

Abstract
Hypoadiponectinemia is a risk factor of gestational diabetes mellitus (GDM). Our previous study reported that adiponectin gene knockout mice (Adipoq−/−) develop GDM due to insulin insufficiency. The main objective of this study was to elucidate the underlying mechanism through which adiponectin controls islet expansion during pregnancy. A significant reduction in β-cell proliferation rates, β-cell areas, and blood insulin concentrations was detected in Adipoq−/− mice at midpregnancy. Surprisingly, conditionally knocking down adiponectin receptor 1 (AdipoR1) or AdipoR2 genes in β-cells during pregnancy did not reduce β-cell proliferation rates or blood insulin concentrations. In vitro adiponectin treatment also failed to show any effect on β-cell proliferation of isolated pancreatic islets. It was reported that placental lactogen (PL) plays a crucial role in pregnancy-induced maternal β-cell proliferation. A significant decrease in phosphorylation of signal transducer and activator of transcription 5, a downstream molecule of PL signaling, was observed in islets from Adipoq−/− dams. The mRNA levels of mouse PL genes were robustly decreased in the placentas of Adipoq−/− dams. In contrast, adiponectin treatment increased PL expression in human placenta explants and JEG3 trophoblast cells. Most importantly, bovine PL injection restored β-cell proliferation and blood insulin concentrations in Adipoq−/− dams. Together, these results demonstrate that adiponectin plays a vital role in pregnancy-induced β-cell proliferation by promoting PL expression in trophoblast cells.
Introduction
Gestational diabetes mellitus (GDM) is a common metabolic disease that complicates ∼17% pregnancies (1,2). Compelling clinical data have demonstrated that GDM not only causes adverse pregnancy outcomes and later-life adverse outcomes in the mother but also has a lifelong impact on the offspring’s health (3–5). Therefore, elucidating the pathological mechanism of GDM should provide valuable information for us to prevent and treat this metabolic disease during pregnancy.
Pregnancy is a unique physiological condition during a woman’s adult life. To ensure the success of gestation, delivery, and lactation, maternal metabolism goes through a series of remarkable adaptations. Among these metabolic adaptations, the reduction in insulin sensitivity in metabolically active tissues and an increase in β-cell mass and blood insulin concentrations have been well-documented in pregnant women and many animal models, particularly rodents (6,7). Like type 2 diabetes (T2D), insulin resistance and insulin insufficiency are the two main underlying mechanisms of GDM (1,8,9). Obesity reduces insulin sensitivity and has been identified as a risk factor of GDM (1,2,8). During normal pregnancy, however, mothers gain a significant amount of body fat (7,10). Therefore, more studies are required to determine the contribution of pregnancy-associated and obesity-induced insulin resistance to the development of GDM. In normal pregnancies without T2D or obesity, there is a significant increase in β-cell proliferation, islet expansion, and insulin secretion (11,12). The failure of these normal β-cell adaptations in pregnant women results in insulin insufficiency that ultimately leads to hyperglycemia and other metabolic defects of GDM (1,8,9).
Despite pregnancy-associated physiological insulin resistance, maternal blood glucose concentrations are usually maintained at a relatively low level in healthy pregnant women and rodents (7,13). Therefore, it is logical to exclude any causal role of glucose in pregnancy-induced β-cell proliferation and islet expansion. Adiponectin is an adipocyte-secreted hormone that not only enhances insulin sensitivity but also directly regulates lipid and energy metabolism (14). Despite the fact that adiponectin expression and blood adiponectin concentrations are reduced in late pregnancy (7,15), clinical studies have revealed that hypoadiponectinemia is a risk factor of GDM (15–18). By using adiponectin gene knockout mice (Adipoq−/−), we demonstrated in our previous study that adiponectin deficiency induces hyperglycemia and other metabolic defects of GDM in pregnant mice (7). Importantly, our study revealed that inadequate β-cell expansion and hypoinsulinemia underlie the development of metabolic abnormalities in pregnant Adipoq−/− mice (7). Furthermore, compelling data have demonstrated that hormones, especially placenta-secreted hormones such as placental lactogen (PL), play a dominant role in promotion of β-cell proliferation and insulin production during pregnancy (19–22) and that PL might be regulated by adiponectin (23,24). Therefore, we performed the current study to further investigate the mechanisms, including PL production, through which adiponectin regulates β-cell expansion during pregnancy.
Our studies reported herein show that adiponectin enhances islet expansion via promoting β-cell proliferation in pregnant mice. However, this regulatory effect could not be reproduced in the cultured islets. Conditional knockdown of adiponectin receptor 1 (AdipoR1) or AdipoR2 genes in β-cells failed to show any impact on maternal β-cell proliferation, blood insulin, and glucose concentrations. Interestingly, remarkable reductions in mRNA levels of some members of prolactin family 3 (Prl3), that encode PL proteins in mice, were observed in the placentas from Adipoq−/− dams. In vitro adiponectin treatment stimulated PL gene expression in JEG3 trophoblast cells and the human placenta explants. Importantly, bovine PL (bPL) injection restored the β-cell proliferation rate and maternal blood insulin and glucose concentrations in Adipoq−/− dams. Together, the results of our study show that adiponectin controls pregnancy-induced β-cell proliferation and islet expansion mainly through PL expression in trophoblast cells.
Research Design and Methods
Materials
Antibodies against insulin and Ki67 were from Abcam (Cambridge, MA). Anti-glucagon antibody was from R&D Systems (Minneapolis, MN). The TRIzol, NuPAGE Gels, SuperScript III Reverse Transcriptase and Oligo(dT)12-18 Primer, and Alexa Fluor–conjugated goat anti-mouse, rabbit, or sheep antibodies were from Invitrogen (Carlsbad, CA). Glucose, glucose oxidase, tamoxifen, BSA, Ficoll, sodium dodecyl sulfate, DMEM, RPMI medium, and F12 medium were from Sigma-Aldrich (St. Louis, MO). The mouse insulin ELISA kit was from Mercodia (Uppsala, Sweden). Full-length mouse adiponectin protein was produced with a system created by Dr. Lily Dong’s laboratory (University of Texas Health Science Center, San Antonio, TX) (25). Adiponectin protein was purified by nickel–nitrilotriacetic acid–agarose beads (QIAGEN). The bPL was from the ProSpec-Tany TechnoGene (Rehovot, Israel). The in situ cell death detection kit was from Roche (Basel, Switzerland).
Experimental Animals
Adipoq−/− mice were imported from Dr. Philipp Scherer’s laboratory (UT Southwestern Medical Center, Dallas, TX) (26). AdipoR1fl/fl and AdipoR2fl/fl mice were created as previously described (27) and had been backcrossed into C57BL/6 for at least 10 generations. The tamoxifen-inducible Ins1CreERT2 mice, which express Cre only in β-cells without any leaking (28), and C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Ten- to 12-week-old nulliparous female mice were randomly selected for mating. Pregnancy was determined by the presence of a vaginal plug and was assigned gestational day (G)0.5. Adipoq−/− female mice were mated with wild-type (WT) males or WT female mice mated with Adipoq−/− males. This crossbreeding produced fetuses that were all Adipoq−/+, which excludes any fetal effect on WT or Adipoq−/− dams. Some pregnant Adipoq−/− mice received bPL injection (5 μg subcutaneously) from G6.5 to G11.5 (29,30). Saline was used for control. The AdipoR1fl/fl or AdipoR2fl/fl mice were crossed with Ins1CreERT2 mice to produce mice with AdipoR1fl/fl;Ins1CreERT2 (inducible β-cell–specific AdipoR1 gene knockout [ibR1ko]) or AdipoR2fl/fl;Ins1CreERT2 (ibR2ko) genotype. Mice with AdipoR1−/−;Ins1CreERT2 or AdipoR2−/−;Ins1CreERT2 genotypes were used as control (Con). C57BL/6 sires were used to mate with ibR1ko, ibR2ko, and Con female mice. Tamoxifen was orally gavaged (1 mg daily in 100 μL corn oil) to pregnant ibR1ko, ibR2ko, and Con mice from G7.5 to G11.5. The tamoxifen dosage had been optimized to minimize side effects on pregnancy (31). The efficiency of AdipoR1 and AdipoR2 gene deletion was determined by real-time PCR with TaqMan probes for the mutated exons (Table 1). Glucose tolerance tests (GTT) were performed after 6 h fasting. Maternal tissues, placentas, and fetuses were collected at G12.5, G15.5, or G18.5. Experiments using mouse models were carried out according to the Association for Assessment and Accreditation of Laboratory Animal Care guidelines with approval from the University of California, San Diego, Animal Care and Use Committee.
Table 1.
Primer sequences for TaqMan real-time PCR
| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) | Probe (5′ to 3′) |
|---|---|---|---|
| AdipoR1 | ACAACGACTACCTGCTACATG | TCGTCAAGATTCCCAGAAAGAG | AGCAGATGTGTCCAGATGTTGCCA |
| AdipoR2 | AGCCTCTATATCACCGGAGC | TTTGAGACTCCGTGGAAGTG | CTTTGTGGTTGCTGGTGCCTTTGT |
Immunohistochemistry, Immunofluorescence, and β-Islet Morphometric Analysis
Pancreases were fixed in 10% neutral-buffered formalin, processed, and paraffin embedded. Some tissues were fixed in 4% paraformaldehyde and then embedded in OCT (31). For immunohistochemistry, tissue sections were blocked with 2% H2O2 in PBS and then heated in 0.1 mol/L pH 6.0 citrate buffer for 15 min at 95°C for antigen retrieval. After second-round blocking, immunostaining of insulin was done with use of an anti-insulin primary antibody (10 μg/mL) or rabbit serum (for a negative control) for 4 h. The sections were visualized with 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA) at room temperature for 1.5 min and counterstained with hematoxylin. β-Cell and pancreas areas were measured using insulin-stained series of sections with the microscope software (BZ-X800E; KEYENCE, Laguna Hills, CA).
For immunofluorescence (IF), the slides were incubated in 1% sodium dodecyl sulfate in PBS for 5 min for induction of antigen retrieval. After blocking with BSA in PBS for 2 h, sections were incubated with anti-insulin, anti-glucagon or anti-Ki67 antibody overnight at 4°C. After rinsing, the secondary antibody conjugated with Alexa Fluor 488 or 568 was applied to the slides and incubated for 2 h at room temperature. After washing, sections were mounted in DAPI Fluoromount-G (SouthernBiotech, Birmingham, AL) and visualized by fluorescent optical microscopy. The percentages of Ki67- and insulin protein–positive cells were calculated with use of IF images. For each dam, 8–12 islets were randomly counted for Ki67- and insulin-positive β-cells from different sections. The average of the percentage of Ki67-positive β-cells from each dam was used for statistical analysis.
The apoptotic cells were detected by the terminal deoxynucleotidyl TUNEL staining. The percentage of apoptotic β-cells was calculated by counting of TUNEL and insulin copositive cells in pancreatic islets.
Pancreatic Islet Isolation, Insulin Secretion, and Proliferation Assay
The pancreatic islets were isolated by collagenase digestion and differential centrifugation through Ficol gradients as we described previously (7). For protein and mRNA extraction, all islets were immediately homogenized in protein lysis buffer or TRIzol. For the insulin secretion assay, islets of similar size were handpicked and incubated overnight in RPMI with 5 mmol/L glucose. One hour before the assay, the culture medium was changed to the Krebs-Ringer medium with 2 mmol/L glucose. Then the islets were stimulated by addition of glucose (20 mmol/L) for 1 h. The medium was collected at the end of stimulation. The insulin left in the islets was extracted (7,32). For study of the direct effect of adiponectin on β-cell proliferation, islets were isolated from 3-month old nonpregnant C57BL/6 mice with use of a previously published procedure (33).
JEG3 Cells and Human Explant Culture
Human JEG3 trophoblast cells were cultured in DMEM with 10% FBS (34). The term human placentas were collected from healthy women after C-section with the approval of the Institutional Review Board at the University of California, San Diego. Placental villi were dissected under a stereomicroscope and cultured in DMEM/F12 (35). Confluent JEG3 cells or placental explants were treated with adiponectin (2 μg/mL) overnight or for 6 h, respectively. BSA was used for control.
Western Blot and Real-time PCR Assays
Protein samples were extracted from cultured cells or islets and separated with use of NuPAGE Gels. Proteins were blotted with the indicated antibodies (details can be found in figure legends). The bands from Western blots were quantified with Quantity One software (Bio-Rad Laboratories). Total RNA was prepared from tissues or cells with use of TRIzol according to the manufacturer’s protocol. cDNA was synthesized with SuperScript III Reverse Transcriptase and Oligo(dT)12-18 Primer. Real-time PCR was performed with a QuantStudio 3 Real-Time PCR System (Invitrogen) and specific primers (Table 2). Expression data were normalized to the amount of 18S rRNA or GAPDH.
Table 2.
Sequences for real-time PCR primers
| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
|---|---|---|
| 18S rRNA | CGAAAGCATTTGCCAAGAAT | AGTCGGCATCGTTTATGGTC |
| CSH1 | GCT CAC CTA GTG GCA ATG G | GGA TAA GGG AAC GGT TTG G |
| CSH2 | TCC AAA CCG TTC CGT TAT CC | CAT GTC CTT CCT GAA GCA GTA G |
| Prl3b1 | GGG CTT CTG GAA GGA CTG A | TGA CCA TGC AGA CCA GAA AG |
| Prl3d1 | ACA GAA CAC TTC CCC TGT GTC | TTG CTT TCA GAA GGT CTT CAG TT |
| Prl3d2 | ACA GAA CAC TTC CCC TGT GTC | TTG CTT TCA GAA GGT CTT CAG TT |
| Prl3d3 | ACA GAA CAC TTC CCC TGT GTC | TTG CTT TCA GAA GGT CTT CAG TT |
| Foxm1 | GTG TGC CTG TTC CCA AGC | CTG TTG TCC AGC GTG CAG |
| Foxd3 | CCC CAA CAC TGA CCA ACA G | GTT TGC TCC GCC AGC TTA |
| Mafa | CTCCAGAGCCAGGTGGAG | GTACAGGTCCCGCTCCTTG |
| Mafb | GCAGGTATAAACGCGTCCAG | TGAATGAGCTGCGTCTTCTC |
| Ccna2 | CTT GGC TGC ACC AAC AGT AA | CAA ACT CAG TTC TCC CAA AAA CA |
| Ccnb2 | CAA CCG TAC CAA GTT CAT CG | GAG GGA TCG TGC TGA TCT TC |
| Ccnd1 | CATCCATGCGGAAAATCG | CAGGCGGCTCTTCTTCAA |
| Ccnd3 | GGGCATCCCGATAGAAGC | GAGCAGCTGGAGTACGGATT |
| Cdk2 | CTGCATCTTTGCTGAAATGG | GATCCGGAAGAGTTGGTCAAT |
| Cdk4 | AGAGCTCTTAGCCGAGCGTA | TTCAGCCACGGGTTCATATC |
| Pdx1 | GAA ATC CAC CAA AGC TCA CG | CGG GTT CCG CTG TGT AAG |
| Nkx6.1 | CCC GGA GTG ATG CAG AGT | GAA CGT GGG TCT GGT GTG TT |
| Tph1 | TTC TGA CCT GGA CTT CTG CG | GGG GTC CCC ATG TTT GTA GT |
Statistical Analysis
Data are expressed as mean ± SEM. Statistical analyses were performed with Student t test or ANOVA, followed by Bonferroni posttests using Prism software. Differences were considered significant at P < 0.05.
Data and Resource Availability
The data sets and reagents generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Results
Adiponectin Gene Knockout Impaired Pregnancy-Induced Maternal β-Cell Proliferation
In our previous study, we observed a significant reduction in both maternal blood insulin concentrations and pancreatic islet mass in Adipoq−/− dams at the end of pregnancy (7). It is known that pregnancy induces maternal islet expansion mainly through increasing β-cell proliferation at early and midpregnancy (19,21). Therefore, we analyzed blood insulin concentrations and β-cell areas in the pancreases of Adipoq−/− and WT dams at G12.5 and G15.5. Results showed that maternal blood insulin concentrations and pancreatic β-cell areas of Adipoq−/− dams were remarkably lower than those of WT dams at both G12.5 (Fig. 1A and B) and G15.5 (Supplementary Fig. 1A and B). Pancreas weights were comparable between WT and Adipoq−/− dams (Supplementary Fig. 1C and D). As expected, significantly elevated blood glucose concentrations were observed in Adipoq−/− dams (Supplementary Fig. 1E). Interestingly, unlike Adipoq−/− dams at G18.5 (7), blood triglyceride and free fatty acid concentrations of Adipoq−/− dams at G12.5 were not significantly altered (Supplementary Fig. 1F and G).
Figure 1.
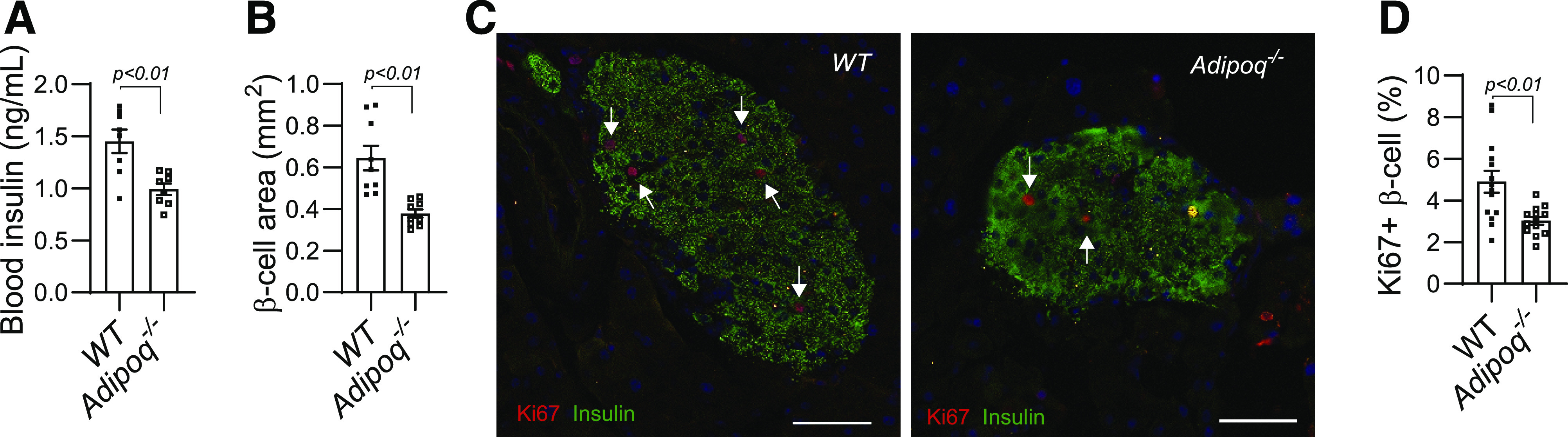
A significant reduction in β-cell proliferation and blood insulin concentrations of Adipoq−/− dams. WT and Adipoq−/− mice were cross-mated for avoidance of any fetal effect. Tissue samples were collected at G12.5 of fed dams. A: Blood insulin concentrations were determined by ELISA. B: We measured β-cell areas using immunochemistry-stained pancreatic sections. Four sections per pancreas were used. C and D: Pancreatic sections were probed by IF with anti-insulin (green) and anti-Ki67 antibody (red, indicated by arrowhead). A total of 8–10 islets was counted for positive cells of each pancreas. The average of the Ki67-positive percentage from each pancreas was calculated and used for statistical comparison (D). White scale bar, 50 μm. Data are presented as mean ± SEM.
Increased β-cell proliferation is the main underlying mechanism for pregnancy-induced islet expansion (11,36). We compared the positive rates of proliferation marker Ki67 protein in pancreatic β-cells between WT and Adipoq−/− dams using IF. Results showed that the positive rates of Ki67 protein in β-cells were significantly lower in Adipoq−/− dams than in WT dams (Fig. 1C and D and Supplementary Fig. 1H). Apoptosis has been suggested to be involved in pregnancy-induced islet expansion (11,36). However, TUNEL-positive β-cell rates were not different between WT and Adipoq−/− dams (data not shown), indicating similar apoptotic rates of β-cells. Together, these results indicate that maternal adiponectin deficiency impairs pregnancy-induced β-cell expansion by reducing β-cell proliferation.
Conditional AdipoR1 and AdipoR2 Gene Knockdown During Pregnancy Showed No Effect on Maternal β-Cell Expansion and Glucose Metabolism
AdipoR1 and AdipoR2 have been identified as two receptors that initiate adiponectin signaling in target cells. Our study showed that both AdipoR1 and AdipoR2 mRNA can be detected in mouse islets but at a relatively low level compared with in liver and skeletal muscle (Fig. 2A and Supplementary Fig. 2A). Interestingly, the expression of the AdipoR1 gene was much more abundant than that of the AdipoR2 gene in mouse islets (Fig. 2A). To determine how adiponectin controls β-cell proliferation during pregnancy, we used the Cre/LoxP system to knock out the AdipoR1 or AdipoR2 gene in β-cells during pregnancy using ibR1ko or ibR2ko mice (detailed breeding approach in Research Design and Methods). After five consecutive days’ tamoxifen gavaging, mRNA levels of the AdipoR1 and AdipoR2 were robustly decreased in islets from ibR1ko and ibR2ko mice compared with their controls (Con) (Fig. 2B and C). Surprisingly, despite the robust knocking down of AdipoR1 and AdipoR2 genes, both maternal blood glucose and insulin concentrations during GTT of ibR1ko and ibR2ko dams were comparable with those of Con, respectively (Fig. 2D–G). Pancreatic β-cell areas and Ki67-positive β-cell rates were the same between ibR1ko or ibR2ko dams and Con (Fig. 2H–M). Of note, we also analyzed blood insulin and glucose concentrations of nonpregnant female ibR1ko, ibR2ko, and Con mice after 1 week of tamoxifen treatment. Like pregnant mice, neither blood glucose nor insulin concentrations were significantly altered in nonpregnant ibR1ko and ibR2ko mice (Supplementary Fig. 2B–E). Furthermore, glucose-stimulated insulin secretion rates of ibR1ko and ibR2ko islets were similar to those of islets from Con (Supplementary Fig. 2F and G). These results suggest that AdipoR1 and AdipoR2 are not essential in adiponectin-regulated β-cell proliferation and islet expansion during pregnancy.
Figure 2.
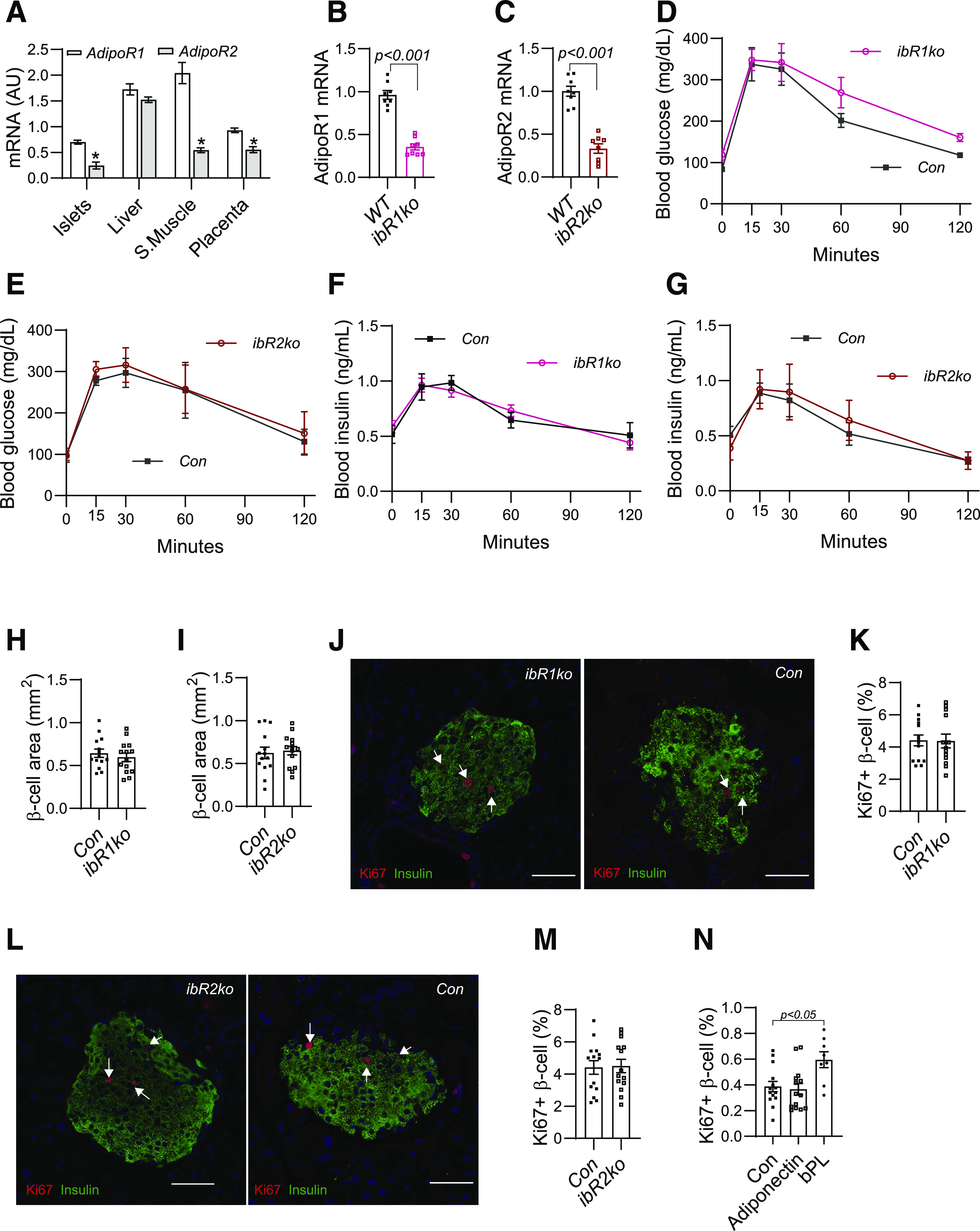
Conditional AdipoR1 or AdipoR2 gene knockout in β-cells exhibited no effect on glucose metabolism and β-cell proliferation during pregnancy. A: mRNA levels of AdipoR1 and AdipoR2 were measured by real-time PCR with samples from C57BL/6 dams at G18.5. B–M: The AdipoR1fl/fl or AdipoR2fl/fl mice were crossed with Ins1CreERT2 mice to produce female mice with AdipoR1fl/fl;Ins1CreERT2 (ibR1ko) or AdipoR2fl/fl;Ins1CreERT2 genotype (ibR2ko). Littermates with AdipoR1−/−;Ins1CreERT2 or AdipoR2−/−;Ins1CreERT2 genotypes were used as control (Con). Tamoxifen was gavaged (1 mg in 100 μL corn oil) from G7.5 to G11.5. B and C: expression levels of AdipoR1 and AdipoR2 were determined by TaqMan PCR with purified islets. D–G: GTT was performed on G12.5 after 6 h fasting (n = 6–8). H and I: β-Cell areas were measured with anti-insulin antibody–probed immunohistochemistry images (image not shown). J–M: Ki67 protein in β-cells were detected by IF with anti-Ki67 antibody (red, indicated with arrowhead) and anti-insulin antibody (green). Nuclei were stained by DAPI (blue); white scale bar, 50 μm. N: Pancreatic islets were isolated from 10-week-old female C57BL/6 mice. After overnight incubation, islets were treated with purified adiponectin (2 μg/mL), bPL (0.1 μg/mL), or BSA for 48 h. The islets were trypsinized, and cells were loaded to coated slides. IF was performed with use of anti-Ki67 and anti-insulin antibody. We calculated the percentages of Ki67-positive β-cells calculated using IF images. Data are presented as mean ± SEM. AU, arbitrary units; S.Muscle, skeletal muscle.
Adiponectin Treatment Exhibited No Effect on β-Cell Proliferation of Cultured Islets
Although AdipoR1 and AdipoR2 are expressed in β-cells, other adiponectin receptors have been proposed (37). The negative results from ibR1ko and ibR2ko mice led us to treat isolated islets with adiponectin to determine whether adiponectin directly enhances β-cell proliferation. Forty-eight hours after adiponectin treatment, trypsinized β-cells were probed with anti-Ki67 and anti-insulin antibody (33). To our surprise, there was no difference in Ki67-positive rates between adiponectin-treated and BSA-treated islets (Fig. 2N), indicating that short-term in vitro adiponectin treatment did not increase β-cell proliferation.
Together, the results from ibR1ko and ibR2ko mice and the in vitro adiponectin treatment assay suggest that adiponectin might regulate pregnancy-induced β-cell proliferation indirectly.
Adiponectin Deficiency Reduced Phosphorylation of STAT5 in Islets and Placental Lactogen Expression in Placentas
PL plays a vital role in mediating pregnancy-induced β-cell proliferation and islet expansion (19–21). Unfortunately, there is no available specific antibody or assay kit to accurately measure mouse blood PL concentration. Given the difference in PL genes between humans and rodents, PLs exert their regulatory effect through prolactin receptor (PRLR). STAT5 (signal transducer and activator of transcription 5) serves as a downstream molecule of PL signaling in both human and rodent cells. We measured the phosphorylation level of STAT5 in isolated islets from Adipoq−/− and Con dams. A significant reduction in STAT5 phosphorylation was observed in the islets from Adipoq−/− dams (Fig. 3A). Similarly, the mRNA levels of Tph1, a PL target gene, and Nkx6.1 were significantly reduced in islets of Adipoq−/− dams (Fig. 3B). The expression of Prlr (Fig. 3B) and genes for cell cycle, including Cdk4 and Ccnd1&3, was slightly reduced without statistical significance (Fig. 3C). Interestingly, the mRNA levels of the key transcription factors for insulin expression, Mafa and Pdx1, were significantly higher in islets from Adipoq−/− dams (38) (Fig. 3B). Since blood insulin concentrations were significantly decreased in Adipoq−/− dams, the increase of Mafa and Pdx1 expression suggests compensatory feedback.
Figure 3.
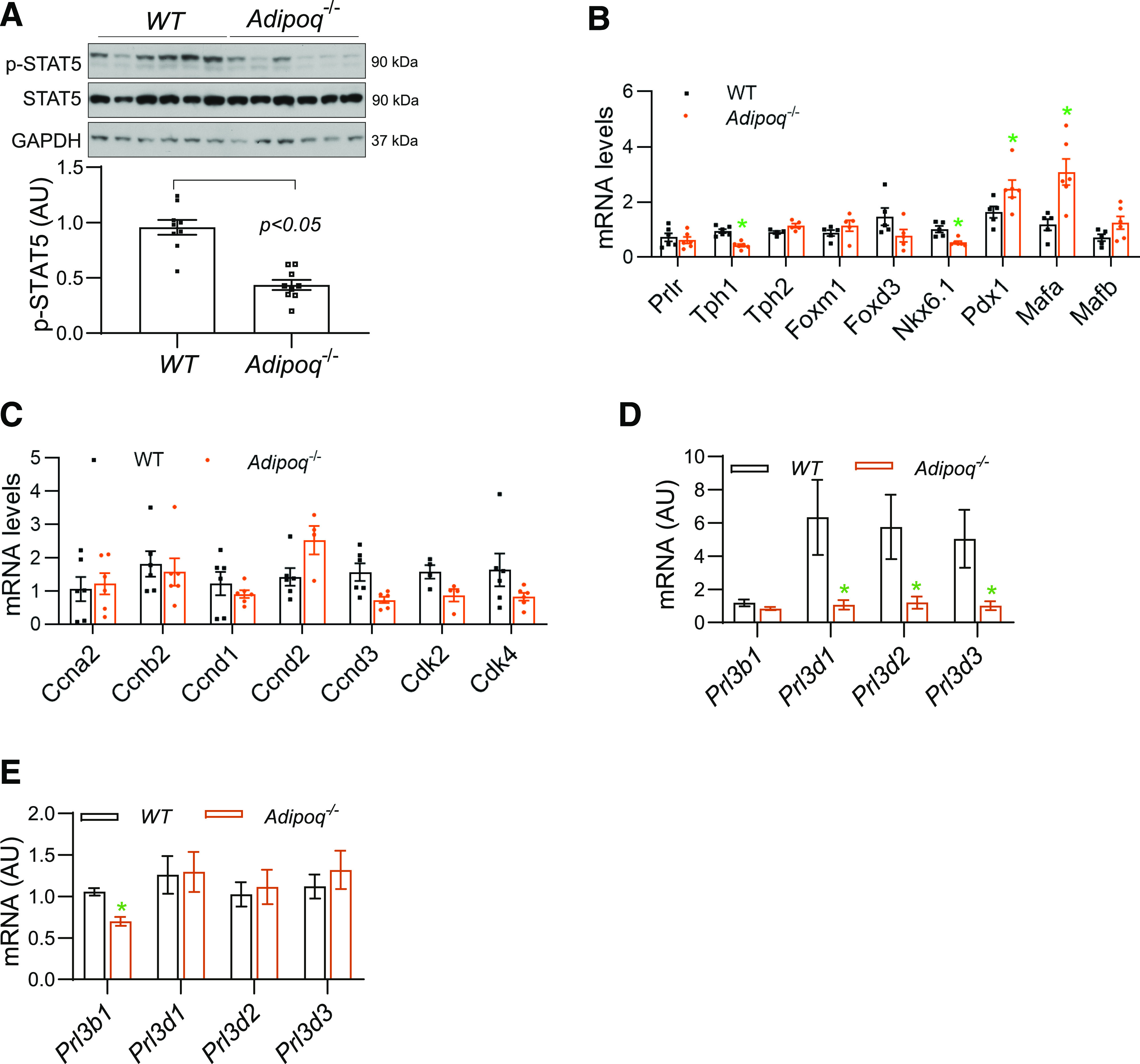
Reduction in PL signaling in islets and PL expression in the placentas of Adipoq−/− dams. A–C: Islets were isolated from pancreases at G12.5 from Adipoq−/− and WT dams. A: The phosphorylation levels of STAT5 were detected by Western blotting with use of protein from isolated islets. B and C: mRNA levels were measured by real-time PCR using samples of isolated islets (n = 8). D and E: Placentas were collected at G12.5 (D) and G18.5 (E) from Adipoq−/− and WT dams. mRNA levels of major members from Prl3 family, analogs of PL, were determined by real-time PCR (n = 18–24, 3–4 placentas per dam). Data are presented as mean ± SEM. AU, arbitrary units; p-STAT5, phosphorylated STAT5.
As in the human, mouse PLs are expressed in trophoblast cells and released into the maternal circulation. There are 23 prolactin/placental lactogen–related genes in mice. However, only the Prl3b1 (also known as PL-II, Csh2) and Prl3d (PL-I, Csh1) gene-encoded proteins bind to the PRLR and exhibit regulatory effects as PRL in target cells (39,40). Mouse PLs are mainly expressed from the Prl3d1, Prl3d2, and Prl3d3 genes in the placenta in early pregnancy, while Prl3b1expressess PL in late pregnancy (39). Despite no significant change in placental weight and structure (7), the mRNA levels of Prl3d1, Prl3d2, and Prl3d3 genes were significantly reduced in placentas from Adipoq−/− dams at G12.5 (Fig. 3D), while Prl3b1 mRNA levels were decreased at G18.5 (Fig. 3E). These data suggest that adiponectin might regulate expression of PL genes in trophoblast cells.
Adiponectin Treatment Increased PL Expression in Human Placental Explants and JEG3 Trophoblast Cells
As in mouse placentas, both AdipoR1 and AdipoR1 are expressed in human placentas and JEG3 trophoblast cells (Supplementary Fig. 3). The human PL is encoded by human chorionic somatomammotropin 1 (CSH1) and CSH2. To determine the effect of adiponectin on PL expression, we treated human placental explants and JEG3 trophoblast cells with adiponectin. Adiponectin treatment significantly increased PL protein and CSH1 and CSH2 mRNA levels in human placental explants (Fig. 4A and B). Similarly, robustly increased CSH1 mRNA levels were observed in JEG3 cells, while CSH2 expression was barely detectable. (Fig. 4C). These results indicate that adiponectin enhances PL expression in trophoblast cells.
Figure 4.
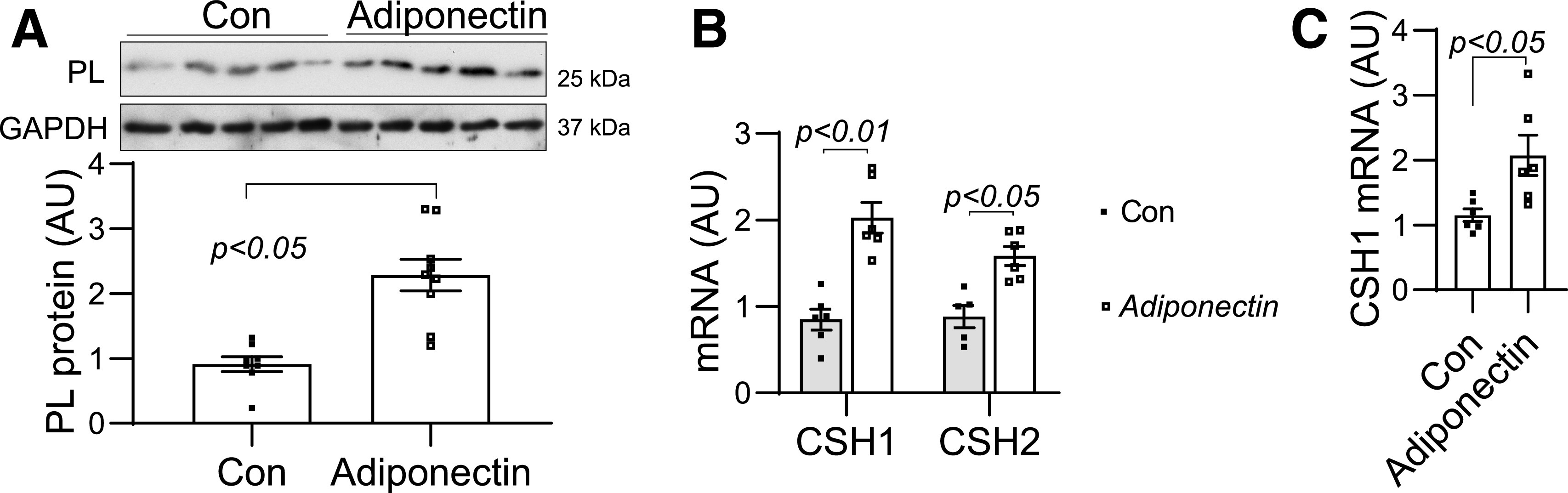
Adiponectin treatment increased PL expression in placental explants and JEG3 cells. Placental villi explants (A and B) and confluent JEG3 cells (C) were treated with adiponectin (2 μg/mL) for 6 h or overnight, respectively. The PL protein levels in the placental explants were measured by Western blotting (A). mRNA of CSH1 and CSH2 was determined by real-time PCR. The expression level of CSH2 is very low in JEG3 cells. Therefore, only CSH1 data of JEG3 cells are presented (C). Data are presented as mean ± SEM. AU, arbitrary units.
bPL Treatment Restored Blood Insulin Concentrations and β-Cell Area in Adipoq−/− Dams
The above studies showed that adiponectin deficiency reduces PL expression in the placenta and might impair pregnancy-induced β-cell proliferation. Therefore, we treated Adipoq−/− dams with bPL to determine whether adiponectin controls β-cell proliferation through PL. As shown in Fig. 5A and B, maternal blood insulin and glucose concentrations were restored to the levels of WT dams with bPL treatment in pregnant Adipoq−/− dams. In parallel, pancreatic β-cell areas and Ki67-positive β-cell rates were also restored (Fig. 5C and D). These results indicate that adiponectin deficiency impairs β-cell proliferation and islet expansion during pregnancy through the reduction of PL production.
Figure 5.
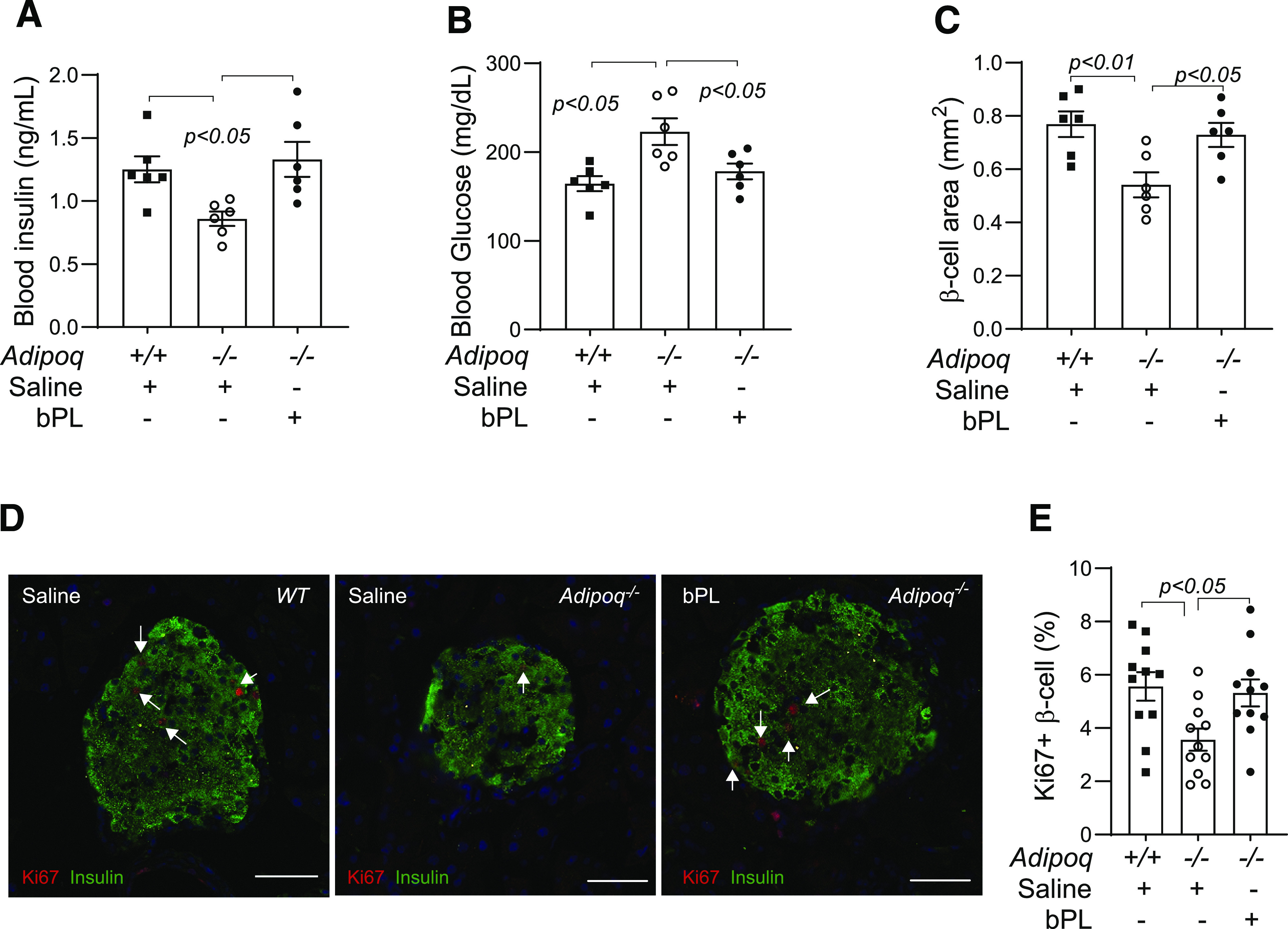
bPL injection restored β-cell proliferation and blood insulin concentration in Adipoq−/− dams. WT and Adipoq−/− dams were injected bPL (5 μg subcutaneously) from G6.5 to G11.5. Saline was used as a control. Tissue samples were collected at G12.5 from fed dams. bPL injection significantly increased blood insulin concentrations (A), β-cell areas (C), and percentage of Ki67-positive β-cells (D and E) (white scale bar, 50 μm) in Adipoq−/− dams. In contrast, maternal blood glucose concentrations were restored to the level of those in WT dams (B). Data are presented as mean ± SEM.
Discussion
Pancreatic islet expansion and hyperinsulinemia are hallmarks of maternal metabolic adaptations to pregnancy (41–44). Appropriate blood insulin concentration is essential in maintaining systemic glucose homeostasis. Like T2D, inadequate β-cell expansion and insulin production during pregnancy cause hyperglycemia and GDM (43–45). The current study demonstrates that adiponectin plays a vital role in pregnancy-induced β-cell proliferation, islet expansion, and insulin production. Our studies demonstrate that adiponectin controls maternal β-cell proliferation mainly through promoting PL gene expression in trophoblast cells.
During pregnancy, maternal blood insulin concentrations progressively increase mainly through enhanced β-cell proliferation and insulin secretory capacity (11,12). Using isolated pancreatic islets from pregnant mice, we demonstrated in our previous study that adiponectin deficiency exhibits no significant effect on basal and glucose-induced insulin secretion rates (7). Similarly, adiponectin treatment exhibited no effects on insulin secretion of nonpregnant human pancreatic islets (46). These results almost exclude the contribution of impaired insulin secretory rate in the development of hypoinsulinemia of Adipoq−/− dams. Therefore, this project focused on the regulatory effect of adiponectin on pregnancy-induced β-cell expansion. Although β-cell neogenesis, α–to–β-cell transdifferentiation, and reduction of β-cell apoptosis have been proposed in pregnancy-induced islet expansion, most experimental data indicate that significantly enhanced β-cell proliferation plays a dominant role in pregnancy-induced islet expansion (19–22). Consistent with these studies, a robust increase in the number of Ki67-positive β-cells was observed in pregnant WT mice at E7.5, G12.5, and G15.5 compared with those of nonpregnant female mice (data not shown). However, the percentages of Ki67-positive β-cells were significantly lower in islets of Adipoq−/− dams than those of WT pregnant mice, while there was no change in apoptotic β-cell number. Together, these studies demonstrate that adiponectin plays a vital role in pregnancy-induced β-cell proliferation. A separate study is underway to determine whether α–to–β-cell transdifferentiation is involved in adiponectin-regulated islet expansion during pregnancy.
Adiponectin improves glucose metabolism, mainly through increasing insulin sensitivity in metabolically active tissues. Interestingly, whether adiponectin has any effect on insulin production is still under debate (46–48). There is almost no information available regarding the effect of adiponectin on β-cell proliferation and development. Three lines of Adipoq−/− mice have been independently created and extensively studied (26,49,50). No abnormalities in islet structure and insulin secretion have been reported in either male or nonpregnant female Adipoq−/− mice (26,49,50). AdipoR1 and AdipoR2 are expressed in β-cells. Systemic AdipoR2 gene deletion did not significantly alter β-cell mass and insulin production in chow-fed mice (51). Similarly, comparable blood insulin concentrations were observed in pregnant AdipoR1 gene knockout mice (Supplementary Fig. 4). Our current study shows that conditional knockdown of the AdipoR1 or the AdipoR2 gene in β-cells exhibited no significant effect on β-cell mass or insulin secretion in both pregnant and nonpregnant mice. By using cultured islets, we show that adiponectin treatment had no effect on β-cell proliferation at a nonpregnant condition (Fig. 2N). Furthermore, the β-cell area and blood insulin and glucose concentrations were comparable between Adipoq−/−, Adipoq−/+, and WT littermates from fetuses (at G18.5 [data not shown]) toward adulthood (7). Together, these studies suggest that adiponectin plays no or only a minor role in islet development and postnatal β-cell function. In contrast, the results of a significant reduction in β-cell proliferation rate, β-cell area, and maternal blood insulin concentrations in Adipoq−/− dams from midpregnancy (Fig. 1) to late pregnancy (7) indicate that adiponectin plays a vital role in pregnancy-induced β-cell proliferation and islet expansion. However, β-cell–specific knockdown of the AdipoR1 or AdipoR2 gene showed no effect on maternal blood glucose and insulin concentrations, indicating that adiponectin indirectly regulates maternal β-cell proliferation and islet expansion. We realize that AdipoR1 or AdipoR2 gene expressions were robustly reduced (knockdown) but not completely deleted in ibR1ko and ibR2ko dams, and an AdipoR1 and AdipoR2 double gene deletion might be required to completely block adiponectin signaling in β-cells. The in vitro adiponectin treatment was relatively short. Therefore, these studies cannot completely rule out the direct effect of adiponectin on β-cell proliferation. However, these data and available information suggest that adiponectin controls β-cell proliferation during pregnancy mainly through an indirect pathway.
The placenta is a transient organ that not only transports nutrients to the fetus but also secrets several hormones into the maternal compartment. PL is a trophoblast cell–secreted hormone that promotes maternal β-cell proliferation and islet expansion through prolactin receptor (20,22,52). In mice, despite the fact that 23 PL-related proteins are expressed in the placenta at different levels during pregnancy, only Prl3b1- and Prl3d-encoded PLs bind and activate the PRL receptor (39,40). Although we did not measure maternal blood PL analog concentrations due to the lack of specific antibodies, a significant reduction in expression of the major Prl3ds and Prl3b1 genes during early and late pregnancy was detected in the placentas of Adipoq−/− dams. Importantly, bPL treatment restored islet mass and blood insulin concentrations in Adipoq−/− dams. Together, these studies indicate that maternal adiponectin regulates β-cell proliferation and islet expansion through PL. The results of adiponectin-induced PL expression in human placental explants and JEG3 cells further support this conclusion. The regulatory effect of adiponectin on PL expression explains why adiponectin promotes β-cell proliferation only during pregnancy. Of note, despite some inconsistency in results, previous in vitro studies revealed a regulatory effect of adiponectin on placental hormone expression (23,24). The current study provides further evidence, specifically from in vivo assays, that confirms the regulatory effect of adiponectin on placental endocrine function. A separate project is studying how adiponectin regulates PL expression and secretion in trophoblast cells.
In summary, by using genetic mouse models, we demonstrate in our current study that adiponectin plays a vital role in pregnancy-induced β-cell proliferation and islet expansion. Interestingly, adiponectin exerts this regulatory effect mainly through PL expression in trophoblast cells. However, future studies are required to verify whether adiponectin has the same regulatory effect on maternal β-cell expansion in humans due to the difference in islet structure and adaptation to pregnancy (6,53). If adiponectin/PL-mediated cross talk among maternal fat, the placenta, and pancreatic islets can be verified in human subjects, it will further support the causal role of hypoadiponectinemia in GDM.
Article Information
Acknowledgments. The authors thank members of the laboratories of Dr. Mana Parast (University of California, San Diego) and Dr. Lily Dong (UT Health San Antonio) for assistance with human placenta explant culture and adiponectin preparation.
Funding. This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, grants DK095132 (to J.S.) and DK113007 (to J.S.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. L.Q., S.S., and C.L. contributed to researching data. W.W.H. contributed to research design and manuscript preparation. G.K. contributed the transgenic mice and to manuscript preparation. L.Q. and J.S. designed the study and wrote the manuscript. J.S. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.13090043.
References
- 1.Johns EC, Denison FC, Norman JE, Reynolds RM. Gestational diabetes mellitus: mechanisms, treatment, and complications. Trends Endocrinol Metab 2018;29:743–754 [DOI] [PubMed] [Google Scholar]
- 2.Sacks DA, Hadden DR, Maresh M, et al.; HAPO Study Cooperative Research Group . Frequency of gestational diabetes mellitus at collaborating centers based on IADPSG consensus panel-recommended criteria: the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study. Diabetes Care 2012;35:526–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catalano PM, McIntyre HD, Cruickshank JK, et al.; HAPO Study Cooperative Research Group . The Hyperglycemia and Adverse Pregnancy Outcome study: associations of GDM and obesity with pregnancy outcomes. Diabetes Care 2012;35:780–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Damm P, Houshmand-Oeregaard A, Kelstrup L, Lauenborg J, Mathiesen ER, Clausen TD. Gestational diabetes mellitus and long-term consequences for mother and offspring: a view from Denmark. Diabetologia 2016;59:1396–1399 [DOI] [PubMed] [Google Scholar]
- 5.Landon MB, Rice MM, Varner MW, et al.; Eunice Kennedy Shriver National Institute of Child Health and Human Development Maternal-Fetal Medicine Units (MFMU) Network . Mild gestational diabetes mellitus and long-term child health. Diabetes Care 2015;38:445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler AE, Cao-Minh L, Galasso R, et al. . Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010;53:2167–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiao L, Wattez JS, Lee S, et al. . Adiponectin deficiency impairs maternal metabolic adaptation to pregnancy in mice. Diabetes 2017;66:1126–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catalano PM Trying to understand gestational diabetes. Diabet Med 2014;31:273–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buchanan TA, Xiang AH. Gestational diabetes mellitus. J Clin Invest 2005;115:485–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiao L, Chu K, Wattez JS, et al. . High-fat feeding reprograms maternal energy metabolism and induces long-term postpartum obesity in mice. Int J Obes 2019;43:1747–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab 2010;21:151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Catalano PM, Tyzbir ED, Roman NM, Amini SB, Sims EA. Longitudinal changes in insulin release and insulin resistance in nonobese pregnant women. Am J Obstet Gynecol 1991;165:1667–1672 [DOI] [PubMed] [Google Scholar]
- 13.Angueira AR, Ludvik AE, Reddy TE, Wicksteed B, Lowe WL Jr., Layden BT. New insights into gestational glucose metabolism: lessons learned from 21st century approaches. Diabetes 2015;64:327–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee B, Shao J. Adiponectin and energy homeostasis. Rev Endocr Metab Disord 2014;15:149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Catalano PM, Hoegh M, Minium J, et al. . Adiponectin in human pregnancy: implications for regulation of glucose and lipid metabolism. Diabetologia 2006;49:1677–1685 [DOI] [PubMed] [Google Scholar]
- 16.Atègbo JM, Grissa O, Yessoufou A, et al. . Modulation of adipokines and cytokines in gestational diabetes and macrosomia. J Clin Endocrinol Metab 2006;91:4137–4143 [DOI] [PubMed] [Google Scholar]
- 17.Hedderson MM, Darbinian J, Havel PJ, et al. . Low prepregnancy adiponectin concentrations are associated with a marked increase in risk for development of gestational diabetes mellitus. Diabetes Care 2013;36:3930–3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lacroix M, Battista MC, Doyon M, et al. . Lower adiponectin levels at first trimester of pregnancy are associated with increased insulin resistance and higher risk of developing gestational diabetes mellitus. Diabetes Care 2013;36:1577–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 1992;130:1459–1466 [DOI] [PubMed] [Google Scholar]
- 20.Banerjee RR, Cyphert HA, Walker EM, et al. . Gestational diabetes mellitus from inactivation of prolactin receptor and MafB in islet β-cells. Diabetes 2016;65:2331–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim H, Toyofuku Y, Lynn FC, et al. . Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med 2010;16:804–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill DJ Placental control of metabolic adaptations in the mother for an optimal pregnancy outcome. What goes wrong in gestational diabetes? Placenta 2018;69:162–168 [DOI] [PubMed] [Google Scholar]
- 23.Benaitreau D, Dos Santos E, Leneveu MC, De Mazancourt P, Pecquery R, Dieudonné MN. Adiponectin promotes syncytialisation of BeWo cell line and primary trophoblast cells. Reprod Biol Endocrinol 2010;8:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDonald EA, Wolfe MW. Adiponectin attenuation of endocrine function within human term trophoblast cells. Endocrinology 2009;150:4358–4365 [DOI] [PubMed] [Google Scholar]
- 25.Galan-Davila AK, Ryu J, Dong K, et al. . Alternative splicing variant of the scaffold protein APPL1 suppresses hepatic adiponectin signaling and function. J Biol Chem 2018;293:6064–6074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nawrocki AR, Rajala MW, Tomas E, et al. . Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem 2006;281:2654–2660 [DOI] [PubMed] [Google Scholar]
- 27.Kajimura D, Lee HW, Riley KJ, et al. . Adiponectin regulates bone mass via opposite central and peripheral mechanisms through FoxO1 [published correction appears in Cell Metab 2014;19:891] Cell Metab 2013;17:901–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thorens B, Tarussio D, Maestro MA, Rovira M, Heikkilä E, Ferrer J. Ins1(Cre) knock-in mice for beta cell-specific gene recombination. Diabetologia 2015;58:558–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goyvaerts L, Lemaire K, Arijs I, et al. . Prolactin receptors and placental lactogen drive male mouse pancreatic islets to pregnancy-related mRNA changes. PLoS One 2015;10:e0121868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh K, Ambler GR, Breier BH, Klempt M, Gluckman PD. Ovine placental lactogen is a potent somatogen in the growth hormone (GH)-deficient rat: comparison of somatogenic activity with bovine GH. Endocrinology 1992;130:2758–2766 [DOI] [PubMed] [Google Scholar]
- 31.Wattez JS, Qiao L, Lee S, Natale DRC, Shao J. The platelet-derived growth factor receptor alpha promoter-directed expression of cre recombinase in mouse placenta. Dev Dyn 2019;248:363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiao L, Wattez JS, Lim L, Rozance PJ, Hay WW Jr., Shao J. Prolonged prepregnant maternal high-fat feeding reduces fetal and neonatal blood glucose concentrations by enhancing fetal β-cell development in C57BL/6 mice. Diabetes 2019;68:1604–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mosser RE, Gannon M. An assay for small scale screening of candidate β cell proliferative factors using intact islets. Biotechniques 2013;55:310–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiao L, Guo Z, Bosco C, et al. . Maternal high-fat feeding increases placental lipoprotein lipase activity by reducing SIRT1 expression in mice. Diabetes 2015;64:3111–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller RK, Genbacev O, Turner MA, Aplin JD, Caniggia I, Huppertz B. Human placental explants in culture: approaches and assessments. Placenta 2005;26:439–448 [DOI] [PubMed] [Google Scholar]
- 36.Banerjee RR Piecing together the puzzle of pancreatic islet adaptation in pregnancy. Ann N Y Acad Sci 2018;1411:120–139 [DOI] [PubMed] [Google Scholar]
- 37.Hug C, Wang J, Ahmad NS, Bogan JS, Tsao T-S, Lodish HF. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U S A 2004;101:10308–10313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao L, Guo M, Matsuoka TA, et al. . The islet beta cell-enriched MafA activator is a key regulator of insulin gene transcription. J Biol Chem 2005;280:11887–11894 [DOI] [PubMed] [Google Scholar]
- 39.Rawn SM, Huang C, Hughes M, Shaykhutdinov R, Vogel HJ, Cross JC. Pregnancy hyperglycemia in prolactin receptor mutant, but not prolactin mutant, mice and feeding-responsive regulation of placental lactogen genes implies placental control of maternal glucose homeostasis. Biol Reprod 2015;93:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simmons DG, Rawn S, Davies A, Hughes M, Cross JC. Spatial and temporal expression of the 23 murine prolactin/placental lactogen-related genes is not associated with their position in the locus. BMC Genomics 2008;9:352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Catalano PM, Huston L, Amini SB, Kalhan SC. Longitudinal changes in glucose metabolism during pregnancy in obese women with normal glucose tolerance and gestational diabetes mellitus. Am J Obstet Gynecol 1999;180:903–916 [DOI] [PubMed] [Google Scholar]
- 42.Catalano PM, Drago NM, Amini SB. Longitudinal changes in pancreatic beta-cell function and metabolic clearance rate of insulin in pregnant women with normal and abnormal glucose tolerance. Diabetes Care 1998;21:403–408 [DOI] [PubMed] [Google Scholar]
- 43.Buchanan TA, Metzger BE, Freinkel N, Bergman RN. Insulin sensitivity and B-cell responsiveness to glucose during late pregnancy in lean and moderately obese women with normal glucose tolerance or mild gestational diabetes. Am J Obstet Gynecol 1990;162:1008–1014 [DOI] [PubMed] [Google Scholar]
- 44.Moyce BL, Dolinsky VW. Maternal β-cell adaptations in pregnancy and placental signalling: implications for gestational diabetes. Int J Mol Sci 2018;19:3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kautzky-Willer A, Prager R, Waldhausl W, et al. . Pronounced insulin resistance and inadequate beta-cell secretion characterize lean gestational diabetes during and after pregnancy. Diabetes Care 1997;20:1717–1723 [DOI] [PubMed] [Google Scholar]
- 46.Staiger K, Stefan N, Staiger H, et al. . Adiponectin is functionally active in human islets but does not affect insulin secretory function or β-cell lipoapoptosis. J Clin Endocrinol Metab 2005;90:6707–6713 [DOI] [PubMed] [Google Scholar]
- 47.Okamoto M, Ohara-Imaizumi M, Kubota N, et al. . Adiponectin induces insulin secretion in vitro and in vivo at a low glucose concentration. Diabetologia 2008;51:827–835 [DOI] [PubMed] [Google Scholar]
- 48.Wijesekara N, Krishnamurthy M, Bhattacharjee A, Suhail A, Sweeney G, Wheeler MB. Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J Biol Chem 2010;285:33623–33631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kubota N, Terauchi Y, Yamauchi T, et al. . Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem 2002;277:25863–25866 [DOI] [PubMed] [Google Scholar]
- 50.Ma K, Cabrero A, Saha PK, et al. . Increased beta -oxidation but no insulin resistance or glucose intolerance in mice lacking adiponectin. J Biol Chem 2002;277:34658–34661 [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, Michael MD, Kash S, et al. . Deficiency of adiponectin receptor 2 reduces diet-induced insulin resistance but promotes type 2 diabetes. Endocrinology 2007;148:683–692 [DOI] [PubMed] [Google Scholar]
- 52.Huang C, Snider F, Cross JC. Prolactin receptor is required for normal glucose homeostasis and modulation of β-cell mass during pregnancy. Endocrinology 2009;150:1618–1626 [DOI] [PubMed] [Google Scholar]
- 53.Chen H, Kleinberger JW, Takane KK, et al. . Augmented Stat5 signaling bypasses multiple impediments to lactogen-mediated proliferation in human β-cells. Diabetes 2015;64:3784–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]