

Alström syndrome (ALMS) is an extremely rare autosomal recessive disorder caused by mutations in ALMS1 (1,2). ALMS was first reported in 1959 but has only been recently recognized as a ciliopathy (3,4). Ciliopathies comprise a group of human genetic diseases associated with primary cilia, microtubule-based organelles extending from the cell surface that transduce signals from the extracellular environment. ALMS patients have a spectrum of clinical features including a neurosensory deficit, renal degeneration, cardiomyopathy, and metabolic dysregulation (5,6). The metabolic phenotypes are especially severe, such that nearly all individuals develop childhood obesity within the first 5 years of life, accompanied by insulin resistance, hyperinsulinemia, hyperleptinemia, hyperlipidemia, and, eventually, type 2 diabetes (6,7) (Fig. 1A). Notably, the degree of insulin resistance is much more severe in ALMS patients compared with other genetic causes of obesity such as Bardet-Biedl syndrome, which is a polygenic ciliopathy (7), thus suggesting that ALMS1 mutation affects a crucial part of the machinery that maintains insulin sensitivity. In addition, since excessive accumulation of subcutaneous fat is one of the traits of ALMS, it has been speculated that adipose tissue plays a key role in its metabolic phenotypes, yet little research has been done on the underlying mechanisms. In this issue of Diabetes, Geberhiwot et al. (8) provide evidence that adipose tissue malfunction is a key contributor to ALMS’s systemic insulin resistance.
Figure 1.
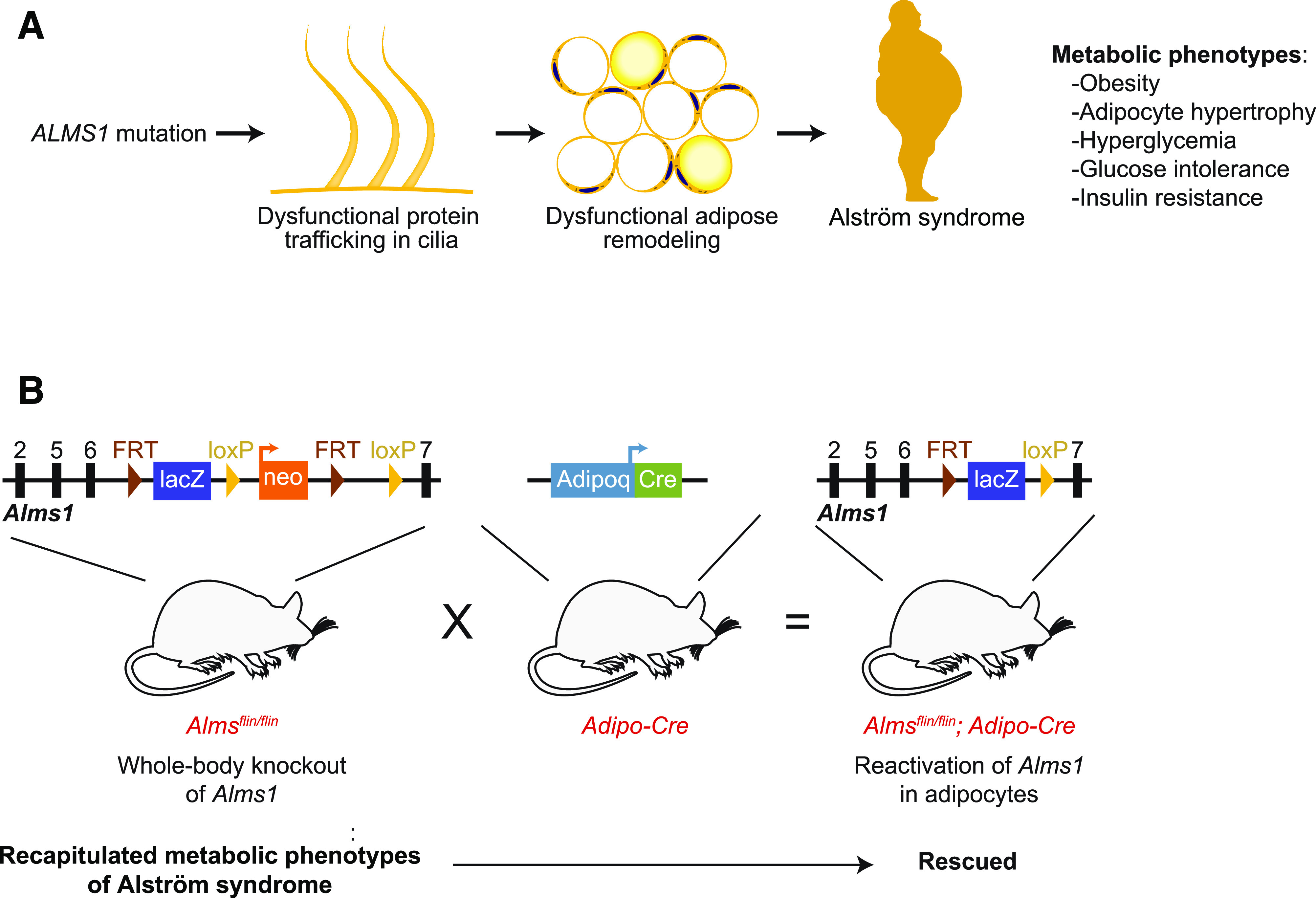
A: ALMS is caused by mutation of ALMS1. Early-onset obesity and severe insulin resistance are among the common clinical features of ALMS (6,7). Geberhiwot et al. (8) propose that metabolic phenotypes of ALMS are mainly derived from adipose dysfunction. B: Geberhiwot et al. support their hypothesis by demonstrating that whole-body knockout of Alms1 recapitulates the metabolic phenotypes of ALMS and that reintroducing ALMS1 to adipocytes fully rescues the phenotype.
Geberhiwot et al. first cataloged insulin resistance in various tissues, finding that ALMS patients had insulin resistance in adipose tissue, hepatic tissue, and skeletal muscle as compared with sex- and age-matched control subjects with a similar BMI. Importantly, the authors noted that subcutaneous fat from ALMS fails to suppress lipolysis in response to insulin, suggesting adipose tissue insulin resistance in ALMS. Moreover, ALMS subcutaneous adipose tissue had larger adipocytes and increased oxidative stress and mitochondrial dysfunction, confirming that the adipose tissue had altered structure and was severely dysfunctional.
The authors sought to determine the contribution of adipose tissue to the metabolic dysfunction of ALMS through mouse genetic studies. First, the authors characterized the fat aussie mice, which bear a spontaneous mutation in the Alms1 gene (9) resulting in premature termination of translation. Similar to previous reports (10,11), the authors observed that the fat aussie mice develop severe insulin resistance accompanied by adipocyte hypertrophy. Importantly, the impairment of insulin-stimulated glucose uptake is limited to adipose tissues. The authors created another whole-body Alms1 knockout (Almsflin/flin) by inserting loxP sites between exon 6 and 7 of Alms1 (Fig. 1B). Much like the fat aussie mice, Almsflin/flin mice became obese by 3 months of age and had adipocyte hypertrophy, hyperglycemia, glucose intolerance, and insulin resistance (Fig. 1B). Critically, these metabolic phenotypes were largely reversed by reintroducing Alms1 into adipose tissue, by way of crossing Alms1flin/flin with adipose-specific adiponectin-Cre mice (Fig. 1B). Lastly, the authors performed ALMS1 loss-of-function studies in human adipocytes, finding that ALMS1 silencing inhibited insulin-stimulated glucose uptake, which provides evidence for a cell autonomous role for ALMS1 in adipocyte insulin resistance. Overall, the authors provide convincing evidence that adipose tissue is the main driver for metabolic dysregulation in ALMS syndrome.
A major strength of the work is the rescue of knockout mice with adipose-specific Alms1 expression. The finding that mice are metabolically normal when they are deficient in Alms1 in every tissue but adipose provides powerful evidence supporting the conclusion that adipose tissue is a key player in the metabolic phenotypes of ALMS. Despite that, we still lack detailed mechanistic insight into how ALMS1 loss of function causes obesity and adipose dysfunction.
ALMS has been recognized as a ciliopathy, as available data suggest that ALMS1 is involved in primary cilium function, particularly intracellular trafficking and protein transport (4,7,12). ALMS1 may participate in the trafficking of proteins from the Golgi apparatus to other parts of the cell, including the cilia. It could also have a role in the intraflagellar transport cargo complex, which moves proteins along the cilium to the membrane (7). Mislocalization or deficient transport of specific receptors within the cilium has deleterious effects on cell signaling (13,14). Thus, a defect in peripheral insulin signaling has been proposed as a possible mechanism, yet this does not seem to be the case, as adipose tissues from human and mouse models of ALMS do not have impaired insulin signaling. Future work is required to determine the precise function of ALMS1 in cilium function and whether it is critical for the regulation of adipocyte insulin sensitivity.
Consistent with previous studies (10,11), the authors noted adipocyte hypertrophy in ALMS models, even when their total fat mass was equivalent to that of obese control subjects. This raises the possibility that the function of ALMS1 is involved in adipocyte remodeling, which occurs when adipocytes change in number, size, or function in response to energy needs. A recent study demonstrated that ciliary cAMP signaling in preadipocytes is required for de novo mouse adipogenesis (15), and, of note, the primary cilium is lost in mature adipocytes (15). This raises the possibility that defective adipogenesis, in ALMS, could trigger unhealthy hypertrophy of existing adipocytes, especially under chronic overnutrition. However, data from the current study showed that ALMS1 silencing did not have a major impact on human adipogenesis. This discrepancy might be due to differences between species. The strong rescuing effect by ALMS1 reactivation in whole-body Alms1 knockout mice by way of adiponectin-Cre, which mainly targets mature adipocytes, suggests it is adipocytes that mediate their expansion and dysfunction intrinsically and/or through cell-to-cell communication in ALMS. Another potential mechanism by which ALMS1 could regulate adipose expansion is by regulating adaptive thermogenesis. Supporting this, the authors observed that ALMS1 silencing reduced UCP1 expression, which is a marker for thermogenesis, in human adipocytes. In fact, ALMS1 expression is more abundant in brown adipose tissue than white adipose tissue (N. Petrovsky, unpublished observations). Further studies are warranted to delineate the initiating cell types and the precise role and mechanistic basis of ALMS1 in the regulation of adipocyte remodeling.
In conclusion, the work by Geberhiwot et al. is novel and significant. It demonstrates that adipose tissue is a major driver of systemic insulin resistance and metabolic dysregulation in Alström syndrome. However, as discussed here and within the study, future studies are warranted to delineate how ALMS1 regulates adipocyte metabolic function and remodeling, how it leads adipose dysfunction, and how this leads to comorbidities. Gaining further mechanistic insight may reveal a potential therapeutic strategy to address the metabolic conditions in Alström and other ciliopathies.
Article Information
Funding. This work was funded by National Institute of Diabetes and Digestive and Kidney Diseases grant R01 DK116008 to S.K.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Footnotes
See accompanying article, p. 364.
References
- 1.Alstrom CH, Hallgren B, Nilsson LB, Asander H. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence-Moon-Bardet-Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand Suppl 1959;129:1–35 [PubMed] [Google Scholar]
- 2.Hearn T, Renforth GL, Spalluto C, et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat Genet 2002;31:79–83 [DOI] [PubMed] [Google Scholar]
- 3.Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 2006;7:125–148 [DOI] [PubMed] [Google Scholar]
- 4.Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet 2009;151C:281–295 [DOI] [PubMed] [Google Scholar]
- 5.Marshall JD, Beck S, Maffei P, Naggert JK. Alström syndrome. Eur J Hum Genet 2007;15:1193–1202 [DOI] [PubMed] [Google Scholar]
- 6.Marshall JD, Bronson RT, Collin GB, et al. New Alström syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med 2005;165:675–683 [DOI] [PubMed] [Google Scholar]
- 7.Girard D, Petrovsky N. Alström syndrome: insights into the pathogenesis of metabolic disorders. Nat Rev Endocrinol 2011;7:77–88 [DOI] [PubMed] [Google Scholar]
- 8.Geberhiwot T, Baig S, Obringer C, et al. Relative adipose tissue failure in Alström syndrome drives obesity-induced insulin resistance. Diabetes 2021;70:364–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arsov T, Silva DG, O’Bryan MK, et al. Fat aussie--a new Alström syndrome mouse showing a critical role for ALMS1 in obesity, diabetes, and spermatogenesis. Mol Endocrinol 2006;20:1610–1622 [DOI] [PubMed] [Google Scholar]
- 10.Haczeyni F, Barn V, Mridha AR, et al. Exercise improves adipose function and inflammation and ameliorates fatty liver disease in obese diabetic mice. Obesity (Silver Spring) 2015;23:1845–1855 [DOI] [PubMed] [Google Scholar]
- 11.Haczeyni F, Poekes L, Wang H, et al. Obeticholic acid improves adipose morphometry and inflammation and reduces steatosis in dietary but not metabolic obesity in mice. Obesity (Silver Spring) 2017;25:155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hearn T, Spalluto C, Phillips VJ, et al. Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes 2005;54:1581–1587 [DOI] [PubMed] [Google Scholar]
- 13.Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci U S A 2008;105:4242–4246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brailov I, Bancila M, Brisorgueil MJ, Miquel MC, Hamon M, Vergé D. Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res 2000;872:271–275 [DOI] [PubMed] [Google Scholar]
- 15.Hilgendorf KI, Johnson CT, Mezger A, et al. Omega-3 fatty acids activate ciliary FFAR4 to control adipogenesis. Cell 2019;179:1289–1305.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]