

Summary
Since the discovery of human induced pluripotent stem cells (hiPSCs), numerous strategies have been established to efficiently derive cardiomyocytes from hiPSCs (hiPSC-CMs). Here, we describe a cost-effective strategy for the subsequent massive expansion (>250-fold) of high-purity hiPSC-CMs relying on two aspects: removal of cell-cell contacts and small-molecule inhibition with CHIR99021. The protocol maintains CM functionality, allows cryopreservation, and the cells can be used in downstream assays such as disease modeling, drug and toxicity screening, and cell therapy.
For complete details on the use and execution of this protocol, please refer to Buikema (2020).
Subject areas: Cell culture, Cell differentiation, Stem cells
Graphical Abstract
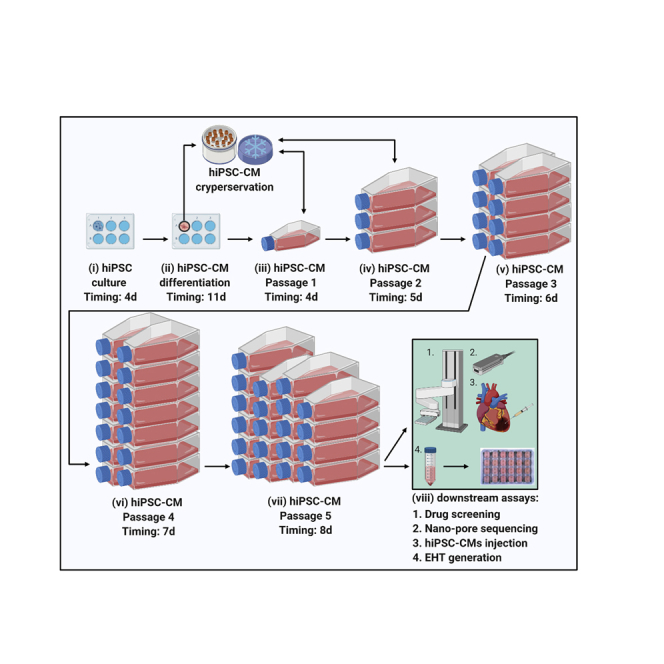
Highlights
-
•
Cost-effective strategy for the massive expansion (>250-fold) of high-purity hiPSC-CMs
-
•
Removing cell-cell contacts and applying CHIR99021 stimulate massive cardiomyocyte (CM) expansion
-
•
Cryopreservation of juvenile CMs allows biobanking of many hiPSC lines
Since the discovery of human induced pluripotent stem cells (hiPSCs), numerous strategies have been established to efficiently derive cardiomyocytes from hiPSCs (hiPSC-CMs). Here, we describe a cost-effective strategy for the subsequent massive expansion (>250-fold) of high-purity hiPSC-CMs relying on two aspects: removal of cell-cell contacts and small-molecule inhibition with CHIR99021. The protocol maintains CM functionality, allows cryopreservation, and the cells can be used in downstream assays such as disease modeling, drug and toxicity screening, and cell therapy.
Before you begin
Timing: 30 min
In a sterile hood, dilute Matrigel to a concentration of 1.2 mg/mL in 4°C RPMI-1640 in a 50-mL tube on ice. Prepare aliquots of 1 mL in 15-mL tubes on ice and store at −20°C. Remove one Matrigel aliquot (1 mL) from the freezer, and add 11 mL of cold RPMI-1640 to a final concentration of 0.1 mg/mL (empirically up to 5× lower concentrations of Matrigel are working). Gently mix the Matrigel solution to thaw and dissolve the Matrigel. Immediately add 1 mL/well Matrigel in RPMI-1640 for 6-well plates, 1.5 mL/flask for T25, 2.5 mL/flask for T75, or 5 mL/flask for T175 flasks. Allow the Matrigel to set for 30 min at 37°C before use. The Matrigel-coated plates and flasks can be stored at 4°C for up to 3 weeks.
Note: Keep Matrigel on ice to avoid solidification during the coating procedure. Boxes with pipet tips can be pre-chilled at −20°C.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-α-actinin, primary mouse monoclonal antibody | Merck | A7811 |
| Anti-Ki67, primary rabbit polyclonal antibody | Abcam | AB833 |
| Goat anti-mouse Alexa Fluor 488 | Invitrogen | A11029 |
| Goat anti-rabbit Alexa Fluor 568 | Invitrogen | A11011 |
| Isotype control, FITC mouse IgM κ isotype | BD | 556652 |
| Chemicals, peptides, and recombinant proteins | ||
| B-27 supplement minus insulin (B27-insulin) | Thermo Fisher | A1895601 |
| B-27 supplement plus insulin (B27+insulin) | Thermo Fisher | 17504-044 |
| Bovine serum albumin fraction V | Roche | 10735086001 |
| CHIR-99021 | Selleckchem | S2924 |
| EDTA | Thermo Fisher | 15575020 |
| Essential 8 (E8) medium | Thermo Fisher | A1517001 |
| Paraformaldehyde solution 4% in 1× PBS, pH 7.0–7.6 | Santa Cruz | SC281692 |
| PBS, pH 7.4 | Thermo Fisher | 10010072 |
| KnockOut (KO) serum replacement | Thermo Fisher | 10828 |
| Matrigel, growth factor reduced, basement membrane matrix | Corning | 356230 |
| Penicillin/streptomycin | Thermo Fisher | 15140 |
| Triton X-100 | Merck | X100-1L |
| TrypLE select enzyme (10×) | Thermo Fisher | A12177 |
| Trypan blue solution, 0.4% | Thermo Fisher | 15250061 |
| RevitaCell | Thermo Fisher | A2644501 |
| ROCK inhibitor Y-27632 | Biorbyt | orb60104 |
| RPMI 1640 medium | Thermo Fisher | 11875 |
| RPMI 1640 medium no glucose | Thermo Fisher | 11879020 |
| STEMdiff cardiomyocyte freezing medium | STEMCELL Technologies | 05030 |
| Wnt-C59 | Tocris | 5148 |
| Experimental models: cell lines | ||
| Human induced pluripotent stem cell lines | (Stanford Cardiovascular Institute (S-CVI) Biobank) | CVI-111 (control 1) |
| Human induced pluripotent stem cell lines | (Stanford Cardiovascular Institute (S-CVI) Biobank) | CVI-114 (control 2) |
| Human induced pluripotent stem cell lines | (Stanford Cardiovascular Institute (S-CVI) Biobank) | CVI-273 (control 3) |
| Other | ||
| 6-well cell culture plates | Greiner | 657160 |
| T25 cell culture flasks | Greiner | 690170 |
| T75 cell culture flasks | Greiner | 658170 |
| T175 cell culture flasks | Greiner | 660160 |
| 15 mL centrifuge tubes | Greiner | 188280 |
| CoolCell cell freezing container | n/a | n/a |
| Cryovials | VWR | 479-6846 |
| Counting slides | n/a | n/a |
| BSL2 biosafety cabinet | n/a | n/a |
| Cell counter | n/a | n/a |
| Cell culture incubator (37°C, 5% CO2, 21% O2) | n/a | n/a |
| 37°C water bath | n/a | n/a |
Materials and equipment
-
•
hiPSC E8 complete medium: In a sterile environment, mix E8 basal medium, E8 supplement (2%, vol/vol) and penicillin/streptomycin (1%, vol/vol).
Note: We recommend to aliquot the E8 supplement (−20°C) and medium (4°C) and prepare weekly aliquots of the complete medium by pre-warming the required amount. The basal medium can be stored at −20°C for up to 1 year. After preparation, hiPSC E8 complete medium can be stored at 4°C for up to 1 month.
-
•
10 mM stock Y27632 (Y): In a sterile environment, add 31.2246 mL DMSO to 100 mg Y27632.
Note: Prepare aliquots of 2–10 μL and store at −80°C for up to 6 months. After thawing the aliquot can be stored at 4°C for 2 weeks.
-
•
E8 + 5 μM Y-27632: In a sterile environment, add 0.05% Y-27632 (1:2,000 dilution of the 10 mM stock) to the hiPSC E8 complete medium.
Note: After preparation, this medium can be stored at 4°C for up to 1 week.
-
•
40 mM stock CHIR99021 (CHIR): In a sterile environment, add 1.2455 mL DMSO to 25 mg CHIR.
Note: Prepare aliquots of 2–10 μL and store at −80°C for up to 1 year. Avoid re-freezing of the aliquots.
-
•
CM differentiation medium (B27): In a sterile environment, mix RPMI 1640, 2% B-27 minus insulin (1:50) and 1% penicillin/streptomycin (1:100).
-
•
CM differentiation medium + 7 μM CHIR: In a sterile environment, add 0.0267% CHIR (1:3,750 dilution of the 40 mM stock) into CM differentiation medium.
Note: After preparation, do not store this medium to avoid repeated freeze-thaw cycles of CHIR.
-
•
CM differentiation medium + 8 μM CHIR: In a sterile environment, add 0.02% CHIR (1:5,000 dilution of the 40 mM CHIR stock) into CM differentiation medium.
Note: After preparation, do not store this medium to avoid repeated freeze-thaw cycles of CHIR.
-
•
Wnt-C59 (20 mM): In a sterile environment, add 1302.2 μL DMSO to 10 mg Wnt-C59.
Note: Prepare aliquots of 50 μL format and store at −80°C for up to 2 years. Avoid re-freezing of the aliquots.
-
•
CM differentiation medium + 2 μM Wnt-C59: In a sterile environment, add 0.01% Wnt-C59 (1:10.000 dilution of the 20 mM Wnt-C59 stock) into CM differentiation medium.
Note: After preparation, do not store this medium to avoid repeated freeze-thaw cycles of Wnt-C59.
-
•
CM culture medium (CM): In a sterile environment mix RPMI 1640, 2% B-27 supplement (1:50) and 1% penicillin/streptomycin (1:100).
-
•
CM purification medium (PM): In a sterile environment mix RPMI 1640 no glucose, 2% B-27 supplement (1:50) and 1% penicillin/streptomycin (1:100).
-
•
CM replating medium (RM): In a sterile environment, add 0.1% Y-27632 (1:1,000 dilution of the 10 mM stock) and 10% KO serum replacement (1:10) into CM culture medium.
Note: After preparation, all cardiac culture mediums can be stored at 4°C for up to 1 month.
-
•
CM expansion medium (EM) (with 2 μM CHIR): In a sterile environment, add 0.005% CHIR (1:20,000 dilution of the 40 mM CHIR stock) into CM culture medium.
Note: After preparation, we do not recommend storing this medium.
-
•
CM splitting medium (SM) (with 2 μM CHIR): In a sterile environment, add 0.005% CHIR (1:20,000 dilution of the 40 mM CHIR stock), 0.1% Y-27632 (1:1,000 dilution of the 10 mM stock), and 10% KO serum replacement (1:10) replacement into CM culture medium.
Note: After preparation, we do not recommend storing this medium.
-
•
CM thawing medium (TM): In a sterile environment, add 1% Revitacell (1:100) and 10% KO serum replacement (1:10) into CM culture medium. Protect medium from light.
CRITICAL: Revitacell is light-sensitive. Protect Revitacell from light.
Note: After preparation, this medium can be stored at 4°C for up to 1 week.
Note: We recommend to prepare aliquots of 100–500 μL Revitcell and store at −20°C for up to 1 year.
-
•
Permeabilization and blocking buffer: This buffer contains PBS, 5% BSA and 0.3% Triton X-100.
-
•
Flow cytometry buffer: This buffer contains 50 mL of PBS and 1% BSA and 0.3% Triton X-100.
-
•
Wash buffer: This buffer contains 50 mL of PBS and 1% BSA.
Note: After preparation, all buffers can be stored at 4°C for up to 1 month.
Step-by-step method details
hiPSC culturing
The hiPSCs are maintained under feeder-free conditions with daily medium changes and kept at 37°C, 5% CO2, 21% O2. When 80%–90% confluent, the hiPSC can be passaged (usually about 3 to 4 days after passaging).
-
1.
Prewarm E8 medium, EDTA Dissociation Buffer, and Matrigel-coated plates to 20°C–25°C.
-
2.
Aspirate the medium, and add 1 mL 0.5 mM EDTA/PBS to each well. At this point, 2.5 to 3 million cells should be present in each well of the 6-well culture plate.
-
3.
Incubate the plate for 3–5 min.
-
4.
Aspirate the EDTA and flush the hiPSC with a 1 mL tip filled with 1 mL E8 + 5 μM Y27632, dispense the medium over the surface of the plate well until all the cells are detached and gently mix 2–5 times using the 1 mL tip.
-
5.
Add the required cell amount from the 1 mL cell suspension into each well of a Matrigel-coated plate containing 2 mL E8 + 5 μM Y27632 (split ratio of 1:13 is performed here).
-
6.
Place the plate in the incubator and make side-to-side movements to equally disperse the cells in every well.
-
7.
Prewarm hiPSC E8 Medium to 20°C–25°C.
-
8.
Each day aspirate the medium and add 2 mL of fresh prewarmed hiPSC E8 complete medium per well.
-
9.
When the hiPSCs are 80%–90% confluent they are ready to start the differentiation. At this point, equal dispersed hiPSC colonies should be present in each well.
Note: Karyotyping of the hiPSC lines is advised. Thereafter, hiPSC can be safely passaged for 20 times before another karyotyping is advised. Passaging over 40 times could lead in spontaneous differentiation of the hiPSC lines, resulting in inefficient differentiations.
Pause point: The hiPSC can be maintained in hiPSC E8 complete medium and passaged every 3–4 days before starting differentiation and hiPSC-CM expansion.
hiPSC-CM differentiation
The cardiac differentiation of hiPSCs can be successfully performed by the steps described below. The cultures of cardiomyocytes typically contract by day 8–9 of differentiation before starting the expansion on day 11 (see Methods Video S1).
-
10.
Day 0: Prepare 3 mL/well of 37°C-preheated CM differentiation medium with the appropriate concentration of CHIR per hiPSC line. Aspire the E8 medium and replace carefully with 3 mL/well with the prepared differentiation medium. For every differentiation apply two concentrations of CHIR for an optimal window concentration per differentiation per plate.
Note: Other differentiation protocols can be used to generate hiPSC-CMs before the expansion. Here, we have used the B27 (Lian, 2012), Heparin (Lin, 2017), and CDM3 (Burridge, 2014) medium, which were all sufficient for the massive expansion of hiPSC-CMs.
Note: Though we identified 7 and 8 μM CHIR in the B27 protocol as the optimal concentration for the three lines that we tested, other lines may respond to CHIR treatment differently. We recommend testing concentrations of 2–8 μM of CHIR in heparin or CDM3 medium and 5–10 μM of CHIR in B27 medium differentiations.
-
11.
Day 1: On top of the original medium, carefully add 2 mL/well of 37°C-preheated CM differentiation medium. Put the plate back into the 37°C, 5% CO2, 21% O2 incubator.
-
12.
Day 2: On top of the original medium, carefully add 1 mL/well of 37°C-preheated CM differentiation medium. Put the plate back into the 37°C, 5% CO2, 21% O2 incubator.
Note: Here we use the addition of medium on day 1 and day 2 to the original medium of step 10. This protocol is effective to gradual decrease the CHIR concentration, mimicking the first steps of embryonic heart development. However, other differentiation protocols such as 2 days of CHIR medium is also possible to generate hiPSC-CMs for the CM expansion.
-
13.
Day 3: Aspirate the old medium and carefully add 3 mL/well of 37°C-preheated CM differentiation medium with 2 μM Wnt-C59 to each well of the 6-well plate. Put the plate back into the 37°C, 5% CO2, 21% O2 incubator.
Note: During differentiation, the cell death rate is high. Remove dead cell debris as much as possible by shaking and aspirating the medium completely.
-
14.
Day 5: Aspirate the medium and carefully add 3 mL/well of 37°C-preheated CM differentiation medium. Put the plate back into the 37°C, 5% CO2, 21% O2 incubator.
-
15.
Day 7: Aspirate the medium and carefully add 3 mL/well of 37°C-preheated CM culture medium. Put the plate back into the 37°C, 5% CO2, 21% O2 incubator. Robust spontaneous contraction should occur by day 8–9.
-
16.
Day 9: Aspirate the medium and add 2 mL/well of 37°C-preheated CM purification medium (PM). Put the plate back into the 37°C, 5% CO2, 21% O2 incubator.
-
17.
Day 11: The cardiomyocytes are now ready for cardiac expansion.
Note:Methods Video S1 shows a typical spontaneous contracting monolayer at day 11.
Replating of differentiated hiPSC-CMs
Replating of hiPSC-derived cardiomyocytes by enzymatic digestion can be performed in order to plate cells into desired culture vessel formats at defined cell numbers if required (e.g., in lower cell density for the maximum expansion capacity; Table 1). Between day 10 and day 14 the hiPSC-derived cardiomyocytes can be used for most successful cardiac expansion. Here, we use monolayers with spontaneous contracting cells on day 11 (Methods Video S1). From a 6 well plate format, select the most optimal well(s) by microscopic observe the amount of beating cardiomyocytes.
-
18.
Aspirate the medium, wash the cells with PBS and incubate hiPSC-CMs with pure TrypLE Select (10×) for 15–45 min at 37°C (0.75 mL of TrypLE per 9.6-cm2 growth surface).
Note: Use the pure TrypLE Select (10×) for efficient dissociation of the hiPSC-CMs. Other reagents such as TrypLE Express or Trypsin will result in an inefficient dissociation and low cell viability.
-
19.
After incubation for ∼15 min, gently shake to detach the hiPSC-CMs. If cells do not detach yet, then repeat incubation for 1 or 2 times (up to 45 min). When cells come off the culture plate, add 1.5 mL per 75-cm2 warm RPMI-1640 per flask and dispense over the surface of the plate until all the cells are detached and gently mix 2–5 times using the 5 mL pipette.
Note: Premature dissociation by pipetting results in shear stress-induced cell death, therefore shaking and longer incubations with TrypLE Select (10×) is advised.
-
20.
Transfer the cell suspension to a 15-mL tube.
-
21.
Centrifuge the cell suspension at 200 × g for 3 min.
-
22.
Aspirate the supernatant and resuspend the cell suspension in 1 mL/flask of 37°C-preheated of the CM replating medium.
-
23.
Count the cell suspension by mixing 10 μL cell suspension with 1:1 Trypan Blue Solution in a counting slide and by using a cell counting machine.
-
24.
Re-plate passage 1 (P1) of hiPSC-CMs in the culture system of choice in a culture surface split ratio of 1:10–20 and add 37°C-preheated CM replating medium (RM) in the volume according to Table 1.
Note: If animal-free and chemical defined is not desired, the knockout serum can be replaced with 10% FBS in the replating medium.
-
25.
Remove the Matrigel coating from the flasks and immediately add the cell solution to the prepared culture flasks/plates.
-
26.
Put the flask into the incubator and distribute the cells by moving the flask in short side-to-side and back-and-forth motions. Incubator conditions should be set to 37°C, 5% CO2, 21% O2, and 90% humidity. Proceed to step 27.
Note:Methods Videos S2 shows a typical morphology of passage 1 after 24 h.
Table 1.
Key culture format cultivation information
| steps | Culture format | Culture formats # | Start amount of cells (104/cm2) | Total amount of cells after each step | Volume medium/format | Timing (days) |
|---|---|---|---|---|---|---|
| hiPSC culturing | 6 wells plate | 1 well | ~2 | ~300 | 2 mL per wells | 4 |
| Cardiac differentiation | 6 wells plate | 1 well | ~30 | ~700 | 3 mL per wells | 11 |
| Passage 1 | T75 flask | 1× | ~2.5 | ~2,100 | 7 mL per T75 flask | ~4 |
| Passage 2 | T175 flask | 3× | ~4 | ~7,000 | 21 mL per T175 flask | ~5 |
| Passage 3 | T175 flask | 8× | ~5 | ~20,000 | 21 mL per T175 flask | ~6 |
| Passage 4 | T175 flask | 14× | ~8 | ~35,000 | 21 mL per T175 flask | ~7 |
| Passage 5 | T175 flask | 20× | ~10 | ~5,000 | 21 mL per T175 flask | ~8 |
hiPSC-CM expansion
The proliferation of hiPSC-CMs can be conducted by the removal of cell-cell contacts and small-molecule inhibition with CHIR99021. The expansion protocol comprise a series of medium changes and splitting as illustrated in the graphical abstract and Figure 2, in between which the cells are kept at 37°C, 5% CO2, 21% O2. The hiPSC-cardiomyocytes can be successfully split for 5 passages with a similar juvenile CM morphology (Figure 1) and contraction (see Methods Videos S2 and S3).
-
27.
Day 1: After attachment and recovery of passage 1 (P1) hiPSC-CM for 24 h start the expansion process by replacing the replating medium with 37°C-preheated cardiac expansion medium according to Table 1 every other day or once every three days. For most cell lines 2 μM CHIR induces efficient expansion of hiPSC-CMs (Methods Video S3).
Note: The protocol for the generation of hiPSC-CMs can be variable. Here, the differentiation methods; B27 (Lian, 2012), Heparin (Lin, 2017), and CDM3 (Burridge, 2014) are used to produce CMs, which can be efficiently expanded (see Figure 1).
-
28.
After 3–5 days, the hiPSC-CMs will reach 70%–80% confluency and should be passaged to inhibit cell-cell contact. Aspirate the medium, wash with PBS. Add 3 mL of 37°C warm TrypLE per 75-cm2growth surface and incubate the hiPSC-CMs for 10–15 min at 37°C.
-
29.
After incubation for ∼15 min, detach the hiPSC-CMs with a 5 or 10 mL tip filled with 6 mL per 75-cm2 37°C-preheated RPMI-1640 and dispense over the surface of the plate well until all the cells are detached and gently mix 2–5 times using the 5 mL pipette.
Note: The dissociation of expanding hiPSC-CMs will be more quickly and easier than the dissociation of d11 differentiated hiPSC-CMs, which sometimes requires prolonged incubation with TrypLE Select (10×).
-
30.
Transfer the cell suspension to a new 15 mL tube.
-
31.
Centrifuge the cell suspension at 200 × g for 3 min.
-
32.
Aspirate the supernatant and resuspend the cell suspension in 1 mL/tube 37°C-preheated of the CM splitting medium supplemented with optimal CHIR concentration.
-
33.
Count the cell suspension.
Note: If cell numbers are high per tube, resuspend hiPSC-CMs in a larger amount of medium before counting the cell suspension (1 mL per 10 million).
-
34.
Add 37°C-preheated CM splitting medium to the hiPSC-CMs, in the density and volume according to Table 1.
-
35.
Remove the Matrigel solution from new flasks and immediately add the cell solution to the prepared culture flasks/plates.
-
36.
Put the flask into the incubator and distribute the cells in the well by moving the flask in short side-to-side and back-and-forth motions. Incubator conditions should be set to 37°C, 5% CO2, 21% O2, and 90% humidity.
-
37.
Refresh the expansion medium every other day in the volume according to Table 1. After 5–8 days, the hiPSC-CMs will reach 70%–80% confluency and should be passaged to inhibit cell-cell contact. Repeat steps 28–36 for passage 2–5. By every passage, split the hiPSC-CMs in a splitting ratio and flasks according to Table 1.
Note: By higher passage numbers hiPSC-CMs proliferation gradually slows down and further passaging then P4–5 usually does not result in increased cell numbers.
Figure 2.

Visualized timeline of hiPSC-CM expansion
(A) Timeline diagram displaying the steps required for expansion and passaging of hiPSC-CMs.
(B) Time-lapse images of cell morphology at the indicated passage numbers and/or culture days. Left: Images of cell confluency on day of passaging for P0-5. Right: Daily consecutive images of P1 hiPSC-CMs demonstrating cell seeding density and growth speed between P1 and P2. Replating at day 15, start of CHIR9021 (CHIR) treatment at day 16, cell cluster formation at day 14–19 is observed the days after and a nearly confluent monolayer before passaging at D18–20. Scale bar, 200 μm. D, day; P, passage.
Figure 1.

Massive expansion of hiPSC-CMs from various differentiation methods
(A) Schematic representation of Wnt-based directed cardiac differentiation and subsequent expansion of hiPSC-CMs generated by three different medium compositions ((B27; Lian, 2012; Heparin; Lin, 2017, and CDM3; Burridge, 2014). Timeline indicating B27-medium with insulin and additional CHIR99021 (CHIR) usage required for expansion. CM, CM culture medium; PM, CM purification medium; RM, CM replating medium; EM, CM expansion medium; SM, CM splitting medium; P, passage.
(B) Graph displaying the hiPSC-CM expansion capacity from hiPSC-CMs generated by the indicated differentiation method. One hiPSC line was used for these experiments. n = 7, n = 2, n = 2 respectively from P0 day 11 to P5. Data are represented as mean ± SD.
(C) Bright-field images at different time points of hiPSC-CM expansion with the input of the indicated differentiation methods. Scale bar, 200 μm.
Figure 4.

Differentiation efficiency and expansion capacity in different control hiPSC lines
(A) Quantitative analysis of small-molecule-based Wnt-modulated directed cardiac differentiation at the passage (P) number P1 and P5. Replicate numbers refer to the number of performed expansion experiments from individual differentiation batches. The percentages of increase in hiPSC-CMs are relative to the input of day 11 hiPSC-CMs. Mean values ± SD are given.
(B) Representative gating strategy for α-actinin positive hiPSC-CMs in a pure population versus negative control, isotype control, and an impure or un-purified hiPSC-CM culture. The number of α-actinin positive analyzed cells is 25 × 105. SSC, side scatter.
(C) Representative immunofluorescence for proliferation assessed by Ki67 expression after 48 h of CHIR treatment. Immunofluorescence: Hoechst (blue), Ki67 (red), and α-actinin (green). Scale bar, 200 μm.
(D) Quantitative graph indicates high proliferation (37%) of cardiomyocytes stimulated with the optimal dose of 2 μM CHIR.
(Please see Figure 2 for representative morphologies during hiPSC-CM passaging)
Note:Methods Video S3 shows a typical morphology of passage 4 after expansion.
-
38.
After substantial expansion of the CM population, CHIR withdrawal from the medium results in the decrease of hiPSC-CMs proliferation, and subsequent regular CM culture medium change intervals can be extended for long-term culturing or downstream assays.
Figure 3.

Cryopreservation and subsequent expansion of beating hiPSC-CMs
(A) Schematic representation of small-molecule-based Wnt-modulated directed cardiac differentiation and subsequent cryopreservation and expansion. hiPSC-CMs can be stored in liquid nitrogen after passage 1–2 and after thawing expanded for 5 passages (P) before downstream assays. TM, CM thawing medium; EM, CM expansion medium; SM, CM splitting medium; P, passage.
(B) Left panels are flow cytometer detection of dead cells with propidium iodide (PI) after detaching or after thawing of cardiomyocytes, cryopreserved on day 15. Right is the quantification of fresh versus thawed PI % (n = 5 per condition, p < 0.05, paired t test.). Data are represented as mean ± SD.
(C) Representative images of the thawed hiPSC-CMs before starting the expansion protocol and during passaging (scale bar, 100 μm).
(D) Quantification of cardiomyocyte expansion curve of fresh versus thawed hiPSC-CMs, n = 7, n = 5 respectively from thawed P1 to P5. Data are represented as mean ± SD.
Freezing of hiPSC-derived CMs
hiPSC-derived cardiomyocytes can be cryopreserved for long-term storage. To ensure pure populations and viable populations after thawing the cells should ideally be frozen after having passed in the expansion protocol. Cryopreservation can be performed from day 15 after the start of differentiation, after passage 1 or after passage 2 (data not shown). After the later passages, the viability of the hiPSC-CMs is less viable (Figure 3).
Note: Volumes are given for one cryovial.
-
39.
Count the CMs after the dissociation as described in the Replating of differentiated hiPSC-CMs section.
-
40.
Transfer the desired cell suspension for freezing to a 15-mL tube.
-
41.
Centrifuge the cell suspension at 200 × g for 3 min.
-
42.
Remove the supernatant and resuspend the CM in STEMdiff Cardiomyocyte Freezing Medium. Use 0.5 mL medium for 0.5–2 × 106 CMs and 1 mL of freezing medium for 2–8 × 106 CMs per vial.
Note: Keep the cell suspension on ice.
-
43.
Freeze the vials at −80°C for minimal 4 h and maximal 12 in a CoolCell or 2-propanol-filled freezing container.
-
44.
Transfer the vials to liquid nitrogen or −150°C for long-term storage.
Thawing of hiPSC-derived CMs
Cryopreserved hiPSC-derived cardiomyocytes can be thawed and recovered into culture as described below.
-
45.
Prepare 9 mL of 37°C-preheated RPMI medium in a 15-mL tube on RT.
-
46.
Prepare a Matrigel-coated T75 per 3–6 × 106 or a T175 for 7–14 × 106 hiPSC-CMs.
-
47.
Collect the vial with hiPSC-CMs from the liquid nitrogen and thaw the cells at 37°C till only a little clump is visible. Do not thaw more than three vials of CMs at one time.
-
48.
Transfer the cell suspension to the 15-mL tube, drop by drop, and swirl the tube while adding the cell suspension. The dropwise addition of medium to the cell suspension is critical to minimize osmotic shock.
-
49.
Centrifuge the cell suspension at 200 × g for 3 min.
-
50.
Remove the supernatant and resuspend the hiPSC-CMs in the desired volume of CM thawing medium.
-
51.
Count the cells within the cell suspension.
-
52.
Aspirate the liquid from the Matrigel-coated flasks, and seed the CMs into the Matrigel-coated flasks, according to the format amount in Table 1.
-
53.
Put the flask into the incubator and distribute the cells in the well by moving the flask in short side-to-side and back-and-forth motions. Incubator conditions should be set to 37°C, 5% CO2, 21% O2, and 90% humidity.
-
54.
Replace the thawing medium with CM expansion medium the next day. The hiPSC-CMs should be attached, beating, and can be used for expansion (step 28) or downstream assays.
(Please see Figure 3 for representative viability, morphology, and proliferation rate after thawing of hiPSC-CM)
Flow cytometry analyses of dissociated hiPSC-CMs
For the quality control and expansion rate analysis of hiPSC-derived cardiomyocytes, the cells can be collected, fixed, and immunocytochemically stained for the expression of a cardiac marker, such as α-actinin, and proliferation marker ki67 (Figure 4).
-
55.
Dissociate the CMs as described in the Replating of differentiated hiPSC-CMs section.
-
56.
After counting the hiPSC-CMs, collect 100,000 CMs from the cell suspension in 1.5 mL tubes.
-
57.
Centrifuge the cell suspension at 200 × g for 3 min.
-
58.
Discard the supernatant and add 50 μL 4% PFA.
-
59.
Incubate 10 min.
-
60.
Centrifuge the cell suspension at 200 × g for 3 min.
-
61.
Discard the supernatant and add 1 mL PBS.
-
62.
Transfer the cell suspension to a FACS tube.
-
63.
Centrifuge the cell suspension at 4°C for 3 min at 200 × g and discard the supernatant.
-
64.
Resuspend 1 × 105 cells in 50 μL of permeabilization buffer containing 5% BSA and 0.3% Triton X-100.
-
65.
Incubate the cells for 30 min at 4°C.
-
66.
Resuspend in 50 μL of flow cytometry buffer containing the α-actinin antibody (1:300 dilution), ki67 antibody (1:200) and in another FACS tube resuspend 1 × 105 cells in 50 μL of flow cytometry buffer with the respective isotype control (e.g., FITC mouse IgM, κ isotype (1:200 dilution)) and 1 × 105 cells in 50 μL of flow cytometry buffer for negative control.
-
67.
Incubate the cells for 30 min at 4°C.
-
68.
Wash the cells with 2.5 mL of flow cytometry buffer and centrifuge at 200 × g for 5 min at 4°C; discard the supernatant and repeat the wash two more times.
-
69.
Resuspend in 50 μL of flow cytometry buffer containing the secondary-antibody Goat-anti-mouse (1:300 dilution) and Goat-anti-rabbit (1:300 dilution).
-
70.
Analyze the cells with a flow cytometer, adjusting the gates according to the standard gating strategy as shown in Figure 4.
Expected outcomes
The efficient expansion of functional CMs (up to a 250-fold increase of CM number within 3–5 weeks) can be obtained from multiple hiPSC lines. For the method described here, hiPSC-CMs derived from three different differentiation protocols were all suitable for expansion in cardiac expansion medium, although the expansion capacity of the hiPSC-CMs differentiated using the B27 differentiation protocol appears to be the most robust (Figure 1).
Before starting the expansion steps, hiPSC-CMs microscopically should have high amounts of beating areas by day 8–9 and should be used between day 11 and day 14 of differentiation (Methods Video S1). After low cell density replating of hiPSC-CMs in culture flasks, addition CHIR to the cardiac culture medium efficiently induces hiPSC-CM proliferation and subsequent passaging (Figure 2). After 3–5 days of expansion, the hiPSC-CMs will reach a near confluent monolayer. Methods Videos S2 and S3 show the required steps and expected morphology of the hiPSC-CMs during expansion.
The cryopreservation steps allow for biobanking of large batches of hiPSC-CMs that can be stored before expansion or after passage 1 or 2. Thawing of cryopreserved hiPSC-CMs is efficient (74% viable hiPSC-CMs), and during subsequent expansion hiPSC-CMs exhibit a similar growth curve when compared to fresh unfrozen hiPSC-CMs (Figure 3). After passage 2, we do not recommend to biobank the expanding hiPSC-CMs due to lower viability upon recovery.
In a successful differentiation batch the expression of cardiac sarcomere protein α-actinin is usually more than 80%, and expanding cells should remain α-actinin positive during subsequent passaging. Upon CHIR optimization, up to 37% of the hiPSC-CMs will be positive for proliferative cell marker Ki67 (Figure 4).
Limitations
Despite high hiPSC-CM differentiation efficiency (85%–99%) in 2D monolayer cultures (85%–99%), yet, batch-to-batch and cell line-to-line variability remains an issue for stable CM production. In our protocol, we provide the possibility to robustly expand and/or further purify hiPSC-CM cultures via serial passaging and cell contact inhibition. This ultimately allows for a 250 fold expansion of day 11 hiPSC-CMs. However, we have noticed that also cell line-to-line and batch-to-batch variabilities exist. Moreover, the expansion capacity decreases when cells are passaged multiple times.
Another limitation of our method is that hiPSC-CMs differentiation should be at least 50% (Flow cytometry positive for α-actinin) to induce an efficient proliferative response at day 11. One explanation for the absence of massive proliferation of unpure hiPSC-CM cultures would be cell-cycle inhibiting paracrine signaling factors secreted by the other cell types. Alternatively, in low efficiency cultures (<50%) the CM are phenotypically different from efficiently differentiated hiPSC-CMs (>70%). Lastly, when it comes to cryopreservation of hiPSC-CMs the cell viability after thawing reduces from P1 to P5 (70% to 28%), which was similar to the non-expanded age-matched CMs. Therefore, cryopreservation hiPSC-CMs is recommended up to passage 2. HiPSC-CMs exceeding passage 2 are suitable for the use in direct downstream assays.
Troubleshooting
Problem 1
There are many dead hiPSC after passaging, is this normal?
Potential solution
The hiPSCs need to be monitored under a microscope and adequate timing of the chemical digestion process of dissociation remains essential for cell survival. Generally, when light sheds through the colonies and the hiPSCs can be easily flushed off the plate, the dissociation is sufficient. Use 0.5 mM EDTA solution and 10 μM Y27632 in the E8 complete medium are necessary.
Problem 2
The hiPSC colonies are too dense in the middle, is this a problem?
Potential solution
The hiPSCs need to be equally divided over the wells to enable full monolayer differentiation with high efficiencies. Mechanically dissociate the detached hiPSC 5–10 times till the clumps are only 3–5 cells big. Move plate more times side-to-side in the incubator and leave cells for 24 h.
Problem 3
The hiPSCs look abnormal or the proliferation speed is slow.
Potential solution
Wait for some passages before starting cardiac differentiation. Perform mycoplasma test and karyotyping to rule out any hiPSC cell line abnormalities.
Problem 4
No spontaneous contraction on day 11.
Potential solution
See solutions concerning problems during the cultivation of hiPSCs. Optimize the CHIR concentrations from 5–10 μM per hiPSC line. Do not re-freeze CHIR aliquots.
Problem 5
Many dead hiPSC-CMs are present upon thawing, is this normal?
Potential solution
We observe a cell viability between 60%–90% upon thawing by using CMs on passage 1–2. Pipette cells carefully with a 5 mL pipette. Place the thawed cells directly into the replating medium. Add 1:100 Revitacell to promote cell survival and 10% KO serum can be added to promote cell survival after thawing.
Problem 6
No hiPSC-CMs are attached to the culture flask after replating.
Potential solution
Allow the Matrigel to incubate at 37°C for at least 1 h. Check culture flask on complete coating coverage. Pipette cells carefully with a 5-mL pipette. Add 1:100 Revitacell to promote cell survival. Add 10% KO serum to promote cell survival. Seed cell number as described in Table 1.
Problem 7
Many dead hiPSC-CMs after replating, is this normal?
Potential solution
Pipette carefully with 5-mL pipette, add Revitacell to promote cell survival, add 10% KO serum to promote cell survival, decrease CHIR concentration, seed cell number as described in Table 1.
Problem 8
HiPSC-CMs capacity is lower than expected.
Potential solution
Optimize the CHIR concentration and add the CHIR every other day. Do not re-freeze CHIR aliquots. Use a lower splitting ratio in the first 3 passages (1:20–1:5) and increase the splitting ratio upon passage 3 and passage 4 to 1:2. Do not split after passage 4. Only use differentiated cell populations with ≥70% α-actinin-positive cells.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sean M. Wu (smwu@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The published article includes all datasets generated or analyzed during this study.
Acknowledgments
This work was supported by a UMC Utrecht Clinical Fellowship, Netherlands Heart Institute Fellowship, and CVON-Dosis young talent grant (to J.W.B.); Stanford Child Health Research Institute Postdoctoral Fellowship and NIH NRSA Postdoctoral Fellowship 5F32HL142205 (to S.L.); R01 HL145676, R01 HL146690, and P01 HL141084 (to J.C.W); NIH (OD004411, HL099776, LM012179) and the Joan and Sanford I. Weill Scholar Fund (to S.M.W.); Netherlands Heart Foundation (CVON-Dosis 2014–40), and Netherlands Organization for Sciences (NWO)-ZonMW (VICI 91818602) (to J.V.); Dutch Research Council (NWO) VENI grant no. 016.176.136 (M.H.); Foundation Leducq (Cure-PLaN) (to R.G.C.M., M.H., and F.A.). R.G.C.M. is supported by a grant of the PLN Foundation. F.W.A. is supported by UCL Hospitals NIHR Biomedical Research Centre; Horizon2020 ERC-2016-COG EVICARE (725229), Horizon 2020 BRAV3 (SC1-BHC-07-2019) and ZonMw-TAS program (no. 116002016) (to J.S.). We thank Prof. Joseph Wu (University of Stanford, USA) for the provision of the SCVI-111, SCVI-114, and SCVI-273 hiPSC lines. The graphical abstract was created with BioRender.com.
Author contributions
R.G.C.M. designed and performed the experiments. R.G.C.M., S.L., and J.W.B. analyzed the data and wrote the manuscript. C.S.B. contributed to the development, analysis, and validation of the flow cytometry. J.P.G.S., S.M.W., and J.W.B. supervised the project. M.H., W.R.G., J.H., P.A.F.M.D., L.W.L., J.V., F.A., and J.C.W. edited and critically reviewed the manuscript.
Declaration of interests
J.W.B. and S.M.W. have filed for a patent with the US Patent and Trademark Office regarding the effect of bioactive lipids plus Wnt signaling activation on hiPSC-CM proliferation/expansion.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100334.
Contributor Information
Sean M. Wu, Email: smwu@stanford.edu.
Jan W. Buikema, Email: j.w.buikema-3@umcutrecht.nl.
References
- Buikema J.W. Wnt activation and reduced cell-cell contact synergistically induce massive expansion of functional human iPSC-derived cardiomyocytes. Cell Stem Cell. 2020;1:50–63. doi: 10.1016/j.stem.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge P. Chemically defined generation of human cardiomyocytes. Nat. Methods. 2014;8:855–860. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical wnt signaling. Proc. Natl. Acad. Sci. U S A. 2012;109:1848–1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. Heparin promotes cardiac differentiation of human pluripotent stem cells in chemically defined albumin-free medium, enabling consistent manufacture of cardiomyocytes. Stem Cells Transl. Med. 2017;6:527–538. doi: 10.5966/sctm.2015-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.