

Abstract
Progression through the cell cycle is driven by bistable switches—specialized molecular circuits that govern transitions from one cellular state to another. Although the mechanics of bistable switches are relatively well understood, it is less clear how cells integrate multiple sources of molecular information to engage these switches. Here, we describe how bistable switches act as hubs of information processing and examine how variability, competition, and inheritance of molecular signals determine the timing of the Rb-E2F bistable switch that controls cell cycle entry. Bistable switches confer both robustness and plasticity to cell cycle progression, ensuring that cell cycle events are performed completely and in the correct order, while still allowing flexibility to cope with ongoing stress and changing environmental conditions.
Keywords: bistability, cell cycle, cell signaling, DNA damage
Early evidence for switch-like behavior
Long before modern tools for manipulating individual genes and proteins were available, the cell cycle was understood primarily in terms of its two primary functions: DNA synthesis and cell division. These two functions are now known to occur during discrete periods of time known as S phase (DNA synthesis) and M phase (mitosis, the process leading up to a visible cell division), respectively. Remarkably, a great deal was learned about these essential activities by making two fairly simple measurements. First, by monitoring single living cells by traditional light microscopy, one could measure the duration of time between consecutive cell divisions, also known as the intermitotic time. Second, by measuring the incorporation of radioactive nucleotides necessary for DNA synthesis, one could understand when, during the intermitotic time, cells were actively synthesizing DNA.
These two tools were sufficient to establish the canonical four-phase cell cycle model with distinct transitions between phases. Autoradiographs of root-tip cells from fava beans [1–3] revealed that a distinct period of time existed after cell division, but before S phase, during which cells did not incorporate radioactive nucleotides. We now know this phase to be called gap phase 1, or G1. Another period of time, immediately following DNA replication, was identified in which cells also showed no incorporation of nucleotides. This phase is now known as gap phase 2, or G2. Importantly, these early studies demonstrated that cells either strongly incorporated radiolabeled nucleotides or contained virtually no signal at all. Thus, even with-out knowledge of the molecular basis of cell cycle phase transitions, these initial observations provided the first hints that cells transition through sudden, switch-like changes between clearly delineated functional states.
With the ability to identify these transitions, one of the first observations was a significant variability in the duration of time individual cells spend in each phase. In fact, much of the early modeling of cell cycle was focused on providing quantitative descriptions to explain this variability [4–12]. We recently showed that the duration of a single cell cycle phase in a given cell has no influence on the duration of the other phases of its cell cycle [13]. That is, they are statistically independent quantities. Mathematical modeling suggests that, in order to achieve this independence, the control mechanisms regulating cell cycle transitions must integrate large and diverse pools of molecular factors that each exerts a small influence on the rate of cell cycle progression. It is important to note that, in each of these studies examining cell cycle phase duration, cell-to-cell variability provides critical clues for understanding general control principles that govern cell cycle progression.
The birth of molecular cloning in the 1970s ushered in a new era of cell cycle research through the identification of individual genes and proteins regulating cell cycle progression. Since that time, the field has acquired a vast catalogue of molecular players that mediate cell cycle progression including cyclins, kinases, phosphatases, and ubiquitin ligases; core DNA replication machinery; cytoskeletal regulators; and a host of signaling components. The sheer complexity of these molecular systems has necessitated the use of computational models to understand how these regulators are coordinated to control progression through the cell cycle. A major theme to emerge from these modeling efforts was the concept of a bistable switch, a molecular circuit that allows rapid transitions between two stable states. Early mathematical modeling of the molecular events that control entry into mitosis in Xenopus oocytes revealed that this transition was controlled by a bistable switch regulating the activation of Cdc2 [14–18]. It now seems that many, if not all, cell cycle phase transitions are governed by bistable switches [17–24]. These bistable switches account for the abrupt transitions observed between cell cycle phases and the irreversibility of most of these transitions under normal physiological conditions.
Bistable switches allow rapid transitions between cell cycle phases
It is well accepted that the transitions between consecutive cell cycle phases are generally rapid and irreversible under normal physiological conditions. How is this behavior accomplished at the molecular level? How do molecular regulators interact to ensure decisive transitions to a new functional state with distinct biochemical activities? To understand this behavior, we need to borrow a concept from the field of nonlinear dynamics known as bistability [25–27]. Bistability is a property of systems that can exist in two distinct configurations or stable steady states. For example, the system may be characterized by either high levels of molecule A and low levels of molecule B or, conversely, low levels of A and high levels of B (Fig. 1). Importantly, a bistable system cannot remain—not for very long, at least—in a molecular configuration that lies between these two states; it will tend strongly toward one of the two stable molecular configurations or attractors. In the same way, a healthy cell does not generally stall between G1 and S, but undergoes a series of rapid, switch-like changes to progress from one state to the next.
Fig. 1.
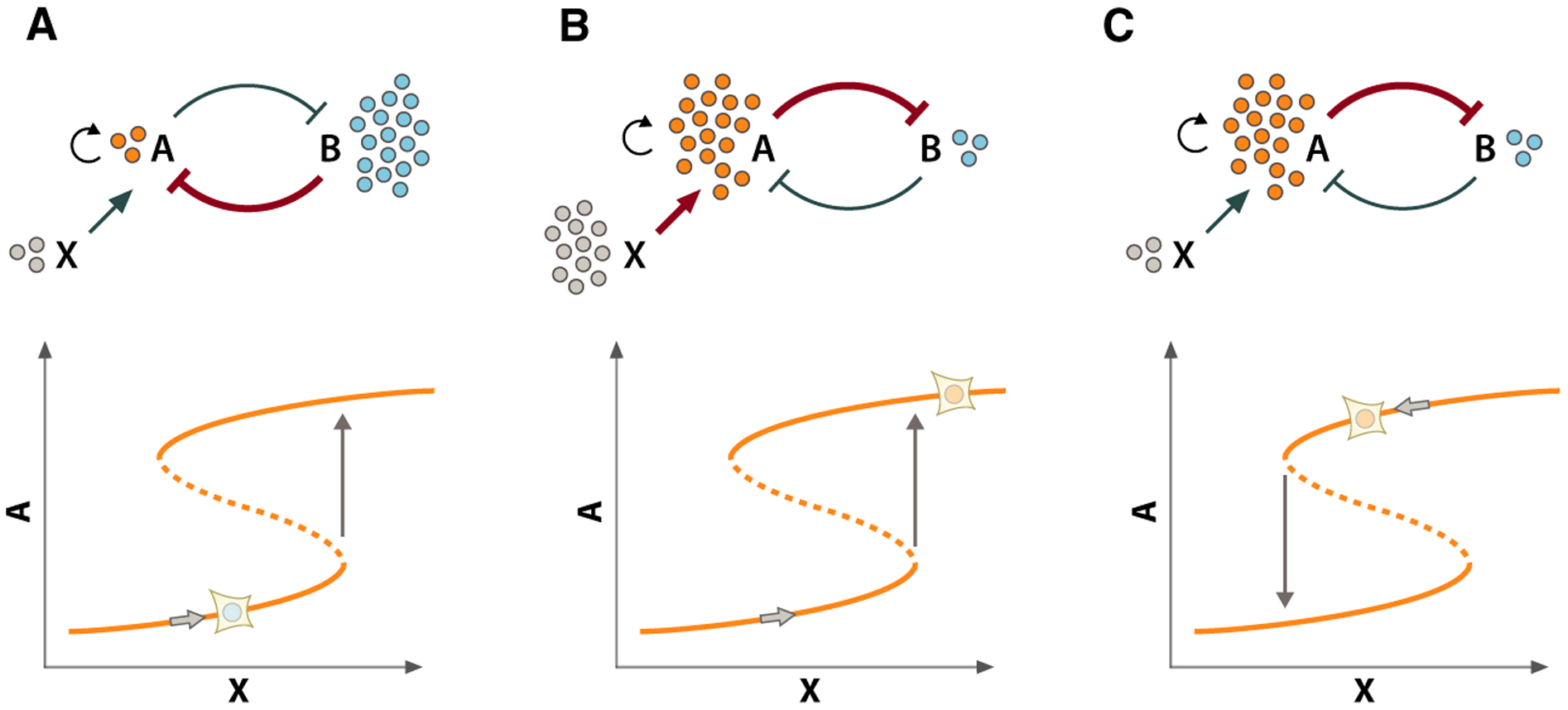
Bistable switches enable abrupt transitions between distinct cellular states. Top panels: schematics of the relationships between A, B, and X. Bottom panels: steady-state plots showing levels of A as a function of changing levels of X (denoted by the position of the cell on the curve). (A) Starting from the Alow/Bhigh steady state (left panel), the system remains in this state until X surpasses a critical threshold value (indicated by the arrow). (B) When X is increased above this threshold (middle panel), the system abruptly and completely transitions to a new steady state characterized by high levels of A and low levels of B (Ahigh/Blow). (C) After this transition, the strong inhibitory effect of A on B in combination with A autoregulation provides stability to this state, preventing the reverse transition even when X decreases below the threshold at which the forward transition occurred. Therefore, the threshold value of X at which the bistable system switches thus depends on the history of the system (i.e., whether it started from the Ahigh/Blow or a Ahigh/Blow state).
Bistability often occurs when a biochemical circuit possesses both positive feedback and ultrasensitivity [27]. This positive feedback can be generated indirectly when two molecular components are inhibitors of one another to form a so-called toggle switch relationship. Figure 1 shows a toggle switch involving molecules A and B (Fig. 1). To introduce ultrasensitivity to the system [27], we will add positive autoregulation to component A. Let us consider how this system transitions from one state to the other, starting from an Alow–Bhigh state and incrementally increasing A using stimulus X. Over a range of small increases in X, the inhibitory effect of B on A dominates and the system remains in the Alow–Bhigh state. However, at some critical value of X, the inhibition of A by B is over-come, and the system transitions to the Ahigh–Blow state. This switch occurs abruptly and completely due to the positive feedback that is generated by the reciprocal inhibition of B by A, which is progressively strengthened in concert with the weakening of B’s inhibition of A. An important characteristic of the toggle switch is that the system cannot settle in a state with intermediate levels of A and B; once the system surpasses this critical threshold of X, it abruptly switches to the Ahigh–Blow state, thus committing to a complete transition.
Another important characteristic of bistable switches is that they possess hysteresis, a property that protects the system from reversing following a state transition. To illustrate how this is possible, consider again our system comprising molecules A and B and stimulus X (Fig. 1). Once the system switches to the Ahigh–Blow state, the inhibitory effect of A on B stabilizes this state (and autoregulation of A reinforces it), preventing the reverse transition to the Alow–Bhigh state even if X is reduced below the critical threshold required to induce the forward transition (Fig. 1B, right panel). Thus, the threshold value of X at which the system transitions between states is determined by the history of the system (i.e., whether it is starting in the Alow–Bhigh state or the Ahigh–Blow state). Depending on the relative rates of the biochemical reactions comprising a bistable switch, some transitions can even be irreversible (e.g., if the reverse transition occurs when X < 0). Bistable switches can therefore provide directionality for transitions between functional states. This property ensures, for example, that a cell proceeds from G1 to S phase and not in reverse. In this sense, bistable switches can provide a productive and reliable temporal ordering of the cellular events required for successful DNA replication and cell division.
Thus, bistable switches provide an excellent mechanistic explanation for the rapid transitions observed between distinct functional states of the cell cycle and the temporal ordering of these states during cell cycle progression. Less clear, however, are the mechanisms that regulate the timing of these switches. In other words, what are the molecular determinants that ensure a cell has completed all the activities required for a given phase of the cell cycle and is adequately prepared to enter the next phase? In the following sections, we will describe how bistable switches integrate multiple diverse molecular inputs, such as mitogenic stimuli and DNA damage, to control cell cycle phase transitions. We examine how differences in these intrinsic or extrinsic factors regulate the timing of bistable switches, generating the heterogeneity in cycle phase durations observed within a population of cells. We will focus specifically on the Rb-E2F switch, which governs re-entry into the cell cycle following mitosis. We discuss how the Rb-E2F switch coordinates with other bistable switches controlling G1 progression to ensure successful DNA replication and cell division.
The Rb-E2F bistable switch (Restriction Point)
Contrary to what its name may suggest, the first ‘gap’ phase of the cell cycle, G1, is far from a static cellular state. During this phase, the cell must sense its external environment and determine whether conditions are amenable to another round of cell division. The cell must also repair any residual DNA damage and prepare new sites for DNA replication through the process of origin licensing [28]. All the while, the cell must increase its physical size and repertoire of molecular machinery in preparation for eventual cell division. Information regarding the timely progress of these activities is communicated to the molecular machinery governing the G1/S transition. One of the major events that must occur prior to this transition in mammalian cells is passage through the restriction point (RP) [29], which is controlled by a bistable switch formed by mutual inhibition between the tumor suppressor retinoblastoma (Rb) protein and the E2F family of transcription factors [19] (Fig. 2A). E2F proteins control the expression of several genes required for DNA synthesis [30–32], thus making this switch a prerequisite for S phase initiation.
Fig. 2.

Integration of analog inputs to the Rb-E2F switch. (A) The Rb-E2F bistable switch is formed by a toggle switch mechanism in which Rb transcriptionally represses E2F family transcription factors, while E2F transcription factors induce their own expression, as well as the expression of cyclins E and A, which activate CDK2 to reciprocally inhibit Rb. Growth factor-induced mitogenic signaling triggers the Rb-E2F switch through a CDK4/6-dependent phosphorylation and inhibition of Rb. Cell–cell contact promotes an increase in p27 expression and decreases cell surface expression of growth factor receptors. DNA damage promotes an increase in p21 and/or p27 expression. Increases in cell size dilute the concentration of Rb. (B) Engagement of the Rb-E2F bistable switch triggers an abrupt increase in E2F expression at a given threshold of mitogenic signaling (left panel). By inhibiting CDK2 activity, cell–cell contact or DNA damage shift this threshold to higher levels of mitogenic signaling (green, right panel). Conversely, increases in cell size shift this threshold to lower levels of mitogenic signaling (purple, right panel). (C) Linear integration of mitogenic signals by ERK-dependent transcription factors determines the timing of the Rb-E2F bistable switch. Increased growth factor concentration increases the rate of mitogenic signaling integration (blue vs gray, left panel), decreasing the time required to surpass the threshold required to engage the Rb-E2F switch (see A, left panel). By decreasing cell surface expression of growth factor receptors, cell–cell contact reduces the rate of mitogenic signal integration, requiring longer integration times to engage the switch (green, left panel). By shifting the threshold of mitogenic signaling required to engage the switch (see B, right panel), cell–cell contact and DNA damage delay the switch (green, right panel), while cell growth expediates the switch (purple, right panel).
Factors influencing the Rb-E2F bistable switch
Extracellular mitogens
Passage through the RP is primarily determined by the presence of extracellular mitogens (i.e., growth factors) that signal to the cell that amenable environmental conditions exist to commence another round of cell division. In the absence of mitogenic stimulation, Rb is unphosphorylated and acts as a repressor of E2F-dependent transcription [33], holding cells in a transiently arrested state called quiescence (Fig. 2A). Mitogenic stimulation promotes Ras/ERK signaling activity and induces expression of cyclin D [34], which generates an active heterodimeric complex with cyclin-dependent kinases 4 and 6 (CDK4/6). It is generally thought that active CDK4/6 complexes promote an initial, but incomplete, phosphorylation of specific Rb residues, which partially alleviates its inhibitory effect on E2F-dependent transcription. These first Rb phosphorylation events initiate a positive feedback loop in which E2F transcription factors induce their own expression [35], as well as the expression of cyclins A and E [36,37]. The subsequent formation of active cyclin-dependent kinase 2 (CDK2)/cyclin E and CDK2/cyclin A complexes then further hyperphosphorylate and inhibit Rb to generate maximal E2F-dependent transcription of additional S phase genes. Recent evidence suggests that the role of cyclin D:CDK4/6-dependent Rb phosphorylation in G1 may in fact be more complex, with distinct monophosphorylated forms of Rb possessing different activities toward E2F regulation as well as other cell cycle regulators, thus calling into question the precise role through which CDK4/6-dependent Rb phosphorylation contributes to growth factor-induced engagement of the Rb-E2F switch [38–40].
At low levels of mitogenic signaling, the suppressive activity of Rb remains sufficiently strong to dampen E2F-dependent transcription. Once mitogenic signaling surpasses a given threshold required to engage the CDK2-promoted positive feedback, however, the system abruptly switches to a state characterized by low Rb and high E2F levels [32] (Fig. 2B). After the switch to a high E2F steady state, elevated CDK2 activity is sufficient to keep Rb hyperphosphorylated and the system remains in the high E2F steady state even if mitogenic stimuli are completely removed. In this way, the Rb-E2F switch is irreversible with respect to mitogenic stimulation under normal physiological conditions [29,32,41,42] (Fig. 2B).
But how is information about the concentration of extracellular growth factors incorporated into the decision of when to engage the Rb-E2F switch? ERK activity increases rapidly (within minutes) following growth factor stimulation [43], with the amplitude and the kinetics of ERK activation increasing with growth factor concentration [44]. Furthermore, over longer periods of sustained growth factor stimulation, ERK activity often fluctuates over time. Since sustained ERK activity is often required to promote cell cycle progression [45–47], the cell must possess mechanisms that integrate this time-varying input into the all-or-none decision regulating cell cycle re-entry. Gillies et al. [48] recently demonstrated that the expression of Fra-1, an ERK target gene controlling cyclin D expression [47], increased over time in proportion to both the amplitude and duration of ERK activation, acting as a linear integrator of total ERK activity. At higher concentrations of growth factor, Fra-1 expression increased more rapidly, while low concentrations induced a slower accumulation of Fra-1. If a given threshold of Fra-1 and/or related ERK target genes regulating cyclin D expression (e.g., c-jun, c-fos, and c-myc) is required to engage the positive feedback that drives the Rb-E2F switch, such an integration mechanism provides a plausible mechanistic solution for how growth factor concentrations might control the timing of the switch: Higher growth factor concentrations would reach this threshold sooner by promoting a more rapid increase in ERK target genes, while lower concentrations would require longer periods of integration time to surpass the threshold of mitogenic signaling required to engage the switch. This ‘dose-to-duration’ mechanism seems to be pervasive in other signaling contexts including the yeast response to environmental cues [49].
Cell–cell contact
In addition to extracellular mitogens, the Rb-E2F switch is also influenced by physical contact with other cells. Cell surface receptors activated upon physical interactions with neighboring cells relay information about the density of the cellular environment, transducing inhibitory signals that block cell cycle progression by arresting cells in G1. In fact, intact contact-inhibitory signaling is an important mechanism of tumor suppression by inhibiting cell cycle progression in crowded cell environments, and the loss of contact inhibition is a hallmark of many cancers [50]. Activation of the cadherin family of cell–cell adhesion receptors, for example, has been shown to increase the expression of p27 [51,52], which binds to and inhibits cyclin/CDK complexes (Fig. 2A). This stoichiometric inhibition of CDK activity antagonizes mitogenic stimulation of the Rb-E2F switch proportionally to the amount of contact a cell is making with its neighbors. Furthermore, contact inhibition can directly inhibit mitogenic signaling by reducing cell surface expression of growth factor receptors [53] (Fig. 2A). Both mechanisms counteract the proliferative effect of growth factors, effectively delaying the timing of the Rb-E2F switch in proportion to the degree of cell–cell contact by increasing the threshold of mitogenic signaling required to engage the switch (Fig. 2C, left panel) or by decreasing the rate at which ERK target genes are expressed, respectively [54] (Fig. 2C, right panel).
DNA damage repair
The decision to commit to another round of cell division is not driven entirely by the cellular environment. The cell must also determine whether its internal state is prepared for another round of DNA synthesis. For example, the cell must check that it has repaired any DNA damage it has incurred to ensure successful replication during S phase. A sophisticated network of molecular surveillance mechanisms monitors the cell for the existence of DNA damage and generally halts cell cycle progression until the damage is repaired [55,56]. DNA damage increases the expression of p21 and/or p27 [57–60], which regulate cell cycle re-entry by binding to and stoichiometrically inhibiting the activity of CDK/cyclin complexes (Fig. 2A). Both p27- and p21-mediated inhibition of CDKs effectively increase the threshold of mitogenic signaling required to engage the Rb-E2F switch [32] (Fig. 2B), thus delaying (perhaps indefinitely) the commitment to cell cycle re-entry until DNA damage has been adequately repaired (Fig. 2C).
Cell size
The Rb-E2F switch is also influenced by cell size and other metabolic measures of a cell’s readiness to divide. Although the precise mechanism through which cell size regulates G1 progression in mammalian cells remains unclear [61], an inhibitor-dilution mechanism has recently been proposed to control cell cycle re-entry in yeast [62,63]. During G1, expression of the yeast G1 cyclin Cln3 scales proportionally with cell size, while the expression of Whi5, a transcriptional inhibitor of cell cycle progression, remains constant. These effectors exert opposing effects on the activity of the SBF transcription complex which promotes the expression of various genes regulate cell cycle progression. Thus, as cell size increases during G1, concentration of the SBF repressor (Whi5) progressively decreases relative to a constant concentration of Cln3 and at a given threshold (‘Start’) engages additional positive feedback (through the SBF-mediated expression of Cln1/2 that further inactivates Whi5), committing the cell to cell cycle re-entry. Evidence from a recent preprint [64] suggests that a similar mechanism may function to dilute Rb in mammalian cells as cells grow larger in G1 (Fig. 2A). Furthermore, inhibition of the p38 MAPK pathway was found to decouple cell size from G1 duration through the activity of p27 [65], indicating yet another mechanism through which cell size might influence the Rb-E2F switch. How cell size regulation is coordinated with mitogenic signaling, cell–cell contact, and internal stresses such as DNA damage remains to be explored.
Integration of internal and external signals by the Rb-E2F Switch
Thus, the core Rb-E2F bistable switch in G1 is embedded within a molecular network that allows it to integrate information about both its external (i.e., mitogen concentration and cell density) and internal (i.e., DNA damage and cell size) states (Fig. 2A), converting these graded inputs into a binary decision controlling cell cycle re-entry (Fig. 2B). How are these signals integrated? What additive, synergistic, antagonistic, or compensatory behaviors do these signals display? Here, we describe the various mechanisms by which these influences are assimilated and processed by the bistable switch.
Competition among input signals
Passage through the RP appears to be driven primarily by mitogen concentration. In the absence of mitogenic stimulation, cells will arrest in G1 even if the antagonistic inputs to the RP bistable switch are removed (e.g., p21 knockout [66]). These antagonistic inputs (i.e., cell–cell contact, DNA damage, cell size) act instead to determine the mitogenic threshold at which the Rb-E2F switch will be triggered and thus control the timing of the switch. For example, in the presence of DNA damage, higher thresholds of mitogenic stimulation are required to overcome the inhibitory effect of p21 [32] (Fig. 2). Conversely, this mechanism allows the cell to actively monitor the progression of DNA damage repair, limiting passage through the RP at a fixed mitogen concentration until DNA damage is repaired and p21 levels return to sufficiently low levels. A similar relationship may exist between mitogenic stimulation and contact inhibition [67]. By increasing p27 expression and/or decreasing cell surface expression of mitogen receptors, increasing amounts of cell–cell contact should require proportionally higher mitogen concentrations or longer integration time to trigger the Rb-E2F switch. At very high cell densities, contact-inhibitory mechanisms may delay activation of the Rb-E2F switch indefinitely, despite the presence of extracellular mitogens, until the surrounding tissue can physically support the addition of a new cell, acting as a homeostatic mechanism to control the number of cells in a given tissue [68]. On the other hand, the dilution of cell cycle inhibitors such as Rb that has been proposed to occur as cell size increases in G1 [64] should reduce the threshold of mitogenic signaling required to engage the Rb-E2F switch, effectively coupling the timing of RP passage to cell size as well. Thus, the precise timing of the Rb-E2F bistable switch in an individual cell is determined by the integration of multiple graded inputs, both internal and external, culminating in a decision of whether and when to re-enter the cell cycle.
Long-term accumulation of inputs
Another mode in which bistable switches can integrate continuous information is through the accumulation of signals, sometimes over multiple cell cycle generations. Several phenomenological studies of cell-to-cell variability in cell cycle durations have proposed that some factor, or group of factors, explained the similarities observed in sister cell phase durations [12,69]. Furthermore, recent studies in human cells have demonstrated that daughter cells can indeed inherit memories of mitogenic stimulation and DNA damage from their mother during the previous mitotic cycle, which influences their subsequent commitment to cell cycle re-entry [70–72]. These studies suggest that cyclin D1 mRNA is produced if mother cells are exposed to mitogens during G2 and inherited by daughter cells following cell division. Similarly, DNA damage accumulated during the previous mitotic cycle can regulate re-entry into the next one through an inheritance of p21 or its upstream regulator p53 from mother to daughter cell [70–73]. These studies indicate that the RP decision may not even occur in G1 in some cells and may instead be predetermined by events that occurred during the previous cell cycle. Indeed, in the presence of continuous growth factor stimulation, some cells never disengage the Rb-E2F switch, giving birth to daughter cells directly into the post-RP state [70]. Interestingly, the timing of the RP also seems to vary between cell types, occurring predominantly during G1 in some cell types (e.g., Swiss 3T3) or in the previous G2 phase in others (MCF10A), and even varying in its precise timing across individual cells of the same cell type [70].
Integration of the Rb-E2F switch with other bistable switches
While its precise timing may vary from cell to cell or between cell types, in most cells under physiological conditions, the Rb-E2F switch seems to be the first G1 bistable switch to be triggered. After passage through the RP, the cell initiates a series of molecular events later in G1 as it completes its preparation for DNA replication. In recent years, several other bistable switches have been proposed to regulate the progression through G1 and into S phase [66,74,75]. While it is not clear at this point whether these switches occur in a fixed temporal order [76], or if all these switches are fundamentally important prior to DNA replication in all cell types, each of them provides mechanisms that allow a cell to ensure it has adequately fulfilled the requirements to begin DNA replication. Theses switches are not entirely independent of the earlier RP switch either, as some of the molecular players that control passage through the RP participate in the control of other G1 bistable switches.
A bistable switch involving the inactivation of the ubiquitin ligase APC/CCdh1 has been shown to regulate the commitment to DNA replication during G1 [75]. APC/CCdh1 is activated immediately following mitosis and promotes the degradation of various G1 cell cycle effectors required for S phase transition. Inactivation of APC/CCdh1 generally occurs late in G1 and is induced by cyclin E/CDK2 complexes, suggesting that engagement of the Rb-E2F switch should be required prior to the APC/CCdh1 switch. Indeed, Cappell et al. demonstrated that APC/CCdh1 inactivation was observed in the vast majority of cells only after prior engagement of the Rb-E2F switch and within 1 h before S phase initiation [75]. Consistent with the idea that this mechanism confers a temporal order to the switches, E2F-dependent expression of Emi1 [77] was also shown to play a crucial role in the kinetics of APC/CCdh1 inactivation and was required to make the switch irreversible, thus preventing reactivation of APC/CCdh1 during S phase. While the RP is irreversible with respect to mitogen signaling, recent evidence suggests that a window of time exists after the Rb-E2F switch but before the APC/CCdh1 switch in which cells can revert to a pre-RP state and become sensitive once again to the presence of mitogens if they acquire new DNA damage [32,75]. Thus, the APC/CCdh1 switch provides an additional safeguard mechanism in G1 capable of detecting DNA damage (and possibly other cell cycle stresses) acquired after RP passage and has been proposed to function as the final ‘point of no return’ in the commitment to DNA replication. This capacity is vitally important given recent observations that the RP decision may be predetermined in the previous G2 phase of mother cells, allowing cells to respond to cellular stress incurred during cell division. This also highlights an important facet of the RP decision: Although engagement of the Rb-E2F switch will commit the cell to DNA replication even if extracellular mitogens are removed, other changes in cellular state that could be detrimental to successful DNA replication or division continue to be monitored throughout the cell cycle, and additional mechanisms exist to halt cell cycle progression in response to new challenges.
At the onset of S phase, the cell undergoes an abrupt transition to a distinct biochemical and functional state that promotes DNA replication. Whereas engagement of the APC/CCdh1 switch normally commits cells to DNA synthesis, the initiation of replication itself is controlled by additional bistable switches that confer irreversibility to S phase through hysteresis. Although progression through the RP occurs when mitogenic signaling surpasses a given threshold that is determined in part by p21 expression, efficient DNA replication requires the complete degradation of p21 due to its ability, to bind to proliferating cell nuclear antigen (PCNA) replication complexes and inhibit DNA replication [32,74,78,79]. Degradation of p21 is mediated by the ubiquitin ligases SCFSkp2 and CRL4Cdt2. Two cooperative double-negative feedback loops between p21 and these ubiquitin ligases generate a bistable switch that is engaged at the onset of DNA replication: (a) CDK2 phosphorylates p21 and targets it for SCFSkp2-mediated degradation, while p21 inhibits CDK2 activity; and (b) p21 is degraded by CRL4Cdt2 following recruitment to PCNA in active replication complexes (aRCs), whereas p21 binds to PCNA and prevents the activation of RCs [74]. The switch-like degradation of p21 relies primarily on the activity of CRL4Cdt2 following its recruitment to aRCs [74] and thus co-occurs with the start of DNA replication. Once this bistable switch is engaged, the persistent activity of CRL4Cdt2 at aRCs during S phase generates hysteresis through the persistent degradation of p21, preventing it from interfering with DNA replication and insulating S phase progression from the influence of DNA damage [56]. In addition to p21, it is likely that other cell cycle regulators that are degraded by CRL4Cdt2 (e.g., Cdt1 [80–82]) should also exhibit hysteresis at the G1/S transition.
Temporal ordering of the G1 bistable switches
As described in the previous section, progression through G1 and into S phase under normal physiological conditions seems to involve a fixed temporal ordering of multiple bistable switches. Engagement of the Rb-E2F switch occurs first, possibly in the previous G2 phase for some cells. Rb hyperphosphorylation and maximal E2F-dependent transcription increase the expression and/or activity of key cell cycle regulators (e.g., cyclin E, CDK2, Emi1, Skp2) that act as inputs or modifiers of the later G1 bistable switches. Furthermore, engagement of the APC/CCdh1 switch may also act as a precursor to the later replication-dependent switches, since inactivation of APC/CCdh1 would increase the expression of Skp2 and cyclin A (and, consequently, CDK2 activity) and promote p21 degradation. Although this exact ordering of the switches (i.e., Rb-E2F, APC/CCdh1, and replication-dependent switches, respectively) may represent the most efficient path through G1 and into S phase, recent evidence suggests that the cell may be able to take detours in its path under certain conditions. For example, a recent preprint [76] has shown that in the presence of a CDK4/6 inhibitor, which prevents the initial mitogen-induced Rb phosphorylation events that engage the Rb-E2F switch, cells will divert to an alternative pathway in which the APC/CCdh1 bistable switch precedes the Rb-E2F switch. This alternative pathway relies instead on a Myc-promoted increase in cyclin E/CDK2 activity to engage the APC/CCdh1 switch, with the Rb-E2F switch being engaged much later in G1 after cyclin E/CDK2 activity is increased to sufficiently high levels. Interestingly, this reversal in the timing of the Rb-E2F and APC/CCdh1 bistable switches also reverses their functional roles in G1 regulation to some extent: The earlier APC/CCdh1 switch is now controlled by mitogenic signaling, and the Rb-E2F switch becomes the point of no return for S phase entry. While this pathway may be a less efficient path through G1 (the duration of G1 is increased by several hours on average), cells exhibited no additional signs of dysfunction throughout the later phases of the cell cycle. These data demonstrate that cell cycle control is not necessarily achieved through a fixed temporal ordering of regulatory events and that considerable plasticity may in fact be built into these regulatory systems to accommodate variability in internal or external cellular states.
Emergence of population heterogeneity in cell cycle progression
Even in a genetically identical population, cells can differ from one another in a variety of ways. Cells can vary in their local density or in size; they may experience local differences in mitogen concentrations due to paracrine signaling; and they can accumulate or repair DNA damage at different rates. The recent studies described in the previous section indicate that even the prior history of a cell can be molecularly encoded to regulate its subsequent cell cycle fate decisions [70–73]. All of these factors (and likely many more) can directly influence the properties of the Rb-E2F bistable switch. Individual cells within a population may exhibit vast differences both in local mitogen environment and in the mitogenic signaling threshold required to engage this switch, and thus will vary in the amount of time spent in a pre-RP state. At extreme values of these factors (e.g., excessive DNA damage and complete contact inhibition), engagement of the Rb-E2F switch may require a level of mitogenic signaling that is unattainable under normal physiological conditions, effectively increasing the mitogenic integration time to infinity, blocking progression past the RP, and inducing cell cycle arrest (‘quiescence’) until these conditions change. Under identical cell culture conditions, these functionally opposed cell states (i.e., cell cycle entry and quiescence) can even coexist within an isogenic population of cells [73], highlighting the fundamental role cell-to-cell variability plays in determining cell cycle fate decisions.
Conclusions
Cell cycle progression relies on a series of interdependent bistable switches that regulate transitions between distinct functional states. This type of decentralized control confers both robustness and plasticity to cell cycle progression. A variety of analog inputs, representing the favorability of intracellular and extracellular conditions, is converted into all-or-none cell fate decisions. The requirement for multiple switches to be activated may ensure that no single upstream signal can overwhelm the system and prematurely activate the initiation of cell proliferation. Although essential cell cycle activities such as DNA synthesis and cell division occur as all-or-none decisions and in a defined temporal order, ancillary cell cycle processes, such as DNA damage signaling, control the timing and sensitivity of these switches. Bistable switches, and their ability to convert multiple, graded inputs into switch-like responses confer the ability to delay progression through the cell cycle until all the requirements for a given cell cycle state are adequately met. In this sense, bistable switches act as both integrators of information and actuators that control the ordered transitions through various functional states. This viewpoint explains why the term checkpoint [83] is so appropriate to describe the dual functionality of bistable switches. Switches function to check cellular conditions through their capacity to integrate, balance, and interpret multiple analog input signals. By virtue of their kinetics, which generate abrupt transitions between molecular states, bistable switches act as the defined points that delineate the phases and subphases of cell cycle progression.
Abbreviations
- aRCs
active replication complexes
- CDK2
cyclin-dependent kinase 2
- CDK4/6
cyclin-dependent kinases 4 and 6
- PCNA
proliferating cell nuclear antigen
- Rb
retinoblastoma
- RP
restriction point
References
- 1.Pelc SR and Spear FG (1950) Autoradiographs of avian fibroblasts in tissue culture made with P 32. Br J Radiol 23, 287–289. [DOI] [PubMed] [Google Scholar]
- 2.Howard A and Pelc SR (1951) Nuclear incorporation of P32 as demonstrated by autoradiographs. Exp Cell Res 2, 178–187. [Google Scholar]
- 3.Howard A and Pelc SR (1951) Synthesis of nucleoprotein in bean root cells. Nature 167, 599–600. [DOI] [PubMed] [Google Scholar]
- 4.Smith JA and Martin L (1973) Do cells cycle? Proc Natl Acad Sci USA 70, 1263–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shields R and Smith JA (1977) Cells regulate their proliferation through alterations in transition probability. J Cell Physiol 91, 345–355. [DOI] [PubMed] [Google Scholar]
- 6.Shields R (1977) Transition probability and the origin of variation in the cell cycle. Nature 267, 704–707. [DOI] [PubMed] [Google Scholar]
- 7.Shields R, Brooks RF, Riddle PN, Capellaro DF and Delia D (1978) Cell size, cell cycle and transition probability in mouse fibroblasts. Cell 15, 469–74. [DOI] [PubMed] [Google Scholar]
- 8.Shields R (1978) Further evidence for a random transition in the cell cycle. Nature 273, 755–758. [DOI] [PubMed] [Google Scholar]
- 9.Castor LN (1980) A G1 rate model accounts for cell-cycle kinetics attributed to ‘transition probability’. Nature 287, 857–859. [DOI] [PubMed] [Google Scholar]
- 10.Cooper S and Fantes P (1987) On G0 and cell cycle controls. BioEssays 7, 220–223. [DOI] [PubMed] [Google Scholar]
- 11.Cooper S (2003) Reappraisal of serum starvation, the restriction point, G0, and G1 phase arrest points. FASEB J 17, 333–340. [DOI] [PubMed] [Google Scholar]
- 12.Lee TJ, Yao G, Bennett DC, Nevins JR and You L (2010) Stochastic E2F activation and reconciliation of phenomenological cell-cycle models. PLoS Biol. 8, e1000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao HX, Fakhreddin RI, Shimerov HK, Kumar RJ, Gupta GP and Purvis JE (2019) Evidence that the human cell cycle is a series of uncoupled, memoryless phases. Mol Syst Biol 15, e8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tyson JJ (1991) Modeling the cell division cycle: cdc2 and cyclin interactions. Proc Natl Acad Sci USA 88, 7328–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Novak B and Tyson JJ (1993) Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos. J Cell Sci 106(Pt 4), 1153–68. [DOI] [PubMed] [Google Scholar]
- 16.Thron CD (1997) Bistable biochemical switching and the control of the events of the cell cycle. Nonlinear Anal Theory Methods Appl 30, 1825–1834. [Google Scholar]
- 17.Sha W, Moore J, Chen K, Lassaletta AD, Yi CS, Tyson JJ and Sible JC (2003) Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts. Proc Natl Acad Sci USA 100, 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pomerening JR, Sontag ED and Ferrell JE (2003) Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat Cell Biol 5, 346–351. [DOI] [PubMed] [Google Scholar]
- 19.Yao G, Lee TJ, Mori S, Nevins JR and You L (2008) A bistable Rb-E2F switch underlies the restriction point. Nat Cell Biol 10, 476–482. [DOI] [PubMed] [Google Scholar]
- 20.Hutter LH, Rata S, Hochegger H and Novak B (2017) Interlinked bistable mechanisms generate robust mitotic transitions. Cell Cycle 16, 1885–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.López-Avilés S, Kapuy O, Novák B and Uhlmann F (2009) Irreversibility of mitotic exit is the consequence of systems-level feedback. Nature 459, 592–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holt LJ, Krutchinsky AN and Morgan DO (2008) Positive feedback sharpens the anaphase switch. Nature 454, 353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novak B, Tyson JJ, Gyorffy B and Csikasz-Nagy A (2007) Irreversible cell-cycle transitions are due to systems-level feedback. Nat Cell Biol 9, 724–728. [DOI] [PubMed] [Google Scholar]
- 24.Verdugo A, Vinod PK, Tyson JJ and Novak B (2013) Molecular mechanisms creating bistable switches at cell cycle transitions. Open Biol 3, 120179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrell JE and Xiong W (2001) Bistability in cell signaling: How to make continuous processes discontinuous, and reversible processes irreversible. Chaos 11, 227. [DOI] [PubMed] [Google Scholar]
- 26.Sobie EA (2011) Bistability in biochemical signaling models. Sci Signal 4, tr10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrell JE and Ha SH (2014) Ultrasensitivity part III: cascades, bistable switches, and oscillators. Trends Biochem Sci 39, 612–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grant GD and Cook JG (2017) The temporal regulation of S phase proteins during G1. Adv Exp Med Biol 1042, 335–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pardee AB (1974) A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci USA 71, 1286–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frolov MV and Dyson NJ (2004) Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci 117, 2173–2181. [DOI] [PubMed] [Google Scholar]
- 31.Novák B and Tyson JJ (2004) A model for restriction point control of the mammalian cell cycle. J Theor Biol 230, 563–579. [DOI] [PubMed] [Google Scholar]
- 32.Heldt FS, Barr AR, Cooper S, Bakal C and Novak B (2018) A comprehensive model for the proliferation-quiescence decision in response to endogenous DNA damage in human cells. Proc Natl Acad Sci USA 115, 2532–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chellappan SP, Hiebert S, Mudryj M, Horowitz JM and Nevins JR (1991) The E2F transcription factor is a cellular target for the RB protein. Cell 65, 1053–1061. [DOI] [PubMed] [Google Scholar]
- 34.Weber JD, Raben DM, Phillips PJ and Baldassare JJ (1997) Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem J 326, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson DG, Ohtani K and Nevins JR (1994) Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev 8, 1514–1525. [DOI] [PubMed] [Google Scholar]
- 36.Shan B, Farmer AA and Lee WH (1996) The molecular basis of E2F–1/DP-1-induced S-phase entry and apoptosis. Cell Growth Differ 7, 689–697. [PubMed] [Google Scholar]
- 37.Duronio RJ and O’Farrell PH (1995) Developmental control of the G1 to S transition in Drosophila: cyclin Eis a limiting downstream target of E2F. Genes Dev 9, 1456–1468. [DOI] [PubMed] [Google Scholar]
- 38.Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P and Dowdy S (2014) Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 3, e02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanidas I, Morris R, Fella KA, Rumde PH, Boukhali M, Tai EC, Ting DT, Lawrence MS, Haas W and Dyson NJ (2019) A code of mono-phosphorylation modulates the function of RB. Mol Cell 73, 985–1000.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Topacio BR, Zatulovskiy E, Cristea S, Xie S, Tambo CS, Rubin SM, Sage J, Kõivomägi M and Skotheim JM (2019) Cyclin D-Cdk 4,6 drives cell-cycle progression via the retinoblastoma protein’s C-terminal helix. Mol Cell 74, 758–770.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zetterberg A, Larsson O and Wiman KG (1995) What is the restriction point? Curr Opin Cell Biol 7, 835–842. [DOI] [PubMed] [Google Scholar]
- 42.Cappell SD, Chung M, Jaimovich A, Spencer SL and Meyer T (2016) Irreversible APC Cdh1 inactivation underlies the point of no return for cell-cycle entry. Cell 166, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peraldi P, Scimeca JC, Filloux C and Van Obberghen E (1993) Regulation of extracellular signal-regulated protein kinase-1 (ERK-1; pp44/mitogen-activated protein kinase) by epidermal growth factor and nerve growth factor in PC12 cells: implication of ERK1 inhibitory activities. Endocrinology 132, 2578–2585. [DOI] [PubMed] [Google Scholar]
- 44.Albeck JG, Mills GB and Brugge JS. (2013) Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell 49, 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dobrowolski S, Harter M and Stacey DW (1994) Cellular ras activity is required for passage through multiple points of the G0/G1 phase in BALB/c 3T3 cells. Mol Cell Biol 14, 5441–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marshall C (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80, 179–185. [DOI] [PubMed] [Google Scholar]
- 47.Balmanno K and Cook SJ (1999) Sustained MAP kinase activation is required for the expression of cyclin D1, p21Cip1 and a subset of AP-1 proteins in CCL39 cells. Oncogene 18, 3085–3097. [DOI] [PubMed] [Google Scholar]
- 48.Gillies TE, Pargett M, Minguet M, Davies AE and Albeck JG (2017) Linear integration of ERK activity predominates over persistence detection in Fra-1 regulation. Cell Syst 5, 549–563.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Behar M, Hao N, Dohlman HG and Elston TC (2008) Dose-to-duration encoding and signaling beyond saturation in intracellular signaling networks. PLoS Comput Biol 4, e1000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 51.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM and Koff A (1994) p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 8, 9–22. [DOI] [PubMed] [Google Scholar]
- 52.Levenberg S, Yarden A, Kam Z and Geiger B (1999) p27 is involved in N-cadherin-mediated contact inhibition of cell growth and S-phase entry. Oncogene 18, 869–876. [DOI] [PubMed] [Google Scholar]
- 53.Swat A, Dolado I, Rojas JM and Nebreda AR (2009) Cell density-dependent inhibition of epidermal growth factor receptor signaling by p38alpha mitogen-activated protein kinase via Sprouty2 downregulation. Mol Cell Biol 29, 3332–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim J-H, Kushiro K, Graham NA and Asthagiri AR (2009) Tunable interplay between epidermal growth factor and cell-cell contact governs the spatial dynamics of epithelial growth. Proc Natl Acad Sci USA 106, 11149–11153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K and Linn S (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73, 39–85. [DOI] [PubMed] [Google Scholar]
- 56.Chao HX, Poovey CE, Privette AA, Grant GD, Chao HY, Cook JG and Purvis JE (2017) Orchestration of DNA damage checkpoint dynamics across the human cell cycle. Cell Syst 5, 445–459.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW and Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–25. [DOI] [PubMed] [Google Scholar]
- 58.el-Deiry WS, Harper JW, O’Connor PM, Velculescu VE, Canman CE, Jackman J, Pietenpol JA, Burrell M, Hill DE and Wang Y et al. (1994) WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res 54, 1169–1174. [PubMed] [Google Scholar]
- 59.Cuadrado M, Gutierrez-Martinez P, Swat A, Nebreda AR and Fernandez-Capetillo O (2009) p27Kip1 stabilization is essential for the maintenance of cell cycle arrest in response to DNA damage. Cancer Res 69, 8726–8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duli c V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ and Reed SI (1994) p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 76, 1013–23. [DOI] [PubMed] [Google Scholar]
- 61.Ginzberg MB, Chang N, D’Souza H, Patel N, Kafri R and Kirschner MW (2018) Cell size sensing in animal cells coordinates anabolic growth rates and cell cycle progression to maintain cell size uniformity. eLife 7, e26957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmoller KM, Turner JJ, Kõivomägi M and Skotheim JM (2015) Dilution of the cell cycle inhibitor Whi5 controls budding-yeast cell size. Nature 526, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heldt FS, Lunstone R, Tyson JJ and Novák B (2018) Dilution and titration of cell-cycle regulators may control cell size in budding yeast. PLoS Comput Biol 14, e1006548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zatulovskiy E, Berenson DF, Topacio BR and Skotheim JM (2018) Cell growth dilutes the cell cycle inhibitor Rb to trigger cell division. bioRxiv 470013 [PREPRINT]. November 19, 2018 [cited 2019 Sept 26]. 10.1101/470013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu S, Ginzberg MB, Patel N, Hild M, Leung B, Li Z, Chen YC, Chang N, Wang Y, Tan C et al. (2018) Size uniformity of animal cells is actively maintained by a p38 MAPK-dependent regulation of G1-length. eLife 7, e26947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barr AR, Heldt FS, Zhang T, Bakal C and Novak B (2016) A Dynamical framework for the all-or-none G1/S transition. Cell Syst 2, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gérard C and Goldbeter A (2014) The balance between cell cycle arrest and cell proliferation: control by the extracellular matrix and by contact inhibition. Interface Focus 4, 20130075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thrasher JD (1966) Chapter 12 analysis of renewing epithelial cell populations. Methods Cell Biol 2, 323–357. [Google Scholar]
- 69.Chao HX, Fakhreddin RI, Shimerov HK, Kedziora KM, Kumar RJ, Perez J, Limas JC, Grant GD, Cook JG, Gupta GP et al. (2018) Evidence that the cell cycle is a series of uncoupled, memoryless phases. bioRxiv 283614 [PREPRINT]. 10.1101/283614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL and Meyer T (2013) The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang HW, Chung M, Kudo T and Meyer T (2017) Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature 549, 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moser J, Miller I, Carter D and Spencer SL (2018) Control of the Restriction Point by Rb and p21. Proc Natl Acad Sci USA 115, E8219–E8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Overton KW, Spencer SL, Noderer WL, Meyer T and Wang CL (2014) Basal p21 controls population heterogeneity in cycling and quiescent cell cycle states. Proc Natl Acad Sci USA 111, E4386–E4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, Novák B and Bakal C (2017) DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat Commun 8, 14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cappell SD, Mark KG, Garbett D, Pack LR, Rape M and Meyer T (2018) EMI1 switches from being a substrate to an inhibitor of APC/C(CDH1) to start the cell cycle. Nature 558, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu C, Konagaya Y, Chung M, Daigh LH, Fan Y, Yang HW, Terai K, Matsuda M and Meyer T (2019) A parallel cell-cycle entry pathway with inverted G1 signaling and alternate point of no return. bioRxiv 600007 [PREPRINT]. April 4, 2019 [cited 2019 Sept 26]. 10.1101/600007. [DOI] [Google Scholar]
- 77.Hsu JY, Reimann JDR, Sørensen CS, Lukas J and Jackson PK (2002) E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APCCdh1. Nat Cell Biol 4, 358–366. [DOI] [PubMed] [Google Scholar]
- 78.Waga S, Hannon GJ, Beach D and Stillman B (1994) The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 369, 574–578. [DOI] [PubMed] [Google Scholar]
- 79.Rousseau D, Cannella D, Boulaire J, Fitzgerald P, Fotedar A and Fotedar R (1999) Growth inhibition by CDK-cyclin and PCNA binding domains of p21 occurs by distinct mechanisms and is regulated by ubiquitin-proteasome pathway. Oncogene 18, 4313–4325. [DOI] [PubMed] [Google Scholar]
- 80.Tsanov N, Kermi C, Coulombe P, Van der Laan S, Hodroj D and Maiorano D (2014) PIP degron proteins, substrates of CRL4Cdt2, and not PIP boxes, interfere with DNA polymerase g and j focus formation on UV damage. Nucleic Acids Res 42, 3692–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leng F, Saxena L, Hoang N, Zhang C, Lee L, Li W, Gong X, Lu F, Sun H and Zhang H (2018) Proliferating cell nuclear antigen interacts with the CRL4 ubiquitin ligase subunit CDT2 in DNA synthesis–induced degradation of CDT1. J Biol Chem 293, 18879–18889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pozo P and Cook J (2016) Regulation and function of Cdt1; a key factor in cell proliferation and genome stability. Genes (Basel) 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hartwell LH and Weinert TA (1989) Checkpoints: controls that ensure the order of cell cycle events. Science 246, 629–634. [DOI] [PubMed] [Google Scholar]