

Abstract
Circulating branched-chain amino acids (BCAAs) are elevated in obesity and diabetes, and recent studies support a causal role for BCAAs in insulin resistance and defective glycemic control. The physiological mechanisms underlying BCAA regulation are poorly understood. Here we show that insulin signaling in the mediobasal hypothalamus (MBH) of rats is mandatory for lowering plasma BCAAs, most probably by inducing hepatic BCAA catabolism. Insulin receptor deletion only in agouti-related protein (AgRP)–expressing neurons (AgRP neurons) in the MBH impaired hepatic BCAA breakdown and suppression of plasma BCAAs during hyperinsulinemic clamps in mice. In support of this, chemogenetic stimulation of AgRP neurons in the absence of food significantly raised plasma BCAAs and impaired hepatic BCAA degradation. A prolonged fasting or ghrelin treatment recapitulated designer receptors exclusively activated by designer drugs–induced activation of AgRP neurons and increased plasma BCAAs. Acute stimulation of vagal motor neurons in the dorsal motor nucleus was sufficient to decrease plasma BCAAs. Notably, elevated plasma BCAAs were associated with impaired glucose homeostasis. These findings suggest a critical role of insulin signaling in AgRP neurons for BCAA regulation and raise the possibility that this control may be mediated primarily via vagal outflow. Furthermore, our results provide an opportunity to closely examine the potential mechanistic link between central nervous system–driven BCAA control and glucose homeostasis.
Introduction
Commonly observed in obesity and type 2 diabetes (T2D), impaired glycemic control and lipotoxicity as well as their underlying mechanisms have been extensively studied (1,2). Although amino acids provide calories and are essential components in metabolic health, including in protein synthesis and maintenance, their role in the development of obesity and insulin resistance/T2D has not been explored in detail until recently. In particular, branched-chain amino acids (BCAAs) have attracted considerable attention in the last 10 years. BCAAs (leucine, isoleucine, and valine) are not synthesized in our body, making them essential amino acids we need to obtain from diet. The observation of elevated plasma BCAA levels in obese compared with lean individuals, first made in the 1960s by Felig et al. (3), has been highlighted and confirmed through the use of more sophisticated metabolomic platforms by multiple investigators (4–8). A positive association between BCAAs and insulin resistance/T2D independent of age, sex, and ethnicity has also been demonstrated (6,9–12). More importantly, emerging studies suggest a causal role of BCAAs in the pathogenesis of obesity and insulin resistance. Supplementation of BCAAs or 3-hydroxyisobutyrate, a metabolite of valine, in diet or drinking water markedly decreases insulin sensitivity and impairs glucose homeostasis in rodents (7,13,14). Interestingly, Roux-en-Y gastric bypass surgery, which often reverses diabetes, can effectively normalize circulating BCAAs and their derived metabolites (15–17), raising the possibility that lowering BCAAs may be in part responsible for correcting glycemic control. In support of this concept, dietary restriction or deficiency of BCAAs leads to restored insulin sensitivity and glucose tolerance in rodents, even when the animals are maintained on a high-fat diet (18–21). Likewise, a recent study by Zhou et al. (22) demonstrated that lowering plasma BCAAs by treating diet-induced obese, insulin-resistant mice with an inhibitor of branched-chain α-keto acid dehydrogenase (BCKDH) kinase, an enzyme that suppresses BCAA breakdown, ameliorates insulin resistance and improves glucose homeostasis. Collectively, these studies suggest not just a correlative but also a causal link between BCAAs and the development of insulin resistance and diabetes. Although many investigators have tried to unravel how BCAAs induce insulin resistance in peripheral tissues, such as muscle, adipose tissue, and liver (7,14,21–24), the physiological mechanism regulating systemic BCAA levels remains poorly understood. Addressing this clinically relevant issue may help identify novel BCAA-related targets for pharmacological and/or dietary intervention before insulin resistance or glucose imbalance sets in.
In rodents, we previously demonstrated that insulin, but not glucose, is an important physiological regulator of BCAA metabolism and that brain insulin action is required for the control of circulating BCAAs through their efficient breakdown, specifically in liver (25). We further showed that insulin infusion directly into the mediobasal hypothalamus (MBH), a classical homeostatic center known to be critical for energy homeostasis (26), is sufficient to regulate BCAA metabolism. What remains unclear, however, is the identity of the neuronal phenotype that mediates hypothalamic insulin action for BCAA control. The MBH harbors proopiomelanocortin- and agouti-related protein (AgRP)–expressing neurons (AgRP neurons), which play an important role in regulating energy balance and nutrient metabolism (26). Insulin receptors (IRs) are abundantly expressed in these neurons (27–29), thus making them attractive neuronal populations in which to study how insulin action in the brain regulates BCAA levels.
The aim of this study was to test if hypothalamic control of BCAA metabolism by insulin is mediated via AgRP neurons. To this end, we used transgenic mice devoid of IRs, specifically in AgRP neurons (AgRP IR knockout [KO]), and a chemogenetic designer receptors exclusively activated by designer drugs (DREADD) approach. We further examined the role of the BCAA regulatory pathway on glucose homeostasis and determined its physiological relevance to nutritional status. The autonomic nervous system (ANS) links the brain to the peripheral organs and is responsible for nutrient partitioning (30). Therefore, we also tested whether the ANS is involved in regulating BCAA metabolism through DREADD-induced manipulation of neuronal activity in the dorsal motor nucleus of the vagus nerve (DMV).
Research Design and Methods
Animals
Male Sprague-Dawley rats (age 3–4 months; Charles River Laboratories) were provided a regular chow diet (Rodent Diet 5001; LabDiet) and water ad libitum. The animals underwent stereotaxic cannula implantation targeting the MBH bilaterally as previously described (25). The cannula coordinates in reference to the bregma were −3.3 mm anteroposterior (AP), 0.4 mm lateral (L), and −9.6 mm dorsoventral (DV). After 5 days of recovery, rats were implanted with indwelling catheters in the carotid artery and jugular vein. Rats were allowed to recover for 4–6 days before undergoing pancreatic clamps.
Mice age 2–3 months were used in this study unless otherwise stated. They were provided a regular chow diet (Rodent Diet 5001; LabDiet) and water ad libitum. AgRP IR KO mice were generated by crossing mice floxed for IR with mice expressing Cre recombinase driven under the AgRP promoter (stock number 012899; The Jackson Laboratory). Floxed mice without Cre recombinase expression were used as controls (wild-type [WT] mice). Mice were initially fasted overnight (∼16 h) and then refed a regular chow diet for 2 h, during which blood was collected through tail nick at baseline (t = 0 h) and postrefeeding (t = 2 h) for measurement of plasma BCAAs (experiment design shown in Fig. 3A). After 1 week, mice were implanted with a jugular catheter followed by a 3- to 5-day recovery period. Then, we conducted hyperinsulinemic euglycemic clamps in these mice as described previously (31). Choline acetyltransferase (ChAT)–Cre mice were generated, in which Cre recombinase expression was driven under the ChAT promoter with the IRES-Cre sequence inserted downstream of the stop codon (32). Phox2b-Cre mice on a C57Bl/6 genetic background were generated as described elsewhere (33). This mouse line was chosen because it expresses Cre in all parasympathetic neurons (both afferent and efferent) but not in sympathetic ganglia or spinal sensory neurons. The Cre allele was detected using the following primers: 5′-CCGTCTCCACATCCATCTTT-3′ (Phox2bF2), 5′-CTACGGACTCTGGTGGT-3′ (Phox2bR2), and 5′-ATTCTCCCACC GTCACTACG-3′ (CreR2).
Figure 3.
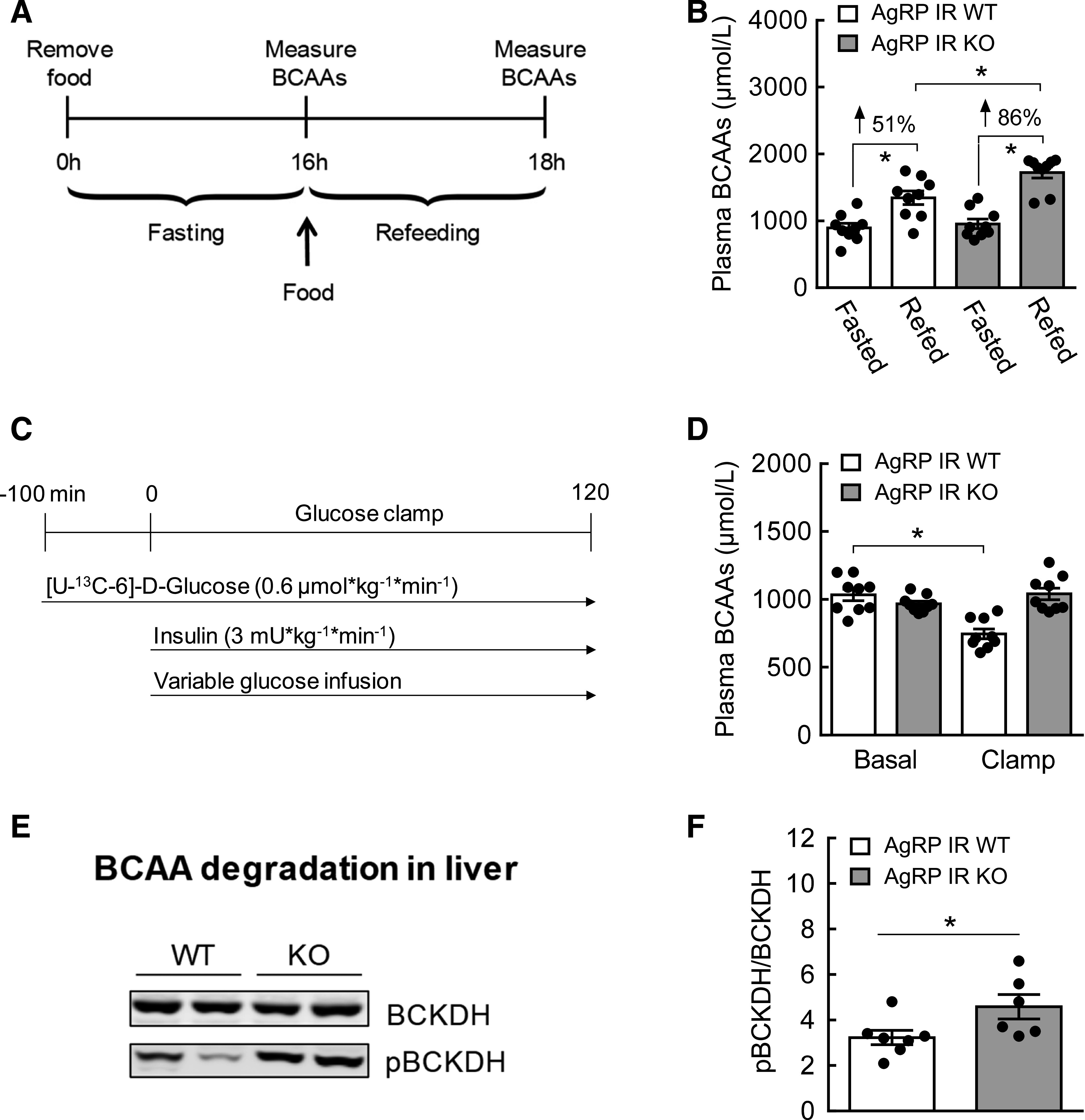
Plasma BCAAs are elevated in AgRP IR KO mice. A: Experimental design of fasting and refeeding. B: Plasma BCAAs after refeeding expressed as percent increase from the fasting baseline. C: Hyperinsulinemic euglycemic clamp schematic in AgRP IR WT and AgRP IR KO mice with food removed 2 h before clamps started. D: Plasma BCAAs during basal and clamp periods. E: Western blots showing hepatic BCKDH and pBCKDH between AgRP IR WT and AgRP IR KO mice at the end of clamps. F: Quantification of Western blot data expressed as the ratio of pBCKDH to BCKDH, an inactivation index of BCKDH activity. N = 6–9/group; values are mean ± SEM; *P < 0.05.
All animals were housed at a constant ambient temperature of 21–23°C, with a 12:12-h light/dark cycle (lights on at 0700 h and off at 1900 h). All studies were conducted in accordance with the guidelines established by the National Institutes of Health, and all protocols were approved by the institutional animal care and use committees at Texas Tech University and Seoul National University and Seoul National University Hospital Institute of Biomedical Research (Seoul, South Korea).
Hyperinsulinemic Euglycemic Clamps
Rats
After MBH cannula and venous catheter implantation, Sprague-Dawley rats were studied through basal insulin or hyperinsulinemic euglycemic clamps (clamp design shown in Fig. 1A) after 2 h of fasting as previously described (25). Briefly, this consisted of primed continuous infusion of [3-3H]-glucose tracer (0.5 μCi/min) at t = 0 that lasted for 4 h. Basal insulin (1 mU/kg/min) or hyperinsulinemic (3 mU/kg/min) clamps began at t = 120 min and lasted for 2 h. Along with insulin, somatostatin (3 μg/kg/min) was coinfused to suppress secretion of endogenous insulin as well as counterregulatory hormones. Euglycemia was maintained with a variable infusion rate of 25% glucose solution. To minimize blood loss, erythrocytes after collection of plasma at multiple time points were resuspended with sterile saline and reinfused. At t = −120 min, artificial cerebrospinal fluid or S961, a specific insulin receptor antagonist, was infused bilaterally into the MBH at a rate of 0.36 μL/h for 6 h.
Figure 1.

Hypothalamic insulin signaling is required for the control of BCAA metabolism. A: Normoinsulinemic and hyperinsulinemic euglycemic clamp schematic in Sprague-Dawley male rats paired with hypothalamic infusion of artificial cerebrospinal fluid (aCSF) or S961, a specific IR antagonist, with the removal of food 2 h before clamps. B: Body weight before pancreatic clamps. C: Plasma BCAAs at the end of clamps measured by spectrophotometric assay. D: Western blots showing total BCKDH, the rate-limiting enzyme of BCAA breakdown, and phosphorylated, inactive pBCKDH protein expression in the liver. E: Quantification of Western blot data expressed as the ratio of pBCKDH to BCKDH, an inactivation index of BCKDH activity. N = 5–6/group; values are mean ± SEM; *P < 0.05.
Mice
Hyperinsulinemic euglycemic clamps were performed on WT and AgRP IR KO mice (2-h fasting; clamp schematic shown in Fig. 3C) as previously described (31). After blood sampling at baseline (t = −100 min), a primed continuous infusion of [U-13C-6]-D-glucose (0.6 μmol/kg/min) was infused until t = 120 min for determination of glucose flux. Starting at t = 0, insulin was first primed and then continuously infused at 3 mU/kg/min for 2 h. This insulin dose was chosen instead of the 4 mU used in our earlier study (31) to more closely match physiological levels of circulating insulin at postprandial state. Blood glucose was monitored every 10 min, and blood was collected at multiple time points. Erythrocytes mixed in saline were infused back into the mice to minimize blood loss.
At the termination of clamps, animals were euthanized, and livers were harvested, snap frozen in liquid nitrogen, and stored at −80°C for further analysis.
Glucose Flux Analysis
Hepatic glucose production (hGP) and whole-body Rd from rat and mouse pancreatic clamps were analyzed as described elsewhere (31,34).
Stereotaxic AAV-DREADD-mCherry Injections
Animals were first anesthetized with i.p. injection of ketamine (80 mg/kg) and xylazine (20 mg/kg). Adeno-associated virus (AAV) carrying a gene construct of stimulatory M3-muscarinic receptor (AAV-hSyn-DIO-hM3D[Gq]-mCherry, serotype 8; University of North Carolina Vector Core, Chapel Hill, NC) was bilaterally injected into the MBH of WT or AgRP-IRES-Cre mice by a Hamilton syringe with a 33-gauge needle (Hamilton Company, Reno, NV). The coordinates were −1.5 mm AP, 0.25 mm L, and −5.8 mm DV in reference to the bregma. ChAT-Cre mice and Phox2b-Cre mice were bilaterally injected with excitatory AAV-hSyn-DIO-hM3D(Gq)-mCherry or inhibitory AAV-hSyn-DIO-hM4D(Gi)-mCherry into the DMV. The coordinates were −7.83 mm AP, 0.25 mm L, and −4.5 mm DV in reference to the bregma. Animals were injected twice per side with 1 μL per injection for 2.5 min, and the needle was left for an extra 5 min after the second injection to ensure adequate diffusion of the virus into the target neurons. Animals were monitored for 4–6 days for recovery after surgery.
Fasting/Ghrelin Experiment
C56Bl/6 WT mice age 5–6 months were fasted overnight (16 h), and blood samples were collected via tail nick at t = 0 before fasting and t = 2, 4, and 16 h after fasting for plasma BCAA measurement. Blood glucose was monitored at each time point via hand-held glucometer (AlphaTrak; Zoetis). Another cohort of WT mice (age 3–4 months) at fed state were injected with either saline or acylated ghrelin (1 mg/kg i.p.) in the absence of food, and blood was collected at t = 0, 30, 60, and 120 min for measurement of plasma BCAAs and blood glucose. The ghrelin dose used here has been shown to achieve plasma ghrelin levels similar to those observed in mice after 16–24 h of fasting and also significantly raise blood glucose compared with saline-injected mice (35,36).
AgRP Neuronal Stimulation
After 2 weeks of AAV-hM3Dq DREADD injection within the MBH in WT and AgRP-IRES-Cre mice, the designer drug clozapine-N-oxide (CNO) was injected (1 mg/kg i.p.) at fed state, and 3-h food intake was assessed for validation of DREADD expression and stimulation of AgRP neurons. AgRP-Cre mice with at least twofold increase in food intake compared with WT mice were used in the study. After 4 days of washout period, blood samples from both groups at fed state were collected at baseline t = 0 min through tail nick. Then, CNO was injected in the absence of food, and blood was collected 1 h after for measurement of plasma BCAAs. Immediately afterward, the animals were euthanized, and brains were harvested for confirmation of DREADD expression via immunostaining. Livers were harvested, snap frozen in liquid nitrogen, and stored at −80°C for further analysis.
Vagal Manipulation Experiment
Both overnight-fasted ChAT-Cre and Phox2b-Cre mice expressing hM3Dq (excitatory) or hM4Di (inhibitory) in the DMV were first injected with either saline or CNO (1 mg/kg i.p.) in the absence of food. After 1 h, animals underwent transcardial perfusion, after which brains were harvested and fixed in 4% paraformaldehyde in PBS, and their blood samples were collected from tail nick for assessment of plasma BCAA levels. Plasma samples from ChAT-Cre were obtained from our previous study (32), in which DREADD-mediated neuronal activity of ChAT-expressing neurons was validated through electrophysiology and immunohistochemistry.
GTT
After overnight fasting (∼16 h), WT and AgRP-Cre mice expressing hM3D(Gq) DREADD in AgRP neurons were first injected with CNO (1 mg/kg i.p.) in the absence of food; 1 h later, a glucose tolerance test (GTT) was performed in which they received an injection of glucose (1.5 g/kg i.p.) and a second injection of CNO simultaneously, as described elsewhere (37). Blood samples from tail nick were obtained at baseline (t = 0) and t = 15, 30, 60, 90, and 120 min after glucose injection. Blood glucose was determined using a hand-held glucometer (AlphaTrak; Zoetis).
Immunohistochemistry
After a single i.p. CNO injection, transcardial perfusion was performed in mice 1 h afterward with cold PBS (pH 7.4). Harvested brains were immersed in 4% paraformaldehyde in PBS for 24 h at 4°C and then transferred to 20% sucrose solution for 1 day before they were embedded in Tissue-Tek OCT compound. Brains were mounted on a freezing microtome (Leica CM3050S; Leica, Wetzlar, Germany) and cut into sections 14 μm in thickness. For immunofluorescence staining, sections were blocked in 3% normal donkey serum (Sigma Aldrich) for 1 h at room temperature and then incubated with rabbit c-Fos antibody (1:1,000, sc-52; Santa Cruz Biotechnology) for 2 days at 4°C. Sections were washed with PBS and incubated with Alexa Fluor 488–labeled goat anti-rabbit secondary antibody (1:500; Invitrogen, Carlsbad, CA) at room temperature for 1 h. c-Fos and mCherry fluorescent images were visualized using an Olympus fluorescence microscope (Olympus, Tokyo, Japan).
BCAA Assay
Total BCAAs (leucine, isoleucine, and valine) from plasma samples were measured by a spectrophotometric assay measuring NADH generated from BCAA oxidation as previously described (25).
Western Blots
Frozen liver samples were processed for Western blots as previously described (25). Briefly, 80 mg of tissue was homogenized in lysis buffer, and supernatants were collected after centrifugation at 13,000 rpm at 4°C for 15 min; 20 ug of protein was loaded in 4–12% NuPAGE gels (Invitrogen). Primary antibodies against total BCKDH and the phosphorylated form of BCKDH (pBCKDH) were obtained from Abcam (Cambridge, MA). Membranes were incubated at 4°C overnight in primary antibodies at a 1:1,000 dilution in blocking solution. After incubation in secondary antibody solution (goat anti-rabbit, 1:4,000 dilution; Thermo Scientific), the membranes were scanned with the LI-COR Odyssey (LI-COR), and BCKDH and pBCKDH were quantified using Odyssey 3.0 software.
Insulin ELISA
Plasma insulin levels were determined with the Mouse Insulin ELISA kit (10–1247–01; Mercodia, Winston Salem, NC) according to the manufacturer’s protocol. Data were analyzed by performing cubic spline regression with GraphPad Prism (GraphPad Software).
Statistics
For the rat clamp experiment, body weight, plasma BCAAs, BCKDH and pBCKDH proteins, hGP, GP suppression (%), Rd, and plasma insulin were analyzed by one-way ANOVA followed by Bonferroni post hoc test. Blood glucose and glucose infusion rate (GIR) were analyzed by two-way repeated measures ANOVA, with treatment as a between-subject factor and time as a within-subject factor, followed by Bonferroni post hoc multiple comparisons. For the mouse clamp experiment, plasma BCAAs, hGP, and plasma insulin were analyzed by two-way ANOVA, with treatment and time (basal vs. clamp) factors followed by Bonferroni post hoc test. pBCKDH/BCKDH, GIR, hGP suppression (%), and Rd were analyzed by t test. Association between plasma BCAAs and hGP was analyzed by regression analysis. Plasma BCAAs in the fasting/refeeding experiment were analyzed by t test. In the AgRP neuronal stimulation experiment, food intake and blood glucose during the GTT were analyzed by two-way ANOVA followed by Bonferroni post hoc test. Plasma BCAAs and BCKDH and pBCKDH proteins were analyzed by t test. In the fasting/ghrelin experiment, body weight was analyzed by t test, and plasma BCAAs and blood glucose were analyzed by paired t test in the fasting experiment, whereas in the ghrelin experiment, plasma BCAAs and blood glucose were analyzed by two-way repeated measures ANOVA followed by Bonferroni multiple comparisons. In the vagal manipulation experiment, plasma BCAAs were analyzed by t test. All data are expressed as mean ± SEM. Statistically significant difference was set at P < 0.05.
Data and Resource Availability
The data sets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request. Any remaining tissues or plasma samples generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Results
Insulin Action in the MBH Is Required for Suppression of Circulating BCAA Levels
Systemic insulin during hyperinsulinemic clamps effectively lowers circulating BCAAs in a dose-dependent manner (25,38). We previously demonstrated that insulin infusion directly into the MBH is sufficient to decrease plasma BCAAs (25). To test if hypothalamic insulin signaling is required to regulate systemic BCAA metabolism, we performed hyperinsulinemic clamps in rats while blocking insulin signaling in the MBH via infusion of S961, a specific insulin receptor antagonist. Using the experimental design shown in Fig. 1A ensured the maintenance of euglycemia during hypothalamic manipulations. Moreover, somatostatin was coinfused to inhibit secretion of insulin and any counterregulatory hormones such as glucagon and corticosterone that could potentially alter glucose metabolism, thereby allowing us to isolate the role of hypothalamic insulin action specifically in BCAA metabolism. Preclamp body weight was similar between groups (Fig. 1B). Animals under hyperinsulinemic clamps (3 mU) displayed lower plasma BCAAs (Fig. 1C) compared with those with basal insulin levels (1 mU). However, systemic hyperinsulinemia failed to suppress plasma BCAAs in rats with inhibition of MBH insulin signaling via S961. BCKDH is the rate-limiting enzyme involved in BCAA degradation (39). Although it is expressed and active in many tissues (40,41), its activity in the liver has been shown to be mainly responsible for insulin-induced breakdown of BCAAs (25). In parallel to insulin-induced reduction of plasma BCAAs, hyperinsulinemic animals displayed a lower inactive state of pBCKDH without affecting its total protein (Fig. 1D and E), indicative of greater catabolic activity. This effect was abolished in rats with a hypothalamic insulin signaling blockade, which likely contributed to the higher levels of circulating BCAAs. These findings provide evidence that hypothalamic insulin action is mandatory for BCAA homeostasis, which is most likely mediated by efficient BCAA oxidation in the liver.
Hypothalamic Control of Systemic BCAAs Is Linked to Glucose Metabolism
Multiple studies have indicated that BCAAs may be causally linked to defective glycemic control (7,13,14,22). To test if hypothalamic regulation of BCAAs is associated with glucose metabolism, we used a tracer dilution technique during hyperinsulinemic clamps to assess insulin action, which is defined as the ability of insulin to suppress hGP and promote glucose disposal. Euglycemia was maintained throughout basal and hyperinsulinemic clamps (Fig. 2A and G). GIR, or the amount of glucose necessary to maintain euglycemia, was significantly higher in hyperinsulinemic animals compared with animals at basal insulin (Fig. 2B and C). However, inhibition of insulin signaling in the MBH markedly reduced GIR by nearly 50%, indicating impaired whole-body insulin sensitivity. This was further reflected by insulin-induced suppression of hGP (43%), but not when insulin signaling in the MBH was abrogated (Fig. 2D and E). Of note, the ability of insulin to enhance Rd was not affected by blocking hypothalamic insulin signaling (Fig. 2F). Importantly, plasma BCAAs were positively correlated with hGP (R2 = 0.54) (Fig. 2H). These results suggest that high circulating BCAAs induced by impaired hypothalamic insulin signaling are associated with defective glucose metabolism, thus highlighting a role of the BCAA regulatory pathway in the control of glucose homeostasis.
Figure 2.
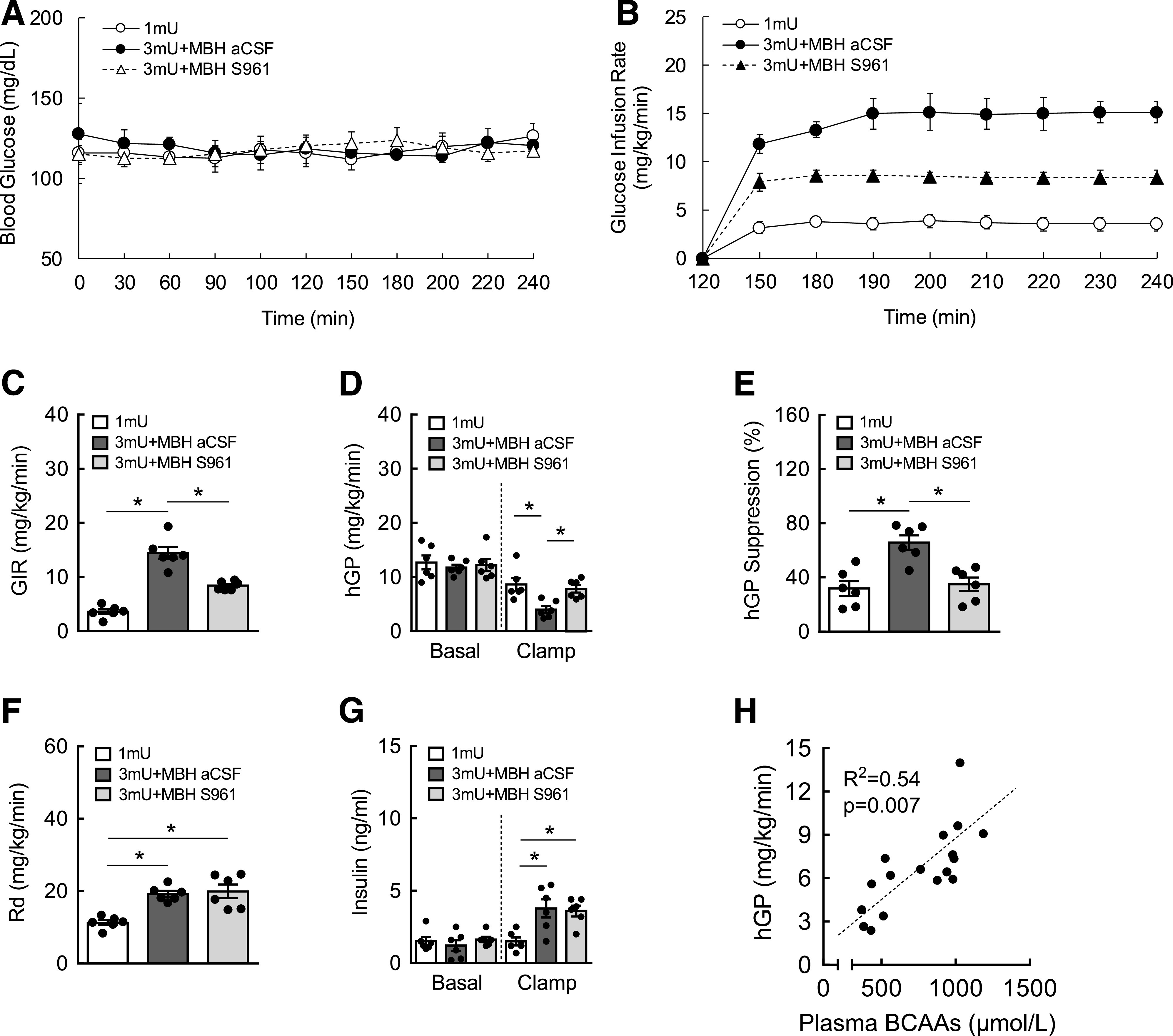
Blockade of MBH insulin signaling impairs suppression of hGP during hyperinsulinemic euglycemic clamps. A: Blood glucose maintained at euglycemia during clamps. B: GIR (mg/kg/min) during clamps. C: Average GIR between groups. D: hGP (mg/kg/min) during basal and clamp periods. E: Suppression of hGP (%) by insulin. F: Whole-body Rd during clamps. G: Plasma insulin levels during basal and clamp periods. H: Regression analysis between plasma BCAAs and hGP during clamps. N = 6/group; values are mean ± SEM; *P < 0.05. aCSF, artificial cerebrospinal fluid.
Insulin Signaling in AgRP Neurons Mediates the Regulation of BCAA Metabolism
AgRP neurons localized in the MBH are known to control energy balance and nutrient partitioning (1,26). Because they express abundant IRs (28,29), we sought to determine if insulin action on AgRP neurons is critical for BCAA metabolism. AgRP-Cre mice were crossed with IR flox mice to generate AgRP IR KO mice (29). To test if circulating BCAAs are regulated in a physiological setting, we used a fasting/refeeding model in which both WT and AgRP IR KO mice were first fasted overnight and then refed for 2 h (Fig. 3A). As expected, both groups displayed higher plasma BCAAs 2 h after refeeding compared with their fasting baseline. However, the percent rise in BCAAs was considerably smaller in WT (51%) compared with AgRP IR KO mice (86%), most likely because of feeding-induced insulin action (Fig. 3B). Next, hyperinsulinemic euglycemic clamps were performed on these mice to isolate the role of insulin signaling in AgRP neurons in BCAA regulation independent of changes in circulating glucose and insulin. Both groups showed similar plasma BCAA levels at preclamp baseline, and WT mice exhibited insulin-dependent suppression of circulating BCAAs (Fig. 3D). In contrast, circulating BCAAs remained elevated in AgRP IR KO mice during hyperinsulinemia. In line with our previous results (25), this was likely a result of impaired hepatic BCAA breakdown, as evidenced by significantly higher inactive pBCKDH without difference in total BCKDH in the livers of AGRP IR KO mice (Fig. 3E and F). To examine the potential relationship between AgRP neuronal control of BCAAs and glucose metabolism, the tracer dilution technique was used, which allowed us to assess glucose flux. Blood glucose was not different between groups throughout the clamps (Fig. 4A). As expected, compared with WT mice, AgRP IR KO mice displayed significantly lower GIR (Fig. 4B and C) and impaired suppression of hGP (Fig. 4D and E), without changes in Rd (Fig. 4F). Our data confirm the achievement of systemic hyperinsulinemia, as shown by markedly increased plasma insulin during the hyperinsulinemic clamp period (Fig. 4G). Consistent with our earlier result in the current study (Fig. 2H), a positive correlation was observed between plasma BCAAs and glucose output (hGP) (Fig. 4H). Collectively, these findings suggest that insulin signaling in AgRP neurons mediate hypothalamic control of BCAA homeostasis and that this particular neuroendocrine pathway is closely associated with glucose metabolism.
Figure 4.
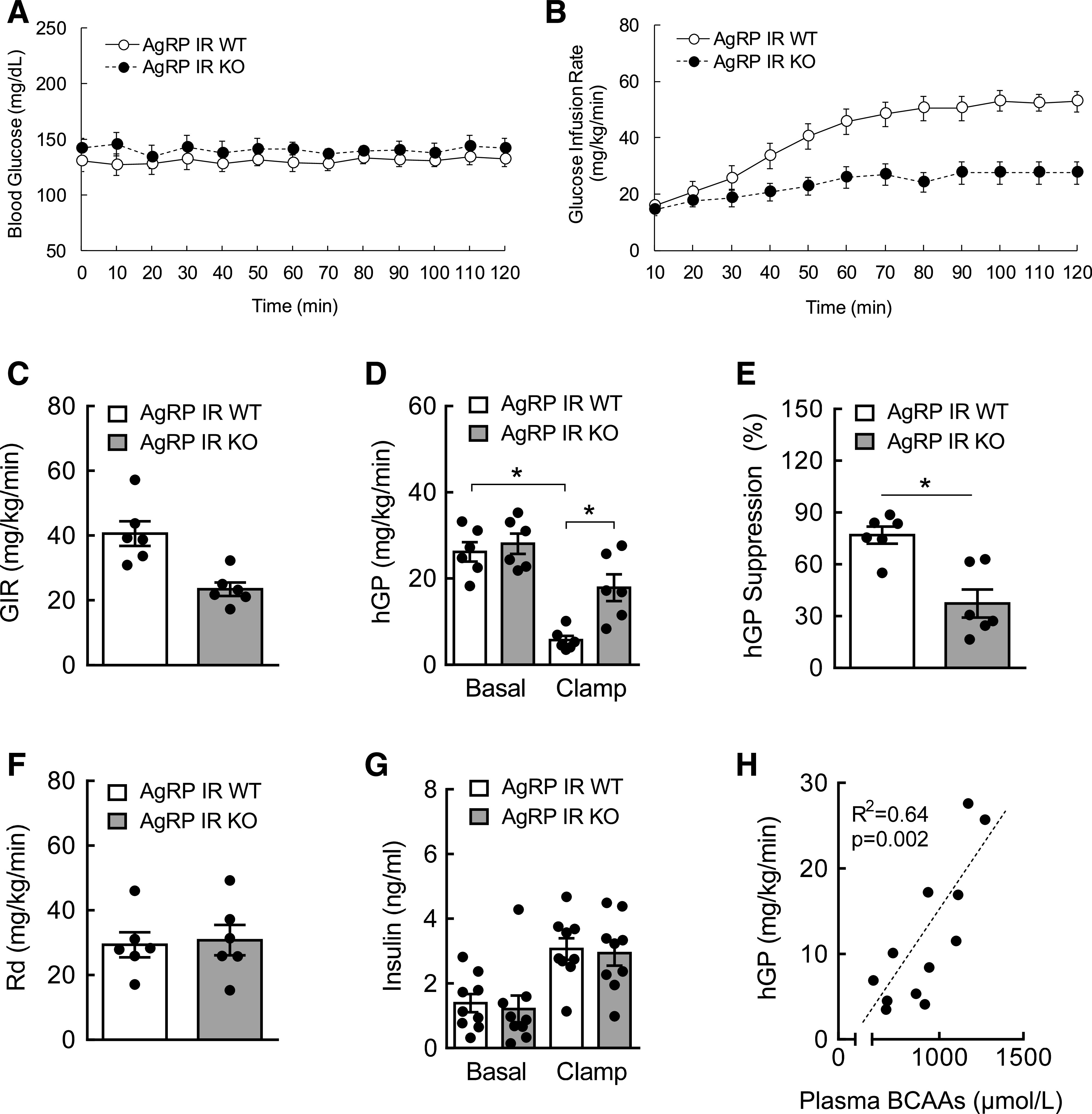
AgRP IR KO mice show defective glucose metabolism during hyperinsulinemia. A: Blood glucose during hyperinsulinemic (3 mU) clamps. B: GIR during clamps. C: Average GIR. D: hGP during basal and clamp periods. E: Suppression of hGP (%) by insulin. F: Whole-body Rd during clamps. G: Plasma insulin levels during basal and clamp periods. H: Regression analysis between plasma BCAAs and hGP. N = 6–9/group; values are mean ± SEM; *P < 0.05.
Acute Activation of AgRP Neurons Increases Plasma BCAAs and Impairs Glucose Tolerance
Insulin hyperpolarizes and inhibits AgRP neurons (28,29), implying that these neurons from AgRP IR KO mice were most likely disinhibited (i.e., remained active) as a result of the lack of insulin signaling, which led to impaired suppression of plasma BCAAs. To test this concept further, we used a chemogenetic DREADD approach to directly activate AgRP neurons. This was accomplished by injecting AAV-hM3Dq-mCherry bilaterally into the MBH of AgRP-Cre mice and administering an i.p. injection of CNO to acutely and selectively stimulate AgRP neurons. WT mice receiving an AAV injection and CNO treatment were used as a control group. Immunostaining confirmed DREADD expression in the MBH of AgRP-Cre mice in which AgRP neurons were localized (Fig. 5A). Because voracious feeding behavior is considered an excellent functional readout of AgRP neuronal activation (42), WT and AgRP-Cre mice were injected with CNO, and food intake was measured over the course of 3 h. In support of DREADD expression, AgRP-Cre mice dramatically increased food intake (+350%), thereby confirming activation of AgRP neurons (Fig. 5B). Next, we tested if acute stimulation of AgRP neurons raises plasma BCAAs. Because feeding can influence circulating BCAA levels, these neurons in AgRP-Cre mice were activated by CNO in the absence of food, and plasma BCAAs were measured before and 1 h after stimulation. Although there was no change in WT mice, plasma BCAAs levels were significantly increased (+50%) after 1 h in AgRP-Cre mice (Fig. 5C). This was associated with higher inactive hepatic pBCKDH protein (Fig. 5D and E). To establish a link between the BCAA regulatory pathway and glucose metabolism, we performed GTT. In parallel with the findings by Steculorum et al. (37), AgRP-Cre mice demonstrated marked glucose intolerance compared with WT mice after acute stimulation of AgRP neurons (Fig. 5F). These results are complementary to our current findings and suggest that the stimulation of AgRP neurons, equivalent to the lack of insulin signaling in AgRP neurons, impairs BCAA metabolism, which is also linked to defective glucose homeostasis.
Figure 5.

Chemogenetic activation of AgRP neurons raises plasma BCAAs and impairs glucose tolerance. AAV-hSyn-DIO-hM3D(Gq)-mCherry was injected bilaterally into the MBH of WT or AgRP-Cre mice. A: Immunostaining of mCherry as confirmation of DREADD expression in the MBH. B: Cumulative food intake after DREADD stimulation of AgRP neurons with CNO (1 mg/kg i.p.) at fed state. C: Plasma BCAAs at baseline (t = 0) and 1 h after activation of AgRP neurons. D: Western blots showing total BCKDH and pBCKDH in the liver 1 h after AgRP neuronal stimulation. E: Quantification of Western blot data expressed as the ratio of pBCKDH to BCKDH. F: GTT (1.5 g/kg i.p.) performed in overnight-fasted mice 1 h after DREADD activation of AgRP neurons with CNO (1 mg/kg i.p.). N = 5–6/group; values are mean ± SEM; *P < 0.05 compared with WT controls.
Fasting or Ghrelin Treatment Elevates Circulating BCAAs
AgRP neurons are activated upon prolonged fasting (43,44). We sought to test if results from the DREADD experiment could be recapitulated via a more natural and physiological approach. To this end, we conducted a fasting experiment in which normal mice were subjected to fasting that would gradually stimulate AgRP neuronal activity as a function of time and sampled blood to measure BCAAs. Fasting did not significantly alter body weight (Fig. 6A). Basal plasma BCAAs were maintained for up to 2 h of fasting (Fig. 6B). However, they were significantly increased by 83% at the end of 4 h of fasting and stayed elevated until the end of the fasting period (16 h). Blood glucose did not change as expected, perhaps through the orchestration of multiple physiological mechanisms to maintain euglycemia. The orexigenic hormone ghrelin is secreted from the stomach in response to fasting and stimulates AgRP neurons and triggers hunger mode to modify energy partitioning (45). To determine if ghrelin-induced activation of AgRP neurons mimics the effect of fasting on circulating BCAAs, WT mice at fed state were injected i.p. with either saline or ghrelin in the absence of food. Although body weight was similar (Fig. 6D), ghrelin-treated mice showed significantly higher plasma BCAAs compared with saline-treated controls at t = 30 min (Fig. 6E). Calculation of percent change from baseline indicated that ghrelin increased plasma BCAAs by 20%, as compared with saline injection, which decreased BCAAs by 23% (Fig. 6F). Interestingly, blood glucose was also markedly increased at t = 30 min after ghrelin injection (55% from baseline), and this persisted for up to t = 120 min (Fig. 6G and H), without any change in saline-treated animals. Altogether, these data suggest that physiological activation of AgRP neurons is sufficient to elevate circulating BCAAs and raise blood glucose, thus further supporting our chemogenetic findings.
Figure 6.
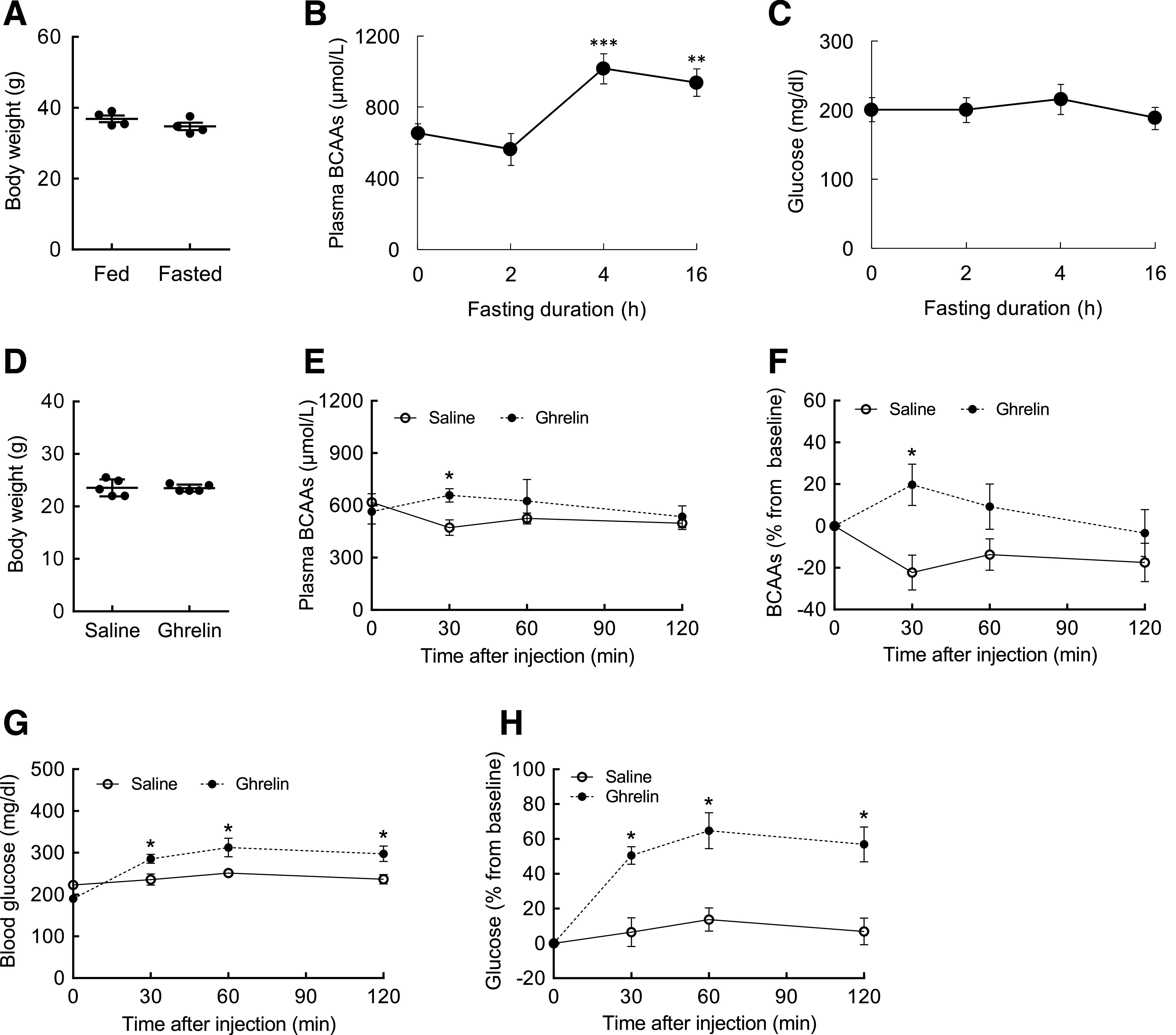
Fasting or ghrelin treatment acutely increases plasma BCAAs and induces hyperglycemia. A: Body weight in WT mice before and after fasting. B: Plasma BCAAs during fasting period (t = 0, 2, 4, and 16 h). C: Blood glucose during fasting period. D: Body weight in WT mice before treatment. E: Plasma BCAA profile for 120 min after a single injection of either saline or ghrelin (1 mg/kg i.p.). F: Plasma BCAA levels expressed as percent change from baseline (t = 0) after saline or ghrelin injection. G: Blood glucose. H: Blood glucose expressed as percent change from baseline (t = 0). N = 4–5/group; values are mean ± SEM; *P < 0.05 compared with saline group; **P < 0.01, ***P < 0.001 compared with baseline (t = 0 h).
Acute Stimulation of the Vagal Outflow Suppresses Plasma BCAAs
The ANS serves as an important conduit that links the central nervous system (CNS) to peripheral organs for execution of involuntary physiological processes, including gastrointestinal and liver functions (30). Because parasympathetic vagal activation is often associated with improved metabolic homeostasis (46), we sought to determine if stimulating vagal outflow could lower plasma BCAAs. To test this, stimulatory AAV-hSyn-hM3D(Gq)-mCherry was injected into the DMV in ChAT- or Phox2b-Cre mice for DREADD expression targeted to preganglionic parasympathetic neurons. These vagal motor neurons were then activated by CNO in the absence of food (Fig. 7A) (activation of ChAT-expressing neurons in the DMV shown in our previous article (32)), and plasma BCAAs were measured 1 h later from tail blood. In a separate cohort, inhibitory AAV-hSyn-DIO-hM4D(Gi)-mCherry was injected into the DMV of ChAT- or Phox2b-Cre mice to test if inhibiting vagal motor neurons leads to changes in plasma BCAAs. Although acute activation of ChAT-expressing neurons in the DMV significantly decreased plasma BCAAs by 47% compared with controls (Fig. 7B), inhibiting these neurons did not alter circulating BCAAs (Fig. 7C). In contrast, no difference was observed after stimulation or inhibition of Phox2b-expressing neurons in the DMV (Fig. 7D and E). These results suggest that induction of parasympathetic outflow through stimulation of ChAT-expressing vagal motor neurons represents a potential mechanism for BCAA regulation.
Figure 7.

Acute stimulation of vagal motor neurons in the DMV significantly lowers plasma BCAAs. AAV-hSyn-DIO-hM3D(Gq)-mCherry or AAV-hSyn-DIO-hM4D(Gi)-mCherry was bilaterally injected into the DMV of overnight-fasted ChaT- or Phox2b-Cre mice. A and B: Plasma BCAAs in ChaT-Cre mice 1 h after saline or CNO (1 mg/kg i.p.) injection. C: Immunostaining of c-Fos and 3Dq mCherry in the DMV of Phox2b-Cre mice 30 min after CNO injection. D and E: Plasma BCAAs in Phox2b-Cre mice 1 h after saline or CNO injection. N = 6–10/group; values are mean ± SEM; *P < 0.05 compared with saline group. CC, central canal.
Discussion
BCAAs are known to provide beneficial effects on protein turnover and muscle wasting in patients with cirrhosis, cancer, or sepsis (47–49), but recent evidence links BCAAs to insulin resistance and impaired glycemic control (13,14,23), key features found in obesity and diabetes. Although the molecular catabolic pathway of BCAAs has been well delineated, the physiological mechanisms that regulate BCAA degradation and hence circulating BCAA levels have remained elusive. The current study reveals that insulin signaling in the MBH, more specifically in AgRP neurons, is required for the control of hepatic BCAA metabolism and lowering of plasma BCAAs. Through physiological and complementary chemogenetic approaches to stimulate AgRP neuronal activity, we further demonstrate that activation of AgRP neurons, analogous to suppressed insulin signaling, impairs hepatic BCAA breakdown and raises plasma BCAAs. Interestingly, this BCAA regulatory pathway was found to be associated with glucose metabolism. Mechanistically, our results show that the CNS control of BCAAs may be mediated in part by vagal outflow.
We previously reported that hypothalamic insulin infusion in rats increases hepatic BCAA breakdown and lowers circulating BCAA levels (25). By demonstrating that blocking MBH insulin signaling by S961 impairs the ability of insulin to suppress plasma BCAAs during hyperinsulinemic clamps, our current findings support the concept that hypothalamic insulin signaling is crucial for regulating BCAA metabolism independently of glucose. Although many tissues, including the liver, white adipose tissue, and muscle, are capable of oxidizing BCAAs (23,24,40), defective hepatic BCAA catabolism in mice with impaired MBH insulin signaling is consistent with our previous report in which the liver, but not white adipose tissue or muscle, enhanced BCAA breakdown in response to insulin (25).
AgRP neurons are well positioned to mediate hypothalamic control of BCAAs because of their localization in the MBH and expression of IRs (28,29). Under a physiological fasting/refeeding setting where endogenous glucose and insulin are not manipulated, our results show that AgRP IR KO mice fail to suppress refeeding-induced rise of plasma BCAAs. Furthermore, the inability of these mice to lower plasma BCAAs, along with impaired hepatic BCAA breakdown under hyperinsulinemic clamps, clearly points to insulin action in AgRP neurons as a prerequisite for BCAA regulation. By mimicking defective insulin signaling in AgRP neurons by directly stimulating AgRP neuronal activity, the chemogenetic approach allowed us to test its effect on BCAA metabolism without inducing developmental changes or permanently altering genes. Similar BCAA-increasing outcomes after AgRP neuronal activation strongly support the concept that AgRP neurons are critical players for the regulation of circulating BCAA levels. Additional studies are needed to test if elevated plasma BCAAs persist after chronic or repeated activation of AgRP neurons by DREADD. It is also important to note that we investigated primarily the role of AgRP neurons in this study; there may be other neuronal populations in the MBH or extrahypothalamic areas in the brain that could mediate hypothalamic insulin action on BCAA homeostasis.
It is well established that fasting activates AgRP neurons (43,44). Our present data demonstrate that activation of AgRP neurons either by fasting or by injection of the fasting-induced hormone ghrelin in fed mice raises plasma BCAAs independently of body weight and food intake. These recapitulate the observations in mice with chemogenetically stimulated AgRP neurons, suggesting that indeed, AgRP neurons physiologically regulate circulating BCAAs. The difference in the magnitude of BCAA excursion between fasting versus ghrelin treatment is most likely explained by more dramatic endocrine and autonomic changes brought about by fasting. Although ghrelin treatment increased blood glucose for up to 2 h, fasting for up to 16 h did not change blood glucose. That is expected, because the animals are able to maintain euglycemia through orchestrating the complex interplay between regulatory and counterregulatory responses.
Insulin action in the MBH has been shown to affect glucose partitioning in rodents (29,34,50). Our observation that hypothalamic infusion of S961 impairs suppression of hGP without affecting whole-body Rd is in agreement with reports by others in which insulin signaling blockade is accomplished by an IR antibody, IR antisense oligonucleotide, or KATP channel blocker (34,50). It is important to note that the inability of insulin to suppress hGP is one of the major features observed in insulin-resistant individuals (1). The positive correlation between plasma BCAAs and hGP shown in this study supports the emerging concept that BCAAs may lower insulin sensitivity and impair glucose metabolism (7,13,14), even in an acute setting and independently of other comorbidities, such as obesity. Our finding is also consistent with the results from humans demonstrating the failure of insulin to lower circulating BCAAs in insulin-resistant individuals (51) and further raises the possibility of the potential mechanistic link between CNS-driven BCAA control and glucose homeostasis. Moreover, the observation of impaired hGP in AgRP IR KO mice or glucose intolerance in AgRP-Cre mice with DREADD-stimulated AgRP neurons extends the possibility of interplay between BCAA and glucose through insulin action in AgRP neurons.
Acute chemogenetic stimulation of ChAT-expressing cholinergic neurons in the DMV significantly reduced plasma BCAAs, suggesting that the vagal outflow can control BCAA metabolism. Sohn et al. (52) showed earlier that MTII, an MC4R agonist, directly inhibits cholinergic neurons in the DMV through the opening of a KATP channel. Another study reported that although deletion of MC4R in cholinergic neurons raises plasma insulin, restoration of MC4R in these neurons lowers insulin in mice (33). These findings imply that stimulation of cholinergic neurons may increase circulating insulin, which would consequently lower plasma BCAAs. This is in agreement with our findings of reduced plasma BCAAs after acute activation of cholinergic neurons in the DMV. In contrast, DREADD-induced inhibition of these neurons failed to produce any change in plasma BCAAs. Although the exact reasons are not clear, nutritional status may have played a role. Because mice were subjected to overnight fasting, the sympathetic nervous system would have been activated to mobilize fuel sources, whereas the parasympathetic, vagal activity would have been dampened. As a result, compared with saline-treated controls, mice with stimulated cholinergic neurons in the DMV may have had restored the vagal outflow to peripheral tissues, including the liver, and most likely had enhanced BCAA breakdown, leading to lower plasma BCAAs. However, inhibition of these neurons in mice that already have low vagal tone induced by overnight fasting is not expected to produce any significant effect beyond what is observed in fasted, saline-treated mice. Examination of the effect of inhibiting these neurons at fed state should address this issue. We also cannot rule out the possibility that the lack of BCAA change after DMV neuronal inhibition may be due to the limited gene transfer and/or transduction efficiency in the targeted neurons. Taken together, along with our recent report showing improved glucose tolerance in mice after stimulation of the same vagal motor neurons (32), these results indicate the possibility of a potential functional link between BCAA and glucose metabolism that is mediated through the parasympathetic nervous system. On the basis of these observations, it is reasonable to speculate that vagal activity may be impaired or suppressed in individuals with obesity/diabetes. This warrants further investigation. Alternatively, sympathetic outflow to the liver may also play a role in the control of BCAAs. Furthermore, in light of the recent study showing active BCAA breakdown in cold-stimulated brown adipose tissue that contributes to systemic BCAA clearance (53), it is possible that insulin-induced inhibition of AgRP neurons could activate sympathetic outflow to the brown adipose tissue, which in turn would induce efficient BCAA breakdown and help lower circulating BCAAs. This would be another novel mechanism by which systemic BCAAs are regulated through AgRP neurons.
Unlike with ChAT-expressing neurons, stimulation of Phox2b-expressing neurons in the DMV did not alter plasma BCAAs. It is important to note that ChAT-expressing, cholinergic neurons in the DMV indeed represent preganglionic vagal motor neurons, whereas Phox2b-expressing neurons do not necessarily possess the enzyme ChAT and therefore do not produce acetylcholine (54). Initially expressed in neural precursors during the development of both sympathetic and parasympathetic ganglia (54), Phox2b may have evolved to be expressed in heterogeneous neuronal populations with different autonomic functions, even within the DMV. Results from Rossi et al. (33) support this notion by demonstrating that in contrast to ChAT-expressing cholinergic neurons, reactivation of MC4R in Phox2b-expressing neurons does not affect body weight or energy expenditure and is not able to lower hGP.
We speculate that hypothalamic insulin resistance, more specifically insulin resistance in AgRP neurons, may be an early event induced by a combination of genetic and environmental factors that promotes elevated BCAAs, which would then causally contribute to systemic insulin resistance and impaired glycemic control, as shown by other investigators (7,13,18,19). Another potential mechanism relates to the ability of BCAAs to hyperactivate the mammalian target of rapamycin pathway, leading to insulin resistance (7,55,56). It is possible that this mechanism also exists at the level of the MBH, which would consequently further hypothalamic insulin resistance and worsen the ability of insulin to regulate glucose homeostasis. In support of this concept, Ono et al. (57) showed that high-fat feeding increases activation of hypothalamic S6 kinase, an enzyme downstream of mammalian target of rapamycin, and impairs suppression of hGP. They further showed that constitutive activation of hypothalamic S6K in chow-fed lean animals leads to similar glucose defects, whereas suppressing S6K restores the ability of hypothalamic insulin to suppress hGP. Additional investigations are warranted to test this hypothesis.
In conclusion, our findings suggest that hypothalamic insulin controls systemic BCAA metabolism through AgRP neurons. Both physiological and chemogenetic stimulations of AgRP neurons are sufficient to elevate circulating BCAAs and glucose, thus providing an opportunity to closely examine the potential mechanistic link between CNS-driven BCAA control and glucose homeostasis. Furthermore, the neuroendocrine BCAA regulatory pathway may be mediated mainly via the parasympathetic nervous system.
Article Information
Funding. This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, grant DK099463; a Presidents’ Collaborative Research Initiative grant from Texas Tech University Systems (A.C.S.); and National Research Foundation of Korea grants 2017R1A6A3A1131814 and 2018R1A5A2025964 (H.J.C.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. R.B.G. and C.N. collected and analyzed data and wrote the manuscript; H.H.R. collected data and edited the manuscript; H.J.C. conceived an experiment, analyzed data, and edited the manuscript; and A.C.S. conceived and supervised the study, collected and analyzed data, and wrote the manuscript. A.C.S. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Ruud J, Steculorum SM, Brüning JC. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat Commun 2017;8:15259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001;414:799–806 [DOI] [PubMed] [Google Scholar]
- 3.Felig P, Marliss E, Cahill GF Jr. Plasma amino acid levels and insulin secretion in obesity. N Engl J Med 1969;281:811–816 [DOI] [PubMed] [Google Scholar]
- 4.Kim JY, Park JY, Kim OY, et al. Metabolic profiling of plasma in overweight/obese and lean men using ultra performance liquid chromatography and Q-TOF mass spectrometry (UPLC-Q-TOF MS). J Proteome Res 2010;9:4368–4375 [DOI] [PubMed] [Google Scholar]
- 5.McCormack SE, Shaham O, McCarthy MA, et al. Circulating branched-chain amino acid concentrations are associated with obesity and future insulin resistance in children and adolescents. Pediatr Obes 2013;8:52–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mihalik SJ, Goodpaster BH, Kelley DE, et al. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity (Silver Spring) 2010;18:1695–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newgard CB, An J, Bain JR, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 2009;9:311–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.She P, Van Horn C, Reid T, Hutson SM, Cooney RN, Lynch CJ. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Metab 2007;293:E1552–E1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams SH, Hoppel CL, Lok KH, et al. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr 2009;139:1073–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang TJ, Larson MG, Vasan RS, et al. Metabolite profiles and the risk of developing diabetes. Nat Med 2011;17:448–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Würtz P, Soininen P, Kangas AJ, et al. Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care 2013;36:648–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahola-Olli AV, Mustelin L, Kalimeri M, et al. Circulating metabolites and the risk of type 2 diabetes: a prospective study of 11,896 young adults from four Finnish cohorts. Diabetologia 2019;62:2298–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jang C, Oh SF, Wada S, et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med 2016;22:421–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang F, Zhao S, Yan W, et al. Branched chain amino acids cause liver injury in obese/diabetic mice by promoting adipocyte lipolysis and inhibiting hepatic autophagy. EBioMedicine 2016;13:157–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laferrère B, Reilly D, Arias S, et al. Differential metabolic impact of gastric bypass surgery versus dietary intervention in obese diabetic subjects despite identical weight loss. Sci Transl Med 2011;3:80re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magkos F, Bradley D, Schweitzer GG, et al. Effect of Roux-en-Y gastric bypass and laparoscopic adjustable gastric banding on branched-chain amino acid metabolism. Diabetes 2013;62:2757–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lips MA, Van Klinken JB, van Harmelen V, et al. Roux-en-Y gastric bypass surgery, but not calorie restriction, reduces plasma branched-chain amino acids in obese women independent of weight loss or the presence of type 2 diabetes. Diabetes Care 2014;37:3150–3156 [DOI] [PubMed] [Google Scholar]
- 18.Cummings NE, Williams EM, Kasza I, et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J Physiol 2018;596:623–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao F, Yu J, Guo Y, et al. Effects of individual branched-chain amino acids deprivation on insulin sensitivity and glucose metabolism in mice. Metabolism 2014;63:841–850 [DOI] [PubMed] [Google Scholar]
- 20.Fontana L, Cummings NE, Arriola Apelo SI, et al. Decreased consumption of branched-chain amino acids improves metabolic health. Cell Rep 2016;16:520–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White PJ, Lapworth AL, An J, et al. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol Metab 2016;5:538–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou M, Shao J, Wu CY, et al. Targeting BCAA catabolism to treat obesity-associated insulin resistance. Diabetes 2019;68:1730–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lerin C, Goldfine AB, Boes T, et al. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol Metab 2016;5:926–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herman MA, She P, Peroni OD, Lynch CJ, Kahn BB. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem 2010;285:11348–11356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin AC, Fasshauer M, Filatova N, et al. Brain insulin lowers circulating BCAA levels by inducing hepatic BCAA catabolism. Cell Metab 2014;20:898–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gautron L, Elmquist JK, Williams KW. Neural control of energy balance: translating circuits to therapies. Cell 2015;161:133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams KW, Margatho LO, Lee CE, et al. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci 2010;30:2472–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu J, Zhang C, Borgquist A, et al. Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab 2014;19:682–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Könner AC, Janoschek R, Plum L, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab 2007;5:438–449 [DOI] [PubMed] [Google Scholar]
- 30.Yi CX, la Fleur SE, Fliers E, Kalsbeek A. The role of the autonomic nervous liver innervation in the control of energy metabolism. Biochim Biophys Acta 2010;1802:416–431 [DOI] [PubMed] [Google Scholar]
- 31.Shin AC, Filatova N, Lindtner C, et al. Insulin receptor signaling in POMC, but not AgRP, neurons controls adipose tissue insulin action. Diabetes 2017;66:1560–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.NamKoong C, Song WJ, Kim CY, et al. Chemogenetic manipulation of parasympathetic neurons (DMV) regulates feeding behavior and energy metabolism. Neurosci Lett 2019;712:134356. [DOI] [PubMed] [Google Scholar]
- 33.Rossi J, Balthasar N, Olson D, et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab 2011;13:195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 2002;8:1376–1382 [DOI] [PubMed] [Google Scholar]
- 35.Chuang JC, Sakata I, Kohno D, et al. Ghrelin directly stimulates glucagon secretion from pancreatic alpha-cells. Mol Endocrinol 2011;25:1600–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao TJ, Sakata I, Li RL, et al. Ghrelin secretion stimulated by beta1-adrenergic receptors in cultured ghrelinoma cells and in fasted mice. Proc Natl Acad Sci U S A 2010;107:15868–15873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steculorum SM, Ruud J, Karakasilioti I, et al. AgRP neurons control systemic insulin sensitivity via myostatin expression in brown adipose tissue. Cell 2016;165:125–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tessari P, Nosadini R, Trevisan R, et al. Defective suppression by insulin of leucine-carbon appearance and oxidation in type 1, insulin-dependent diabetes mellitus. Evidence for insulin resistance involving glucose and amino acid metabolism. J Clin Invest 1986;77:1797–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harris RA, Joshi M, Jeoung NH, Obayashi M. Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J Nutr 2005;135(Suppl.):1527S–1530S [DOI] [PubMed] [Google Scholar]
- 40.Neinast MD, Jang C, Hui S, et al. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metab 2019;29:417–429.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hutson SM, Sweatt AJ, Lanoue KF. Branched-chain [corrected] amino acid metabolism: implications for establishing safe intakes [published correction appears in J Nutr 2005;135:2009]. J Nutr 2005;135(Suppl.):1557S–1564S [DOI] [PubMed] [Google Scholar]
- 42.Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci 2011;14:351–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takahashi KA, Cone RD. Fasting induces a large, leptin-dependent increase in the intrinsic action potential frequency of orexigenic arcuate nucleus neuropeptide Y/Agouti-related protein neurons. Endocrinology 2005;146:1043–1047 [DOI] [PubMed] [Google Scholar]
- 44.Liu T, Kong D, Shah BP, et al. Fasting activation of AgRP neurons requires NMDA receptors and involves spinogenesis and increased excitatory tone. Neuron 2012;73:511–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Q, Liu C, Uchida A, et al. Arcuate AgRP neurons mediate orexigenic and glucoregulatory actions of ghrelin. Mol Metab 2013;3:64–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berthoud HR, Neuhuber WL. Vagal mechanisms as neuromodulatory targets for the treatment of metabolic disease. Ann N Y Acad Sci 2019;1454:42–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsien C, Davuluri G, Singh D, et al. Metabolic and molecular responses to leucine-enriched branched chain amino acid supplementation in the skeletal muscle of alcoholic cirrhosis. Hepatology 2015;61:2018–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choudry HA, Pan M, Karinch AM, Souba WW. Branched-chain amino acid-enriched nutritional support in surgical and cancer patients. J Nutr 2006;136(Suppl.):314S–318S [DOI] [PubMed] [Google Scholar]
- 49.García-de-Lorenzo A, Ortíz-Leyba C, Planas M, et al. Parenteral administration of different amounts of branch-chain amino acids in septic patients: clinical and metabolic aspects. Crit Care Med 1997;25:418–424 [DOI] [PubMed] [Google Scholar]
- 50.Pocai A, Lam TK, Gutierrez-Juarez R, et al. Hypothalamic K(ATP) channels control hepatic glucose production. Nature 2005;434:1026–1031 [DOI] [PubMed] [Google Scholar]
- 51.Caballero B, Wurtman RJ. Differential effects of insulin resistance on leucine and glucose kinetics in obesity. Metabolism 1991;40:51–58 [DOI] [PubMed] [Google Scholar]
- 52.Sohn JW, Harris LE, Berglund ED, et al. Melanocortin 4 receptors reciprocally regulate sympathetic and parasympathetic preganglionic neurons. Cell 2013;152:612–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoneshiro T, Wang Q, Tajima K, et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature 2019;572:614–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Udit S, Gautron L. Molecular anatomy of the gut-brain axis revealed with transgenic technologies: implications in metabolic research. Front Neurosci 2013;7:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newgard CB Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab 2012;15:606–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Um SH, Frigerio F, Watanabe M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004;431:200–205 [DOI] [PubMed] [Google Scholar]
- 57.Ono H, Pocai A, Wang Y, et al. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J Clin Invest 2008;118:2959–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]