

Abstract
The prognosis of recurrent malignant peripheral nerve sheath tumors (MPNST) is dismal, with surgical resection being the only definitive salvage therapy. Treatment with chemoradiation approaches has not significantly improved patient outcomes. Similarly, trials of therapies targeting MPNST genomic drivers have thus far been unsuccessful. Improved understanding of the molecular pathogenesis of MPNST indicates frequent activation of the mitogen-activated protein kinase (MAPK) cell signaling pathway. MEK inhibitors have shown activity in preclinical studies; however, their clinical efficacy has not been reported to date. We describe here a case of sustained complete response to MEK inhibition in an adolescent patient with a recurrent metastatic MPNST with multiple alterations in the MAPK pathway, guided by a precision oncology approach.
Subject terms: Paediatric cancer, Computational biology and bioinformatics, Molecular medicine
Introduction
Malignant peripheral nerve sheath tumor (MPNST) is a rare, aggressive soft-tissue sarcoma with poor overall survival of <50%1. The only definitive therapy is complete resection, with >90% survival rate for patients who achieve gross total resection with clear margins1. In comparison, patients with unresectable or metastatic tumors have a survival rate of only 25%1,2. While neoadjuvant chemotherapy and focal radiation therapy are used, neither have a significant impact on patient outcomes. An improved understanding of the molecular drivers of MPNST may lead to new avenues of therapy and precision medicine approaches. Importantly, MPNST is associated with a germline alteration of neurofibromatosis type 1 (NF1) in 40–50% of cases3, with frequent somatic aberrations in the mitogen-activated protein kinase (MAPK) pathway. Foremost are recurrent mutations in NF1 as well as other drivers, including TP53, EED, SUZ12, and CDKN2A4,5. Preclinical studies have shown MPNST dependence on the MAPK pathway6, but clinical efficacy with inhibition of MAPK kinase (MAP2K or MEK) has not been reported to date. We describe here a clinical case of successful treatment of an MPNST with a MEK inhibitor (MEKi). Our patient had a recurrent, metastatic MPNST and was enrolled on a precision medicine trial that identified multiple aberrations in the MAPK pathway. She was treated with the MEKi trametinib, with a sustained complete response of >15 months. To our knowledge, this represents evidence of a potentially effective systemic targeted therapy for MPNST.
Results
A previously well 14-year-old female, with no history or features of NF1, presented to a peripheral hospital with a painless, rapidly growing 10-cm left breast mass. The mass was incompletely removed. Histological examination demonstrated a highly cellular tumor with closely packed spindle cells with moderate nuclear pleomorphism, hyperchromatic nucleoli, frequent mitoses (>10 per high-power field), and multifocal necrosis but no epithelial component. Immunohistochemistry revealed diffusely positive CD99, CD56, and TLE1, and moderate FLI1. The tumor equivocally stained for MDM2 and CDK4 and negatively for S100, with loss of H3K27me3. Ki67 ranged between 30 and 50%. Fluorescent in situ hybridization (FISH) analysis for SS18 and EWSR1 rearrangements was negative. The overall features were consistent with the diagnosis of a high-grade MPNST. She was transferred to our unit for further care. Staging with F18-fluorodeoxyglucose positron emission tomography/computerized tomography (FDG-PET/CT) confirmed localized disease. She underwent a skin-sparing mastectomy and implant reconstruction with a complete microscopic surgical clearance. She subsequently underwent fertility preservation with an egg harvest, followed by 5550 cGy of focal radiation in 30 fractions and four cycles of ifosfamide/doxorubicin chemotherapy. Post-treatment surveillance was performed using CT/MRI (magnetic resonance imaging) and FDG-PET/CT. She had a clinically detected 35-mm local recurrence within the surgical and radiation field, 12 months from diagnosis, 4 months after completion of multimodal therapy, and underwent extensive surgical clearance, including chest wall excision with clear surgical margins. Histological examination demonstrated spindled cells, high-grade atypia, hyperchromatic nuclei with frequent mitoses up to 60 per 10 high-power field, and absence of glandular or squamoid differentiation, with Ki67 ranged between 30 and 80%. The overall features were consistent with the diagnosis of a high-grade MPNST and were identical to the original diagnostic specimen. The relapse sample was submitted for comprehensive genomic profiling on the Australian PRecISion Medicine for Children (PRISM, NCT03336931) trial, to identify potential targetable molecular aberrations7.
Whole-genome (paired tumor and germline) sequencing, transcriptomic (tumor) sequencing, and DNA methylation analysis was performed. The analyses demonstrated tumor purity of 98% and low tumor mutation burden (3 mutations/megabase). A somatic pathogenic TP53 p.R273H mutation with copy-neutral loss of heterozygosity was identified. The tumor demonstrated a complex genomic profile with multiple structural and copy number events (Fig. 1A). They included homozygous deletion of 17q11.2 resulting in biallelic deletion of NF1 and SUZ12 with low RNA expression (Fig. 1B–D), biallelic loss of CDKN2A/2B with low RNA expression, ALK amplification (ten copies) with high RNA expression, and EGFR copy gain (four copies) with high RNA expression (Fig. 1E, F). DNA methylation (Illumina Infinium Human Methylation 450 Array) analysis performed using the DKFZ sarcoma classifier (https://www.molecularneuropathology.org/mnp/classifier/3) classified the tumor as an MPNST (score 0.99, German Cancer Research Center DKFZ). Neither NF1 nor other known cancer predisposition germline mutations were identified.
Fig. 1. Results of genomic profiling.

A Circos plot illustrating complex genomic profile. Outermost circle: chromosomes; Second circle: purity-adjusted allelic frequency of all observed somatic SNV, colored according to its Cosmic signature (http://cancer.sanger.ac.uk/cosmic/signatures); Third circle: small (<50 bp) insertions (yellow) and deletions; Fourth circle: copy number changes, including deletions45 and amplifications. If the absolute copy number is >6, it is shown as 6 with a green dot; the fifth circle: minor allele copy number, where the loss of heterozygosity is shown in orange and amplification of the minor allele shown in blue. Innermost circle: structural variants with translocations in blue, deletions in red, insertions in yellow, tandem duplications in green, and inversions in black. B–G RNA abundance was assessed for ALK, EGFR, NF1, SUZ12, CDKN2A, and CDKN2B using RNAseq and represented as transcripts per million (TPM). The red dot and arrow represent this case, relative to n = 5 unrelated MPNST tumors and n = 133 high-risk pediatric tumors from the PRISM cohort (black).
The tumor harbored multiple aberrations in the MAPK pathway (Fig. 2). NF1 encodes neurofibromin 1, a RAS–GTPase-activating protein. Its negative regulatory function is attributed to a central GAP-related domain (GRD) region that is similar to ras–guanosine-triphosphate (GTP)ase activation proteins (GAPs). GAPs inactivate RAS by accelerating the conversion of active Ras–GTP to its inactive guanosine diphosphate (GDP)-bound form, preventing downstream pathway activation8. Hence, biallelic deletion of NF1 leads to loss of NF1 function and increased RAS signaling9,10. SUZ12, a core component of the PRC2 complex, is essential to epigenetic regulation11. SUZ12 loss potentiates the effects of NF1 loss by amplifying downstream RAS activity through the loss of H3K27me3 and aberrant transcriptional activation4,12.
Fig. 2. Schematic of somatic molecular aberrations.
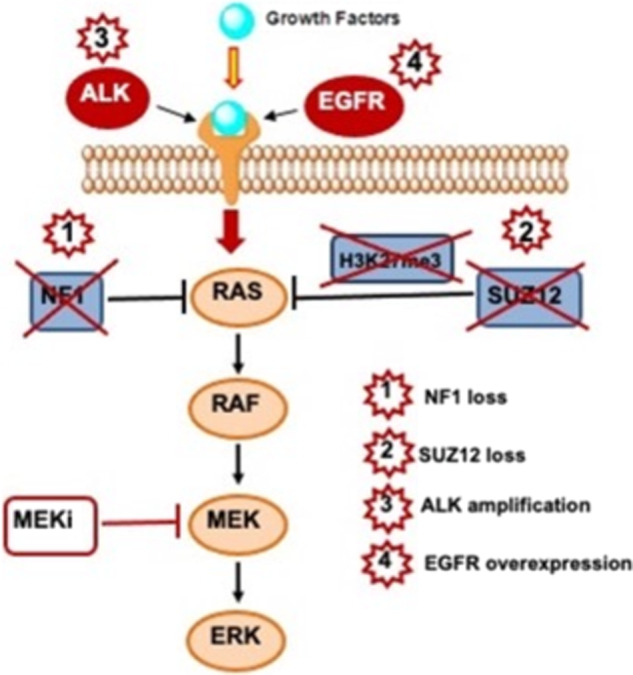
WGS and transcriptomic sequencing demonstrated multiple aberrations in the MAPK pathway, supporting downstream targeting with a MEK inhibitor (MEKi).
ALK activation can induce cellular differentiation through the MAPK pathway13 but could conversely confer resistance to ALK inhibitor monotherapy14. EGFR activation is known to induce MAPK activation through G-protein-coupled receptor kinase 2 (GPRK2)15. While the biallelic loss of NF1 is a well-established activating event in syndromic NF1 and its associated malignancies like glioma, leukemia, plexiform neurofibroma, MPNST, and melanoma16–18, the impact of ALK amplification, low EGFR copy number gain, and SUZ12 biallelic deletion on both MAPK pathway activation and sensitivity to MEK inhibitors is less certain.
Biallelic loss of CDKN2A (p16-INK4A) results in p16-mediated cell cycle promotion through the cyclin D-CDK4/6-pRb phosphorylation route19. This tumor carried wild-type RB1, which is essential for CDK4/6 inhibitor sensitivity20. Co-inactivation of p16 and TP53 has been observed in ~75% of MPNSTs, leading to functional consequences on cell growth control and apoptosis, with potential impact on CDK4/6 inhibitor efficacy21.
Following the second resection, the patient underwent 6 months of radiological surveillance but experienced a second metastatic relapse in the left 4th anterior intercostal region, adjacent to the previous chest wall resection. This 6.5-mm lesion was not biopsied and treatment was guided by the genomic profiling of the first relapse sample. Given the multiple MAPK pathway aberrations, she was commenced on trametinib 2 mg daily, achieving a complete remission of the metastatic recurrence in 3 months. Serial disease response assessments by CT and FDG-PET/CT imaging showed complete resolution of the metastatic lesion (Fig. 3) with an ongoing complete metabolic response more than 15 months after the second relapse. There has been no recurrence of the primary chest lesion since the second surgery or at any other site. The only side effect of the treatment was Grade 2 acneiform rash.
Fig. 3. Comparative fused FDG-PET/CT images of the left 4th intercostal space nodule.

a Focal avid disease (red circle) before commencing trametinib in May 2019. b Complete metabolic response after 15 months of treatment in September 2020 (with physiological brown fat activity in paravertebral and costovertebral regions). Comparative high-resolution CT images demonstrating (c) 6.5-mm lesion before commencing trametinib and (d) complete radiological response after 6 months of treatment with trametinib in October 2019.
Discussion
MPNST is an aggressive nonrhabdomyomatous soft-tissue sarcoma arising from perineural and Schwann cells of the peripheral nerve sheath, with a high rate of recurrence and metastasis. They occur sporadically or in association with a germline alteration of NF1 in ~50% of cases22. Patients with NF1 germline mutations carry ~10% lifetime risk of developing MPNST3. While resectable tumors with clear margins carry good outcomes, survival for all other patients remains poor, with the majority ultimately succumbing to the disease1,2. The prognosis for relapsed pediatric MPNST is exceedingly poor, with a median survival of 11 months and 5-year overall survival of 15%23. Relapsed tumors are insensitive to cytotoxic therapies and second-line treatments other than aggressive surgery have limited value in improving outcomes23. There is thus an urgent need for novel therapeutic approaches.
Trials of targeted agents have all thus far failed in MPNST, including targeted monotherapy with erlotinib, sorafenib, imatinib, dasatinib, and alisertib24–27. Similarly, combination trials of everolimus and bevacizumab or ganetespib and sirolimus have not shown clinical activity in recurrent MPNST28,29.
The genomic landscape of MPNST includes recurrent alterations in NF1 (87.5%), CDKN2A (75%), SUZ12 (56%), TP53 (40%), and EED (32.5%)5. The striking feature of multilevel MAPK pathway activation makes clinical translation with MAPK-targeted agents attractive. The range of somatic aberrations identified in this case offered multiple potential targets and targeted therapy options. EGFR inhibition with erlotinib has shown promising preclinical anti-MPNST activity but lacked clinical efficacy in adults with metastatic or unresectable MPNST24,30,31. There is no current preclinical or clinical data for ALK monotherapy activity in pediatric MPNST and EGFR and ALK targeting could potentially encounter therapy resistance due to NF1 and SUZ12 loss-of- function mutations14.
In this RB1 wild-type tumor, the biallelic loss of CDKN2A was potentially targetable with CDK4/6 inhibitors. Anti-tumor activity has been observed in preclinical models of RB1-preserved MPNST when treated with palbociclib20,32. However, CDK4/6 inhibitors are generally inactive clinically as single agents.
In contrast, MAPK signaling suppression has been shown in in vitro MPNST models with MEK inhibitors such as trametinib and selumetinib33. Trametinib treatment resulted in tumor growth reduction in murine models of NF1-null MPNST34. Mirdametinib (PD0325901), a highly selective MEK inhibitor, demonstrated a reduction in cell proliferation in vitro and prolonged mice survival in in vivo NF1-negative MPNST models6. Clinical efficacy of MEK inhibitor monotherapy in MPNST has not been reported to date, but a Phase 2 trial is testing the combination of selumetinib and sirolimus (mTOR inhibitor) in MPNST (NCT03433183). Selumetinib has demonstrated clinical activity with objective responses in 71% of children with NF1-driven plexiform neurofibromas in Phase 1 clinical trial9. A recent Phase 2 trial in children with NF1-driven plexiform neurofibromas showed 70% partial tumor response with meaningful improvement in pain and quality-of-life parameters with selumetinib35. Similarly, trametinib has shown a reduction in tumor volume in recurrent pediatric low-grade gliomas and NF1-driven plexiform neurofibromas36–38.
Therapeutic decision-making was challenging in this case, but factors favoring the choice of trametinib were the biological rationale, its ability to target downstream molecular abnormalities and alleviate therapy resistance, accessibility in Australia outside a clinical trial setting, and experience in monitoring and managing drug toxicity. The patient has tolerated trametinib well, apart from moderate acne, which is improving on cessation of the drug and has remained in continuous complete remission for more than 15 months. To our knowledge, objective response to a MEK inhibitor in relapsed MPNST has not been described in the literature to date. We provide evidence of the utility of a personalized approach for these rare, aggressive tumors, minimizing off-target toxicity, and positively impacting quality of life.
It is important to note that there were some unusual clinical features in this case. MPNST is uncommon in childhood and cancer is more often seen in young and middle-aged adults, accounting for 5% of soft-tissue sarcomas in patients less than 20 years of age18. Although primary MPNSTs of the breast have been sporadically reported in adults39,40, they are rare in the pediatric age group41, particularly in the absence of any clinical or germline features of NF1. Moreover, high-grade MPNSTs are usually associated with distant than local metastatic recurrence42. The genomic changes, however, are consistent with those described in both NF1- associated and sporadic cases of MPNST. H3K27 trimethylation loss is associated with high-grade MPNSTs and correlates with poor survival43,44. This patient’s disease has shown an objective response in the presence of these unusual and high-risk features. The clinical response seen in this patient needs to be confirmed in other patients, ideally through evaluation in a clinical trial, to determine whether the results can be replicated.
Improving our understanding of the molecular basis of MPNST through genomic, transcriptomic, drug screening responses, and tumor microenvironment approaches is critical to the discovery of new effective therapy for this challenging disease. Given the high proportion of MPNST tumors with aberrations in the MAPK pathway, treatment with MEK inhibitors may represent an effective therapy for many patients with the appropriate genotype and merits further prospective evaluation through clinical trials.
Methods
Whole-genome sequencing
DNA was extracted from tumor: normal matched-pair tissue and libraries were prepared using KAPA Hyper PCR-Free Library Preparation Kit (Roche Inc). Both normal and tumor libraries were sequenced on Illumina HiSeq X Ten to generate 2 × 150-bp reads at a target coverage of >30× for germline and >90× for tumor samples. WGS was conducted at the Kinghorn Centre for Clinical Genomics, Garvan Institute of Medical Research (Australia). WGS data analysis was conducted as previously described7. Raw fastq files were aligned to the hs37d5 reference genome using BWA-MEM (v0.17.10-r789), with the resulting BAM files merged and duplicate reads marked using Novosort (v1.03.01, default settings), and read alignment improved using GATK Indel Realignment (v3.3). Germline SNVs and short (<50-bp) indels were identified using GATK HaplotypeCaller, GenotypeVCFs, and VQSR (all v3.3), annotated with VEP (v87), converted into a GEMINI (v0.11.0) database, and imported into Seave for filtration and prioritization. Somatic SNVs and short indels (<50 bp) were identified using Strelka (v2.0.17). Somatic variants were annotated using SnpEff (v4_3t). Tumor purity, ploidy, and somatic copy number alteration were identified using PURPLE (v2.39), and somatic SVs were identified using GRIDSS (v2.7.2) and then annotated using Ensembl genes. Somatic and germline variant curation was conducted as described7.
RNA sequencing
RNAseq was conducted at the Murdoch Children’s Research Institute (Australia) and performed with the TruSeq Stranded mRNA Preparation Kit. Libraries were pooled, and sequencing runs were performed in paired-end mode using the NextSeq 500 platform generating at least 80 million reads. RNA data analysis was performed as previously described7. Initial RNAseq quality control (QC) checks included an evaluation of GC and per-base sequence content using FastQC (v0.11.5). Reads were aligned to the human genome assembly (build hg19) using the STAR (v2.5) two-pass method with quantMode parameters set to TranscriptomeSAM for alignments translated into transcript coordinates. Alignments were sorted with SAMTools (v1.3.1), duplicates were marked with Picard Tools (v2.4.1), reads were split and trimmed, and mapping qualities were reassigned with the Genome Analysis Toolkit (v3.6) using the methods SplitNCigarReads and ReassignOneMappingQuality, respectively. Post-alignment QC required at least 70% of reads to be uniquely aligned, assessed using STAR alignment statistics (100% of samples passed). Raw gene counts and transcripts per kilobase million (TPM) were calculated using RSEM (v1.2.31). All RNAseq expression values are represented as TPM. Variants were called on RNA with GATK HaplotypeCaller (v3.6), and ANNOVAR (v20190929) was used for variant annotation. Fusions were identified using three methods: STAR-Fusion (v1.3.1), JAFFA (v1.09), and Arriba (v1.1.0, https://github.com/suhrig/arriba/).
DNA methylation
DNA methylation analysis was conducted at the Australian Genome Research Facility (Australia). Tumor DNA was extracted followed by bisulfite conversion using Zyma EZ DNA Methylation kit. Samples were hybridized on the Illumina 850 K Infinium Methylation EPIC BeadChip. Methylation data were uploaded into the DKFZ methylation analysis portal for sarcoma classification (https://www.molecularneuropathology.org/mnp/classifier/3).
Ethics approval
Ethical approval for the PRISM trial was granted by the Hunter New England Human Research Ethics Committee, New South Wales, Australia, in accordance with the National Health and Medical Research Council’s National Statement on Ethical Conduct in Human Research (2007) (National Statement) and the CPMP/ICH Note for Guidance on Good Clinical Practice (Ref: HREC/17/HNE/29). Written informed consent was provided by the parents prior to enrolment on the PRISM trial.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank Murdoch Children’s Research Institute (MCRI), Australian Genome Research Facility (AGRF), and the Garvan Institute of Medical Research for performing comprehensive sequencing services. We thank our funding supporters: The Kids Cancer Alliance (KCA), Cancer Therapeutics Cooperative Research Centre (CTx CRC), Australian Cancer Research Foundation (ACRF), NSW and Australian Department of Health, Tour de Cure, Lions Club International Foundation (LCIF), Australian Lions Children’s Cancer Research Foundation (ALCCRF), The Kids Cancer Project (TKCP), University of New South Wales, the Minderoo Foundation Collaborate Against Cancer Initiative, and the Zero Childhood Cancer Capacity Campaign, a joint initiative of Children’s Cancer Institute and Sydney Children’s Hospital Foundation. We thank the extensive ZERO Childhood Cancer Team and the patient and family for consenting to publish this report.
Author contributions
S.N. and L.M.S.L. contributed equally. Drafting of the paper: S.N., D.S.Z., A.A., and L.M.S.L. Clinical care of patient and provision of clinical data: A.A. and P.C. Radiology analysis: E.A.W. Molecular analysis and curation: L.M.S.L., P.B., M.W., A.S., P.G.E., M.J.C., and C.M. All authors reviewed and edited the paper.
Data availability
The data generated and analyzed during this study are described in the following data record: 10.6084/m9.figshare.1346900145. Whole-genome sequencing, RNA sequencing, and DNA methylation data are available from the European Genome-phenome Archive under dataset accession https://identifiers.org/ega.dataset:EGAD00001006793 (study accession: https://identifiers.org/ega.study:EGAS00001004899)46. The data are controlled access but are publicly available after Data Access Agreement consent.
Code availability
Software and scripts related to this publication are available at https://github.com/CCICB/2020-hrPC-landscape.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Sumanth Nagabushan, Loretta M. S. Lau
Supplementary information
The online version contains supplementary material available at 10.1038/s41698-021-00145-8.
References
- 1.van Noesel MM, et al. Outcome and prognostic factors in pediatric malignant peripheral nerve sheath tumors: an analysis of the European Pediatric Soft Tissue Sarcoma Group (EpSSG) NRSTS-2005 prospective study. Pediatr. blood cancer. 2019;66:e27833. doi: 10.1002/pbc.27833. [DOI] [PubMed] [Google Scholar]
- 2.Carli M, et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J. Clin. Oncol. 2005;23:8422–8430. doi: 10.1200/JCO.2005.01.4886. [DOI] [PubMed] [Google Scholar]
- 3.McCaughan JA, Holloway SM, Davidson R, Lam WW. Further evidence of the increased risk for malignant peripheral nerve sheath tumour from a Scottish cohort of patients with neurofibromatosis type 1. J. Med. Genet. 2007;44:463–466. doi: 10.1136/jmg.2006.048140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee W, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014;46:1227–1232. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brohl AS, Kahen E, Yoder SJ, Teer JK, Reed DR. The genomic landscape of malignant peripheral nerve sheath tumors: diverse drivers of Ras pathway activation. Sci. Rep. 2017;7:14992. doi: 10.1038/s41598-017-15183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jessen WJ, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J. Clin. Investig. 2013;123:340–347. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong, M. et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat. Med.10.1038/s41591-020-1072-4 (2020). [DOI] [PubMed]
- 8.Yap YS, et al. The NF1 gene revisited—from bench to bedside. Oncotarget. 2014;5:5873–5892. doi: 10.18632/oncotarget.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dombi E, et al. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N. Engl. J. Med. 2016;375:2550–2560. doi: 10.1056/NEJMoa1605943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll SL, Stonecypher MS. Tumor suppressor mutations and growth factor signaling in the pathogenesis of NF1-associated peripheral nerve sheath tumors: II. The role of dysregulated growth factor signaling. J. Neuropathol. Exp. Neurol. 2005;64:1–9. doi: 10.1093/jnen/64.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell. 2004;15:57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 12.De Raedt T, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–251. doi: 10.1038/nature13561. [DOI] [PubMed] [Google Scholar]
- 13.Souttou B, Carvalho NB, Raulais D, Vigny M. Activation of anaplastic lymphoma kinase receptor tyrosine kinase induces neuronal differentiation through the mitogen-activated protein kinase pathway. J. Biol. Chem. 2001;276:9526–9531. doi: 10.1074/jbc.M007333200. [DOI] [PubMed] [Google Scholar]
- 14.Hrustanovic G, Bivona TG. RAS-MAPK in ALK targeted therapy resistance. Cell Cycle. 2015;14:3661–3662. doi: 10.1080/15384101.2015.1096103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao J, Li J, Ma L. Regulation of EGF-induced ERK/MAPK activation and EGFR internalization by G protein-coupled receptor kinase 2. Acta biochimica et. biophysica Sin. 2005;37:525–531. doi: 10.1111/j.1745-7270.2005.00076.x. [DOI] [PubMed] [Google Scholar]
- 16.Laycock-van Spyk S, Thomas N, Cooper DN, Upadhyaya M. Neurofibromatosis type 1-associated tumours: their somatic mutational spectrum and pathogenesis. Hum. Genomics. 2011;5:623–690. doi: 10.1186/1479-7364-5-6-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Investig. 2017;97:146–157. doi: 10.1038/labinvest.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Philpott C, Tovell H, Frayling IM, Cooper DN, Upadhyaya M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genomics. 2017;11:13. doi: 10.1186/s40246-017-0109-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 20.Kohlmeyer, J. L. et al. RABL6A is an essential driver of MPNSTs that negatively regulates the RB1 pathway and sensitizes tumor cells to CDK4/6 inhibitors. Clin. Cancer Res.10.1158/1078-0432.Ccr-19-2706 (2020). [DOI] [PMC free article] [PubMed]
- 21.Perrone F, et al. p15INK4b, p14ARF, and p16INK4a inactivation in sporadic and neurofibromatosis type 1-related malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2003;9:4132–4138. [PubMed] [Google Scholar]
- 22.Kolberg M, et al. Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro Oncol. 2013;15:135–147. doi: 10.1093/neuonc/nos287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergamaschi, L. et al. Salvage rates and prognostic factors after relapse in children and adolescents with malignant peripheral nerve sheath tumors. Pediatric Blood Cancer65, 10.1002/pbc.26816 (2018). [DOI] [PubMed]
- 24.Albritton KH, et al. Phase II study of erlotinib in metastatic or unresectable malignant peripheral nerve sheath tumors (MPNST) J. Clin. Oncol. 2016;24:9518–9518. doi: 10.1200/jco.2006.24.18_suppl.9518. [DOI] [Google Scholar]
- 25.Maki RG, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J. Clin. Oncol. 2009;27:3133–3140. doi: 10.1200/JCO.2008.20.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chugh R, et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J. Clin. Oncol. 2009;27:3148–3153. doi: 10.1200/JCO.2008.20.5054. [DOI] [PubMed] [Google Scholar]
- 27.Schuetze SM, et al. SARC009: phase 2 study of dasatinib in patients with previously treated, high-grade, advanced sarcoma. Cancer. 2016;122:868–874. doi: 10.1002/cncr.29858. [DOI] [PubMed] [Google Scholar]
- 28.Widemann BC, et al. Targeting sporadic and neurofibromatosis type 1 (NF1) related refractory malignant peripheral nerve sheath tumors (MPNST) in a phase II study of everolimus in combination with bevacizumab (SARC016) Sarcoma. 2019;2019:7656747. doi: 10.1155/2019/7656747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim A, et al. Targeting refractory sarcomas and malignant peripheral nerve sheath tumors in a phase I/II study of sirolimus in combination with ganetespib (SARC023) Sarcoma. 2020;2020:5784876. doi: 10.1155/2020/5784876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu J, et al. EGFR-STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene. 2014;33:173–180. doi: 10.1038/onc.2012.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holtkamp N, et al. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro Oncol. 2008;10:946–957. doi: 10.1215/15228517-2008-053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perez M, Muñoz-Galván S, Jiménez-García MP, Marín JJ, Carnero A. Efficacy of CDK4 inhibition against sarcomas depends on their levels of CDK4 and p16ink4 mRNA. Oncotarget. 2015;6:40557–40574. doi: 10.18632/oncotarget.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kahen EJ, et al. Neurofibromin level directs RAS pathway signaling and mediates sensitivity to targeted agents in malignant peripheral nerve sheath tumors. Oncotarget. 2018;9:22571–22585. doi: 10.18632/oncotarget.25181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peacock JD, et al. Genomic status of MET potentiates sensitivity to MET and MEK inhibition in NF1-related malignant peripheral nerve sheath tumors. Cancer Res. 2018;78:3672–3687. doi: 10.1158/0008-5472.CAN-17-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gross AM, et al. Selumetinib in children with inoperable plexiform neurofibromas. N. Engl. J. Med. 2020;382:1430–1442. doi: 10.1056/NEJMoa1912735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kondyli M, et al. Trametinib for progressive pediatric low-grade gliomas. J. neuro-Oncol. 2018;140:435–444. doi: 10.1007/s11060-018-2971-9. [DOI] [PubMed] [Google Scholar]
- 37.Miller C, Guillaume D, Dusenbery K, Clark HB, Moertel C. Report of effective trametinib therapy in 2 children with progressive hypothalamic optic pathway pilocytic astrocytoma: documentation of volumetric response. J. Neurosurg. Pediatrics. 2017;19:319–324. doi: 10.3171/2016.9.PEDS16328. [DOI] [PubMed] [Google Scholar]
- 38.McCowage GB, et al. Trametinib in pediatric patients with neurofibromatosis type 1 (NF-1)–associated plexiform neurofibroma: a phase I/IIa study. J. Clin. Oncol. 2018;36:10504–10504. doi: 10.1200/JCO.2018.36.15_suppl.10504. [DOI] [Google Scholar]
- 39.Catania S, Pacifico E, Zurrida S, Cusumano F. Malignant schwannoma of the breast. Eur. J. Surg. Oncol. 1992;18:80–81. [PubMed] [Google Scholar]
- 40.Dhingra KK, Mandal S, Roy S, Khurana N. Malignant peripheral nerve sheath tumor of the breast: case report. World J. Surg. Oncol. 2007;5:142. doi: 10.1186/1477-7819-5-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shuayb M, Begum R. Unusual primary breast cancer - malignant peripheral nerve sheath tumor: a case report and review of the literature. J. Med. Case Rep. 2017;11:161. doi: 10.1186/s13256-017-1332-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watson KL, et al. Patterns of recurrence and survival in sporadic, neurofibromatosis Type 1-associated, and radiation-associated malignant peripheral nerve sheath tumors. J. Neurosurg. 2017;126:319–329. doi: 10.3171/2015.12.JNS152443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu VM, et al. H3K27 trimethylation loss in malignant peripheral nerve sheath tumor: a systematic review and meta-analysis with diagnostic implications. J. Neuro-Oncol. 2019;144:433–443. doi: 10.1007/s11060-019-03247-3. [DOI] [PubMed] [Google Scholar]
- 44.Cleven AH, et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod. Pathol. 2016;29:1113. doi: 10.1038/modpathol.2016.103. [DOI] [PubMed] [Google Scholar]
- 45.Nagabushan, S. et al. Metadata record for the manuscript: Personalised therapy with MEK inhibition leads to a sustained complete response in an adolescent patient with a recurrent malignant peripheral nerve sheath tumor. Figshare 10.6084/m9.figshare.13469001 (2020).
- 46.European Genotype-phenotype Archive https://identifiers.org/ega.dataset:EGAD00001006793 (2020).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated and analyzed during this study are described in the following data record: 10.6084/m9.figshare.1346900145. Whole-genome sequencing, RNA sequencing, and DNA methylation data are available from the European Genome-phenome Archive under dataset accession https://identifiers.org/ega.dataset:EGAD00001006793 (study accession: https://identifiers.org/ega.study:EGAS00001004899)46. The data are controlled access but are publicly available after Data Access Agreement consent.
Software and scripts related to this publication are available at https://github.com/CCICB/2020-hrPC-landscape.