

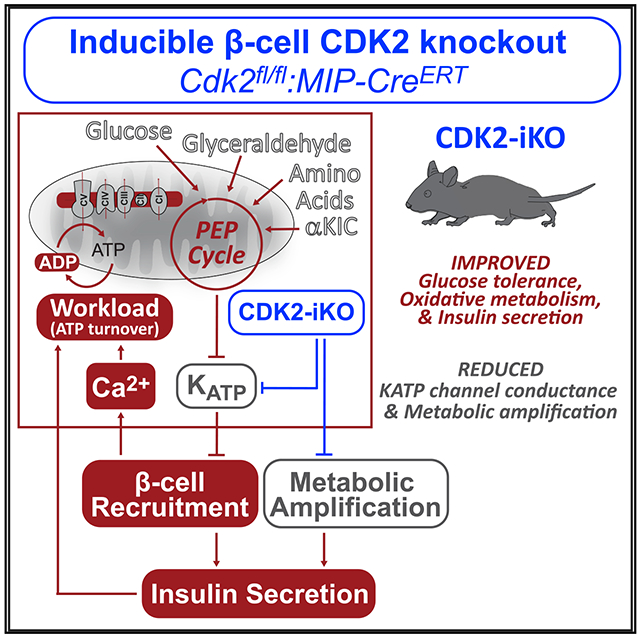
SUMMARY
Hallmarks of mature β cells are restricted proliferation and a highly energetic secretory state. Paradoxically, cyclin-dependent kinase 2 (CDK2) is synthesized throughout adulthood, its cytosolic localization raising the likelihood of cell cycle-independent functions. In the absence of any changes in β cell mass, maturity, or proliferation, genetic deletion of Cdk2 in adult β cells enhanced insulin secretion from isolated islets and improved glucose tolerance in vivo. At the single β cell level, CDK2 restricts insulin secretion by increasing KATP conductance, raising the set point for membrane depolarization in response to activation of the phosphoenolpyruvate (PEP) cycle with mitochondrial fuels. In parallel with reduced β cell recruitment, CDK2 restricts oxidative glucose metabolism while promoting glucose-dependent amplification of insulin secretion. This study provides evidence of essential, non-canonical functions of CDK2 in the secretory pathways of quiescent β cells.
Graphical Abstract
In Brief
Despite loss of proliferative capacity with age, mature β cells continually synthesize CDK2. Sdao et al. demonstrate that CDK2 depletion in adult β cells improves glucose tolerance in vivo. By augmenting PEP cycle-dependent KATP channel closure, CDK2 inactivation lowers the set point for membrane depolarization, augmenting oxidative metabolism and insulin secretion.
INTRODUCTION
Diabetes is associated with loss of functional β cell mass, and there is growing interest in the therapeutic potential of re-initiating the β cell mitogenic program (Ackeifi et al., 2020; Wang et al., 2019), which is typically active only in the first few months of life in mice and a year or two in humans (Cozar-Castellano et al., 2006). However, cyclin-dependent kinases (CDKs) and their cognate cyclins, having a half-life of hours, are re-synthesized continuously in β cells of adult mice and humans for their entire lifespan (Fiaschi-Taesch et al., 2013), suggesting that they perform non-canonical, non-cell cycle functions.
Most adult β cells remain quiescent in the G0/G1 phase of the cell cycle and express CDK2, CDK4, and CDK6 (Cozar-Castellano et al., 2006; Fiaschi-Taesch et al., 2013), which governs the CDK-retinoblast protein (RB)-E2F pathway (Aguilar and Fajas, 2010; Buchakjian and Kornbluth, 2010; DeBerardinis et al., 2008). E2F transcription factors, normally bound by RB, are released in response to RB phosphorylation by the CDKs. The CDK-RB-E2F pathway has been linked genetically with changes in mitochondrial mass, morphology, and bioenergetics (Blanchet et al., 2011; Dali-Youcef et al., 2007; Goto et al., 2006; Hsieh et al., 2008; Sakamaki et al., 2006), and multiple genetic deletion studies have reported changes in insulin secretion (Annicotte et al., 2009; Kim et al., 2017). In addition to transcriptional programs, cell cycle regulators have been reported to regulate extranuclear metabolic processes that may be unrelated to proliferation (Gregg et al., 2019; Lagarrigue et al., 2016; Lee et al., 2014; Lopez-Mejia et al., 2017; Wang et al., 2014). In support of this idea, many if not most cell cycle regulators localize in the cytosol rather than in the nucleus of adult β cells (Fiaschi-Taesch et al., 2013).
Here, we targeted CDK2 to examine its relationship with the β cell metabolic and secretory pathways. Deletion of the Cdk2 gene from the pancreatic endoderm in mice (i.e., Cdk2-floxed:Pdx1-Cre) results in glucose intolerance because of reduced insulin secretion (Kim et al., 2017). Importantly, this and other previous studies of the CDK-RB-E2F pathway utilized embryonic deletion models (i.e., Ccnd1−/−, Cdk4−/−, E2f1−/−) (Annicotte et al., 2009; Blanchet et al., 2011; Dali-Youcef et al., 2007; Goto et al., 2006; Hsieh et al., 2008; Kim et al., 2017; Sakamaki et al., 2006; Xue et al., 2019), imposing chronic developmental manipulations that may not be relevant in adults. To observe CDK2 signaling in the context of a healthy adult β cell, we generated an inducible β cell-specific Cdk2 deletion mouse model (CDK2-iKO, Cdk2-floxed:MIP-CreERT). Unlike the developmental knockout, insulin secretion and glucose tolerance are enhanced in CDK2-iKO mice. Using this model, we demonstrate pleotropic regulation of β cell excitability, ion channels, oxidative metabolism, and insulin secretion by CDK2 in adult β cells.
RESULTS
Restricting CDK2 in adult β cells improves glucose homeostasis and insulin secretion
To understand the role of CDK2 in adult β cells, we generated CDK2-iKO mice by breeding Cdk2fl/fl mice (Kim et al., 2017; Jayapal et al., 2015) with mice expressing tamoxifen-inducible Cre recombinase using the mouse insulin promoter (MIP-CreERT) (Tamarina et al., 2014; Figure 1A). Recombination was induced at 10 weeks of age, and mice were analyzed at 14 weeks of age. In isolated islets, Cdk2 mRNA was reduced by 70% in CDK2-iKO islets compared with MIP-CreERT controls (Figure 1B). In sectioned pancreas, we observed CDK2 primarily in the cytosol of mature β cells (Figure 1C), corroborating previous findings in human β cells (Fiaschi-Taesch et al., 2013). We rarely observed β cells expressing CDK2 in CDK2-iKO islets, confirming the efficacy of the MIP-CreERT transgene (Figure 1D). In vivo, CDK2-iKO mice exhibited normal fasting blood glucose, and glucose tolerance was improved significantly (Figure 1E). Consistently, islets isolated from CDK2-iKO mice secreted more insulin when challenged with glucose, whereas no effect on insulin secretion was observed at basal glucose levels (Figure 1F). No changes in α or β cell mass (Figures 1G and 1H), proliferation (Figure 1I), maturity (Figures 1J–1L), or insulin content (data not shown) were observed. Thus, the glucose homeostasis and insulin secretory phenotype in mice with short-term CDK2 restriction is opposite of what was observed in Cdk2fl/fl:Pdx1-Cre mice (Kim et al., 2017), indicating that gain-of-function CDK2-iKO is a more appropriate genetic model for understanding the role of CDK2 in adult β cells.
Figure 1. Short-term CDK2 restriction enhances insulin secretion from mouse islets and improves glucose tolerance.
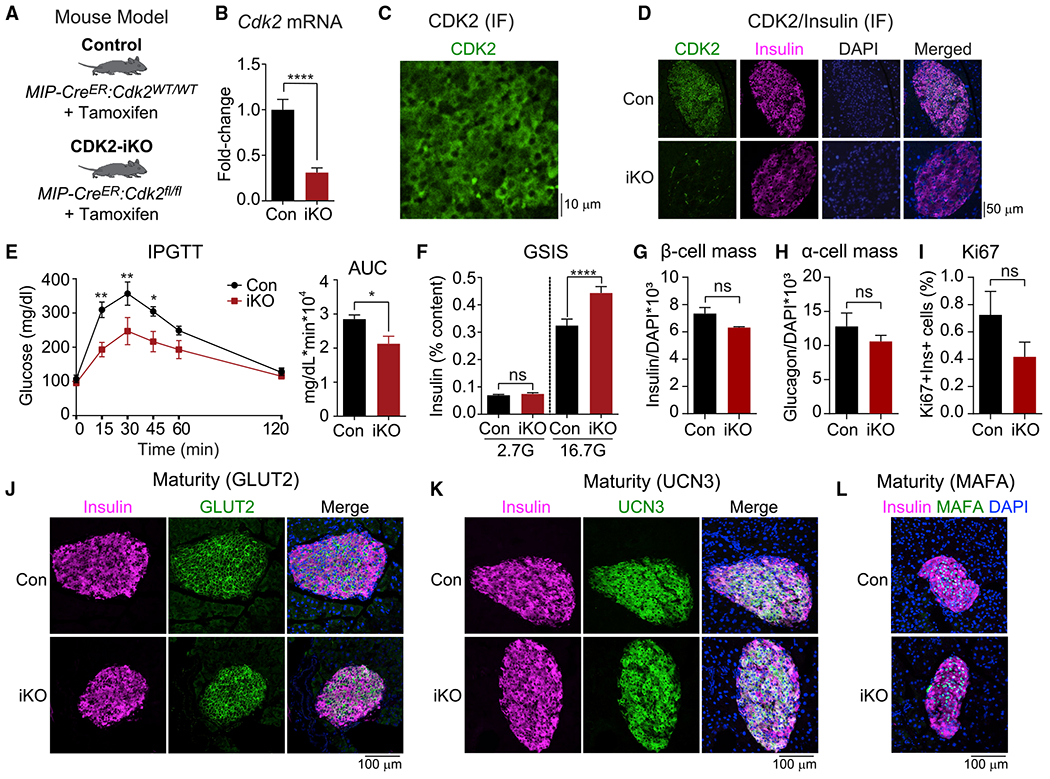
(A) Mouse model used to inducibly delete CDK2 from adult β cells. Tamoxifen was injected intraperitoneally into Cdk2fl/fl-MIPCreER (CDK2-iKO) and MIP-CreERT controls (Con) at 10 weeks of age. Mice were given 4 weeks to clear tamoxifen and phenotyped at 14 weeks of age.
(B) Cdk2 mRNA expression measured by qPCR in pancreatic islets isolated from Con (n = 15) and CDK2-iKO (n = 16) mice.
(C) CDK2 immunofluorescence (green) in a mouse pancreatic section.
(D) CDK2 (green) and insulin (pink) immunofluorescence in pancreatic sections from Con and CDK2-iKO mice.
(E) Glucose tolerance test (GTT) in Con (n = 5) and CDK2-iKO (n = 4) mice, quantified by area under the curve (AUC).
(F) Ex vivo glucose-stimulated insulin secretion (GSIS) normalized to insulin content, measured in isolated islets from Con (n = 3) and CDK2-iKO (n = 4) mice.
(G–I) Quantification of β cell mass (G), α cell mass (H), and Ki67-positive β cells (I) in Con (n = 4) and CDK2-iKO (n = 5) pancreatic sections.
(J–L) Insulin (pink) and maturation marker (green) GLUT2 (J), UCN3 (K), and MAFA (L) immunofluorescence in pancreatic sections from Con and CDK2-iKO mice.
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001 by t test.
CDK2 is essential for glucose-dependent amplification of insulin secretion
In β cells, glucose is a strong driver of several metabolic amplifying pathways of insulin secretion (Prentki et al., 2013). Consistent with a role of CDK2 in these pathways, we observed a severe defect in insulin granule exocytosis in CDK2-iKO islet β cells compared with controls (Figure 2A). This secretion defect was not due to differences in calcium channel activity (Figure 2B), nor was it observed in α cells from CDK2-iKO islets, which retained full function (Figures 2C and 2D). Importantly, we conducted these assays in the presence of 5 mM glucose, which is sufficient in capacitance assays to engage the β cell metabolic amplifying pathways at 80% maximum (Ferdaoussi et al., 2015), while avoiding electrical activity in neighboring β cells that would otherwise confound the capacitance measurements. KCl-stimulated insulin secretion was also reduced significantly in CDK2-iKO islets at a high glucose concentration without any significant change at low glucose (Figure 2E). These findings demonstrate that CDK2 is essential for glucose-dependent amplification of insulin secretion; however, they cannot explain the enhanced glucose tolerance or glucose-stimulated insulin secretion observed in CDK2-iKO mice.
Figure 2. Reduced metabolic amplifying pathway in CDK2-iKO β cells.
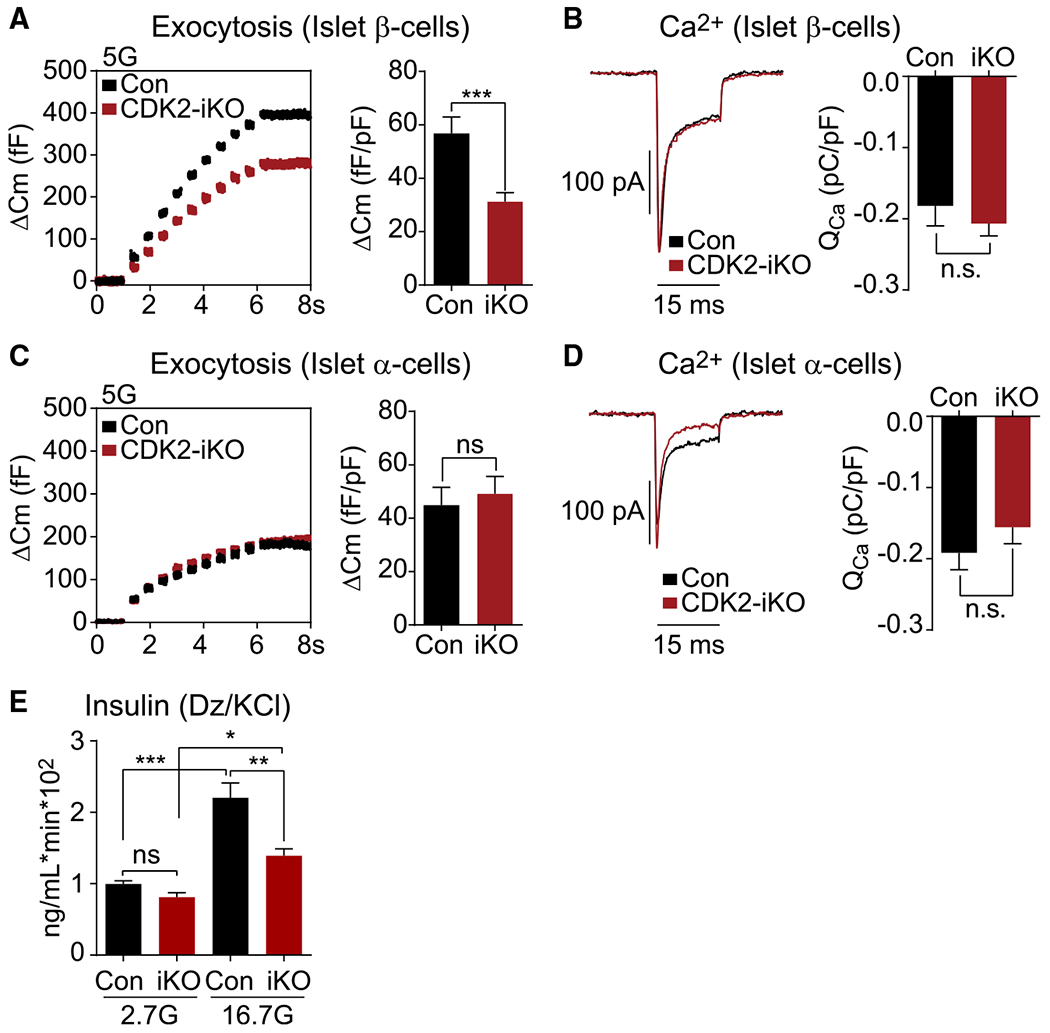
(A and C) Capacitance increases in response to 10 step depolarizations from −70 mV to 0 mV in (A) β cells (Con, n = 19; CDK2-iKO, n = 39) and (C) α cells (Con, n = 14; CDK2-iKO, n = 14) isolated from 4 Con and 3 CDK2-iKO mice.
(B and D) Averaged leak-subtracted calcium current (Ica) and influx (Qca) from β cells (B) and α cells (D), measured during a 15-ms step depolarization from −70 mV to 0 mV for each cell above.
(E) Ex vivo KCl-stimulated insulin secretion normalized to DNA content, measured in isolated islets from Con (n = 3) and CDK2-iKO (n = 4) mice, quantified by AUC.
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by t test (B and D) or one-way ANOVA (E).
CDK2 controls KATP channel activity at the β cell plasma membrane
The CDK-RB-E2F pathway, through E2F1-dependent transcription, has been shown to directly regulate Kir6.2, which encodes the pore-forming subunit of the KATP channel (Annicotte et al., 2009). In pharmacologic and genetic models of CDK2 restriction, we observed a significant decrease in Kir6.2 mRNA (Figures 3A and 3C). To determine whether CDK2 affects KATP channel function, we measured β cell KATP conductance (GKATP) using patch-clamp electrophysiology. GKATP was calculated from voltage ramps in 10 mM glucose and again in the presence of the KATP channel opener diazoxide (200 μM) (Figures 3B and 3D). As a first approach to CDK2 blockade, we used the small-molecule CDK2 inhibitor SU9516 (CDK2i), which exhibits 2- and 10-fold selectivity over CDK1 and CDK4, respectively (Lane et al., 2001). GKATP was increased significantly by addition of diazoxide, as expected, and reduced in CDK2i-treated β cells relative to vehicle controls (Figure 3B).
Figure 3. Islets with short-term CDK2 restriction have reduced Kir6.2 mRNA, resulting in reduced KATP channel activity.
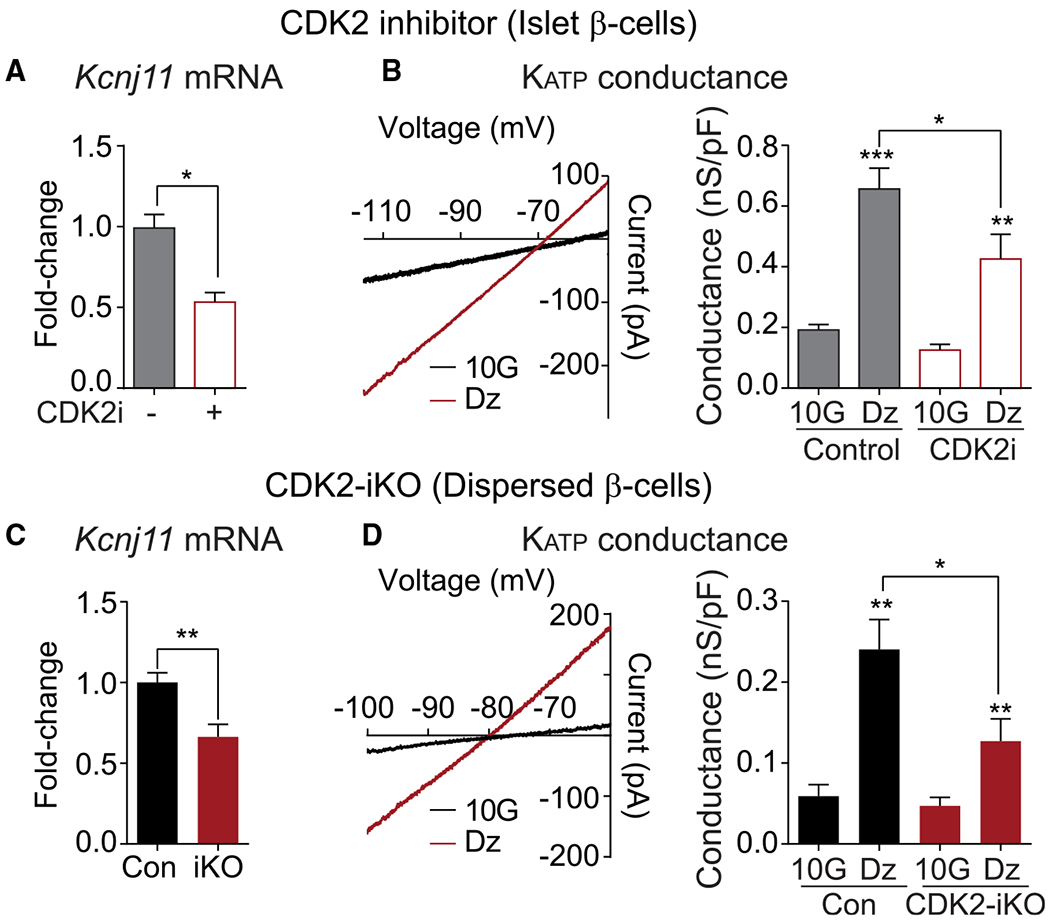
(A and C) Kcnj11 expression measured by qPCR in pancreatic islets treated with vehicle Con or CDK2i (n = 3 mice) (A) or islets isolated from Con and CDK2-iKO mice (n = 3 mice per genotype) (C).
(B and D) Measurements of KATP conductance in islets treated with vehicle Con or CDK2i (B) or CDK2-iKO islets (C). Left: representative current-voltage relationship collected during a voltage ramp, showing conductance (slope) changes in β cells measured in 10 mM glucose (10G) and after treatment with 200 μM diazoxide (DZ). Right: KATP conductance was reduced in CDK2i-treated (n = 7) relative to vehicle-treated Con (n = 11) β cells (B) and in CDK2-iKO β cells relative to Cons treated with Ad-Cre (n = 11 cells from 3 mice each genotype) (D).
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by t test (A and C) or one-way ANOVA (B and D).
A limitation of using the MIP-CreERT recombination strategy is incomplete penetrance of the Cre-mediated recombination (Figure 1B). To optimize our chances of recording from β cells with restricted CDK2 signaling, we took advantage of adenovirally delivered Cre recombinase. Islets were isolated from Cdk2fl/fl:MIPCreERT or Cdk2wt/wt:MIPCreERT mice that had not been injected with tamoxifen, dispersed into single cells, and infected with Ad-Cre-IRES-GFP. GKATP was recorded from GFP-expressing cells of both genotypes. Irrespective of the treatment condition, GKATP was substantially lower in dispersed β cells (Figure 3D) than in β cells within intact islets (Figure 3B). Despite this difference, GKATP was reduced significantly in adenovirally treated Cdk2fl/fl:MIPCreERT cells relative to Cdk2wt/wt:MIPCreERT controls (Figure 3D), again demonstrating the functional decrease in KATP channel activity associated with CDK2 deficiency.
Enhanced β cell recruitment in models of CDK2 blockade
The proportion of β cells that are electrically active, termed recruitment, increases with glucose concentration as metabolically derived ATP/ADP closes KATP channels to reach the threshold required for membrane depolarization (Jonkers and Henquin, 2001; Lewandowski et al., 2020). Consequently, as KATP channel activity is reduced, membrane depolarization and calcium influx occur at a lower glucose concentration (Grapengiesser et al., 1990; Gregg et al., 2016; Jonkers et al., 2001). We measured cytosolic calcium to determine the extent to which the reduced GKATP in CDK2-iKO islets caused a reduction in the glucose threshold for β cell recruitment. Reflecting a lower threshold, the duration of glucose-dependent calcium oscillations was increased strongly in CDK2-iKO and CDK2i-treated islets relative to controls (Figures 4A and 4E). An effect of CDK2 on β cell recruitment was evidenced by failure of islet calcium oscillations to switch off at subthreshold glucose concentrations in CDK2-iKO (Figure 4B) or CDK2i-treated islets (Figure 4F). We also performed a more rigorous quantification of glucose-stimulated calcium oscillations by measuring the oscillatory duty cycle, the fractional time spent in the active, secretory phase of an oscillation relative to the full cycle (Henquin, 2009; Lewandowski et al., 2020). We observed an increase in the duty cycle of calcium oscillations from CDK2-iKO and CDK2i islets relative to controls (Figures 4C and 4G) together with increased oscillation frequency (Figures 4D and 4H). These observations persisted when islets were provided with fuels downstream of glucokinase, including glyceraldehyde (Figures 4I–4K), and the mitochondrial fuel α-ketoisocap-roate (αKIC) (Figures 4L–4N). These data indicate β cell recruitment as a mechanism that offsets the reduction in glucose-dependent amplifying pathways in CDK2-iKO islets, contributing to enhanced glucose tolerance and glucose-stimulated insulin secretion in this model.
Figure 4. Increased islet recruitment underlies enhanced insulin secretion following short-term CDK2 restriction.
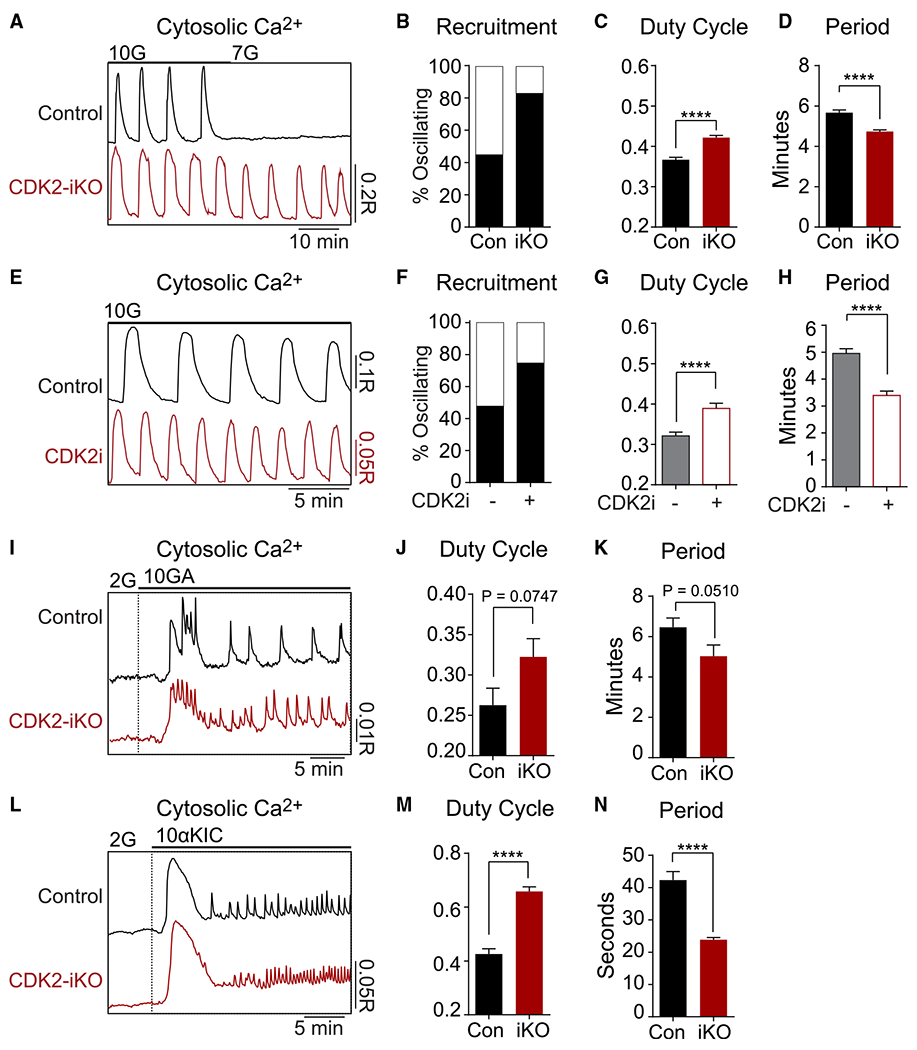
(A) Representative recordings of cytosolic calcium oscillations in islets isolated from 4 Con and 4 CDK2-iKO mice stimulated by 10G or 7 mM glucose (7G).
(B) Percentage of islets that continued oscillating at 7G (Con, n = 89; CDK2-iKO, n = 95).
(C and D) CDK2-iKO increased the duty cycle (C) and reduced the period (D) of glucose-stimulated calcium oscillations (Con, n = 168 islets; CDK2-iKO, n = 182 islets).
(E) Representative recordings of cytosolic calcium oscillations in vehicle- or CDK2i-treated islets isolated from 2 wild-type (WT)/B6J mice stimulated by 10G.
(F) Percentage of islets that oscillated at 6 mM glucose (vehicle Con, n = 30; CDK2i, n = 32).
(G and H) CDK2i increased the duty cycle (G) and reduced the period (H) of glucose-stimulated calcium oscillations (vehicle Con, n = 40; CDK2i, n = 44).
(I) Representative recordings of cytosolic calcium response to 10 mM glyceraldehyde (10GA) at 2 mM glucose (2G) in islets isolated from 3 Con and 3 CDK2-iKO mice.
(J and K) CDK2-iKO increased the duty cycle (J) and reduced the period (K) of glyceraldehyde-stimulated calcium oscillations (Con, n = 34; CDK2-iKO, n = 29).
(L) Representative recordings of cytosolic calcium response to 10 mM α-ketoisocaproate (10aKIC) at 2G in islets isolated from 2 Con and 2 CDK2-iKO mice.
(M and N) CDK2-iKO increased the duty cycle (M) and reduced the period (N) of keto-isocaproate-stimulated calcium oscillations (Con, n = 29; CDK2-iKO, n = 24).
Data are shown as mean ± SEM. #p < 0.1, ****p < 0.0001 by t test.
Independent of glycolysis, amino acid-stimulated phosphoenolpyruvate (PEP) cycling is sufficient to enhance recruitment in CDK2-defcient β cells
Recently, pyruvate kinase (PK)-dependent PEP cycling was demonstrated to generate the ATP/ADP that locally closes KATP channels (Lewandowski et al., 2020). Importantly, there are two sources of PEP in β cells: glycolytic enolase and mitochondrial PCK2 (Figure 5A). At low glucose, when glycolytic enolase is inactive, mitochondrial PEP production by PCK2 is required for amino acid-stimulated, PK-dependent KATP closure and calcium influx (Abulizi et al., 2020). Using this approach, calcium influx in response to amino acids was found to be increased in CDK2-iKO islets (Figures 5B and 5C). This effect persisted in the presence of a pharmacologic PK activator (Figures 5B and 5C) that maximally activates the PEP cycle (Abulizi et al., 2020; Lewandowski et al., 2020). These findings, matching the effects of the leucine analog αKIC (Figures 4L–4N), indicate that amino acid-stimulated PEP cycling contributes to enhanced β cell recruitment in the CDK2-iKO model independent of glucose metabolism.
Figure 5. Amino acid-stimulated PEP cycling is sufficient to enhance islet recruitment in CDK2-iKO islets.
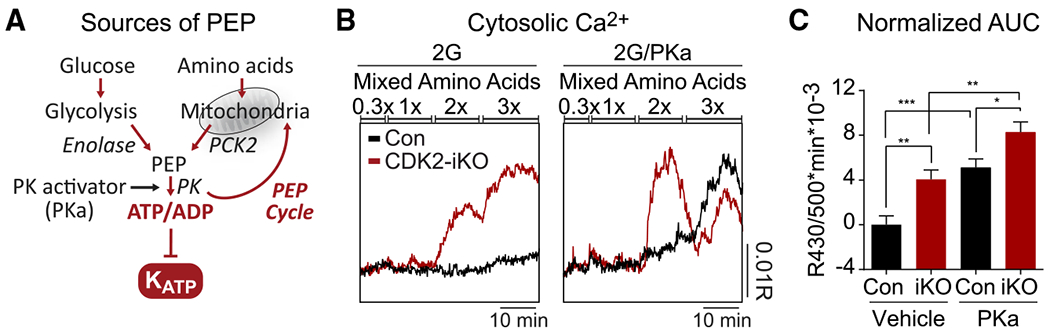
(A) PEP can be generated from glycolytic enolase or from mitochondrial oxaloacetate conversion via PCK2 and export to the cytosol. Addition of amino acids fuels the TCA cycle and activates the PEP cycle, and pyruvate kinase activator (PKa) also activates the PEP cycle.
(B) Representative recordings of cytosolic calcium elevation in response to an amino acid ramp at 2G and in the absence (left) or presence (right) of PKa in islets isolated from 3 Con and 3 CDK2-iKO mice. At 1×, physiological amino acids (PAA) is (in μM) 2,100 alanine, 600 glutamine, 700 glycine, 550 valine, 500 leucine, 350 serine, 200 arginine, 218 lysine, and 121 threonine.
(C) Quantification of (B) by AUC at 2× mixed amino acids, normalized to Con/vehicle. (Con/vehicle, n = 44; CDK2-iKO/vehicle, n = 48; Con/PKa, n = 38; CDK2-iKO/PKa, n = 44).
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA.
CDK2 depletion enhances oxidative glucose metabolism
Any mechanism that increases β cell workload (i.e., ATP consumption), including the increased calcium influx and insulin secretion observed in CDK2-iKO islets, is expected to increase oxidative glucose metabolism (Lewandowski et al., 2020), which we measured in islet β cells from CDK2-iKO mice and controls using three distinct assays.
NADH utilization is increased in CDK2-deficient islets
To investigate mitochondrial metabolism in the CDK2-iKO model, we performed NAD(P)H fluorescence lifetime imaging (FLIM) using the assay we developed to estimate NADH utilization by the electron transport chain in intact islets (Gregg et al., 2016, 2019). NAD(P)H lifetime is strongly dependent on the microenvironment of each coenzyme as it binds to protein, yielding a spatial display of islet redox metabolism (Figure 6A). With this technique, relative fractional compositions of bound: free NAD(P)H can be measured using the phasor approach (Digman et al., 2008) as the peak position along the 1-g axis (Gregg et al., 2016). In the presence of low and high glucose, the phasor peak was lower along the 1-g axis in CDK2-iKO islets relative to controls (Figure 6C), reflecting increased NADH consumption, measured as a decrease in the fraction of bound:free NAD(P)H.
Figure 6. Enhanced metabolism in CDK2-iKO islets.
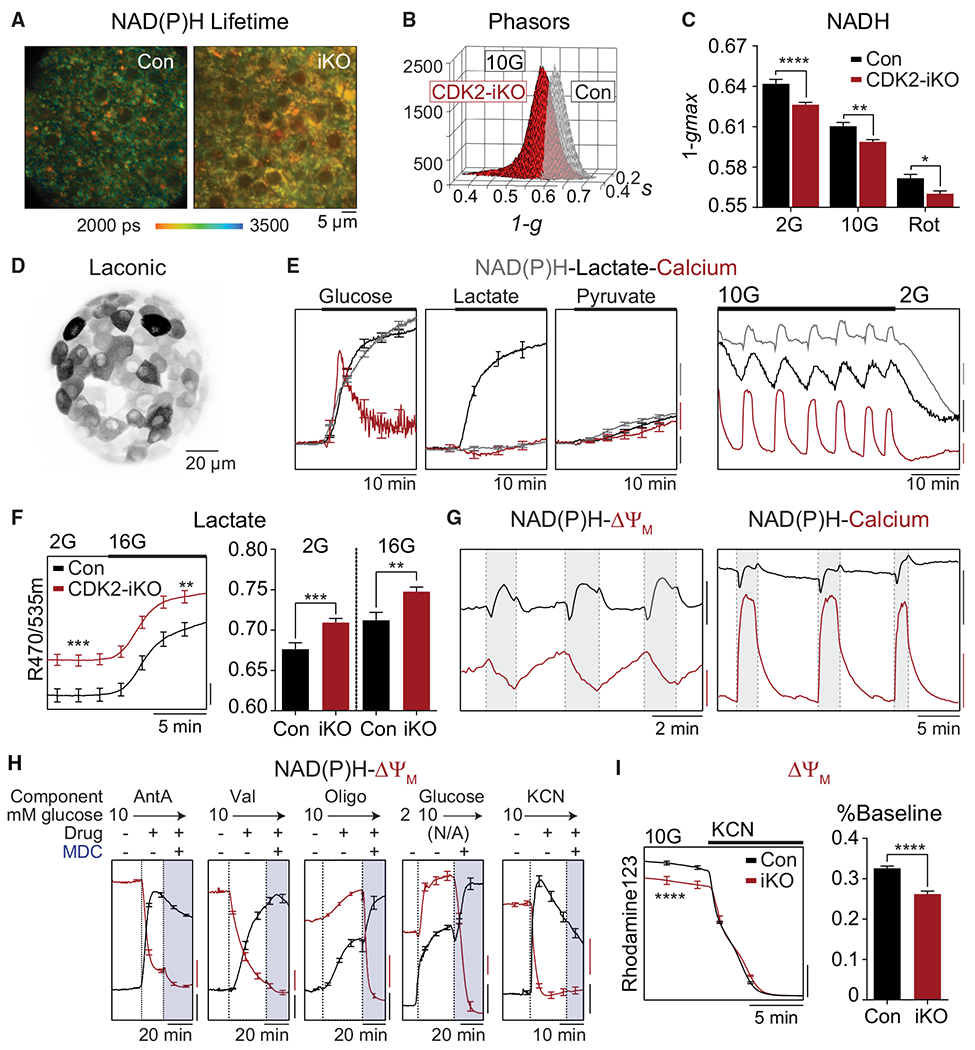
(A) Representative NAD(P)H fluorescence lifetime images from Con and CDK2-iKO islets imaged in 10G. Scale bar, 5 μM.
(B) Phasor histograms showing the frequency distribution of NAD(P)H lifetimes (1-g, s) in islets isolated from Con (n = 4) and CDK2-iKO (n = 4) mice imaged in 10G.
(C) Projection of the phasor histogram peak along the 1-g axis was used to quantify NAD(P)H utilization in the presence of 2G, 10G, and complex I inhibitor (5 μM rotenone [Rot], 15 min) as indicated (n = 10 islets per mouse for each condition).
(D) Maximum intensity projection of intact islet expressing Ad-RIP-Laconic in β cells imaged using a 2-photon Nikon TE-300 inverted confocal microscope.
(E) Left: response of each metabolite to 10G (n = 10 islets), 20 mM lactate (n = 12 islets), or 20 mM pyruvate (n = 10 islets) in islets isolated from 1 WT/B6J mouse, normalized to baseline signal at 2G. Scale bars, 2% baseline. Right: representative traces demonstrating the phase relationship between oscillations in NAD(P)H (endogenous, blue trace; scale bar, 200 IU), cytosolic lactate (Ad-RIP-Laconic, black trace; scale bar, 0.02 R470/535 m), and cytosolic calcium (FuraRed, red trace; scale bar, 0.05R 430/500×) at 10G and 2G.
(F) Increased lactate levels in CDK2-iKO islets (n = 59 islets from 3 mice) relative to Con (n = 72 islets from 4 mice) at 2G and 10G. Scale bar, 0.01 R470/535 m.
(G) Phase relationships between oscillations in NAD(P)H and ΔΨm (left) and cytosolic calcium (right). NAD(P)H: black scale bar, 100 IU (left) or 500 IU (right). ΔΨm: red scale bar, 50 IU. FuraRed (Ca2+): red scale bar, 0.2 R430/500×.
(H) Response of ΔΨm (red traces) and NAD(P)H (black traces) to the ETC inhibitors antimycin A(AntA; 1 μM, n = 10 islets), valinomycin (Val; 1 μM, n = 11 islets), and oligomycin (Oligo; 2 μM, n = 12 islets); to 10G (n = 11 islets); and to 5 mM cyanide (n = 11 islets) in islets isolated from 1 WT/B6J mouse. ΔΨm is normalized to fluorescence after depolarization with MDC. NAD(P)H is normalized to fluorescence at the beginning of the recording. Scale bars, 0.1 normalized IU.
(I) ΔΨm is depolarized in CDK2-iKO islets (n = 70 islets from 3 mice) relative to Con islets (n = 85 islets from 4 mice), normalized to fluorescence after depolarization with cyanide, quantified as percentage of baseline. Scale bar, 0.2 normalized IU.
Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way AVOVA (C) or t test (F and I).
Lactate production is increased in CDK2-deficient β cells
Lactate production is understood to be counterproductive in β cells, where low levels of lactate dehydrogenase prevent glucose utilization from accelerating under anaerobic conditions (i.e., the Pasteur effect) to facilitate tight coupling of cytosolic and mitochondrial fluxes (MacDonald, 1981; Schuit et al., 1997; Zhao et al., 2001). Because there is no mechanism of lactate consumption aside from pyruvate regeneration by lactate dehydrogenase, lactate levels are dependent on the glycolytic rate of NADH production (by GAPDH) and, thus, appropriate for testing whether β cell CDK2 depletion affects glycolytic flux, keeping in mind that mitochondrial substrate utilization may accelerate glycolysis (Civelek et al., 1996)(McKenna et al., 2016). To assess the cytosolic lactate levels specifically in islet β cells, we used an adenovirus harboring the insulin promoter to express the fluorescence resonance energy transfer (FRET)-based lactate sensor Laconic (San Martín et al., 2013; Figure 6D). In response to 10 mM glucose, steady-state oscillations in lactate were in phase with cytosolic Ca2+, measured simultaneously, and glucose reduction was accompanied by a reduction in lactate (Figure 6E). Lactate production is therefore highest after membrane depolarization and calcium influx, when NADH and oxygen consumption are also maximal (Jung et al., 2000; Lewandowski et al., 2020). Application of high glucose or lactate to the bath solution increased cytosolic lactate, although only glucose was sufficient to trigger a rise in NAD(P)H, membrane depolarization, and calcium influx (Figure 6E). In comparison, application of pyruvate induced only a modest increase in cytosolic lactate, NAD(P)H, and calcium (Figure 6E), consistent with low levels of lactate dehydrogenase and monocarboxylate transporters in β cells, in addition to an unfavorable NADH/NAD+ ratio for pyruvate conversion to lactate. Based on these control data, the enhancement of cytosolic lactate levels in CDK2-iKO islet β cells relative to MIP-CreERT controls (Figure 6F) is consistent with an increased glycolytic rate in CDK2-iKO β cells at low and high glucose, matching the NAD(P)H-FLIM assay. Given that glucose and glyceraldehyde (which enters glycolysis downstream of glucokinase) have similar effects on calcium in CDK2-iKO islets (Figure 4), the high lactate flux in these β cells more likely reflects the workload-dependent pull of glycolytic substrate into mitochondria rather than an increased push of excess fuel through glucokinase.
Reduced mitochondrial membrane potential (ΔΨm) in CDK2-deficient islets
As a third direct measurement of oxidative metabolism in β cells, we measured ΔΨm using Rhodamine-123 in non-quenching mode, which utilizes low (nanomolar) dye concentrations so that hyperpolarized (more negative) mitochondria will have higher dye concentrations and fluorescence (Perry et al., 2011). Although TMRM has been used to calculate absolute ΔΨm in dispersed β cells, this dye was avoided because of its lower dynamic range (ΔΨm oscillations were poorly resolved), and because of the fast redistribution of TMRM across membranes is confounded by plasma membrane depolarization (which must be monitored independently) (Gerencser, 2015; Gerencser et al., 2016). Although Rhodamine-123 must be used cautiously in quench mode because changes in its quench limit in response to ATP synthase inhibition with oligomycin (Gerencser et al., 2016), we validated non-quench mode Rhodamine-123 in intact islets using the same cocktail of inhibitors.
Phase-locked oscillations in ΔΨm and NAD(P)H were observed in the presence of 10 mM glucose (Figure 6G). Using parallel measurements of NAD(P)H and calcium to relate ΔΨm to plasma membrane depolarization, we observed that ΔΨm builds up (hyperpolarizes) prior to plasma membrane depolarization and is consumed (depolarized) shortly after plasma membrane depolarization (Figure 6G), consistent with maximal oxygen consumption during periods of high calcium (Jung et al., 2000). As a further validation of the ΔΨm measurements, we then evaluated each component of a mitochondrial depolarization cocktail (MDC) containing 2 μM oligomycin (a complex V inhibitor), 1 μM antimycin A (a complex III inhibitor), and 1 μM valinomycin (a potassium ionophore) (Gerencser et al., 2016). We also observed the expected hyperpolarization of ΔΨm in response to high glucose and depolarization in response to KCN to a similar extent as to the MDC (Figure 6H). Because KCN induced an equivalent depolarization as the MDC, it was used to normalize the Rhodamine-123 signal measured in control and CDK2-iKO islets. In the presence of stimulatory glucose, CDK2-iKO islets were significantly more depolarized than control islets (Figure 6I), consistent with a more active electron transport chain. The NAD(P)H FLIM, cytosolic lactate, and ΔΨm measurements identify a multi-site increase in oxidative glucose metabolism in CDK2-deficient β cells as necessary to support the observed increases in glucose-stimulated calcium influx and insulin secretion.
DISCUSSION
These data provide genetic and pharmacologic evidence for regulation of metabolism, ion channels, and insulin secretion by CDK2 in adult β cells. Overall, we find that CDK2 suppresses oxidative glucose metabolism and insulin secretion and plays an essential role in supporting the metabolic signals associated with fuel surfeit known as the metabolic amplifying pathways. It is unlikely that these mechanisms are secondary to β cell decline because our studies were conducted in the context of enhanced insulin secretion and improved glucose tolerance in the CDK2-iKO model. We further demonstrate that CDK2 blockade increases β cell recruitment and identify two interdependent mechanisms of this regulation, restricted KATP conductance and enhanced mitochondrial PEP cycling, revealing that CDK2 signaling influences β cell sensing of glucose and amino acids.
The genetic link between CDK2, KATP channels, and secretion is supported by prior studies in E2f1−/− mice, where Kir6.2 was identified as a transcriptional target of the CDK-RB-E2F signaling pathway (Annicotte et al., 2009), as well as in Cdk2fl/fl:Pdx1-Cre mice (Kim et al., 2017). We also observed a reduction in Kir6.2 mRNA levels in CDK2-iKO islets and islets treated overnight with a CDK2i, in parallel with a reduction in KATP conductance and increased calcium influx. However, in contrast with developmental knockout, deleting CDK2 from mature β cells improved glucose tolerance because of increased insulin secretion. These data raise the possibility that embryonic deletion of CDK2 induces a time-dependent or developmental defect in the islet, whereas CDK2 signaling in adult β cells suppresses insulin secretion. Hyperexcitability, which can be found in all CDK2 depletion models, including Cdk2fl/fl:Pdx1-Cre (unpublished data), may explain the ultimate decline in insulin secretion in these mice as well as in E2f1−/− mice.
In our models of short-term CDK2 restriction, we observe that the consequences of reducing KATP channel conductance include enhanced glucose-stimulated β cell recruitment and, thus, an elevated calcium duty cycle. It is possible that other CDK2 targets not identified in this study contribute to β cell recruitment. Indeed, if reduced GKATP was the sole effector of CDK2 depletion, then the calcium oscillation period would be expected to increase in a fashion similar to glucose elevation. Instead, CDK2 depletion strongly increased the cycling frequency. A recently discovered mechanism of β cell recruitment that increases cycling frequency is PEP and ADP conversion to pyruvate and ATP by PK, which can raise mean calcium provided a net synthesis of PEP is generated via mitochondrial anaplerosis (Lewandowski et al., 2020). Mitochondria-derived PEP produced by PCK2 is required for full insulin secretion in INS1 cells and mice (Abulizi et al., 2020; Stark et al., 2009). In CDK2-iKO islets, enhanced β cell recruitment persisted when β cells were provided with glucose or glyceraldehyde to generate glycolytic PEP or, orthogonally, anaplerotic fuels (here, αKIC or amino acids) that increase mitochondrial PEP production and cytosolic PK-driven KATP closure at low glucose (Abulizi et al., 2020; Lewandowski et al., 2020). When PEP cycling was driven maximally by pharmacological PK activation and amino acids, enhanced PEP cycling in CDK2-deficient cells synergized with the reduction in GKATP, driving a further increase in fuel sensitivity. In vivo, amino acids would be expected to augment β cell recruitment by glucose and contribute to the enhanced glucose tolerance observed in CDK2-iKO mice.
Multi-site measurements of lactate, NAD(P)H, and ΔΨm in living β cells indicate that CDK2 depletion augments oxidative glucose metabolism. Increased glucokinase flux (a “push” mechanism) is unlikely to explain these effects because downstream glycolytic and mitochondrial fuels (glyceraldehyde and αKIC) had effects on calcium oscillations comparable with glucose. A more compelling alternative (Figure 7) is that ADP-dependent oxidative phosphorylation is driven up in CDK2-deficienct β cells by the increased workload (ATP turnover) associated with elevated calcium and secretion, consuming ΔΨm. In this case, mitochondrial NADH and cytosolic lactate depletion are dependent on increased mitochondrial substrate utilization, as needed to maintain ΔΨm (Civelek et al., 1996; McKenna et al., 2016). To validate this interpretation of our oxidative metabolic assays, we used glucose-stimulated calcium oscillations as a tool to understand the relationships between oxygen consumption, ΔΨm, and lactate. Our observations fit prior studies demonstrating that oxidative phosphorylation is controlled by ADP availability in β cells (Civelek et al., 1997; Jung et al., 2000; Lewandowski et al., 2020), produced by an increase in ATP turnover in the cell during periods of high calcium (Ainscow and Rutter, 2002; Doliba et al., 2003; Panten et al., 1986; Sweet et al., 2004; Affourtit et al., 2018). Here we demonstrated consumption of ΔΨm during periods of high calcium during glucose-stimulated oscillations and further measured a depolarized ΔΨm in CDK2-iKO islets, corroborating previous studies connecting ΔΨm to CDK2 signaling in proliferating cells (Mitra et al., 2009; Schieke et al., 2008).
Figure 7. Model of CDK2 regulation of β cell metabolism.
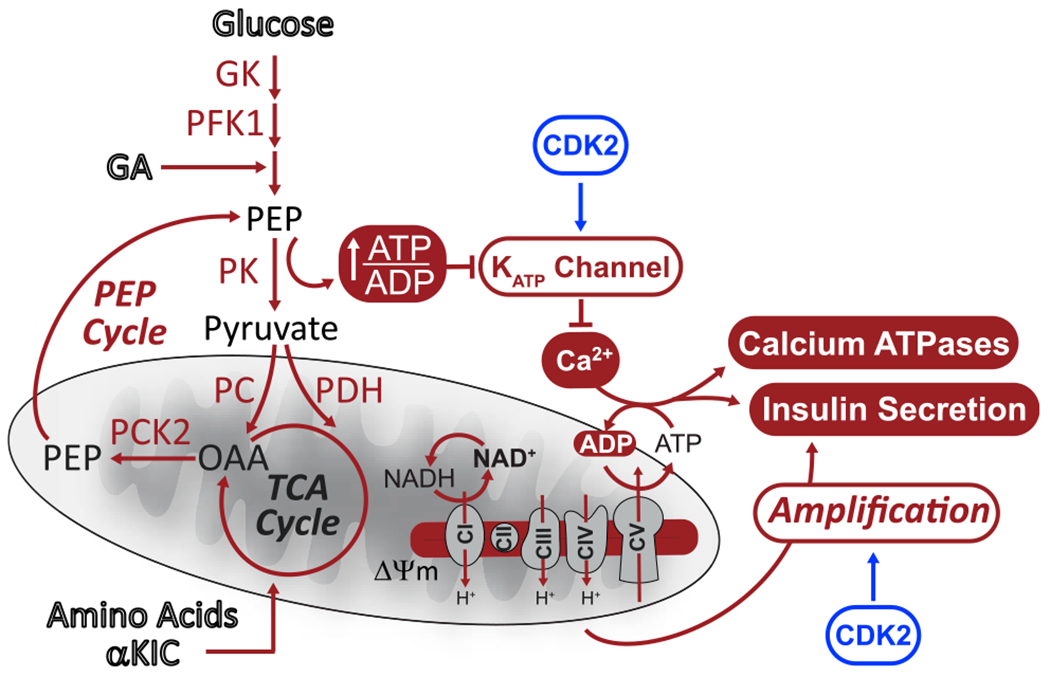
CDK2 regulates β cell excitability, oxidative metabolism, and insulin secretion. CDK2 activates KATP channel conductance independent of the PEP source. KATP channel activation limits β cell excitability and calcium influx, which alleviates ATP consumption by exocytosis and calcium removal through calcium ATPases, limiting oxidative metabolism. Additionally, CDK2 promotes metabolic amplification of insulin secretion.
The ability of β cell mitochondria to function as a demand-driven system may also explain why glucose fails to fully amplify secretion when CDK2 is depleted. Although other cell cycle regulators have been shown to act directly on exocytotic machinery (Bembenek et al., 2007; Xin et al., 2004), such a generalizable defect cannot explain our findings because CDK2 deletion had no effect KCl-stimulated insulin secretion at low glucose. Rather, the disconnect between triggering and amplification in the CDK2-iKO model highlights the fundamental difference between oxidative metabolism driven by calcium influx (a metabolic “pull”) and oxidative metabolism stimulated by excess fuel (a metabolic “push”) that supports amplification. Fuel surfeit, which would not be supported by the low ΔΨm and NADH found in CDK2-deficient β cells, is necessary to generate metabolic coupling factors (e.g., reactive oxygen species) that potentiate secretion (Leloup et al., 2009; Prentki et al., 2013). Loss of KATP channel conductance in CDK2-iKO β cells may induce a calcium-driven workload (ATP turnover) that consumes metabolic substrates and reduces the metabolic capacitance that benefits amplification (Figure 7).
In addition to CDK2, CDK1 (Gregg et al., 2019), CDK5 (Wei et al., 2005), and CDK8 (Xue et al., 2019) have been found to regulate glucose-stimulated insulin secretion. Additionally, the CDK regulator p16/INK4a (Helman et al., 2016; Zheng et al., 2013) and the effector molecule c-MYC (Puri et al., 2018; Rosselot et al., 2019) have demonstrated significant control over insulin secretion in addition to established canonical cell cycle control of proliferation. Our results provide further evidence that CDK2 can modify the metabolic and secretory state independent of maturation defects reflected by MAFA, GLUT2, or UCN3 (Blum et al., 2012; Hang et al., 2014; van der Meulen et al., 2015).
Limitations of study
It is likely that the phenotypes observed here are the result of complex, CDK2-dependent transcriptional regulation and post-translational modification that were not identified in this study. Although CDK2 may act through the RB-E2F pathway to transcriptionally regulate Kir6.2 mRNA levels, further experiments are necessary to confirm this mechanism in adult β cells, where CDK2 was found primarily in the cytosol. Although the CDK2 ablation results in gain of function, it remains possible that, in the CDK2-iKO model, there is enhanced, compensatory activity of other cell cycle regulators (Fiaschi-Taesch et al., 2013). Our findings highlight the importance of CDK2 regulation of metabolism, ion channel function, and insulin secretion in mature β cells.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthew Merrins (mmerrins@medicine.wisc.edu).
Materials availability
This study did not generate new reagents.
Data and code availability
This study did not generate new code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experiments performed with laboratory animals were performed according to the guidelines of the Institutional Animal Care and Use Committees of the University of Wisconsin-Madison and the William S. Middleton Memorial Veterans Hospital, and followed the NIH Guide for the Care and Use of Laboratory Animals (8th ed. The National Academies Press. 2011.). Cdk2fl/fl mice (Kim et al., 2017; Jayapal et al., 2015) were crossed with MIP-CreERT mice (Jax 024709) (Tamarina et al., 2014) to generate a β-cell specific Cdk2 deletion mouse (CDK2-iKO). To properly control for the ectopic expression of human growth hormone (hGH) and CreERT in β-cells (Brouwers et al., 2014; Oropeza et al., 2015), and the independent effects of tamoxifen (Ahn et al., 2019; Carboneau et al., 2016), all mice studied were CreERT-positive and tamoxifen-injected. While Ins1-CreER mice (Jax 026802) are also β-cell specific and lack hGH (Thorens et al., 2015), they failed to catalyze IoxP excision and we continued with the MIP-CreERT strain. Male mice were housed 1-4 per cage at 21-23°C, maintained on a 12 hour light/dark cycle (light on 6:00AM to 6:00PM), with chow diet and water provided ad libitum. 10 week old mice received intraperitoneal injections of tamoxifen (4 mg in 200 μL sunflower seed oil) three times over five days to induce Cre-mediated recombination. Mice were analyzed four weeks after the final injection to allow tamoxifen clearance. Cre-mediated deletion of Cdk2 exons 4-5 was confirmed by qPCR (primers are listed below).
METHOD DETAILS
Glucose Tolerance Test
Glucose tolerance was measured after an overnight fast by intraperitoneal injection of 2 g/kg glucose. Blood glucose was measured using a Bayer Contour glucometer at 0, 15, 30, 45, 60, and 120 minutes post-injection.
Pancreas Fixation
Dissected pancreata from 12 week old mice were fixed in 4% PFA diluted in PBS for 3 hours at 4C. Fixed pancreata were then washed in PBS for 1 hour at room temperature. Following the wash, pancreata were equilibrated in 30% (w/v) sucrose in PBS for 1 hour, followed by embedding in OCT with subsequent freezing on dry ice.
Immunostaining
Slides with 10 μm thick pancreas sections were blocked in 10% Normal Donkey Serum (Jackson) diluted in 0.2% PBST (PBS, 0.2% Triton X-100) for one hour at room temperature. Following block, slides where incubated with primary antibodies diluted in 10% Normal Donkey Serum overnight at 4C. Slides were then washed 3 × 10 mins with 0.2% PBST and then incubated with secondary antibodies diluted in 10% Normal Donkey Serum for one hour at room temperature. Slides were then washed 3 × 10 minutes with 0.2% PBST. Primary antibodies used were Guinea pig anti-Insulin (1:6 Agilent), Rabbit anti-glucagon (1:200 CST), Rabbit anti-MafA (1:100, CST), Rabbit anti-Ucn3 (1:500, Phoenix), Rabbit anti-Glut2 (1:400, Sigma), and Rabbit anti-Ki67 (1:400; CST). Secondary antibodies used were Donkey anti Rabbit-488 (Jackson), and Donkey anti-Guinea pig 647 (Jackson).
α-Cell and β-cell Mass Measurement
For each mouse (n = 4-5 per genotype), eight 10 μm thick whole pancreas sections, each 150 μm apart, were stained with Guinea pig anti-insulin (Agilent), Rabbit anti-glucagon (CST), and DAPI. Whole sections were then imaged on a Nikon A1RS confocal microscope at 10x magnification. The area of insulin, glucagon, and DAPI staining was measured in μm2 using FIJI’s auto thresholding and Anal-zeParticles functions. β-Cell and α-cell mass were determined for each animal by dividing the sum of insulin or glucagon area across all sections imaged by the sum of the area of DAPI across those same sections.
Cell Proliferation Analysis
For each mouse (n = 4-5 per genotype) four to five 10 μm thick whole pancreas sections, each 150 μm apart, were stained with Guinea pig anti-insulin (Agilent), Rabbit anti-ki67 (CST), and DAPI. 18-26 islets with at least 3 coming from each section were imaged using a Nikon A1RS confocal microscope at 20x magnification. Ki-67 +/Insulin+ cells in all islets from each mouse were counted and divided by the sum of insulin area across the same islets to determine the relative beta cell proliferation in each mouse.
Islet isolation
Mouse pancreas was inflated with 3-5 mL of 0.67mg/mL collagenase (Sigma) and 0.2 mg/mL BSA in Hanks Buffered Salt Solution (HBSS) (Invitrogen). After excision, pancreas was incubated in a glass vial of 5mL collagenase/BSA/HBSS for 5 minutes on an orbital shaking water bath at 250 rpm. At the 6th minute of incubation, pancreas was agitated for 20 s at 350 rpm every 2 minutes until 24 minutes of incubation. Pancreatic digests were washed three times with 30 mL ice cold BSA/HBSS solution by pelleting at 50 g for 2 minutes, aspirating 20-25mL supernatant, and resuspending in 5-10 mL of remaining BSA/HBSS solution by vortexing at medium speed. Islets were hand-picked from acinar tissue in 40 mL ice cold BSA/HBSS solution and finally picked into RPMI 1640 (Sigma Aldrich) supplemented with 10% FBS (v/v) (Thermo Fisher), 100 units/mL penicillin, and 100 ug/mL streptomycin (Fisher Scientific). For experiments requiring adenovirus, islets were infected immediately post-isolation for 2 hours at 37°C, then moved to fresh medium overnight. Generation of Ad-RIP-Laconic was described previously (Lewandowski et al., 2020) and Ad-Cre-IRES-GFP is commercially available (Vector Biolabs 1710). For experiments requiring CDK2i, islets were incubated in media containing 5 μM SU9516 (Enzo) or 0.1% DMSO vehicle overnight.
Quantitative PCR
Following a 16 hour recovery post-isolation, islets were collected and washed with PBS. RNA was isolated using the QIAGEN RNeasy RNA extraction kit. 100 ug of RNA was reverse transcribed into cDNA using a cDNA reverse transcriptase kit (Applied Biosystems). Quantitative PCR was performed in triplicate using the following primers: Cdk2: F (5′- AATTCTTCTGGGCTGCAAGTA-3′), R (5′-GGGTACACACTAGGTGCATTT-3′); Kcnj11: F (5′- GTGTCCAAGAAAGGCAACTG –3′), R (5′- GCACAGGAAGGACATGGTG –3′); normalized to a reference gene β-Actin: F (5′-GAGACCTTCAACACCCC-3′), R (5′GTGGTGGTGAAGCTGTAGCC-3′). SYBR Green PCR Master Mix (Rox) (Roche) was used with 1 μL of cDNA per reaction with a final primer concentration of 6 pM. Reactions were run on a StepOne Plus Real-Time PCR system (Applied Biosystems). PCR product specificity was determined by melt-curve analysis and gel electrophoresis. Data was reported as ΔΔCt.
Glucose-Stimulated Insulin Secretion
Glucose-stimulated insulin secretion was measured in DMEM (Sigma D-5030) supplemented with 4 mM L-glutamine, 44 mM sodium bicarbonate, 10 mM HEPES and 0.2% BSA at 37°C in 5% CO2. 60 islets per mouse were preincubated for 45 minutes in 2 mL of DMEM containing 3.3 mM glucose. Six groups of 10 islets were then transferred to 12-well plates (CellTreat 229112) containing 1 mL of DMEM/3.3 mM glucose per well and were incubated for 45 minutes. Islets were then transferred to a new 12 well plate containing 1 mL of DMEM containing 16.7 mM glucose, and were incubated for 45 minutes. Islets were then collected for content measurements in 500 μL cell lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 1% Triton-X. Secretion media was also collected and all samples were frozen at −80°C prior to insulin measurement by ELISA.
Insulin ELISA
ELISA was used to measure insulin secretion as a percentage of total islet insulin content. 96-well high-binding plates (Corning 3690) were coated overnight with 3 μg/mL (50 μL/well) of anti-insulin primary antibody (Fitzgerald Industries International Research 10R-I136a) diluted 1:2500 in PBS. Plates were blocked for 1 hour with 100 μL/well PBS containing 4% BSA (Sigma A-7888). Plates were then emptied and 25 μL/well of insulin standards (Millipore 24304391/8013-k, 0.1-10ng/mL), secretion media, or islet lysate were added to the plate and incubated for 1 hour. 25 μL/well of secondary antibody (Fitzgerald Industries International Research 10R-I136bBt) diluted 1:1000 in PBS with 1% BSA was added to each well. Plates were gently mixed and incubated for 1 hour. Plates were then emptied and rinsed three times with 100 μL/well wash buffer containing 50 mM Tris and 0.2% Tween-20, pH 8.0. Wells were incubated with 50 μL/well of 1 μg/mL of streptratavidin-HRP (Pierce 21126) in PBS with 0.1% BSA for 30 minutes. Plates were rinsed three times with wash buffer before addition of 50 μL/well of 16 μM/mL of o-phenylenediamine (Sigma P-5412) dissolved in 0.1 M citrate-phosphate, 0.03% H2O2 at pH 5.0. The plate was allowed to develop for 3-5 minutes before quenching the reaction with 50 μL/well of 18 mM sulfuric acid. Absorbance at 492 nm was measured by plate reader (TECAN Infinite M1000 Pro). Insulin content was calculated by comparison to known standards. All products unlisted are from Sigma.
Timelapse Imaging
Biosensors were introduced for the following metabolites as follows: cytosolic calcium, 45-minute pre-incubation in 2.5 μM FuraRed (Molecular Probes); mitochondrial membrane potential, 5-minute pre-incubation in 0.83 μM Rhodamine-123 (Sigma); lactate, 2 hour infection with 1 μL high-titer adenovirus immediately following isolation, then 2 days incubation in fresh media. Islets were then placed in a glass-bottom chamber (Warner Instruments) on a Nikon Eclipse Ti-E inverted microscope equipped with Super Flour 10x/0.5 NA objectives (Nikon Instruments). The chamber was perfused with standard external solution containing (in mM): 135 NaCl, 4.8 KCl, 2.5 CaCl2, 1.2 MgCl2, 20 HEPES (pH 7.35) with metabolic fuels as indicated in the figures and figure legends. The flow rate was 0.3 mL/min, and temperature was maintained at 33°C using inline solution and chamber heaters (Warner Instruments). Excitation was provided by a SOLA SEII 365 (Lumencor) set to < 20% output. Fluorescence emission was collected with a Hamamatsu ORCA-Flash4.0 V2 Digital CMOS camera at 0.125-0.2 Hz. A single region of interest was used to quantify the average response of each islet using Nikon NIS-Elements. For cytosolic calcium recordings, excitation (430/20x and 500/20x) and emission (630/70m) filters (ET type, Chroma Technology Corporation) were used in combination with an FF444/521/608-Di01 dichroic (Semrock) and reported as an excitation ratio (R430/500x). The same dichroic mirror was used for laconic sensor FRET imaging, with CFP excitation provided by a 430/24x filter and emission filters 470/24m and 535/30m reported as an emission ratio (R470/535m). For mitochondrial membrane potential recordings using Rhod123, the same dichroic mirror was used, with excitation provided by a 500/20x filter and a 535/30m filter was used for emission light. A custom MATLAB script calculated cytosolic calcium R430/500x oscillation parameters including the average duty cycle of each islet. Lactate measurements were normalized to baseline values in low glucose. Mitochondrial membrane potential values were normalized to baseline following depolarization.
NAD(P)H Fluorescence Lifetime Imaging
Islets were imaged in #1.5 glass-bottom dishes on a custom-built multiphoton laser scanning system based around a Nikon TE-300 inverted microscope equipped with a PlanApo 60X/1.4NA oil immersion objective (Nikon Instruments) in a standard external solution (above). Temperature was maintained at 35°C using a LiveCell incubator (Pathology Devices). NAD(P)H was excited with Mai Tai DeepSee Ti:Sapphire laser (Spectra-Physics) at 740 nm with a 450/70 m bandpass emission filter (Chroma) before collection by a Hamamatsu H7422P-40 GaAsP photomultiplier tube. FLIM images were collected at 256×256 resolution with 120 s collection at 1 Hz using SPC-830 Time-Correlated Single Photon Counting (TCSPC) electronics (Becker & Hickl GbmH). In each experiment, urea crystals were used to define the instrument response function with a 370/10m bandpass emission filter (Chroma), and coumarin (Sigma) was used as a reference for lifetime (2.5 ns) using a 450/70m bandpass emission filter (Chroma). For analysis, raw SDT files were imported into MATLAB (MathWorks), and a custom script was used to generate phasor histograms for each treatment using the equations in Digman et al. (2008). All data were reported as the phasor histogram peak (1-gmax). Glucose reduces 1-gmax reflecting mitochondrial NADH consumption, as measured by an increase in NAD(P)H production and larger pool of free NAD(P)H. Addition of the positive control, rotenone, blocks NADH consumption by complex I of the electron transport chain, reducing the fraction of bound:free NAD(P)H and thus further reducing 1-gmax.
KATP conductance
β-cell membrane potential and KATP conductance were measured as in Ren et al. (2013). Briefly, a Sutter MP-225 micromanipulator was used together with a HEKA EPC10 patch-clamp amplifier in the perforated patch-clamp configuration to record membrane potential from intact islets perfused with standard external solution (above). Pipette tips were filled with an internal solution (in mM: 28.4 K2SO4, 63.7 KCl, 11.8 NaCl, 1 MgCl2, 20.8 HEPES, 0.5 EGTA; 40 sucrose; pH 7.2) containing 0.36 mg/ml amphotericin B. Islet β-cells were identified by the presence of slow oscillations in 10 mM glucose. The amplifier was then switched into voltage clamp mode. Conductance changes were determined from the current-voltage relation (I-V) using 2 s voltage ramps from −120 to −50 mV every 20 s during the silent phase of bursting and following application of 200 μm diazoxide (Sigma).
Exocytosis
β-cell Ca2+ current and exocytosis were measured as in Merrins and Stuenkel (2008) with minor changes. Briefly, a Sutter MP-225 micromanipulator was used together with a HEKA EPC10 patch-clamp amplifier (Heka Instruments, Bellmore, NY) in the whole cell patch-clamp configuration to record Ca2+ current from intact islets perfused with standard external solution (above). Pipette tips were filled with an internal solution (in mM: 125 Cs-glutamate, 10 CsCl, 10 NaCl, 1 MgCl2·6H2O, 0.05 EGTA, 5 HEPES, 0.1 cAMP, 3 MgATP; pH 7.15 with CsOH). After membrane rupture, cell size was used to distinguish islet β-cells (> 5.5 pF) from α-cells (< 5 pF), and after 1 min Ca2+ current was quantified from a 15 ms depolarization from −70 to 0 mV followed by a P/4 leak subtraction protocol. After 1 additional min, exocytosis was stimulated by activating VDCCs with a series of ten 500 ms membrane depolarizations from −70 to 0 mV. Capacitance responses (fF) and Ca2+ currents (pA) were normalized to initial cell size (pF).
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical details of experiments can be found in the figure legends. Data are expressed as means ± SEM. Statistical significance was determined using one- or two-way ANOVA with Sidak multiple-comparisons test post hoc or Student’s t test as appropriate. Differences were considered to be statistically significant at p < 0.05. Statistical calculations were performed with GraphPad Prism.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea Pig Polyclonal Anti-Insulin | Agilent | Cat#A056401-2; RRID: AB_2617169 |
| Rabbit Polyclonal Anti-Glucagon | Cell Signaling Technology | Cat#2760; RRID: AB_659831 |
| Rabbit Monoclonal Anti-MAFA | Cell Signaling Technology | Cat#79737; RRID: AB_2799938 |
| Rabbit Polyclonal Anti-Urocortin III | Phoenix Pharmaceuticals | Cat#H-019-29 |
| Rabbit Polyclonal Anti-GLUT-2 | Sigma-Aldrich | Cat#07-1402; RRID: AB_1587076 |
| Rabbit Monoclonal Anti-Ki-67 | Cell Signaling Technology | Cat#9129; RRID: AB_2687446 |
| Alexa Fluor 488 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat#711-545-152; RRID: AB_2313584 |
| Alexa Fluor 647 AffiniPure Donkey Anti-Guinea Pig IgG (H+L) | Jackson ImmunoResearch | Cat#706-605-148; RRID: AB_2340476 |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI-1640 cell culture media | Sigma-Aldrich | Cat#R8758 |
| Fetal bovine serum | Thermo Fisher | Cat#A31605 |
| Penicillin-Streptomycin (10,000U/mL) | Fisher Scientific | Cat#5140122 |
| SU9516 | Enzo Life Sciences | Cat#ALX-270-400; CAS: 377090-84-1 |
| SYBR Green PCR Master Mix | Thermo Fisher | Cat#4364346 |
| DMEM | Sigma-Aldrich | Cat#D5030 |
| Rat/Mouse Insulin Standard | Millipore | Cat#E8013-k |
| Streptavidin protein, HRP | Thermo Scientific | Cat#21126 |
| o-phenylenediamine | Millipore Sigma | Cat#P5412; CAS: 95-54-5 |
| FuraRed | Invitrogen | Cat#F3020; CAS: 179732-62-7 |
| Rhodamine-1,2,3 | Invitrogen | Cat#R302; CAS: 62669-70-9 |
| Amphotericin B | Sigma-Aldrich | Cat#A4888; CAS: 1397-89-3 |
| Diazoxide | Sigma-Aldrich | Cat#D9035; CAS: 364-98-7 |
| Accutase | Sigma-Aldrich | Cat#A6964 |
| Normal Donkey Serum | Jackson ImmunoResearch | Cat#017-000-121; RRID: AB_2337258 |
| TEPP-46 (PKa) | MilliporeSigma | Cat#50-548-70001; CAS: 1221186-53-3 |
| Antimycin A | Sigma-Aldrich | Cat#A8674; CAS: 1397-94-0 |
| Valinomycin | Sigma-Aldrich | Cat#V0627; CAS: 2001-95-8 |
| Oligomycin | Sigma-Aldrich | Cat#75351; CAS: 579-13-5 |
| Potassium Cyanide | Sigma-Aldrich | Cat#60178; CAS: 151-50-8 |
| Coumarin | Sigma-Aldrich | Cat#C4261; CAS: 91-64-5 |
| Rotenone | Sigma-Aldrich | Cat#R8875; CAS: 83-79-4 |
| Critical commercial assays | ||
| RNeasy RNA extraction kit | QIAGEN | Cat#74104 |
| High-capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| Experimental models: organisms/strains | ||
| B6.Cg-Tg(Ins1-cre/ERT)1Lphi/J mice | The Jackson Laboratory | JAX: 024709 |
| Cdk2fl/fl mice | Jayapal et al., 2015 | N/A |
| C57BL/6J mice | The Jackson Laboratory | JAX: 000664 |
| Oligonucleotides | ||
| Primer: Cdk2 Forward: AATTCTTCTGGGCTGCAAGTA | This paper | N/A |
| Primer: Cdk2 Reverse: GGGTACACACTAGGTGCATTT | This paper | N/A |
| Primer: Kcnj11 Forward: GTGTCCAAGAAAGGCAACTG | This paper | N/A |
| Primer: Kcnj11 Reverse: GCACAGGAAGGACATGGTG | This paper | N/A |
| Primer: β-Actin Forward: GAGACCTTCAACACCCC | Bhalla et al., 2014 | N/A |
| Primer: β-Actin Reverse: GTGGTGGTGAAGCTGTAGCC | Bhalla et al., 2014 | N/A |
| Recombinant DNA | ||
| β-cell specific Laconic Lactate biosensor | Lewandowski et al., 2020 | N/A |
| Ad-CMV-Cre-IRES-GFP | Vector Biolabs | Cat#1710 |
| Software and algorithms | ||
| NIS-Elements | Nikon Instruments | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| MATLAB Software | Mathworks | https://www.mathworks.com/products/matlab.html |
| FIJI | ImageJ | https://imagej.net/Fiji |
| Axon pClamp 10 software | Axon Instruments/Molecular Devices | https://support.moleculardevices.com/s/article/Axon-pCLAMP-10-Electrophysiology-Data-Acquisition-Analysis-Software-Download-Page |
| GraphPad Prism 7.0 | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| StepOne Plus Real-Time PCR System | Applied Biosystems | Cat#4376600 |
| Plate Reader Infinite M1000 Pro | TECAN | Cat#30063849 |
| 10X/0.5NA SuperFluor Objective | Nikon Instruments | Cat#MRF00100 |
| 60X/1.4NA Plan Apochromat Objective | Nikon Instruments | Cat#MRD01605 |
| SOLA SE-5-LCR-VA | Lumencor | N/A |
| FF444/521/608-Di01 dichroic beamsplitter | Semrock | Cat#FF444/521/608-Di01-25x36 |
| ORCA-Flash4.0 V2 | Hamamatsu | Cat#C11440-22CU |
| HEKA EPC10 patch-clamp amplifier | Heka | N/A |
| MaiTai HP DeepSee TI:Sapphire Laser | Spectra-Physics | N/A |
| Time-Correlated Single Photon Counting module | Becker & Hickl | Cat#SPC-830 |
| Photo Sensor Module | Hamamatsu | Cat#H7422P-40 |
Highlights.
Cyclin-dependent kinase 2 (CDK2) switches off the β cell secretory state
CDK2 augments KATP conductance and limits PEP cycle-dependent β cell recruitment
CDK2 attenuates oxidative glucose metabolism by reducing Ca2+-driven metabolic workload
CDK2 KO in mature β cells enhances insulin secretion and improves glucose tolerance
ACKNOWLEDGMENTS
We would like to thank Brian Schmidt and Kara Mortensen for technical assistance and the University of Wisconsin-Madison BRMS Breeding Core and Research Services. The Merrins laboratory gratefully acknowledges support from the American Diabetes Association (1-16-IBS-212), the NIH/NIDDK (K01DK101683 and R01DK113103), the NIH/NIA (R21AG050135 and R01AG062328), the Wisconsin Partnership Program, and the Central Society for Clinical and Translational Research. This work utilized facilities and resources from the William S. Middleton Memorial Veterans Hospital and does not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abulizi A, Cardone RL, Stark R, Lewandowski SL, Zhao X, Hillion J, Ma L, Sehgal R, Alves TC, Thomas C, et al. (2020). Multi-Tissue Acceleration of the Mitochondrial Phosphoenolpyruvate Cycle Improves Whole-Body Metabolic Health. Cell Metab. 32, 751–766. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackeifi C, Wang P, Karakose E, Manning Fox JE, González BJ, Liu H, Wilson J, Swartz E, Berrouet C, Li Y, et al. (2020). GLP-1 receptor agonists synergize with DYRK1A inhibitors to potentiate functional human β cell regeneration. Sci. Transl. Med 12, eaaw9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affourtit C, Alberts B, Barlow J, Carré JE, and Wynne AG (2018). Control of pancreatic β-cell bioenergetics. Biochem. Soc. Trans 46, 555–564. [DOI] [PubMed] [Google Scholar]
- Aguilar V, and Fajas L (2010). Cycling through metabolism. EMBO Mol. Med 2, 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S-H, Granger A, Rankin MM, Lam CJ, Cox AR, and Kushner JA (2019). Tamoxifen suppresses pancreatic β-cell proliferation in mice. PLoS ONE 14, e0214829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainscow EK, and Rutter GA (2002). Glucose-stimulated oscillations in free cytosolic ATP concentration imaged in single islet beta-cells: evidence for a Ca2+-dependent mechanism. Diabetes 51 (Suppl 1), S162–S170. [DOI] [PubMed] [Google Scholar]
- Annicotte J-S, Blanchet E, Chavey C, lankova I, Costes S, Assou S, Teyssier J, Dalle S, Sardet C, and Fajas L (2009). The CDK4-pRB-E2F1 pathway controls insulin secretion. Nat. Cell Biol 11, 1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bembenek JN, Richie CT, Squirrell JM, Campbell JM, Eliceiri KW, Poteryaev D, Spang A, Golden A, and White JG (2007). Cortical granule exocytosis in C. elegans is regulated by cell cycle components including separase. Development 134, 3837–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla K, Liu W-J, Thompson K, Anders L, Devarakonda S, Dewi R, Buckley S, Hwang B-J, Polster B, Dorsey SG, et al. (2014). Cyclin D1 represses gluconeogenesis via inhibition of the transcriptional coactivator PGC1α. Diabetes 63, 3266–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet E, Annicotte J-S, Lagarrigue S, Aguilar V, Clapé C, Chavey C, Fritz V, Casas F, Apparailly F, Auwerx J, and Fajas L (2011). E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol 13, 1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum B, Hrvatin S, Schutz C, Bonal C, Rezaniaand A, and Melton DA (2012). Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat. Biotechnol 30, 261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers B, de Faudeur G, Osipovich AB, Goyvaerts L, Lemaire K, Boesmans L, Cauwelier EJG, Granvik M, Pruniau VPEG, Van Lommel L, et al. (2014). Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab. 20, 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchakjian MR, and Kornbluth S (2010). The engine driving the ship: metabolic steering of cell proliferation and death. Nat. Rev. Mol. Cell Biol 11, 715–727. [DOI] [PubMed] [Google Scholar]
- Carboneau BA, Le TDV, Dunn JC, and Gannon M (2016). Unexpected effects of the MIP-CreER transgene and tamoxifen on β-cell growth in C57Bl6/J male mice. Physiol. Rep 4, e12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelek VN, Deeney JT, Fusonie GE, Corkey BE, and Tornheim K (1997). Oscillations in oxygen consumption by permeabilized clonal pancreatic beta-cells (HIT) incubated in an oscillatory glycolyzing muscle extract: roles of free Ca2+, substrates, and the ATP/ADP ratio. Diabetes 46, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelek VN, Deeney JT, Shalosky NJ, Tornheim K, Hansford RG, Prentki M, and Corkey BE (1996). Regulation of pancreatic beta-cell mitochondrial metabolism: influence of Ca2+, substrate and ADP. Biochem. J 318, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocaña A, Vasavada R, and Stewart AF (2006). Molecular control of cell cycle progression in the pancreatic β-cell. Endocr. Rev 27, 356–370. [DOI] [PubMed] [Google Scholar]
- Dali-Youcef N, Mataki C, Coste A, Messaddeq N, Giroud S, Blanc S, Koehl C, Champy M-F, Chambon P, Fajas L, et al. (2007). Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc. Natl. Acad. Sci. USA 104, 10703–10708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, and Thompson CB (2008). The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20. [DOI] [PubMed] [Google Scholar]
- Digman MA, Caiolfa VR, Zamai M, and Gratton E (2008). The phasor approach to fluorescence lifetime imaging analysis. Biophys. J 94, L14–L16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doliba NM, Vatamaniuk MZ, Buettger CW, Qin W, Collins HW, Wehrli SL, Carr RD, and Matschinsky FM (2003). Differential effects of glucose and glyburide on energetics and Na+ levels of betaHC9 cells: nuclear magnetic resonance spectroscopy and respirometry studies. Diabetes 52, 394–402. [DOI] [PubMed] [Google Scholar]
- Ferdaoussi M, Dai X, Jensen MV, Wang R, Peterson BS, Huang C, Ilkayeva O, Smith N, Miller N, Hajmrle C, et al. (2015). Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J. Clin. Invest 125, 3847–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiaschi-Taesch NM, Kleinberger JW, Salim FG, Troxell R, Wills R, Tanwir M, Casinelli G, Cox AE, Takane KK, Scott DK, and Stewart AF (2013). Human pancreatic β-cell G1/S molecule cell cycle atlas. Diabetes 62, 2450–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerencser AA (2015). Bioenergetic Analysis of Single Pancreatic β-Cells Indicates an Impaired Metabolic Signature in Type 2 Diabetic Subjects. Endocrinology 156, 3496–3503. [DOI] [PubMed] [Google Scholar]
- Gerencser AA, Mookerjee SA, Jastroch M, and Brand MD (2016). Measurement of the Absolute Magnitude and Time Courses of Mitochondrial Membrane Potential in Primary and Clonal Pancreatic Beta-Cells. PLoS ONE 11, e0159199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Hayashi R, Kang D, and Yoshida K (2006). Acute loss of transcription factor E2F1 induces mitochondrial biogenesis in HeLa cells. J. Cell. Physiol 209, 923–934. [DOI] [PubMed] [Google Scholar]
- Grapengiesser E, Gylfe E, and Hellman B (1990). Sulfonylurea mimics the effect of glucose in inducing large amplitude oscillations of cytoplasmic Ca2+ in pancreatic beta-cells. Mol. Pharmacol 37, 461–467. [PubMed] [Google Scholar]
- Gregg T, Poudel C, Schmidt BA, Dhillon RS, Sdao SM, Truchan NA, Baar EL, Fernandez LA, Denu JM, Eliceiri KW, et al. (2016). Pancreatic β-Cells From Mice Offset Age-Associated Mitochondrial Deficiency With Reduced KATP Channel Activity. Diabetes 65, 2700–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg T, Sdao SM, Dhillon RS, Rensvold JW, Lewandowski SL, Pagliarini DJ, Denu JM, and Merrins MJ (2019). Obesity-dependent CDK1 signaling stimulates mitochondrial respiration at complex I in pancreatic β-cells. J. Biol. Chem 294, 4656–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang Y, Yamamoto T, Benninger RKP, Brissova M, Guo M, Bush W, Piston DW, Powers AC, Magnuson M, Thurmond DC, and Stein R (2014). The MafA transcription factor becomes essential to islet β-cells soon after birth. Diabetes 63, 1994–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, Swisa A, Condiotti R, Granit RZ, Nevo Y, et al. (2016). p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med 22, 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC (2009). Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 52, 739–751. [DOI] [PubMed] [Google Scholar]
- Hsieh MCF, Das D, Sambandam N, Zhang MQ, and Nahlé Z (2008). Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J. Biol. Chem 283, 27410–27417. [DOI] [PubMed] [Google Scholar]
- Jayapal SR, Wang CQ, Bisteau X, Caldez MJ, Lim S, Tergaonkar V, Osato M, and Kaldis P (2015). Hematopoiesis specific loss of Cdk2 and Cdk4 results in increased erythrocyte size and delayed platelet recovery following stress. Haematologica 100, 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers FC, and Henquin JC (2001). Measurements of cytoplasmic Ca2+ in islet cell clusters show that glucose rapidly recruits beta-cells and gradually increases the individual cell response. Diabetes 50, 540–550. [DOI] [PubMed] [Google Scholar]
- Jonkers FC, Guiot Y, Rahier J, and Henquin JC (2001). Tolbutamide stimulation of pancreatic beta-cells involves both cell recruitment and increase in the individual Ca(2+) response. Br. J. Pharmacol 133, 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SK, Kauri LM, Qian WJ, and Kennedy RT (2000). Correlated oscillations in glucose consumption, oxygen consumption, and intracellular free Ca(2+) in single islets of Langerhans. J. Biol. Chem 275, 6642–6650. [DOI] [PubMed] [Google Scholar]
- Kim SY, Lee J-H, Merrins MJ, Gavrilova O, Bisteau X, Kaldis P, Satin LS, and Rane SG (2017). Loss of Cyclin-dependent Kinase 2 in the Pancreas Links Primary β-Cell Dysfunction to Progressive Depletion of β-Cell Mass and Diabetes. J. Biol. Chem 292, 3841–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarrigue S, Lopez-Mejia IC, Denechaud P-D, Escoté X, Castillo-Armengol J, Jimenez V, Chavey C, Giralt A, Lai Q, Zhang L, et al. (2016). CDK4 is an essential insulin effector in adipocytes. J. Clin. Invest 126, 335–348. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lane ME, Yu B, Rice A, Lipson KE, Liang C, Sun L, Tang C, McMahon G, Pestell RG, and Wadler S (2001). A novel cdk2-selective inhibitor, SU9516, induces apoptosis in colon carcinoma cells. Cancer Res. 61, 6170–6177. [PubMed] [Google Scholar]
- Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, Chim H, Lim J-H, Ruan H-B, Yang X, et al. (2014). Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 510, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leloup C, Tourrel-Cuzin C, Magnan C, Karaca M, Castel J, Carneiro L, Colombani A-L, Ktorza A, Casteilla L, and Pénicaud L (2009). Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 53, 673–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski SL, Cardone RL, Foster HR, Ho T, Potapenko E, Poudel C, VanDeusen HR, Sdao SM, Alves TC, Zhao X, et al. (2020). Pyruvate Kinase Controls Signal Strength in the Insulin Secretory Pathway. Cell Metab. 32, 736–750. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Mejia IC, Lagarrigue S, Giralt A, Martinez-Carreres L, Zanou N, Denechaud P-D, Castillo-Armengol J, Chavey C, Orpinell M, Delacuisine B, et al. (2017). CDK4 Phosphorylates AMPKα2 to Inhibit Its Activity and Repress Fatty Acid Oxidation. Mol. Cell 63, 336–349. e6. [DOI] [PubMed] [Google Scholar]
- MacDonald MJ (1981). High content of mitochondrial glycerol-3-phosphate dehydrogenase in pancreatic islets and its inhibition by diazoxide. J. Biol. Chem 256, 8287–8290. [PubMed] [Google Scholar]
- McKenna JP, Ha J, Merrins MJ, Satin LS, Sherman A, and Bertram R (2016). Ca2+ Effects on ATP Production and Consumption Have Regulatory Roles on Oscillatory Islet Activity. Biophys. J 110, 733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrins MJ, and Stuenkel EL (2008). Kinetics of Rab27a-dependent actions on vesicle docking and priming in pancreatic beta-cells. J. Physiol 536, 5367–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K, Wunder C, Roysam B, Lin G, and Lippincott-Schwartz J (2009). A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 106, 11960–11965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oropeza D, Jouvet N, Budry L, Campbell JE, Bouyakdan K, Lacombe J, Perron G, Bergeron V, Neuman JC, Brar HK, et al. (2015). Phenotypic Characterization of MIP-CreERT1Lphi Mice With Transgene-Driven Islet Expression of Human Growth Hormone. Diabetes 64, 3798–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panten U, Zünkler BJ, Scheit S, Kirchhoff K, and Lenzen S (1986). Regulation of energy metabolism in pancreatic islets by glucose and tolbutamide. Diabetologia 29, 648–654. [DOI] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Barbieri J, Brown EB, and Gelbard HA (2011). Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques 50, 98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M, Matschinsky FM, and Madiraju SRM (2013). Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 18, 162–185. [DOI] [PubMed] [Google Scholar]
- Puri S, Roy N, Russ HA, Leonhardt L, French EK, Roy R, Bengtsson H, Scott DK, Stewart AF, and Hebrok M (2018). Replication confers β cell immaturity. Nat. Commun 9, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Sherman A, Bertram R, Goforth PB, Nunemaker CS, Waters CD, and Satin LS (2013). Slow oscillations of KATP conductance in mouse pancreatic islets provide support for electrical bursting driven by metabolic oscillations. Am. J. Physiol. Endocrinol. Metab 305, E805–E817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosselot C, Kumar A, Lakshmipathi J, Zhang P, Lu G, Katz LS, Prochownik EV, Stewart AF, Lambertini L, Scott DK, and Garcia-Ocaña A (2019). Myc Is Required for Adaptive β-Cell Replication in Young Mice but Is Not Sufficient in One-Year-Old Mice Fed With a High-Fat Diet. Diabetes 68, 1934–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamaki T, Casimiro MC, Ju X, Quong AA, Katiyar S, Liu M, Jiao X, Li A, Zhang X, Lu Y, et al. (2006). Cyclin D1 determines mitochondrial function in vivo. Mol. Cell. Biol 26, 5449–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Martín A, Ceballo S, Ruminot I, Lerchundi R, Frommer WB, and Barros LF (2013). A genetically encoded FRET lactate sensor and its use to detect the Warburg effect in single cancer cells. PLoS ONE 3, e57712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieke SM, McCoy JP Jr., and Finkel T (2008). Coordination of mitochondrial bioenergetics with G1 phase cell cycle progression. Cell Cycle 7, 1782–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuit F, De Vos A, Farfari S, Moens K, Pipeleers D, Brun T, and Prentki M (1997). Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J. Biol. Chem 272, 18572–18579. [DOI] [PubMed] [Google Scholar]
- Stark R, Pasquel F, Turcu A, Pongratz RL, Roden M, Cline GW, Shulman GI, and Kibbey RG (2009). Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J. Biol. Chem 284, 26578–26590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet IR, Cook DL, DeJulio E, Wallen AR, Khalil G, Callis J, and Reems J (2004). Regulation of ATP/ADP in pancreatic islets. Diabetes 53, 401–409. [DOI] [PubMed] [Google Scholar]
- Tamarina NA, Roe MW, and Philipson L (2014). Characterization of mice expressing Ins1 gene promoter driven CreERT recombinase for conditional gene deletion in pancreatic β-cells. Islets 6, e27685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorens B, Tarussio D, Maestro MA, Rovira M, Heikkilä E, and Ferrer J (2015). Ins1(Cre) knock-in mice for beta cell-specific gene recombination. Diabetologia 58, 558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Donaldson CJ, Cáceres E, Hunter AE, Cowing-Zitron C, Pound LD, Adams MW, Zembrzycki A, Grove KL, and Huising MO (2015). Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med 21, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fan M, Candas D, Zhang T-Q, Qin L, Eldridge A, Wachsmann-Hogiu S, Ahmed KM, Chromy BA, Nantajit D, et al. (2014). Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev. Cell 29, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Karakose E, Liu H, Swartz E, Ackeifi C, Zlatanic V, Wilson J, González BJ, Bender A, Takane KK, et al. (2019). Combined Inhibition of DYRK1A, SMAD, and Trithorax Pathways Synergizes to Induce Robust Replication in Adult Human Beta Cells. Cell Metab. 29, 638–652. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F-Y, Nagashima K, Ohshima T, Saheki Y, Lu Y-F, Matsushita M, Yamada Y, Mikoshiba K, Seino Y, Matsui H, and Tomizawa K (2005). Cdk5-dependent regulation of glucose-stimulated insulin secretion. Nat. Med 11, 1104–1108. [DOI] [PubMed] [Google Scholar]
- Xin X, Ferraro F, Bäck N, Eipper BA, and Mains RE (2004). Cdk5 and Trio modulate endocrine cell exocytosis. J. Cell Sci 117, 4739–4748. [DOI] [PubMed] [Google Scholar]
- Xue J, Scotti E, and Stoffel M (2019). CDK8 Regulates Insulin Secretion and Mediates Postnatal and Stress-Induced Expression of Neuropeptides in Pancreatic β Cells. Cell Rep. 23, 2892–2904. e7. [DOI] [PubMed] [Google Scholar]
- Zhao C, Wilson MC, Schuit F, Halestrap AP, and Rutter GA (2001). Expression and distribution of lactate/monocarboxylate transporter isoforms in pancreatic islets and the exocrine pancreas. Diabetes 50, 361–366. [DOI] [PubMed] [Google Scholar]
- Zheng Y-L, Li C, Hu Y-F, Cao L, Wang H, Li B, Lu X-H, Bao L, Luo H-Y, Shukla V, et al. (2013). Cdk5 inhibitory peptide (CIP) inhibits Cdk5/p25 activity induced by high glucose in pancreatic beta cells and recovers insulin secretion from p25 damage. PLoS ONE 8, e63332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate new code.