small CPP2 and lower T50 with the reference group (small CPP2 and high T50) in Model 4. CPP2 and T50 categories were not associated with TAC or PWV.
Exploratory Analysis of CPP2 size and T50 with risk of all-cause mortality
Median follow-up was 3.5 years (IQR: 1.7–6.2). Participants were followed until December 31, 2017 (n=115), death (n=195), transplant (n=61), transfer to peritoneal dialysis (n=18), or loss to follow-up (n=11). Participants with higher CPP2 at baseline had higher risk of mortality (Table 3). After adjusting for participant demographics and comorbidities, the hazard ratio (HR) was 1.17 (95% CI: 1.05, 1.31, p=0.004) per 100 nm higher CPP2. The association between CPP2 and mortality remained significant after adjusting for additional covariates in all models, whereas there was no association between T50 and risk of mortality.
Table 3.
Exploratory analysis of CPP2 size and T50 with all-cause mortality
| CPP2 size (per 100 nm) | T50 (per 100 min) | |||||||
|---|---|---|---|---|---|---|---|---|
| Participants (n) | Death (n) | HR (95% CI) | p | Participants (n) | Death (n) | HR (95% CI) | p | |
| Unadjusted | 402 | 188 | 1.14 (1.04, 1.24) | 0.01 | 388 | 182 | 0.94 (0.83, 1.06) | 0.32 |
| Model 1 | 372 | 169 | 1.17 (1.05, 1.31) | 0.004 | 358 | 163 | 0.93 (0.82, 1.07) | 0.33 |
| Model 2 | 367 | 167 | 1.14 (1.01, 1.28) | 0.03 | 353 | 161 | 0.98 (0.85, 1.13) | 0.81 |
| Model 3 | 367 | 167 | 1.21 (1.07, 1.38) | 0.003 | 353 | 161 | 0.97 (0.84, 1.13) | 0.74 |
| Model 4 | 351 | 158 | 1.14 (1.02, 1.28) | 0.02 | 337 | 152 | 0.94 (0.81, 1.08) | 0.38 |
| Model 5 | 370 | 168 | 1.17 (1.05, 1.31) | 0.005 | 356 | 162 | 0.94 (0.82, 1.07) | 0.35 |
Cox proportional hazard models were used. A total of 195 individuals died after a median follow up of 3.5 years. Abbreviation: HR, Hazard ratio
Model 1: adjusted for demographics (age, sex, race), comorbidities (diabetes, coronary artery disease)
Model 3: adjusted for demographics, comorbidities, serum markers of mineral metabolism (calcium, phosphorous, FGF-23)
Model 4: adjusted for demographics, comorbidities, serum albumin, fetuin-A
Model 5: adjusted for demographics, comorbidities, dp-MGP, osteoprotegerin
Model 6: adjusted for demographics, comorbidities, C-reactive protein
DISCUSSION
In patients treated by maintenance hemodialysis, arterial calcification is prevalent and independently predicts mortality.1,35 In the past decade, calciprotein particles and characteristics of their transformation have emerged as potential therapeutic targets and biomarkers for arterial calcification. In prevalent hemodialysis patients, the time of primary to secondary CPP transformation, T50, may be a marker of mortality,18 but data on the relationship of CPP2 size and T50 with arterial calcification are limited. Using our dynamic light scattering assay,11 we measured CPP2 size in 402 and T50 in 388 PACE study participants. Contrarily to prior studies,21,36 we did not find evidence that the CPP parameters were associated with arterial calcification and arterial stiffness.
There are four possible explanations for our findings. First, a relationship between CPP parameters (CPP2 size and T50) and medical calcification may exist but we were unable to detect it because arterial calcification measured by CT scan is not specific to medial calcification alone. Patients with CKD can develop calcification in both arterial intima and media. While intimal calcification is an indicator of atherosclerosis, medial calcification is characterized by diffuse calcium and phosphate deposition.37,38 When exposed to excess mineral, such as hyperphosphatemia, VSMCs in the arteria media undergo phenotypic transition to cells that resemble osteoblasts and initiate mineralization.8,39,40 Since CPPs may mediate the effect of phosphate on arterial calcification,14 and may induce oxidative stress in VSMCs leading to mineralization,15 CPPs may affect medial calcification. Experimental animal studies suggest that CPP may also contribute to intimal calcification by promoting inflammation.41,42 Because coronary CT does not differentiate between intimal and medial calcification, we were unable to fully test the relationship of CPP parameters with medial calcification.
Second, our finding of no association between CPP2 size and T50 with PWV and ABI suggest that there may be no relationship between CPP parameters and arterial stiffness, which is a consequence of medial calcification. Our results differ from a prior report that found that among pre-dialysis CKD patients, lower T50 was associated with aortic PWV and progression of aortic stiffness. However, this analysis lacked adjustment for DM, an important risk factor for arterial calcification.17,43 Similar to our findings, a study of kidney transplant recipients (n=1,435), reported no association of T50 with PWV.19 Taken together, it is possible that the CPP2 size and T50 are not associated with arterial stiffness. We speculate that this lack of relationship could be due to the measured CPP parameters (CPP2 size and T50) not reflecting the in vivo CPP transformation process. The predominant form of circulating CPP is CPP1.10–12 Transformation of primary to secondary CPP occurs in vitro.16 In cultured VSMCs, only CPP2, but not CPP1, induced mineralization.15 In addition, in vitro and animal studies used synthetic CPP,9,15 and their findings may not be translated into the human studies.
Third, differences between our findings and those of prior studies may reflect differences in study populations and covariate adjustment. Prior studies that examined the relationship between CPP and arterial calcification were performed in patients with pre-dialysis CKD,21,36 and residual confounding may be present in these studies. In a study of patients with pre-dialysis CKD (n=73), higher fetuin-A containing CPPs level, quantified using fetuin-A sedimentation assay, was correlated with higher CAC score.36 The study was limited to univariate analysis, and did not account for important confounders such as age and kidney function.11,35,44 In another study of patients with pre-dialysis CKD, low T50 was not associated with the prevalence of CAC (n=1274 cross-sectional) nor incident CAC (n=780 longitudinal).21 Among participants with prevalent CAC at baseline, low T50 was associated with the severity and progression of CAC, but these associations were not significant among those without prevalent CAC. Compared to prior studies, our study population is more homogenous as they were all incident to HD for less than 6 months and all had hypertension. Lastly, PACE may be underpowered to detect a significant association between CPP and arterial calcification; however, it is the most readily available cohort with cardiovascular measures in an incident dialysis population.
We identified some correlations of CPP2 size and T50 with circulating inhibitors of arterial calcification including fetuin-A, serum albumin, dp-ucMGP and osteoprotegerin. We found that larger CPP2 and lower T50 were significantly correlated with lower fetuin-A and lower serum albumin. Fetuin-A inhibits arterial calcification by coalescing with calcium and phosphate to form CPP, thus preventing the growth and aggregation of calcium phosphate particles.45,46 Serum albumin can also become a component of CPP to inhibit arterial calcification, but it is less potent than fetuin-A.13,29 Our findings reflect a higher consumption of fetuin-A and albumin to form CPP, and provide support that fetuin-A has a higher affinity for calcium phosphate particles than albumin.13 We also found that CPP2 size was positively correlated with osteoprotegerin, and T50 was inversely correlated with dp-ucMGP level. Selective deletion of osteoprotegerin, a cytokine receptor, in mice results in early-onset osteoporosis and medial calcification of arteries.32,47 A positive correlation between fetuin-A containing CPP and osteoprotegerin levels has also previously been identified in pre-dialysis CKD patients.36 Dp-ucMGP increases in CKD and is associated with the severity of aortic calcification.30,31 Gla-rich protein has been shown to be a constitutive component of circulating CPP.48
In an exploratory analysis, we found that after a median follow-up of 3.5 years, larger CPP2 was independently associated with all-cause mortality in our study population. While additional well-powered studies are needed to confirm our finding, we speculate that the association of larger CPP2 with mortality may reflect the relationship between CPP and inflammation.9,41 In cultured macrophages, synthetic CPP triggered inflammation via the secretion of inflammasome-dependent IL-1β9 and upregulation of scavenger receptors.41 In our study, we found that CPP2 size was positively correlated with an inflammatory marker, C-reactive protein,49 supporting the relationship between serum CPP and inflammation. Further investigation of the role of serum CPP on inflammation may elucidate the pathophysiology underlying the high mortality seen in CKD.
We were unable to detect an association between T50 and mortality, but may have been limited by insufficient statistical power. Previous studies have examined such relationship in CKD patients, and the results were inconsistent. In patients with pre-dialysis CKD, T50 was associated with mortality, but the association became nonsignificant after adjusting for eGFR and proteinuria.44 In the HD cohort from the Evaluation of Cinacalcet Therapy to Lower Cardiovascular Events trial, lower T50 at baseline predicted mortality.18 This finding was not demonstrated in another HD cohort,50 in which worsening T50, but not T50 at baseline, predicted mortality.
Our study has several limitations. As in every observational study, residual confounding can exist despite adjustment for covariates. Our analyses of arterial calcification and arterial stiffness could be underpowered, and longitudinal analysis of PWV was limited to two time points (baseline and year 1). The CVs of our CPP assays were relatively high, indicating possible imprecision in measurement. In addition, we did not have repeated measures of CPP2 size and T50. Longitudinal trajectories of these parameters may enhance their ability to predict outcomes such as worsening arterial stiffness and mortality. African Americans have a disproportionate burden of ESRD;51 they were well-represented in our study cohort and had a lower prevalence of CAC than whites. However, these limit the generalizability of our findings as other incident dialysis populations may have different racial compositions. Lastly, the analyses between CPP parameters and mortality were exploratory, so the findings need to be confirmed in future large studies.
Our study has several strengths. Previous studies only measured T50 as a parameter of CPP transformation. We measured the hydrodynamic radius of CPP2 in addition to T50. We found that size of CPP2 aggregates was associated with mortality, a new finding that highlights the complexity of arterial calcification and mortality in dialysis. We were able to account for a large number of vascular measures of calcification and stiffness as well as study a number of potential confounders that were previously reported as independent factors associated with mortality. Importantly, our findings support the relationship between CPP and inflammation, a potential new direction. Finally, since different regions of the arterial tree may have a differential susceptibility to calcification,52 we evaluated arterial calcification, in the coronaries, thoracic aorta, and central and peripheral vessels with standardized protocols.25
In our cohort of incident HD patients, CPP2 size and T50 were correlated with certain serum markers of mineral mineralization and circulating inhibitors of calcification. However, we found no associations of CPP2 size or T50 with arterial calcification and stiffness. While current imaging modalities for arterial calcification may lack the specificity to detect medial calcification, it is possible that the circulating form of CPPs alone does not directly contribute to arterial calcification. In an exploratory analysis, we found an association between CPP2 size and mortality, suggesting that CPP2 size may be a potential marker of mortality in patients on HD. These findings need to be confirmed in future large studies.
Supplementary Material
Acknowledgements:
We thank participants, nephrologists, and staff of the DaVita and MedStar dialysis units in the Baltimore region who contributed to the PACE study.
Support: The PACE Study was funded by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK, R01 DK072367), National Kidney Foundation of Maryland, and by NIDDK (DK090070). W.C. is supported by the American Society of Nephrology Carl W. Gottschalk Research Grant and NIDDK (K23 DK114476). D.A.B is supported by NIDDK (R01 DK075462). RSP is supported by the Canada Research Chair in chronic kidney disease epidemiology. The Dynamic Light Scattering DynaPro Plate Reader II was supported by National Center for Research Resources grants 1S10 RR026501 and 1S10 RR027241, as well as National Institute of Allergy and Infectious Diseases P30 AI078495 and the School of Medicine and Dentistry, University of Rochester. The funding agencies did not have a role in study design, data collection, analysis, reporting, or the decision to submit for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosure: DAB is a consultant for Relyspa/Vifor/Fresenius, Amgen, Sanofi/Genzyme, and Tricida and has an equity interest in Amgen and Tricida. BLM is a consultant for Dodo Omnidata. The remaining authors declare that they have no relevant financial interests.
Peer Review: Received December 12, 2019. Evaluated by 3 external peer reviewers, with direct editorial input from a Statistics/Methods Editor and an Associate Editor, who served as Acting Editor-in-Chief. Accepted in revised form May 20, 2020. The involvement of an Acting Editor-in-Chief was to comply with AJKD’s procedures for potential conflicts of interest for editors, described in the Information for Authors & Journal Policies.
REFERENCES
- 1.London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2003;18(9):1731–1740. [DOI] [PubMed] [Google Scholar]
- 2.Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension. 2001;38(4):938–942. [DOI] [PubMed] [Google Scholar]
- 3.Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness on survival in end-stage renal disease. Circulation. 1999;99(18):2434–2439. [DOI] [PubMed] [Google Scholar]
- 4.Blacher J, Pannier B, Guerin AP, Marchais SJ, Safar ME, London GM. Carotid arterial stiffness as a predictor of cardiovascular and all-cause mortality in end-stage renal disease. Hypertension. 1998;32(3):570–574. [DOI] [PubMed] [Google Scholar]
- 5.Wilson PW, Kauppila LI, O’Donnell CJ, et al. Abdominal aortic calcific deposits are an important predictor of vascular morbidity and mortality. Circulation. 2001;103(11):1529–1534. [DOI] [PubMed] [Google Scholar]
- 6.Gorriz JL, Molina P, Cerveron MJ, et al. Vascular Calcification in Patients with Nondialysis CKD over 3 Years. Clinical journal of the American Society of Nephrology : CJASN. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.London GM. Mechanisms of arterial calcifications and consequences for cardiovascular function. Kidney international supplements. 2013;3(5):442–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. American journal of physiology Renal physiology. 2014;307(8):F891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koppert S, Buscher A, Babler A, et al. Cellular Clearance and Biological Activity of Calciprotein Particles Depend on Their Maturation State and Crystallinity. Frontiers in immunology. 2018;9:1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miura Y, Iwazu Y, Shiizaki K, et al. Identification and quantification of plasma calciprotein particles with distinct physical properties in patients with chronic kidney disease. Scientific reports. 2018;8(1):1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, Anokhina V, Dieudonne G, et al. Patients with advanced chronic kidney disease and vascular calcification have a large hydrodynamic radius of secondary calciprotein particles. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2019;34(6):992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith ER, Hewitson TD, Cai MMX, et al. A novel fluorescent probe-based flow cytometric assay for mineral-containing nanoparticles in serum. Scientific reports. 2017;7(1):5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heiss A, DuChesne A, Denecke B, et al. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. The Journal of biological chemistry. 2003;278(15):13333–13341. [DOI] [PubMed] [Google Scholar]
- 14.Ter Braake AD, Eelderink C, Zeper LW, et al. Calciprotein particle inhibition explains magnesium-mediated protection against vascular calcification. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aghagolzadeh P, Bachtler M, Bijarnia R, et al. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-alpha. Atherosclerosis. 2016;251:404–414. [DOI] [PubMed] [Google Scholar]
- 16.Pasch A, Farese S, Graber S, et al. Nanoparticle-based test measures overall propensity for calcification in serum. Journal of the American Society of Nephrology : JASN. 2012;23(10):1744–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith ER, Ford ML, Tomlinson LA, et al. Serum calcification propensity predicts all-cause mortality in predialysis CKD. Journal of the American Society of Nephrology : JASN. 2014;25(2):339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pasch A, Block GA, Bachtler M, et al. Blood Calcification Propensity, Cardiovascular Events, and Survival in Patients Receiving Hemodialysis in the EVOLVE Trial. Clinical journal of the American Society of Nephrology : CJASN. 2017;12(2):315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dahle DO, Asberg A, Hartmann A, et al. Serum Calcification Propensity Is a Strong and Independent Determinant of Cardiac and All-Cause Mortality in Kidney Transplant Recipients. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2016;16(1):204–212. [DOI] [PubMed] [Google Scholar]
- 20.Keyzer CA, de Borst MH, van den Berg E, et al. Calcification Propensity and Survival among Renal Transplant Recipients. Journal of the American Society of Nephrology : JASN. 2016;27(1):239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bundy JD, Cai X, Scialla JJ, et al. Serum Calcification Propensity and Coronary Artery Calcification Among Patients With CKD: The CRIC (Chronic Renal Insufficiency Cohort) Study. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2019;73(6):806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dekker M, Pasch A, van der Sande F, et al. High-Flux Hemodialysis and High-Volume Hemodiafiltration Improve Serum Calcification Propensity. PloS one. 2016;11(4):e0151508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bressendorff I, Hansen D, Schou M, Pasch A, Brandi L. The Effect of Increasing Dialysate Magnesium on Serum Calcification Propensity in Subjects with End Stage Kidney Disease: A Randomized, Controlled Clinical Trial. Clinical journal of the American Society of Nephrology : CJASN. 2018;13(9):1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kendrick J, Shah P, Andrews E, et al. Effect of Treatment of Metabolic Acidosis on Vascular Endothelial Function in Patients with CKD: A Pilot Randomized Cross-Over Study. Clinical journal of the American Society of Nephrology : CJASN. 2018;13(10):1463–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parekh RS, Meoni LA, Jaar BG, et al. Rationale and design for the Predictors of Arrhythmic and Cardiovascular Risk in End Stage Renal Disease (PACE) study. BMC nephrology. 2015;16:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaar BG, Zhang L, Chembrovich SV, et al. Incidental findings on cardiac computed tomography in incident hemodialysis patients: the predictors of arrhythmic and cardiovascular events in end-stage renal disease (PACE) study. BMC nephrology. 2014;15:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohler ER 3rd. Peripheral arterial disease: identification and implications. Archives of internal medicine. 2003;163(19):2306–2314. [DOI] [PubMed] [Google Scholar]
- 28.Hendriks EJ, Westerink J, de Jong PA, et al. Association of High Ankle Brachial Index With Incident Cardiovascular Disease and Mortality in a High-Risk Population. Arteriosclerosis, thrombosis, and vascular biology. 2016;36(2):412–417. [DOI] [PubMed] [Google Scholar]
- 29.Garnett J, Dieppe P. The effects of serum and human albumin on calcium hydroxyapatite crystal growth. The Biochemical journal. 1990;266(3):863–868. [PMC free article] [PubMed] [Google Scholar]
- 30.Schurgers LJ, Barreto DV, Barreto FC, et al. The circulating inactive form of matrix gla protein is a surrogate marker for vascular calcification in chronic kidney disease: a preliminary report. Clinical journal of the American Society of Nephrology : CJASN. 2010;5(4):568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386(6620):78–81. [DOI] [PubMed] [Google Scholar]
- 32.Bucay N, Sarosi I, Dunstan CR, et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes & development. 1998;12(9):1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schafer C, Heiss A, Schwarz A, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. The Journal of clinical investigation. 2003;112(3):357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reilly MP, Wolfe ML, Localio AR, Rader DJ. Coronary artery calcification and cardiovascular risk factors: impact of the analytic approach. Atherosclerosis. 2004;173(1):69–78. [DOI] [PubMed] [Google Scholar]
- 35.Goodman WG, Goldin J, Kuizon BD, et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. The New England journal of medicine. 2000;342(20):1478–1483. [DOI] [PubMed] [Google Scholar]
- 36.Hamano T, Matsui I, Mikami S, et al. Fetuin-mineral complex reflects extraosseous calcification stress in CKD. Journal of the American Society of Nephrology : JASN. 2010;21(11):1998–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shroff RC, McNair R, Figg N, et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118(17):1748–1757. [DOI] [PubMed] [Google Scholar]
- 38.Hassan NA, D’Orsi ET, D’Orsi CJ, O’Neill WC. The risk for medial arterial calcification in CKD. Clinical journal of the American Society of Nephrology : CJASN. 2012;7(2):275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Oliveira RB, Okazaki H, Stinghen AE, Drueke TB, Massy ZA, Jorgetti V. Vascular calcification in chronic kidney disease: a review. Jornal brasileiro de nefrologia : ‘orgao oficial de Sociedades Brasileira e Latino-Americana de Nefrologia. 2013;35(2):147–161. [DOI] [PubMed] [Google Scholar]
- 40.Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circulation research. 2011;109(6):697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith ER, Hanssen E, McMahon LP, Holt SG. Fetuin-A-containing calciprotein particles reduce mineral stress in the macrophage. PloS one. 2013;8(4):e60904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herrmann M, Schafer C, Heiss A, et al. Clearance of fetuin-A--containing calciprotein particles is mediated by scavenger receptor-A. Circulation research. 2012;111(5):575–584. [DOI] [PubMed] [Google Scholar]
- 43.Katz R, Budoff MJ, O’Brien KD, Wong ND, Nasir K. The metabolic syndrome and diabetes mellitus as predictors of thoracic aortic calcification as detected by non-contrast computed tomography in the Multi-Ethnic Study of Atherosclerosis. Diabet Med. 2016;33(7):912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bundy JD, Cai X, Mehta RC, et al. Serum Calcification Propensity and Clinical Events in CKD. Clinical journal of the American Society of Nephrology : CJASN. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heiss A, Eckert T, Aretz A, et al. Hierarchical role of fetuin-A and acidic serum proteins in the formation and stabilization of calcium phosphate particles. The Journal of biological chemistry. 2008;283(21):14815–14825. [DOI] [PubMed] [Google Scholar]
- 46.Holt SG, Smith ER. Fetuin-A-containing calciprotein particles in mineral trafficking and vascular disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2016;31(10):1583–1587. [DOI] [PubMed] [Google Scholar]
- 47.Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–319. [DOI] [PubMed] [Google Scholar]
- 48.Viegas CSB, Santos L, Macedo AL, et al. Chronic Kidney Disease Circulating Calciprotein Particles and Extracellular Vesicles Promote Vascular Calcification: A Role for GRP (Gla-Rich Protein). Arteriosclerosis, thrombosis, and vascular biology. 2018;38(3):575–587. [DOI] [PubMed] [Google Scholar]
- 49.Pepys MB, Hirschfield GM. C-reactive protein: a critical update. The Journal of clinical investigation. 2003;111(12):1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lorenz G, Steubl D, Kemmner S, et al. Worsening calcification propensity precedes all-cause and cardiovascular mortality in haemodialyzed patients. Scientific reports. 2017;7(1):13368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi AI, Rodriguez RA, Bacchetti P, Bertenthal D, Hernandez GT, O’Hare AM. White/black racial differences in risk of end-stage renal disease and death. The American journal of medicine. 2009;122(7):672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schlieper G Vascular calcification in chronic kidney disease: not all arteries are created equal. Kidney international. 2014;85(3):501–503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.