

Abstract
Objective
To evaluate the usefulness of biomarkers to predict the evolution of patients suspected of systemic lupus erythematosus (SLE), designated as probable SLE (pSLE), into classifiable SLE according to the American College of Rheumatology (ACR) classification criteria.
Methods
Patients suspected of SLE were enrolled by lupus experts if they fulfilled three ACR criteria for SLE and were followed for approximately 1‐3 years to evaluate transition into ACR‐classifiable SLE. Individual cell‐bound complement activation products (CB‐CAPs), serum complement proteins (C3 and C4), and autoantibodies were measured by flow cytometry, turbidimetry, and enzyme‐linked immunosorbent assay, respectively. Blood levels of hydroxychloroquine (HCQ) were measured by mass spectrometry. A multianalyte assay panel (MAP), which includes CB‐CAPs, was also evaluated. A MAP of greater than 0.8 reflected the optimal cutoff for transition to SLE. Time to fulfillment of ACR criteria was evaluated by Kaplan‐Meier analysis and Cox proportional hazards model.
Results
Of the 92 patients with pSLE enrolled, 74 had one or two follow‐up visits 9‐35 months after enrollment for a total of 128 follow‐up visits. Overall, 28 patients with pSLE (30.4%) transitioned to ACR‐classifiable SLE, including 16 (57%) in the first year and 12 (43%) afterwards. A MAP score of greater than 0.8 at enrollment predicted transition to classifiable SLE during the follow‐up period (hazard ratio = 2.72; P = 0.012), whereas individual biomarkers or fulfillment of Systemic Lupus International Collaborating Clinics criteria did not. HCQ therapy was not associated with the prevention of transition to SLE.
Conclusion
Approximately one‐third of patients with pSLE transitioned within the study period. MAP of greater than 0.8 predicted disease evolution into classifiable SLE.
Significance & Innovations.
A total of 30.4% of patients with probable lupus (three American College of Rheumatology [ACR] criteria including positive antinuclear antibodies) transitioned to ACR‐classifiable systemic lupus erythematosus (SLE) (four or more criteria) within 3 years.
Cell‐bound complement activation products measured at baseline using a multianalyte assay panel predicted transition from probable lupus to SLE.
Traditional serum biomarkers, including complement levels and anti–double‐stranded DNA antibodies, did not predict transition.
Clinical features, including fulfillment of Systemic Lupus Collaborating Clinics classification criteria, modified European League Against Rheumatism/ACR classification criteria, lupus severity index, and family history, did not predict transition.
INTRODUCTION
Numerous studies have shown the evolving nature of systemic lupus erythematosus (SLE). Often, patients present with nonspecific signs and symptoms, such as arthralgia, arthritis, myalgia, rashes, fatigue, unexplained fever, and positive antinuclear antibodies (ANAs) (1, 2, 3, 4, 5). Some patients may develop classifiable SLE within a few years; however, many patients with undifferentiated connective tissue disease may still not fulfill the classification criteria of a specific disease after 5 or 6 years (6, 7).
Anti–double‐stranded DNA (dsDNA) antibodies and low levels of the complement protein C3 in serum have been identified as potential biomarkers of “at high risk patients” (7); however, clear predictors of transition to SLE are still lacking. The ability to predict disease evolution is important, as appropriate early intervention may prevent lupus flares and more serious organ inflammation (1, 8, 9).
Lambers at al (10) reviewed studies of patients with lupus symptoms not fulfilling any of the current classification criteria for SLE and proposed a definition of incomplete lupus. In addition to positive ANAs, they identified clinical and immunologic features as risk factors for developing SLE. Interesting, a family history of autoimmune disease was also included among the risk factors.
We have shown previously that complement activation, measured reliably by cell‐bound complement activation products (CB‐CAPs)—in particular C4d bound to erythrocytes (EC4d) and to B lymphocytes (BC4d)—can be detected in SLE with greater frequency than the high titer of anti‐dsDNA antibodies and low serum complement proteins (11, 12, 13). Sensitivity and specificity of CB‐CAPs are further increased when these biomarkers are incorporated in a multianalyte assay panel (MAP) (11, 12, 14). The improved performance characteristics of CB‐CAPs and MAP compared with traditional lupus biomarkers—such as complement proteins (C3 and C4), anti‐C1q, anti‐dsDNA, anti‐Smith antibodies—have also been demonstrated in a cohort of patients suspected of SLE but not fulfilling American College of Rheumatology (ACR) classification criteria at enrollment (probable SLE [pSLE]) (14).
We also demonstrated that a MAP score of greater than 0.8 predicted disease evolution of pSLE to classifiable SLE over a short period of time (up to 18 months), whereas other biomarkers were not predictive (14). These results suggest that a combination of biomarkers, such as those that are part of the MAP, has greater ability to predict disease evolution and transition to classifiable SLE than individual analytes. In addition, fulfillment of Systemic Lupus Collaborating Clinics (SLICC) classification criteria at enrollment was not predictive of transition to SLE in our study (14), suggesting that certain biological pathways, not necessarily only historical clinical features scored in the classification criteria, drive disease evolution and new clinical manifestations of SLE.
This study adds to our previous report by continuing to follow the patients with pSLE described in Ramsey‐Goldman at al (14) to better determine whether more patients transitioned to classifiable SLE and whether the MAP score retained its ability to predict this transition. We also evaluated other clinical and laboratory features, including family history, the lupus severity index (LSI) (15, 16), and a modification of the newly proposed European League Against Rheumatism (EULAR)/ACR classification criteria (17), to determine the predictive value of these measures. In addition, we evaluated whether the use of hydroxychloroquine (HCQ) may have an impact on disease progression in this patient population.
METHODS
Study populations
Adult patients were enrolled from 2015 to 2017 in compliance with the Helsinki Declaration. Central or internal review boards at seven academic institutions approved the study, and all subjects provided informed consent. Patients were recruited from the lupus cohorts and faculty practices overseen by an experienced SLE investigator.
Patients with SLE fulfilled both the ACR (18) and the SLICC (19) classification criteria for SLE at enrollment. Patients with pSLE were enrolled if they fulfilled ANA and two additional ACR criteria (18)—irrespective of whether they fulfilled the SLICC criteria (19)—and if the investigator had a high suspicion of the diagnosis of lupus. Inclusion and exclusion criteria of the patients with pSLE have been described previously (14). In particular, renal disease was an exclusion criterion in the pSLE group. As the study was initiated before the publication of the EULAR/ACR criteria (17), these criteria were not collected. We calculated a modified score without fever and delirium and scored the other manifestations on the basis of the definition of the SLICC or ACR criterion that was more similar to the definition of the EULAR/ACR criterion.
Patients with pSLE were followed prospectively. At every study visit, the investigators determined whether patients met additional ACR criteria and the approximate date that classifiable SLE occurred, either at or before clinical evaluation. Medication use was collected at every visit. Immunosuppressants are defined as described previously (16) and include methotrexate, azathioprine, mycophenolate, belimumab, rituximab, oral ciclosporin, cyclophosphamide, or intravenous immunoglobulins.
Disease activity was evaluated with the Safety of Estrogens in Lupus Erythematosus National Assessment (SELENA) version of the SLE Disease Activity Index (SLEDAI) (20). The LSI was computed from the ACR classification criteria and subcriteria as described previously (15, 16) (see supplemental material).
Case report forms (CRFs) were reviewed and adjudicated as described previously (14). CRFs of the follow‐up visits of the subjects with pSLE were adjudicated by KK (n = 54) and AW (n = 74).
Biomarker analysis
Specimens of venous blood in EDTA‐containing tubes and serum separator tubes were collected at all visits and shipped overnight to Exagen Inc, for diagnostic immunology testing. Autoantibodies and serum complement proteins C3 and C4 were measured by enzyme‐linked immunosorbent assay and turbidimetry, respectively, as described previously (14).
Individual CB‐CAPs, EC4d and BC4d, were measured by quantitative flow cytometry and expressed as net mean fluorescence intensity (MFI), as described (12, 14, 21).
The MAP with algorithm, which includes EC4d, BC4d, ANA, anti‐dsDNA, anti‐Smith, and other lupus and nonlupus autoantibodies, was determined as described in detail elsewhere (12, 21, 22). These tests were performed using the reagents, instruments, and algorithm identical to those used for the AVISE Lupus commercial test manufactured by Exagen.
Blood levels of HCQ in patients who were prescribed this medication were measured by mass spectrometry as described (23), with the difference that volumes of reagents for the sample treatment procedure were 10‐fold higher than those reported in Qu et al (23) to take into account the larger starting volume of blood (100 μl in the present study vs. 10 μl in the previous study). One HCQ value was not measured.
Statistical analysis
Statistical comparisons were done by paired or unpaired t test, Mann‐Whitney test, Fisher’s exact test, or McNemar's test, as appropriate (GraphPad Software and MedCalc Software).
Follow‐up data of the subjects with pSLE were analyzed by Kaplan‐Meier with log‐rank test and Cox proportional hazards model (R software, version 3.1‐12) for time to fulfillment of the fourth ACR criterion. Initial decision analysis (Analyse‐it software, Capterra) (14) with Youden Index showed that MAP greater than 0.8 and EC4d greater than 20 MFI at enrollment reflected the optimal cutoffs for transition to ACR‐classifiable SLE at follow‐up; the same cutoffs were used for analysis of all follow‐up visits.
RESULTS
Study populations
A total of 246 patients were included in this study: 92 with pSLE, 53 with SLE, and the remainder with primary Sjogren syndrome and other rheumatic diseases (14).
The demographic characteristics at enrollment of the entire study population are reported in Ramsey‐Goldman at al (14), and the comparison of patients with pSLE to patients with SLE is shown in Table 1. Of the 92 patients with pSLE, 74 had one or two follow‐up visits 9 to 35 months after enrollment, for a total of 128 follow‐up visits.
Table 1.
Demographic characteristics at enrollment of subjects with SLE and pSLE (all or only those who had at least one follow‐up visit) included in the study (14)
| Disease | n | Age [Mean ± SD (Range), Yr] | Female Sex (%) | Time Since Diagnosis [Mean ± SD (Range), Yr] | Race/Ethnicity (%) | |||
|---|---|---|---|---|---|---|---|---|
| White | Black | Asian | Other | |||||
| SLE | 53 | 39 ± 14 (19‐72) | 91 |
9.6 ± 9.4 (0‐33) |
34 | 38 | 8 | 21 |
| pSLE (total) | 92 | 43 ± 15 (19‐78) | 88 |
3.6 ± 4.9 (0‐29) |
61 | 16 | 5 | 17 |
| pSLE (with at least one follow‐up visit) | 74 | 43 ± 15 (19‐78) | 89 |
4.0 ± 5.2 (0‐29) |
62 | 16 | 5 | 16 |
pSLE, probable SLE; SLE, systemic lupus erythematosus.
Other races include individuals of Hispanic ethnicity.
Disease activity in the patients with pSLE was mild at all visits. SELENA‐SLEDAI scores were available for 72, 71, and 63 patients at baseline, first, and second follow‐up visits, respectively, and average scores were 1.71, 1.20, and 1.40 at the baseline, first, and second follow‐up visits, respectively. This compares with a SELENA‐SLEDAI of 4.02 at the baseline visit of the 52 patients with SLE in this study for whom SELENA‐SLEDAI scores were available (14).
Overall, 28 patients with pSLE (30.4% of the initial cohort) transitioned to ACR‐classifiable SLE during the study. Of these, 16 (57%) transitioned in the first year, and 12 (43%) transitioned afterward. The 16 patients with pSLE who fulfilled ACR criteria within 1 year (≤365 days) accrued a total of 22 new ACR criteria, with hematological criteria being the most common. The 12 patients with pSLE who fulfilled ACR criteria after the first year (>365 days) accrued a total of 15 criteria, with hematological criteria again being the most common (Table 2). A total of 17 patients with pSLE accrued hematological criteria during the study, with 11 as the only criterion and six as one of the new criteria; 15 accrued lymphopenia, one accrued leukopenia, and one accrued both lymphopenia and leukopenia; none accrued hemolytic anemia or thrombocytopenia. During the adjudication process, the adjudicator and investigators concluded that the cytopenias were not drug related. In addition, a minority of the 17 subjects who accrued lymphopenia and/or leukopenia were on immunosuppressants (six at enrollment, five at the first follow‐up visit, and three at the second follow‐up visit). Also, the percentage of subjects on immunosuppressants remained stable during the study in the patients with pSLE who transitioned to classifiable SLE (21%, 25%, and 21% at the enrollment, first follow‐up visit, and second follow‐up visit, respectively; Table 3). A few patients were on prednisone, and there were no differences between the two groups of patients with pSLE during the study (data not shown). In addition, the use of HCQ and immunosuppressants at all visits was similar in the patients with pSLE who did and did not transition to SLE (Table 3). In particular, the use of HCQ at the enrollment visit was similar in the patients with pSLE who transitioned and those who did not transition (P = 0.603) (Table 3).
Table 2.
Clinical and laboratory features leading to the transition of pSLE to SLE
| New ACR Criteria From Baseline to 12 Mo | New ACR Criteria From 13 to 26 Mo | |
|---|---|---|
| Hematological | 10 | 7 |
| Ulcers | 4 | 2 |
| Pleuritis or pericarditis | 2 | 0 |
| Immunological | 2 | 4 |
| Arthritis | 2 | 1 |
| Discoid rash | 1 | 0 |
| Photosensitivity | 1 | 2 |
| Renal | 0 | 1 |
ACR, American College of Rheumatology; pSLE, probable SLE; SLE, systemic lupus erythematosus.
Sixteen individuals fulfilled ACR criteria within 1 year (≤365 days) (22 new ACR criteria), and 12 fulfilled ACR criteria after the first year of follow‐up (>365 days) (15 new criteria). Of note, two subjects who fulfilled ACR criteria within 1 year accrued additional criteria after this time point (an immunological criterion and ulcers); these criteria are included in the table (17 new criteria in total).
Table 3.
Use of HCQ and immunosuppressants at enrollment, first follow‐up visit, and second follow‐up visit in the patients with pSLE who fulfilled (n = 28) or did not fulfill (n = 46) ACR classification criteria during the study
| Patients With pSLE Who Transitioned During the Study | Patients With pSLE Who Did Not Transition During the Study | |
|---|---|---|
| HCQ use at enrollment, n (%) | 21 (75) | 31 (67) |
| HCQ use at first follow‐up, n (%) | 21 (75) | 33 (72) |
| HCQ use at second follow‐up, n (%) | 17 (61) | 22 (48) |
| Immunosuppressant use at enrollment, n (%) | 6 (21) | 10 (22) |
| Immunosuppressant use at first follow‐up, n (%) | 7 (25) | 15 (33) |
| Immunosuppressant use at second follow‐up, n (%) | 6 (21) | 8 (17) |
ACR, American College of Rheumatology; HCQ, hydroxychloroquine; pSLE, probable systemic lupus erythematosus.
We measured blood levels of HCQ to corroborate the finding that the use of HCQ did not prevent fulfillment of additional ACR criteria in our patient population. As reported in Table 3, 52 patients overall (70%) were taking HCQ at enrollment. Of the 51 patients for whom HCQ levels were measured, 36 (71%) had blood levels greater than 500 ng/ml and, thus, were above the minimum therapeutic level (24), whereas 15 (29%) had subtherapeutic blood levels. Of the 36 individuals with HCQ blood levels greater than 500 ng/ml, 16 (44%) transitioned to classifiable SLE; of the 15 with lower HCQ levels, 5 (33%) transitioned. This difference was not statistically significant (P = 0.543).
One patient who had a negative MAP score at enrollment and at both follow‐up visits (−2.3, −1.9, and −2.1, respectively) fulfilled the criterion of proteinuria during the study. Urine protein:creatinine ratio (UPCR) was 505 mg/g approximately 2 weeks before the last study visit. As the investigator could not attribute it to any comorbidity or medication, it was scored in the ACR and SLICC criteria but not in the SELENA‐SLEDAI. Proteinuria improved spontaneously (UPCR = 328 mg/g at the study visit), a biopsy was not performed, and the patient was not prescribed immunosuppressant medications. Interestingly, in the 2 years after the end of the study, this patient presented with mild proteinuria at several visits; however, UPCR never exceeded 500 mg/g, and the patient did not require a biopsy or antirheumatic medications other than HCQ.
Because family history has been recently included in the definition of incomplete lupus as a risk factor for the development of SLE (10), we evaluated the family history of the patients with pSLE included in our study. We found no association of family history of autoimmune rheumatic diseases (SLE, rheumatoid arthritis, psoriatic arthritis, or Sjogren syndrome; n = 30) or family history of SLE only (n = 19) (Table 4) with the transition to classifiable SLE (P = 0.470 and P = 0.785, respectively).
Table 4.
Family history of SLE and other rheumatic diseases of the patients with pSLE who fulfilled (n=28) or did not fulfill (n = 46) ACR classification criteria during the study
| Patients With pSLE Who Transitioned During the Study (n = 28) | Patients With pSLE Who Did Not Transition During the Study (n = 46) | |||
|---|---|---|---|---|
| Family history of rheumatic diseases, n (%) | 13 (46) | 17 (38) | ||
| SLE only, n | 5 | Three with first‐degree relatives with SLE | 10 | Five with first‐degree relatives with SLE |
| Two with second‐degree relatives with SLE | Five with second‐degree relatives with SLE | |||
| Rheumatoid arthritis only, n | 5 | — | 5 | — |
| Psoriatic arthritis only, n | 0 | — | 1 | — |
| SLE and other rheumatic diseases, n | 3 | One with first‐degree relative with SLE | 1 | One with first‐degree relative with SLE |
| Two with second‐degree relatives with SLE | — | |||
ACR, American College of Rheumatology; pSLE, probable SLE; SLE, systemic lupus erythematosus.
We calculated the LSI in the subjects with pSLE to evaluate whether a higher LSI at baseline was predictive of a transition to SLE. As expected, the subjects with pSLE who had at least one follow‐up visit (n = 74) had a lower LSI at enrollment than the subjects with SLE (n = 53) (average 5.09, 95% confidence interval [CI] 4.89‐5.28 vs. 6.49, 95% CI 6.10‐6.89, respectively; P < 0.0001). The LSI at enrollment was similar in the subjects who transitioned (n = 28) and those who did not (n = 46) (5.15, 95% CI 4.82‐5.49 vs. 5.04, 95% CI 4.79‐5.29, respectively; P = 0.586). The LSI did not increase significantly during the study in the 28 patients with pSLE who transitioned to SLE (average 5.34, 95% CI 4.96‐5.73 at the last follow‐up vs. 5.15, 95% CI 4.82‐5.49 at enrollment; P = 0.169) and remained lower than the LSI of the patients with SLE (P < 0.0001), consistent with the mild disease manifestations accrued by most of the patients with pSLE who transitioned.
As reported previously with fewer transitioned patients (14), fulfillment of SLICC criteria at enrollment did not predict (Table 5) and was not associated with the fulfillment of ACR criteria during the study (P = 0.085).
Table 5.
HR of variables at enrollment in predicting fulfillment of ACR classification criteria in the pSLE population
| HR | 95% CI | P Value | |
|---|---|---|---|
| SLICC criteria | 1.82 | 0.86‐3.84 | 0.116 |
| Low C3 and/or C4 | 1.02 | 0.24‐4.29 | 0.984 |
| Anti‐dsDNA (IFA) | 2.04 | 0.71‐5.91 | 0.188 |
| Positive CB‐CAPs (EC4d and/or BC4d) | 1.36 | 0.61‐3.01 | 0.454 |
| EC4d > 20 MFI | 2.07 | 0.86‐4.94 | 0.103 |
| MAP > 0.8 | 2.72 | 1.25‐5.93 | 0.012 |
CB‐CAP= cell‐bound complement activation product; CI, confidence interval; dsDNA, double‐stranded DNA; IFA, Immunofluorescence assay; HR, hazard ratio; MAP, multianalyte assay panel; MFI, mean florescence intensity; SLICC, Systemic Lupus Collaborating Clinics.
We also attempted to evaluate the association of the newly described EULAR/ACR criteria with disease progression. As described in the methods, we used a modified score that did not include fever or delirium and scored manifestations according to the definitions of ACR or SLICC criteria. As expected, the vast majority of the patients with SLE (96%) had a modified EULAR/ACR score of 10 or greater (n = 51; average 21.27, 95% CI 19.32‐23.23, median 21). The two patients with SLE with a score of less than 10 both had a score of 8 and fulfilled arthritis, hematological, immunological, and ANA criteria. Of the 92 patients with pSLE, half (n = 46) had a modified EULAR/ACR score of 10 or greater (average 12.85, 95% CI 12.25‐13.44, median 12) and half had a score of less than 10 (average 6.50, 95% CI 5.81‐7.19, median 6) at enrollment. Of the 74 patients with pSLE who had at least one follow‐up visit, the modified score of 10 or greater at enrollment was not associated with the fulfillment of ACR criteria during the study (P = 0.341). In addition, the modified EULAR/ACR score at enrollment was similar in the subjects who transitioned (n = 28) and in those who did not (n = 46) (average 10.54, 95% CI 8.89‐12.18 vs. 9.46, 95% CI 8.35‐10.56, respectively; P = 0.255).
Biomarkers commonly measured in patients with SLE did not predict transition to SLE (Table 5). Consistent with previous analysis, only MAP greater than 0.8 had significantly high hazard ratio for transition to SLE (Table 5). Figure 1 shows the Kaplan‐Meier survival curves of the two pSLE groups. Log‐rank test showed a statistically significant difference between the groups, and Cox proportional hazards regression analysis resulted in hazard ratio of 2.72 (95% CI 1.25‐5.93; P = 0.012). Anti‐dsDNA and EC4d greater than 20 MFI showed higher hazard ratios (2.04; P = 0.188 and 2.07; P = 0.103, respectively) than low complement proteins (1.02; P = 0.984); however, MAP greater than 0.8 outperformed all the other measured biomarkers (Table 5). Logistic regression analysis confirmed that MAP greater than 0.8 at enrollment was significantly associated with the fulfillment of ACR criteria during the study (odds ratio [OR] = 0.59; P = 0.0314), whereas MAP of 0.8 or less was significantly associated with remaining pSLE during the study (OR = 3.11; P < 0.0001).
Figure 1.
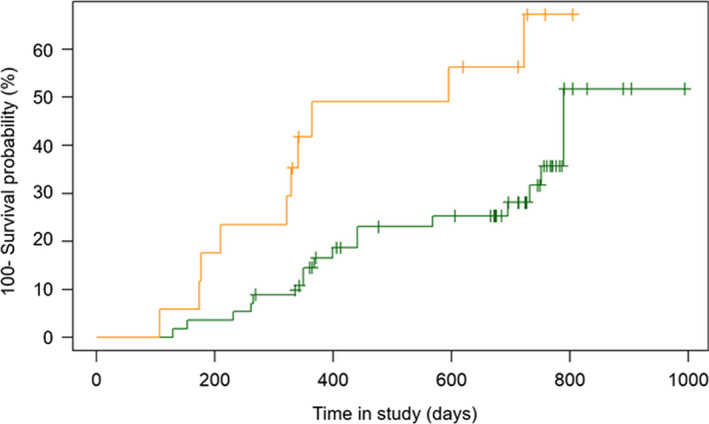
Kaplan‐Meier survival estimates in the cohort of patients with probable systemic lupus erythematosus (pSLE) who had at least a follow‐up visit during the study. Kaplan‐Meier survival curves showing the percentage (100‐survival probability) of patients with pSLE who fulfilled American College of Rheumatology classification criteria during the study. Data for the 73 patients for whom a multianalyte assay panel (MAP) score could be calculated are plotted. The orange line represents the 17 patients with pSLE with a MAP > 0.8 at enrollment, and the green line represents the 56 patients with pSLE with a MAP ≤ 0.8 at enrollment.
DISCUSSION
We continued to follow the patients with pSLE described in Ramsey‐Goldman et al (14) to further evaluate transition over time to classifiable SLE based on ACR classification criteria. During this extension study, eight more patients with pSLE transitioned to classifiable SLE, making the total 28 in all. Slightly more than half of the patients with pSLE transitioned in the first year (16 patients; 57%) and the reminder (12 patients; 43%) transitioned after that time up to 35 months. This implies that longer follow‐up should not be neglected in this patient population.
Although disease activity was mild at enrollment and during the study, more than one‐third of patients with pSLE acquired one or more additional ACR criteria during the study. Hematological criteria (lymphopenia and leukopenia) were most often the fourth criterion fulfilled by the patients who transitioned to classifiable SLE (Table 2). The use of immunosuppressants at enrollment was not different between the patients with pSLE who acquired new criteria and those who did not (Table 3). This finding, combined with the adjudication of each visit, suggests that the cytopenias were likely due to the disease rather than the use of medications.
Also, the use of HCQ at enrollment was similar in the two subgroups of patients with pSLE (Table 3), suggesting that prescription and use of HCQ did not prevent the occurrence of new disease manifestations in this group of patients, even when blood levels of HCQ were above the therapeutic levels (>500 ng/ml). Numerous studies have demonstrated the beneficial effects of HCQ in preventing flares, decreasing thrombosis, improving lipid levels, treating joint and skin manifestations, and overall decreasing organ damage, lowering cardiovascular events, and improving survival in established SLE over time (24). Our data show that patients suspected of SLE and fulfilling three ACR classification criteria acquired new classification criteria over a short period of time even when appropriately treated with HCQ and with HCQ reaching blood levels that are considered therapeutic; however, new disease manifestations were not severe (Table 2).
The benign disease course in our pSLE cohort is substantiated by the low SELENA‐SLEDAI score at all visits and by the LSI, which is calculated by weighting ACR criteria and subcriteria (see supplemental material). Mucocutaneous involvement, arthritis, and hematological criteria (excluding haemolytic anemia) have a negative or very low positive score in the calculation of the LSI. In our pSLE group that transitioned to SLE, the LSI remained similar at the end of the study (P = 0.169) and remained lower than the LSI of the SLE cohort (P < 0.0001). In addition, the LSI at enrollment was similar in the subjects who transitioned and those who did not (P = 0.586), suggesting that higher severity of disease in pSLE did not predispose to the accrual of additional ACR criteria, at least within approximately 1 to 3 years.
Only one patient developed renal disease; however, proteinuria was not severe and did not require a biopsy or treatment with immunosuppressants either during the study or after. Interestingly, this patient had a negative MAP score throughout the study. Thus, in this patient, fulfillment of a new important classification criterion could not be predicted by the MAP score at enrollment. It remains unclear whether this one episode of increased proteinuria reflects lupus nephritis.
Although overall the patients with pSLE in our study did not acquire severe disease manifestations, identification at an early stage of patients more likely to have true incipient SLE can be important for appropriate patient management.
In agreement with previous data (14), MAP greater than 0.8 predicted fulfillment of ACR criteria during the study, whereas biomarkers commonly measured in SLE, such as anti‐dsDNA and complement proteins C3 and C4, did not. Also, EC4d greater than 20 MFI did not reach statistical significance in this analysis (Table 5), indicating that a panel provides more information than individual biomarkers.
Confirming previous results (14), fulfillment of SLICC criteria at enrollment was not associated with fulfillment of additional ACR criteria during the study. A modification of the EULAR/ACR classification was also not associated with disease progression. The inability to score fever in these criteria represents a limitation of this analysis, as does the use of the ACR and SLICC definitions for scoring. However, the observation that more patients with pSLE had a modified EULAR/ACR score of 10 or more than fulfilled SLICC criteria at enrollment (50% vs. 38%) (14) is consistent with the high sensitivity of the new criteria. The modified EULAR/ACR score was not associated with disease progression during the study (P = 0.255). Likewise, a modified score of 10 or greater at enrollment was not associated with transition, which is in line with the results obtained with the SLICC criteria (P = 0.341 and P = 0.085, respectively). These data strengthen our hypothesis that certain biological pathways, rather than only historical clinical features scored in the classification criteria, drive disease evolution and new clinical manifestations of SLE.
We also evaluated whether family history of autoimmune diseases or specifically SLE was associated with the transition to classifiable SLE and did not find a significant association. Thus, although family history is important and has been proposed as a risk factor for the development of SLE in patients with incomplete SLE (10), it did not associate with transition to classifiable SLE in our cohort.
This study has several limitations, including the small sample size and the loss to follow‐up. Although 18 patients with pSLE were lost to follow‐up, there were no differences between the patients who remained in the study and those who did not, as described in Ramsey‐Goldman at al (14). Although the relatively short follow‐up period is a limitation, more than one‐third of patients fulfilled new classification criteria during this time, and patients with MAP greater than 0.8 at enrollment did so sooner than patients with a lower score. Of note, individual biomarkers did not predict disease progression. Another limitation is that we did not collect data on the newly developed EULAR/ACR classification criteria because they were published after we had collected baseline data on the patients in the study. Therefore, we used an unvalidated modified criteria based on the data we had collected with the ACR and SLICC criteria. Finally, our results may not be generalizable to all incomplete lupus. In fact, the patients with pSLE in our study were very specifically selected as having three ACR criteria (ANA and other two criteria) in addition to a high likelihood of having lupus, as judged by a lupus expert. Thus, our results are important for this subset of patients with incomplete lupus.
In conclusion, these data show that, over time, additional patients transitioned from pSLE to SLE by ACR criteria compared with our previous report. Thus, these data confirm our previous findings (14) and support the hypothesis that biological processes that lead to complement activation and/or formation of autoantibodies play a role in disease progression in patients suspected of SLE and fulfilling three ACR classification criteria. Recognition of these patients and institution of early appropriate management may potentially slow disease progression (9, 25).
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Roberta Alexander had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Ramsey‐Goldman, Alexander, Arriens, Narain, Massarotti, Wallace, Collins, Saxena, Putterman, Weinstein.
Acquisition of data
Ramsey‐Goldman, Alexander, Arriens, Narain, Massarotti, Wallace, Collins, Saxena, Putterman, Brady, Kalunian, Weinstein.
Analysis and interpretation of data
Ramsey‐Goldman, Alexander, Conklin, Brady, Kalunian, Weinstein.
ROLE OF THE STUDY SPONSOR
Exagen, Inc, had no role in the study design or in the collection, analysis, or interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript for publication. Publication of this article was not contingent upon approval by Exagen, Inc.
The research team at Exagen Inc. facilitated the study design along with the investigators. The investigators independently collected the data and the case adjudication was done independently. The research team at Exagen Inc. analyzed the data and with the investigators, interpreted the results. The manuscript was primarily written and critiqued by the senior investigators (RR‐G, RVA, AW) and the decision to submit the manuscript was made by all the authors and was approved by Exagen Inc.
Supporting information
Supplementary Material
Acknowledgments
The authors thank the research coordinators and the patients who participated in the study. They also thank Armida Sace, Rowena LaFon, and JoAnne Ligayon for technical assistance in biomarker analysis. They thank Thierry Dervieux for his role in the study design and Claudia Ibarra for the management and supervision of the clinical laboratory.
Supported by Exagen Inc.
Drs. Alexander, Conklin, and Mr. Brady are employees of Exagen Inc. Dr. Weinstein is a consultant to Exagen Inc. All other authors have received research support from Exagen Inc. No other disclosures relevant to this article were reported.
REFERENCES
- 1. Robertson JM, James JA. Preclinical systemic lupus erythematosus. Rheum Dis Clin North Am 2014;40:621–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doria A, Mosca M, Gambari PF, Bombardieri S. Defining unclassifiable connective tissue diseases: Incomplete, undifferentiated, or both? [Editorial]. J Rheumatol 2005;32:213–5. [PubMed] [Google Scholar]
- 3. Doria A, Zen M, Canova M, Bettio S, Bassi N, Nalotto L, et al. SLE diagnosis and treatment: when early is early. Autoimmun Rev 2010;10:55–60. [DOI] [PubMed] [Google Scholar]
- 4. Aberle T, Bourn RL, Munroe ME, Chen H, Roberts VC, Guthridge JM, et al. Clinical and serological features in patients with incomplete lupus classification versus systemic lupus erythematosus patients and controls. Arthritis Care Res 2017;69:1780–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mosca M, Costenbader KH, Johnson SR, Lorenzoni V, Sebastiani GD, Hoyer BF, et al. Brief report: how do patients with newly diagnosed systemic lupus erythematosus present? A multicenter cohort of early systemic lupus erythematosus to inform the development of new classification criteria. Arthritis Rheumatol 2019;71:91–8. [DOI] [PubMed] [Google Scholar]
- 6. Bodolay E, Csiki Z, Szekanecz Z, Ben T, Kiss E, Zeher M, et al. Five‐year follow‐up of 665 Hungarian patients with undifferentiated connective tissue disease (UCTD). Clin Exp Rheumatol 2003;21:313–20. [PubMed] [Google Scholar]
- 7. Costenbader KH, Schur PH. We need better classification and terminology for “people at high risk of or in the process of developing lupus”. Arthritis Care Res 2015;67:593–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bruce IN, O’Keeffe AG, Farewell V, Hanly JG, Manzi S, Su L, et al. Factors associated with damage accrual in patients with systemic lupus erythematosus: results from the Systemic Lupus International Collaborating Clinics (SLICC) Inception Cohort. Ann Rheum Dis 2015;74:1706–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruce IN, Urowitz M, van Vollenhoven R, Aranow C, Fettiplace J, Oldham M, et al. Long‐term organ damage accrual and safety in patients with SLE treated with belimumab plus standard of care. Lupus 2016;25:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lambers WM, Westra J, Jonkman MF, Bootsma H, de Leeuw K. Incomplete systemic lupus erythematosus: what remains after application of American College of Rheumatology and Systemic Lupus International Collaborating Clinics criteria? [Review ]. Arthritis Care Res 2020;72:607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalunian KC, Chatham WW, Massarotti EM, Reyes‐Thomas J, Harris C, Furie RA, et al. Measurement of cell‐bound complement activation products enhances diagnostic performance in systemic lupus erythematosus. Arthritis Rheum 2012;64:4040–7. [DOI] [PubMed] [Google Scholar]
- 12. Putterman C, Furie R, Ramsey‐Goldman R, Askanase A, Buyon JP, Kalunian K, et al. Cell‐bound complement activation products in systemic lupus erythematosus: comparison with anti‐double‐stranded DNA and standard complement measurements. Lupus Sci Med 2014;1:e000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramsey‐Goldman R, Li J, Dervieux T, Alexander RV. Cell‐bound complement activation products in SLE. Lupus Sci Med 2017;4:e000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramsey‐Goldman R, Alexander RV, Massarotti EM, Wallace DJ, Narain S, Arriens C, et al. Complement activation in patients with probable systemic lupus erythematosus and ability to predict progression to American College of Rheumatology–classified systemic lupus erythematosus. Arthritis Rheumatol 2020;72:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bello GA, Brown MA, Kelly JA, Thanou A, James JA, Montgomery CG. Development and validation of a simple lupus severity index using ACR criteria for classification of SLE. Lupus Sci Med 2016;3:e000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arriens C, Alexander RV, Narain S, Saxena A, Collins CE, Wallace DJ, et al. Cell‐bound complement activation products associate with lupus severity in SLE. Lupus Sci Med 2020;7:e000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey‐Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol 2019;71:1400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40:1725. [DOI] [PubMed] [Google Scholar]
- 19. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the systemic lupus international collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012;64:2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petri M, Kim MY, Kalunian KC, Grossman J, Hahn BH, Sammaritano LR, et al. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005;353:2550–8. [DOI] [PubMed] [Google Scholar]
- 21. Dervieux T, Conklin J, Ligayon JA, Wolover L, O’Malley T, Alexander RV, et al. Validation of a multi‐analyte panel with cell‐bound complement activation products for systemic lupus erythematosus. J Immunol Methods 2017;446:54–9. [DOI] [PubMed] [Google Scholar]
- 22. Wallace DJ, Silverman SL, Conklin J, Barken D, Dervieux T. Systemic lupus erythematosus and primary fibromyalgia can be distinguished by testing for cell‐bound complement activation products. Lupus Sci Med 2016;3:e000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qu Y, Brady K, Apilado R, O’Malley T, Reddy S, Chitkara P, et al. Capillary blood collected on volumetric absorptive microsampling (VAMS) device for monitoring hydroxychloroquine in rheumatoid arthritis patients. J Pharm Biomed Anal 2017;140:334–41. [DOI] [PubMed] [Google Scholar]
- 24. Durcan L, Clarke WA, Magder LS, Petri M. Hydroxychloroquine blood levels in systemic lupus erythematosus: clarifying dosing controversies and improving adherence. J Rheumatol 2015;42:2092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lamichhane D, Weinstein A. Probable systemic lupus erythematosus with cell‐bound complement activation products (CB‐CAPS). Lupus 2016;25:1050–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material