

Abstract
Background:
Family history of prostate cancer (PCa) is a well-known risk factor, and both common and rare genetic variants are associated with the disease.
Objective:
To detect new genetic variants associated with PCa, capitalizing on the role of family history and more aggressive PCa.
Design, setting, and participants:
A two-stage design was used. In stage one, whole-exome sequencing was used to identify potential risk alleles among affected men with a strong family history of disease or with more aggressive disease (491 cases and 429 controls). Aggressive disease was based on a sum of scores for Gleason score, node status, metastasis, tumor stage, prostate-specific antigen at diagnosis, systemic recurrence, and time to PCa death. Genes identified in stage one were screened in stage two using a custom-capture design in an independent set of 2917 cases and 1899 controls.
Outcome measurements and statistical analysis:
Frequencies of genetic variants (singly or jointly in a gene) were compared between cases and controls.
Results and limitations:
Eleven genes previously reported to be associated with PCa were detected (ATM, BRCA2, HOXB13, FAM111A, EMSY, HNF1B, KLK3, MSMB, PCAT1, PRSS3, and TERT), as well as an additional 10 novel genes (PABPC1, QK1, FAM114A1, MUC6, MYCBP2, RAPGEF4, RNASEH2B, ULK4, XPO7, and THAP3). Of these 10 novel genes, all but PABPC1 and ULK4 were primarily associated with the risk of aggressive PCa.
Conclusions:
Our approach demonstrates the advantage of gene sequencing in the search for genetic variants associated with PCa and the benefits of sampling patients with a strong family history of disease or an aggressive form of disease.
Patient summary:
Multiple genes are associated with prostate cancer (PCa) among men with a strong family history of this disease or among men with an aggressive form of PCa.
Keywords: Whole-exome sequencing, Custom-capture sequencing, Familial prostate cancer, Genetic risk variants
1. Introduction
It is estimated that there were 1 276 106 new cases of prostate cancer (PCa) worldwide in 2018 [1]. In the USA, 174 650 new cases of PCa and 31 620 deaths due to PCa were predicted to occur during 2019 [2]. The well-established risk factors for PCa are older age, African-American ancestry [3,4], and a family history of the disease. Variants in the genes BRCA2 and HOXB13 are well established as having a strong association with PCa risk. ATM and the genes involved in mismatch repair (BRCA1, MLH1, PMS2, MSH2, and MSH6) have also been implicated in PCa risk. In addition, genome-wide association studies have identified approximately 170 common genetic variants spread across all autosomes and the X chromosome that account for an estimated 28.4% of the familial PCa (FPC) risk [5]. As much of the hereditary risk remains unexplained, identification of additional genes and genetic variants associated with PCa risk can potentially aid in early detection, discernment of aggressive disease, and possibly provide targets for therapy.
To search for genes involved in hereditary or more aggressive PCa, we capitalized on pedigrees and samples available from the International Consortium for Prostate Cancer Genetics (ICPCG), an international collaboration that has conducted numerous family-based studies for almost 20 yr [6–15].
2. Patients and methods
We used a two-stage design in which the first stage was used to screen for genes with suggestive evidence of association with PCa, followed by a second-stage case-control study that conducted a more rigorous evaluation of the candidate genes suggested by the first stage. Study participants for both stages were recruited from the 15 ICPCG groups (see the Supplementary material). Analyses focused on men with European ancestry because of a limited number of samples with other ancestries. As a guide, Figure 1 provides details on the samples used in the two stages and why some samples were excluded. All participants gave informed consent to utilize samples for research purposes, and the study was approved by the respective institutional review boards.
Fig. 1 –
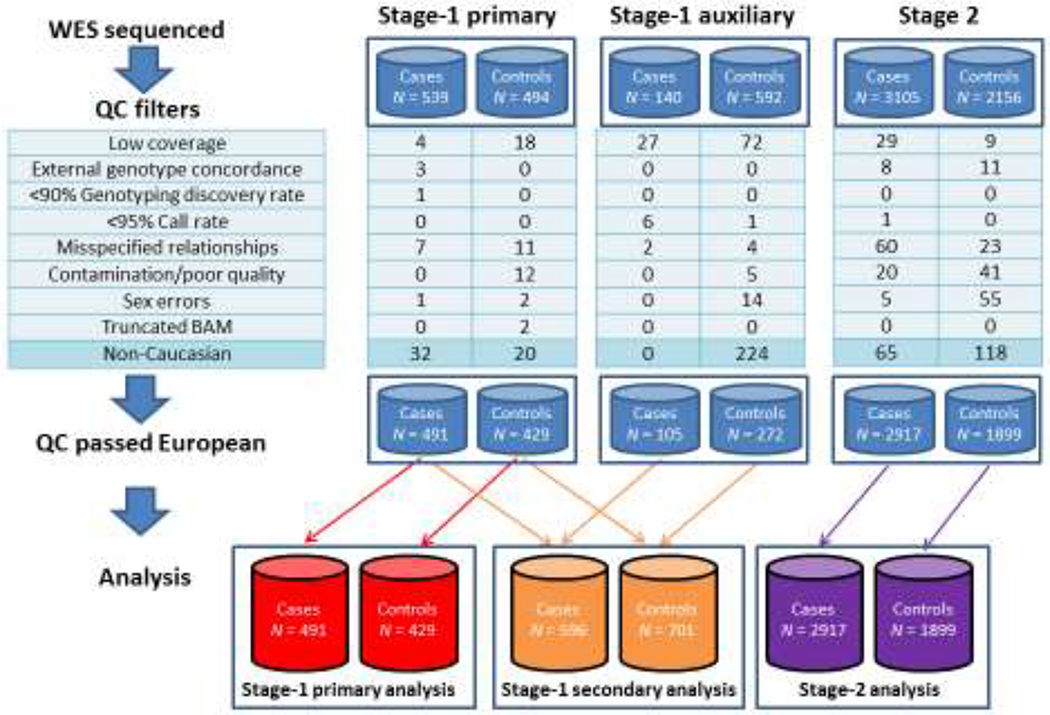
Flow of samples excluded from stage one (primary and auxiliary samples) and stage two, resulting in sample sizes used in analyses at the bottom of the figure. QC = quality control; WES = whole-exome sequencing.
2.1. Stage-one samples
Families with FPC, defined as families with three or more affected first-degree relatives with PCa, were reviewed to select 539 PCa cases from 366 pedigrees for whole-exome sequencing (WES) at the Mayo Clinic. Controls were 494 samples from noncancer studies with WES conducted at the Mayo Clinic. To increase power, we obtained auxiliary WES data for 140 unrelated cases and 592 unrelated controls. Secondary analyses included the cases and controls used in the primary analyses, pooled with the auxiliary data (see the Supplementary material for details).
2.2. Stage-one WES
For all cases and controls sequenced at the Mayo Clinic, exome capture was performed using Agilent SureSelect Human All Exon 50Mb or V4 + UTR capture kits. Samples were pooled after the capture and sequenced three to a lane using the Illumina HiSeq. All cases sequenced at the Mayo Clinic were also genotyped on the Illumina Infinium OmniExpress-12 array. Externally sequenced samples were sequenced using a variety of capture kits, including Illumina TruSeq and Nimblegen SeqCap (see Supplementary Table 1 and the Supplementary material).
2.3. Stage-one statistical analyses
The association of PCa with genetic variants, either as a single variant or as a group of variants in a gene, was assessed by association analyses (comparing cases with controls) and cosegregation of genetic variants within pedigrees (eg, excess sharing of alleles among relatives). All variants with minor allele frequency (MAF) >0.001 were included in single-variant association analyses. To screen for less common variants, all variants with MAF ≤5% were included in gene-based tests. Single-variant and gene-level associations were evaluated using burden (ie, sum of alternate alleles for variants within a gene) and kernel statistics that allow for known pedigree relationships [16]. As variants are likely to differ in terms of functional effect on traits, a variety of weighting schemes were used to evaluate simultaneously all variants within a gene (see the Supplementary material). For single-variant tests, we considered only tier-1 (likely to cause protein truncation) and tier-2 (nonsynonymous coding variants and in-frame indels) variants when selecting genes to carry forward to stage two. For primary analyses, five principal components and capture kits (V3 vs V4 + UTR) were included as covariates. For secondary analyses, 12 principal components were included. We used a liberal significance threshold of p < 0.01 to select genes that showed some minimal association with PCa to carry forward to stage two.
2.4. Stage-two samples
In stage two, 3105 unrelated PCa cases were selected for targeted sequencing. Of these cases, 2145 were selected from the remaining FPC pedigrees that were not included in stage one, with only one individual selected per family. Cases with DNA available were selected based on the strongest family history and the most aggressive PCa. In pedigrees with multiple aggressive cases, the individual with the youngest age at diagnosis was chosen. Beyond these cases with a family history of PCa, 960 men with more aggressive disease were selected because of the clinical importance of this phenotype and because more aggressive cancer might be more enriched for causative genetic factors. These aggressive cases were unrelated to the other stage-one or stage-two patients, and some lacked a family history of PCa. The initial criteria for selecting aggressive cases were based on the ICPCG criteria used in stage one (see the Supplementary material). We subsequently refined a score for aggressive disease based on the sum of scores for the following clinical factors: Gleason score 8–10 (+2 points), node status N1+ (+2 points), metastasis Ml (+2 points), tumor stage T4 (+2 points), prostate-specific antigen (PSA) at diagnosis (PSA >20, +1 point; PSA >50, +2 points), systemic recurrence (+2 points), and time to PCa death (<5 yr, +2 points; 5–10 yr, +1 point). For our analyses, cases with a score of at least 2 were categorized as aggressive. Owing to missing clinical data, some cases could have a score of 0–1 and were categorized as nonaggressive.
A set of 2156 stage-two controls was identified from 12 ICPCG member groups and was restricted to unrelated men with no personal history of cancer, preferably those with no family history of PCa, who were unrelated to cases used in stages one and two, and were of European descent. Each of the ICPCG groups selected controls by frequency matching with their contributed cases based on birth year. Controls selected from the Mayo Biobank were required to be free of PCa and at least 70 yr old.
2.5. Stage-two custom-capture resequencing
A custom-capture sequencing array was designed to characterize all genes meeting stage-one significance criteria. The custom array was designed by Agilent to target 1202 genes (5.9 Mb) at an average coverage of 100×. Coverage of the targeted genes included the exons, ±30 bp surrounding each exon, and 1 kb of the 5’ UTR. In addition, we created a custom genotyping panel consisting of 29 highly polymorphic single nucleotide polymorphisms to provide sample identity verification.
2.6. Stage-two statistical analyses
Gene-level association analyses were conducted by burden tests and kernel statistics as implemented in SKAT-O [17]. Single-variant associations were performed using PLINK [18]. Covariates associated with PCa status included as adjusting covariates were a factor representing the ICPCG member group, log(missing call rate), and log(Qubit concentration); see the Supplementary material for details.
3. Results
3.1. Stage one: screening by WES
Stage-one primary analyses included 491 cases and 429 controls (see Table 1 for clinical characteristics of cases). In total, 720 genes met the significance threshold of p < 0.01 and were selected for targeted sequencing in stage two. An additional 482 candidate genes were nominated by ICPCG collaborating members, resulting in 1202 genes for targeted sequencing in stage two (see the Supplementary material for details of stage-one results and Supplementary Table 5 for genes sequenced in stage two).
Table 1 –
Clinical characteristics of stage-one and stage-two cases and controls
| Stage-one cases | Stage-two cases | Stage-two controls | ||||
|---|---|---|---|---|---|---|
| 491 | % | 2917 | % | 1899 | % | |
| Family history | 491 | 100.0 | 1993 | 68.3 | 0 | 0 |
| Positive | ||||||
| Negative | 0 | 0 | 924 | 31.7 | 1218 | 64.1 |
| Unknown | 0 | 0 | 0 | 0 | 681 | 35.9 |
| Age at diagnosis (yr) | ||||||
| ≤50 | 26 | 5.3 | 233 | 8.0 | 170 | 9 |
| 51–65 | 264 | 53.8 | 1683 | 57.7 | 772 | 40.7 |
| 66–75 | 165 | 33.6 | 826 | 28.3 | 509 | 26.8 |
| >75 | 30 | 6.1 | 157 | 5.4 | 374 | 19.7 |
| Unknown | 6 | 1.2 | 18 | 0.6 | 74 | 3.9 |
| Gleason score | ||||||
| <7 | 129 | 26.3 | 840 | 28.8 | ||
| 7 | 108 | 22.0 | 695 | 23.8 | ||
| >8 | 59 | 12.0 | 698 | 23.9 | ||
| Unknown | 195 | 39.7 | 684 | 23.4 | ||
| Stage | ||||||
| T1 | 21 | 4.3 | 368 | 12.6 | ||
| T2 | 79 | 16.1 | 836 | 28.7 | ||
| T3 | 56 | 11.4 | 722 | 24.8 | ||
| T4 | 6 | 1.2 | 101 | 3.5 | ||
| Unknown | 318 | 64.8 | 890 | 30.5 | ||
| Metastasis | ||||||
| N0 | 52 | 10.6 | 1018 | 34.9 | ||
| N1 | 7 | 1.4 | 329 | 11.3 | ||
| N2 | 0 | 0.0 | 4 | 0.1 | ||
| NX | 432 | 88.0 | 1566 | 53.7 | ||
| M0 | 68 | 13.8 | 1096 | 37.6 | ||
| M1 | 12 | 2.4 | 306 | 10.5 | ||
| MX | 411 | 83.7 | 1515 | 51.9 | ||
| PSA | ||||||
| <4 | 25 | 5.1 | 233 | 8.0 | ||
| 4–19 | 157 | 32.0 | 979 | 33.6 | ||
| 20–99 | 60 | 12.2 | 403 | 13.8 | ||
| ≥100 | 11 | 2.2 | 194 | 6.7 | ||
| Unknown | 238 | 48.5 | 1108 | 38.0 | ||
| ICPCG aggressiveness | ||||||
| Insignificant | 2 | 0.4 | 45 | 1.5 | ||
| Moderate | 155 | 31.6 | 704 | 24.1 | ||
| Aggressive | 252 | 51.3 | 1905 | 65.3 | ||
| Unknown | 82 | 16.7 | 263 | 9.0 | ||
| Aggressiveness score a | ||||||
| Missing all clinical data | 138 | 28.1 | 153 | 5.2 | ||
| 0 | 223 | 45.4 | 1390 | 47.7 | ||
| 1 | 36 | 7.3 | 116 | 4.0 | ||
| 2 | 55 | 11.2 | 457 | 15.7 | ||
| 3 | 11 | 2.2 | 176 | 6.0 | ||
| 4 | 17 | 3.5 | 295 | 10.1 | ||
| 5 | 3 | 0.6 | 89 | 3.1 | ||
| 6 | 7 | 1.4 | 130 | 4.5 | ||
| 7 | 1 | 0.2 | 26 | 0.9 | ||
| 8 | 0 | 0.0 | 60 | 2.1 | ||
| 9 | 0 | 0.0 | 10 | 0.3 | ||
| 10 | 0 | 0.0 | 12 | 0.4 | ||
| 12 | 0 | 0.0 | 3 | 0.1 | ||
ICPCG = International Consortium for Prostate Cancer Genetics; PCa = prostate cancer; PSA = prostate-specific antigen.
Aggressiveness score is the sum of scores for the following clinical factors: Gleason score 8–10 (+2 points), node status N1+ (+2 points), metastasis M1 (+2 points), tumor stage T4 (+2 points), PSA at diagnosis (PSA >20, +1 point; PSA >50, +2 points), systemic recurrence (+2 points), and time to PCa death (<5 yr, +2 points; 5–10 yr, +1 point).
3.2. Stage-two association results
After extensive quality control (QC; see Fig. 1, and Supplementary Table 3) and restriction to European ancestry, 2917 unrelated cases and 1899 unrelated controls were used in statistical analyses. Clinical characteristics of the cases are presented in Table 1 (see Supplementary Table 4 for more details). Of the 1202 sequenced genes, 1188 had at least one variant that passed QC (total of 29 838 variants for single-variant analyses), and 1116 genes had at least one tier-1 or tier-2 variant that passed QC for gene-level analyses.
3.3. Single-variant associations
The results of analyzing 29 838 single variants are summarized in Figure 2 using Manhattan plots for the four types of comparisons: all cases versus controls (Fig. 2A), familial cases versus controls (Fig. 2B), aggressive cases versus controls (Fig. 2C), and aggressive cases versus nonaggressive cases (Fig. 2D). We used a Bonferroni correction for multiple testing based on a p-value threshold of 1e–6 (red horizontal line in Fig. 2). For the comparisons of all cases and familial cases with controls, 15 variants were statistically significant (summarized in Table 2). The missense variant in HOXB13 (rs138213197) showed the greatest risk when comparing all cases with controls (odds ratio [OR] = 21.01; 95% confidence interval [CI]: 6.58, 67.15; Table 2) and familial cases with controls (OR = 22.44, 95% CI: 6.99, 71.97; Table 2). Two variants in the MSMB and TERT genes were also associated with an increase in PCa risk, where the alternate allele was more frequent in cases than in controls (Table 2). In contrast, the remaining variants in the HNF1B, KLK3, MSMB, PCAT1, and ULK4 genes were associated with reduced PCa risk, where the alternate alleles were significantly less frequent in cases than in controls. To place our results in context of the findings from a recent large-scale genome-wide association study, we compared our results with those from the PRACTICAL consortium, which analyzed >140 000 cases and controls and had 20 370 946 genotyped or high-quality imputed variants [19]. Our results from comparing all cases or familial cases with controls were consistent with those from PRACTICAL, in terms of direction of OR and significant p values, for 14 of the 15 variants (Table 2) corresponding to seven genes: HNF1B, HOXB13, KLK3, MSMB, PCAT1, TERT, and ULK4.
Fig. 2 –
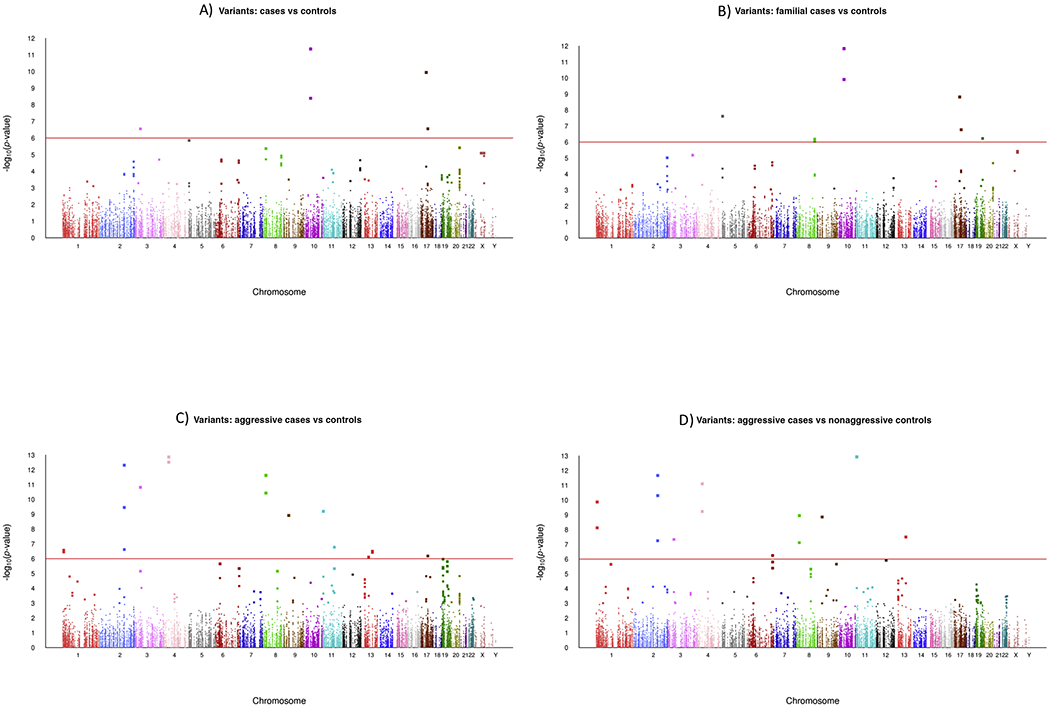
Manhattan plots of stage-two associations of single variants with PCa. The x axes show the chromosomal positions and the y axes show the −log10 p value. (A) All PCa cases versus controls. (B) PCa cases with a positive family history versus controls. (C) Aggressive PCa cases versus controls; (D) Aggressive PCa cases versus nonaggressive PCa cases. Statistical analyses based on log-additive effects of alternate alleles. PCa = prostate cancer.
Table 2 –
All cases and familial cases: stage-two single-variant analyses that detected significant associations, illustrating the gene and summary statistics for variants
| Genea | rsID | Allele | All cases (N = 2817) vs controls (N = 1899) | PRACTICALb | Familial cases (N = 1993) vs controls (N = 1899) | ALT allele frequencies | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| REF | ALT | OR | 95% CI | p value | OR | p value | OR | 95% CI | p value | All cases | Familial cases | Controls | ||
| HNF1B | rs3216929 | G | GCAGA | 0.74 | 0.68, 0.81 | 1.13E-10 | 0.85 | 1.55E-81 | 0.74 | 0.67, 0.82 | 1.52E-09 | 0.3145 | 0.3084 | 0.3775 |
| HOXB13 | rs138213197 | C | T | 21.01 | 6.58, 67.15 | 2.78E-07 | 3.85 | 9.17E-63 | 22.44 | 6.99, 71.97 | 1.69E-07 | 0.0135 | 0.0161 | 0.0008 |
| KLK3 | rs17632542 | T | C | 0.72 | 0.61, 0.86 | 1.70E-04 | 0.74 | 6.69E-81 | 0.61 | 0.50, 0.74 | 5.96E-07 | 0.0591 | 0.0489 | 0.0777 |
| MSMB (56 bp) | rs10993994 | A | G | 0.74 | 0.68, 0.80 | 4.41E-12 | 0.81 | 2.29E-147 | 0.71 | 0.65, 0.78 | 1.46E-12 | 0.5329 | 0.5258 | 0.6124 |
| MSMB (238 bp) | rs12770171 | G | A | 1.38 | 1.24, 1.54 | 4.05E-09 | 1.18 | 3.71E-58 | 1.46 | 1.30, 1.63 | 1.23E-10 | 0.2384 | 0.2481 | 0.1854 |
| PCAT1 | rs1551515 | T | A | 0.82 | 0.75, 0.90 | 3.93E-05 | 0.88 | 1.55E-48 | 0.77 | 0.70, 0.86 | 7.67E-07 | 0.2691 | 0.2561 | 0.3099 |
| rs1551513 | T | C | 0.82 | 0.75, 0.90 | 3.31E-05 | 0.88 | 1.17E-48 | 0.77 | 0.70, 0.85 | 6.53E-07 | 0.2691 | 0.2561 | 0.3104 | |
| rs4473999 | C | T | 0.82 | 0.75, 0.90 | 3.93E-05 | 0.88 | 2.16E-48 | 0.77 | 0.70, 0.86 | 7.67E-07 | 0.2691 | 0.2561 | 0.3099 | |
| rs9656964 | C | G | 0.82 | 0.75, 0.90 | 3.93E-05 | 0.88 | 2.80E-48 | 0.77 | 0.70, 0.86 | 7.67E-07 | 0.2691 | 0.2561 | 0.3099 | |
| rs17762938 | T | C | 0.82 | 0.75, 0.90 | 3.31E-05 | 0.88 | 3.42E-48 | 0.77 | 0.70, 0.85 | 6.53E-07 | 0.2691 | 0.2561 | 0.3104 | |
| rs7823297 | C | T | 0.82 | 0.75, 0.90 | 3.31-05 | 0.88 | 2.12E-48 | 0.77 | 0.70, 0.85 | 6.53E-07 | 0.2691 | 0.2561 | 0.3104 | |
| rs6651240 | A | T | 0.82 | 0.75, 0.90 | 4.44E-05 | 0.88 | 1.62E-48 | 0.77 | 0.70, 0.86 | 9.02E-07 | 0.2691 | 0.2561 | 0.3096 | |
| PCAT1 (135 bp) | rs4573233 | G | A | 0.82 | 0.75, 0.90 | 3.93E-05 | 0.88 | 1.65E-48 | 0.77 | 0.70, 0.86 | 7.67E-07 | 0.2691 | 0.2561 | 0.3099 |
| TERT (909 bp) | rs7712562 | A | G | 1.35 | 1.20, 1.53 | 1.40E-06 | 1.18 | 2.24E-48 | 1.47 | 1.29, 1.69 | 2.45E-08 | 0.8768 | 0.8861 | 0.8420 |
| ULK4 | rs202114865 | G | A | 0.62 | 0.52, 0.75 | 2.81E-07 | NA | NA | 0.80 | 0.66, 0.96 | 0.02 | 0.0502 | 0.0686 | 0.0796 |
CI = confidence interval; NA = genetic variants from this current study were not genotyped in PRACTICAL; OR = odds ratio.
Variants outside a gene have their base-pair distance to the nearest gene in parentheses, based on Genome Reference Consortium Human Build 38.
PRACTICAL OR and p value provided by the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) consortium, based on genome-wide analyses of >140 000 men [19].
When comparing the aggressive cases with unaffected controls (Table 3), the variant rs138213197 in HOXB13 showed the largest risk (OR = 23.36; 95% CI: 6.75, 80.87; Table 3), followed by the variant rs764430438 in the PRSS3 gene (OR = 2.53; 95% CI: 1.88, 3.42) and the variant rs374242789 in the MYCBP2 gene (OR = 1.73; 95% CI: 1.40, 2.13). The remaining variants in Table 3 showed significantly less frequent alternate alleles among aggressive cases compared with controls or nonaggressive cases. These included variants in the following genes: EMSY, FAM114A1, MUC6, MYCBP2, RAPGEF4, RNASEH2B, THAP3, ULK4, and XPO7.
Table 3 –
Aggressive cases: stage-two single-variant analyses that detected significant associations, illustrating the gene and summary statistics for variants
| Gene a | rsID | Allele | Aggressive cases (N = 1258) vs controls (N = 1899) | Aggressive cases (N = 1258) vs Nonaggressive cases (N = 1659) | ALT allele frequency | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| REF | ALT | OR | 95% CI | p value | OR | 95% CI | p value | Aggressive cases | Controls | ||
| EMSY | rs200165356 | C | T | 0.20 | 0.11, 0.36 | 1.68E-07 | 0.29 | 0.16, 0.55 | 1.00E-04 | 0.0056 | 0.0266 |
| FAM114A1 | rs754808360 | C | T | 0.35 | 0.27, 0.47 | 3.07E-13 | 0.40 | 0.30, 0.53 | 5.92E-10 | 0.0318 | 0.0882 |
| rs778907323 | C | T | 0.32 | 0.23, 0.43 | 1.35E-13 | 0.33 | 0.24, 0.46 | 7.88E-12 | 0.0250 | 0.0790 | |
| HOXB13 | rs138213197 | C | T | 23.36 | 6.75, 80.87 | 6.56E-07 | 0.94 | 0.56, 1.58 | 0.81 | 0.0119 | 0.0008 |
| MUC6 | rs770247761 | A | C | 0.43 | 0.33, 0.57 | 6.35E-10 | 0.36 | 0.27, 0.47 | 1.19E-13 | 0.0362 | 0.0956 |
| MYCBP2 | rs374242789 | TA | T | 1.73 | 1.40, 2.13 | 3.12E-07 | 1.59 | 1.27, 1.99 | 4.37E-05 | 0.1222 | 0.0691 |
| rs751238365 | T | TA | 0.36 | 0.24, 0.53 | 3.76E-07 | 0.32 | 0.21, 0.48 | 3.19E-08 | 0.0146 | 0.0508 | |
| PRSS3 | rs764430438 | C | T | 2.53 | 1.88, 3.42 | 1.21E-09 | 2.70 | 1.96, 3.73 | 1.41E-09 | 0.0656 | 0.0247 |
| RAPGEF4 | rs746414802 | A | T | 0.25 | 0.17, 0.36 | 4.88E-13 | 0.25 | 0.17, 0.37 | 2.16E-12 | 0.0151 | 0.0629 |
| rs758902129 | C | T | 0.30 | 0.20, 0.43 | 3.49E-10 | 0.28 | 0.19, 0.41 | 4.93E-11 | 0.0155 | 0.0569 | |
| rs747279958 | G | T | 0.29 | 0.18, 0.46 | 2.41E-07 | 0.27 | 0.16, 0.43 | 5.74E-08 | 0.0099 | 0.0377 | |
| RNASEH2B | rs1172291060 | C | T | 0.50 | 0.38, 0.66 | 7.74E-07 | 0.53 | 0.39, 0.71 | 2.08E-05 | 0.0335 | 0.0795 |
| THAP3 (603 bp) | rs1369585326 | C | T | 0.48 | 0.37, 0.64 | 3.50E-07 | 0.40 | 0.30, 0.53 | 1.34E-10 | 0.0338 | 0.0764 |
| THAP3 (600 bp) | rs1228553175 | C | T | 0.39 | 0.27, 0.56 | 2.64E-07 | 0.35 | 0.24, 0.50 | 7.57E-09 | 0.0187 | 0.0529 |
| ULK4 | rs202114865 | G | A | 0.34 | 0.25, 0.46 | 1.52E-11 | 0.40 | 0.29, 0.55 | 4.67E-08 | 0.0231 | 0.0796 |
| XPO7 | rs771633865 | C | T | 0.32 | 0.23, 0.45 | 3.70E-11 | 0.39 | 0.27, 0.55 | 7.70E-08 | 0.0219 | 0.0706 |
| rs760440285 | G | T | 0.29 | 0.21, 0.41 | 2.37E-12 | 0.33 | 0.23, 0.47 | 1.13E-09 | 0.0191 | 0.0690 | |
CI = confidence interval; OR = odds ratio.
Variants outside a gene have their base-pair distance to the nearest gene in parentheses based on Genome Reference Consortium Human Build 38.
3.4. Gene-level associations
The results from 1116 SKAT-O gene-level analyses are summarized in Figure 3 using Manhattan plots for each of the four comparisons. Genes detected by burden analyses were a subset of those detected by SKAT-O, except for the ATM gene that was borderline significant in the SKAT-O analyses yet crossed the significance threshold in the burden analyses. When considering all analyses, there were six genes that achieved statistical significance based on Bonferroni threshold p < 5e–5: ATM (156 variants), BRCA2 (172 variants), FAM111A (35 variants), HOXB13 (15 variants), PABPC1 (29 variants), and QK1 (11 variants). Details on the gene-level and single-variant analyses for the variants that had an allele frequency of at least 0.001 are provided in Supplementary Tables 6–8.
Fig. 3 –
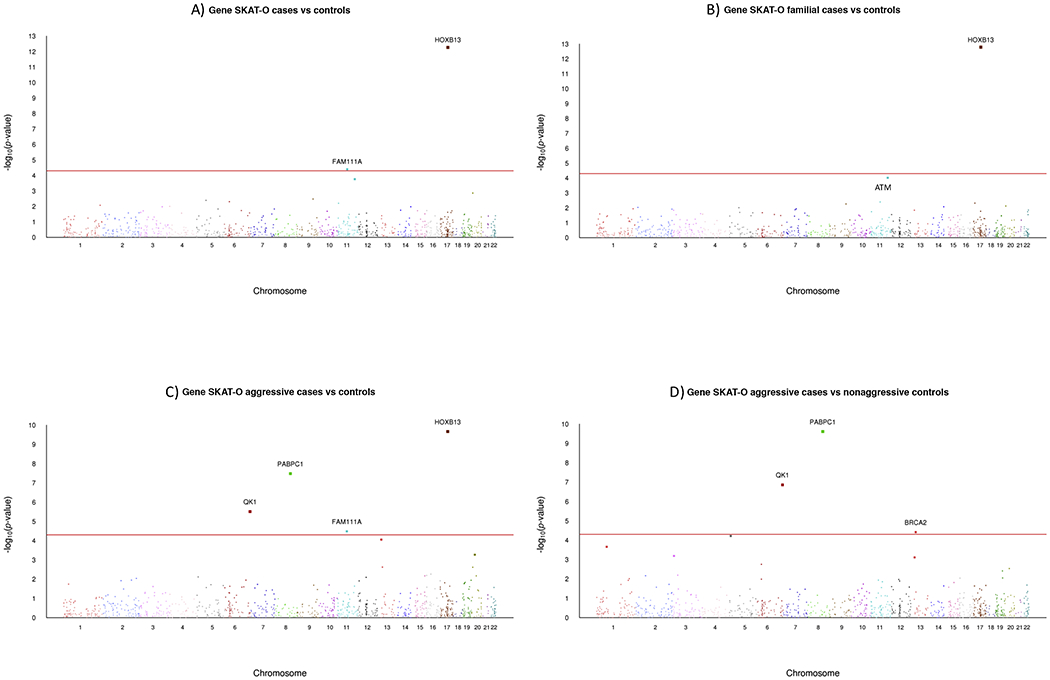
Manhattan plots of stage-two SKAT-O gene-level associations with PCa. The x axes show the chromosomal positions and the y axes show the −log10 p value. (A) All PCa cases versus controls. (B) PCa cases with a positive family history versus controls. (C) Aggressive PCa cases versus controls. D) Aggressive PCa cases versus nonaggressive PCa cases. PCa = prostate cancer.
4. Discussion
Although multiple studies have demonstrated that PCa risk has a significant genetic component, it has been difficult to identify the contributing genes. Large-scale genome-wide association studies have suggested as many as 170 distinct risk loci, with many contributing a very small amount of risk [5]. To enrich for genetic causes of PCa, our study focused on cases with a strong family history of the disease selected from the ICPCG resource, which has collected high-risk families for several decades as well as cases with a more aggressive disease.
We successfully identified a genetic variant (rs138213197) in the HOXB13 gene—our strongest association with PCa. This rare nonconservative substitution (G84E) has previously been reported and validated in multiple studies and different populations [20]. In addition, we found evidence for 10 genes previously reported to be associated with PCa: ATM, BRCA2, and FAM111A (detected by gene-level analyses), and EMSY, HNF1B, KLK3, MSMB, PCAT1, PRSS3, and TERT (detected by single-variant analyses). Background information on these genes is provided in Supplementary Table 8. Further, we detected 10 genes whose association with PCa has not been reported previously: PABPC1 and QK1 (identified by gene-level analyses), and FAM114A1, MUC6, MYCBP2, RAPGEF4, RNASEH2B, ULK4, XPO7, and THAP3 (identified by single-variant analyses). Background information on these genes is provided in Supplementary Table 9. Of these 10 novel genes, all but PABPC1 and ULK4 were primarily associated with the risk of aggressive PCa.
A major strength of our study is the number of PCa cases with a strong family history of disease or those with an aggressive phenotype. Our two-stage approach was a cost-efficient way to screen for less common variants that might be associated with PCa risk. Some limitations of our study include limited power to detect rare variants due to sample size restrictions [21] and the mixture of different capture kits in our stage-one samples. Although the controls we used for stage one were a mix of deidentified samples from prior WES studies conducted at the Mayo Clinic, with no information on PCa screening or family history of cancers, using them would not likely increase the chance of false associations, albeit power to detect genetic associations at stage one could have been diminished. For stage two, ICPCG groups selected controls by frequency matching age with their contributed cases, thereby adjusting for age, although missing detailed family history of some controls limited control for family history. Despite these limitations, we detected 11 genes previously known to be associated with PCa, thus validating our study approach, as well as 10 novel genes that have not been reported previously. It will be important to replicate our novel findings, to assure that they are not false positives, as well as conduct functional studies to evaluate thoroughly the clinical relevance of our detected genes and their potential as therapeutic targets.
It is intriguing that many of our discovered genes have variants whose alternate allele is less frequent among aggressive PCa cases compared with controls, giving the impression that the alternate allele is protective and also implying that the common reference allele is a risk allele. However, it is possible that the analyzed variant with the common risk allele is negatively correlated with a high-risk unmeasured variant, in which case the unmeasured less common allele is the actual risk allele. We recognize that our definition of aggressive disease was pragmatic based on available clinical data and included some variables dependent on the type of treatment administered (ie, recurrence and time to death), which could introduce hidden biases. Additional studies linking factors related to aging and lifespan may aid in better understanding of our findings.
5. Conclusions
We identified 11 genes previously reported to be associated with PCa (ATM, BRCA2, HOXB13, FAM111A, EMSY, HNF1B, KLK3, MSMB, PCAT1, PRSS3, and TERT) and an additional 10 novel genes (PABPC1, QK1, FAM114A1, MUC6, MYCBP2, RAPGEF4, RNASEH2B, ULK4, XPO7, and THAP3). Of these 10 novel genes, all but PABPC1 and ULK4 were primarily associated with the risk of aggressive PCa. Our results are consistent with other large-scale genomic studies that find multiple genes associated with the risk of PCa. Replication of our findings is needed to determine the value of these genes for screening purposes, and functional studies are needed to determine the therapeutic potential of these genes.
Supplementary Material
Acknowledgments
Funding/Support and role of the sponsor: This research was supported by the U.S. Public Health Service, National Institutes of Health, contract grant U01CA08960 (Stephen N. Thibodeau, ICPCG). Additional grant support for collection of samples and personnel efforts are as follows: NIH CA89600 (William Isaacs); CA080122, CA056678, CA082664, CA092579, and P30-CA015704 (Janet L. Stanford); Intramural Research Program of the National Human Genome Research Institute (Joan E. Bailey-Wilson and Elaine A. Ostrander); National Health and Medical Research Council (APP IDs 940394, 126402, 209057, APP1028280, and APP1074383); and WES datasets for the ARIC study (dbGaP accession study number phs000398.v1.p1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial disclosures: Daniel J. Schaid certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: None.
References
- [1].IARC. Prostate fact sheet. G.C. Observatory; 2018. http://gco.iarc.fr/today/data/factsheets/cancers/27-Prostate-fact-sheet.pdf
- [2].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- [3].Bunker CH, Patrick AL, Konety BR, et al. High prevalence of screening-detected prostate cancer among Afro-Caribbeans: the Tobago Prostate Cancer Survey. Cancer Epidemiol Biomarkers Prev 2002;11:726–9. [PubMed] [Google Scholar]
- [4].Howlader N, Noone AM, Krapcho M, editors. SEER cancer statistics review, 1975–2014. Bethesda, MD: N.C. Institute; 2017. [Google Scholar]
- [5].Benafif S, Kote-Jarai Z, Eeles RA. A review of prostate cancer genome-wide association studies (GWAS). Cancer Epidemiol Biomarkers Prev 2018;27:845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Xu J Combined analysis of hereditary prostate cancer linkage to 1q24-25: results from 772 hereditary prostate cancer families from the International Consortium for Prostate Cancer Genetics. Am J Hum Genet 2000;66:945–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Schaid DJ, Chang BL. Description of the International Consortium For Prostate Cancer Genetics, and failure to replicate linkage of hereditary prostate cancer to 20q13. Prostate 2005;63:276–90. [DOI] [PubMed] [Google Scholar]
- [8].Xu J, Dimitrov L, Chang BL, et al. A combined genomewide linkage scan of 1,233 families for prostate cancer-susceptibility genes conducted by the international consortium for prostate cancer genetics. Am J Hum Genet 2005;77:219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schaid DJ, McDonnell SK, Zarfas KE, et al. Pooled genome linkage scan of aggressive prostate cancer: results from the International Consortium for Prostate Cancer Genetics. Hum Genet 2006;120:471–85. [DOI] [PubMed] [Google Scholar]
- [10].Camp NJ, Cannon-Albright LA, Farnham JM, et al. Compelling evidence for a prostate cancer gene at 22q12.3 by the International Consortium for Prostate Cancer Genetics. Hum Mol Genet 2007;16:1271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Christensen GB, Baffoe-Bonnie AB, George A, et al. Genome-wide linkage analysis of 1,233 prostate cancer pedigrees from the International Consortium for Prostate Cancer Genetics using novel sumLINK and sumLOD analyses. Prostate 2010;70:735–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jin G, Lu L, Cooney KA, et al. Validation of prostate cancer risk-related loci identified from genome-wide association studies using family-based association analysis: evidence from the International Consortium for Prostate Cancer Genetics (ICPCG). Hum Genet 2012;131:1095–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bailey-Wilson JE, Childs EJ, Cropp CD, et al. Analysis of Xq27-28 linkage in the International Consortium for Prostate Cancer Genetics (ICPCG) families. BMC Med Genet 2012;13:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xu J, Lange EM, Lu L, et al. HOXB13 is a susceptibility gene for prostate cancer: results from the International Consortium for Prostate Cancer Genetics (ICPCG). Hum Genet 2013;132:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Teerlink CC, Thibodeau SN, McDonnell SK, et al. Association analysis of 9,560 prostate cancer cases from the International Consortium of Prostate Cancer Genetics confirms the role of reported prostate cancer associated SNPs for familial disease. Hum Genet 2014;133:347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schaid DJ, McDonnell SK, Sinnwell JP, Thibodeau SN. Multiple genetic variant association testing by collapsing and kernel methods with pedigree or population structured data. Genet Epidemiol 2013;37:409–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics (Oxford, England) 2012;13:762–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pu reel I S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schumacher FR, Al Olama AA, Berndt SI, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet 2018;50:928–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ewing CM, Ray AM, Lange EM, et al. Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med 2012;366:141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet 2014;95:5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.