

Abstract
Cholesterol is a crucial component of membrane bilayers that determines their physical and functional properties. Cells largely satify their need for cholesterol through the novo synthesis from acetyl-CoA and this demand is particularly critical for cancer cells to sustain dysregulated cell proliferation. However, the association between serum or tissue cholesterol levels and cancer development is not well established as epidemiologic data do not consistently support this link. While most preclinical studies focused on the role of total celular cholesterol, the specific contribution of the mitochondrial cholesterol pool to alterations in cancer cell biology has been less explored. Although low compared to other bilayers, the mitochondrial cholesterol content plays an important physiological function in the synthesis of steroid hormones in steroidogenic tissues or bile acids in the liver and controls mitochondrial function. In addition, mitochondrial cholesterol metabolism generates oxysterols, which in turn, regulate multiple pathways, including cholesterol and lipid metabolism as well as cell proliferation. In the present review, we summarize the regulation of mitochondrial cholesterol, including its role in mitochondrial routine performance, cell death and chemotherapy resistance, highlighting its potential contribution to cancer. Of particular relevance is hepatocellular carcinoma, whose incidence in Western countries had tripled in the past decades due to the obesity and type II diabetes epidemic. A better understanding of the role of mitochondrial cholesterol in cancer development may open up novel opportunities for cancer therapy.
Keywords: Mevalonate pathway, free cholesterol, hepatocellular carcinoma, oxysterols, StARD1, chemotherapy
1. Introduction.
The incidence of cancer is a leading cause of death in the world and has escalated to become a global health concern. Solid tumors, including pancreatic cancer or hepatocellular carcinoma (HCC), are particularly aggresive and consitute one of major causes of cancer-related deaths. HCC is the end stage of chronic liver disease caused by nonalcoholic fatty liver disease and nonalcoholic steatohepatitis (NASH) and one of the most frequent cancers due to its association with obesity and type II diabetes pandemic. Cancer cells exhibit important metabolic alterations and are under anabolic pressure for the synthesis of membrane lipids to sustain dysregulated cell proliferation. In this regard, increased fatty acid synthesis has been reported to support cancer cell growth, including HCC [1]. Moreover, consistent with its crucial role in determining physical properties and function of membrane bilayers, increased cholesterol levels and de novo synthesis in the endoplasmic reticulum (ER) has been considered as an important player in cancer pathogenesis. However, the impact of cholesterol in cancer development is not fully understood. In this regard, epidemiologic studies suggest a positive association between elevated serum cholesterol level and the risk to develop certain types of cancer, such as prostate cancer, and the use of statins to block cholesterol levels was linked to lowered risk of melanoma, non-Hodgkin lymphoma, endometrial and breast cancers [2–7]. Indeed, the use of statins has been associated with a reduction in cancer-related mortality for different cancer types [8]. Despite these studies, there have been a growing number of epidemiologic observations that suggest no association between cholesterol and cancer [9–11]. In line with these observational studies, preclinical models have also pictured the controversial role of cholesterol in cancer cell biology [12–17]. Although cholesterol is predominantly present in the plasma membrane of cells where it regulates a plethora of signaling pathways, cholesterol undergoes intracellular trafficking to different intracellular organelles, including mitochondria. Most studies related to the role of cholesterol in cancer focused on the levels of cholesterol in serum or tissues; however, the impact of mitochondrial cholesterol in cancer cell biology has been less explored. Given the relevance of mitochondria in life/death fate and metabolic homeostasis, alterations in the regulation of mitochondrial cholesterol may have an important impact in cancer cell biology involving more than changes in mitochondrial function, including metabolic reprogramming or apoptosis/chemotherapy resistance among others. Further understanding the role of mitochondrial cholesterol in cancer development may uncover new targets for treatment. In the present review, we briefly summarize key aspects of mitochondrial cholesterol homeostasis, including intracellular trafficking, metabolism and impact in cancer.
2. Mitochondrial cholesterol: from de novo synthesis in the ER to mitochondrial trafficking.
Cholesterol is present in all cells mainly localized in membrane bilayers where it plays a crucial role in the determination of their physical properties (e.g. fluidity and membrane curvature) [18, 19]. Rather than being uniformly distributed in the bilayer, cholesterol is segregated in discrete domains, called lipid rafts, in close association with glycosphingolipids where a variety of intricate signaling pathways are located and generate intracellular signals. As such, cholesterol-mediated regulation of membrane fluidity can have far-reaching effects in cell functions by modulating the action of specific membrane proteins (e.g. receptor, carriers). Compared to other membrane bilayers, such as plasma membrane, the content of cholesterol in mitochondrial membranes is rather low, with estimates of around 3% of the total cellular cholesterol content vs 25–30% present in plasma membranes [19, 20]. Due to the impact of cholesterol in the regulation of bilayerś physical properties, the trafficking of cholesterol to mitochondrial membranes, particularly the mitochondrial inner membrane (MIM) is a highly-regulated process controlled by specific proteins.
2.1. De novo cholesterol synthesis in the mevalonate pathway
All cells meet their need for cholesterol via de novo synthesis from acetate in the ER in the so-called mevalonate pathway [21, 22]. This complex process is initiated by the conversion of acetyl-CoA into 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) and then irreversibly metabolized to mevalonate by the rate-limiting enzyme, HMG-CoA reductase (HMGR). A series of enzymatic reactions sequentially convert mevalonate into farnesyl pyrophosphate, squalene, 2,3-oxidosqualene, lanosterol, and finally cholesterol, involving two distinct but related pathways (Figure 1). The Block and Kandutsch-Russell pathways use desmosterol and 7-dehydrocholesterol as the immediate precursors of cholesterol through their reduction by the 24-dehydrocholesterol reductase (DHCR24) and the 7-dehydrocholesterol reductase (DHCR7), respectively. In addition to its biotransformation into cholesterol, UV light can convert 7-dehydrocholesterol into vitamin D3, limiting the generation of cholesterol in the Kandutsch-Russell pathway, which has an important impact in cancer biology [23]. The generation of farnesyl pyrophosphate also serves as the precursor of other types of lipid intermediates, including isoprenoids, that regulate multiple proteins by farnesylation or geranylgeranylation. The de novo cholesterol synthesis is a highly-regulated process controlled by an efficient feedback mechanism. The increase in the cholesterol content of the ER above a threshold level triggers the ubiquitination of HMGR by Insig-1 and Insig-2 and the subsequent proteasome-mediated degradation of HMGR. In addition, HMGR gene expression is regulated by ER-bound membrane transcription factor sterol regulatory element protein 2 (SREBP2), whose activation depends on the sterol-sensitive SREBP cleavage activating protein (SCAP). The content of cholesterol present in the ER is sensed by SCAP and retains full-length SREBP2 at the ER. Upon cholesterol depletion, SCAP interacts with SREBP2 to exit the ER and move to the Golgi, where SREBP2 is sequentially cleaved by the specific proteases S1P and S2P. This event releases the soluble N terminus domain of SREBP2, which travels to the nucleus to induce the transcription of HMGR. Although this feedback inhibition of cholesterol synthesis by cholesterol aims to avoid superfluous cholesterol synthesis, a recent refractory mechanism involving a novel TNFR1-Caspase-2-S1P-SREBP2 axis has been described whereby increased cholesterol levels associates with continued cholesterol synthesis [24]. This mechanism overrides the inhibition of de novo cholesterol synthesis in the face of high cholesterol levels, as indicated in states of chronic liver disease preceding HCC development such as nonalcoholic steatohepatitis [25, 26]. Blocking cholesterol synthesis at the HMGCR step with statins results not only in cholesterol inhibition but also prevents farnesyl pyrophosphate generation, which can have a wide range of effects through alterations of multiple proteins that are regulated by farnesylation or geranylgeranylation. However, inhibition of squalene synthase (e.g. YM5361) or squalene epoxidase (e.g. terbinafine) ensures a more specific prevention of cholesterol synthesis.
Fig 1. De novo synthesis and regulation of cholesterol.
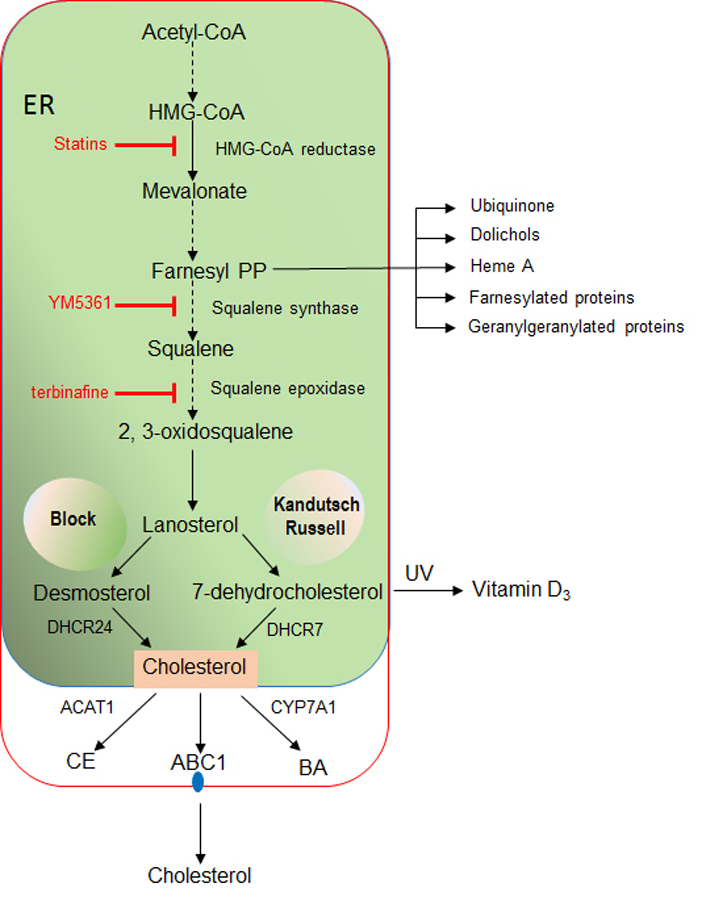
Cholesterol is synthesized from acetyl-CoA in the mevalonate pathway in the ER. Sequential conversion of acetyl-CoA into farnesyl pyrophosphate (Farnesyl PP) branches the pathway into cholesterol synthesis from squalene or the generation of ubiquinone, dolichols, heme A or isoprenoids, which are involved in the modification of proteins via farnesylation or geranylgeranylation. Squalene metabolism into lanosterol ensures cholesterol synthesis through both the Block and Kandutsch-Russell pathways from desmosterol or 7-dehydrocholesterol mediated by DHCR24 and DHCR7, respectively. Once synthesized, cholesterol can be esterified by ACAT, metabolized into BAs, predominantly through the classic pathway catalyzed by CYP7A1 or exported out of cells via cholesterol pumps present in plasma membrane, such as ABC1. The rate-limiting step in the cholesterol synthesis is the conversion of HMG-CoA into mevalonate catalyzed by HMGCR, which is inhibited by statins. Inhibition of squalene synthase or squalene epoxidase ensures a more specific approach to block cholesterol synthesis with minor effects in the levels of farnesyl PP and hence isoprenylation of proteins.
The level of cellular cholesterol is not only determined by the de novo synthesis but also by its disposition via different pathways and mechanisms (Figure 1). Cholesterol can be exported from cancer cells via specific transporters belonging to the ABC family, which are overexpressed in different types of cancers, and more predominantly in cancer stem cells [27, 28]. As shown in certain types of cancer cells, ABC1 overexpression in colon cancer cells determines mitochondrial cholesterol steady levels and their susceptibility to cell death [28]. In addition, cholesterol can be esterified with long chain fatty acyl-CoA to cholesterol esters in a reaction catalyzed by acyl-CoA cholesterol acyltransferase (ACAT). ACAT has been reported to be overexpressed in different types of cancers, including glioma, pancreatic and liver cancer [29, 30; Human protein Atlas, TCGA database], although its role in liver cancer is not well understood with reports indicating a lower expression of ACAT in tumor areas from patients with HCC [31]. Moreover, the metabolism of cholesterol into bile acids (BAs) predominantly via the classic (neutral) pathway regulated by CYP7A1 is an important mechanism to downregulate cholesterol levels, although the resulting BAs act as signaling molecules and have been reported to modulate HCC and cholangiocarcinoma development [32, 33]. Besides the neutral pathway of BA synthesis, cholesterol metabolism in mitochondria constitutes an alternative mechanism of BAs generation whose contribution to HCC or cholangiocarcinoma has been less explored. Findings from our lab indicate that stimulated trafficking of cholesterol to mitochondria and subsequent BAs generation from the alternative mitochondria acidic pathway promote HCC (unpublished observations). Moreover, oxysterol generated from mitochondrial cholesterol metabolism has been shown to contribute to the development of some types of cancers (see below).
2.2. Mechanisms of mitochondrial cholesterol trafficking
Once cholesterol is synthesized in the ER or taken up from the diet and delivered to endolysosomes, it is distributed to discrete membrane bilayers by different mechanisms, such as vesicle-mediated inter-organelle cholesterol transport and protein-mediated, non-vesicular transfer [34]. The latter is a predominant mechanism of transport accross organelles and requires sterol transfer proteins marked with the steroidogenic acute regulatory protein-related lipid transfer (START) domain [21, 34–37]. START proteins encompass a growing family of members that bind diverse ligands, such as cholesterol, oxysterols, phospholipids, sphingolipids, and possibly fatty acids. Among them, the members that bind and traffic cholesterol are STARD1, STARD3 (also known as MLN64) and STARD4 subfamily (Figure 2). STARD4 subfamily, which encompass STARD4, STARD5 and STARD6, is closely related to the STARD1 with 20% homology and is regulated by SREBP2. STARD4 colocalizes with ER and ACAT, and exposure of isolated microsomes with recombinant STARD4 results in increased cholesterol ester synthesis [35]. STARD1 (also known as StAR), the founding member of the STARD family, is of particular relevance for the mitochondrial transport of cholesterol between the mitochondrial outer membrane (MOM) and MIM as inferred from the outcome that mutations in the human STARD1 gene are the most common cause of congenital lipoid adrenal hiperplasia (CAH) [38, 39]. Moreover, mice with global Stard1 deletion die within 7–10 days after birth due to lethal CAH [40]. These findings suggest that STARD1 is essential in providing cholesterol to MIM for metabolism and that it cannot be replaced by other members of the START family. STARD1 is a nuclear-encoded phosphoprotein that contains a mitochondria-targeting sequence, an amino-terminal amphipathic helix that directs the protein to the mitochondria. It is synthesized in the cytosol as a 37 kDa precursor protein, and following mitochondrial import it is processed to a 32 kDa intermediate product and a mature 30 kDa form that is localized within the matrix [41]. STARD1 function has been mostly characterized in steroidogenic cells and its expression in other tissues is rather low. In the liver, the basal expression of STARD1 is low but increases several fold in acute liver injury and chronic liver disease, such as NASH, one of most prevalent causes of HCC due to its association with obesity and type II diabetes [25, 42]. To further support the crucial role of STARD1 in the transport of cholesterol to MIM in extra steroidogenic tissues and given the premature death of mice with global Stard1 deletion, we have recently generated Stard1 floxed mice to produce different mouse lines with tissue specific STARD1 deletion [43]. In this regard, hepatocyte-specific Stard1 knockout (Stard1ΔHep) mice exhibit defective cholesterol transport into mitochondria in response to acetaminophen and are resistant to drug-induced liver failure [43]. Besides STARD1, MLN64, a late endosomal cholesterol-binding protein, has also been proposed to mediate transport of cholesterol to mitochondria. Overexpression of MLN64 in mice liver increased mitochondrial cholesterol levels and resulted in increased superoxide anion generation, decreased membrane potential and defective ATPase activity [44]. Although these findings suggest a role for MLN64 in the intramitochonrial transport of cholesterol, targeted mutations in MLN64 (StARD3) have been shown to cause minor alterations in metabolism and intracellular distribution of cholesterol [37, 45]. Importantly, Stard1ΔHep mice exhibit defective cholesterol transport to mitochondria despite unaltered MLN64 expression [43]. These outcomes suggest that MLN64 provides cholesterol to the MOM from endosomes and together with STARD1 work in tandem in the net import of cholesterol to the MIM for metabolism. Similar to MLN64, STARD4 appears to move cholesterol to MOM when overexpressed in primary mouse hepatocytes, although it also stimulates cholesterol ester synthesis, suggesting that STARD4 is potentially involved not only in the transport of cholesterol to mitochondria but also to the ER for cholesterol esterification [35].
Fig 2. Mitochondrial cholesterol trafficking and regulation.
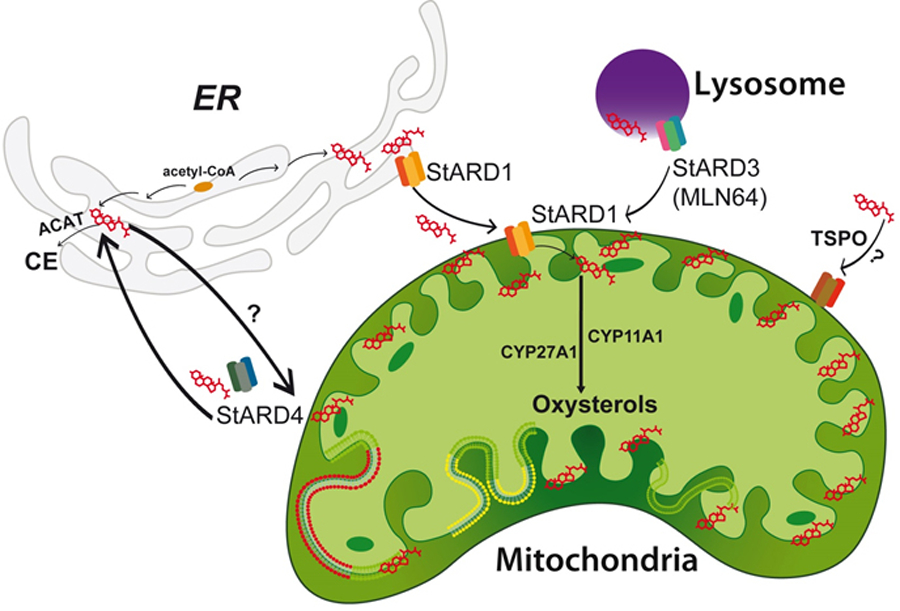
Intracellular cholesterol is translocated to different membrane bilayers, including mitochondria. Several members with the START domain bind cholesterol, such as STARD1, STARD3 and STARD4. STARD1 plays a prominent role in the translocation of cholesterol to mitochondrial inner membrane for further metabolism into oxysterols and steroid hormones or bile acids. STARD1 is synthesized as a 37 kDa precursor protein that following mitochondrial import it is processed to a 32 kDa intermediate product and a mature 30 kDa form that catalyzes the transfer of cholesterol to mitochondrial inner membrane. STARD3 (MLN64) is a late endosomal cholesterol-binding protein that is thought to transfer cholesterol to STARD1 at the mitochondrial outer membrane for movement to the inner membrane. The role of STARD4 in the transfer of cholesterol to mitochondria is less understood and still not fully estabablished although it can mediate transfer of cholesterol to the ER for esterification by ACAT. The contribution of TSPO to cholesterol transfer to mitochondria inner membrane is controversial and not definitively demonstrated.
Besides these proteins, translocator protein (TSPO), a protein particularly abundant in steroidogenic tissues and primarily localized in the MOM, has been suggested to play an important role in steroidogenesis via the transport of cholesterol to the MIM [46, 47]. However, recent studies using cell-specific genetic deletion of TSPO in MA-10 and primary Leydig cells revealed uncompromised steroidogenesis, suggesting that the lack of TSPO in these cells does not interfere with the transport of cholesterol to mitochondria for metabolism to steroid hormones [48–50]. While these findings question the relevance of TSPO in cholesterol trafficking to MIM using pharmacological ligands and inhibitors, TSPO seems to be crucial for the preimplantation embryo development associated with steroidegenesis [50].
2.3. Metabolism and physiological role
Once cholesterol is delivered to MIM, it is metabolized by specific cytochromes and serves as the precursor for steroid hormones and bile acids (BAs) in steroidogenic tissues and hepatocytes, respectively, and acts as the immediate precursor for oxysterols. In steroidogenic tissues, cholesterol at the MIM is metabolized by CYP11A1 generating pregnenolone, the precursor of steroid hormones and neurosteroids in steroidogenic tissues and neurons, respectively. In the liver, the sterol 27-hydroxylase (encoded by CYP27A1) is localized at the MIM and metabolizes cholesterol into 27-hydroxycholesterol (27-OH-Chol) followed by 25-hydroxycholesterol 7-α-hydroxylase (encoded by CYP7B1), which then proceeds with the synthesis of the primary BA chenodeoxycholic acid in the alternative acidic pathway. Compared to the predominant classical pathway of BA synthesis in hepatocytes, which is controlled by the rate-limiting enzyme 7-α-hydroxylase (encoded by CYP7A1), the alternative mitochondrial pathway of BA synthesis is considered a minor component of the total hepatic BA pool and its physiological relevance is still poorly understood. Studies in hepatocytes and the human hepatoma cell line HepG2 indicated that the overexpression of STARD1 resulted in the stimulation of BA generation that exceeded that caused by CYP27A1 overexpression, indicating that the STARD1-dependent cholesterol trafficking to MIM is the rate-limiting step in the alternative pathway of BA synthesis [51, 52]. Moreover, a recent report described a crucial role for CYP7B1 in adaptive thermogenesis whereby BAs generated in the alternative pathway were further metabolized in the gut to modulate cold adaptation [53]. Although the physiological relevance of the alternative pathway is not fully understood and is considered to contribute to a minor extent to the total pool of BA, the alternative mitochondrial pathway may compensate for the defects of the classical pathway of BAs synthesis to maintain BAs homeostasis. This is illustrated in the case of disruption of the Cyp7a1 gene in mice. While mutant mice exhibit a complex phenotype with increased rate of postnatal death, fat malabsorption, wasting, skin abnormalities and vision problems, a small significant proportion of null mice survive, and exhibit an indistinguishable phenotype from their wild-type littermates, indicating that the alternative acidic pathway of BA synthesis compensates for the loss of Cyp7a1 [54].
3. Cholesterol and mitochondrial function in cancer cells.
Although cholesterol trafficking to mitochondria is biotransformed into steroid hormones or BAs, as described above, an estimulated cholesterol transport mediated by STARD1 overexpression can exceed mitochondrial cholesterol metabolism and result in the accumulation of cholesterol in mitochondrial membranes, which has far-reaching consequences besides alterations of mitochondrial function and physiology (Figure 3). This process has been involved in several human pathologies, including Alzheimeŕs disease, lysosomal storage disorders and cancer [43, 55–57]. In this regard, unphysiologic accumulation of mitochondrial cholesterol has been described in solid tumors, such as in rats bearing transplanted Morris hepatomas and the levels of mitochondrial cholesterol correlated with the degree of tumor growth and malignancy [58, 59]. Importantly, the degree of cholesterol accumulation in mitochondria from HCC cells correlated with the expression of STARD1 [56, 60]. Increased mitochondrial cholesterol accumulation was also described in colon cancer cells and inversely correlated with the expression of the ABC1 pump, which catalyzes the egress of cholesterol out of cancer cells [28].
Fig 3. Consequences of mitochondrial cholesterol accumulation.
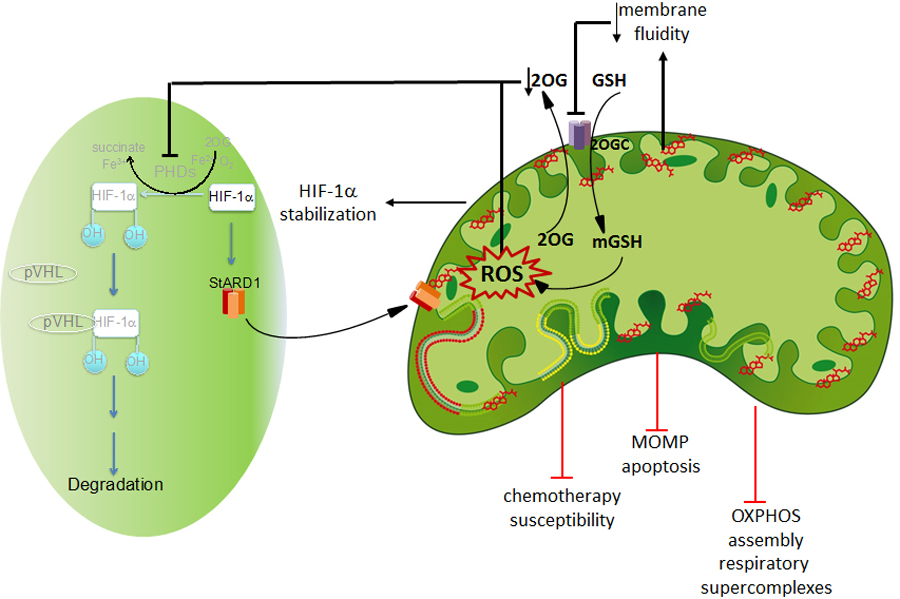
Once cholesterol accumulates in mitochondria of cancer cells it triggers a wide range of effects starting with the disruption of mitochondrial membrane fluidity. This outcome, in turn, impacts specific membrane carriers (e.g. 2OGC) and impairs the transport of GSH into mitochondrial matrix, resulting in increased ROS generation. ROS can signal the activation of HIF-1α pathway by inactivating PHDs, which results in the HIF-1α stabilization. Iin addition, decreased 2-oxoglutarate levels in cytosol may also contribute to PHD inhibition and HIF-1α stabilization. Besides these effects, mitochondrial cholesterol-mediated ROS generation can also regulate the assembly of respiratory supercomplexes resulting in impaired oxidative phosphorylation that often marks the metabolic changes of cancer cells. Through the changes in mitochondrial membrane fluidity, cholesterol accumulation in mitochondria has been shown to impair MOMP and mitochondrial apoptosis and contributes to chemotherapy resistance.
Given the crucial role of cholesterol in membrane physical properties, accumulation of cholesterol in mitochondria results in the decrease of membrane fluidity. For instance, in chronic liver disease induced by alcohol consumption or NASH the increase in mitochondrial content results in the decrease in membrane fluidity determined by fluorescence anisotropy from diphenylhexatriene-labeled mitochondria and partition of di-4-ANEPPDHQ [61, 62], and similar results have been reported in HCC cancer cells [60, 63]. As as consequence of increased mitochondrial membrane order by cholesterol accumulation specific mitochondrial carriers become defective, leading to altered transport rates and equilibrium between mitochondrial matrix and cytosol. This is best illustrated in the case of mitochondrial GSH transport from cytosol, where GSH is synthesized from its constituents aminoacids. Indeed, it has been shown that the 2-oxoglutarate carrier (OGC, SLC25A11) is sensitive to cholesterol-mediated changes in membrane fluidity, and its inhibition due to cholesterol accumulation leads to mitochondrial GSH (mGSH) depletion in primary cells, which hence sensitized to oxidative stress-mediated cell death [64, 65]. The inhibitory effect of mitochondrial cholesterol on SLC25A11 seems to be specific, as the transport of other solutes, such as S-adenosyl-L-methionine into mitochondria is insensitive to cholesterol accumulation [66].
Of relevance for cancer biology, mitochondria from cancer cells exhibit increased cholesterol accumulation, which paradoxically does not lead to mGSH depletion due to the compensated overexpression of SLC25A11 carrier that overcomes the inhibitory effect of cholesterol, as characterized in HCC [63]. This adaptive response of mitochondria from HCC cancer cells in exhibiting increased mitochondrial cholesterol loading while maintaining unrestricted mGSH is of advantage for tumor growth, as cholesterol protects cancer cells from undergoing apoptosis, as discussed below. Moreover, increased antioxidant defense promotes tumor growth as demonstrated by an emerging role of antioxidants as tumor promoters that accelerate lung cancer progression and melanoma metastasis [67, 68]. In addition, increased expression of SLC25A11 in non-small lung cancer and melanoma facilitates NADPH supply to maintain mitochondrial function, while in HCC cells SLC25A11 has been shown to promote cell growth by sustaining ATP generation through oxidative phosphorylation and glycolysis [63, 69]. The ability of cancer cells to maintain unrestricted oxidative phosphorylation despite increased cholesterol trafficking is quite remarkable in light of recent observations showing that cholesterol accumulation in mitochondria from primary hepatocytes results in defective assembly of respiratory chain supercomplexes by an oxidative stress-dependent mechanism [70]. Overall, mitochondria from different cancer cells exhibit increased cholesterol accumulation. While this outcome may account for the recognized mitochondrial dysfunction and role in cancer chemotherapy [71, 72], the adaptive overexpression of SLC25A11 may be advantageous for promoting cancer growth leading to the maintenance of mitochondrial ATP synthesis, resistance to mitochondrial outer membrane permeabilization (MOMP) and chemotherapy insensitivity.
4. Mitochondrial cholesterol in cell death and chemotherapy sensitivity of cancer cells.
A hallmark of cancer is the development of mechanisms that cause apoptosis resistance [73]. Given the central role of mitochondria in the orchestration of apoptotic signals, including caspase activation, resistance to mitochondrial apoptosis can also result in chemotherapy refractoriness, particularly to chemotherapeutic agents targeting mitochondria [72]. Mitochondrial cholesterol-mediated depletion of antioxidant defense sensitizes primary cells to oxidative stress and cell death, and this has been best documented in preneoplastic stages of chronic liver diseases, such as alcoholic and NASH [74, 75]. However, the associated depletion of mGSH by cholesterol masks the antiapoptotic role of cholesterol through its effects in increasing the membrane order of mitochondrial membranes. The mitochondrial cholesterol accumulation described in HCC, colon cancer cells or HeLa cells has been shown to impair MOMP triggered by Bax oligomerization and hence the release of cytochrome c leading to a defective assembly of the apoptosome [28, 60, 76, 77]. In this regard, increased mitochondrial cholesterol accumulation associated with lower ABC1 leves in human colon cancer cells has been shown to decreased sensitivity to Ca2+ triggered mitochondrial permeability transition pore opening (MPT) and release of cytochrome c, whereas knockdown of lanosterol cyclase, which catalyzes the conversion of oxidosqualene to lanosterol, decreases mitochondrial cholesterol loading and increases Ca2+-mediated MPT and cytochrome c, further confirming the crucial role of mitochondrial cholesterol in regulation of mitochondrial engagement in cell death [28]. Cholesterol extraction by methyl-β-cyclodextrin (MCD), restored the ability of Bax to induce MOMP. In this regard, the initiation of apoptosis by the steroid saponin Rh2 is exacerbated by removal of cholesterol from mitochondrial membranes from human lung epithelial adenocarcinoma, monocytic leukemia and histiocytic lymphoma [78]. More importantly, fluidization of mitochondria from HCC cells with 2-(2-methoxyethoxy)ethyl 8-(cis-2-noctylcyclopropyl)octanoate (A2C) restored Bax-mediated MOMP and the release of cytochrome c [56, 60], indicating that cholesterol-mediated changes in membrane dynamics rather than cholesterol itself regulates MOMP and hence apoptosome activation. An unbiassed approach to decipher the role of cholesterol on the permeabilization of membrane bilayers was addressed using liposomes of defined lipid composition containing cholesterol and analyzed for susceptibility to release entrapped fluorescent dextrans. The presence of cholesterol in the composition of liposomes abrogated the ability of Bax to permeabilize liposomes, which was reversed by A2C fluidization of liposomes or cholesterol extraction by MCD, indicating that membrane lipid composition can be a critical regulator of MOMP [60, 76, 77]. In addition to cholesterol, cardiolipin (CL), an acidic phospholipid present exclusively in MIM, is also known to modulate MOMP [76]. Due to the presence of four unsaturated fattyl acyl chains in the carbon backbone, CL is susceptible to peroxidation and the resulting peroxidized CL (CLOOH) has been shown to reverse the inhibitory effect of cholesterol in the permeabilization of liposomes [63]. Thus, in premalignant stages mitochondrial cholesterol accumulation favors CLOOH formation via mGSH depletion in a scenario in which both cholesterol and CLOOH engage in two opposite effects on MOMP, with the final outcome being likely determined by the severity of mGSH depletion and relative CL/CLOOH generated. In established cancer cells, such as human hepatoblastoma Hep3B cells, the observed accumulation of cholesterol, however, does not result in mGSH depletion due to the overexpression of SLC25A11, which preserves CL in intact reduced state, leaning the balance towards apoptosis resistance [63].
Given the central role of mitochondria in cell death regulation and in mediating chemotherapy, the accumulation of cholesterol has been reported to lead to mitochondria-targeted chemotherapy resistance in cancer cells. Both rat H35 and human HepG2 HCC cells have been shown to be resistant to arsenic trioxide, lonidamine, thapsigargin or doxorubicin, antineoplastic drugs that target mitochondria and induce MPT by different mechanisms, including the Ca2+ overload and ROS generation [79–82]. These agents induce cell death with apoptotic characteristics, including caspase 3 activation and chromatin disruption. Interestingly, the resistance to these chemotherapeutic agents was reversed upon treatment of H35 or HepG2 cells with lovastatin to inhibit de novo cholesterol synthesis by inhibiting HMGCoAR, which prevented the accumulation of cholesterol in mitochondrial membranes. More importantly, sensitization to these chemotherapeutic agents was also observed with the squalene synthase inhibitor YM-53601, which does not affect isoprenoid metabolism (Figure 1). Moreover, the susceptibility of H35 cell to doxorubicin by lovastatin pretreatment was prevented by mevalonate independently of the inhibition of farnesyltransferase, whereas mevalonate failed to restore resistance to doxorubicin after squalene synthase inhibition. Thus, the trafficking of cholesterol to mitochondria of neoplastic cells plays a predominant antiapoptotic role and protects cancer cells against the efficacy of chemotherapeutical agents, particularly those acting in mitochondria. Maneouvers aimed to decrease the accumulation of cholesterol in mitochondria may thus result in a more efficient approach for cancer treatment. In this regard, there have been studies suggesting the combination of statins as coadjuvants to increase the efficacy of chemotherapy [83, 84].
5. Mitochondrial cholesterol and activation of HIF pathway.
Hypoxia is a characteristic feature of cancer, particularly in solid tumors, whose impact in tumor development is well recognized. Hypoxia in growing tumors develops as a consequence of the disorganized structure and architecture of tumor vasculature, resulting in irregular and inefficient oxygen delivery. The onset of hypoxia is considered a negative prognostic factor for response to treatment and survival of cancer patients [85–87]. A central pathway in oxygen sensing that mediates the gene expression reprogramming and the adaptation to this stressful condition is the hypoxia-inducible transcription factor 1 (HIF-1), which activates a genetic program and the regulation of key pathways such as angiogenesis, glucose metabolism or invasion, contributing to the survival of cancer cells despite low oxygen environment [88–90]. HIF-1 comprises a stable β subunit (HIF-1β/Arnt) and a labile α subunit (HIF-1α), which encompasses three family members, HIF-1α, HIF-2α, and HIF-3α. In normoxia HIF-1α is degraded by the proteasome upon ubiquitination by Von Hippel Landau (VHL) tumor suppressor, and this process is preceded by the hydroxylation of HIF-1 in prolyl residues catalyzed by prolylhydroxylases (PHDs), of which there are three isoforms. Although PHDs are dependent on iron and 2-oxoglutarate (2-OG), these enzymes primarily function as oxygen sensors so that in normoxia PHDs become activated and hence mediate HIF-1α hydroxylation, which initiates its degradation via VHL-mediated proteasome degradation. In low oxygen conditions, PHDs are inactivated and therefore HIF-1α is stabilized and translocates to the nucleus where heterodimerizes with HIF-1β/Arnt to regulate gene expression [91, 92]. Besides oxygen, PHDs are also regulated by oxidative stress and reactive oxygen species (ROS), which during hypoxia originate from mitochondria [93, 94]. Therefore, HIF-1α can be regulated during normoxia in the context of ROS generation by modulating the activity of PHDs.
HIF-1α is overexpressed in human cancers as a result of intratumoral hypoxia as well as genetic alterations, such as gain-of-function mutations in oncogenes (for example, ERBB2) and loss-of-function mutations in tumour-suppressor genes (for example, VHL and PTEN). Due to its fundamental role as a tumor promoter, there has been a great level of interest to identify small molecules that inhibit HIF-1α for cancer treatment [85, 86]. Several such molecules have been developed to antagonize HIF-1α at different levels, including inhibition HIF-1α mRNA expression, HIF-1α protein translation, HIF-1α protein degradation, HIF-1α DNA binding and HIF-1α transcriptional activity [95]. An additional mechanism of HIF-1α activation has been linked to the increase in cholesterol by a mechanism requiring ROS generation from mitochondria [96, 97]. Quite interestingly, cholesterol-mediated HIF-1α stabilization was observed in normoxia and required increased nitric oxide levels and mitochondrial ROS production. Moreover, cholesterol-induced HIF-1α activation was abolished upon mitochondrial DNA depletion, further confirming the mitochondrial origin of ROS generation. Besides cholesterol-mediated mitochondrial ROS generation, cholesterol accumulation in normoxia can lead to decreased 2-OG in cytosol because of the inhibition of OGC by loss of membrane fluidity [63], which in turn can result in impaired PHDs activity and subsequent HIF-1α statibilization. In line with this, it has been shown that the disruption of the 2-oxoglutarate dehydrogenase complex controls HIF-1α stability in aerobic conditions [98]. This outcome can be extended to other intermediates of the TCA cycle, including succinate, as potential inhibitors of PHDs [99]. Moreover, the 2-OG analog 3-oxoglutarate has been shown to decrease normoxic HIF1-α in cancer cells, resulting in the induction of cell death and the potentiation of chemotherapy and reduction of tumor growth in vivo [100]. Thus, 3-oxoglutarate and vincristine synergistically induced apoptosis in different cancer cell types and reduced in vivo tumor growth. These findings suggest that STARD1-mediated cholesterol trafficking to mitochondria results in HIF-1α stabilization by mechanisms involving mitochondrial ROS generation and changes in TCA metabolites (e.g. 2-OG) affecting PHD-induced HIF-1α hydroxylation and stabilization. Interestingly, STARD1 expression is regulated by HIF-1α, consistent with the well-recognized stimulating effect of hypoxia in steroidogenesis [101–104]. Chromatin immunoprecipitation assays demonstrated the presence of hypoxia response elements (HRE) in the STARD1 promoter, and the association of HIF-1α with the STARD1 promoter was potentiated under reduced oxygen tension compared to normoxic condition [103]. Thus, based on these observations it is conceivable that STARD1 and HIF-1α engage in a mutual upregulation, leading to a self-amplifying cycle favoring mitochondrial cholesterol accumulation and sustained HIF-1α stabilization. These findings suggest the possiblity that targeting STARD1 and HIF-1α may be of potential relevance in cancer biology and therapy.
6. Mitochondrial oxysterols and cancer.
Oxysterols represent a pool of oxygenated derivatives of cholesterol that have a wide range of biological actions and regulate multiple pathways, such as cholesterol and lipid metabolism, membrane fluidity or cell proliferation and inflammation through the modulation of a cadre of proteins and receptors (e.g. liver X receptors or oxysterol-binding protein-related proteins, ORPs). They can arise either as a consequence of cholesterol metabolism within mitochondria (e.g. 25-OH-Chol or 27-OH-Chol) or as byproducts of cholesterol oxidation in non-enzymatic reactions (7β-hydroxycholesterol or 7-ketocholesterol) [105, 106]. Given the stimulated trafficking of cholesterol to mitochondria in cancer cells, as discussed above, the relevant pool of oxysterols linked to cancer biology derives from the metabolism of cholesterol initiated by CYP27A1. The acidic pathway of cholesterol metabolism by CYP27A1 is considered more than an alternative pathway of BA synthesis, and represents a regulated pathway for the generation of oxysterols, which in turn, can have far-reaching consequences in lipid metabolism and cell death/proliferation [107].
CYP27A1 is located in the MIM and unlike liver-specific CYP7A1, which is expressed exclusively in hepatocytes, CYP27A1 is widely distributed in all tissues [108, 109]. CYP27A1 is the first enzyme in the acidic pathway of mitochondrial cholesterol metabolism and catalyzes the introduction of a hydroxyl group in cholesterol at carbon 27, yielding 27-OH-Chol, while in the classic pathway CYP27A1 generates 5β-cholestan-3α,7α,26-triol and 5β-cholestan-3α,7α,12α,26-tetrol. In humans, deficiency of CYP27A1 results in a devastating disease called cerebrotendinous xantomatosis (CTX), a rare lipid storage disorder characterized by dementia, ataxia, cataracts and xantomas of the tendoms and the nervous system [110, 111]. Oxysterols, including CYP27A1 products 27-OH-Chol and 3β-hydroxy-5-cholestanoic acid, are LXR ligands, and activate genes involved in cholesterol/lipid metabolism, including STARD1, which in turn stimulates cholesterol trafficking to mitochondria to generate further 27-OH-Chol, establishing a feed forward loop [107, 112].
The effects of oxysterols in cancer cell biology and development is complex and not fully elucidated. While there is evidence indicating that oxysterols act as tumor promoters associated to their proliferative effects, modulation of inflammation and diverse signaling pathways (e.g. Wnt or Hedgehog) or through ORPs [113–115], there have been reports indicating that oxysterols have a pro-apoptotic and cytotoxic effects on tumor cells by mechanisms linked to ROS generation and disrupted Ca2+ homeostasis mediated by intrinsic mitochondrial pathways and extrinsic death receptor-dependent pathway [116–118]. Despite this complexity, several mitochondrial derived oxysterols have been linked to cancer development. For instance, 27-OH-Chol has been reported to modulate breast cancer development through its binding to the estrogen receptor, which affects its transcriptional activity and the action of the estrogen receptor protein [119]. In addition, 27-OH-Chol is the first identified endogenous selective estrogen receptor modulators, which are used for breast cancer treatment. Besides 27-OH-Chol, 25-OH-Chol acts as a agonistic estrogen receptor ligand [120], and accordingly both 25-OH-Chol and 27-OH-Chol increased the transcription of estrogen receptor target genes in breast cancer cell lines MCF7 and HCC1428, suggesting that both oxysterols can substitute estrogen in the activation of estrogen receptor-mediated expression and can play a potential role in resistance to therapy [121]. Further evidence for the tumor promoter role of 27-OH-Chol in breast cancer progression derives from the findings that Cyp7B1 knockout mice, which lack the enzymatic step involved in 27-OH-Chol catabolism, exhibit an accelerated rate of breast tumors development than Cyp27A1 null mice [122]. Similar to breast cancer, oxysterols have been shown to modulate prostate cancer progression by regulating androgen action via androgen receptor (AR). AR triggers proliferation of prostate cancer cells and a high level of AR has been associated to poor survival in patients and 27-OH-Chol has been shown to increase AR transcriptinal activity [123]. Moreover, 27-OH-Chol can suppress docetaxel-induced apoptosis, indicating a role for this oxysterol in chemotherapy resistance [124]. As STARD1 is the rate-limiting step in mitochondrial cholesterol trafficking, its stimulation in prostate cancer can exceed the ability of CYP27A1 to catalyze 27-OH-Chol resulting in mitochondrial cholesterol loading. Besides these hormone-dependent tumors, 27-OH-Chol has been reported to modulate colon cancer as increased expression of CYP27A1 was associated with poor outcome while increased CYP7B1 levels was linked to good prognosis in patients with colorectal cancer [125]. Intriguingly, despite its potential prevalence due to its association with obesity and type II diabetes pandemic, the role of oxysterols in HCC remains poorly explored, with evidence for elevated levels of 4β-hydroxycholesterol, 7α-hydroxycholesterol and 25-OH-Chol in an experimental HCC model and in a cohort of patients with a high risk of HCC development, such as chronic hepatitis type C infection [126, 127]. Further research in this particular type of cancer is warranted in future investigations to determine whether the levels of oxysterols in HCC are linked to mitochondrial cholesterol trafficking.
7. Conclusions and future directions.
Given the key role of cholesterol in the maintenance of structural and functional properties in membrane bilayers, cancer cells need a constant supply of cholesterol to sustain dysregulated proliferation, most of which is ensured through stimulated synthesis de novo from acetyl-CoA in the ER. In line with this notion, a substantial amount of evidence associates increased cholesterol levels with cancer development. Less recognized and investigated, however, has been the contribution of the specific mitochondrial cholesterol pool to cancer biology and progression. Consistent with the key role of mitochondria in cell death pathways and metabolism and the alterations of mitochondria in cancer, cancer cells exhibit increased trafficking and accumulation of cholesterol in mitochondrial membranes, which can result in far-reaching consequences other than affecting mitochondria physiology (Figure 3). The enhanced loading of cholesterol in mitochondria implies a synchronized stimulation of cholesterol availability through the novo synthesis and increased expression of STARD1 to efficiently translocate cholesterol to mitochondria. Blocking cholesterol synthesis or antagonizing STARD1 would be expected to ameliorate mitochondrial cholesterol accumulation. Unfortunately, there are no described STARD1 inhibitors that have shown efficient action in preventing the mitochondrial cholesterol trafficking, although some compounds have been described that may serve as model lead compounds for the design of more specific and potent STARD1 inhibitors [128]. Statins, on the other hand, although widely available and well characterized are expected to not only inhibit HMGCR and hence cholesterol synthesis, but they also disrupt the generation of farnesyl pyrophosphate in the mevalonate pathway and hence alter multiple proteins that are regulated via prenylation, accounting for the pleiotropic effects of statins beyond inhibition of just cholesterol synthesis. In this regard, targeting enzymatic steps downstream of HMGCR, such as squalene synthase or squalene epoxidase may be more specific in preventing cholesterol synthesis without significant alterations in the generation of isoprenoids. Since cholesterol is a crucial determinant of bilayers physical properties, accumulation of cholesterol in mitochondrial membranes in cancer cells can disrupt mitochondrial membrane fluidity, which in turn, can have a significant impact in protecting MOM from permeabilization and hence protect against apoptosis. Moreover, mitochondrial cholesterol-mediated changes in membrane fluidity can contribute to the resistance of cancer cells against mitochondrial-targeted chemotherapy. Besides these effects, mitochondrial cholesterol metabolism into oxysterols can also contribute to the alterations of cancer metabolism and regulation of cell death/proliferation. Overall, further understanding the biology of mitochondrial cholesterol regulation in cancer cells may constitute an attractive target for intervention in cancer cell biology and open up the opportunity to design novel treatments for cancer.
Acknowledgements
We acknowledge the support from grants PID2019–111669RB, SAF2017–85877R and SAF2015–73579-JIN from Plan Nacional de I+D funded by the Agencia Estatal de Investigación (AEI) and the Fondo Europeo de Desarrollo Regional (FEDER) and from the CIBEREHD; the center grant P50AA011999 Southern California Research Center for ALPD and Cirrhosis funded by NIAAA/NIH; as well as support from AGAUR of the Generalitat de Catalunya SGR-2017–1112, European Cooperation in Science & Technology (COST) ACTION CA17112 Prospective European Drug-Induced Liver Injury Network, the “ER stress-mitochondrial cholesterol axis in obesity-associated insulin resistance and comorbidities”-Ayudas FUNDACION BBVA, the Red Nacional 2018–102799-T de Enfermedades Metabólicas y Cáncer and the Project 201916/31 Contribution of mitochondrial oxysterol and bile acid metabolism to liver carcinogenesis 2019 by Fundació Marato TV3.
ABBREVIATIONS
- AR
Androgen receptor
- BA
Bile acids
- CAH
Congenital lipoid adrenal hiperplasia
- CTX
Cerebrotendenous xanthomatosis
- ER
Endoplasmic reticulum
- HCC
Hepatocellular carcinoma
- 27-OH-Chol
27-hydroxycholesterol
- HIF-1
Hypoxia inducing factor-1
- HMGR
3-hydroxy-3-methylglutaryl-CoA reductase
- MCD
methyl-β-cyclodextrine
- MIM
Mitochondrial inner membrane
- MOM
Mitochondrial outer membrane
- MOMP
Mitochondrial outer membrane permeabilization
- NASH
Nonalcoholic steatohepatitis
- ORPs
oxysterol-binding protein-related proteins
- StARD1
Steroidogenic acute regulatory protein 1
- SREBP2
Sterol regulatory element protein 2
- SCAP
SREBP cleavage activating protein
- START
Steroidogenic acute regulatory protein-related lipid transfer
- TSPO
Translocator protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interests
Authors declare no conflict of interests.
References
- [1].Che L, Chi W, Qiao Y, Zhang J, Song X, Liu Y, Li L, Jia J, Pilo MG, Wang J, Cigliano A, Ma Z, Kuang W, Tang Z, Zhang Z, Shui G, Ribback S, Dombrowski F, Evert M, Pascale RM, Cossu C, Pes GM, Osborne TF, Calvisi DF, Chen X, Chen L, Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans, Gut 69(1) (2020) 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shafique K, McLoone P, Qureshi K, Leung H, Hart C, Morrison DS, Cholesterol and the risk of grade-specific prostate cancer incidence: evidence from two large prospective cohort studies with up to 37 years’ follow up, BMC cancer 12 (2012) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pelton K, Freeman MR, Solomon KR, Cholesterol and prostate cancer, Current opinion in pharmacology 12(6) (2012) 751–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Allott EH, Howard LE, Cooperberg MR, Kane CJ, Aronson WJ, Terris MK, Amling CL, Freedland SJ, Serum lipid profile and risk of prostate cancer recurrence: Results from the SEARCH database, Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 23(11) (2014) 2349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jacobs EJ, Newton CC, Thun MJ, Gapstur SM, Long-term use of cholesterol-lowering drugs and cancer incidence in a large United States cohort, Cancer Res 71(5) (2011) 1763–71. [DOI] [PubMed] [Google Scholar]
- [6].Murtola TJ, Visvanathan K, Artama M, Vainio H, Pukkala E, Statin use and breast cancer survival: a nationwide cohort study from Finland, PLoS One 9(10) (2014) e110231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cardwell CR, Hicks BM, Hughes C, Murray LJ, Statin use after colorectal cancer diagnosis and survival: a population-based cohort study, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 32(28) (2014) 3177–83. [DOI] [PubMed] [Google Scholar]
- [8].Nielsen SF, Nordestgaard BG, Bojesen SE, Statin use and reduced cancer-related mortality, The New England journal of medicine 367(19) (2012) 1792–802. [DOI] [PubMed] [Google Scholar]
- [9].Ravnskov U, Rosch PJ, McCully KS, Statins do not protect against cancer: quite the opposite, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 33(7) (2015) 810–1. [DOI] [PubMed] [Google Scholar]
- [10].Bjerre LM, LeLorier J, Do statins cause cancer? A meta-analysis of large randomized clinical trials, Am J Med 110(9) (2001) 716–23. [DOI] [PubMed] [Google Scholar]
- [11].Pedersen TR, Wilhelmsen L, Faergeman O, Strandberg TE, Thorgeirsson G, Troedsson L, Kristianson J, Berg K, Cook TJ, Haghfelt T, Kjekshus J, Miettinen T, Olsson AG, Pyorala K, Wedel H, Follow-up study of patients randomized in the Scandinavian simvastatin survival study (4S) of cholesterol lowering, The American journal of cardiology 86(3) (2000) 257–62. [DOI] [PubMed] [Google Scholar]
- [12].Bakiri L, Hamacher R, Grana O, Guio-Carrion A, Campos-Olivas R, Martinez L, Dienes HP, Thomsen MK, Hasenfuss SC, Wagner EF, Liver carcinogenesis by FOS-dependent inflammation and cholesterol dysregulation, The Journal of experimental medicine 214(5) (2017) 1387–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liang JQ, Teoh N, Xu L, Pok S, Li X, Chu ESH, Chiu J, Dong L, Arfianti E, Haigh WG, Yeh MM, Ioannou GN, Sung JJY, Farrell G, Yu J, Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling, Nat Commun 9(1) (2018) 4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu D, Wong CC, Fu L, Chen H, Zhao L, Li C, Zhou Y, Zhang Y, Xu W, Yang Y, Wu B, Cheng G, Lai PB, Wong N, Sung JJY, Yu J, Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target, Science translational medicine 10(437) (2018). [DOI] [PubMed] [Google Scholar]
- [15].Qin WH, Yang ZS, Li M, Chen Y, Zhao XF, Qin YY, Song JQ, Wang BB, Yuan B, Cui XL, Shen F, He J, Bi YF, Ning G, Fu J, Wang HY, High Serum Levels of Cholesterol Increase Anti-tumor Functions of Nature Killer Cells and Reduce Growth of Liver Tumors in Mice, Gastroenterology (2020). [DOI] [PubMed]
- [16].Zhao Z, Zhong L, He K, Qiu C, Li Z, Zhao L, Gong J, Cholesterol attenuated the progression of DEN-induced hepatocellular carcinoma via inhibiting SCAP mediated fatty acid de novo synthesis, Biochemical and biophysical research communications 509(4) (2019) 855–861. [DOI] [PubMed] [Google Scholar]
- [17].Yang Z, Qin W, Chen Y, Yuan B, Song X, Wang B, Shen F, Fu J, Wang H, Cholesterol inhibits hepatocellular carcinoma invasion and metastasis by promoting CD44 localization in lipid rafts, Cancer letters 429 (2018) 66–77. [DOI] [PubMed] [Google Scholar]
- [18].Maxfield FR, Tabas I, Role of cholesterol and lipid organization in disease, Nature 438(7068) (2005) 612–21. [DOI] [PubMed] [Google Scholar]
- [19].Mesmin B, Maxfield FR, Intracellular sterol dynamics, Biochim Biophys Acta 1791(7) (2009) 636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jefcoate C, High-flux mitochondrial cholesterol trafficking, a specialized function of the adrenal cortex, J Clin Invest 110(7) (2002) 881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ikonen E, Cellular cholesterol trafficking and compartmentalization, Nature reviews. Molecular cell biology 9(2) (2008) 125–38. [DOI] [PubMed] [Google Scholar]
- [22].Garcia-Ruiz C, Mari M, Colell A, Morales A, Caballero F, Montero J, Terrones O, Basanez G, Fernandez-Checa JC, Mitochondrial cholesterol in health and disease, Histol Histopathol 24(1) (2009) 117–32. [DOI] [PubMed] [Google Scholar]
- [23].Carlberg C, Munoz A, An update on vitamin D signaling and cancer, Seminars in cancer biology (2020). [DOI] [PubMed]
- [24].Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, Kaufman RJ, Saltiel AR, Karin M, ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P, Cell 175(1) (2018) 133–145 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Caballero F, Fernandez A, De Lacy AM, Fernandez-Checa JC, Caballeria J, Garcia-Ruiz C, Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH, J Hepatol 50(4) (2009) 789–96. [DOI] [PubMed] [Google Scholar]
- [26].Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, Kellum J, Warnick R, Contos MJ, Sanyal AJ, Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease, Cell Metab 15(5) (2012) 665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Begicevic RR, Falasca M, ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance, Int J Mol Sci 18(11) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Smith B, Land H, Anticancer activity of the cholesterol exporter ABCA1 gene, Cell reports 2(3) (2012) 580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gu P, Wang Y, Bisht KK, Wu L, Kukova L, Smith EM, Xiao Y, Bailey SM, Lei M, Nandakumar J, Chang S, Pot1 OB-fold mutations unleash telomere instability to initiate tumorigenesis, Oncogene 36(14) (2017) 1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bemlih S, Poirier MD, El Andaloussi A, Acyl-coenzyme A: cholesterol acyltransferase inhibitor Avasimibe affect survival and proliferation of glioma tumor cell lines, Cancer Biol Ther 9(12) (2010) 1025–32. [DOI] [PubMed] [Google Scholar]
- [31].Zheng Y, Liu Y, Zhao S, Zheng Z, Shen C, An L, Yuan Y, Large-scale analysis reveals a novel risk score to predict overall survival in hepatocellular carcinoma, Cancer Manag Res 10 (2018) 6079–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gadaleta RM, van Mil SW, Oldenburg B, Siersema PD, Klomp LW, van Erpecum KJ, Bile acids and their nuclear receptor FXR: Relevance for hepatobiliary and gastrointestinal disease, Biochim Biophys Acta 1801(7) (2010) 683–92. [DOI] [PubMed] [Google Scholar]
- [33].Sun L, Beggs K, Borude P, Edwards G, Bhushan B, Walesky C, Roy N, Manley MW Jr., Gunewardena S, O’Neil M, Li H, Apte U, Bile acids promote diethylnitrosamine-induced hepatocellular carcinoma via increased inflammatory signaling, Am J Physiol Gastrointest Liver Physiol 311(1) (2016) G91–G104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Maxfield FR, van Meer G, Cholesterol, the central lipid of mammalian cells, Current opinion in cell biology 22(4) (2010) 422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Clark BJ, The mammalian START domain protein family in lipid transport in health and disease, The Journal of endocrinology 212(3) (2012) 257–75. [DOI] [PubMed] [Google Scholar]
- [36].Elustondo P, Martin LA, Karten B, Mitochondrial cholesterol import, Biochim Biophys Acta Mol Cell Biol Lipids 1862(1) (2017) 90–101. [DOI] [PubMed] [Google Scholar]
- [37].Miller WL, Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol transporter, Biochim Biophys Acta 1771(6) (2007) 663–76. [DOI] [PubMed] [Google Scholar]
- [38].Lin D, Sugawara T, Strauss JF 3rd, Clark BJ, Stocco DM, Saenger P, Rogol A, Miller WL, Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis, Science 267(5205) (1995) 1828–31. [DOI] [PubMed] [Google Scholar]
- [39].Bose HS, Sugawara T, Strauss JF 3rd, Miller WL, International C Congenital Lipoid Adrenal Hyperplasia, The pathophysiology and genetics of congenital lipoid adrenal hyperplasia, The New England journal of medicine 335(25) (1996) 1870–8. [DOI] [PubMed] [Google Scholar]
- [40].Caron KM, Soo SC, Wetsel WC, Stocco DM, Clark BJ, Parker KL, Targeted disruption of the mouse gene encoding steroidogenic acute regulatory protein provides insights into congenital lipoid adrenal hyperplasia, Proc Natl Acad Sci U S A 94(21) (1997) 11540–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stocco DM, StAR protein and the regulation of steroid hormone biosynthesis, Annual review of physiology 63 (2001) 193–213. [DOI] [PubMed] [Google Scholar]
- [42].Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, Karin M, Preclinical Models for Studying NASH-Driven HCC: How Useful Are They?, Cell Metab 29(1) (2019) 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Torres S, Baulies A, Insausti-Urkia N, Alarcon-Vila C, Fucho R, Solsona-Vilarrasa E, Nunez S, Robles D, Ribas V, Wakefield L, Grompe M, Lucena MI, Andrade RJ, Win S, Aung TA, Kaplowitz N, Garcia-Ruiz C, Fernandez-Checa JC, Endoplasmic Reticulum Stress-Induced Upregulation of STARD1 Promotes Acetaminophen-Induced Acute Liver Failure, Gastroenterology (2019). [DOI] [PubMed]
- [44].Balboa E, Castro J, Pinochet MJ, Cancino GI, Matias N, Saez PJ, Martinez A, Alvarez AR, Garcia-Ruiz C, Fernandez-Checa JC, Zanlungo S, MLN64 induces mitochondrial dysfunction associated with increased mitochondrial cholesterol content, Redox biology 12 (2017) 274–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kishida T, Kostetskii I, Zhang Z, Martinez F, Liu P, Walkley SU, Dwyer NK, Blanchette-Mackie EJ, Radice GL, Strauss JF 3rd, Targeted mutation of the MLN64 START domain causes only modest alterations in cellular sterol metabolism, J Biol Chem 279(18) (2004) 19276–85. [DOI] [PubMed] [Google Scholar]
- [46].Papadopoulos V, Miller WL, Role of mitochondria in steroidogenesis, Best practice & research. Clinical endocrinology & metabolism 26(6) (2012) 771–90. [DOI] [PubMed] [Google Scholar]
- [47].Miller WL, Steroid hormone synthesis in mitochondria, Mol Cell Endocrinol 379(1–2) (2013) 62–73. [DOI] [PubMed] [Google Scholar]
- [48].Morohaku K, Pelton SH, Daugherty DJ, Butler WR, Deng W, Selvaraj V, Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis, Endocrinology 155(1) (2014) 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chung JY, Chen H, Papadopoulos V, Zirkin B, Cholesterol accumulation, lipid droplet formation, and steroid production in Leydig cells: Role of translocator protein (18-kDa), Andrology 8(3) (2020) 719–730. [DOI] [PubMed] [Google Scholar]
- [50].Fan J, Campioli E, Midzak A, Culty M, Papadopoulos V, Conditional steroidogenic cell-targeted deletion of TSPO unveils a crucial role in viability and hormone-dependent steroid formation, Proc Natl Acad Sci U S A 112(23) (2015) 7261–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pandak WM, Ren S, Marques D, Hall E, Redford K, Mallonee D, Bohdan P, Heuman D, Gil G, Hylemon P, Transport of cholesterol into mitochondria is rate-limiting for bile acid synthesis via the alternative pathway in primary rat hepatocytes, J Biol Chem 277(50) (2002) 48158–64. [DOI] [PubMed] [Google Scholar]
- [52].Ren S, Hylemon PB, Marques D, Gurley E, Bodhan P, Hall E, Redford K, Gil G, Pandak WM, Overexpression of cholesterol transporter StAR increases in vivo rates of bile acid synthesis in the rat and mouse, Hepatology 40(4) (2004) 910–7. [DOI] [PubMed] [Google Scholar]
- [53].Worthmann A, John C, Ruhlemann MC, Baguhl M, Heinsen FA, Schaltenberg N, Heine M, Schlein C, Evangelakos I, Mineo C, Fischer M, Dandri M, Kremoser C, Scheja L, Franke A, Shaul PW, Heeren J, Cold-induced conversion of cholesterol to bile acids in mice shapes the gut microbiome and promotes adaptive thermogenesis, Nature medicine 23(7) (2017) 839–849. [DOI] [PubMed] [Google Scholar]
- [54].Schwarz M, Lund EG, Setchell KD, Kayden HJ, Zerwekh JE, Bjorkhem I, Herz J, Russell DW, Disruption of cholesterol 7alpha-hydroxylase gene in mice. II. Bile acid deficiency is overcome by induction of oxysterol 7alpha-hydroxylase, J Biol Chem 271(30) (1996) 18024–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Arenas F, Castro F, Nunez S, Gay G, Garcia-Ruiz C, Fernandez-Checa JC, STARD1 and NPC1 expression as pathological markers associated with astrogliosis in post-mortem brains from patients with Alzheimer’s disease and Down syndrome, Aging 12(1) (2020) 571–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Montero J, Mari M, Colell A, Morales A, Basanez G, Garcia-Ruiz C, Fernandez-Checa JC, Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death, Biochim Biophys Acta 1797(6–7) (2010) 1217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ribas V, Garcia-Ruiz C, Fernandez-Checa JC, Mitochondria, cholesterol and cancer cell metabolism, Clin Transl Med 5(1) (2016) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Feo F, Canuto RA, Garcea R, Gabriel L, Effect of cholesterol content on some physical and functional properties of mitochondria isolated from adult rat liver, fetal liver, cholesterol-enriched liver and hepatomas AH-130, 3924A and 5123, Biochim Biophys Acta 413(1) (1975) 116–34. [DOI] [PubMed] [Google Scholar]
- [59].Crain RC, Clark RW, Harvey BE, Role of lipid transfer proteins in the abnormal lipid content of Morris hepatoma mitochondria and microsomes, Cancer Res 43(7) (1983) 3197–202. [PubMed] [Google Scholar]
- [60].Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basanez G, Antonsson B, Prieto J, Garcia-Ruiz C, Colell A, Fernandez-Checa JC, Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma, Cancer Res 68(13) (2008) 5246–56. [DOI] [PubMed] [Google Scholar]
- [61].Colell A, Garcia-Ruiz C, Lluis JM, Coll O, Mari M, Fernandez-Checa JC, Cholesterol impairs the adenine nucleotide translocator-mediated mitochondrial permeability transition through altered membrane fluidity, J Biol Chem 278(36) (2003) 33928–35. [DOI] [PubMed] [Google Scholar]
- [62].Bosch M, Mari M, Herms A, Fernandez A, Fajardo A, Kassan A, Giralt A, Colell A, Balgoma D, Barbero E, Gonzalez-Moreno E, Matias N, Tebar F, Balsinde J, Camps M, Enrich C, Gross SP, Garcia-Ruiz C, Perez-Navarro E, Fernandez-Checa JC, Pol A, Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility, Curr Biol 21(8) (2011) 681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Baulies A, Montero J, Matias N, Insausti N, Terrones O, Basanez G, Vallejo C, Conde de La Rosa L, Martinez L, Robles D, Morales A, Abian J, Carrascal M, Machida K, Kumar DBU, Tsukamoto H, Kaplowitz N, Garcia-Ruiz C, Fernandez-Checa JC, The 2-oxoglutarate carrier promotes liver cancer by sustaining mitochondrial GSH despite cholesterol loading, Redox biology 14 (2018) 164–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Coll O, Colell A, Garcia-Ruiz C, Kaplowitz N, Fernandez-Checa JC, Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion, Hepatology 38(3) (2003) 692–702. [DOI] [PubMed] [Google Scholar]
- [65].Ribas V, Garcia-Ruiz C, Fernandez-Checa JC, Glutathione and Mitochondria, Frontiers in Pharmacology 5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Fernandez A, Colell A, Caballero F, Matias N, Garcia-Ruiz C, Fernandez-Checa JC, Mitochondrial S-adenosyl-L-methionine transport is insensitive to alcohol-mediated changes in membrane dynamics, Alcohol Clin Exp Res 33(7) (2009) 1169–80. [DOI] [PubMed] [Google Scholar]
- [67].Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO, Antioxidants accelerate lung cancer progression in mice, Science translational medicine 6(221) (2014) 221ra15. [DOI] [PubMed] [Google Scholar]
- [68].Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, Dalin MG, Akyurek LM, Lindahl P, Nilsson J, Bergo MO, Antioxidants can increase melanoma metastasis in mice, Science translational medicine 7(308) (2015) 308re8. [DOI] [PubMed] [Google Scholar]
- [69].Lee JS, Lee H, Lee S, Kang JH, Lee SH, Kim SG, Cho ES, Kim NH, Yook JI, Kim SY, Loss of SLC25A11 causes suppression of NSCLC and melanoma tumor formation, EBioMedicine 40 (2019) 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Solsona-Vilarrasa E, Fucho R, Torres S, Nunez S, Nuno-Lambarri N, Enrich C, Garcia-Ruiz C, Fernandez-Checa JC, Cholesterol enrichment in liver mitochondria impairs oxidative phosphorylation and disrupts the assembly of respiratory supercomplexes, Redox biology 24 (2019) 101214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Baggetto LG, Role of mitochondria in carcinogenesis, European journal of cancer 29A(1) (1992) 156–9. [DOI] [PubMed] [Google Scholar]
- [72].Galluzzi L, Larochette N, Zamzami N, Kroemer G, Mitochondria as therapeutic targets for cancer chemotherapy, Oncogene 25(34) (2006) 4812–30. [DOI] [PubMed] [Google Scholar]
- [73].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell 144(5) (2011) 646–74. [DOI] [PubMed] [Google Scholar]
- [74].Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, Garcia-Ruiz C, Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis, Cell Metab 4(3) (2006) 185–98. [DOI] [PubMed] [Google Scholar]
- [75].Colell A, Garcia-Ruiz C, Miranda M, Ardite E, Mari M, Morales A, Corrales F, Kaplowitz N, Fernandez-Checa JC, Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor, Gastroenterology 115(6) (1998) 1541–51. [DOI] [PubMed] [Google Scholar]
- [76].Lucken-Ardjomande S, Montessuit S, Martinou JC, Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes, Cell death and differentiation 15(3) (2008) 484–93. [DOI] [PubMed] [Google Scholar]
- [77].Christenson E, Merlin S, Saito M, Schlesinger P, Cholesterol effects on BAX pore activation, Journal of molecular biology 381(5) (2008) 1168–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Verstraeten SL, Albert M, Paquot A, Muccioli GG, Tyteca D, Mingeot-Leclercq MP, Membrane cholesterol delays cellular apoptosis induced by ginsenoside Rh2, a steroid saponin, Toxicology and applied pharmacology 352 (2018) 59–67. [DOI] [PubMed] [Google Scholar]
- [79].Le Bras M, Borgne-Sanchez A, Touat Z, El Dein OS, Deniaud A, Maillier E, Lecellier G, Rebouillat D, Lemaire C, Kroemer G, Jacotot E, Brenner C, Chemosensitization by knockdown of adenine nucleotide translocase-2, Cancer Res 66(18) (2006) 9143–52. [DOI] [PubMed] [Google Scholar]
- [80].Don AS, Kisker O, Dilda P, Donoghue N, Zhao X, Decollogne S, Creighton B, Flynn E, Folkman J, Hogg PJ, A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells, Cancer cell 3(5) (2003) 497–509. [DOI] [PubMed] [Google Scholar]
- [81].Denmeade SR, Isaacs JT, The SERCA pump as a therapeutic target: making a “smart bomb” for prostate cancer, Cancer Biol Ther 4(1) (2005) 14–22. [DOI] [PubMed] [Google Scholar]
- [82].Kumar D, Kirshenbaum L, Li T, Danelisen I, Singal P, Apoptosis in isolated adult cardiomyocytes exposed to adriamycin, Ann N Y Acad Sci 874 (1999) 156–68. [DOI] [PubMed] [Google Scholar]
- [83].Ahmadi Y, Karimian R, Panahi Y, Effects of statins on the chemoresistance-The antagonistic drug-drug interactions versus the anti-cancer effects, Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 108 (2018) 1856–1865. [DOI] [PubMed] [Google Scholar]
- [84].Lee JS, Roberts A, Juarez D, Vo TT, Bhatt S, Herzog LO, Mallya S, Bellin RJ, Agarwal SK, Salem AH, Xu T, Jia J, Li L, Hanna JR, Davids MS, Fleischman AG, O’Brien S, Lam LT, Leverson JD, Letai A, Schatz JH, Fruman DA, Statins enhance efficacy of venetoclax in blood cancers, Science translational medicine 10(445) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Giaccia A, Siim BG, Johnson RS, HIF-1 as a target for drug development, Nature reviews. Drug discovery 2(10) (2003) 803–11. [DOI] [PubMed] [Google Scholar]
- [86].Semenza GL, Targeting HIF-1 for cancer therapy, Nature reviews. Cancer 3(10) (2003) 721–32. [DOI] [PubMed] [Google Scholar]
- [87].Melillo G, Inhibiting hypoxia-inducible factor 1 for cancer therapy, Mol Cancer Res 4(9) (2006) 601–5. [DOI] [PubMed] [Google Scholar]
- [88].Pugh CW, Ratcliffe PJ, Regulation of angiogenesis by hypoxia: role of the HIF system, Nature medicine 9(6) (2003) 677–84. [DOI] [PubMed] [Google Scholar]
- [89].Safran M, Kaelin WG Jr., HIF hydroxylation and the mammalian oxygen-sensing pathway, J Clin Invest 111(6) (2003) 779–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kaelin WG Jr., ROS: really involved in oxygen sensing, Cell Metab 1(6) (2005) 357–8. [DOI] [PubMed] [Google Scholar]
- [91].Nath B, Szabo G, Hypoxia and hypoxia inducible factors: diverse roles in liver diseases, Hepatology 55(2) (2012) 622–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Rosmorduc O, Housset C, Hypoxia: a link between fibrogenesis, angiogenesis, and carcinogenesis in liver disease, Semin Liver Dis 30(3) (2010) 258–70. [DOI] [PubMed] [Google Scholar]
- [93].Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS, Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation, Cell Metab 1(6) (2005) 409–14. [DOI] [PubMed] [Google Scholar]
- [94].Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT, Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing, Cell Metab 1(6) (2005) 401–8. [DOI] [PubMed] [Google Scholar]
- [95].Onnis B, Rapisarda A, Melillo G, Development of HIF-1 inhibitors for cancer therapy, Journal of cellular and molecular medicine 13(9A) (2009) 2780–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Anavi S, Hahn-Obercyger M, Madar Z, Tirosh O, Mechanism for HIF-1 activation by cholesterol under normoxia: a redox signaling pathway for liver damage, Free radical biology & medicine 71 (2014) 61–9. [DOI] [PubMed] [Google Scholar]
- [97].Tirosh O, Hypoxic Signaling and Cholesterol Lipotoxicity in Fatty Liver Disease Progression, Oxidative medicine and cellular longevity 2018 (2018) 2548154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Burr SP, Costa AS, Grice GL, Timms RT, Lobb IT, Freisinger P, Dodd RB, Dougan G, Lehner PJ, Frezza C, Nathan JA, Mitochondrial Protein Lipoylation and the 2-Oxoglutarate Dehydrogenase Complex Controls HIF1alpha Stability in Aerobic Conditions, Cell Metab 24(5) (2016) 740–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Bailey PSJ, Nathan JA, Metabolic Regulation of Hypoxia-Inducible Transcription Factors: The Role of Small Molecule Metabolites and Iron, Biomedicines 6(2) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Koivunen P, Fell SM, Lu W, Rabinowitz JD, Kung AL, Schlisio S, The 2-oxoglutarate analog 3-oxoglutarate decreases normoxic hypoxia-inducible factor-1alpha in cancer cells, induces cell death, and reduces tumor xenograft growth, Hypoxia (Auckl) 4 (2016) 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kerkela R, Karsikas S, Szabo Z, Serpi R, Magga J, Gao E, Alitalo K, Anisimov A, Sormunen R, Pietila I, Vainio L, Koch WJ, Kivirikko KI, Myllyharju J, Koivunen P, Activation of hypoxia response in endothelial cells contributes to ischemic cardioprotection, Mol Cell Biol 33(16) (2013) 3321–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Redding GP, Bronlund JE, Hart AL, Mathematical modelling of oxygen transport-limited follicle growth, Reproduction 133(6) (2007) 1095–106. [DOI] [PubMed] [Google Scholar]
- [103].Kowalewski MP, Gram A, Boos A, The role of hypoxia and HIF1alpha in the regulation of STAR-mediated steroidogenesis in granulosa cells, Mol Cell Endocrinol 401 (2015) 35–44. [DOI] [PubMed] [Google Scholar]
- [104].Fadhillah, Yoshioka S, Nishimura R, Yamamoto Y, Kimura K, Okuda K, Hypoxia-inducible factor 1 mediates hypoxia-enhanced synthesis of progesterone during luteinization of granulosa cells, The Journal of reproduction and development 63(1) (2017) 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kloudova A, Guengerich FP, Soucek P, The Role of Oxysterols in Human Cancer, Trends Endocrinol Metab 28(7) (2017) 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Olkkonen VM, Beaslas O, Nissila E, Oxysterols and their cellular effectors, Biomolecules 2(1) (2012) 76–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Pandak WM, Kakiyama G, The acidic pathway of bile acid synthesis: Not just an alternative pathway(), Liver Res 3(2) (2019) 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Javitt NB, Cholesterol, hydroxycholesterols, and bile acids, Biochemical and biophysical research communications 292(5) (2002) 1147–53. [DOI] [PubMed] [Google Scholar]
- [109].Russell DW, The enzymes, regulation, and genetics of bile acid synthesis, Annual review of biochemistry 72 (2003) 137–74. [DOI] [PubMed] [Google Scholar]
- [110].Lorbek G, Lewinska M, Rozman D, Cytochrome P450s in the synthesis of cholesterol and bile acids--from mouse models to human diseases, The FEBS journal 279(9) (2012) 1516–33. [DOI] [PubMed] [Google Scholar]
- [111].Bjorkhem I, Leitersdorf E, Sterol 27-hydroxylase deficiency: a rare cause of xanthomas in normocholesterolemic humans, Trends Endocrinol Metab 11(5) (2000) 180–3. [DOI] [PubMed] [Google Scholar]
- [112].Song C, Liao S, Hypolipidemic effects of selective liver X receptor alpha agonists, Steroids 66(9) (2001) 673–81. [DOI] [PubMed] [Google Scholar]
- [113].Yoon JH, Canbay AE, Werneburg NW, Lee SP, Gores GJ, Oxysterols induce cyclooxygenase-2 expression in cholangiocytes: implications for biliary tract carcinogenesis, Hepatology 39(3) (2004) 732–8. [DOI] [PubMed] [Google Scholar]
- [114].Nagano K, Imai S, Zhao X, Yamashita T, Yoshioka Y, Abe Y, Mukai Y, Kamada H, Nakagawa S, Tsutsumi Y, Tsunoda S, Identification and evaluation of metastasis-related proteins, oxysterol binding protein-like 5 and calumenin, in lung tumors, International journal of oncology 47(1) (2015) 195–203. [DOI] [PubMed] [Google Scholar]
- [115].de Weille J, Fabre C, Bakalara N, Oxysterols in cancer cell proliferation and death, Biochemical pharmacology 86(1) (2013) 154–60. [DOI] [PubMed] [Google Scholar]
- [116].Kulig W, Cwiklik L, Jurkiewicz P, Rog T, Vattulainen I, Cholesterol oxidation products and their biological importance, Chemistry and physics of lipids 199 (2016) 144–160. [DOI] [PubMed] [Google Scholar]
- [117].Appukuttan A, Kasseckert SA, Kumar S, Reusch HP, Ladilov Y, Oxysterol-induced apoptosis of smooth muscle cells is under the control of a soluble adenylyl cyclase, Cardiovascular research 99(4) (2013) 734–42. [DOI] [PubMed] [Google Scholar]
- [118].Lee T, Chau L, Fas/Fas ligand-mediated death pathway is involved in oxLDL-induced apoptosis in vascular smooth muscle cells, American journal of physiology. Cell physiology 280(3) (2001) C709–18. [DOI] [PubMed] [Google Scholar]
- [119].Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, Korach KS, Shaul PW, Mangelsdorf DJ, 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen, Nature medicine 13(10) (2007) 1185–92. [DOI] [PubMed] [Google Scholar]
- [120].Lappano R, Recchia AG, De Francesco EM, Angelone T, Cerra MC, Picard D, Maggiolini M, The cholesterol metabolite 25-hydroxycholesterol activates estrogen receptor alpha-mediated signaling in cancer cells and in cardiomyocytes, PLoS One 6(1) (2011) e16631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Simigdala N, Gao Q, Pancholi S, Roberg-Larsen H, Zvelebil M, Ribas R, Folkerd E, Thompson A, Bhamra A, Dowsett M, Martin LA, Cholesterol biosynthesis pathway as a novel mechanism of resistance to estrogen deprivation in estrogen receptor-positive breast cancer, Breast cancer research : BCR 18(1) (2016) 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, Umetani M, Geradts J, McDonnell DP, 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology, Science 342(6162) (2013) 1094–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Lee D, High androgen receptor levels are predictive of decreased survival in prostate cancer, Clin Prostate Cancer 2(1) (2003) 13–4. [DOI] [PubMed] [Google Scholar]
- [124].Raza S, Meyer M, Schommer J, Hammer KD, Guo B, Ghribi O, 27-Hydroxycholesterol stimulates cell proliferation and resistance to docetaxel-induced apoptosis in prostate epithelial cells, Medical oncology 33(2) (2016) 12. [DOI] [PubMed] [Google Scholar]
- [125].Swan R, Alnabulsi A, Cash B, Alnabulsi A, Murray GI, Characterisation of the oxysterol metabolising enzyme pathway in mismatch repair proficient and deficient colorectal cancer, Oncotarget 7(29) (2016) 46509–46527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Hirayama T, Honda A, Matsuzaki Y, Miyazaki T, Ikegami T, Doy M, Xu G, Lea M, Salen G, Hypercholesterolemia in rats with hepatomas: increased oxysterols accelerate efflux but do not inhibit biosynthesis of cholesterol, Hepatology 44(3) (2006) 602–11. [DOI] [PubMed] [Google Scholar]
- [127].Ikegami T, Honda A, Miyazaki T, Kohjima M, Nakamuta M, Matsuzaki Y, Increased serum oxysterol concentrations in patients with chronic hepatitis C virus infection, Biochemical and biophysical research communications 446(3) (2014) 736–40. [DOI] [PubMed] [Google Scholar]
- [128].Akula N, Midzak A, Lecanu L, Papadopoulos V, Identification of small-molecules inhibitors of the steroidogenic acute regulatory protein (STARD1) by structure-based design, Bioorg Med Chem Lett 22(12) (2012) 4139–43. [DOI] [PubMed] [Google Scholar]