

Abstract
Purpose:
Triple-negative breast cancer (TNBC) is the most challenging and aggressive subtype of breast cancer with limited treatment options because of tumor heterogeneity, lack of druggable targets and therapy resistance. TNBCs are characterized by overexpression of growth factor receptors such as epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), and platelet derived growth factor receptor (PDGFR) making them promising therapeutic targets. Regorafenib is an FDA approved oral multi-kinase inhibitor that blocks the activity of multiple protein kinases including those involved in the regulation of tumor angiogenesis [VEGFR1-3, TIE2], tumor microenvironment [PDGFR-β, FGFR] and oncogenesis (KIT, RET, RAF-1, BRAF). In the current study, we examined the radiosensitizing effects of Regorafenib on TNBC cell lines and explored the mechanism by which Regorafenib enhances radiosensitivity.
Methods:
MDA-MB-231 and SUM159PT (human TNBC cell lines) and MCF 10a (human mammary epithelial cell line) were treated with Regorafenib, ionizing radiation or a combination of both. Following treatment with Regorafenib and radiation we conducted clonogenic assay to determine radiosensitivity, immunoblot analysis to assess the effect on key signaling targets, tube formation to evaluate effect on angiogenesis and comet assay as well as western blot for γH2AX to assess DNA damage response (DDR).
Results:
Regorafenib reduced cell proliferation and enhanced radiosensitivity of MDA-MB-231 and SUM159PT cell lines but had no effect on the MCF 10a cells. Clonogenic survival assays showed that the surviving fraction at 2 Gy for both MDA-MB-231 and SUM159PT was reduced from 66.4 ± 8.9 and 88.2 ± 1.7 in controls to 38.1 ± 4.9 and 75.1 ± 1.1 following a 24 hr pretreatment with 10μM and 5 μM Regorafenib, respectively. A marked reduction in the expression of VEGFR, PDGFR, EGFR and the downstream target, ERK, was observed with Regorafenib treatment alone or in combination with radiation. We also observed a significant inhibition of VEGF-A production in the TNBC cell lines following treatment with Regorafenib. Further, the addition of conditioned medium from Regorafenib-treated tumor cells onto human umbilical vein endothelial cells (HUVEC) suppressed tube formation, indicating an inhibition of tumor angiogenesis. Regorafenib also decreased migration of TNBC cells and suppressed radiation-induced DNA damage repair in a time-dependent manner.
Conclusions:
Our findings demonstrate that Regorafenib enhanced radiosensitivity of breast cancer cells by inhibiting the expression of multiple receptor tyrosine kinases, VEGF-mediated angiogenesis and DNA damage response in TNBC. Therefore, combining Regorafenib with radiation and antiangiogenic agents will be beneficial and effective in controlling TNBC.
Keywords: Regorafenib, VEGF, angiogenesis, DNA damage, radiotherapy, breast cancer
1. Introduction
Triple-negative breast cancer (TNBC) is a breast cancer subtype characterized by the absence of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (Her2/Neu) and associated with aggressive disease and poor overall prognosis (Bianchini et al. 2016). TNBC treatment poses clinical challenges due to significant heterogeneity within the subtypes and unique molecular features leading to aggressive behavior and distinct metastatic patterns (Bianchini et al. 2016; Collignon et al. 2016). The success rate of conventional therapy regimens, including radiation therapy, is limited in TNBC patients because of the development of resistance to therapy and high risk of relapse and recurrence (Collignon et al. 2016). Treatment of TNBC is also stymied by the lack of effective targeted therapy. Therefore, new therapeutic strategies are urgently needed for TNBC treatment.
Several receptor tyrosine kinases (RTKs) such as vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), and colony-stimulating factor-1 receptor (CSF1-R) are highly activated and overexpressed in the TNBC subtype. Overexpression of these RTKs and their associated signaling pathways results in aggressive breast cancer growth, metastasis, initiation of tumor neoangiogenesis, resistance to therapy and poor prognosis rendering them as promising candidates (Butti et al. 2018). Recent reports have shown that targeting RTKs is beneficial in reverting resistance induced by conventional therapies and in improving the disease-free survival in metastatic breast cancer patients (Perez et al. 2011; Li et al. 2013; Kwon et al. 2015). Regorafenib (STIVARGA®), is an FDA approved oral multi-targeted tyrosine kinase inhibitor that targets the activity of several kinases involved in the regulation of tumor angiogenesis [VEGFR and Tie-2], tumor microenvironment [PDGFR-β and FGFR], and oncogenesis [c-KIT, RET, RAF-1, BRAF, and BRAFV600E] (Strumberg and Schultheis 2012; Davis et al. 2013; Ettrich and Seufferlein 2014; Goel 2018). Regorafenib was approved for the treatment of patients with metastatic colorectal cancer and advanced gastrointestinal stromal tumors by the FDA in 2012 (Crona et al. 2013; Sirohi et al. 2014; de la Fouchardière 2018). Phase-III studies showed moderate improvement in overall survival in metastatic colorectal cancer patients treated with Regorafenib (de la Fouchardière 2018). While preclinical and clinical studies reported Regorafenib to suppress tumor growth, metastasis and angiogenesis, there is paucity of data on the effect of Regorafenib on TNBCs. A recent study demonstrated that Regorafenib suppressed the metastatic potential of TNBC cells through a SHP-1/p-STAT3/VEGF-A dependent mechanism (Su et al. 2016). Interestingly, Regorafenib combined with other agents and modalities is suggested to achieve enhanced therapeutic outcome against certain cancers (Daudigeos-Dubus et al. 2015; Belli et al. 2017; Lin et al. 2018). However, the combinatorial therapeutic effect of Regorafenib and radiation against TNBC has not been reported.
In this study, we investigated the therapeutic effect of Regorafenib alone and in combination with ionizing radiation against human TNBC cell lines. We demonstrated that Regorafenib inhibited not only constitutive expression but also radiation-induced activation of RTKs and their associated cell signaling pathways and significantly inhibited the colony formation ability of TNBCs when used in combination with ionizing radiation. Additionally, significant inhibition of migration and invasion, reduced vascular endothelial growth factor-A (VEGF-A) secretion and inhibition of angiogenesis, as measured by disruption of endothelial tube formation, was observed following treatment with Regorafenib alone or in combination with radiation. Regorafenib treatment also induced persistent DNA damage and inhibited DNA double strand break (DSB) repair pathway which correlated with enhanced radiosensitivity. Taken together, our findings suggest that Regorafenib radiosensitizes TNBC cells by inhibiting RTKs, suppressing cell migration and angiogenesis, and inducing DNA damage.
2. Materials and methods
2.1. Cell culture
Cell culture conditions of the human TNBC cell lines - MDA-MB-231 and SUM159PT as well as the normal mammary epithelial cell line, MCF 10a, were as previously described (Mehta et al. 2016). Human umbilical vein endothelial cells (HUVECs) were obtained from Clonetics (San Diego, CA, USA) and maintained in complete EGM-2 medium (Clonetics, San Diego, CA, USA) as per manufacturer’s instructions. All cultures were maintained at 37 °C in an atmosphere of 5% CO2.
2.2. Chemicals
Regorafenib was obtained from Selleck Chemicals (Houston, TX, USA) as a 10 mM stock solution in DMSO, aliquoted and stored at −80 °C till further use.
2.3. Clonogenic survival
Clonogenic cell survival assay was performed on human TNBC cell lines (MDA-MB-231 and SUM159PT) and normal mammary epithelial cell line (MCF 10a) as previously described (Munshi et al. 2006). Vehicle-treated control cells or cells pretreated with Regorafenib (5 μM or 10 μM) for 24 h were irradiated with a high dose rate 137Cs unit at room temperature, trypsinized, counted and reseeded in known numbers onto 60-mm dishes in triplicate. The cells were allowed to form colonies over a 12- to 14-day incubation period and were then fixed and stained with 0.5% gentian violet solution in methanol. The number of colonies with a cutoff of 50 viable cells per colony were counted and surviving fraction was determined. Data presented here is the mean of at least three independent experiments, each done in triplicate.
2.4. Cell cycle analysis
Alterations in cell cycle were determined by using flow cytometric analysis. MDA-MB-231, SUM159PT and MCF 10a cell lines were seeded in 35 mm dishes at a density of 1 × 105 and incubated with Regorafenib for 2h and then irradiated at 6 Gy. The cells were harvested 24 h later, fixed in ice cold 70% ethanol at 4 °C overnight and treated with PI/RNAse staining buffer at room temperature in the dark for 30 min. Finally, samples were analyzed on a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA) to detect cell cycle distribution (Muralidharan et al. 2017).
2.5. Western blot analysis
Total cell lysates were used to assess protein expression levels and were prepared using RIPA buffer (25 mM Tris HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS with Halt Protease Inhibitor Cocktail and Halt Phosphatase Inhibitor Cocktail [Pierce, Thermo Scientific, IL]). Protein concentration was measured using the BCA protein assay kit (Pierce, Thermo Scientific, IL). SDS-PAGE and immunoblotting was performed using standard protocol as described previously (Mehta et al. 2016). Antibodies for p-ERK, ERK, PDGFR-β, p-PDGFR-β, VEGFR2, p-VEGFR2, EGFR, p-EGFR, Actin and γ-H2AX were obtained from Cell Signaling (Boston, MA).
2.6. Transwell migration assay
Transwell chambers containing polycarbonate filters with a pore size of 8 μm (BD Biosciences, Bedford, MA, USA) were used to perform the cell migration assay as previously described (Panneerselvam et al. 2015). Briefly, cells in 2% FBS containing medium were loaded into the upper chamber of the insert and placed in a six-well plate containing serum-free medium in the lower chamber. After placing at 37 °C for 24 h, the culture medium in the six-well plate was replaced with fresh medium containing 20% FBS and the upper chamber was filled with 2% FBS containing medium with or without Regorafenib. At 24 and 48 h following incubation the inserts were removed and the non-migrated cells on the top surface of the membrane were wiped with a cotton swab and the cells passing through the membrane and located on the bottom (invasive cells) were fixed and stained with crystal violet solution in methanol. After rinsing with PBS, images were taken using a Nikon fluorescence microscope and the number of cells migrated to the lower part of the insert were counted using the NIS-Elements imaging software (Nikon Instruments, NY). The result of each group was expressed as the average number of migrated cells per microscopic field.
2.7. Wound-healing assay
Migration of the TNBC cells following treatment with Regorafenib was measured using the wound healing assay. Briefly, MDA-MB-231 and SUM159PT cells were seeded uniformly into 6-well dishes and grown to near 90% confluency in complete medium. Cells were wounded by scratching with a 200 μl pipette tip. After scratching, each well was washed with PBS to remove suspended cells and debris. Cells in complete medium were then mock-treated with DMSO or with the appropriate dose of Regorafenib for 24, 48 and 72 h. Imaging of the wounded area was performed at the specific time at the same position. Results are presented as the percentage of the total distance of the original wound enclosed by cells.
2.8. Comet assay
Comet assay was used to determine DNA damage level of cells treated with Regorafenib in the presence and absence of radiation using a Comet Assay kit (Trevigen) as previously described (Mehta et al. 2016). Regorafenib-treated cells (5 or 10 μM for 24 h) were either irradiated with 20 Gy or not, following which they were harvested and resuspended in ice-cold PBS. A mixture of cells and low-melting-point agarose at a ratio of 1:10 (v/v) was prepared and spread onto glass slides (Trevigen). The slides were then incubated in ice-cold neutral lysis buffer (Trevigen) at 4 °C overnight. Subsequently, the slides were placed in an electrophoresis chamber filled with 1X TBE buffer and electrophoresed at 1.0V/cm for 45 min. Slides were then fixed with 70% ethanol and nuclei stained with SYBR Green. Slides were dried for 15–20 minutes at room temperature and stored overnight in a dessicator. Comet images were captured with a Nikon microscope using the NIS-Elements imaging software (Nikon Instruments, NY) and olive Tail Moment was analyzed using the Casplab comet assay software for at least 50 comets in each sample.
2.9. VEGF ELISA assay
The levels of VEGF in the cell culture supernatants were detected using an ELISA kit (R&D Systems, Minneapolis, MN) following the manufacturer’s instructions. SUM159PT and MDA-MB-231 cells seeded in 10 cm dishes were treated 24 h later with 5 or 10 μM Regorafenib respectively, in 2% serum containing media. Next day the cells were irradiated at 5 Gy and the media was replaced with fresh 2% serum containing media without any Regorafenib. Culture supernatants from these cells were collected at 2, 4 and 24 h post-irradiation, spun at 1500 rpm to remove any debris and stored at −80 °C till ready to use. The levels of VEGF in the supernatants were measured in accordance with the manufacturer’s instructions. Each sample was analyzed in triplicate.
2.10. Endothelial tube formation assay
The capillary tube formation assay was used to assess the effect of Regorafenib on HUVEC angiogenesis in vitro as previously described (Malinda et al. 1999). In brief, HUVECs were seeded in Matrigel-coated 96-well plates at a density of 2 × 104 cells/100 μl/well and incubated for 1–2h at 37 °C to allow for capillary-like structure formation. HUVECs were then treated with various concentrations of Regorafenib. After incubating for 18 h at 37 °C the formation of tube-like structures was examined using an inverted Nikon microscope. The number of capillary tubules from five random microscopic fields were photographed and counted. Mean and standard error of two experiments was calculated for each treatment group.
In a separate experiment, HUVEC cells seeded on Matrigel-coated plates, were treated with 100 μl of the conditioned media collected at different time points from control and Regorafenib-treated MDA-MB-231 and SUM159PT cells. Tube formation was assessed 18h later and the number of the tubes quantified from five random fields as described above.
2.11. VEGF neutralizing antibody assay
To assess the contribution of secreted VEGF in angiogenesis, supernatant from Regorafenib treated MDA-MB-231 cells was incubated with VEGF neutralizing antibody at a ratio of 1:1, 1:2 and 1:5 at 4 °C on a shaker for 2-3 h. HUVEC cells were seeded in Matrigel coated (50 μl/well) 96-well plates and then layered either with vehicle control or 100 μl of the supernatant-antibody mix. Tube formation was assessed 18 h later and the number of tubes was counted and data plotted as described above.
2.12. Immunofluorescence
MDA-MB-231 cells were cultured on coverslips placed in 35-mm dishes and treated with Regorafenib and/or radiation (2 Gy) as indicated. At specified times, medium was aspirated and cells were fixed with 1% paraformaldehyde for 10 min at room temperature. Cells were then permeabilized for 10 min with 70% ethanol at room temperature followed by treatment with 0.1% NP40 in PBS for 20 minutes. Following PBS rinses, the cells were incubated in blocking buffer (5% BSA in PBS) for 30 min at room temperature. The cells were then incubated in primary antibody (γ-H2AX, Millipore) overnight at 4 °C with gentle shaking. After washing with PBS, primary antibody was visualized with Alexa Fluor-488 conjugated secondary antibody. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) in PBS, and the coverslips were mounted on slides using Prolong Antifade Reagent (Molecular Probes). Images were then acquired using a Nikon Eclipse Ti fluorescent microscope (Nikon Instruments, NY) and imported into Image J (NIH) analysis software. To quantify γ-H2AX foci, minimum of 50 nuclei were evaluated.
2.13. Statistical analysis
Study results were analyzed using the t-test (Sigma Plot 5.02v, Richmond, CA). Data were expressed as mean ± standard error of the mean and p ≤ .05 was considered to be statistically significant.
3. Results
3.1. Regorafenib enhances radiosensitivity of human TNBC cells
The survival of MDA-MB-231 and SUM159PT cell lines treated with radiation alone or a combination of radiation and Regorafenib was determined using clonogenic assays and compared to that of normal human mammary epithelial cell line (MCF 10a) (Figure 1). Cells pretreated with either, 5 μM or 10 μM Regorafenib for 24 h were irradiated and plated for clonogenic cell survival. Regorafenib significantly (p ≤ .05) suppressed the clonogenic survival in both the TNBC cell lines compared to the vehicle control (DMSO), whereas no effect on survival was observed in the MCF 10a cells. Surviving fraction at 2 Gy (SF2) was reduced from 66.4%±8.9 in the control to 38.1% ± 4.9 (p ≤ .05) in Regorafenib-treated MDA-MB-231 cells (Figure 1(A)). In the SUM159PT cells, the SF2 values were 88.2%±1.7 in the control and 75.1%±1.1 (p ≤ .03) in Regorafenib treated group (Figure 1(B)). Dose enhancement factor (DEF) at 10% (for MDA-MB-231) or 50% (for SUM159PT) cell survival was 1.25 and 1.26, respectively (Figure 1(D)). MCF 10a cells were not radiosensitized upon Regorafenib treatment (Figure 1(C)), indicating the selectivity of Regorafenib for TNBC cells.
Figure 1.
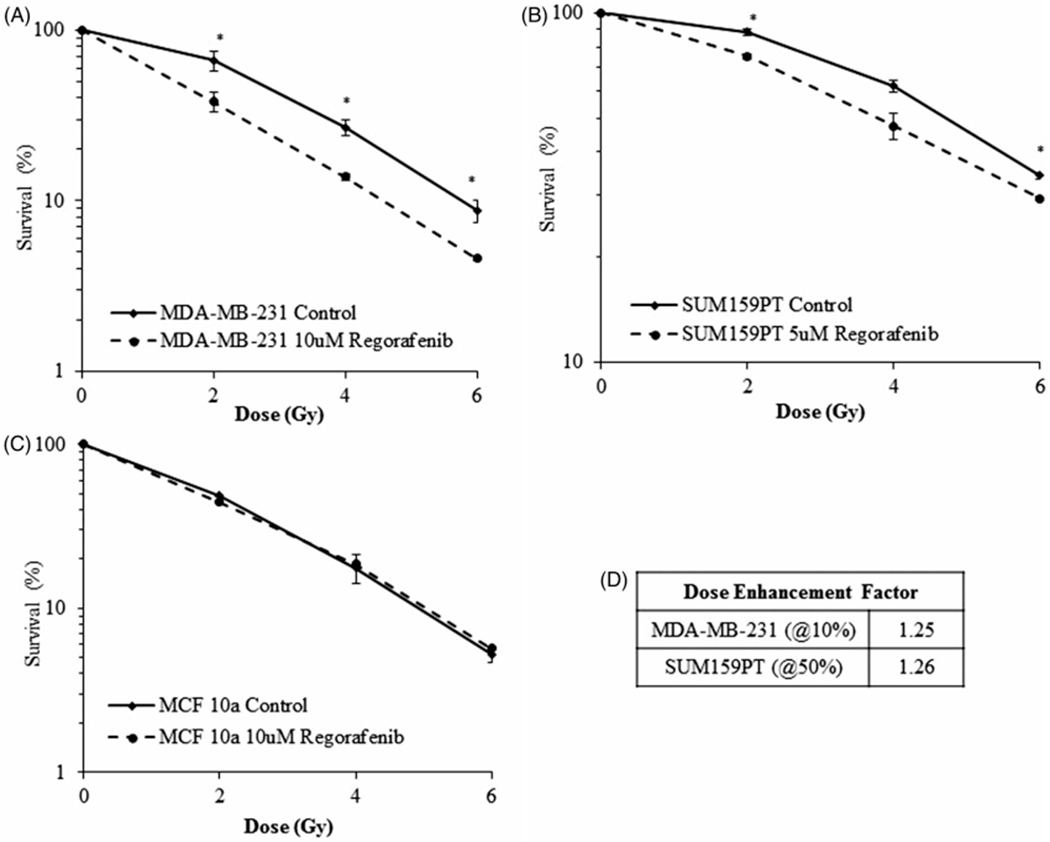
Regorafenib radiosensitizes breast cancer cells but not normal cells as assessed by clonogenic cell survival assay. Regorafenib treatment shows a significant reduction in the surviving fraction compared with the DMSO controls in (A). MDA-MB-231 (B). SUM159PT cells while having no effect on (C). MCF 10a normal cells. (D). Dose enhancement factor (DEF) values calculated from the survival curves shown in panel A & B. DEF was calculated by dividing the radiation dose that produced 10 or 50% cell survival in control cells by that of the treated cells. Values shown are the means±SE of three independent experiments. *p ≤ .05.
3.2. Regorafenib induces G1 phase arrest in TNBC cells
The effect of Regorafenib on cell cycle distribution was detected in the TNBC and normal cell lines after 24 h of treatment. As revealed in Table 1, Regorafenib induced a G1 phase arrest in both MDA-MB-231 and SUM159PT cell lines. Following a 24-h treatment Regorafenib increased the G1 fraction by 12% in MDA-MB-231 and 10% in SUM159PT cells compared to vehicle-treated control cells (Table 1). Only a 4% increase in G1 fraction was noted in MCF 10a cells, which was not further enhanced upon combination with radiation. Both the TNBC cell lines, MDA-MB-231 and SUM159PT, demonstrated a decrease in the S phase in all three treatment groups (radiation alone, Regorafenib alone and combination) when compared to the control. Regorafenib in combination with radiation led to an increase in the G2 phase of the cell cycle in all three cell lines compared to Regorafenib alone (Table 1).
Table 1.
Regorafenib treatment arrests cells in G1 phase of cell cycle.
| MDA-MB-231 | Control | 5Gy | Regorafenib | Regorafenib + 5Gy |
|---|---|---|---|---|
| G1 | 51.32 | 43.6 | 63.73 | 62.62 |
| G2 | 13.45 | 23.93 | 8.55 | 15.98 |
| S | 35.23 | 21.4 | 27.72 | 21.4 |
| SUM159PT | ||||
| G1 | 31.11 | 20.46 | 41.02 | 39.5 |
| G2 | 47.76 | 63.79 | 44.01 | 52.83 |
| S | 21.13 | 15.75 | 14.97 | 7.67 |
| MCF 10a | ||||
| G1 | 69.25 | 80.32 | 73.84 | 63.86 |
| G2 | 6.9 | 8.4 | 5.38 | 13.03 |
| S | 23.85 | 11.3 | 20.77 | 23.11 |
3.3. Regorafenib blocks constitutive and radiation-induced activation of RTKs in human TNBC cells
Constitutive and aberrant activation of RTKs is a common molecular event in a variety of human malignancies including breast cancer and has been reported to be associated with radioresistance. We examined the effect of Regorafenib on the RTKs -EGFR, VEGFR2, and PDGFR-β, and the associated downstream signaling molecule, ERK, in the TNBC cell lines under investigation. A significant inhibition of phosphorylation (p) of EGFR, VEGFR2, and PDGFR-β in the TNBC cell lines was observed following Regorafenib treatment (Figure 2). Radiation activated pEGFR in SUM159PT cells but not in MDA-MB-231 cells, and this activation was suppressed by Regorafenib. No further decrease in the levels of pEGFR, pVEGFR2 and pPDGFR-β was detected when Regorafenib was used in combination with radiation. Consistent with a decrease in the levels of pEGFR, pVEGFR2, and pPDGFR-β, a significant inhibition of the downstream signaling molecule pERK1/2 (Thr202/Tyr204), was observed in both the cell lines (Figure 2). Radiation activated pERK1/2 in both the cell lines and this activation was suppressed by Regorafenib. Total protein levels of EGFR, VEGFR2, PDGFR-β, and ERK remained unchanged.
Figure 2.
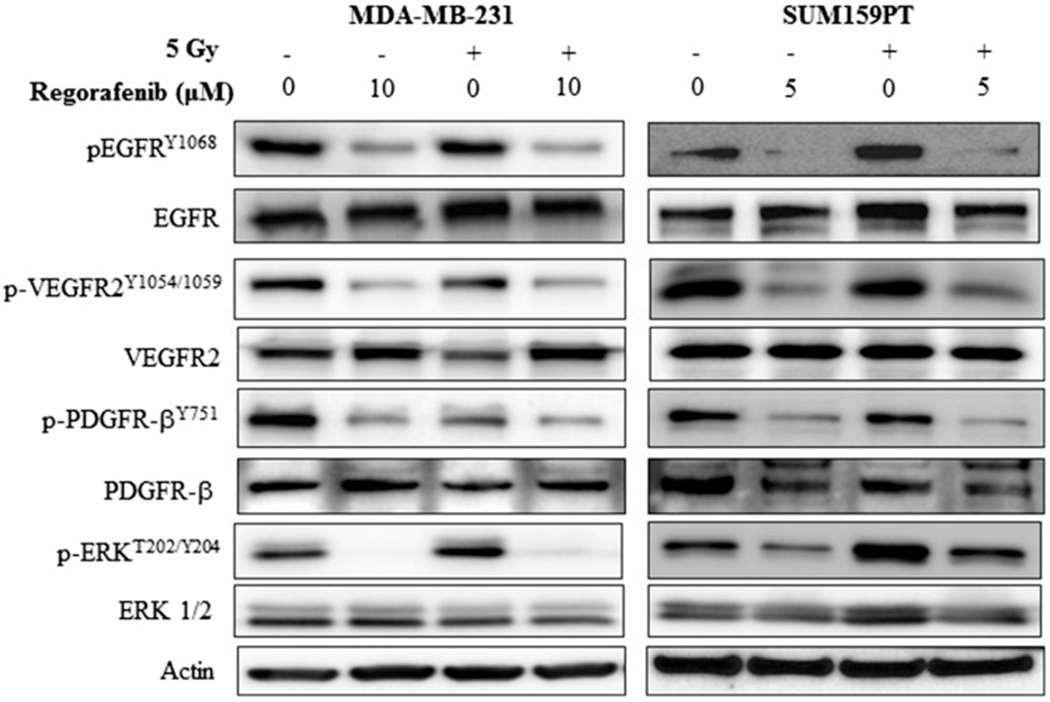
Effect of Regorafenib on receptor tyrosine kinases. MDA-MB-231 and SUM159PT cells were treated with DMSO control or Regorafenib for 24 h (10 and 5 μM respectively) followed by 5 Gy radiation. Two hours post radiation, cell lysates were evaluated by Western blot using the respective antibodies. β-Actin was used as an internal loading control.
3.4. Regorafenib inhibits VEGF secretion and endothelial cell differentiation
To investigate whether Regorafenib exhibits anti-angiogenic activities, we evaluated its effect on VEGF expression using ELISA. MDA-MB-231 and SUM159PT cells were pretreated with Regorafenib for either 24 h or 48 h followed by exposure to radiation for 2 h or 24 h respectively. As shown in Figure 3, Regorafenib treatment for 24 h, either as a single agent or in combination with radiation, significantly decreased VEGF secretion, compared to the respective controls (Figure 3(A,B); p < .01). Radiation exposure for 2h caused no significant reduction in VEGF secretion, though reduced VEGF secretion was seen upon combined Regorafenib and radiation treatment. A sharp increase in VEGF was observed in both the untreated control cell lines at 48 h. However, Regorafenib treatment, when administered as a single agent or in combination with radiation (5 Gy/24 h; 48 h post-Regorafenib treatment), led to a sharp decrease in VEGF secretion (pg/ml) in both the TNBC cell lines. These results demonstrate that Regorafenib inhibits VEGF secretion in both the TNBC cell lines and this suppression was further enhanced upon combination with radiation.
Figure 3.
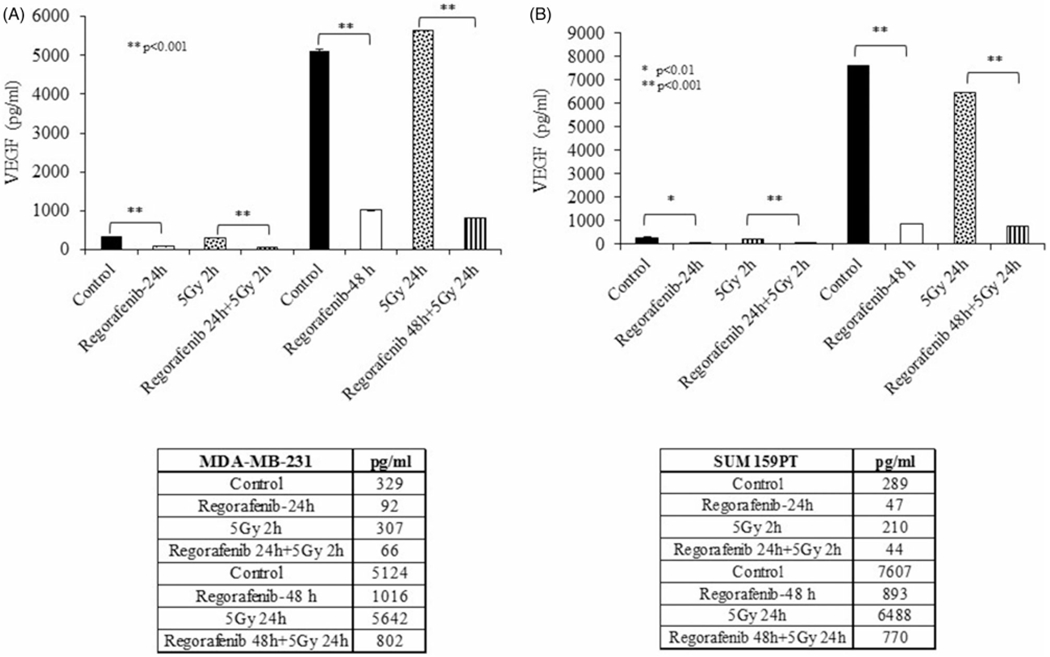
Regorafenib inhibits vascular endothelial growth factor (VEGF) secretion in TNBC cell lines. (A). MDA-MB-231 and (B). SUM159PT cells were irradiated after 24-h pretreatment with Regorafenib and VEGF (pg/ml) was measured in the culture media supernatant at 2-h or 24-h postirradiation. *p ≤ .01, **p ≤ .001.
Since endothelial cell proliferation and tube formation are critical steps in angiogenesis and VEGF is an important growth factor for endothelial cell proliferation and survival, we sought to determine whether Regorafenib-mediated VEGF inhibition abrogated HUVEC cell proliferation and capillary tube formation. As shown in Figure 4(A), HUVECs became elongated and formed capillary tube-like structures in the DMSO treated MDA-MB-231 control cells. However, a 24 h treatment with Regorafenib (50 nM and 75 nM) significantly inhibited tube formation by HUVECs in a dose-dependent manner compared to the control (Figure 4(A)). Furthermore, we performed a cell viability assay to assess the effect of Regorafenib on the proliferation of HUVECs. Consistent with the tube formation assay, Regorafenib significantly decreased HUVEC proliferation at both 50 nM and 75 nM doses compared to the DMSO control group (Figure 4(B)). These results further suggest that Regorafenib inhibits angiogenesis in TNBC cells.
Figure 4.
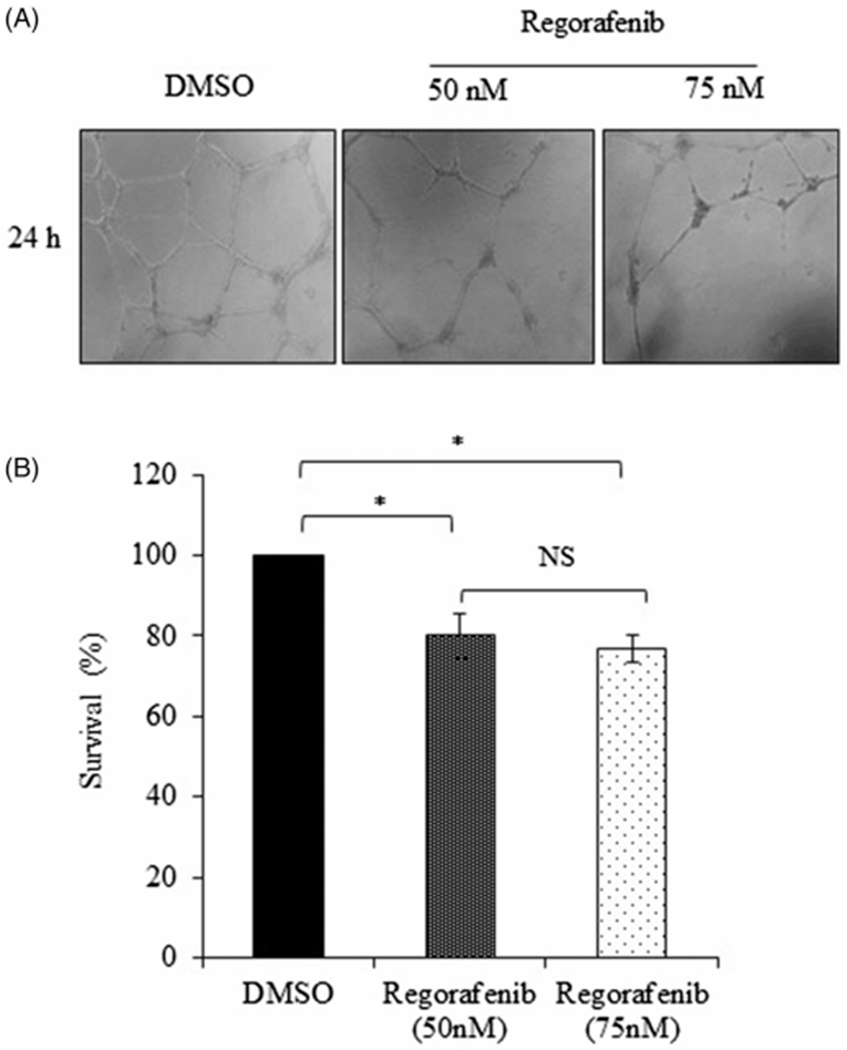
Regorafenib inhibits tube formation in Human Umbilical Vein Endothelial Cells (HUVECs). (A). Microscopic images of HUVECs showing the changes in tube formation when incubated with or without Regorafenib (50 nM or 75 nM) for 24 h. Distinct disruption of tube network can be seen from the images of HUVECs treated with Regorafenib. (B). Cell viability of HUVECs at 24-h post-Regorafenib treatment (50 nM or 75 nM) compared to DmSO control. *p ≤ .05.
Next we determined whether Regorafenib mediated decrease in VEGF expression in tumor cells affected endothelial tube formation. For this purpose, MDA-MB-231 and SUM159PT cell lines were treated with Regorafenib for 24 h, conditioned media was collected and utilized for tube formation assay. Incubation of HUVECs with conditioned media from the DMSO control MDA-MB-231 and SUM159PT cells resulted in the formation of elongated and tube-like structures, whereas conditioned media from Regorafenib-treated MDA-MB-231 and SUM159PT cells, which is deprived of secreted VEGF, effectively and significantly reduced the number of tubes formed (p ≤ .001, Figure 5). Together, these findings suggest that Regorafenib significantly suppressed VEGF-mediated angiogenesis.
Figure 5.
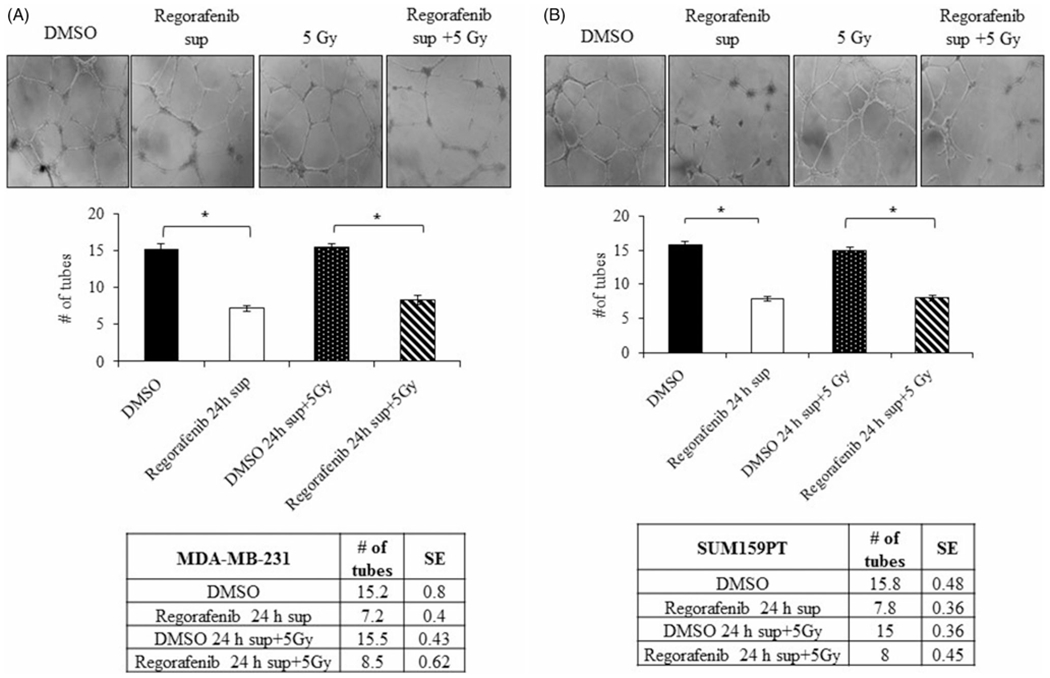
Conditioned media from Regorafenib-treated TNBC cells inhibits tube formation of HUVEC cells. Conditioned media collected from TNBC cells following treatment with Regorafenib for 24 h (10 μM in MDA-MB-231 and 5 μM in SUM159PT), radiation (5 Gy) or combination was added on HUVECs and analyzed for tube formation. Microscopic images and graphical representation of tube formation in HUVECs incubated in conditioned media from (A). MDA-MB-231 (B). SUM159PT cells treated with Regorafenib, radiation (5 Gy) or a combination of both. *p ≤ .001.
As it has been shown that VEGF promotes endothelial tube formation, using a neutralizing antibody against VEGF should decrease the length of the tubes. To confirm the above results, we evaluated the capacity of a neutralizing antibody against VEGF to induce tube formation in HUVECs. Conditioned media from MDA-MB-231 cells treated with Regorafenib alone inhibited HUVEC tube formation by about 50% compared to vehicle control (p ≤ .001, Figure 6). However, treatment of HUVECs with a mixture of conditioned media from MDA-MB-231 cells treated with Regorafenib and VEGF blocking antibody significantly enhanced the inhibitory effects of Regorafenib on tube formation in a dose-dependent manner (p ≤ .001, Figure 6), indicating that the inhibition of tube formation is due to inhibition of VEGF present in the conditioned media. Collectively, these findings validate a clear role of Regorafenib in modulating tumor angiogenesis.
Figure 6.
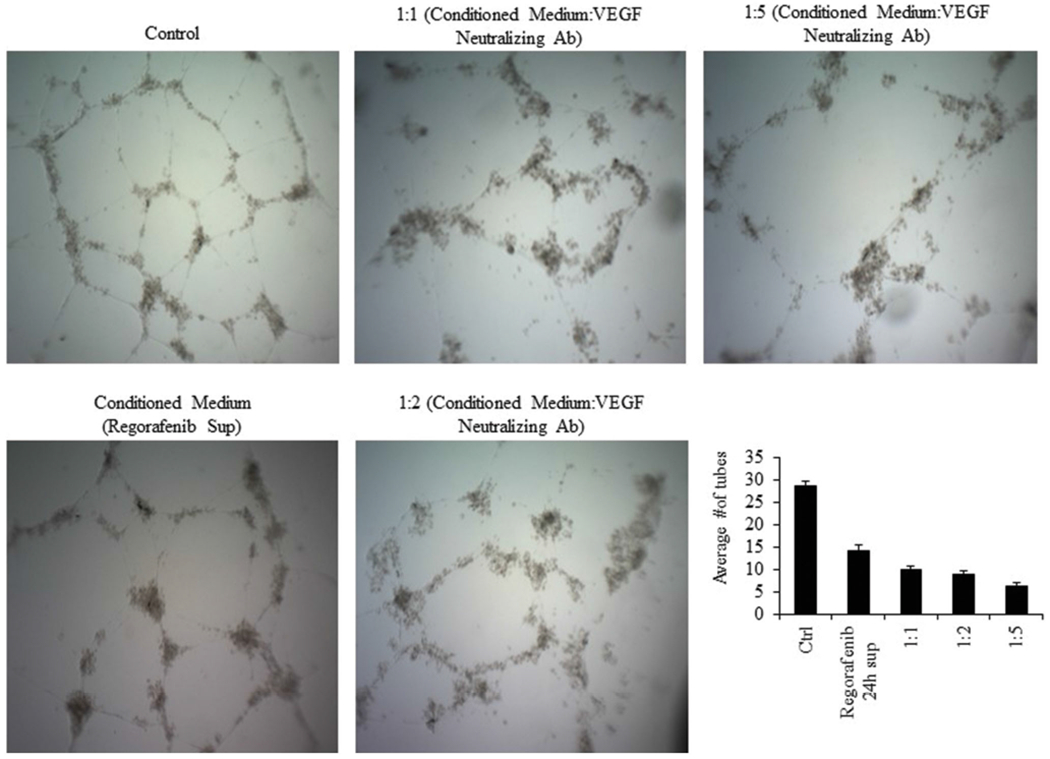
Blocking of secreted VEGF leads to disruption of tube formation. Conditioned media from Regorafenib treated cells inhibited HUVEC tube formation. Exogenous addition of various concentrations of VEGF neutralizing antibody produced greater inhibition of tube formation.
3.5. Regorafenib impairs cancer cell migration
The effect of Regorafenib on tumor cell migration was assessed using the wound healing and transwell migration assays. Treatment with Regorafenib dramatically slowed down the closure of the wound scratched into the confluent monolayer of MDA-MB-231 and SUM159PT cells as observed by the inability to fill the gap at 24, 48 and 72 h compared to the DMSO control (Figure 7(A,B)). Transwell assay also showed a significant reduction (~ 50%; p ≤ .01) in the migration potential compared to the untreated control cells at 24 and 48 h, in the Regorafenib treated MDA-MB-231 and SUM159PT cells (Figure 7(C–F)).
Figure 7.
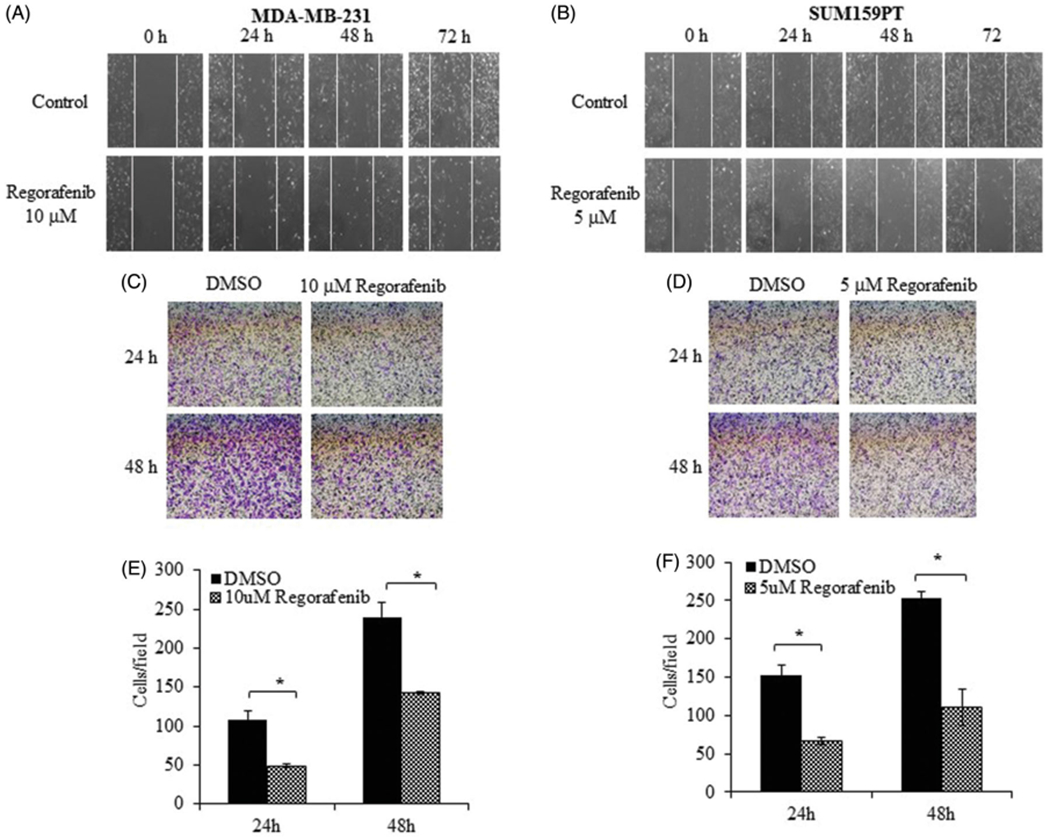
Regorafenib inhibits TNBC cell migration and invasion. Wound healing assays of (A). MDA-MB-231 and (B). SUM159PT cells at 0 h, 24 h, 48 h and 72 h treatment with Regorafenib (10 μM or 5 μM respectively) compared to control (DMSO alone) is shown. Graphical representation shows the relative percentage of wound closure in TNBC cells. Images show the transwell analysis of (C). MDA-MB-231 and (D). SUM159PT cells at 24 and 48 h treatment with Regorafenib or DMSO alone. (E, F). The number of migrated cells per field is shown in the graphical representation of respective cell lines. *p ≤ .01.
3.6. Regulation of DNA repair by regorafenib may be responsible for mediating radiosensitivity
Exposure of cells to ionizing radiation produces a variety of DNA lesions of which DNA double-strand breaks (DSBs) are particularly toxic as they result in reduced integrity of the genome (Santivasi and Xia 2014). Since phosphorylation of the core histone protein H2AX is the earliest cellular response to DSBs we determined the effect of Regorafenib on DNA damage signaling by evaluating γH2AX expression in MDA-MB-231 and SUM159PT cells (Figure 8(A,B)). Regorafenib treatment alone induced phosphorylation of γH2AX in both the cell lines. Exposure to a combination of Regorafenib and radiation further exacerbated this effect (Figure 8(A,B)), indicating that Regorafenib in combination with radiation causes sustained DNA damage response as indicated by activation of γH2AX in both the cell lines. We further validated this observation in MDA-MB-231 cells by evaluating the kinetics of γ-H2AX foci by immunofluorescent staining. Regorafenib treated cells were immunostained and γ-H2AX foci were assessed at 2h and 24 h after 2 Gy (Supplementary Figure 2). The average number of γ-H2AX foci/cell in cells receiving the combined Regorafenib and radiation treatment was significantly greater than in the radiation only group at both 2 h and 24 h time points, p ≤ 1.65E-06 and p ≤ .01, respectively (Supplementary Figure 2). Treatment with Regorafenib alone also produced a significant induction of γ-H2AX foci (p ≤ .0003), indicating that inhibition of RTKs and their downstream targets can induce DNA damage.
Figure 8.
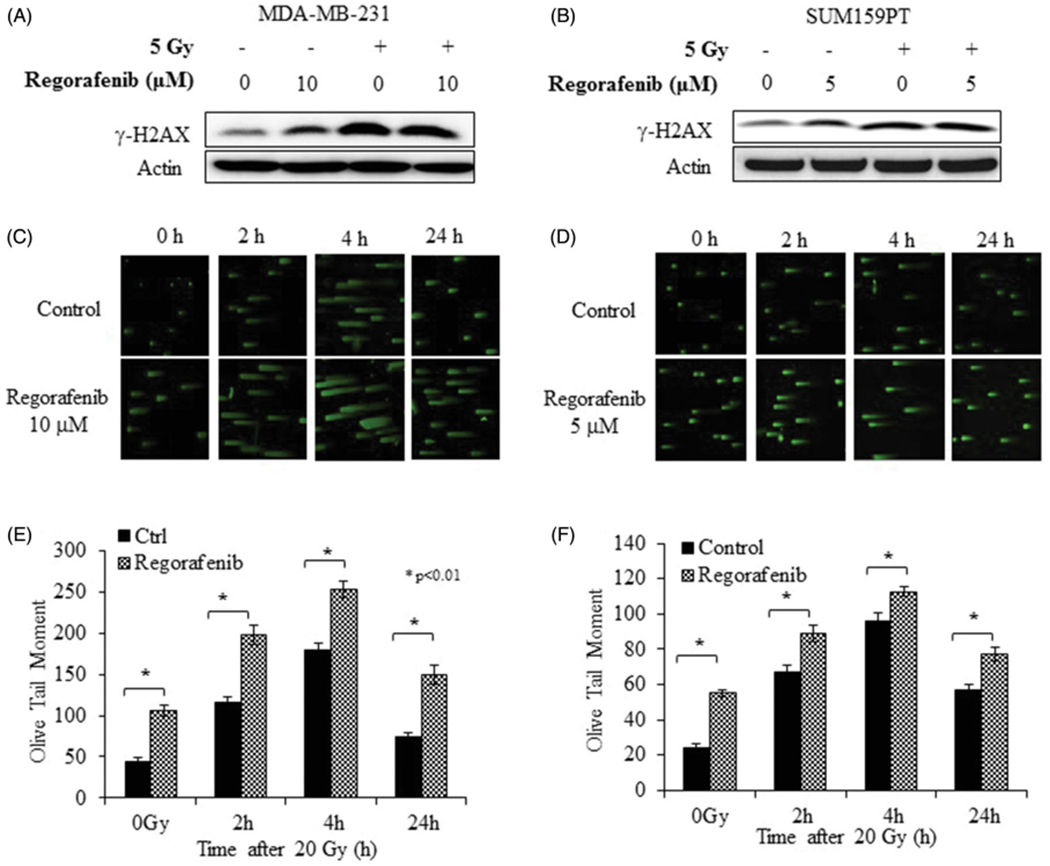
Regorafenib engages the DNA damage response in TNBC cells. (A). MDA-MB-231 and (B). SUM159PT were treated with Regorafenib, irradiated, and lysates analyzed for γ-H2AX expression. DNA damage was also assessed by neutral comet assay (C). MDA-MB-231 (D). SUM159PT. Fluorescent microscopy images were obtained at 2 h, 4 h and 24 h post irradiation (20 Gy). Graphical representation shows the Olive Tail Moments of control versus treatment groups in (E). MDA-MB-231 (F). SUM159PT. *p ≤ .01.
To further substantiate whether combination of Regorafenib and radiation enhances DNA damage, comet tail moments were measured at various time points after treatment with radiation. Both, MDA-MB-231 (Figure 8(C,E)) and SUM159PT cells (Figure 8(D,F)) exhibited greater tail moment upon Regorafenib-treatment compared to the DMSO-treated control cells not receiving radiation. However, combination of Regorafenib with radiation (20 Gy) resulted in higher DNA damage, as evidenced by longer comet tails compared to the radiation treatment alone at all three-time points tested (Figure 8(C,D); p ≤ .005), suggesting that Regorafenib treatment coupled with radiation inhibits DNA repair thereby resulting in radiosensitization of tumor cells.
4. Discussion
Regorafenib is an FDA approved oral multiple kinase inhibitor for treating patients with colorectal cancer, gastrointestinal stromal tumors and refractory hepatocellular carcinoma (Strumberg and Schultheis 2012; Davis et al. 2013; Ettrich and Seufferlein 2014; Goel 2018; Crona et al. 2013; Sirohi et al. 2014; de la Fouchardière 2018). Data exist demonstrating that RTKs play a key role in radiation resistance of tumors and that disruption of RTK signaling enhances the damaging effects of ionizing radiation (Abdollahi et al. 2003; Williams et al. 2008; De Bacco et al. 2011; Guryanova and Bao 2011; Sano et al. 2011). The mechanism by which RTKs contribute to radioresistance has been shown to occur by promoting DNA repair and inhibition of apoptosis (Cosaceanu et al. 2007; Liccardi et al. 2011; Bai et al. 2012; Chou et al. 2014; Mahajan and Mahajan 2015). Since Regorafenib potently inhibits a number of RTKs, combining it with anticancer agents, including radiation is likely to enhance the therapeutic outcome. In this study we investigated the combinatorial effect of Regorafenib and radiation treatment on radiosensitization of TNBC cells. In vitro clonogenic assays revealed that TNBC cell lines, but not the normal mammary epithelial cell line, were significantly sensitized to subsequent doses of radiation when pretreated with Regorafenib (Figure 1). As has been previously reported in the literature Regorafenib induces G0/G1 cell cycle arrest in multiple cancer types (Ribatti et al. 2016; Hu et al. 2018). In accordance with these reports, the effects of Regorafenib on cell cycle in TNBC and normal cells were evaluated. TNBC cells underwent a G1 phase arrest in response to Regorafenib treatment, alone as well as in combination with radiation (Figure 2). A G1 phase arrest in response to DNA damage prevents the defective cells from entering into S phase thereby providing time for the cells to repair the damage and proceed with the cell cycle or enter into apoptosis (Norbury and Zhivotovsky 2004; Cann and Hicks 2007). A significant decrease in S phase was also observed in TNBC cells following treatment with Regorafenib alone and in combination with radiation, suggesting the occurrence of DNA damage in the treated cells.
Since RTKs are expressed at high levels in TNBC they are promising drug targets. We evaluated the effects of Regorafenib on the expression levels of the EGFR, PDGFR, VEGFR as well as the downstream effector ERK. Potent inhibition of all three RTKs was observed when Regorafenib was used as a single agent or in combination with radiation (Figure 2). Furthermore, Regorafenib suppressed radiation-induced pERK in MDA-MB-231 and to some extent in the SUM159PT cells. Since the ERK pathway, through its ability to up-regulate the transcription of DNA repair genes, contributes to radiation resistance its suppression is therefore expected to mediate radiosensitizing effects (Dent et al. 2003). Thus, ERK inhibition by Regorafenib likely contributes to the radiosensitization observed.
Radiation therapy is a vital component of cancer treatment and studies have shown that its efficacy can be enhanced in combination with anti-angiogenic agents (Wachsberger et al. 2003). As VEGF is a growth factor that plays a pivotal role in human tumorigenesis and angiogenesis of cancer we investigated the impact of Regorafenib on tumor angiogenesis by measuring VEGF secretion. We observed a significant decrease in VEGF production upon Regorafenib treatment, suggesting an inhibitory role of Regorafenib on VEGF signaling. The combination of Regorafenib with radiation did not produce a significant reduction in VEGF secretion compared to Regorafenib alone but showed a significant decrease when compared to radiation treatment alone, suggesting that Regorafenib inhibits VEGF production even in the absence of radiation. Next, we investigated whether Regorafenib can modulate the tumor microenvironment by inhibiting VEGF, a potential target for increasing the tumor radiosensitivity (Karar and Maity 2009). For this purpose, we evaluated the impact of Regorafenib treatment on inhibition of neo-angiogenesis using HUVECs as a model. Regorafenib inhibited VEGF-stimulated tubular network formation by HUVEC which showed a clear indication of inhibitory activity of Regorafenib on VEGF, its activity in directly controlling the VEGF secretion, and its associated anti-angiogenic role in TNBC cells. Apart from inhibition of VEGF secretion, suppression of NF-κB activation is also implicated as an antiangiogenesis mechanism induced by Regorafenib (Liu et al. 2017). However, the contribution of NF-κB modulation by Regorafenib in radiosensitivity of TNBC was not investigated in the present study and warrants further investigation. Our data suggests that Regorafenib in combination with radiation impacts VEGF production by TNBC which in turn reduces the ability of TNBC to support neo-angiogenesis resulting in an antiangiogenic effect.
A major mechanism that governs response to radiotherapy is enzyme-mediated repair of DNA-DSBs. Accordingly, inhibition of DNA-DSB repair could be plausible strategy for radio-potentiation. To confirm the inhibition of DSB repair, we carried out the neutral comet assay which under neutral pH conditions detects DNA fragments that occur due to DSBs (Olive et al. 1991). Consistently larger and longer tails were observed in Regorafenib treated cells compared with control cells alone or in combination with radiation (Figure 8), thereby providing strong evidence of a link between Regorafenib and the DNA damage response and suggesting that Regorafenib sensitizes TNBCs to radiation by suppressing the ability of the cells to repair radiation-induced DNA damage.
In summary, our data demonstrated that Regorafenib radiosensitizes TNBC by inhibiting RTKs, inducing DNA damage and suppressing angiogenesis, thereby implying that Regorafenib could be a promising targeted therapeutic strategy for TNBC. Our findings concur with published literature demonstrating that the signaling pathways downstream of growth factor receptors intersect with, and regulate DSB repair mechanisms to modulate cellular responses to ionizing radiation. The possible mechanisms by which receptor signaling modulates DNA repair activity include direct physical interaction of receptor molecules with repair proteins, regulation of repair protein activation and function through specific phosphorylation events, and by governing the transcription of genes that encode various repair proteins (Meyn et al. 2009). Examination of these interactions in greater detail may divulge additional strategies to radiosensitize human tumor cells and help uncover biomarkers to identify patients who may benefit from the combination of molecular targeting agents and radiotherapy.
Supplementary Material
Acknowledgments
Funding
This study was supported in part by grants received from an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences [P20 GM103639] of the National Institutes of Health (NIH) (AM, RR), a pilot grant from the Stephenson Cancer Center funded by the NCI Cancer Center Support Grant [P30 CA225520] awarded to the University of Oklahoma Stephenson Cancer Center, and the OUHSC Department of Radiation Oncology Research Development Funds (AM). RR is an Oklahoma TSET Research Scholar and holds the Jim and Christy Everest Endowed Chair in Cancer Developmental Therapeutics. The content is solely the responsibility of the authors and does not represent the official views of any grant-awarding agency.
Biographies
Notes on contributors
Meghna Mehta, MS is a Research Associate in the Department of Radiation Oncology. She received her Master’s degree in Microbiology from the University of Oklahoma. Her research interests include studies combining drug treatment with radiation to radio sensitize tumor cells and evaluating DNA damage response. Other areas of interests include investigating the role of mitochondrial metabolism in metabolic reprogramming of cancer cells.
James Griffith, BS is a Research Assistant at the University of Oklahoma Health Sciences Center. He received his Bachelor of Science in Microbiology from the University of Oklahoma at Norman. He is currently a member of a research laboratory in the Department of Surgery at OUHSC. The laboratory focuses on wound healing, fibrosis, and the progression of metastasis in colon cancer.
Janani Panneerselvam, PhD is a Senior Research Associate for Center for Cancer Prevention and Drug Development and Division of Medical Oncology at the University of Oklahoma Health Sciences Center. Her research mainly focuses on molecular characterization of cancer cells, chemoprevention, target identification, and validation and drug development at the functional and molecular level. She is also assessing the role of immune cells in cancer development and progression and for anti-tumor immunity.
Anish Babu is Post-Doctoral research fellow at the Stephenson Cancer Center, University of Oklahoma Health Science Center, Oklahoma City, USA. He holds a PhD degree in Biology in the area of anti-cancer therapy and nano-drug delivery from Madurai Kamaraj University, India. His current research is focused on identifying the mechanism(s) and devising therapeutic strategies to overcome therapy resistance in cancer.
Jonathan Mani, MD is a resident physician for the Department of Radiation Oncology at the University of Oklahoma. His interests include the interactions between drugs and tissue effects with ionizing radiation.
Terence Herman, MD, is Professor and Chairman of the Department of Radiation Oncology and an Adjunct Professor of Internal Medicine (Hematology/Oncology) at the University of Oklahoma Health Sciences Center. He specializes in drug/radiation interactions in patients with gastrointestinal, sarcoma, breast and lymphomatous cancers. Dr. Herman is a reviewer for several cancer related journals and is the Principle Investigator for the Radiation Therapy Oncology Group studies at the Stephenson Cancer Center and University of Oklahoma.
Rajagopal Ramesh, PhD, is a Professor in the Department of Pathology at the University of Oklahoma Health Sciences Center and Co-Leader of the Cancer Biology Program and Co-Director of the Nanomedicine Program at the Stephenson Cancer Center. His research is focused on developing novel gene-based therapeutics using viral and non-viral vectors, and nanomedicine with emphasis on translational cancer research. His research has been supported by the National Institutes of Health, Department of Defense and the Department of Veterans Affairs.
Anupama Munshi, PhD, is an Associate Professor in the Department of Radiation Oncology at the University of Oklahoma Health Sciences Center. Her research focuses on understanding the specific molecular mechanisms and biological processes that govern tumor response to radiation and other anticancer agents. A major emphasis of her research is to develop innovative and effective approaches to sensitize tumor cells to radiation and DNA damage and study the molecular mechanism of resistance to radiation-induced cell death.
Footnotes
Supplemental data for this article can be accessed here.
Disclosure statement
The authors declare no conflict of interest.
References
- Abdollahi A, Lipson KE, Han X, Krempien R, Trinh T, Weber KJ, Hahnfeldt P, Hlatky L, Debus J, Howlett AR, et al. 2003. SU5416 and SU6668 attenuate the angiogenic effects of radiation-induced tumor cell growth factor production and amplify the direct anti-endothelial action of radiation in vitro. Cancer Res. 63:3755–3763. [PubMed] [Google Scholar]
- Bai J, Guo XG, Bai XP. 2012. Epidermal growth factor receptor-related DNA repair and radiation-resistance regulatory mechanisms: a mini-review. Asian Pac J Cancer Prev. 13:4879–4881. [DOI] [PubMed] [Google Scholar]
- Belli V, Sforza V, Cardone C, Martinelli E, Barra G, Matrone N, Napolitano S, Morgillo F, Tuccillo C, Federico A, et al. 2017. Regorafenib in combination with Silybin as a novel potential strategy for the treatment of metastatic colorectal cancer. Oncotarget. 8: 68305–68316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. 2016. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 11:674–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butti R, Das S, Gunasekaran VP, Yadav AS, Kumar D, Kundu GC. 2018. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol Cancer. 17:34–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann KL, Hicks GG. 2007. Regulation of the cellular DNA double-strand break response. Biochem Cell Biol. 85:663–674. [DOI] [PubMed] [Google Scholar]
- Chou RH, Wang YN, Hsieh YH, Li LY, Xia W, Chang WC, Chang LC, Cheng CC, Lai CC, Hsu JL, et al. 2014. EGFR modulates DNA synthesis and repair through Tyr phosphorylation of histone H4. Dev Cell. 30:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collignon J, Lousberg L, Schroeder H, Jerusalem G. 2016. Triple-negative breast cancer: treatment challenges and solutions. Breast Cancer (Dove Med Press). 8:93–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosaceanu D, Budiu RA, Carapancea M, Castro J, Lewensohn R, Dricu A. 2007. Ionizing radiation activates IGF-1R triggering a cytoprotective signaling by interfering with Ku-DNA binding and by modulating Ku86 expression via a p38 kinase-dependent mechanism. Oncogene. 26:2423–2434. [DOI] [PubMed] [Google Scholar]
- Crona DJ, Keisler MD, Walko CM. 2013. Regorafenib: a novel multi-targeted tyrosine kinase inhibitor for colorectal cancer and gastrointestinal stromal tumors. Ann Pharmacother. 47:1685–1696. [DOI] [PubMed] [Google Scholar]
- Daudigeos-Dubus E, Le Dret L, Lanvers-Kaminsky C, Bawa O, Opolon P, Vievard A, Villa I, Pagès M, Bosq J, Vassal G, et al. 2015. Regorafenib: antitumor activity upon mono and combination therapy in preclinical pediatric malignancy models. PLoS One. 10: e0142612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SL, Eckhardt SG, Messersmith WA, Jimeno A. 2013. The development of Regorafenib and its current and potential future role in cancer therapy. Drugs Today. 49:105–115. [DOI] [PubMed] [Google Scholar]
- De Bacco F, Luraghi P, Medico E, Reato G, Girolami F, Perera T, Gabriele P, Comoglio PM, Boccaccio C. 2011. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J Natl Cancer Inst. 103:645–661. [DOI] [PubMed] [Google Scholar]
- de la Fouchardière C 2018. Regorafenib in the treatment of metastatic colorectal cancer. Future Oncol. 14:2239–2246. [DOI] [PubMed] [Google Scholar]
- Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. 2003. MAPK pathways in radiation responses. Oncogene. 22:5885–5896. [DOI] [PubMed] [Google Scholar]
- Ettrich TJ, Seufferlein T. 2014. Regorafenib – recent results. Recent Results Cancer Res. 201:185–196. [DOI] [PubMed] [Google Scholar]
- Goel G 2018. Evolution of Regorafenib from bench to bedside in colorectal cancer: is it an attractive option or merely a “me too” drug? Cancer Manag Res. 10:425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guryanova OA, Bao S. 2011. How scatter factor receptor c-MET contributes to tumor radioresistance: ready, set, scatter! J Natl Cancer Inst. 103:617–619. [DOI] [PubMed] [Google Scholar]
- Hu X, Wu LW, Zhang ZY, Chen ML, Li YL, Zhang C. 2018. The antitumor effect of Regorafenib in lung squamous cell carcinoma in vitro. Biochem Biophys Res Commun. 503:1123–1129. [DOI] [PubMed] [Google Scholar]
- Karar J, Maity A. 2009. Modulating the tumor microenvironment to increase radiation responsiveness. Cancer Biol Ther. 8:1994–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YS, Chun SY, Nam KS, Kim S. 2015. Lapatinib sensitizes quiescent MDA-MB-231 breast cancer cells to doxorubicin by inhibiting the expression of multidrug resistance-associated protein-1. Oncol Rep. 34:884–890. [DOI] [PubMed] [Google Scholar]
- Li P, Veldwijk MR, Zhang Q, Li ZB, Xu WC, Fu S. 2013. Co-inhibition of epidermal growth factor receptor and insulin-like growth factor receptor 1 enhances radiosensitivity in human breast cancer cells. BMC Cancer. 13:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liccardi G, Hartley JA, Hochhauser D. 2011. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res. 71:1103–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Lin TH, Chen CC, Chen MC, Chen CP. 2018. Combination chemotherapy with Regorafenib in metastatic colorectal cancer treatment: a single center, retrospective study. PLoS One. 13:e0190497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Wu RH, Wang WS. 2017. Regorafenib diminishes the expression and secretion of angiogenesis and metastasis associated proteins and inhibits cell invasion via NF-κB inactivation in SK-Hep1 cells. Oncol Lett. 14:461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K, Mahajan NP. 2015. Cross talk of tyrosine kinases with the DNA damage signaling pathways. Nucleic Acids Res. 43: 10588–10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinda KM, Nomizu M, Chung M, Delgado M, Kuratomi Y, Yamada Y, Kleinman HK, Ponce ML. 1999. Identification of laminin a1 and b1 chain peptides active for endothelial cell adhesion, tube formation, and aortic sprouting. FASEB J. 13:53–62. [PubMed] [Google Scholar]
- Mehta M, Basalingappa K, Griffith JN, Andrade D, Babu A, Amreddy N, Muralidharan R, Gorospe M, Herman T, Ding WQ, et al. 2016. HuR silencing elicits oxidative stress and DNA damage and sensitizes human triple-negative breast cancer cells to radiotherapy. Oncotarget. 7:64820–64835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyn RE, Munshi A, Haymach J, Milas L, Ang KK. 2009. Receptor signaling as a regulatory mechanism of DNA repair. Radiotherapy and Oncol. 92:316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi A, Tanaka T, Hobbs ML, Tucker SL, Richon VM, Meyn RE. 2006. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther. 5:1967–1974. [DOI] [PubMed] [Google Scholar]
- Muralidharan R, Mehta M, Ahmed R, Roy S, Xu L, Aubé J, Chen A, Zhao YD, Herman T, Ramesh R, et al. 2017. HuR-targeted small molecule inhibitor exhibits cytotoxicity towards human lung cancer cells. Sci Rep. 7:9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury CJ, Zhivotovsky B. 2004. DNA damage-induced apoptosis. Oncogene. 23:2797–2808. [DOI] [PubMed] [Google Scholar]
- Olive PL, Wlodek D, Banáth JP. 1991. DNA double-strand breaks measured in individual cells subjected to gel electrophoresis. Cancer Res. 51:4671–4676. [PubMed] [Google Scholar]
- Panneerselvam J, Jin J, Shanker M, Lauderdale J, Bates J, Wang Q, Zhao YD, Archibald SJ, Hubin TJ, Ramesh R. 2015. IL-24 inhibits lung cancer cell migration and invasion by disrupting the SDF-1/CXCR4 signaling axis. PLoS One. 10:e0122439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EA, Romond EH, Suman VJ, Jeong JH, Davidson NE, Geyer CE, Martino S Jr, Mamounas EP, Kaufman PA, Wolmark N. 2011. Four-year follow-up of trastuzumab plus adjuvant chemotherapy for operable human epidermal growth factor receptor 2-positive breast cancer: joint analysis of data from NCCTG N9831 and NSABP B-31. J Clin Oncol. 29:3366–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribatti D, Nico B, Ruggieri S, Tamma R, Simone G, Mangia A. 2016. Angiogenesis and Antiangiogenesis in Triple-Negative Breast cancer. Transl Oncol. 9:453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano D, Matsumoto F, Valdecanas DR, Zhao M, Molkentine DP, Takahashi Y, Hanna EY, Papadimitrakopoulou V, Heymach J, Milas L, et al. 2011. Vandetanib restores head and neck squamous cell carcinoma cells’ sensitivity to cisplatin and radiation in vivo and in vitro. Clin Cancer Res. 17:1815–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santivasi WL, Xia F. 2014. Ionizing radiation-induced DNA damage, response, and repair. Antioxid Redox Signal. 21:251–259. [DOI] [PubMed] [Google Scholar]
- Sirohi B, Philip DS, Shrikhande SV. 2014. Regorafenib in gastrointestinal stromal tumors. Future Oncol. 10:1581–1587. [DOI] [PubMed] [Google Scholar]
- Strumberg D, Schultheis B. 2012. Regorafenib for cancer. Expert Opin Investig Drugs. 21:879–889. [DOI] [PubMed] [Google Scholar]
- Su JC, Mar AC, Wu SH, Tai WT, Chu PY, Wu CY, Tseng LM, Lee TC, Chen KF, Liu CY, et al. 2016. Disrupting VEGF-A paracrine and autocrine loops by targeting SHP-1 suppresses triple negative breast cancer metastasis. Sci Rep. 6:28888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachsberger P, Burd R, Dicker AP. 2003. Tumor response to ionizing radiation combined with antiangiogenesis or vascular targeting agents: exploring mechanisms of interaction. Clin Cancer Res. 9: 1957–1971. [PubMed] [Google Scholar]
- Williams KJ, Telfer BA, Shannon AM, Babur M, Stratford IJ, Wedge SR. 2008. Inhibition of vascular endothelial growth factor signaling using Cediranib (RECENTIN; AZD2171) enhances radiation response and causes substantial physiological changes in lung tumor xenografts. Br J Radiol. 81:S21–S27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.