

Abstract
R‐loops represent an abundant class of large non‐B DNA structures in genomes. Even though they form transiently and at modest frequencies, interfering with R‐loop formation or dissolution has significant impacts on genome stability. Addressing the mechanism(s) of R‐loop‐mediated genome destabilization requires a precise characterization of their distribution in genomes. A number of independent methods have been developed to visualize and map R‐loops, but their results are at times discordant, leading to confusion. Here, we review the main existing methodologies for R‐loop mapping and assess their limitations as well as the robustness of existing datasets. We offer a set of best practices to improve the reproducibility of maps, hoping that such guidelines could be useful for authors and referees alike. Finally, we propose a possible resolution for the apparent contradictions in R‐loop mapping outcomes between antibody‐based and RNase H1‐based mapping approaches.
Keywords: DNA hybrid, DRIP, R‐loops, RNA, RNase H1, S9.6 antibody
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination; RNA Biology
With the increasing appreciation of R‐loops as frequent non‐B DNA structures with large impact on genome function and stability, this commentary proposes general guidelines for their reproducible study with antibody or RNase H1‐based approaches.
Glossary
- DRIP
DNA‐RNA Immunoprecipitation. An S9.6 antibody‐based mapping strategy of DNA:RNA hybrids. Various iterations of the method (DRIPc, sDRIP, qDRIP) have been published that differ in the way the genomic DNA is sheared before immunoprecipitation or whether the DNA moiety or the RNA moiety of the hybrid is sequenced
- dRNase H1
defective RNase H1. a catalytically inactive form of RNase H1 able to recognize but not to process RNA:DNA hybrids
- R‐ChIP
dRNase H1‐mediated Chromatin Immunoprecipitation. A DNA:RNA hybrid mapping strategy that relies on the ability of an inactive RNase H1 to recognize but not process DNA:RNA hybrids
- RDIP
RNA‐DNA Immunoprecipitation. An alternative S9.6 antibody‐based mapping strategy that differs from DRIP in that it includes a first step of nuclei purification. RDIP also only sequence the RNA moiety of DNA:RNA hybrids
- RNase H1
an endonuclease that degrades the RNA moiety of RNA:DNA hybrids
- S9.6
a monoclonal antibody directed against DNA:RNA hybrids
- SMRF‐seq
Single‐Molecule R‐loop Footprinting. Bisulfite‐based mapping of the extruded single strand DNA within an R‐loop adapted for single‐molecule long‐read sequencing technologies
Introduction
When a single‐stranded RNA invades the DNA double helix and hybridizes with a complementary template DNA strand, a three‐stranded structure called an R‐loop is formed. Although some evidence supports the idea that R‐loops could sometimes form in trans (Wahba et al, 2013; Cloutier et al, 2016; Ariel et al, 2020), it is widely accepted that the vast majority of R‐loops form co‐transcriptionally in cis behind the advancing RNA polymerase (RNAP), and that high rates of transcription increase the likelihood of their formation. For example, the stimulation of transcription at intronic immunoglobulin switch regions upon cytokine‐induced B cell activation triggers the formation of R‐loops that contribute to antibody class‐switch recombination (CSR) (Yu et al, 2003; Yu & Lieber, 2019). The involvement of R‐loops in CSR was one of the first described examples of how “programmed” R‐loops can participate in normal cell physiology. However, work from a number of laboratories has also suggested that minimizing R‐loop formation to low levels is important to preserve genome integrity, particularly during DNA replication. As thoroughly discussed by a number of recent reviews (Crossley et al, 2019; García‐Muse & Aguilera, 2019; Niehrs & Luke, 2020), dedicated R‐loop‐destabilizing enzymes, proper packaging of nascent RNA, proper assembly of chromatin, and the relief of transcription‐associated topological stress have been implicated in limiting the formation and stability of R‐loops, and thereby maintaining genome stability.
Current models propose that a variety of genetic and/or pharmacological perturbations can lead to the formation of so‐called “harmful” R‐loops (García‐Muse & Aguilera, 2019). Despite their rise in prominence, such harmful R‐loops have not yet been precisely defined at the genomic level, and major questions remain concerning their molecular characteristics. Do they arise de novo in regions that normally do not form R‐loops (“aberrant” R‐loops)? Are they normal R‐loops that now arise at higher frequency (“excessive” R‐loops)? Do they form at the “wrong” time with respect to the cell cycle (“unscheduled” R‐loops)? Do they possess a longer dwell time [“persistent” R‐loops (García‐Rubio et al, 2018; Costantino & Koshland, 2018)] or are they otherwise larger in size (“extended” R‐loops)? Addressing these questions will require efficient and reproducible R‐loop‐mapping procedures at both the population and single‐molecule levels in a number of carefully controlled cellular models of proposed R‐loop dysfunction. Technical improvements that permit the measurement of dynamic R‐loop turnover will also be required to distinguish and quantify stable and transient R‐loops.
In addition to this, a key challenge in the field is to disentangle the direct contribution of R‐loops to a phenotype of interest from the contribution of perturbed nascent transcription itself. This challenge is all the more relevant, in that most known instances of genome‐destabilizing R‐loops are observed upon alterations of co‐transcriptional RNA processes such as splicing, termination, and nuclear export [for example, see Huertas & Aguilera, 2003; Paulsen et al, 2009; Wahba et al, 2011; Stirling et al, 2012)]. A large body of work shows that such processes are intimately coupled to transcription elongation through a variety of feedback mechanisms (Zhong et al, 2009; Hsin & Manley, 2012; Bentley, 2014; Herzel et al, 2017; Core & Adelman, 2019). Thus, altered transcription resulting from defective co‐transcriptional processing might itself affect genome stability independently of R‐loops (Salas‐Armenteros et al, 2019). This key challenge will require the development of methods to precisely and specifically manipulate R‐loop levels. Currently, the main strategy to manipulate R‐loop levels is to over‐express RNase H1, an endonuclease that degrades the RNA moiety of RNA:DNA hybrids (Hyjek et al, 2019), in the nucleus, but the results of this approach need to be carefully interpreted, as discussed below.
As outlined below, we came to the conclusion that the elucidation of the physiological and pathological roles of R‐loops currently suffers from a lack of available datasets describing “harmful” R‐loops, from a lack of clear tools to manipulate R‐loops in cells, and from significant inconsistencies between the results of different R‐loop mapping studies, sometimes even in the context of similar mapping strategies. In our opinion, this highlights the urgent need for objective, unbiased criteria to increase rigor and reproducibility. Here we propose simple guidelines to evaluate the quality of R‐loop maps and offer possible explanations to harmonize and reconcile apparent inconsistencies observed between R‐loop mapping methods.
Commonly used approaches to map R‐loops
As discussed in previous reviews (Vanoosthuyse, 2018; Crossley et al, 2019), two main types of methods are currently used to provide a population‐average view of R‐loop distribution and abundance. The first relies on the so‐called S9.6 antibody that displays a strong affinity for the RNA:DNA hybrid moiety of R‐loops (Boguslawski et al, 1986), permitting DNA:RNA immunoprecipitation (DRIP) followed by high‐throughput DNA sequencing (Ginno et al, 2012). Various iterations of the DRIP methodology have now been published and can achieve R‐loop mapping at various degrees of resolution and strandedness through the sequencing of DNA‐ or RNA‐derived R‐loop signals (Nojima et al, 2018; Sanz & Chédin, 2019; preprint: Smolka et al, 2020; Crossley et al, 2020). The second approach relies on the often transient and inducible expression of a tagged, catalytically inactive form of RNase H1 (called thereafter dRNase H1 for defective RNase H1), which is able to recognize but not to process RNA:DNA hybrids (Wu et al, 2001). Mapping in vivo dRNase H1 binding sites by chromatin immunoprecipitation (ChIP) approaches (in this case referred to as R‐ChIP) was proposed as an alternative method for revealing R‐loop patterns (Chen et al, 2017). dRNase H1 has also been used, in a CUT&RUN‐based approach termed “MapR”, for targeting micrococcal nuclease (MNase) to R‐loops, allowing to solubilize them out of chromatin (Yan et al, 2019).
A third approach, which was developed first but whose application is not yet as widespread, maps the single‐stranded DNA moiety of R‐loops using bisulfite‐induced deamination of exposed cytosines. Under non‐denaturing conditions, R‐loops are associated with strand‐specific and RNase H‐sensitive bisulfite‐mediated patches of C‐to‐T base conversions (Yu et al, 2003; Malig et al, 2020). This approach not only demonstrates the presence of genuine three‐stranded R‐loop structures, it also provides an accurate view of the position of individual R‐loop footprints at single‐molecule resolution. Owing to recent improvements with the single‐molecule R‐loop footprinting (SMRF‐seq) strategy, this approach is well‐suited to allow R‐loop characterization on multi‐kilobase‐size, single‐molecule amplicons at ultra‐deep coverage and at a range of specific loci (Malig et al, 2020). However, it remains currently less amenable to genome‐wide studies than the other two alternatives.
Reconciling discrepancies in R‐loop distribution patterns obtained by S9.6‐ and dRNase H1‐based approaches
At a broad level, RNase H1‐based and S9.6‐based approaches produce strikingly disparate R‐loop distribution patterns. DRIP‐based studies consistently show that R‐loops principally form along transcribed gene bodies, with hotspots along GC‐skewed CpG island promoters and terminal genic regions, particularly for closely spaced genes (Sanz et al, 2016). By contrast, R‐ChIP‐based studies identify only a subset of the loci highlighted by S9.6‐based methods, with a clear bias toward G‐rich loci associated with promoter‐proximal pausing of RNA polymerase II (Chen et al, 2017). Only few dRNase H1 binding sites are found in gene bodies, and terminal genic regions are depleted in the datasets. Even at loci identified by both strategies, the position of the signal can differ strikingly. For example, at the classic R‐loop‐forming gene ACTB, one report showed formation of R‐loops primarily in the 5′‐end (first intron) of the gene using dRNase H1‐based approaches, and primarily in the 3′‐end of the gene using S9.6‐based approaches (Tan‐Wong et al, 2019). By contrast, another study reported association of dRNase H1 with the 3′‐end of the ACTB gene (Nguyen et al, 2017), while several independent S9.6‐based DRIP studies indicated R‐loop formation throughout the ACTB transcription unit (Skourti‐Stathaki et al, 2011; Sanz et al, 2016; Crossley et al, 2020). Thus, the broad dissimilarities between dRNase H1‐based and S9.6‐based R‐loop patterns are further compounded by apparent discrepancies within each approach, which may be caused by technical variability as well as insufficient systematic quantitative and qualitative controls. Below, we propose guidelines to improve reproducibility and standardize approaches, in order to reduce the current issues with variability, particularly as they relate to the more commonly used DRIP‐based methods.
It has been argued that RNase H1‐based approaches are more specific than S9.6‐based approaches because RNase H1 exclusively hydrolyses RNA:DNA hybrids in vitro. While the specificity of RNase H1’s catalytic activity toward RNA:DNA hybrids is not in question, we note that the more pertinent parameter when using a catalytically inactive dRNase H1 protein as an R‐loop reporter is its binding affinity (K d). We could not find data quantifying the affinity of the full‐length human RNase H1 for RNA:DNA hybrids, let alone for dRNase H1. We note however that the isolated hybrid‐binding domain (HBD) of human RNase H1 showed a 0.2 µM K d for RNA:DNA hybrids (Nowotny et al, 2008), nearly a thousand‐fold higher than S9.6 (0.6 nM; Phillips et al, 2013). Furthermore, S9.6 and the RNase H1 HBD appear similarly challenged in their ability to discriminate between RNA:DNA hybrid and double‐stranded RNA (dsRNA) substrates, with each showing only a roughly 20‐fold lower affinity for dsRNA. Based on the limited biochemical studies available, it is therefore unclear if dRNase H1 is intrinsically more specific for RNA:DNA hybrids than S9.6.
A second argument put forward in favor of dRNaseH1‐based approaches is that they capture sites of native R‐loop formation in cells, while DRIP‐based methods only query those ex vivo, after nucleic acid extraction and deproteinization. A follow‐up argument is that R‐loops may artificially and spontaneously form during DNA extraction, explaining their increased recovery in DRIP‐based approaches. However, as previously argued (Sanz & Chédin, 2019; Malig et al, 2020), artificial R‐loop formation ex vivo is very unlikely to occur due to strong energy barriers and even less likely to generate the robust patterns of long R‐loop hotspots that are commonly observed. On the contrary, the opposite problem can clearly be observed, namely loss of short, unstable R‐loops, which are stabilized in vivo by DNA topology, upon DNA fragmentation prior to DRIP (Sanz & Chédin, 2019; Stolz et al, 2019). It is also worth noting that the over‐expression of dRNase H1 may exert a dominant‐negative effect on catalytically active endogenous RNase H1 and thereby interfere with the physiological turnover and stability of R‐loops (Kabeche et al, 2018; Li et al, 2020). This could potentially alter the distribution patterns of R‐loops and contribute to discrepancies with S9.6‐based approaches.
A recent in‐depth comparison over 24 human single‐gene loci showed an excellent agreement between bisulfite‐based and S9.6‐based techniques, and a poor agreement with dRNase H1‐based approaches (Malig et al, 2020). The results of bisulfite footprinting experiments have been further validated by direct AFM‐based visualization of R‐loops (Carrasco‐Salas et al, 2019) and by mathematical modeling (Stolz et al, 2019). Importantly, the use of non‐denaturing bisulfite approaches independent of S9.6 enrichment confirmed that R‐loops are a prevalent feature of the 3′‐end of genes, although they remain largely undetected by dRNase H1 (Malig et al, 2020). The specificity of DRIP‐based signals is further demonstrated by their sensitivity to exogenous RNase H treatment, something that is not usually tested in the context of dRNase H1‐based strategies (with the notable exception of fission yeast studies, where dRNase H1 signals were shown to be sensitive to the over‐expression of the catalytically active RnhA enzyme from Escherichia coli (Rivosecchi et al, 2019; Legros et al, 2014)). Overall, the bulk of these observations support that loci identified by S9.6‐based approaches but missed by dRNase H1 correspond to genuine R‐loops and strongly suggest that S9.6‐ and bisulfite‐based strategies provide a more exhaustive distribution pattern for R‐loops. It is possible, however, that these approaches might miss a subset of small R‐loops that are stabilized in cells by negative topological stress. We therefore suggest that alternative explanations for the smaller number of R‐loops identified by RNase H1‐based approaches should be considered, recognizing the biological interest of elucidating patterns of RNase H1 binding in vivo.
One possibility is that RNase H1‐based strategies only identify a subset of RNA:DNA hybrids formed in specific contexts. In support of this, we note that the dRNase H1‐based approach proved helpful to characterize R‐loop formation at RNA Polymerase III‐transcribed genes in fission yeast, but was a poor reporter of R‐loop formation elsewhere in the genome (Legros et al, 2014). One hypothesis to account for the possibility that dRNase H1 might capture a specific class of R‐loops that is poorly identified by other strategies is that RNase H1 may require ancillary factors to efficiently recognize R‐loops in vivo. Consistent with this, the ssDNA‐binding protein RPA was shown to be necessary for RNase H1 to recognize and process R‐loops (Petzold et al, 2015; Nguyen et al, 2017). Thus, accordingly, RNase H1 may recognize a subclass of RPA‐bound R‐loops. This, in turn, suggests that many R‐loops, for reasons that remain unclear, may escape RPA binding and therefore, RNase H1. There is currently no data available to evaluate the genome‐wide overlap between the localizations of RPA, dRNase H1, and R‐loops, which would determine which R‐loops are more likely to be targeted by the RPA/RNase H1 complex.
A second, complementary hypothesis is that dRNase H1 may have a particular specificity for R‐loops associated with RNA polymerase pausing. dRNase H1 recognizes highly G‐skewed loci associated with promoter‐proximal RNAP pausing (Chen et al, 2017; Tan‐Wong et al, 2019). Furthermore, dRNase H1 recruitment to promoters is functionally and dynamically coupled to RNAP pausing (Chen et al, 2017). It is thus plausible that dRNaseH1 is particularly attracted to paused RNAPs to deal with short R‐loops that may form there. Paused transcription complexes, which occur at thousands of genes (Adelman & Lis, 2012; Core & Adelman, 2019), represent an ideal environment for R‐loop formation for several reasons. First, the relative stability of paused complexes provides a kinetic window of opportunity for the RNA to engage with its template DNA. Second, this engagement is further facilitated by the confluence of two R‐loop‐favoring characteristics: (i) optimal sequence (sequences located immediately downstream of the transcription start sites are highly GC‐skewed (Ginno et al, 2012; Hartono et al, 2015)); and (ii) proximity to the nascent RNA transcript's 5′‐end (Roy et al, 2008; Chen et al, 2017; Stolz et al, 2019). Finally, the capping enzyme, which is immediately bound to the emerging transcript, was proposed to facilitate R‐loop formation, at least in vitro (Kaneko et al, 2007). The hypothesis that short R‐loops formed around paused transcription complexes represent a major landing pad for dRNAseH1 is attractive. We believe that such short R‐loops would be difficult to detect via DRIP‐based or non‐denaturing bisulfite‐based approaches. This might explain why R‐ChIP and DRIPc‐seq signals do not overlap around transcription start sites (TSS) (Fig 1A). R‐loops associated with paused RNA polymerases are expected to be small (~ 60 bp at most given the lengths of RNA transcripts at pause sites (Adelman & Lis, 2012)) and therefore probably unstable during genome fragmentation in DRIP‐based approaches. Furthermore, their high G‐content on a short displaced strand would make them invisible to bisulfite conversion approaches, which require stretches of cytosines to be able to make confident calls. As a result, such small promoter‐associated R‐loops may be best captured by in situ approaches relying on crosslinking such as R‐ChIP. Alternatively, we note that paused RNA polymerases are often backtracked (Sheridan et al, 2019), and it was recently proposed that small so‐called “anterior R‐loops” can form ahead of backtracked RNA polymerases (Zatreanu et al, 2019). It is possible that dRNase H1 and RPA have a particular affinity for those “anterior R‐loops” (Fig 1B). Additional work will be necessary to clarify the mechanisms underlying RNase H1 targeting and elucidate why dRNase H1 and/or RPA are relatively depleted from longer “posterior” R‐loops commonly identified by S9.6‐based and other approaches. Overall, we suggest that dRNase H1 may be primarily driven to bind to short, possibly RPA‐bound, R‐loops that preferentially form around promoter‐proximal regions due to RNAP pausing. We note that the physiological relevance of RNase H1 binding to mechanisms of pause release, if any, remains unclear.
Figure 1. Discrepancies in R‐loop distribution patterns obtained by S9.6‐ and dRNase H1‐based approaches.
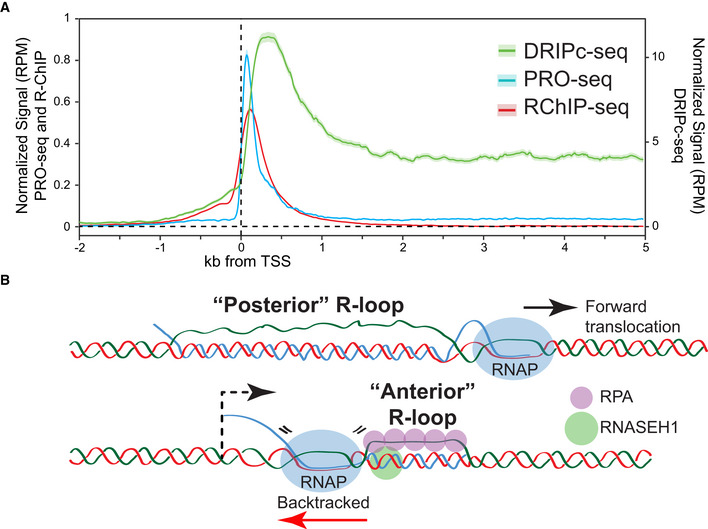
(A) Population‐average distributions of dRNase H1 (measured by R‐ChIP‐seq in HEK293 cells (Chen et al, 2017)), nascent transcription (measured by PRO‐seq in HeLa cells), and R‐loops (measured by DRIPc‐seq in HeLa cells) around the TSS of genes positive for R‐ChIP‐seq signals. (B) Schematic of “posterior” R‐loops and “anterior” R‐loops that may form around the TSS of genes upon RNA polymerase backtracking. Speculative binding of RPA and RNASEH1 to posterior R‐loops are shown.
Weaknesses of S9.6‐based approaches and practical guidelines for improved reproducibility and rigor
While the S9.6 antibody is currently the best existing tool to map and quantify RNA:DNA hybrids, it is nevertheless important to be aware of its limitations. For instance, the affinity of S9.6 for its targets might be influenced by their sequence, at least as documented for short synthetic RNA:DNA hybrids in vitro (König et al, 2017). While this had no measurable impact on the composition of sequences recovered in DRIP data (Sanz & Chédin, 2019), the recovery of short hybrids, could, in principle, be affected by such preference. S9.6 is commonly used in three different types of approaches: imaging, dot blots, and DRIP. Each of these approaches presents a particular set of challenges, as detailed below.
Risk of inaccuracies associated with S9.6‐based imaging applications
S9.6 has probably been used most frequently for visualization and quantification of in situ RNA:DNA hybrid formation using immunofluorescence (IF) microscopy. Mounting evidence suggests that S9.6‐based imaging approaches are challenged by the significant residual affinity of S9.6 for dsRNA species (Phillips et al, 2013; Hartono et al, 2018; Tan‐Wong et al, 2019). Given that RNA‐derived materials are more abundant in cells than RNA:DNA hybrid‐derived signals by orders of magnitude, this residual affinity is highly problematic and might interfere with the reliable quantification of genuine RNA:DNA hybrids in situ.
While RNA:DNA hybrids are expected to show an exclusively nuclear and mitochondrial (as well as—in plants—chloroplastic) distribution, most studies report that the majority of the IF signals detected by S9.6 antibodies is cytoplasmic. Recent results (preprint: Smolka et al, 2020) show that, under normal conditions in human cells, the S9.6 signal does not overlap with mitochondria, in agreement with prior work (Koo et al, 2015; Silva et al, 2018). In addition, this cytoplasmic signal was found not to be sensitive to exogenous RNase H treatment (Silva et al, 2018; preprint: Smolka et al, 2020), even under conditions where RNase H was proven effective against transfected in vitro labeled RNA:DNA hybrids (preprint: Smolka et al, 2020). Instead, the sensitivity of the cytoplasmic signal to RNA‐specific ribonucleases T1 and III implicated that the majority of the S9.6 signal in standard IF microscopy studies derives from RNA, but not from RNA:DNA hybrids (Silva et al, 2018; preprint: Smolka et al, 2020). These observations do raise substantive concerns about the results of past S9.6‐based imaging studies. The odds that S9.6 IF microscopy have led to inaccurate claims of R‐loop fluctuations are significant and further enhanced by the fact that many of the conditions tested involved perturbations of co‐ and post‐transcriptional processes. Such perturbations could have profound effects on nascent transcription and RNA transport, triggering cascading effects on mRNA pools and their translation, and therefore potentially affecting the levels and distribution of many types of RNAs in both the cytoplasm and the nucleus. Such changes in RNA quantities and sub‐cellular distribution may significantly confound the detection and quantification of genuine RNase H‐sensitive RNA:DNA hybrids by IF.
We note that some studies have reported the cytoplasmic S9.6 signal to be at least partially sensitive to exogenous RNase H treatment, often as measured under perturbed cellular conditions [for example, see Koo et al, 2015; Silva et al, 2018; Prendergast et al, 2020; Wu et al, 2020)]. However, the origin of such RNase H‐sensitive cytoplasmic RNA:DNA hybrid species remains still largely unexplained, especially considering that this signal is often more intense than the nuclear signal. It is also of paramount importance that images obtained with and without RNase H treatment are collected under the exact same conditions, as anecdotal evidence suggests that buffers, incubation times and temperatures have a strong impact on the S9.6 IF microscopy signal. Finally, we note that in addition to issues of specificity, it is possible that S9.6‐based imaging may lack the sensitivity needed for detecting genuine R‐loops. As mentioned above, several groups have reported that S9.6 IF microscopy failed to detect mitochondrial signals in unperturbed cells (Koo et al, 2015; Silva et al, 2018; preprint: Smolka et al, 2020), although these organelles were shown to carry stable R‐loops (for review Holt, 2019).
Practical guidelines
Overall, while S9.6 IF microscopy approaches should probably be avoided whenever possible, any protocol should incorporate steps to destroy non‐hybrid RNA molecules and demonstrate the RNase H sensitivity of the proposed hybrid signal using exact mock conditions as a control. We also encourage investigators to make efforts to eliminate other confounding variables such as cell cycle perturbations or modifications to the nascent transcriptome (see discussion below). It is important to note that those recommendations apply equally if imaging were to be performed using dRNaseH1‐based reagents instead of the S9.6 antibody.
The large amount of RNA‐derived and RNase H‐resistant S9.6 signal must first be reduced through RNase T1 and RNase III treatments. The elimination of this contaminating signal is paramount to the ability to detect the much less abundant R‐loop and RNA:DNA hybrid signals (estimated to 300 per cell at steady‐state; (Crossley et al, 2020)).
It needs to be verified that any S9.6 signal claimed to be due to genuine RNA:DNA hybrids is broadly RNase H‐sensitive. The use of transfected labeled hybrids (Rigby et al, 2014; preprint: Smolka et al, 2020) provides a useful control for RNase H activity in situ. This should be shown visually on fields of cells, and the S9.6 signal should be carefully quantified. We note that this key control should be performed using exogenous RNase H treatments after cell fixation instead of in vivo over‐expression of RNase H1. As discussed above, questions remain regarding the cellular function of RNase H1 particularly in mammalian cells, and its over‐expression may cause considerable changes to the nascent transcriptome and perhaps also to the proteome (see below).
Considering the technical hurdles associated with the IF strategy, even reproducible evidence of genuine RNase H1‐sensitive nuclear signals obtained by IF should be validated by a complementary approach, such as DRIP‐qPCR with adequate positive and negative controls.
Difficulties with the interpretation of S9.6‐based dot blots
S9.6‐based dot immuno‐blots are often performed to measure overall R‐loop levels and classically performed on total nucleic acids. In general, dot blots do not appear to suffer from interference with non‐hybrid RNA. We speculate that the RNA species recognized by the S9.6 antibody in imaging applications are depleted during nucleic acid extraction in favor of chromosomal DNA. Nevertheless, RNase H and loading controls should always be included, accompanied with quantifications of independent biological replicates. It is important to emphasize that changes observed in total R‐loop loads may not necessarily be attributable to changes in genic R‐loops. Instead, these changes could be driven by fluctuations in mitochondrial R‐loops, or in RNA:DNA hybrids forming over repetitive portions of the genome (telomeres and pericentromeric regions) that are not easily accessible using short‐read sequencing strategies (see below). Because dot blots measure summary R‐loop loads, they would also miss more nuanced events of concurrent R‐loop gains and losses over distinct genomic regions, as observed for instance upon DNA topoisomerase I depletion (Manzo et al, 2018). In addition, dot blots are often not highly sensitive and difficult to quantify precisely and as a result can thus only offer limited concrete information. Caution is therefore required when interpreting their results.
Addressing variability in DRIP‐seq experiments
S9.6‐based R‐loop mapping approaches have been broadly used. A recent analysis of 110 different DRIP‐based datasets from human cells (Pan et al, 2020) suggests that significant variation exists between results of DRIP experiments. To move the field forward, it is important to standardize protocols, both experimental (Sanz & Chédin, 2019) and computational, and to develop objective criteria to evaluate the quality of datasets. For human cells, numerous DRIP‐seq datasets have by now been reported and thus allow for systematic comparisons to be performed. A large ensemble of datasets exhibited strong concordance as measured through signal correlation analysis (Fig 2A). As expected, results from “classic” DRIP‐seq methods, which have lower resolution and lack strandedness, are more congruent with each other than with results from more recently established strand‐specific, high‐resolution DRIP‐based approaches. Nonetheless, a clear correlation between all such datasets is evident, even with data from multiple cell lines and obtained in various different laboratories being compared. This correlation between datasets is also apparent when visualized as genome browser screenshots, defining clear, consistent patterns of R‐loop distribution throughout the genome (Fig 2B). These results support the view that R‐loop formation occurs through transcribed genic regions over conserved R‐loop hotspots (Sanz et al, 2016). In particular, R‐loop formation over housekeeping genes offers a clear template to compare datasets across cell types. Such simple correlation analysis also identifies a number of discordant datasets (Fig 2A) that, similarly to RNase H‐treated negative controls, showed little to no correlation with any other dataset. Visual inspection confirmed that the signal from such studies lacked the widely appreciated features of R‐loop distribution patterns (Fig 2B), mandating caution when interpreting these datasets. To enable data comparison and quality control, we encourage users to exploit the UCSC genome browser (http://genome.ucsc.edu/s/fredericchedinlab/hg19_DRIP_Correlation), which comes pre‐loaded with numerous datasets from both S9.6‐based and dRNaseH1‐based mapping studies.
Figure 2. Addressing variability in DRIP‐seq experiments.
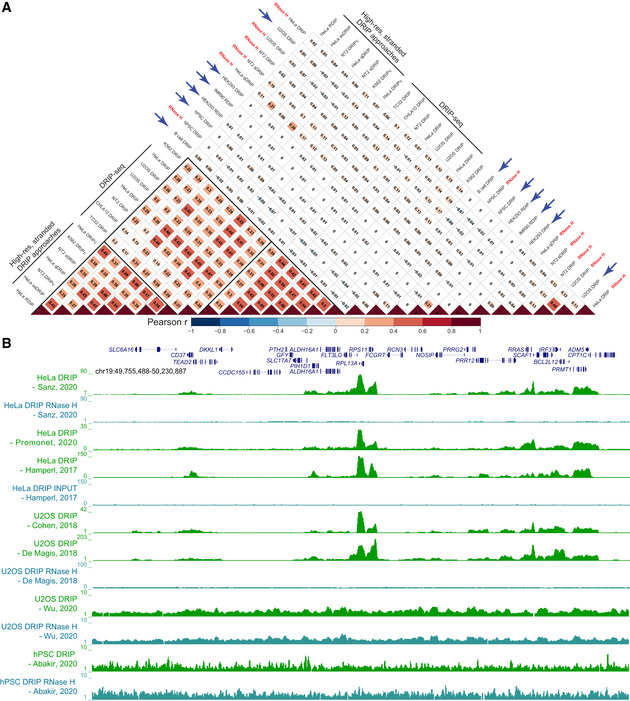
(A) Dataset comparison using Pearson’s correlation analysis. Pearson’s correlation values were calculated pairwise on normalized, log‐transformed signals from the indicated datasets and reported as a heatmap. Correlation was calculated over a set of test regions consisting of “gold standard” genes recently characterized using bisulfite‐based single‐molecule R‐loop footprinting, SMRF‐seq (Malig et al, 2020). In the example shown here, each “gold standard” region was extended by 10 kb on each side before correlation analysis. Similar results were obtained if the regions were extended by 100 kb instead, or if other genic regions chosen at random were used. When a dataset included multiple replicates, correlation analysis was performed on each replicate and then averaged. “RNase H” indicates that a sample had been pre‐treated with RNase H to destroy R‐loops prior to DRIP. Blue arrows highlight discordant datasets. (B) Genome browser screenshot over a large region centered around the standard “gold standard” housekeeping gene RPL13A. For simplicity, only a subset of DRIP‐seq datasets are shown, along with two discordant datasets identified here (bottom). Datasets are identified by their respective publication (first author and year). See Dataset EV1 for a detailed list.
Practical guidelines for S9.6‐based mapping via DNA:RNA immunoprecipitation (DRIP)
Consistency between DRIP datasets requires further harmonization of experimental procedures. Detailed experimental protocols for conducting “standard” DRIP, DRIP‐qPCR, and DRIP‐seq are available (Sanz & Chédin, 2019) and should be followed carefully, especially as it concerns DNA extraction and fragmentation methods. We note that while restriction enzyme fragmentation is often viewed as imposing resolution limits and potential biases, restriction enzyme cocktails can be adapted as needed (Ginno et al, 2013), including the use of four‐base‐pair cutters, which can result in high‐resolution and mostly strand‐specific R‐loop maps [ssDRIP‐seq; Xu et al, 2017; Yang et al, 2019)]. Approaches using sonication for genome fragmentation are now also available [qDRIP‐seq (Crossley et al, 2020) and sDRIP‐seq (preprint: Smolka et al, 2020)], allowing high‐resolution, strand‐specific R‐loop mapping. Owing to the fact that sonication breaks R‐loops down to two‐stranded RNA:DNA hybrids, close attention to library building strategies should be paid in this case: It is necessary to adopt techniques that can convert such two‐stranded RNA:DNA hybrids back into dsDNA, either through an ssDNA ligation step as in qDRIP‐seq (Crossley et al, 2020), or by introducing a second strand‐synthesis step prior to adapter ligation, as in sDRIP‐seq (preprint: Smolka et al, 2020). Results from both methods show strong agreement (Fig 2A).
Consistency between DRIP datasets also requires further harmonization of computational pipelines. We advocate increased transparency in the way authors call and filter peaks, especially when RNase H‐resistant peaks are filtered out. Considering that not all RNA:DNA hybrids seem to be equally sensitive to RNase H (Crossley et al, 2020), filtering out peaks that are only partially resistant to RNase H might discard genuine, biologically relevant hybrids from the analysis. This also underscores the need for standardizing RNase H treatments. Ultimately, we believe that the community should work toward a single analysis pipeline optimized against a series of robust datasets.
Since free RNA, particularly dsRNA, could interfere with the capture of RNA:DNA hybrids by S9.6 antibodies (Hartono et al, 2018), it may be necessary to apply enzymatic treatments to eliminate contaminant RNA, such as RNase T1 and RNase III treatment (note that the use of RNase A is not recommended, as this enzyme possesses significant RNase H‐like activity that is hard to control for). This is particularly important when sequencing libraries are built from RNA‐derived materials (see below). However, even with DNA‐based mapping strategies, the presence of unusually high levels of RNA can sequester S9.6 antibody away, as shown in vitro, and interfere with the quantitative recovery of hybrids (Zhang et al, 2015; Hartono et al, 2018). In pull‐down experiments with the S9.6 antibody, the ratio of genomic DNA to antibody should therefore be standardized, to ensure the quantitative recovery of hybrids (Sanz & Chédin, 2019). Note that the optimal ratio might vary between organisms, because of different degrees of genome complexity and total RNA levels. Spike‐in controls can also be implemented to validate IP efficiency (see below).
RNase H‐treated controls are mandatory and should be optimized to eliminate even the most refractory RNA:DNA hybrids (Crossley et al, 2020).
-
For DRIP‐qPCR studies:
Raw data (shown as “% of input”) rather than normalized data should be provided. Normalized data do not give an indication of the absolute strength of the signal and therefore of the yield of the IP. If necessary, normalized data may be presented in addition to the raw data.
Positive and negative control loci should be queried systematically. One positive control applicable to all organisms could be a site within ribosomal DNA (rDNA). In human cells, other positive controls would be “gold standard” sites that have already been validated by S9.6‐independent approaches in (Malig et al, 2020). Such controls should help to determine the efficiency and specificity of S9.6‐mediated immunoprecipitation. Spike‐in controls, such as transcribed R‐loop plasmids or synthetic RNA:DNA hybrids, should also be included to afford more rigorous quantifications and comparison across experiments or studies (see below).
-
For DRIP‐sequencing studies:
The purpose of conducting R‐loop mapping is often to measure the impact of a genetic or pharmacological disruption on R‐loop patterns. To obtain quantitative data, spike‐in controls should again be developed to normalize sequencing data across samples, as in Crossley et al (2020). Such spike‐in controls could be known amounts of synthetic RNA:DNA hybrids (Crossley et al, 2020) or of exogenous genomes (e.g., Drosophila, Šviković et al, 2019). Obviously, when validating the use of spike‐in controls, great care should be taken to demonstrate that the same amount of spike‐in material has been added to each sample before R‐loop enrichment. To properly call regions of differential signals, a strict minimum of two biological replicates is required and, in all cases, a subset of the regions exhibiting differential R‐loop profiles should be validated using DRIP‐qPCR on independent biological replicates.
Optimized RNase H‐treated controls must be provided to verify the RNA:DNA hybrid dependency of the data. Showing input samples are highly recommended to assess possible sequencing biases and facilitate downstream computational analysis.
Datasets need to be compared to other published high‐quality datasets, now that clear congruent DRIP‐based datasets have been identified (Fig 2). The simple signal correlation analysis shown above is one possible method by which to measure the “fit” of datasets. Results of such comparisons should be reported, focusing on control or mock‐treated conditions. Similarly, screenshots of the data over key “gold standard” regions should be provided as supplemental data to allow assessing of data quality. We hope the UCSC genome browser link provided here will facilitate this process.
When building sequencing libraries from RNA‐derived materials such as in DRIPc‐seq (Sanz et al, 2016) and RDIP‐seq (Nojima et al, 2018), great care should be taken to avoid contamination from RNA sources, as work in fission yeast has demonstrated that such contaminations are a real possibility (Hartono et al, 2018). A close inspection of the RDIP‐seq signal reported in (Nojima et al, 2018) shows widespread patterns that would seem most consistent with contamination by spliced RNA pools, as indicated by clear exon‐delimited peaks over highly expressed genes (Fig 3A). Such patterns are not seen in other high‐resolution mapping approaches including sonication‐derived methods (preprint: Smolka et al, 2020; Crossley et al, 2020), DRIPc‐seq mapping (Sanz et al, 2016; Data ref: Sanz et al, 2020) and single‐molecule R‐loop footprinting (SMRF, Malig et al, 2020). In addition, we noted striking enrichment of RDIP‐seq signal over sequence repeats along transcribed gene bodies, in particular short interspersed elements (SINEs) such as Alu elements, which are well‐known to form abundant dsRNA structures (Fig 3B). Such patterns may well have resulted from contamination with nascent, unspliced transcripts, given their strandedness and distribution over transcribed regions. Similar patterns of possible exon or Alu element contamination from RNA pools can also be observed in other recent DRIPc‐seq datasets (Pérez‐Calero et al, 2020) (Fig 3C). Thus, RNA contamination is a significant problem when mapping R‐loops via the RNA moiety of RNA:DNA hybrids. RNA‐derived material can easily overwhelm rare events derived from R‐loops and lead to erroneous conclusions. To help validate such experiment, we encourage investigators to ascertain that their signals are not abnormally enriched over exons and Alu elements. We note that building sequencing libraries from DNA, as in sDRIP or qDRIP, remedies concerns about RNA contamination while permitting high‐resolution, strand‐specific R‐loop mapping, this making it a viable alternative.
It is important to note that even though R‐loop formation is believed to play a biological role in repetitive regions such as centromeres or telomeres (see for example Graf et al, 2017; Kabeche et al, 2018), current genome‐wide approaches are limited in their ability to map R‐loops in such regions, primarily due to the use of short‐read sequencing technologies. The use of long‐read technologies, such as PacBio (Malig et al, 2020) and nanopore sequencing, combined with the ability to mark R‐loops using non‐denaturing bisulfite probing, may provide future avenues by which to tackle this technically challenging question.
Figure 3. RNA contamination is a significant problem when mapping R‐loops via the RNA moiety of RNA:DNA hybrids.
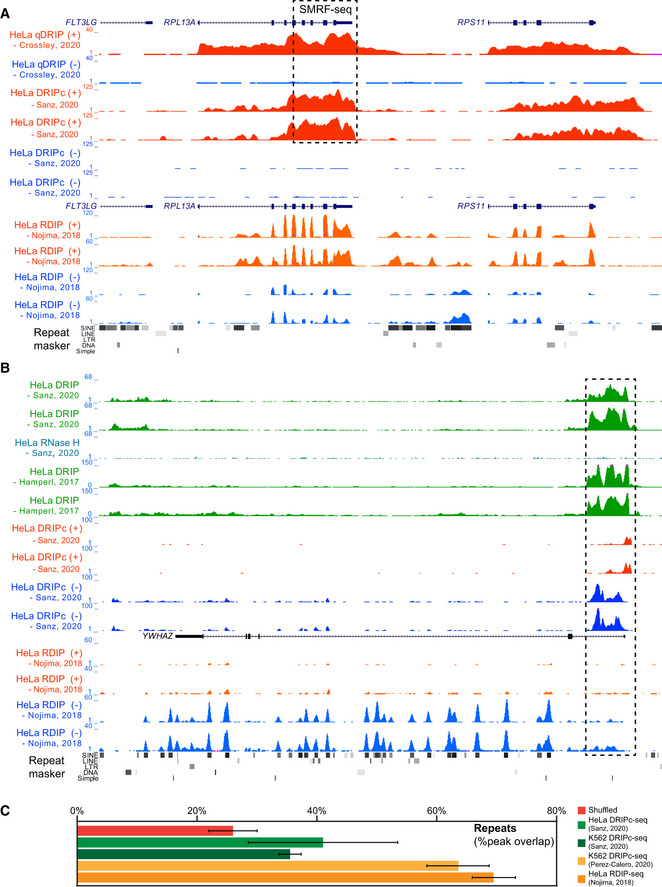
(A) Genome browser screenshot over the RPL13A “gold standard” region showing results from DNA‐based (qDRIP‐seq; (Crossley et al, 2020)) and RNA‐based high‐resolution mapping (DRIPc‐seq and RDIP‐seq; (Nojima et al, 2018; Data ref: Sanz et al, 2020) in HeLa cells. The boxed region was also profiled using SMRF‐seq (Malig et al, 2020). Red and blue colors indicate R‐loops forming on the positive and negative strands, respectively. RDIP‐seq bears striking resemblance to results expected from RNA‐seq data, with a clear exon‐delineated pattern visible genome‐wide for highly expressed genes. (B) R‐loops form over the YWHAZ gene promoter region according to multiple independent datasets in HeLa cells (top, boxed). RDIP‐seq data instead displays striking enrichments over SINE repeats (highlighted by the repeat masker track), which include dsRNA‐generating Alu elements. (C) Overlap of peak calls from various human RNA‐derived R‐loop datasets with repeat elements; some datasets show significantly higher repeat overlap. In all cases, Alu elements are responsible for the overwhelming majority of the repeat overlap (80–97%) and the increased overlap in some datasets is entirely driven by excess Alu elements, as exemplified in Fig 3B. “Shuffled” indicates the extent of repeat overlap expected at random and was measured by arbitrarily moving peaks in each dataset 2 kb to the left before determining repeat overlap (similar results were observed if peaks were moved by 1 or 2 kb to the right or left). Results are shown as average with standard deviation calculated over biological replicates available for each dataset.
Considerations for careful interpretation of R‐loop mapping experiments
Differential gains or losses of RNA:DNA hybrids or R‐loops observed in rigorous R‐loop mapping experiments still have to be carefully interpreted. Changes in R‐loop patterns can result from a variety of sources, including perturbations to the cell cycle, transcriptional alterations affecting the nascent transcriptome, and the inactivation of factors that directly modulate the homeostasis of RNA:DNA hybrids and/or R‐loops. It is important to take these possibilities into consideration when comparing R‐loop formation in different cell populations.
Changes in R‐loop patterns may reflect alterations in cell cycle progression. Given that a number of genes are cell cycle‐controlled, one expects at least a subset of R‐loops to exhibit cell cycle dependency. It was further reported that DNA replication modulates R‐loop formation (Hamperl et al, 2017), and that the bulk of RNA:DNA formation occurs during S phase (Teloni et al, 2019; Šviković et al, 2019). In addition, RNA:DNA hybrids were shown to accumulate upon replication catastrophe, characterized by the nucleus‐wide exhaustion of RPA and the accumulation of DNA damage (Teloni et al, 2019). It is therefore conceivable that changes in the length and efficiency of S phase could impact the average amount of R‐loops at the population scale. To make meaningful comparisons of R‐loop formation in different cell populations, it is therefore important to eliminate the possibility that a change in cell cycle dynamics could underlie the observed changes in the amount or distribution of given R‐loops, e.g., by ensuring that the cell cycle profiles of the compared cell populations are similar. Alternatively, synchronized cell populations may allow a more accurate comparison of genomic R‐loop patterns (Manzo et al, 2018).
Because R‐loops are mainly by‐products of ongoing transcription, R‐loop formation is exquisitely sensitive to perturbations in nascent transcription. Nascent transcription must therefore be monitored to reach rigorously supported conclusions. For instance, the observation that knockdown of a given “factor X” increases R‐loop levels is by itself insufficient to claim that this “factor X is involved in R‐loop resolution”. “Factor X” depletion may instead increase nascent transcription levels, which in turn might entirely account for the observed higher R‐loop accumulation. Generally, R‐loops may accumulate in specific conditions either when (a) the probability R‐loop formation at each transcription cycle remains unchanged but there are more transcription cycles (e.g., R‐loop gains upon estrogen induction (Stork et al, 2016)); (b) transcription is unchanged but each transcription cycle is more likely to produce an R‐loop; or (c) R‐loops form with the same probability but their disassembly is inefficient (which might eventually interfere with nascent transcription). Situations (b) and (c) are predicted to be associated with unchanged or lowered levels of nascent transcription, while situation (a) is predicted to correlate with increased levels of nascent transcripts. Situations (b) and (c) are not easy to distinguish experimentally.
Manipulating R‐loop levels by the strong expression of RNase H1
As mentioned above, cells tend to experience DNA damage and genome instability in conditions where R‐loop levels increase, which often involve altering transcription itself or the co‐transcriptional processing of nascent RNAs. To demonstrate that R‐loops (rather than the transcriptomic alterations associated with such manipulations) indeed account for these phenotypes, a common strategy is to induce the strong nuclear expression of RNase H1, which often suppresses genome instability phenotypes associated with R‐loop‐increasing manipulations to some extent. In most studies, the subsequent conclusion that accumulation/stabilization of R‐loops above a certain threshold indeed underlies an observed phenotype of interest, solely relies on this single experimental approach, and the lack of a complementary confirmatory approach is a limitation in the field. Another problem comes from the fact that most studies lack verification that R‐loop levels are indeed reduced after RNase H1 over‐expression; as notable exceptions, expression of E. coli RNase H was shown to reduce R‐loop levels in fission yeast (Hartono et al, 2018), and human RNase H1 over‐expression was shown to reduce R‐loop levels at specific loci by DRIP‐qPCR in mouse embryonic stem cells (Skourti‐Stathaki et al, 2019) and genome‐wide in HeLa cells (Tan‐Wong et al, 2019) (within the above‐mentioned RDIP‐seq‐associated caveats). In addition, conclusions regarding the toxicity of certain R‐loops for genome stability suffer from the fact that “harmful” R‐loops have thus far not been convincingly identified. Thus, the amelioration of genome instability upon RNase H1 expression has never been conclusively linked to the disappearance of a class of proposed harmful R‐loops.
Although it is relatively simple to implement, some important drawbacks of the described RNase H1 over‐expression strategy are frequently ignored (Vanoosthuyse, 2018). First, excess RNase H1 is often toxic to vertebrate cells (Paulsen et al, 2009; Britton et al, 2014; Salas‐Armenteros et al, 2017; Shen et al, 2017; Barroso et al, 2019; Chang et al, 2019) with the molecular reasons for this toxicity not yet being understood. A second possible side effect of this strategy is again dose‐dependent indirect perturbations to the nascent transcriptome and proteome, the extent of which may vary in different genetic backgrounds (Hartono et al, 2018). In human cells for example, the over‐expression of RNase H1 was shown to impact the production of antisense transcripts (Tan‐Wong et al, 2019) and the stability of a number of DNA damage response factors (Shen et al, 2017), while in mouse ESCs it was shown to derepress Polycomb‐repressed genes (Skourti‐Stathaki et al, 2019). As a result of these transcriptome and proteome alterations, the over‐expression of RNase H1 might only indirectly modulate the phenotype of interest.
In addition, as mentioned above, RPA associates with and stimulates the activity of RNase H1 toward R‐loops and, while the over‐expression of wild‐type RNase H1 could suppress genome instability in several contexts, the over‐expression of a catalytically active RNase H1 mutant that cannot bind RPA failed to do so (Nguyen et al, 2017). It is therefore conceivable that increased RNase H1 nuclear levels might at least partially titrate away RPA and complicate the interpretation of such experiments. To circumvent this issue, it may be advisable to over‐express a bacterial (E. coli RnhA) rather than human enzyme, which lacks the domain required for RPA interaction (Nguyen et al, 2017). Moreover, to limit unwanted indirect effects on the transcriptome, it is advisable to use cell cycle‐regulated promoters and degrons to restrict the over‐expression of RnhA to specific cell cycle phases, as described recently (Lockhart et al, 2019; Šviković et al, 2019). It is also worth further exploring the recently described the possibility of targeting active RNase H1 to specific loci using dCas9 (Li et al, 2020; Abraham et al, 2020) in order to achieve local R‐loop degradation and interrogate R‐loop function. To fully validate this strategy, it will however be important to demonstrate that the presence of a pre‐existing RNA:DNA hybrid does not interfere with the targeting of dCas9. In addition, the ability of dCas9‐RNASEH1 fusion proteins to effectively and selectively degrade the targeted R‐loops in the absence of transcriptional consequences remains to be better established. We suggest that SMRF‐seq assays (Malig et al, 2020) would be ideally suited to determine if such targeted approaches can lead to the full degradation of the long R‐loops (500–1,000 bp) that are frequently encountered in the chromosomes of living cells.
Finally, to strengthen the demonstration that R‐loops contribute directly to the phenotype of interest (for example DNA damage), investigators are encouraged to demonstrate the spatial and temporal co‐localization between R‐loops and DNA damage at the genome‐wide scale and to show that RNase H1‐sensitive damaged loci form exclusively over R‐loops that are stabilized. For example, while the RNase H1‐sensitive genome instability associated with a splicing deficiency has been reported by several studies (Li & Manley, 2005; Chen et al, 2018; Chakraborty et al, 2018), it has yet to be demonstrated that R‐loops and DNA damage form on incorrectly spliced transcripts.
Conclusions
R‐loops have risen to prominence as a major type of transcription‐driven non‐B DNA structure in all genomes. Correctly addressing the possible impact of R‐loop formation on adaptive and maladaptive cellular processes requires well‐controlled, standardized, and carefully interpreted methodologies. This commentary was motivated by the desire to describe best practices for mapping, visualizing, and manipulating R‐loops, to resolve discrepancies in the field, and to promote careful interpretation and (where needed) re‐evaluation of existing datasets. We hope that these recommendations will be useful for authors, referees, and editors alike and will help improve the reliability of future R‐loop‐related studies.
A puzzling observation in the field has been the major disparity between results from dRNase H1‐ and S9.6‐based R‐loop maps. We propose here that endogenous dRNase H1 may only recognize a subset of RNA:DNA hybrids associated with promoter‐paused RNA polymerases. Future studies will determine the molecular underpinnings of such specificity, probe if such short R‐loops have biological significance, and whether endogenous RNase H1 participates in resolving them in the context of physiological pause‐release mechanisms. The thorough identification of harmful R‐loops in a variety of cellular models will also be critical to test the popular notion that a subset of R‐loops may negatively interfere with genome stability. Strategies to evaluate the stability of different R‐loops in vivo will further be important to determine whether altered R‐loop stability, in addition to altered distribution, is an important determinant of genome instability (García‐Rubio et al, 2018). Existing strategies using transcription inhibitors (Sanz et al, 2016; Crossley et al, 2020) are limited in determining the turnover of R‐loops that form at the 3′ end of genes. This is a limitation considering that such terminal R‐loops have been associated with DNA replication stress (Costantino & Koshland, 2018; Promonet et al, 2020). Strategies to accurately determine the stability of individual R‐loops throughout the cell cycle or in response to stress will be invaluable to understand the full impact of R‐loop formation on genome stability.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Dataset EV1
Acknowledgements
Work in the Chedin laboratory is supported by the National Institutes of Health (R01 GM120607). The work of V. Vanoosthuyse is funded by a grant (19‐CE12‐0016‐04) from the Agence Nationale de la Recherche (ANR). We thank our colleagues Drs Karlene Cimprich and Philippe Pasero for sharing datasets prior to their publication.
The EMBO Journal (2021) 40: e106394.
See the Glossary for abbreviations used in this article.
Contributor Information
Frédéric Chédin, Email: flchedin@ucdavis.edu.
Vincent Vanoosthuyse, Email: vincent.vanoosthuyse@ens-lyon.fr.
Data availability
The UCSC genome browser comparison of datasets from published S9.6‐based and dRNaseH1‐based mapping studies is available at: http://genome.ucsc.edu/s/fredericchedinlab/hg19_DRIP_Correlation
References
- Abraham KJ, Khosraviani N, Chan JNY, Gorthi A, Samman A, Zhao DY, Wang M, Bokros M, Vidya E, Ostrowski LA et al (2020) Nucleolar RNA polymerase II drives ribosome biogenesis. Nature 585: 298–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman K, Lis JT (2012) Promoter‐proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13: 720–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariel F, Lucero L, Christ A, Mammarella MF, Jegu T, Veluchamy A, Mariappan K, Latrasse D, Blein T, Liu C et al (2020) R‐loop mediated trans action of the APOLO long noncoding RNA. Mol Cell 77: 1055–1065 [DOI] [PubMed] [Google Scholar]
- Barroso S, Herrera‐Moyano E, Muñoz S, García‐Rubio M, Gómez‐González B, Aguilera A (2019) The DNA damage response acts as a safeguard against harmful DNA‐RNA hybrids of different origins. EMBO Rep 20: e47250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL (2014) Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15: 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguslawski SJ, Smith DE, Michalak MA, Mickelson KE, Yehle CO, Patterson WL, Carrico RJ (1986) Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J Immunol Methods 89: 123–130 [DOI] [PubMed] [Google Scholar]
- Britton S, Dernoncourt E, Delteil C, Froment C, Schiltz O, Salles B, Frit P, Calsou P (2014) DNA damage triggers SAF‐A and RNA biogenesis factors exclusion from chromatin coupled to R‐loops removal. Nucleic Acids Res 42: 9047–9062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco‐Salas Y, Malapert A, Sulthana S, Molcrette B, Chazot‐Franguiadakis L, Bernard P, Chédin F, Faivre‐Moskalenko C, Vanoosthuyse V (2019) The extruded non‐template strand determines the architecture of R‐loops. Nucleic Acids Res 47: 6783–6795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty P, Huang JTJ, Hiom K (2018) DHX9 helicase promotes R‐loop formation in cells with impaired RNA splicing. Nat Commun 9: 4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EY‐C, Tsai S, Aristizabal MJ, Wells JP, Coulombe Y, Busatto FF, Chan YA, Kumar A, Dan Zhu Y, Wang AY‐H et al (2019) MRE11‐RAD50‐NBS1 promotes Fanconi Anemia R‐loop suppression at transcription‐replication conflicts. Nat Commun 10: 4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen J‐Y, Zhang X, Gu Y, Xiao R, Shao C, Tang P, Qian H, Luo D, Li H et al (2017) R‐ChIP using inactive RNase H reveals dynamic coupling of r‐loops with transcriptional pausing at gene promoters. Mol Cell 68: 745–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen J‐Y, Huang Y‐J, Gu Y, Qiu J, Qian H, Shao C, Zhang X, Hu J, Li H et al (2018) The augmented R‐loop is a unifying mechanism for myelodysplastic syndromes induced by high‐risk splicing factor mutations. Mol Cell 69: 412–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier SC, Wang S, Ma WK, Al Husini N, Dhoondia Z, Ansari A, Pascuzzi PE, Tran EJ (2016) Regulated formation of lncRNA‐DNA hybrids enables faster transcriptional induction and environmental adaptation. Mol Cell 61: 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core L, Adelman K (2019) Promoter‐proximal pausing of RNA polymerase II: a nexus of gene regulation. Genes Dev 33: 960–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, Koshland D (2018) Genome‐wide map of R‐loop‐induced damage reveals how a subset of R‐loops contributes to genomic instability. Mol Cell 71: 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley MP, Bocek M, Cimprich KA (2019) R‐Loops as cellular regulators and genomic threats. Mol Cell 73: 398–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley MP, Bocek MJ, Hamperl S, Swigut T, Cimprich KA (2020) qDRIP: a method to quantitatively assess RNA‐DNA hybrid formation genome‐wide. Nucleic Acids Res 48: e84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Muse T, Aguilera A (2019) R loops: from physiological to pathological roles. Cell 179: 604–618 [DOI] [PubMed] [Google Scholar]
- García‐Rubio M, Aguilera P, Lafuente‐Barquero J, Ruiz JF, Simon M‐N, Geli V, Rondón AG, Aguilera A (2018) Yra1‐bound RNA‐DNA hybrids cause orientation‐independent transcription‐replication collisions and telomere instability. Genes Dev 32: 965–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, Chédin F (2012) R‐loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 45: 814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lim YW, Lott PL, Korf I, Chédin F (2013) GC skew at the 5’ and 3’ ends of human genes links R‐loop formation to epigenetic regulation and transcription termination. Genome Res 23: 1590–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf M, Bonetti D, Lockhart A, Serhal K, Kellner V, Maicher A, Jolivet P, Teixeira MT, Luke B (2017) Telomere length determines TERRA and R‐loop regulation through the cell cycle. Cell 170: 72–85 [DOI] [PubMed] [Google Scholar]
- Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA (2017) Transcription‐replication conflict orientation modulates R‐loop levels and activates distinct DNA damage responses. Cell 170: 774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartono SR, Korf IF, Chédin F (2015) GC skew is a conserved property of unmethylated CpG island promoters across vertebrates. Nucleic Acids Res 43: 9729–9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartono SR, Malapert A, Legros P, Bernard P, Chédin F, Vanoosthuyse V (2018) The affinity of the S9.6 antibody for double‐stranded RNAs impacts the accurate mapping of R‐loops in fission yeast. J Mol Biol 430: 272–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzel L, Ottoz DSM, Alpert T, Neugebauer KM (2017) Splicing and transcription touch base: co‐transcriptional spliceosome assembly and function. Nat Rev Mol Cell Biol 18: 637–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ (2019) The mitochondrial R‐loop. Nucleic Acids Res 47: 5480–5489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin J‐P, Manley JL (2012) The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev 26: 2119–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Aguilera A (2003) Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription‐associated recombination. Mol Cell 12: 711–721 [DOI] [PubMed] [Google Scholar]
- Hyjek M, Figiel M, Nowotny M (2019) RNases H: structure and mechanism. DNA Repair 84: 102672 [DOI] [PubMed] [Google Scholar]
- Kabeche L, Nguyen HD, Buisson R, Zou L (2018) A mitosis‐specific and R loop‐driven ATR pathway promotes faithful chromosome segregation. Science 359: 108–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Chu C, Shatkin AJ, Manley JL (2007) Human capping enzyme promotes formation of transcriptional R loops in vitro . Proc Natl Acad Sci USA 104: 17620–17625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- König F, Schubert T, Längst G (2017) The monoclonal S9.6 antibody exhibits highly variable binding affinities towards different R‐loop sequences. PLoS One 12: e0178875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo CX, Kobiyama K, Shen YJ, LeBert N, Ahmad S, Khatoo M, Aoshi T, Gasser S, Ishii KJ (2015) RNA polymerase III regulates cytosolic RNA:DNA hybrids and intracellular microRNA expression. J Biol Chem 290: 7463–7473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legros P, Malapert A, Niinuma S, Bernard P, Vanoosthuyse V (2014) RNA processing factors Swd2.2 and Sen1 antagonize RNA Pol III‐dependent transcription and the localization of condensin at Pol III genes. PLoS Genet 10: e1004794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Manley JL (2005) Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 122: 365–378 [DOI] [PubMed] [Google Scholar]
- Li Y, Song Y, Xu W, Li Q, Wang X, Li K, Wang J, Liu Z, Velychko S, Ye R et al (2020) R‐loops coordinate with SOX2 in regulating reprogramming to pluripotency. Sci Adv 6: eaba0777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart A, Pires VB, Bento F, Kellner V, Luke‐Glaser S, Yakoub G, Ulrich HD, Luke B (2019) RNase H1 and H2 Are differentially regulated to process RNA‐DNA Hybrids. Cell Rep 29: 2890–2900 [DOI] [PubMed] [Google Scholar]
- Malig M, Hartono SR, Giafaglione JM, Sanz LA, Chedin F (2020) Ultra‐deep coverage single‐molecule R‐loop footprinting reveals principles of R‐loop formation. J Mol Biol 2161: 209–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzo SG, Hartono SR, Sanz LA, Marinello J, De Biasi S, Cossarizza A, Capranico G, Chedin F (2018) DNA Topoisomerase I differentially modulates R‐loops across the human genome. Genome Biol 19: 100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Yadav T, Giri S, Saez B, Graubert TA, Zou L (2017) Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol Cell 65: 832–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C, Luke B (2020) Regulatory R‐loops as facilitators of gene expression and genome stability. Nat Rev Mol Cell Biol 21: 167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Tellier M, Foxwell J, Ribeiro de Almeida C, Tan‐Wong SM, Dhir S, Dujardin G, Dhir A, Murphy S, Proudfoot NJ (2018) Deregulated expression of mammalian lncRNA through loss of SPT6 induces R‐loop formation, replication stress, and cellular senescence. Mol Cell 72: 970–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny M, Cerritelli SM, Ghirlando R, Gaidamakov SA, Crouch RJ, Yang W (2008) Specific recognition of RNA/DNA hybrid and enhancement of human RNase H1 activity by HBD. EMBO J 27: 1172–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Jin M, Ghadiyaram A, Kaur P, Miller HE, Ta HM, Liu M, Fan Y, Mahn C, Gorthi A et al (2020) Cohesin SA1 and SA2 are RNA binding proteins that localize to RNA containing regions on DNA. Nucleic Acids Res 48: 5639–5655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee M‐C, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow‐Cordero DE et al (2009) A genome‐wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35: 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Calero C, Bayona‐Feliu A, Xue X, Barroso SI, Muñoz S, González‐Basallote VM, Sung P, Aguilera A (2020) UAP56/DDX39B is a major cotranscriptional RNA‐DNA helicase that unwinds harmful R loops genome‐wide. Genes Dev 34: 898–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold C, Marceau AH, Miller KH, Marqusee S, Keck JL (2015) Interaction with single‐stranded DNA‐binding protein stimulates Escherichia coli ribonuclease HI enzymatic activity. J Biol Chem 290: 14626–14636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DD, Garboczi DN, Singh K, Hu Z, Leppla SH, Leysath CE (2013) The sub‐nanomolar binding of DNA‐RNA hybrids by the single‐chain Fv fragment of antibody S9.6. J Mol Recognit JMR 26: 376–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast L, McClurg UL, Hristova R, Berlinguer‐Palmini R, Greener S, Veitch K, Hernandez I, Pasero P, Rico D, Higgins JMG et al (2020) Resolution of R‐loops by INO80 promotes DNA replication and maintains cancer cell proliferation and viability. Nat Commun 11: 4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promonet A, Padioleau I, Liu Y, Sanz L, Biernacka A, Schmitz A‐L, Skrzypczak M, Sarrazin A, Mettling C, Rowicka M et al (2020) Topoisomerase 1 prevents replication stress at R‐loop‐enriched transcription termination sites. Nat Commun 11: 3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby RE, Webb LM, Mackenzie KJ, Li Y, Leitch A, Reijns MAM, Lundie RJ, Revuelta A, Davidson DJ, Diebold S et al (2014) RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J 33: 542–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivosecchi J, Larochelle M, Teste C, Grenier F, Malapert A, Ricci EP, Bernard P, Bachand F, Vanoosthuyse V (2019) Senataxin homologue Sen1 is required for efficient termination of RNA polymerase III transcription. EMBO J 38: e101955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy D, Yu K, Lieber MR (2008) Mechanism of R‐loop formation at immunoglobulin class switch sequences. Mol Cell Biol 28: 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas‐Armenteros I, Pérez‐Calero C, Bayona‐Feliu A, Tumini E, Luna R, Aguilera A (2017) Human THO‐Sin3A interaction reveals new mechanisms to prevent R‐loops that cause genome instability. EMBO J 36: 3532–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas‐Armenteros I, Barroso SI, Rondón AG, Pérez M, Andújar E, Luna R, Aguilera A (2019) Depletion of the MFAP1/SPP381 splicing factor causes R‐loop‐independent genome instability. Cell Rep 28: 1551–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, Chédin F (2016) Prevalent, dynamic, and conserved R‐loop structures associate with specific epigenomic signatures in mammals. Mol Cell 63: 167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz LA, Chédin F (2019) High‐resolution, strand‐specific R‐loop mapping via S9.6‐based DNA‐RNA immunoprecipitation and high‐throughput sequencing. Nat Protoc 14: 1734–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz LA, Hartono SR, Chédin F (2020) Gene Expression Omnibus GSE158480 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158480) [DATASET]
- Shen W, Sun H, De Hoyos CL, Bailey JK, Liang X‐H, Crooke ST (2017) Dynamic nucleoplasmic and nucleolar localization of mammalian RNase H1 in response to RNAP I transcriptional R‐loops. Nucleic Acids Res 45: 10672–10692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan RM, Fong N, D’Alessandro A, Bentley DL (2019) Widespread backtracking by RNA Pol II is a major effector of gene activation, 5’ pause release, termination, and transcription elongation rate. Mol Cell 73: 107–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva S, Camino LP, Aguilera A (2018) Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc Natl Acad Sci USA 115: 11024–11029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti‐Stathaki K, Proudfoot NJ, Gromak N (2011) Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2‐dependent termination. Mol Cell 42: 794–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti‐Stathaki K, Torlai Triglia E, Warburton M, Voigt P, Bird A, Pombo A (2019) R‐loops enhance polycomb repression at a subset of developmental regulator genes. Mol Cell 73: 930–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolka JA, Sanz LA, Hartono SR, Chédin F (2020) Recognition of cellular RNAs by the S9.6 antibody creates pervasive artefacts when imaging RNA:DNA hybrids. bioRxiv 10.1101/2020.01.11.902981 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling PC, Chan YA, Minaker SW, Aristizabal MJ, Barrett I, Sipahimalani P, Kobor MS, Hieter P (2012) R‐loop‐mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev 26: 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz R, Sulthana S, Hartono SR, Malig M, Benham CJ, Chedin F (2019) Interplay between DNA sequence and negative superhelicity drives R‐loop structures. Proc Natl Acad Sci USA 116: 6260–6269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chédin F, Swigut T, Cimprich KA (2016) Co‐transcriptional R‐loops are the main cause of estrogen‐induced DNA damage. Elife 5: e17548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šviković S, Crisp A, Tan‐Wong SM, Guilliam TA, Doherty AJ, Proudfoot NJ, Guilbaud G, Sale JE (2019) R‐loop formation during S phase is restricted by PrimPol‐mediated repriming. EMBO J 38: e99793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan‐Wong SM, Dhir S, Proudfoot NJ (2019) R‐loops promote antisense transcription across the mammalian genome. Mol Cell 76: 600–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teloni F, Michelena J, Lezaja A, Kilic S, Ambrosi C, Menon S, Dobrovolna J, Imhof R, Janscak P, Baubec T et al (2019) Efficient pre‐mRNA cleavage prevents replication‐stress‐associated genome instability. Mol Cell 73: 670–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoosthuyse V (2018) Strengths and weaknesses of the current strategies to map and characterize R‐loops. Non‐Coding RNA 4: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, Vuica‐Ross M (2011) RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell 44: 978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Gore SK, Koshland D (2013) The homologous recombination machinery modulates the formation of RNA‐DNA hybrids and associated chromosome instability. Elife 2: e00505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Lima WF, Crooke ST (2001) Investigating the structure of human RNase H1 by site‐directed mutagenesis. J Biol Chem 276: 23547–23553 [DOI] [PubMed] [Google Scholar]
- Wu W, Bhowmick R, Vogel I, Özer Ö, Ghisays F, Thakur RS, Sanchez de Leon E, Richter PH, Ren L, Petrini JH et al (2020) RTEL1 suppresses G‐quadruplex‐associated R‐loops at difficult‐to‐replicate loci in the human genome. Nat Struct Mol Biol 27: 424–437 [DOI] [PubMed] [Google Scholar]
- Xu W, Xu H, Li K, Fan Y, Liu Y, Yang X, Sun Q (2017) The R‐loop is a common chromatin feature of the Arabidopsis genome. Nat Plants 3: 704–714 [DOI] [PubMed] [Google Scholar]
- Yan Q, Shields EJ, Bonasio R, Sarma K (2019) Mapping native R‐loops genome‐wide using a targeted nuclease approach. Cell Rep 29: 1369–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Liu Q‐L, Xu W, Zhang Y‐C, Yang Y, Ju L‐F, Chen J, Chen Y‐S, Li K, Ren J et al (2019) m6A promotes R‐loop formation to facilitate transcription termination. Cell Res 29: 1035–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh C‐L, Wilson TE, Lieber MR (2003) R‐loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 4: 442–451 [DOI] [PubMed] [Google Scholar]
- Yu K, Lieber MR (2019) Current insights into the mechanism of mammalian immunoglobulin class switch recombination. Crit Rev Biochem Mol Biol 54: 333–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatreanu D, Han Z, Mitter R, Tumini E, Williams H, Gregersen L, Dirac‐Svejstrup AB, Roma S, Stewart A, Aguilera A et al (2019) Elongation factor TFIIS prevents transcription stress and R‐loop accumulation to maintain genome stability. Mol Cell 76: 57–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZZ, Pannunzio NR, Hsieh C‐L, Yu K, Lieber MR (2015) Complexities due to single‐stranded RNA during antibody detection of genomic rna:dna hybrids. BMC Res Notes 8: 127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X‐Y, Wang P, Han J, Rosenfeld MG, Fu X‐D (2009) SR proteins in vertical integration of gene expression from transcription to RNA processing to translation. Mol Cell 35: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Sanz LA, Hartono SR, Chédin F (2020) Gene Expression Omnibus GSE158480 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158480) [DATASET]
Supplementary Materials
Dataset EV1
Data Availability Statement
The UCSC genome browser comparison of datasets from published S9.6‐based and dRNaseH1‐based mapping studies is available at: http://genome.ucsc.edu/s/fredericchedinlab/hg19_DRIP_Correlation