

Keywords: cancer hallmarks, epigenetics, de-differentiation, microbiome, neuronal signalling
Abstract
Diagnosis and treatment of disease demand a sound understanding of the underlying mechanisms, determining any Achilles' heel that can be targeted in effective therapies. Throughout history, this endeavour to decipher the origin and mechanism of transformation of a normal cell into cancer has led to various theories—from cancer as a curse to an understanding at the level of single-cell heterogeneity, meaning even among a single sub-type of cancer there are myriad molecular challenges to overcome. With increasing insight into cancer genetics and biology, the disease has become ever more complex to understand. The complexity of cancer as a disease was distilled into key traits by Hanahan and Weinberg in their seminal ‘Hallmarks of Cancer' reviews. This lucid conceptualization of complex cancer biology is widely accepted and has helped advance cancer therapeutics by targeting the various hallmarks but, with the advancement in technologies, there is greater granularity in how we view cancer as a disease, and the additional understanding over the past decade requires us to revisit the hallmarks of cancer. Based on extensive study of the cancer research literature, we propose four novel hallmarks of cancer, namely, the ability of cells to regress from a specific specialized functional state, epigenetic changes that can affect gene expression, the role of microorganisms and neuronal signalling, to be included in the hallmark conceptualization along with evidence of various means to exploit them therapeutically.
1. A historical perspective on cancer
Ancient Egyptians believed cancer to be a curse, as seen from evidence as old as 3000 BC from the Edwin Smith Papyrus which described breast cancer [1] and Ebers Papyrus dated 1500 BC which described skin, uterus and other types of tumours [2]. Hippocrates proposed the idea of an excess of black bile to be the reason for cancer, the idea was further developed by Greek physician Galen, revered physician to the Emperor Marcus Aurelius, who suggested black bile caused incurable types of cancer while yellow bile caused curable variants of cancer [3]. This was refuted in the sixteenth century by the renowned anatomist Andreas Vesalius, who disproved the existence of black bile [4]. In the sixteenth century, Paracelsus identified the first correlation between cancer and the environment, showing deposits of arsenic salts and sulfur in the blood of mine workers were associated with cancer. This laid the foundation for later work by others, namely Percival Pott (chimney sweeps), John Hill (snuff) and Ludwig Rehn (aniline dyes) ([5]. In 1914, Theodor Boveri was the first to hypothesize that abnormal segregation of chromosomes to daughter cells can lead to tumour development in ‘Zur Frage der Entstehung Maligner Tumoren' [6].
Fast forward to 2000, Douglas Hanahan and Robert Weinberg compiled the key concepts surrounding cancer into the hallmarks of cancer, discussing the various mechanisms that underpin tumour development (figure 1).
Figure 1.

Hallmark flower.
2. Hallmarks of cancer
The challenges presented by multiple roadblocks, which are in place to prevent excessive cell proliferation and the development of tumours, leads to the daunting complexity of cancer. Tumour cells do not invent new mechanisms, but rather manipulate existing molecular and cellular pathways to circumvent protective mechanisms which are in place to prevent the formation of a tumour.
These conceptually distinct capabilities of tumour cells have a powerful resonance in the field of cancer therapeutics. Despite our knowledge of specific mutations in tumour cells generated through global sequencing efforts, such as the International Cancer Genome Consortium, the reductionist view would be to just focus on the cancer cell. However, we are actually dealing with a complex heterotypic tumour microenvironment where the tumour cells are only the foundation of cancer as a disease but not its complete manifestation.
We propose four novel hallmarks of cancer, justify their importance in tumourigenesis and argue the need to incorporate them in the mainstream hallmark conceptualization (figure 2).
Figure 2.
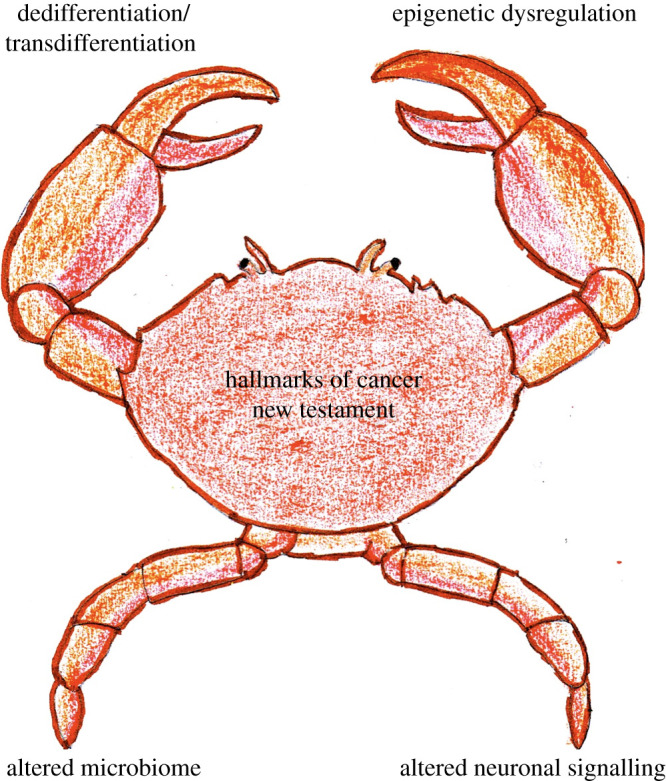
Novel hallmarks of cancer.
3. New hallmark 1: dedifferentiation and transdifferentiation
In 1957, Conrad Waddington proposed the unidirectional developmental model, wherein pluripotent stem cells at the top of the hill progressively lose their pluripotency as they follow developmental pathways and end up among different valleys in a terminally differentiated state [7] (figure 3a). However, the concept of tumour cell plasticity goes against the Waddington landscape, where dedifferentiation allows non-cancer stem cells to acquire stem cell-like features.
Figure 3.

Tumour defiance of Waddington landscape. (a) Waddington landscape depicting the unidirectional nature of differentiation, adapted based on the concept of [8]. (b) The forgotten hallmark Dedifferentiation: Dedifferentiation from terminally differentiated neuron, astrocyte and oligodendrocyte, as well as transdifferentiation of neuron and astrocyte to endothelial cells.
In 1962, Sir John Gurdon challenged the unidirectional dogma of development with his ground-breaking study which showed the formation of a fully functional tadpole clone even when the nucleus of a frog zygote was replaced with a nucleus harvested from a terminally differentiated tadpole intestinal cell [9]. This proved his hypothesis that the genome of a mature specialized cell has all the information required to develop into the different cell types of an organism. However, Gurdon's experiment involved physical removal and transfer of cell nuclei and as such the question remained whether such a hypothesis could be replicated in intact cells. Forty years later, this question was answered with a proof of concept study which defied the Waddington landscape in intact cells. Introducing just four genes, the Yamanaka factors: c-MYC, Kruppel-like factor 4 (KLF4), Sex-determining region Y-box 2 (Sox2) and Octamer-binding transcription factor 3/4 (Oct-3/4); Takashi and Yamanaka were able to develop what they termed induced pluripotent stem cells (iPSCs), which had the ability to differentiate into any of the cell lineages, endodermal, ectodermal and mesodermal [10]. This forms the basis of the hypothesis that tumour cells, which are champion survivors, will hijack any mechanism in order to survive; as such, dedifferentiation is a lucrative hallmark for them to achieve immortality.
Cancer stem cells (CSCs) are a unique subpopulation which possesses the cardinal property of self-renewal. This population can underpin tumour heterogeneity and resistance to cancer therapeutics, leading to relapse. The dedifferentiation of non-CSCs to CSC gives a survival advantage to cancers. Turning the clock back in time to a stem cell progenitor state is not a mere manifestation of the existing hallmark but a pivotal hallmark in itself and it further confers the ability to switch lineages, as lineage plasticity enables resistance against therapeutics. Let us consider some key examples that reiterate dedifferentiation as an integral hallmark of cancer.
3.1. Evidence of dedifferentiation in glioblastoma
The interconvertible nature of cancer stem cells and non-cancer stem cells can be seen within glioblastoma multiforme, a highly lethal sub-type of brain cancer. In 2002, a study reported that even mature astrocytes and neurons can be the cell of origin in certain brain tumours. EGFR activation and dual inactivation of p16INK4a and p19ARF cause astrocytes to undergo dedifferentiation to a multipotent progenitor state dictating the emergence of the high-grade phenotype of gliomas [11]. The extent of dedifferentiation of astrocytes is radical enough to give rise to pluripotent cells which have the ability to differentiate into glia as well as neurons, as evidenced by expression of neuronal marker TUJ1 among such tumours which arise from dedifferentiated astrocytes [11]. Indeed, the majority of mature differentiated cells in the central nervous system, given the right permissive microenvironment, can undergo dedifferentiation to a progenitor state, generating a neural stem cell that can perpetuate tumour progression as well as tumour heterogeneity and resistance to treatment [12] (figure 3b). Tumour plasticity allows for vascular mimicry via the transdifferentiation of glioblastoma cells into vascular endothelial cells [13] and even pericytes, which can support the maintenance of tumour vessel function [14] (figure 3b).
3.2. Evidence of dedifferentiation in intestinal tumours
Tumour initiating cells formed via dedifferentiation have also been reported in intestinal tumours. Enhanced NF-κB signalling leads to activation of β-catenin/TCF transcription via stabilization of β-catenin, inducing dedifferentiation of non-stem intestinal epithelial cells to intestinal epithelial cells with tumour initiating stem-like properties [15]. If Wnt activity plays a role in dedifferentiation of non-stem intestinal epithelial cells to tumour initiating cells, then a further investigation into whether this activity is mediated by the tumour microenvironment is warranted. In colon cancer, myofibroblasts in the tumour niche orchestrate high Wnt activity via β-catenin localization through hepatocyte growth factor secretion, which facilitates the reprogramming of the colon cancer cells to a stem cell-like progenitor state [16].
3.3. The pliability of cell fate in pancreatic cancer via dedifferentiation
There is a dynamic equilibrium between stem-like state and non-stem differentiated state. An activating mutation of the small GTPase KRAS is identified in about 90% of pancreatic tumours [17]. In a proof of concept study in pancreatic ductal adenocarcinoma, KRAS and its downstream target MYC were shown to rapidly reprogram differentiated mature cells to a stem cell-like state, poised to become malignant. Generation of metastatic pancreatic tumour cells with self-renewing capability is particularly shown to be controlled via MYC, which functions as a built-in amplifier [18].
Another study has shown that the major mechanism of initiation of pancreatic ductal adenocarcinoma lies in the synergism between the transcription factor SOX9 and activated KRAS, leading to the dedifferentiation of pancreatic acinar cells through a duct-like phenotype and the subsequent formation of pancreatic intraepithelial neoplasia [19]. Such genetic mutation induced dedifferentiation explains the differences in the kinetics of dedifferentiation of cells, at different states of differentiation in a tumour, which reiterates the need to target both cancer stem cells, and non-stem cancer cells to prevent re-initiation of tumourigenesis following therapy.
3.4. Therapy resistance via lineage plasticity through dedifferentiation
Despite achieving remission in metastatic melanoma with adoptive cell transfer therapies, there is frequent relapse. Relapse may be due to the secretion of the proinflammatory cytokine tumour necrosis factor (TNF)-α by T cells and macrophages in the tumour microenvironment, which results in reversible dedifferentiation of melanoma cells and thereby a loss of melanocytic antigens [20]. If dedifferentiation can aid in evasion from T cell immunotherapy, it raises the possibility of dedifferentiation as an enabling hallmark for immune evasion.
Dedifferentiation is also linked to resistance to targeted therapies in melanoma, for example, resistance to BRAF inhibition is conferred by the downregulation of microphthalmia-associated transcription factor (MITF), which plays a key role in melanocyte differentiation, and the upregulation of the receptor tyrosine kinase AXL, platelet-derived growth factor receptor and EGFR [21]. Dedifferentiation also provides cues for a potential susceptibility target, for example, the ability to induce ferroptosis, a form of iron-dependent necrotic cell death, in dedifferentiated melanoma cells [22]. As such, it can provide an option to induce a form of synthetic lethality by the combination of ferroptosis inducing drugs along with targeted therapies or immunotherapy in melanoma patients. Dedifferentiation-based changes have been found among melanoma patients even within the first week of targeted therapy treatment [23], suggesting a combination regimen with ferroptosis inducing drugs could possibly be initiated up-front to prevent escape via dedifferentiation.
Dedifferentiation has also been implicated in therapeutic resistance among prostate and breast cancers. A gain and loss of function study identified that upregulation of the reprogramming transcription factor Sox2 can confer reversible lineage plasticity by switching prostate cancer cells to a neuroendocrine phenotype, in the context of concomitant loss of tumour suppressors p53 and Rb [24]. Earlier studies showed that multipotent adipose-derived stem cells (ASCs), used in soft tissue reconstruction following mastectomy, undergo a phenotypic alteration via myofibroblastic differentiation, leading to contraction and enhanced stiffness, ultimately promoting tumourigenesis [25]. The impact of physical stresses on the tumour microenvironment leading to tumour progression has also been shown to involve triggering dedifferentiation. When a tumour grows, it causes compression of the surrounding tissue. Physical stress caused by mammary adenocarcinoma via compression of surrounding adipocytes triggers Wnt/β-catenin signalling and their subsequent dedifferentiation to myofibroblasts, which then interact with breast cancer cells leading to enhanced tumour proliferation [26].
3.5. Reflecting upon the Yamanaka factors' relationship to oncogenesis
A definitive proof of the importance of re-programming in cancer ontogeny is that each of the four Yamanaka factors capable of playing a role in dedifferentiation has an ascertained role in oncogenesis among multiple cancers. Oct4 is a biomarker for seminomas [27] and has also been attributed to the maintenance of the undifferentiated cell population with proliferative capacity by blockage of progenitor cell differentiation [28]. Sox2 is a key driver towards a stem cell fate among Ewing's sarcoma, breast and brain tumours [29,30]. Aberrant MYC expression has been strongly linked to several cancers [31] and KLF4 has been linked to colorectal cancer [32]. Though challenging targets themselves, Yamanaka factors may provide insight for the development of more targeted therapies.
The loss of APC maintains a progenitor state, following which oncogenic mutations such as KRAS can be acquired, driving tumourigenesis [33]. So, does dedifferentiation act as an enabling hallmark to grant time to acquire additional mutations to progress in the path towards tumour development? Or does dedifferentiation allow for lineage plasticity for tumour cells to alter their cell fate to a lineage more resistant to therapeutics?
In the case of acute promyelocytic leukaemia (APML), translocation results in promyelocytic leukaemia protein (PML) and retinoic acid receptor α (RARα) fusion. The expression of the PML-RARα fusion gene blocks the terminal differentiation of granulocytes, resulting in the maintenance of neoplastic cells in the promyelocytic progenitor stage, but all-trans retinoic acid has been successful in overcoming the differentiation block by inducing differentiation of neoplastic cells into granulocytes [34]. The abrogation of terminal differentiation, as seen in APML, in order to maintain a progenitor-like state, supports the hypothesis of dedifferentiation as a logical hallmark. Even if cancer cells proceed to, or develop from, a state of terminal differentiation, they can revert back to their progenitor state and maintain their stemness via dedifferentiation. The Waddington landscape has been defied by cancer, providing tumour cells the plasticity to choose their fate by pushing the ball uphill against the landscape, to maintain cancer stem cells and underpin the basis of cancer as a lethal disease.
3.6. Therapeutic interventions based on the hallmark of dedifferentiation
An important aspect of hallmarks of cancer conceptualisation is to aid in advancing therapeutic strategies, so an understanding of the nuances of the hallmark of dedifferentiation is important. There are three therapeutic modalities that can be targeted towards the tumour cell plasticity conferred by dedifferentiation:
-
1.
Blocking dedifferentiation via combination therapies. Target the differentiated cell lineage along with drugs that block dedifferentiation in order to prevent early resistance to therapeutics as a result of lineage plasticity conferred by dedifferentiation.
-
2.
Target dedifferentiation with differentiation therapy towards a permanently differentiated state. Initially attempted in the context of teratoma [35], but a proof of concept study for this approach was treating APML with all-trans retinoic acid therapy [34]. Other studies also reported a differentiation therapeutic approach aimed at the conversion of dedifferentiated tumour cells into epithelial cells that are more sensitive to chemotherapy [36,37].
-
3.
Go with the flow and use the tumour plasticity to target the dedifferentiated cancer stem cells with transcription factors or small molecules to differentiate them into harmless cell lineages which lack tumourigenic potency. This final therapeutic approach necessitates an in-depth understanding of the hallmark of dedifferentiation. Recent work proved the efficacy of this approach by switching malignant breast cancer cells into harmless post-mitotic adipocytes. Combination of PPARγ agonist Rosiglitazone, an anti-diabetic drug, with a MEK inhibitor was used to force the breast tumour cells towards adipogenesis, resulting in harmless post-mitotic functional adipocytes [38] (figure 4).
Figure 4.

Transdifferentiation approach to therapy. Following combination of PPARγ agonist rosiglitazone (an anti-diabetic drug) + MEK inhibitor—cancer cells are converted to functional adipocytes, adapted based on [38].
These studies argue strongly that the dedifferentiation of tumour cells along a developmental pathway towards a progenitor or stem cell-like state among various cancers is a forgotten hallmark, a discrete acquired capability of cancer and certainly warrants further investigation for a better understanding of this novel trait of cancer cells. Hanahan and Weinberg's Hallmarks of Cancer had generic nature as one of the features of every hallmark, as something that is prevalent in the majority of cancers despite the heterogenetic nature of the disease. Dedifferentiation certainly qualifies as a generic hallmark distinct from the other hallmarks of cancer. The reported interplay between transcription regulators Sox2 and Sox9 as an epigenetic switch between high proliferation and high invasiveness [39] leads to our next hallmark of cancer that influences tumourigenesis—epigenetic dysregulation (figure 5).
Figure 5.

The road better not taken: intertwined nature of epigenetic instability and genetic instability in riding over the road of hallmarks towards tumourigenesis.
4. New hallmark 2: epigenetic dysregulation
Dedifferentiation to a progenitor state is a rate-limiting step in melanoma formation but it is underpinned by epigenetic machinery [40]. Although Yamanaka factors provide the possibility of reprogramming differentiated somatic cells to a pluripotent state, the blockage of histone H3 lysine 9 (H3K9) methylation has been shown to enhance this reprogramming capability [41,42]. Similarly, in the context of DNA methylation, another key epigenetic alteration, the promotion of DNA demethylation via stimulation of TET (ten-eleven-translocation) enzymes using vitamin C enhances reprogramming to a pluripotent state [43]. Epigenetics can also regulate the process of winding back the clock to a pluripotent state on the basis of chromatin state and the expression levels of chromatin-modifying enzymes [44], providing a conceptual link with our first hallmark of dedifferentiation.
4.1. What is epigenetics?
Among several phenomenal works, Theodor Boveri laid the foundation of the role of epigenetics in cancer through his observation of abnormal chromatin structures in tumour cells, described over 90 years ago [45]. The term ‘epigenetics’ was first coined by Conrad Waddington, defining it as ‘the branch of biology which studies the causal interactions between genes and their products which bring the phenotype into being' [8]. Vogelstein and Feinberg, in an attempt to dissect the mechanism underlying the higher frequency of mutations among tumours, compared normal tissue with tumour tissue and revealed the loss of DNA methylation in a substantial proportion of tumour tissues, positing that hypomethylation of CpG islands could lead to oncogene activation in cancer [46] and revealing the prevalence of global hypomethylation among tumour genomes.
Holliday refined the definition of epigenetics as heritable changes in the gene expression without alteration in the DNA sequence, that is altering the phenotype without altering the genotype [47]. With epigenetics playing a fundamental role in the development and progression of various cancers via modification of gene expression, such as hypermethylation of tumour suppressor genes in retinoblastoma [48] and epigenetic silencing of microRNAs [49], it is a pivotal hallmark of cancer.
4.2. Epigenetic fingerprints as the ‘midas touch' driving tumourigenesis—the hallmark of hallmarks
As discussed in the features of a hallmark, epigenetic dysregulation is an active functional capability, it is a unique feature among cancer cells and there are epigenetic fingerprints on tumour cells which reflect its chronic nature. Epigenetic dysregulation viewed as a bystander would be a relegation of its active role in tumourigenesis, as several studies have pointed out its role in tumour initiation. In 2006, Feinberg proposed the epigenetic progenitor model of tumourigenesis, wherein epigenetic dysregulations of the progenitor cell population give rise to tumours [50] (figure 6).
Figure 6.

Epigenetic crossroads: multitude of studies supporting the epigenetic progenitor model.
4.3. Sustained proliferative signalling
Many tumours display a gain of function mutation of isocitrate dehydrogenase (IDH) [51,52], leading to the generation of the oncometabolite 2-hydroxyglutarate, which disrupts the function of hydroxylases such as TET — a key catalyst in the process of DNA demethylation [53,54]. The result is a hypermethylated phenotype as seen with the CpG island methylator phenotype (G-CIMP) in IDH mutant glioma [55]. This alters the binding affinity of the DNA binding protein CTCF (CCCTC-binding factor) which is very sensitive to methylation states [56]. CTCF has a critical function as an insulator, setting the boundaries which limit the interactions between an enhancer and a gene in the context of topologically associated domains (TADs) [56]. This insulation is lost as a result of the reduced binding of CTCF, facilitating aberrant interactions between promiscuous enhancers and genes as a result of the altered chromosomal topology caused by epigenetic dysregulation [57]. In this context, Platelet-derived growth factor receptor A (PDGFRA), a predominant oncogene among gliomas [58] is activated as a consequence of the epigenetic dysregulation-led decrease in CTCF insulation, with a potent promiscuous enhancer driving constitutive PDGFRA expression, driving sustained proliferation in gliomas [57]. The loss of CTCF insulation may even be preserved in subsequent cell divisions, compromising the genomic topology otherwise maintained by this insulation, leading to further oncogene activation not limited to just PDGFRA [59].
This mechanism is not limited to gliomas, as CTCF sites which are adjacent to oncogenes have been reported as mutational hotspots and are frequently mutated in multiple tumours, such as endometrial [60], colorectal (CRCs), oesophageal and liver cancer [59].
4.4. Evading growth suppressors
Cyclin-dependent kinase inhibitor 2A (CDKN2A) encodes a potent tumour suppressor p16INK4a, that binds to cyclin-dependent kinase 4/6 (CDK4/6), which leads to an allosteric conformational change inhibiting the cyclin D–CDK4/6 complex formation. As a result of the lack of this complex, retinoblastoma protein (Rb) is maintained in a hypophosphorylated state, promoting the formation of Rb/E2F repressive complex. This leads to the suppression of growth, as a result of cell cycle arrest in G1 [61]. Epigenetic silencing of tumour suppressors such as p16INK4a via promoter hypermethylation mediates evasion of growth suppression, as evident from multiple studies on epigenetic alterations enumerated below (table 1).
Table 1.
Epigenetic instability mediating evasion of growth suppressors.
Similarly, the hyperactivity of enhancer of Zeste homologue 2 (EZH2), a catalytic subunit of polycomb repressive complex 2 (PRC2) involved in the trimethylation of histone H3 lysine 27 to form H3K27me3, is implicated in the evasion of growth suppression via CDKN2A repression [65–67] reiterating the role of epigenetic dysregulation in facilitating the hallmarks [62].
4.5. Invasion and metastasis
An integral component of the hallmark of invasion and metastasis is a reversible epithelial–mesenchymal transition (EMT), orchestrated by the interaction between epigenetic modulators of chromatin configuration and EMT inducing transcription factors. Expression of E-cadherin, a key coordinator of epithelial phenotype, is lost during EMT. Epigenetic repression of CDH1, which encodes E-cadherin, is mediated by the recruitment of EMT inducing transcription factor Snail to the CDH1 promoter, leading to a repressive mark H3K27me3 [68]. Further to this, Snail can associate with Mi-2-nucleosome remodelling and deacetylase (NuRD) repressive complex, which can repress CDH1 activity via deacetylation of CDH1 promoter [69].
4.6. Replicative immortality
Alternative telomere lengthening (ALT) is a telomerase-independent homologous recombination-based pathway which cancer cells use to overcome the Hayflick limit to maintain telomere length [70]. An interplay between epigenetics and genetic mutations leads to perturbations of histone variant H3.3 and its specific chaperone proteins α-thalassemia X-linked mental retardation protein (ATRX) and death domain associated protein (Daxx) which impairs the incorporation of the histone variant H3.3 at telomeres, disrupting their heterochromatic state and facilitating ALT [71].
4.7. Inducing angiogenesis
Epigenetics plays a key role in angiogenesis. Histone deacetylases have been shown to downregulate the expression of von Hippel–Lindau (VHL) and p53, but promote an increase in hypoxia-inducible factor-1α and vascular endothelial growth factor (VEGF), thereby stimulating angiogenesis by the suppression of hypoxia-responsive tumour suppressor genes [72,73]. Choriocarcinoma, a highly vascular tumour derived from trophoblasts, displays epigenetic silencing of FLT1 via promoter hypermethylation. Normal placental trophoblasts express abundant levels of an anti-angiogenic factor, Soluble Fms-like tyrosine kinase-1 (sFLT1) from the FLT1 locus. Epigenetic silencing of FLT1 blocks expression of this negative regulator, thereby facilitating angiogenesis in choriocarcinoma [74].
4.8. Resisting cell death
Glioblastoma multiforme is a very aggressive cancer with a dismal prognosis. Nevertheless, a promising therapeutic strategy is to induce tumour cell death via tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-based therapy that binds to human death receptor 4 (DR4). However, epigenetic silencing via promoter methylation of DR4 attenuates TRAIL/DR4-mediated apoptosis [75]. Further evidence of epigenetics mediating resistance to cell death is resistance to anthracycline therapy in acute myeloid leukaemia (AML), due to impaired DNA damage response from defective nucleosome remodelling, as a result of mutation of epigenetic regulator DNA methyltransferase 3A [76]. CXCL14, a chemokine which can influence apoptosis, is shown to be a frequent candidate for epigenetic silencing among lung tumours. Tumour specific methylation of the CXC-subfamily of chemokines was observed in 75% of lung adenocarcinomas [77].
4.9. Immune evasion
Epigenetics is fundamental to the normal functioning of immune cells. Antigen presentation through MHC class I is pivotal for CD8+ T cells activity. The class I transactivator NLRC5 is a transcriptional regulator of the MHC class I genes, but the promoter region of NLRC5 is frequently methylated among cancers, resulting in the reduction of MHC class I gene expression [78].
4.10. Deregulating cellular energetics
In order to adapt to a hostile microenvironment and to satisfy their high metabolic needs, cancer cells can use glycolysis, instead of oxidative phosphorylation to metabolize glucose, even in aerobic conditions. The central activators implicated in the glycolytic phenotype are the PI3 K/AKT/mTOR pathway along with MYC and HIF-1 signalling [79].
Tumour suppressors that repress this pathway, namely PTEN [80], VHL [81,82], LKB1 [83] and prolyl hydroxylases [84] are epigenetically silenced via promoter hypermethylation, contributing to deregulation of cellular energetics.
4.11. Genomic instability and mutation
Faithful genome replication and the maintenance of genomic integrity are underpinned by epigenetic mechanisms. Transposable elements (TE) are highly repetitive sequences of DNA in the human genome, and have their own regulatory sequence, allowing for independent expression and ability to alter the expression of neighbouring genes. Since TE activity has a high propensity to disrupt genomic integrity, these are usually silenced epigenetically, but this regulation is lost in cancer [85].
4.12. Tumour promoting inflammation
DNA demethylation triggers transcription of inflammation-related genes, including chemokine receptor 4 (CXCR4) and serum amyloid A (SAA) in advanced clear cell renal cell carcinoma (ccRCC), contributing to tumour promoting cancer cell-intrinsic inflammation via epigenetic remodelling [86].
The above studies highlight an epigenetic foundation for each of the established hallmarks of cancer provides compelling evidence of the indispensable nature of epigenetic dysregulation as a pivotal enabling hallmark of cancer. However, just as cancer involves more than just tumour cells, so our bodies are more than simply an assemblage of human cells. This sobering thought leads us to our third hallmark, the microbiome.
5. New hallmark 3: altered microbiome
The concept of the human body being a vessel for other microorganisms is well established—the microbial metagenome in our body outnumbers our genome by at least 100-fold [87]. Microorganisms first appeared around 3.25 billion years ago [88] and over the 1.25 billion years of co-existence with multicellular eukaryotes [89] (figure 7), the interaction with microbes has shaped evolution, as illustrated by the microbial control of host homeostasis [90]. It has been estimated that nearly half of the metabolites in plasma are of microbiotal origin [91], but the human microbiome plays a duplicitous role. Helicobacter pylori is nearly ubiquitous among humans, colonizing about 50% of the world population, having co-evolved with humans in an association spanning over 50 000 years [92]. H. pylori colonization has been shown to decrease the risk of gastroesophageal reflux disease and its subsequent sequela, oesophageal carcinoma [93]. It may also confer protection against asthma [94], demyelinating diseases such as multiple sclerosis [95], tuberculosis [96] and inflammatory bowel disease [97]. H. pylori also has been shown to modulate energy homeostasis via cooperation with gut microbiota, impacting on circulating metabolic gut hormones [98].
Figure 7.

Coexistence.
In contrast with these beneficial roles, as a component of the gut microbiome it is also linked to 90% of gastric cancers [99]. The carcinogenicity of H. pylori is associated with the expression of vacuolating cytotoxin gene A (vacA) and cytotoxin-associated gene A (CagA) [100,101]. CagA positive H. pylori promotes genetic instability via perturbation of the mitotic spindle checkpoint, causing chromosomal instability [102] and epigenetic instability. Increased levels of DNA methyltransferases (DNMTs) [103] lead to hypermethylation of MLH1, a key DNA mismatch repair gene [104], and have been suggested to mediate a mutator phenotype in a hit and run fashion, promoting tumourigenesis [101]. Given these data, it is worthwhile to postulate whether Escherichia coli has a role in cancer, being among the first bacteria to colonize the gastrointestinal tract of neonates [105] and advocated to promote gut health in multiple off the shelf probiotics [106].
Commensal E. coli, within few days following birth, establish a favourable anaerobic environment in the gut facilitating colonization of other species including Bifidobacterium, Clostridium and Bacteroides [107]. Certain strains of E. coli harbour a gene cluster hybrid non-ribosomal peptide synthetase-polyketide synthase (pks) island which produces genotoxic colibactin [108]. Colibactin has been described as a bacterial ‘warhead', forming bulky unstable DNA adducts via alkylation [109]. Among epithelial cell lines, pks+ E. coli has been shown to induce double-stranded DNA breaks [110] and interstrand cross-links [111].
Whole-genome sequencing studies have described mutational signatures from colorectal crypts of healthy individuals where, in a subset of crypts, an unknown mutagenic agent caused co-occurrence of single-base substitution-A (SBS-A) and insertion/deletion A (ID-A). The two motifs were described to be from mutagenic insult which occurs in early childhood [112]. The cause of these two motifs surfaced while investigating the long-term effect of colibactin using single-cell-derived organoids, with pks-mutational signature strongly matching the two motifs SBS-A and ID-A [113]. Since pks+ E. coli is the mutagenic agent responsible, and the study was performed on organoids that cannot precisely mimic inflammation or the immune environment, the inference is that colibactin can directly initiate tumourigenesis via mutations [113]. Interestingly, the impact of these data reaches beyond the gut, suggesting a similar role in head and neck as well as urogenital cancers [113]. So, perhaps one should reconsider probiotics containing genotoxic E.coli and consider screening for pks+ E. coli in the context of colorectal cancer prevention.
Arguing for a microbiotal impact merely on the hallmark of genomic instability as being sufficient for its contribution to tumourigenesis is to grossly underestimate the role of the microbiome. Paget's seed (cancer cells) and soil (tumour microenvironment) hypothesis [114] is highly relevant to the role of the microbiome in tumourigenesis. The microbiome can exploit the inflammatory milieu to a pro- or anti-tumour state, cultivating the soil which is apt for sowing the seeds of tumourigenesis. This can be clearly substantiated by findings from a study using the first identified oncovirus [115], where Rous sarcoma virus does not induce tumours in sterile embryos despite expression of the v-Src oncogene [116].
In 1990, Fearon & Vogelstein [117] proposed the Vogelgram model of multi-step colon cancer pathogenesis. A key reason for the success of colon screening in CRC prevention is due to the long latency period from tumour initiation to overt clinically detectable CRC. Here, we consider this long latency in the context of the proposed hallmark of microbiome dysbiosis.
5.1. Microbiome tug-of-war hypothesis
The tug-of-war between microbiome species may underlie the long latency in CRC. Enterotoxigenic Bacteroides fragilis (ETBF) promotes the colonization of pks+ E. coli, together with leading to genetic and epigenetic instability. Following this, colonization by pro-tumourigenic Fusobacterium nucleatum further promotes tumourigenesis by aiding in the development of an immunosuppressive microenvironment and seeding metastasis, whereas anti-tumourigenic bacteria act to prevent malignancy. The long latency period, which may ultimately lead to subsequent accumulation of genetic/epigenetic mutations and overt malignancy, depends on the balance between pro/anti-tumourigenic microbes (figure 8).
Figure 8.

(a,b) Microbiome tug-of-war hypothesis for CRC latency: improvization of the Vogelgram [117], with an explanation of the role of bacteria in multi-step CRC progression.
ETBF secretes a 20 kDa zinc-dependent metalloprotease toxin, B. fragilis toxin (BFT). BFT degrades E-cadherin, leading to increased intestinal epithelial cell proliferation and permeability of the intestinal barrier [118]. BFT further leads to activation of β-catenin signalling and induces STAT3 (signal transducer and activator of transcription 3) activation [119] and the T helper 17 (TH17) immune response [120]. ETBF modulates the colonic niche to select for bacteria with a colonization advantage, inducing upregulation of antimicrobial peptide lipocalin 2 [121] that causes sequestration of bacterial siderophores. Siderophores are iron-binding complexes that are pivotal for bacteria to thrive in iron limiting environments, hence bacteria which are resilient to lipocalin 2, such as E. coli, begin to thrive along with ETBF [122]. Therefore, the first hit in our hypothesis of CRC tumourigenesis is orchestrated by ETBF followed by the co-colonization of ETBF along with pks+ E. coli, following which Fusobacterium nucleatum comes into play.
An anaerobic Gram-negative bacterium, Fusobacterium nucleatum is typically resident in the oropharynx, participating in dental biofilm formation [123]. Its virulence factor FadA adhesin binds to the extracellular domain of E-cadherin and promotes tumourigenesis via β-catenin/Wnt signalling [124]. F. nucleatum is also immunosuppressive, causing inhibition of T cell responses while allowing for the expansion of tumour promoting myeloid-derived immune cells [125]. Fap2 protein of F. nucleatum binds directly to the inhibitory receptor—T cell immunoglobulin and ITIM domain (TIGIT), and inhibits natural killer (NK) cell activity, leading to immune evasion [126]. In support of this hypothesis, F. nucleatum did not initiate tumour formation in vivo [127], but sustained the pro-tumourigeneic momentum in the latter part of multi-step CRC tumourigenesis and facilitated metastasis [128]. Intriguingly, a study based on biopsies from CRC patients and mouse xenografts revealed that F. nucleatum can accompany primary colorectal adenocarcinoma cells to distant metastatic sites, being maintained among patient-derived xenografts of CRC even through multiple passages. Moreover, treatment with metronidazole, an antibiotic to reduce F. nucleatum load, resulted in reduced tumour growth [128], suggesting that tumour cells are rewarded for carrying F. nucleatum by its modulation of the microenvironment at the distant metastatic site in favour of tumour growth.
Meanwhile, there is a subset of anti-carcinogenic bacteria including Faecalibacterium, Roseburia and Slackia spp. which generate catabolites such as short-chain fatty acids (SCFAs), for example, butyrate [129] and the antioxidant equol [130]. Butyrate downregulates proinflammatory gene expression and suppresses tumour growth via inhibition of histone deacetylases [131]. The outcome of the tug-of-war depends on epigenetic factors such as diet which will give the final upper hand aiding in colonization by either pro- or anti-carcinogenic bacteria.
The majority of pancreatic ductal adenocarcinoma (PDAC) patients have a dismal prognosis, but a small subset of patients survive longer than 5 years [132]. Intriguingly, long-term survivors have higher tumour microbial diversity with distinct tumour microbial signatures compared to short-term survivors [133]. The tumour microbial diversity was shown to exert an immune-activating effect via improved immune cell infiltration to the tumour milieu. Furthermore, colonization of pancreatic tumours by gut microbiota was identified, with 25% of PDAC microbial composition matching that of the gut. Preclinical data from the same study showed that faecal microbial transplant (FMT), from patients who were long-term survivors, into tumour-bearing mice led to immunoactivation in the murine tumour microenvironment and a significant reduction in tumour growth, reiterating the role of tumour microbiome in disease progression and outcome, as well as the potential of FMT in treating PDAC [133].
5.2. The microbiome is more than bacteria
We need to consider more than simply bacteria and viruses. Fungal infiltration from the gut to the pancreas was shown to occur via the sphincter of Oddi (figure 9a), which serves as a direct link between the pancreatic duct and the gut. Taxonomic diversity analysis identified the dominance of the genus Malassezia in PDAC tissues compared to that of the gut, in mouse models. Comparison of sequencing data of PDAC patient faecal samples to that of paired tumour tissue corroborated these findings. Antifungal ablation with amphotericin B mitigated pancreatic dysplasia in mouse models and was shown to work synergistically with gemcitabine in reducing tumour burden [135]. Through repopulation experiments, Malassezia globosa was identified as being responsible for PDAC disease progression, via fungal-mediated activation of the mannose-binding lectin (MBL)–C3 cascade (figure 9b). MBL is a protein of the innate immune system which serves as an opsonin. Upon binding to the sugar motifs on the fungal wall, it triggers the complement cascade, in particular C3, a pivotal component downstream of MBL [135]. Based on the inference from the study, we can speculate that diagnostic assay using the taxonomic composition of stool samples may be appropriate for early detection of PDAC, and that antifungal therapy may be efficacious.
Figure 9.

Tumour promoting inflammation triggered by mycobiome. (a) Sphincter of Oddi. (b) The mannose-binding lectin (MBL) pathway of complement activation, adapted based on [134].
More than 85% of human papillomaviruses (HPV) are cleared spontaneously [136], so why can the remaining 15% mediate progression to cervical neoplasia? The answer lies with the vaginal microbiome, dysbiosis of which plays an important role even in HPV-related cervical cancers [137]. Lactobacillus species are dominant in the vaginal niche and are characteristic of vaginal health [138]. They maintain the vaginal microenvironment in an acidic state (pH < 4.5) via the production of lactic acid [138] and protect against invading pathogens such as herpes simplex virus [139], human immunodeficiency virus [140], Neisseria gonorrhoeae [141] and even E. coli [142]. The depletion of Lactobacillus species has been linked to an increased risk of acquisition of HPV infection and its reduced clearance [137], and reduction in Lactobacillus dominance and increased vaginal microbiome diversity correlated strongly with cervical neoplasia severity [137,143].
The microbiome also plays an important role in deciding the outcome of both conventional chemotherapies and immunotherapeutic interventions. It can alter the bioavailability of drugs [144], and DNA damage induced by platinum-based regimens is severely attenuated in the absence of commensal microbiota [145]. Oral administration of Bifidobacterium in mice controlled melanoma growth on a par with checkpoint blockade using programmed cell death ligand 1 (PD-L1) specific antibody and co-administration resulted in near eradication of tumour growth [146]. Furthermore, the efficacy of blocking CTLA-4, a major negative regulator of T cell activation, depends on Bacteroides species and tumours in axenic or antibiotic-treated mice do not respond to CTLA-4 blockade [147]. The microbiome also has a role in immunosurveillance, as seen with the hygiene hypothesis which links an increase in the incidence of some cancers to decrease in exposure to certain microbes [148,149].
In conclusion, the microbiome exerts both beneficial and nefarious effects over the human body. We argue that it has a role in each of the triumvirate of immunoediting [150], namely elimination, equilibrium and escape during tumourigenesis and as such is a pivotal enabling hallmark of cancer. Antibiotic mediated alteration of gut microbiota has been shown to alter the cerebral tumour microenvironment, thus affecting glioma progression [151], which bring us to our final enabling hallmark of cancer—nerves/neuronal signalling.
6. New hallmark 4: altered neuronal signalling
Vesalius, in his book De corporis humani fabrica libri septem, described the tandem nature of blood vessels and nerves [4,198] (figure 10), innervation and blood supply being indispensable for growth and survival. Since angiogenesis has an established role, it is enticing to delve further into the role of nerves in cancer, a topic that is often overlooked. Perhaps the reason might be the difficulty involved in observing nerves during routine histology of tumour specimens, but nerves are one of the most significant aspects of tumour progression. Metastasis involving the central nervous system/peripheral nervous system results in manifold increased morbidity/mortality.
Figure 10.
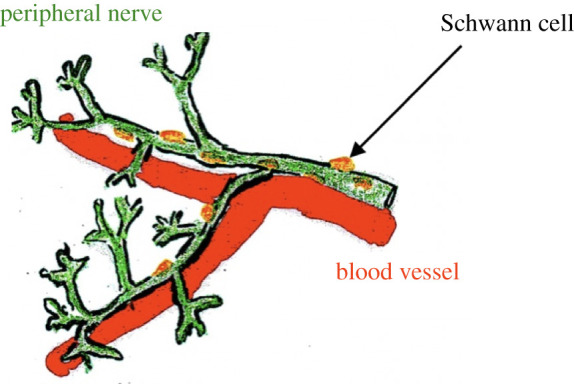
Blood vessels and nerves in tandem.
In 1840, surgeons attempted to transect the trigeminal nerve, which runs along the face, and the accompanying blood vessels, in order to cure tumour of the lips. It provided symptomatic control but failed to cure the patients and eventually mandated complete resection of the tumour [152]. However, with recent advances in the understanding of the function of the nervous system, its role in tumour initiation and progression can be better elucidated, to derive therapeutic benefits. The density of nerve fibres in tumour tissue correlates with the aggressiveness of the disease among multiple cancers, including breast [153], lung [154], colorectal [155] and prostate cancers [156]. Based on these observations, one might advocate neuronal transection to control tumour progression. However, in a PDAC mouse model, sub-diaphragmatic vagotomy, targeting the vagus nerve—a mixed nerve with both sensory and parasympathetic components, resulted in enhanced tumour growth and reduction in survival [157]. Contrastingly, transection of the same nerve in gastric cancer models resulted in suppression of tumourigenesis [158]. Instead of the radical approach of transection, an alternative approach is to use chemical denervation, as performed in specific targeting of the sensory nerves in ductal carcinoma, and the use of capsaicin inhibited pancreatic ductal adenocarcinoma (PDAC) progression [159]. Another approach is via injection of botulinum toxin A (Botox), a neurotoxin, into the gastric wall. This inhibited progression to overt adenocarcinoma among preneoplastic models and inhibited disease progression in advanced gastric cancer models [158].
6.1. β-blockers for inhibiting tumour progression
Sympathetic nerves are implicated in blood vessel patterning during early development [160]. Sympathetic nerves release noradrenaline, the circulating levels of which increase during chronic stress [161]. β-adrenergic receptors mediate most of the effects of noradrenaline. Chronic stress has long been attributed as a risk factor for cancer [162]. Reproduction of the effect of chronic stress in transgenic mouse models of breast cancer via long-term administration of isoprenaline, a non-selective β-adrenergic agonist, resulted in increased lymph node metastasis, while inhibition of adrenergic signalling with propranolol, a non-selective β-blocker, resulted in inhibition of metastasis to the lymph node [163]. Reiterating the role of β-adrenergic receptors, a similar effect was also observed in pancreatic cancer models of chronic stress, with a reduction of tumour volume following administration of propranolol [164].
The pro-tumourigenic effects exerted by sympathetic nerves in response to stress are mediated by β-adrenergic receptors. This was demonstrated elegantly in work showing that an adrenergic nerve-derived signal-mediated activation of an angiogenic switch in a transgenic mouse model of prostate cancer [165].
Sympathetic nerves in prostate tumours release noradrenaline which, via the β2-adrenergic receptor on endothelial cells, triggers an angiogenic switch by inducing a change in endothelial cell metabolism from oxidative phosphorylation towards aerobic glycolysis, driving angiogenesis and fuelling tumour progression. Blockade of β-adrenergic receptor signalling reverts endothelial cell metabolism from aerobic glycolysis towards oxidative phosphorylation through cytochrome C oxidase assembly factor 6 (Coa6) activity, thereby inhibiting angiogenesis and curtailing tumour progression [165].
6.2. Perineural invasion in pancreatic ductal adenocarcinoma
Perineural invasion is linked to worse prognosis in PDAC [166], with PDAC cells recruiting nerves via nerve growth factor (NGF) [167]. In murine PDAC models, chronic stress-dependent sympathetic nerve signalling triggers tumour growth via a feedforward loop, wherein adrenergic signalling stimulates NGF, which promotes further innervation of tumour cells via axogenesis, resulting in increased noradrenaline accumulation in the tumour microenvironment, inducing β2-adrenergic receptor-dependent PDAC progression [168] (figure 11).
Figure 11.

Chronic stress-dependent sympathetic nerve signalling triggering tumour growth via a feedforward loop.
Blockade of the β2-adrenergic receptor, or the NGF receptor tropomyosin-receptor kinase A (TRKA), disrupts this feedforward loop and inhibits tumour progression [167,168]. Clinical studies have reported improved survival among PDAC patients with the use of β-blocker [169]. This provides a window of opportunity to treat patients with pancreatic intraepithelial neoplasias (PanINs) with a non-selective β-blocker regimen to potentially prevent progression to overt PDAC, although the challenge of early detection remains. This might be facilitated using a diagnostic assay based on the taxonomic composition of stool samples, as discussed earlier.
While β-adrenergic signalling is pro-tumourigenic in the aforementioned solid cancers, there is the caveat of an opposite effect adding to the complexity of targeting the hallmark of nerves/neuronal signalling. Noradrenaline-mediated sympathetic nerve signalling has been linked to the maintenance of the steady-state condition of haematopoietic stem cells (HSCs) in the bone marrow niche in a circadian manner [170]. Attrition of the β-adrenergic signalling leads to an increased propensity of myeloproliferative neoplasms [171,172], therefore the implementation of β-blocker targeting sympathetic signalling in malignancy is context-dependent.
6.3. Putting the cart before the horse: whether nerves migrate towards tumours or the tumour cells migrate towards nerves?
Schwann cells, the glial cells responsible for myelinating peripheral nerves, are key to neural homeostasis, participating in Wallerian degeneration, neural repair and regeneration [173]. In an ex vivo model using rat sciatic nerve, Schwann cells displayed a high affinity towards pancreatic and colon tumour cells, but not normal cells, migrating towards tumour cells, thereby outlining a pathway for tumour driven neurogenesis [174]. Nerve growth factor (NGF), and its receptors TRKA and p75NTR are critical regulators of gland innervation and neurite outgrowth. They are also implicated in neural tracking [175], the ability of tumour cells to migrate along axons.
Pro-NGF, the precursor of NGF, serves as a reservoir for NGF [176]. Immunohistochemical studies in prostate cancer suggested that pro-NGF production by tumour cells might drive axonogenesis [177]. Thus, there is an element of reciprocal interaction between nerves and tumour cells driving tumourigenesis; Schwann cells migrate towards tumour cells while prostate tumour cells in turn recruit nerves via pro-NGF.
6.4. Synaptic interaction between brain tumour cells and neurons
Clues to the interaction between tumour cells and neurons come from the study of synapses between neurons and oligodendrocyte precursor cells, demonstrating a neuron to non-neuron synapse [178], as well as the finding that glutamate secretion confers a growth advantage to glioma cells [179]. These preliminary studies were bolstered by the identification of functional synapses between neurons and glioma cells, with transcriptomic analysis further confirming that the glioma cells express GluA2, a subunit of the ionotropic glutamate receptor, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR). Treatment with an AMPAR antagonist inhibited glioma progression, suggesting glioma cells can co-opt glutamatergic signalling to facilitate invasion and tumour progression [180,181] (figure 12). Based on both of these studies, one can speculate whether anti-epileptic drugs that act presynaptically, such as levetiracetam [182], might inhibit glioma progression. An alternative approach could be a non-competitive AMPAR antagonist such as perampanel, which has good penetration to the brain, for use in glioma treatment [183]. Key to note is that the AMPARs mentioned in both the studies [180,181] are calcium permeable, which means the target of the drug candidate must be calcium permeable AMPAR
Figure 12.

Synaptic interaction between presynaptic neuron and glioma via AMPA receptor.
Metastasis to the brain presents a checkmate scenario to clinicians but a breakthrough finding, deciphering the interaction between neurons and metastatic cells [184], can now pave way for new therapeutic approaches.
6.5. Parasitic tripartite synapse
Triple-negative breast cancer (TNBC) carries a poor prognosis as it lacks the expression of targetable hormone receptors and human epidermal growth factor receptor 2, coupled with a propensity to metastasize to the brain [185]. N-Methyl-d-aspartate receptor (NMDAR), a type of glutamate receptor, plays a key role in the synaptic plasticity of the central nervous system, but has also been implicated in ovarian and pancreatic tumour progression [186]. Transcriptomic data identified higher expression of NMDAR among basal sub-types of breast cancers such as (TNBC), in particular the NMDAR GluN2B subunit, which contains phosphorylation sites critical for NMDAR signalling. An autocrine source of glutamate-mediated NMDAR signalling in the breast to brain metastasis (B2BM) microenvironment was excluded, with B2BM cells found to express neuroligin-2 [184], the expression of which by non-neuronal cells has been shown to induce presynaptic differentiation and trigger de novo formation of pseudo-synapses [187,188].
Microscopic analysis of mouse B2BM models revealed a pseudo-tripartite synapse phenomenon. Finger-like projections emanated from the B2BM cells towards excitatory synapses, forming a fake tripartite synapse [184]. In normal neurophysiology, glutamate released from presynaptic neurons is endocytosed by postsynaptic neurons that express the glutamate receptor NMDAR, as well as by astrocytes which are located adjacent to the synaptic cleft [189]. This tripartite phenomenon is mimicked by the B2BM cells, which take the position of the astrocyte next to the synaptic cleft and use the glutamate from the presynaptic neuron to promote further metastasis and colonization in the brain (figure 13).
Figure 13.

Parasitic tripartite synapse: B2BM colonization of brain using glutamate from the fake tripartite synapse, adapted based on [184].
Modulation of GluN2B expression demonstrated that NMDAR signalling was not necessary for the initial seeding of breast tumour cells to the brain but, rather, was critical for the proliferation of B2BM cells [184]. Thus, B2BM cells effectively tune the neural niche to their advantage without disrupting the existing synaptic infrastructure. Tumour cell–astrocyte gap junctions can also be co-opted to promote brain metastasis via 2′3′-cyclic GMP-AMP (cGAMP)-mediated signalling. This can potentially be disrupted by the gap junction modulator meclofenamate, which has oral bioavailability and can pass through the blood–brain barrier [190]. Similarly, based on the inference that B2BM cells co-opt NMDAR signalling for metastatic progression in the brain [184], one could exploit the parasitic tripartite synapse for therapeutic and diagnostics purposes. One approach might be to repurpose memantine, an NMDAR antagonist used for treating Alzheimer's disease, to curtail the progression of B2BM in TNBC patients. Furthermore, radiolabelled glutamine can potentially be used for imaging triple-negative breast cancer brain metastasis [191].
6.6. Nerves and the tumour microenvironment
Nerves can also play a role in immune evasion during tumourigenesis by orchestrating an immune-suppressive tumour microenvironment. β2-adrenergic receptor signalling by adrenergic nerves can inhibit lymphocyte egress, effectively reducing the recruitment of antigen primed T cells [192]. Manipulation of autonomic nerves using a novel viral-vector based neuro-engineering technique revealed accelerated breast cancer progression with sympathetic nerve stimulation in tumours, while local sympathetic denervation curtailed tumour growth and reduced the expression of immune checkpoint molecules, such as programmed death-1 (PD-1) and PD-L1, as well as FOXP3, that mediates immunosuppression [193]. Such a strategy of the localized intervention targeting neural input, using genetic neuro-engineering techniques, may hold promise to stimulate the immune system while offsetting the deleterious side-effects of the systemic use of checkpoint inhibitors.
Nerves and neuronal signalling are an indispensable part of tumourigenesis, playing an active role in modulating the tumour microenvironment. They are involved in the recruitment of blood vessels to the tumour, control constriction/relaxation of blood vessels, alter the expression of immune checkpoint molecules and provide cues for proliferation to tumour cells, yet the nervous system has been largely disregarded in cancer therapeutics. Nerves and neuronal signalling are an enabling hallmark of cancer that provides tumours with a means of interacting with its microenvironment to facilitate metastatic progression. Future treatment regimens must work around the neural circuit to offer better control over tumour progression.
7. Conclusion
The understanding of cancer from a curse, to that of a heterogeneous group of diseases that lack the fundamental ability to respond to principal signals regulating proliferation, differentiation, and cell death is a phenomenal leap of understanding. From multiple resections without anaesthesia in ancient times, to targeted cancer therapeutics is certainly a remarkable feat of achievement. The Hallmarks of Cancer [194] marked the Millenium era for cancer researchers, laying the framework for honing our understanding of cancer as a disease. We present four novel hallmarks, the traits of which are the language which cancer cells use to interact with the microenvironment to facilitate proliferation and survival. We consider two additional core hallmarks: dedifferentiation/transdifferentiation and epigenetic dysregulation, alongside two enabling hallmarks: altered microbiome and altered neuronal signalling.
Seminal studies, we have discussed, overturned the unidirectional landscape of differentiation [9,10], yet the hallmark of dedifferentiation has long been ignored in the field of cancer therapeutics. The lineage plasticity conferred by the proposed hallmark of dedifferentiation, hijacked by tumour cells, can also be used for targeting tumour cells at their most vulnerable state to potentially transdifferentiate them to lineages which lack metastatic potential.
Two of the hallmarks proposed to confer a vantage point for therapeutic manipulation due to their reversible nature: epigenetic dysregulation and the microbiome. Epigenetic dysregulation provides numerous opportunities to intervene in cancer progression and development. For example, dietary factors can influence serum methionine levels, which in turn can affect histone methylation [195]. Microbiome dysbiosis can be manipulated by enhancing our ability to identify anti-carcinogenic (friend) and pro-carcinogenic (foe) among the microbiome. Microbiome composition must be integrated and used as a tool to enhance the outcome of therapeutics.
Finally, the hallmark of altered neuronal signalling consists of multiple clues to halt metastasis. The two factors which cancer cells use to design their microenvironment to their advantage are microbiome and nerves. Tumour cells use nerves to establish blood vessels and garner proliferative cues. Cancer can be associated with excruciating pain, a key being that cancer cells recruit numerous nerves, a trait that can be intercepted for pain management. Two modalities for managing the hallmark of altered neuronal signalling are either to include resection of nerves in surgical protocols for tumour management (significantly more challenging than resecting lymph nodes), or to target the nerve growth factor/localized intervention of neuronal signalling within the tumour microenvironment. Future studies may look into possibilities of targeting artemin which has an established role in the migration of sympathetic precursors [196,197].
Considering cancer as the conductor of a malign symphony and the hallmarks as the musicians, we need to tune our hearing to appreciate every key nuance of the piece. By identifying new performers, we can adapt our interventions, re-educating the orchestra and re-establishing the rhythm of life.
Contributor Information
Sasi S. Senga, Email: authordrsashi@gmail.com.
Richard P. Grose, Email: r.p.grose@qmul.ac.uk.
Data accessibility
This article has no additional data.
Authors' contributions
S.S. conceived the study and analysed literature, under the guidance of R.G. S.S. designed all figures and both authors wrote the manuscript.
Competing interests
We declare we have no competing interests.
Funding
S.S. expresses his gratitude to Her Majesty the Queen for providing the Commonwealth Scholarship to fund his MSc studies (CSC ref INCD-2018-121) and dedicates this in loving memory of his mother, Kalavathi.
References
- 1.Breasted J. 1930. The edwin smith surgical papyrus, published in facsimile and hieroglyphic transliteration With translation and commentary in Two volumes. Chicago, IL: University of Chicago, Oriental Institute. [Google Scholar]
- 2.Ebbell B. 1937. The papyrus ebers, The greatest Egyptian medical document, Translated By B. Ebbell. Copenhagen, Denmark: Levin & Munksgaard. [Google Scholar]
- 3.Galen, Linacre T, Siberch J. 1521. Galeni pergamensis De temperamentis, Et De inaequali intemperie libri tres. Impressum apud præclaram Cantabrigiam: Per Ioannem Siberch. Cambridge, UK: University of Cambridge. [Google Scholar]
- 4.Vesalius A. 1543a. De humani corporis Fabrica (The Fabric of the Human Body). Basel, Switzerland: Johannes Oporinus. [Google Scholar]
- 5.Henschen F. 1968. Yamagiwa's tar cancer and its historical significance. From Percival Pott to Katsusaburo Yamagiwa. Gan. 59, 447-451. [PubMed] [Google Scholar]
- 6.Boveri T. 1914. Zur frage der entstehung maligner tumoren. Jena, Germany: Gustav Fisher. [Google Scholar]
- 7.Waddington CH. 1957. The strategy of the genes. London, UK: George Allen & Unwin. [Google Scholar]
- 8.Waddington CH. 1942. Endeavour 1, 18-20. Reprinted in Int. J. Epidemiol. 2012; 41, 10–13. [Google Scholar]
- 9.Gurdon J. 1962. Adult frogs derived from the nuclei of single somatic cells. Dev. Biol. 4, 256-273. ( 10.1016/0012-1606(62)90043-X) [DOI] [PubMed] [Google Scholar]
- 10.Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663-676. ( 10.1016/j.cell.2006.07.024) [DOI] [PubMed] [Google Scholar]
- 11.Bachoo R, et al. 2002. Epidermal growth factor receptor and Ink4a/Arf. Cancer Cell 1, 269-277. ( 10.1016/S1535-6108(02)00046-6) [DOI] [PubMed] [Google Scholar]
- 12.Friedmann-Morvinski D, Bushong E, Ke E, Soda Y, Marumoto T, Singer O, Ellisman M, Verma I. 2012. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338, 1080-1084. ( 10.1126/science.1226929) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soda Y, et al. 2011. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl Acad. Sci. USA 108, 4274-4280. ( 10.1073/pnas.1016030108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng L, et al. 2013. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153, 139-152. ( 10.1016/j.cell.2013.02.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwitalla S, et al. 2013. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25-38. ( 10.1016/j.cell.2012.12.012) [DOI] [PubMed] [Google Scholar]
- 16.Vermeulen L, et al. 2010. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 12, 468-476. ( 10.1038/ncb2048) [DOI] [PubMed] [Google Scholar]
- 17.Jones S, et al. 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801-1806. ( 10.1126/science.1164368) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ischenko I, Zhi J, Moll U, Nemajerova A, Petrenko O. 2013. Direct reprogramming by oncogenic Ras and Myc. Proc. Natl Acad. Sci. USA 110, 3937-3942. ( 10.1073/pnas.1219592110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopp J, et al. 2012. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 22, 737-750. ( 10.1016/j.ccr.2012.10.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landsberg J, et al. 2012. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 490, 412-416. ( 10.1038/nature11538) [DOI] [PubMed] [Google Scholar]
- 21.Muller J, et al. 2014. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 5, 5712. ( 10.1038/ncomms6712) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsoi J, et al. 2018. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell 33, 890-904. ( 10.1016/j.ccell.2018.03.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwong L, et al. 2015. Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J. Clin. Invest. 125, 1459-1470. ( 10.1172/JCI78954) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mu P, et al. 2017. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355, 84-88. ( 10.1126/science.aah4307) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chandler E, et al. 2012. Implanted adipose progenitor cells as physicochemical regulators of breast cancer. Proc. Natl Acad. Sci. USA 109, 9786-9791. ( 10.1073/pnas.1121160109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, et al. 2020. Compression-induced dedifferentiation of adipocytes promotes tumor progression. Sci. Adv. 6, eaax5611. ( 10.1126/sciadv.aax5611) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gidekel S, Pizov G, Bergman Y, Pikarsky E. 2003. Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell 4, 361-370. ( 10.1016/S1535-6108(03)00270-8) [DOI] [PubMed] [Google Scholar]
- 28.Hochedlinger K, Yamada Y, Beard C, Jaenisch R. 2005. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 121, 465-477. ( 10.1016/j.cell.2005.02.018) [DOI] [PubMed] [Google Scholar]
- 29.Riggi N, et al. 2010. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 24, 916-932. ( 10.1101/gad.1899710) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarkar A, Hochedlinger K. 2013. The Sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell 12, 15-30. ( 10.1016/j.stem.2012.12.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shachaf C, et al. 2008. Genomic and proteomic analysis reveals a threshold level of MYC required for tumor maintenance. Cancer Res. 68, 5132-5142. ( 10.1158/0008-5472.CAN-07-6192) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halim S, Markert E, Vazquez A. 2018. Analysis of cell proliferation and tissue remodelling uncovers a KLF4 activity score associated with poor prognosis in colorectal cancer. Br. J. Cancer 119, 855-863. ( 10.1038/s41416-018-0253-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phelps R, et al. 2009. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell 137, 623-634. ( 10.1016/j.cell.2009.02.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tallman M, et al. 1997. All-trans-retinoic acid in acute promyelocytic leukemia. New Engl. J. Med. 337, 1021-1028. ( 10.1056/NEJM199710093371501) [DOI] [PubMed] [Google Scholar]
- 35.Strickland S, Mahdavi V. 1978. The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell 15, 393-403. ( 10.1016/0092-8674(78)90008-9) [DOI] [PubMed] [Google Scholar]
- 36.Wielenga M, et al. 2015. ER-stress-induced differentiation sensitizes colon cancer stem cells to chemotherapy. Cell Rep. 13, 489-494. ( 10.1016/j.celrep.2015.09.016) [DOI] [PubMed] [Google Scholar]
- 37.Mueller E, Sarraf P, Tontonoz P, Evans R, Martin K, Zhang M, Fletcher C, Singer S, Spiegelman B. 1998. Terminal differentiation of human breast cancer through PPARγ. Mol. Cell 1, 465-470. ( 10.1016/S1097-2765(00)80047-7) [DOI] [PubMed] [Google Scholar]
- 38.Ishay-Ronen D, et al. 2019. Gain fat—lose metastasis: converting invasive breast cancer cells into adipocytes inhibits cancer metastasis. Cancer Cell 35, 17-32.e6. ( 10.1016/j.ccell.2018.12.002) [DOI] [PubMed] [Google Scholar]
- 39.Lin S, Chou Y, Jiang S, Chang J, Chung C, Kao Y, Chang I, Wu C. 2016. Epigenetic switch between SOX2 and SOX9 regulates cancer cell plasticity. Cancer Res. 76, 7036-7048. ( 10.1158/0008-5472.CAN-15-3178) [DOI] [PubMed] [Google Scholar]
- 40.Kaufman C, et al. 2016. A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 351, 2197-2197. ( 10.1126/science.aad2197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J, Huang Z, Duan Y, Xiao X, Jiang J, Zhang R. 2012. Aberrant DNA methylation of P16, MGMT, and hMLH1 genes in combination with MTHFR C677 T genetic polymorphism and folate intake in esophageal squamous cell carcinoma. Asian Pac. J. Cancer Prev. 13, 5303-5306. ( 10.7314/APJCP.2012.13.10.5303) [DOI] [PubMed] [Google Scholar]
- 42.Chen J, et al. 2012b. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 45, 34-42. ( 10.1038/ng.2491) [DOI] [PubMed] [Google Scholar]
- 43.Blaschke K, et al. 2013. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500, 222-226. ( 10.1038/nature12362) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tee W, Reinberg D. 2014. Chromatin features and the epigenetic regulation of pluripotency states in ESCs. Development 141, 2376-2390. ( 10.1242/dev.096982) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boveri T. 1914. [Zur Frage Der Entstehung Der Malignen Tumoren.] Concerning the Origin of Malignant Tumours. Translated and annotated by Henry Harris. J. Cell Science 2008. 121, 1-84. ( 10.1242/jcs.025742) [DOI] [PubMed] [Google Scholar]
- 46.Feinberg A, Vogelstein B. 1983. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301, 89-92. ( 10.1038/301089a0) [DOI] [PubMed] [Google Scholar]
- 47.Holliday R. 1987. The inheritance of epigenetic defects. Science 238, 163-170. ( 10.1126/science.3310230) [DOI] [PubMed] [Google Scholar]
- 48.Greger V, Passarge E, Hopping W, Messmer E, Horsthemke B. 1989. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum. Genet. 83, 155-158. ( 10.1007/BF00286709) [DOI] [PubMed] [Google Scholar]
- 49.Saito Y, Liang G, Egger G, Friedman J, Chuang J, Coetzee G, Jones P. 2006. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9, 435-443. ( 10.1016/j.ccr.2006.04.020) [DOI] [PubMed] [Google Scholar]
- 50.Feinberg A, Ohlsson R, Henikoff S. 2006. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 7, 21-33. ( 10.1038/nrg1748) [DOI] [PubMed] [Google Scholar]
- 51.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan Wet al. 2009. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shlush L, et al. 2014. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506, 328-333. ( 10.1038/nature13038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dang L, et al. 2009. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739-744. ( 10.1038/nature08617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Figueroa M, et al. 2010. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553-567. ( 10.1016/j.ccr.2010.11.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turcan S, et al. 2012. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479-483. ( 10.1038/nature10866) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hark A, Schoenherr C, Katz D, Ingram R, Levorse J, Tilghman S. 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405, 486-489. ( 10.1038/35013106) [DOI] [PubMed] [Google Scholar]
- 57.Flavahan W, Drier Y, Liau B, Gillespie S, Venteicher A, Stemmer-Rachamimov A, Suvà M, Bernstein B. 2015. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110-114. ( 10.1038/nature16490) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verhaak R, et al. 2010. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98-110. ( 10.1016/j.ccr.2009.12.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hnisz D, et al. 2016. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454-1458. ( 10.1126/science.aad9024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marshall A, et al. 2017. CTCF genetic alterations in endometrial carcinoma are pro-tumorigenic. Oncogene 36, 4100-4110. ( 10.1038/onc.2017.25) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Serrano M. 1997. The tumor suppressor protein p16INK4a. Exp. Cell Res. 237, 7-13. ( 10.1006/excr.1997.3824) [DOI] [PubMed] [Google Scholar]
- 62.Jiao L, Zhu J, Hassan M, Evans D, Abbruzzese J, Li D. 2007. K-ras mutation and p16 and preproenkephalin promoter hypermethylation in plasma DNA of pancreatic cancer patients. Pancreas 34, 55-62. ( 10.1097/01.mpa.0000246665.68869.d4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kostaki M, et al. 2014. High-frequency p16INK4A promoter methylation is associated with histone methyltransferase SETDB1 expression in sporadic cutaneous melanoma. Exp. Dermatol. 23, 332-338. ( 10.1111/exd.12398) [DOI] [PubMed] [Google Scholar]
- 64.Robaina M, Faccion R, Arruda V, de Rezende L, Vasconcelos G, Apa A, Bacchi C, Klumb C. 2015. Quantitative analysis of CDKN2A methylation, mRNA, and p16INK4a protein expression in children and adolescents with Burkitt lymphoma: biological and clinical implications. Leuk. Res. 39, 248-256. ( 10.1016/j.leukres.2014.11.023) [DOI] [PubMed] [Google Scholar]
- 65.Bracken A. 2003. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 22, 5323-5335. ( 10.1093/emboj/cdg542) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bracken A, et al. 2007. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 21, 525-530. ( 10.1101/gad.415507) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim K, Roberts C. 2016. Targeting EZH2 in cancer. Nat. Med. 22, 128-134. ( 10.1038/nm.4036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Herranz N, et al. 2008. Polycomb complex 2 Is required for E-cadherin repression by the Snail1 transcription factor. Mol. Cell Biol. 28, 4772-4781. ( 10.1128/MCB.00323-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Von Burstin J, et al. 2009. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 137, 361-371, 371.e1–5. ( 10.1053/j.gastro.2009.04.004) [DOI] [PubMed] [Google Scholar]
- 70.Cesare A, Reddel R. 2010. Alternative lengthening of telomeres: models, mechanisms and implications. Nat. Rev. Genet. 11, 319-330. ( 10.1038/nrg2763) [DOI] [PubMed] [Google Scholar]
- 71.Schwartzentruber J, et al. 2012. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226-231. ( 10.1038/nature10833) [DOI] [PubMed] [Google Scholar]
- 72.Kim M, et al. 2001. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 7, 437-443. ( 10.1038/86507) [DOI] [PubMed] [Google Scholar]
- 73.Kim S, Jeong J, Park J, Lee J, Seo J, Jung B, Bae M, Kim K. 2007. Regulation of the HIF-1α stability by histone deacetylases. Oncol. Rep. 17, 647-651. [PubMed] [Google Scholar]
- 74.Sasagawa T, Jinno-Oue A, Nagamatsu T, Morita K, Tsuruga T, Mori-Uchino M, Fujii T, Shibuya M. 2020. Production of an anti-angiogenic factor sFLT1 is suppressed via promoter hypermethylation of FLT1 gene in choriocarcinoma cells. BMC Cancer 20, 112. ( 10.1186/s12885-020-6598-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Elias A, Siegelin M, Steinmuller A, von Deimling A, Lass U, Korn B, Mueller W. 2009. Epigenetic silencing of death receptor 4 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in gliomas. Clin. Cancer Res. 15, 5457-5465. ( 10.1158/1078-0432.CCR-09-1125) [DOI] [PubMed] [Google Scholar]
- 76.Guryanova O, et al. 2016. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat. Med. 22, 1488-1495. ( 10.1038/nm.4210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tessema M, Klinge D, Yingling C, Do K, Van Neste L, Belinsky S. 2010. Re-expression of CXCL14, a common target for epigenetic silencing in lung cancer, induces tumor necrosis. Oncogene 29, 5159-5170. ( 10.1038/onc.2010.255) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoshihama S, et al. 2016. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl Acad. Sci. USA 113, 5999-6004. ( 10.1073/pnas.1602069113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Duvel K, et al. 2010. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171-183. ( 10.1016/j.molcel.2010.06.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.García J, et al. 2004. Promoter methylation of the PTENgene is a common molecular change in breast cancer. Genes. Chromosomes Cancer 41, 117-124. ( 10.1002/gcc.20062) [DOI] [PubMed] [Google Scholar]
- 81.Schmitt A, et al. 2009. VHL inactivation is an important pathway for the development of malignant sporadic pancreatic endocrine tumors. Endocr. Relat. Cancer 16, 1219-1227. ( 10.1677/ERC-08-0297) [DOI] [PubMed] [Google Scholar]
- 82.Vanharanta S, et al. 2012. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat. Med. 19, 50-56. ( 10.1038/nm.3029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Esteller M, Avizienyte E, Corn P, Lothe R, Baylin S, Aaltonen L, Herman J. 2000. Epigenetic inactivation of LKB1 in primary tumors associated with the Peutz–Jeghers syndrome. Oncogene 19, 164-168. ( 10.1038/sj.onc.1203227) [DOI] [PubMed] [Google Scholar]
- 84.Place T, Fitzgerald M, Venkataraman S, Vorrink S, Case A, Teoh M, Domann F. 2011. Aberrant promoter CpG methylation is a mechanism for impaired PHD3 expression in a diverse set of malignant cells. PLoS ONE 6, 14617. ( 10.1371/journal.pone.0014617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ross J, Rand K, Molloy P. 2010. Hypomethylation of repeated DNA sequences in cancer. Epigenomics 2, 245-269. ( 10.2217/epi.10.2) [DOI] [PubMed] [Google Scholar]
- 86.Nishida J, Momoi Y, Miyakuni K, Tamura Y, Takahashi K, Koinuma D, Miyazono K, Ehata S. 2020. Epigenetic remodelling shapes inflammatory renal cancer and neutrophil-dependent metastasis. Nat. Cell Biol. 22, 465-475. ( 10.1038/s41556-020-0491-2) [DOI] [PubMed] [Google Scholar]
- 87.Huttenhower C, et al. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486, 207-214. ( 10.1038/nature11234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Allwood A, Walter M, Kamber B, Marshall C, Burch I. 2006. Stromatolite reef from the Early Archaean era of Australia. Nature 441, 714-718. ( 10.1038/nature04764) [DOI] [PubMed] [Google Scholar]
- 89.Butterfield N, Knoll A, Swett K. 1990. A bangiophyte red alga from the Proterozoic of arctic Canada. Science 250, 104-107. ( 10.1126/science.11538072) [DOI] [PubMed] [Google Scholar]
- 90.Backhed F, Ley R, Sonnenburg JL, Peterson DA, Gordon JI. 2005. Host–bacterial mutualism in the human intestine. Science 307, 1915-1920. ( 10.1126/science.1104816) [DOI] [PubMed] [Google Scholar]
- 91.Martin F, et al. 2007. A top-down systems biology view of microbiome–mammalian metabolic interactions in a mouse model. Mol. Syst. Biol. 3, 112. ( 10.1038/msb4100153) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Amieva M, Peek R. 2016. Pathobiology of Helicobacter pylori-induced gastric cancer. Gastroenterology 150, 64-78. ( 10.1053/j.gastro.2015.09.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Blaser M, Atherton J. 2004. Helicobacter pylori persistence: biology and disease. J. Clin. Invest. 113, 321-333. ( 10.1172/JCI20925) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen C, Xun P, Tsinovoi C, He K. 2017. Accumulated evidence on Helicobacter pylori infection and the risk of asthma. Annals of Allergy. Asthma Immunol. 119, 137-145. ( 10.1016/j.anai.2017.05.021) [DOI] [PubMed] [Google Scholar]
- 95.Kira J, Isobe N. 2019. Helicobacter pylori infection and demyelinating disease of the central nervous system. J. Neuroimmunol. 329, 14-19. ( 10.1016/j.jneuroim.2018.06.017) [DOI] [PubMed] [Google Scholar]
- 96.Perry S, et al. 2010. Infection with Helicobacter pylori is associated with protection against tuberculosis. PLoS ONE 5, e8804. ( 10.1371/journal.pone.0008804) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Piovani D, Danese S, Peyrin-Biroulet L, Nikolopoulos G, Lytras T, Bonovas S. 2019. Environmental risk factors for inflammatory bowel diseases: an umbrella review of meta-analyses. Gastroenterology 157, 647-659. ( 10.1053/j.gastro.2019.04.016) [DOI] [PubMed] [Google Scholar]
- 98.Khosravi Y, et al. 2016. Helicobacter pylori and gut microbiota modulate energy homeostasis prior to inducing histopathological changes in mice. Gut. Microbes 7, 48-53. ( 10.1080/19490976.2015.1119990) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.De Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, Plummer M. 2012. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 13, 607-615. ( 10.1016/S1470-2045(12)70137-7) [DOI] [PubMed] [Google Scholar]
- 100.Ki M, et al. 2014. Role of vacuolating cytotoxin VacA and cytotoxin-associated antigen CagA of Helicobacter pylori in the progression of gastric cancer. Mol. Cell. Biochem. 396, 23-32. ( 10.1007/s11010-014-2138-8) [DOI] [PubMed] [Google Scholar]
- 101.Hatakeyama M. 2014. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe 15, 306-316. ( 10.1016/j.chom.2014.02.008) [DOI] [PubMed] [Google Scholar]
- 102.Umeda M, Murata-Kamiya N, Saito Y, Ohba Y, Takahashi M, Hatakeyama M. 2009. Helicobacter pylori CagA causes mitotic impairment and induces chromosomal instability. J. Biol. Chem. 284, 22 166-22 172. ( 10.1074/jbc.M109.035766) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang F, Chan A, Rashid A, Wong D, Cho C, Yuen M. 2012. Helicobacter pylori induces promoter methylation of E-cadherin via interleukin-1β activation of nitric oxide production in gastric cancer cells. Cancer 118, 4969-4980. ( 10.1002/cncr.27519) [DOI] [PubMed] [Google Scholar]
- 104.Leung SY, Yuen ST, Chung LP, Chu KM, Chan AS, Ho JC. 1999. hMLH1 promoter methylation and lack of hMLH1 expression in sporadic gastric carcinomas with high-frequency microsatellite instability. Cancer Res. 59, 159-164. [PubMed] [Google Scholar]
- 105.Mackie R, Sghir A, Gaskins H. 1999. Developmental microbial ecology of the neonatal gastrointestinal tract. Am. J. Clin. Nutr. 69, 1035s-1045s. ( 10.1093/ajcn/69.5.1035s) [DOI] [PubMed] [Google Scholar]
- 106.Zimmer C, Dorea C. 2019. Enumeration of Escherichia coli in probiotic products. Microorganisms 7, 437. ( 10.3390/microorganisms7100437) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Payros D, et al. 2014. Maternally acquired genotoxic Escherichia coli alters offspring's intestinal homeostasis. Gut Microbes 5, 313-512. ( 10.4161/gmic.28932) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Arthur J, et al. 2012. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120-123. ( 10.1126/science.1224820) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vizcaino M, Crawford J. 2015. The colibactin warhead crosslinks DNA. Nat. Chem. 7, 411-417. ( 10.1038/nchem.2221) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nougayrede J. 2006. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313, 848-851. ( 10.1126/science.1127059) [DOI] [PubMed] [Google Scholar]
- 111.Bossuet-Greif N, Vignard J, Taieb F, Mirey G, Dubois D, Petit C, Oswald E, Nougayrède J. 2018. The colibactin genotoxin generates DNA interstrand cross-links in infected cells. mBio 9 e02393-17. ( 10.1128/mBio.02393-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee-Six H, et al. 2019. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532-537. ( 10.1038/s41586-019-1672-7) [DOI] [PubMed] [Google Scholar]
- 113.Pleguezuelos-Manzano C, et al. 2020. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 580, 269-273. ( 10.1038/s41586-020-2080-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Paget S. 1889. The distribution of secondary growths in cancer of the breast. Lancet 133, 571-573. ( 10.1016/S0140-6736(00)49915-0) [DOI] [PubMed] [Google Scholar]
- 115.Rous P. 1911. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 13, 397-411. ( 10.1084/jem.13.4.397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dolberg D, Bissell M. 1984. Inability of Rous sarcoma virus to cause sarcomas in the avian embryo. Nature 309, 552-556. ( 10.1038/309552a0) [DOI] [PubMed] [Google Scholar]
- 117.Fearon E, Vogelstein B. 1990. A genetic model for colorectal tumorigenesis. Cell 61, 759-767. ( 10.1016/0092-8674(90)90186-I) [DOI] [PubMed] [Google Scholar]
- 118.Wu S, Rhee K, Zhang M, Franco A, Sears C. 2007. Bacteroides fragilis toxin stimulates intestinal epithelial cell shedding and -secretase-dependent E-cadherin cleavage. J. Cell Sci. 120, 1944-1952. ( 10.1242/jcs.03455) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wick E, et al. 2014. Stat3 activation in murine colitis induced by enterotoxigenic Bacteroides fragilis. Inflamm. Bowel Dis. 20, 821-834. ( 10.1097/MIB.0000000000000019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wu S, et al. 2009. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016-1022. ( 10.1038/nm.2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yoo D, Ko S, Jung J, Kim Y, Kim J, Kim J. 2013. Bacteroides fragilis enterotoxin upregulates lipocalin-2 expression in intestinal epithelial cells. Lab. Invest. 93, 384-396. ( 10.1038/labinvest.2013.1) [DOI] [PubMed] [Google Scholar]
- 122.Skaar E. 2010. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 6, e1000949. ( 10.1371/journal.ppat.1000949) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Okuda T, Kokubu E, Kawana T, Saito A, Okuda K, Ishihara K. 2012. Synergy in biofilm formation between Fusobacterium nucleatum and Prevotella species. Anaerobe 18, 110-116. ( 10.1016/j.anaerobe.2011.09.003) [DOI] [PubMed] [Google Scholar]
- 124.Rubinstein M, Wang X, Liu W, Hao Y, Cai G, Han Y. 2013. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 14, 195-206. ( 10.1016/j.chom.2013.07.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nosho K. 2016. Association of Fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J. Gastroenterol. 22, 557. ( 10.3748/wjg.v22.i2.557) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gur C, et al. 2015. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42, 344-355. ( 10.1016/j.immuni.2015.01.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tomkovich S, et al. 2017. Locoregional effects of microbiota in a preclinical model of colon carcinogenesis. Cancer Res. 77, 2620-2632. ( 10.1158/0008-5472.CAN-16-3472) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bullman S, et al. 2017. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443-1448. ( 10.1126/science.aal5240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Marchesi J, Dutilh B, Hall N, Peters W, Roelofs R, Boleij A, Tjalsma H. 2011. Towards the human colorectal cancer microbiome. PLoS ONE 6, e20447. ( 10.1371/journal.pone.0020447) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jin J, Kitahara M, Sakamoto M, Hattori M, Benno Y. 2009. Slackia equolifaciens sp. nov., a human intestinal bacterium capable of producing equol. Int. J. Syst. Evol. Biol. 60, 1721-1724. ( 10.1099/ijs.0.016774-0) [DOI] [PubMed] [Google Scholar]
- 131.Chriett S, Dąbek A, Wojtala M, Vidal H, Balcerczyk A, Pirola L. 2019. Prominent action of butyrate over β-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 9, 742. ( 10.1038/s41598-018-36941-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dal Molin M, et al. 2015. Very long-term survival following resection for pancreatic cancer is not explained by commonly mutated genes: results of whole-exome sequencing analysis. Clin. Cancer Res. 21, 1944-1950. ( 10.1158/1078-0432.CCR-14-2600) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Riquelme E, et al. 2019. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 178, 795-806.e12. ( 10.1016/j.cell.2019.07.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Heja D, Kocsis A, Dobo J, Szilagyi K, Szasz R, Zavodszky P, Pal G, Gal P. 2012. Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proc. Natl Acad. Sci. USA 109,10 498-10 503. ( 10.1073/pnas.1202588109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Aykut B, et al. 2019. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 574, 264-267. ( 10.1038/s41586-019-1608-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shulzhenko N, Lyng H, Sanson G, Morgun A. 2014. Ménage à trois: an evolutionary interplay between human papillomavirus, a tumor, and a woman. Trends Microbiol. 22, 345-353. ( 10.1016/j.tim.2014.02.009) [DOI] [PubMed] [Google Scholar]
- 137.Łaniewski P, Barnes D, Goulder A, Cui H, Roe D, Chase D, Herbst-Kralovetz M. 2018. Linking cervicovaginal immune signatures, HPV and microbiota composition in cervical carcinogenesis in non-Hispanic and Hispanic women. Sci. Rep. 8, 7593. ( 10.1038/s41598-018-25879-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Martin D, Marrazzo J. 2016. The vaginal microbiome: current understanding and future directions. J. Infect. Dis. 214(Suppl 1), S36-S41. ( 10.1093/infdis/jiw184) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Conti C, Malacrino C, Mastromarino P. 2009. Inhibition of herpes simplex virus type 2 by vaginal lactobacilli. J. Physiol. Pharmacol. 60(Suppl 6), 19-26. [PubMed] [Google Scholar]
- 140.Tyssen D, et al. 2018. Anti-HIV-1 activity of lactic acid in human cervicovaginal fluid. mSphere 3, e00055-18. ( 10.1128/mSphere.00055-18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Graver M, Wade J. 2011. The role of acidification in the inhibition of Neisseria gonorrhoeae by vaginal lactobacilli during anaerobic growth. Ann. Clin. Microbiol. Antimicrob. 10, 8. ( 10.1186/1476-0711-10-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cadieux PA, Burton J, Devillard E, Reid G. 2009. Lactobacillus by-products inhibit the growth and virulence of uropathogenic Escherichia coli. J. Physiol. Pharmacol. 60(Suppl. 6), 13-18. [PubMed] [Google Scholar]
- 143.Mitra A, et al. 2015. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 5, 16865. ( 10.1038/srep16865) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Li H, Jia W. 2013. Cometabolism of microbes and host: implications for drug metabolism and drug-induced toxicity. Clin. Pharmacol. Ther. 94, 574-581. ( 10.1038/clpt.2013.157) [DOI] [PubMed] [Google Scholar]
- 145.Lida N, et al. 2013. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342, 967-970. ( 10.1126/science.1240527) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sivan A, et al. 2015. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084-1089. ( 10.1126/science.aac4255) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vetizou M, et al. 2015. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079-1084. ( 10.1126/science.aad1329) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rook G, Dalgleish A. 2011. Infection, immunoregulation, and cancer. Immunol. Rev. 240, 141-159. ( 10.1111/j.1600-065X.2010.00987.x) [DOI] [PubMed] [Google Scholar]
- 149.Oikonomopoulou K, Brinc D, Kyriacou K, Diamandis E. 2013. Infection and cancer: revaluation of the hygiene hypothesis. Clin. Cancer Res. 19, 2834-2841. ( 10.1158/1078-0432.CCR-12-3661) [DOI] [PubMed] [Google Scholar]
- 150.Zitvogel L, Tesniere A, Kroemer G. 2006. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat. Rev. Immunol. 6, 715-727. ( 10.1038/nri1936) [DOI] [PubMed] [Google Scholar]
- 151.D'Alessandro G, Antonangeli F, Marrocco F, Porzia A, Lauro C, Santoni A, Limatola C. 2020. Gut microbiota alterations affect glioma growth and innate immune cells involved in tumor immunosurveillance in mice. Eur. J. Immunol. 50, 705-711. ( 10.1002/eji.201948354) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jobert M. 1840. New treatment of cancer. Lancet 34, 65-112. ( 10.1016/S0140-6736(02)98476-X) [DOI] [Google Scholar]
- 153.Huang D, et al. 2014. Nerve fibers in breast cancer tissues indicate aggressive tumor progression. Medicine 93, e172. ( 10.1097/MD.0000000000000172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shao J, Wang B, Yao Y, Pan Z, Shen Q, Zhou J. 2016. Autonomic nervous infiltration positively correlates with pathological risk grading and poor prognosis in patients with lung adenocarcinoma. Thorac. Cancer 7, 588-598. ( 10.1111/1759-7714.12374) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Albo D, Akay C, Marshall C, Wilks J, Verstovsek G, Liu H, Agarwal N, Berger D, Ayala G. 2011. Neurogenesis in colorectal cancer is a marker of aggressive tumor behavior and poor outcomes. Cancer 117, 4834-4845. ( 10.1002/cncr.26117) [DOI] [PubMed] [Google Scholar]
- 156.Magnon C, Hall S, Lin J, Xue X, Gerber L, Freedland S, Frenette P. 2013. Autonomic nerve development contributes to prostate cancer progression. Science 341,1 236 361-1 236 361. ( 10.1126/science.1236361) [DOI] [PubMed] [Google Scholar]
- 157.Partecke L, et al. 2017. Subdiaphragmatic vagotomy promotes tumor growth and reduces survival via TNFα in a murine pancreatic cancer model. Oncotarget 8,22 501-22 512. ( 10.18632/oncotarget.15019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhao C, et al. 2014. Denervation suppresses gastric tumorigenesis. Sci. Transl. Med. 6, 250ra115. ( 10.1126/scitranslmed.3009569) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Saloman J, Albers K, Li D, Hartman D, Crawford H, Muha E, Rhim A, Davis B. 2016. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc. Natl Acad. Sci. USA 113, 3078-3083. ( 10.1073/pnas.1512603113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.James J, Mukouyama Y. 2011. Neuronal action on the developing blood vessel pattern. Semin. Cell Dev. Biol. 22, 1019-1027. ( 10.1016/j.semcdb.2011.09.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Thaker P, et al. 2006. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 12, 939-944. ( 10.1038/nm1447) [DOI] [PubMed] [Google Scholar]
- 162.Lillberg K. 2003. Stressful life events and risk of breast cancer in 10,808 women: a cohort study. Am. J. Epidemiol. 157, 415-423. ( 10.1093/aje/kwg002) [DOI] [PubMed] [Google Scholar]
- 163.Le C, et al. 2016. Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nat. Commun. 7, 10634. ( 10.1038/ncomms10634) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Partecke L, et al. 2016. Chronic stress increases experimental pancreatic cancer growth, reduces survival and can be antagonised by β-adrenergic receptor blockade. Pancreatology 16, 423-433. ( 10.1016/j.pan.2016.03.005) [DOI] [PubMed] [Google Scholar]
- 165.Zahalka A, Arnal-Estapé A, Maryanovich M, Nakahara F, Cruz C, Finley L, Frenette P. 2017. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 358, 321-326. ( 10.1126/science.aah5072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Liebl F, et al. 2014. The impact of neural invasion severity in gastrointestinal malignancies. Ann. Surg. 260, 900-908. ( 10.1097/SLA.0000000000000968) [DOI] [PubMed] [Google Scholar]
- 167.Bapat A, Munoz R, Von Hoff D, Han H. 2016. Blocking nerve growth factor signaling reduces the neural invasion potential of pancreatic cancer cells. PLoS ONE 11, e0165586. ( 10.1371/journal.pone.0165586) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Renz B, et al. 2018. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell 33, 75-90.e. ( 10.1016/j.ccell.2017.11.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Udumyan R, Montgomery S, Fang F, Almroth H, Valdimarsdottir U, Ekbom A, Smedby K, Fall K. 2017. Beta-blocker drug use and survival among patients with pancreatic adenocarcinoma. Cancer Res. 77, 3700-3707. ( 10.1158/0008-5472.CAN-17-0108) [DOI] [PubMed] [Google Scholar]
- 170.Mendez-Ferrer S, Lucas D, Battista M, Frenette P. 2008. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452, 442-447. ( 10.1038/nature06685) [DOI] [PubMed] [Google Scholar]
- 171.Arranz L, et al. 2014. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 512, 78-81. ( 10.1038/nature13383) [DOI] [PubMed] [Google Scholar]
- 172.Maryanovich M, et al. 2018. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 24, 782-791. ( 10.1038/s41591-018-0030-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gaudet A, Popovich P, Ramer M. 2011. Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J. Neuroinflammation 8, 110. ( 10.1186/1742-2094-8-110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Demir I, et al. 2014. Investigation of Schwann cells at neoplastic cell sites before the onset of cancer invasion. J. Natl Cancer Inst. 106, dju184. ( 10.1093/jnci/dju184) [DOI] [PubMed] [Google Scholar]
- 175.Geldof A, De Kleijn M, Ramanath Rao B, Don Newling W. 1997. Nerve growth factor stimulates in vitro invasive capacity of DU145 human prostatic cancer cells. J. Cancer Res. Clin. Oncol. 123, 107-112. ( 10.1007/BF01269888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lee R, Kermani P, Teng K, Hempstead B. 2001. Regulation of cell survival by secreted proneurotrophins. Science 294, 1945-1948. ( 10.1126/science.1065057) [DOI] [PubMed] [Google Scholar]
- 177.Pundavela J, et al. 2014. ProNGF correlates with gleason score and is a potential driver of nerve infiltration in prostate cancer. Am. J. Pathol. 184, 3156-3162. ( 10.1016/j.ajpath.2014.08.009) [DOI] [PubMed] [Google Scholar]
- 178.Bergles D, Roberts J, Somogyi P, Jahr C. 2000. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405, 187-191. ( 10.1038/35012083) [DOI] [PubMed] [Google Scholar]
- 179.Takano T, Lin J, Arcuino G, Gao Q, Yang J, Nedergaard M. 2001. Glutamate release promotes growth of malignant gliomas. Nat. Med. 7, 1010-1015. ( 10.1038/nm0901-1010) [DOI] [PubMed] [Google Scholar]
- 180.Venkatesh H, et al. 2019. Electrical and synaptic integration of glioma into neural circuits. Nature 573, 539-545. ( 10.1038/s41586-019-1563-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Venkataramani V, et al. 2019. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 573, 532-538. ( 10.1038/s41586-019-1564-x) [DOI] [PubMed] [Google Scholar]
- 182.Vogl C, Mochida S, Wolff C, Whalley B, Stephens G. 2012. The synaptic vesicle glycoprotein 2A ligand levetiracetam inhibits presynaptic Ca2+ channels through an intracellular pathway. Mol. Pharmacol. 82, 199-208. ( 10.1124/mol.111.076687) [DOI] [PubMed] [Google Scholar]
- 183.Izumoto S, Miyauchi M, Tasaki T, Okuda T, Nakagawa N, Nakano N, Kato A, Fujita M. 2018. Seizures and tumor progression in glioma patients with uncontrollable epilepsy treated with perampanel. Anticancer Res. 38, 4361-4366. ( 10.21873/anticanres.12737) [DOI] [PubMed] [Google Scholar]
- 184.Zeng Q, et al. 2019. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 573, 526-531. ( 10.1038/s41586-019-1576-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Niwinska A, Murawska M, Pogoda K. 2010. Breast cancer brain metastases: differences in survival depending on biological subtype, RPA RTOG prognostic class and systemic treatment after whole-brain radiotherapy (WBRT). Ann. Oncol. 21, 942-948. ( 10.1093/annonc/mdp407) [DOI] [PubMed] [Google Scholar]
- 186.Li L, Hanahan D. 2013. Hijacking the neuronal NMDAR signaling circuit to promote tumor growth and invasion. Cell 153, 86-100. ( 10.1016/j.cell.2013.02.051) [DOI] [PubMed] [Google Scholar]
- 187.Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. 2000. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101, 657-669. ( 10.1016/S0092-8674(00)80877-6) [DOI] [PubMed] [Google Scholar]
- 188.Fu Z, Washbourne P, Ortinski P, Vicini S. 2003. Functional excitatory synapses in HEK293 cells expressing neuroligin and glutamate receptors. J. Neurophysiol. 90, 3950-3957. ( 10.1152/jn.00647.2003) [DOI] [PubMed] [Google Scholar]
- 189.Danbolt N. 2001. Glutamate uptake. Prog. Neurobiol. 65, 1-105. ( 10.1016/S0301-0082(00)00067-8) [DOI] [PubMed] [Google Scholar]
- 190.Chen Q, et al. 2016. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493-498. ( 10.1038/nature18268) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wu Z, Zha Z, Li G, Lieberman B, Choi S, Ploessl K, Kung H. 2014. [18F](2S,4S)-4-(3-Fluoropropyl) glutamine as a tumor imaging agent. Mol. Pharm. 11, 3852-3866. ( 10.1021/mp500236y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Nakai A, Hayano Y, Furuta F, Noda M, Suzuki K. 2014. Control of lymphocyte egress from lymph nodes through β2-adrenergic receptors. J. Exp. Med. 211, 2583-2598. ( 10.1084/jem.20141132) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kamiya A, et al. 2019. Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat. Neurosci. 22, 1289-1305. ( 10.1038/s41593-019-0430-3) [DOI] [PubMed] [Google Scholar]
- 194.Hanahan D, Weinberg R. 2000. The hallmarks of cancer. Cell 100, 57-70. ( 10.1016/S0092-8674(00)81683-9) [DOI] [PubMed] [Google Scholar]
- 195.Mentch S, et al. 2015. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 22, 861-873. ( 10.1016/j.cmet.2015.08.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Honma Y, Araki T, Gianino S, Bruce A, Heuckeroth R, Johnson E, Milbrandt J. 2002. Artemin is a vascular-derived neurotropic factor for developing sympathetic neurons. Neuron 35, 267-282. ( 10.1016/S0896-6273(02)00774-2) [DOI] [PubMed] [Google Scholar]
- 197.Nishino J, et al. 1999. GFRα3, a component of the artemin receptor, is required for migration and survival of the superior cervical ganglion. Neuron 23, 725-736. ( 10.1016/S0896-6273(01)80031-3) [DOI] [PubMed] [Google Scholar]
- 198.Hochedlinger K. 2004. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev. 18, 1875-1885. ( 10.1101/gad.1213504) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sachs L. 1986. Hematopoietic growth and differentiation factors and the reversibility of malignancy: cell differentiation and by-passing of genetic defects in leukemia. Med. Oncol. Tumor Pharmacother. 3, 165-176. ( 10.1007/978-1-4612-4594-0_1) [DOI] [PubMed] [Google Scholar]
- 200.Lotem J, Sachs L. 2002. Epigenetics wins over genetics: induction of differentiation in tumor cells. Semin. Cancer Biol. 12, 339-346. ( 10.1016/S1044-579X(02)00054-8) [DOI] [PubMed] [Google Scholar]
- 201.Holst CR, et al. 2003. Methylation of p16INK4a promoters occurs in vivo in histologically normal human mammary epithelia. Cancer Res. 63, 1596-1601. [PubMed] [Google Scholar]
- 202.Crawford Y, Gauthier M, Joubel A, Mantei K, Kozakiewicz K, Afshari C, Tlsty T. 2004. Histologically normal human mammary epithelia with silenced p16INK4a overexpress COX-2, promoting a premalignant program. Cancer Cell 5, 263-273. ( 10.1016/S1535-6108(04)00023-6) [DOI] [PubMed] [Google Scholar]
- 203.Hu M, Yao J, Cai L, Bachman K, van den Brûle F, Velculescu V, Polyak K.. 2005. Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet. 37, 899–905.. ( 10.1038/ng1596) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.