

Abstract
The recent outbreak of coronavirus disease (COVID‐19) in China caused by the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has led to worldwide human infections and deaths. The nucleocapsid (N) protein of coronaviruses (CoVs) is a multifunctional RNA binding protein necessary for viral RNA replication and transcription. Therefore, it is a potential antiviral drug target, serving multiple critical functions during the viral life cycle. This study addresses the potential to repurpose antiviral compounds approved or in development for treating human CoV induced infections against SARS‐CoV‐2 N. For this purpose, we used the docking methodology to better understand the inhibitory mechanism of this protein with the existing 34 antiviral compounds. The results of this analysis indicate that rapamycin, saracatinib, camostat, trametinib, and nafamostat were the top hit compounds with binding energy (−11.87, −10.40, −9.85, −9.45, −9.35 kcal/mol, respectively). This analysis also showed that the most common residues that interact with the compounds are Phe66, Arg68, Gly69, Tyr123, Ile131, Trp132, Val133, and Ala134. Subsequently, protein‐ligand complex stability was examined with molecular dynamics simulations for these five compounds, which showed the best binding affinity. According to the results of this study, the interaction between these compounds and crucial residues of the target protein were maintained. These results suggest that these residues are potential drug targeting sites for the SARS‐CoV‐2 N protein. This study information will contribute to the development of novel compounds for further in vitro and in vivo studies of SARS‐CoV‐2, as well as possible new drug repurposing strategies to treat COVID‐19 disease.
Keywords: COVID‐19, CoVs, drug repurposing, MD simulations, molecular docking, N protein, RNA binding domain, SARS‐CoV‐2
1. INTRODUCTION
Coronaviruses (CoVs) that are the subject of the main research of this study are large, enveloped and single‐stranded (positive‐sense) RNA viruses. 1 , 2 , 3 , 4 , 5 , 6 Until 2019, only six CoVs have been observed to cause disease in humans: (a) HCoV‐229E, (b) HCoV‐OC43, (c) HCoV‐NL63, (d) HCoV‐HKU1, (e) Middle East Respiratory Syndrome Coronavirus (MERS‐CoV), and (f) Severe Acute Respiratory Syndrome Coronavirus (SARS‐CoV). 7 , 8 In late 2019 and early 2020 in Wuhan, China, a novel coronavirus was discovered to be the cause of a rapidly spreading outbreak of the respiratory disease.
The newly discovered severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) was characterized as a betacoronavirus and considered the seventh discrete coronavirus species that can cause a human disease. 9 The disease caused by the virus was officially named Coronavirus Disease 2019 (COVID‐19) by the World Health Organization (WHO). The emerging global epidemic spread rapidly with 42,512,186 confirmed cases and 1,147,301 deaths across 218 countries (COVID‐19 situation Report WHO, October 25, 2020). Given the serious nature of the 2019‐nCoV outbreaks, an urgent need for a new drug against COVID‐19 has arisen.
The amino acid sequence of the SARS‐CoV‐2 shows a high similarity to both SARS‐CoV and MERS‐CoV. The sequence has approximately 82% similarity in its amino acid sequence to SARS‐CoV 9 , 10 overall, and more than 90% sequence identity with respect to various essential enzymes and structural proteins. 11 Especially, the nucleocapsid protein of SARS‐CoV‐2 has a 90.52% sequence identity compared to SARS‐CoV.
In this case, medical chemistry efforts for new therapeutic options for human CoVs can be very helpful to identify potential treatments for SARS‐CoV‐2. 12 , 13 Therefore, drug repurposing studies for coronavirus infections are used as an alternative approach that can help discover potential antiviral molecules quickly and reliably. Since the molecules considered in these studies proceed through several stages and have well‐defined profiles, they would be excellent candidates in case of disease emergencies or outbreaks, without the need for long‐term preclinical studies. 14 , 15
Coronaviruses contain a very large RNA genome of about 30 kb in length, and a unique replication strategy. These viruses encode two overlapping open‐reading frames (ORF) that transform into two polyproteins, (a) pp1a and (b) pp1ab. These polyproteins are also processed to generate four main structural proteins (a) Spike (S), (b) Membrane (M), (c) Envelope glycoproteins (E), and (d) Nucleocapsid (N) and 16 non‐structural proteins (nsps) as shown as in Figure 1. 16
FIGURE 1.
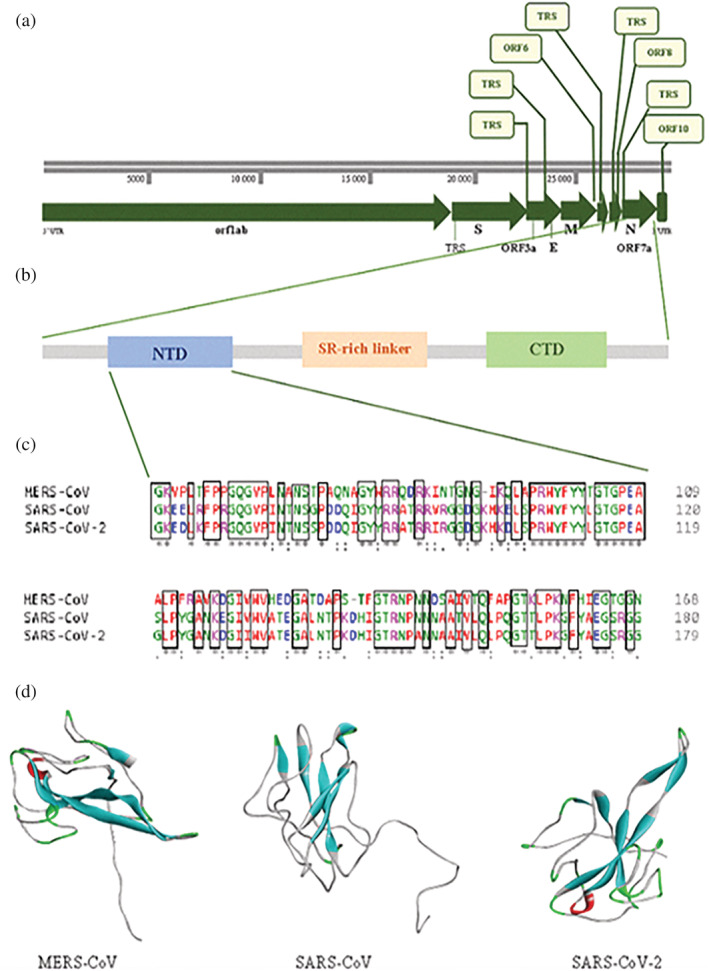
Sequence features and structures of CoVs nucleocapsid protein. (a) Complete genome of SARS‐CoV‐2. (b) Domain architectures of coronavirus nucleocapsid protein. (c) Multiple sequence alignment of CoVs N‐NTD analysis with Clustal Omega Service (https://www.ebi.ac.uk/Tools/msa/clustalo/). (d) 3D structures of CoVs N‐NTD (MERS‐CoV: PDB code: 4UD1, SARS‐CoV: PDB code: 1SSK, SARS‐CoV‐2: PDB code: 6VYO)
The N protein of coronaviruses (CoVs) plays a pivotal role in the viral structure in addition to, the replication and transcription of CoVs via interactions with the large positive‐strand RNA viral genomes. 17 RNA chaperones are nonspecific nucleic acid binding proteins that facilitate folding of RNA and protein substrates into their correct functional structures. 18 The main function of the N protein of CoVs is to bind to the viral RNA genome to promote the correct folding of the hammerhead ribozyme avoiding unproductive RNA conformations and make them into a helical capsid structure or ribonucleoprotein (RNP) complex, whose packaging is critical to viability. 19 , 20 Because of this function, it is suggested that the COV's N protein is an RNA chaperone and conducts a special step in viral genomic RNA replication. 21 Furthermore, in vitro studies showed that the N protein promotes the binding of DNA and viral transcriptional regulatory sequence (TRS) RNAs, confirming its role as an RNA chaperon in a coronavirus associated system. 18 The N protein also plays a major role in increasing the yield of transcription and assembly during virion assembly by interacting with the M protein. 22 , 23 In addition, the N protein is involved in the regulation of cellular processes, such as actin reorganization, host cell cycle progression, and apoptosis. 24 , 25 Besides, the N protein has been shown to induce protective immune responses against CoV and is a significant antigen to develop a sensitive diagnostic assay. 26 Thus, it is one of the potential antiviral drug targets, serving critical functions throughout the viral life cycle.
The CoVs N protein has two main domains: (a) the RNA binding domain (N‐terminal domain [NTD]) and the (b) C‐terminal dimerization domain (CTD). The NTD domain is known to play an important role in CoV replication and transcription by binding to the 3′ end of the viral RNA genome due to electrostatic interactions of positive amino acids. 19 , 27 Some in vitro, in vivo, and in silico studies have revealed that several critical amino acids play a role in RNA binding and virus infectivity via the NTD of CoV N proteins. Specifically, they indicated that several conserved aromatic residue groups in the SARS‐CoV N‐NTD were identified for RNA binding and virus infectivity. 27 , 28 , 29 , 30 Therefore, SARS‐CoV N‐NTD is considered an important drug target, as processes involving virus viability, such as viral replication, transcription, and assembly begin with the binding of NTD to RNA. 22 , 31
In recent times, three‐dimensional (3D) crystal structures of CoVs N‐NTD solved in isolation or complex with other molecules are available in the protein database (PDB) as shown in Figure 1. However, these important roles in their biological mechanisms are largely unknown at the molecular level. Understanding these aspects will make it easier to develop agents that specifically inhibit CoV genome replication, transcription, and viral assembly.
In light of this information, the current study aimed to identify the structural mechanism of SARS‐CoV‐2 N protein with approved drugs and antiviral agents and elucidated the residues of this enzyme that play an important role in the inhibition of CoV genome replication at the molecular level. Accordingly, a computational molecular modeling approach was applied for the repurpose of these agents, which is approved or under development to treat infections caused by human CoVs, against SARS‐CoV‐2 N protein. Computational approaches such as structure‐based virtual screening provide significant savings in experimental cost and time in drug discovery. These study results will be a guide for identifying drug molecules that can be directly tested for in vitro and in vivo studies to deal with a global threat of COVID‐19.
2. MATERIALS AND METHODS
Our study was conducted using structure‐based drug design (SBDD) methods. In the first stage, structural analyses of the SARS‐CoV‐2 N protein (target protein) and antiviral compounds (ligand compounds) were performed. Next, molecular docking was applied to study the mechanism of interaction between target protein and ligand compounds. In the last stage, the stability analysis of the important contact interaction between these compounds and the target enzyme was investigated by molecular dynamic (MD) simulations (see in Figure 2). The visualization of the results was carried out with the help of BIOVIA Discovery Studio.
FIGURE 2.
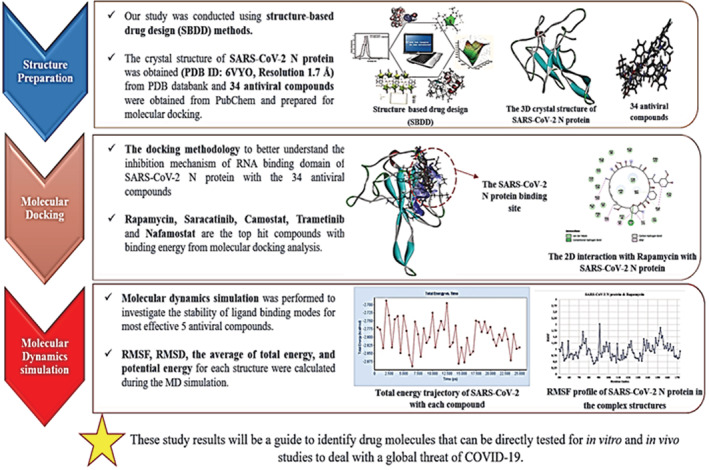
Flowchart showing the steps to screen antiviral compounds for the SARS‐CoV‐2 N protein
2.1. Structure preparation of protein and ligands
The crystal structure of the NTD of the SARS‐CoV‐2 N protein was obtained from the PDB web site at http://www.rcsb.org/pdb (PDB ID: 6VYO, Resolution 1.7 Å). This structure contains a range of 50–173 amino acids. Water and ion molecules were removed from this crystal structure. The structure was then prepared for continuous electrostatic calculations by reconstructing the missing atoms in the crystal structure with the APBS‐PDB2PQR 32 that added hydrogens, assigned atomic charges and radii from specified force fields. Antiviral compounds for molecular docking studies were obtained from PubChem as SDF form and the 3D versions of these compounds were drawn in Marvin Sketch (Marvin 18.12, ChemAxon (https://www.chemaxon.com). To make sure that all values of the heavy atoms were satisfied, all of the hydrogen atoms were added these structures, geometrically cleaned, and then converted to .pdb format with BIOVIA Discovery Studio. 33
2.2. Molecular docking
Blind docking is used in cases in which the ligand binding site of the protein is not known. It can be used to predict the possible ligand binding sites on the whole protein target, and it is anticipated that each ligand will bind to the site with the lowest calculated docking energy. In this study, the ligand binding site of the NTD domain of the target SARS‐CoV‐N protein is not known yet. Therefore, blind docking studies were performed to estimate the binding energies of antiviral compounds to the therapeutic protein targets of SARS‐CoV‐2 N protein using the AutoDock 4.2. 34 The PDB files of the protein and ligands prepared in the previous step were converted to the PDBQT format. All molecular docking studies were performed with a total number of 200 runs with the Lamarckian Genetic algorithm and grid box dimensions of 126, 126, and 126 points in x, y, and z directions, respectively, which were set with grid spacing of 0.375 Å between them. The default settings were applied for all other parameters.
2.3. Molecular dynamic simulations
MD simulations were performed for structural stability, conformational changes, and other critical aspects of protein function analysis of the protein‐ligand complex. This analysis was carried out using the NAMD module of the BIOVIA Discovery Studio 33 for SARS‐CoV‐2 with the top five compounds selected as a result of molecular docking. First, energy minimization (1,000 steps) was carried out with Steepest Descent algorithm, and then with the Conjugate Gradient algorithm (1,000 numsteps), provided that a low‐energy starting point is provided to subsequent dynamic stages. Heating and equilibration were performed distribute the energy in the system in accordance with all degrees of freedom and to ensure that the system reached thermal equilibrium at the target temperature of 300 K. The production run was carried out at 25 ns with both a constant temperature and volume canonical ensemble (NVT) for each complex. During this run, the timesteps were set to 2 fs. The root mean square deviation and root mean square fluctuation (RMSD and RMSF, respectively) were examined using MD trajectories during the complete MD simulation.
3. RESULTS AND DISCUSSIONS
3.1. Identification of SARS‐CoV‐N protein binding mechanism with antiviral compounds via molecular docking
In this study, we used molecular docking to focus on 34 approved antiviral agents used to treat human CoVs infections against the SARS‐CoV‐2 N protein (Table S1). Based on results, we found that rapamycin has the best binding affinity to the SARS‐CoV‐2 N protein (Binding energy: −11.87 kcal/mol with low micromolar K i values (K i : <0.001 μM) among the 34 compounds (see Table 1, Table S1). Besides, saracatinib (−10.40 kcal/mol), camostat (−9.85 kcal/mol), trametinib (−9.45 kcal/mol), and nafamostat (−9.35 kcal/mol) also exhibited significant binding affinity.
TABLE 1.
Two‐dimensional (2D) structures, interaction residues, indication, lowest binding energy and inhibition constant (K i ) values of the top five compounds from each docking simulation. Bold colors indicate common interactions in the binding site of SARS‐CoV‐2 N‐NTD domain
| Ligand name | Binding energy (kcal/mol) | Inhibition Constant (μM) | SARS‐CoV‐2 N residues interacting with ligands | Compound structure | Indication |
|---|---|---|---|---|---|
| Rapamycin | −11.87 | 0.001 | Lys65, Phe66, Pro67, Arg68, Gly69, Gln70, Ile84, Pro122, Tyr123, Gly124, Ala125, Asn126, Ile130, Ile131, Trp132, Val133, Ala134, Thr135, Glu136, Gly137, Ala138, Asn140 |
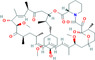
|
Antiviral drug used for the treatment of MERS‐CoV. 35 |
| Saracatinib | −10.40 | 0.023 | Lys65, Phe66, Pro67, Arg68, Gly69, Gln70, Tyr123, Gly124, Ile131, Trp132, Val133, Ala134, Thr135, Glu136, Gly137, Ala138 |
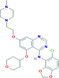
|
Antiviral drug used for the treatment of MERS‐CoV. 36 |
| Camostat | −9.85 | 0.060 | Lys65, Phe66, Arg68, Gly69, Tyr123, Gly124, Ala125, Asn126, Ile130, Ile131, Trp132, Val133, Ala134, Thr135, Glu136, Gly137, Ala138 |
|
Antiviral drug used for the treatment of SARS‐CoV, MERS‐CoV, HCoV‐229E 37 |
| Trametinib | −9.45 | 0.118 | Phe66, Pro67, Arg68, Gly69, Gln70, Tyr123, Ile131, Trp132, Val133, Ala134 |
|
Antiviral drug used for the treatment of SARS‐CoV, MERS‐CoV. 35 |
| Nafamostat | −9.35 | 0.140 | Phe66, Pro67, Arg68, Gly69, Gln70, Gly71, Tyr123, Gly124, Ala125, Asn126, Lys127, Asp128, Gly129, Ile130, Ile131, Trp132, Val133, Ala134 |
|
Antiviral drug used for the treatment of 2019‐nCoV, MERS‐CoV 38 , 39 |
Previous studies have indicated that the catalytic regions of SARS‐CoV‐2 proteins are highly conserved, sharing more than 90% sequence similarity with the corresponding SARS‐CoV and MERS‐CoV proteins. 40 Accordingly, sequence identities between the subfamilies of the receptor and, target complexes are expected to share the same binding mode (drug binding pockets) and exhibit similar activities. The high efficiency of these drug molecules against various viruses, especially MERS‐CoV and SARS‐CoV has already been proven by many experimental studies. For instance, rapamycin has been shown to be a key factor in regulating the replication of various viruses, including Andes orthohantavirus and CoVs. Rapamycin has also been reported to effectively block viral protein expression and virion release. 41 Also, in an in vitro study, it was reported that rapamycin at a 10 μM inhibited MERS‐CoV infection by 61% and at the lowest concentration tested (0.1 μM) by 24%. The other clinical trial reported that saracatinib, which is an inhibitor of the Src/abl family of kinases, exhibited significant antiviral activity against MERS‐COV with an EC50 = 2.9 μM and CC50 > 50 μM, resulting in selectivity (SI) of about >17. Moreover, Shin et al also indicated that saracatinib showed broad antiviral activity against hCoV‐OC43 with an EC50 = 5.1 μM. 36 Furthermore, nafamostat was the most potent inhibitor (IC50: 0.1 μM) based on its capability of blocking MERS‐CoV infection 42 and inhibition of SARS‐CoV‐2 at a low‐micromolar drug concentration in vitro with an EC50 = 2.12 μM; CC50 > 35.53 μM, and SI > 16.76. 38 Likewise, trametinib and camostat demonstrated inhibitory activity against MERS‐CoV and SARS‐CoV infection at low concentrations. 35 , 43 As a result, our in silico study results are compatible with available experimental data.
In addition, in this study, the energetically favorable binding mechanism of the compounds was predicted and a better understanding of this mechanism at the molecular level was provided. Accordingly, rapamycin, the compound with the best binding affinity, was observed to bind to the residues Gly124, Ala125, Ile131, and Trp132 of SARS‐CoV‐2 N protein with a hydrogen bond and Lys65, Ala125, Ala134, and Val133 with an alkyl bond (see Table 2 and Figure 3). Besides, other significantly effective compounds mainly common interacted with Phe66, Arg68, Gly69, Tyr123, Ile131, Trp132, Val133, and Ala134 (see Table 1), which are conserved amino acids in the MERS‐CoV, SARS‐CoV, and SARS‐CoV‐2 N‐NTD domains (see Figure 1c). The results of this study showed that these compounds bind with high affinity to conserved sites in human CoVs, such as MERS‐CoV and SARS‐CoV. The conserved sites in the N‐NTD is responsible for RNA binding and virus infectivity.
TABLE 2.
The non‐covalent interaction analysis of SARS‐CoV‐2 N protein with top five compounds initial and final stage in MD simulation. The stable interactions are indicated in bold color
| Initial stage | Final stage | |||||
|---|---|---|---|---|---|---|
| Complexes | H‐bond interaction | H‐bond length | Hydrophobic interaction | H‐bond interaction | H‐bond length | Hydrophobic interaction |
| SARS‐CoV‐2 N‐NTD & Rapamycin | Trp132 , Ile131, Gln124, Ala125 | 2.81, 1.76, 2.73, 2.68 | Lys65 , Ala125, Ile131, Ala134, Val133 | Asn126, Gly124, Ala125 , Tyr123, Phe66 | 1.94, 2.36, 2.95, 2.41, 2.97 | Lys65, Ala134 , Ile131, Arg68, Ile84 |
| SARS‐CoV‐2 N‐NTD & Saracatinib | Ala134, Lys65, Gln124, Gln70, Ala138, Glu136, Gly69 | 2.02, 2.19, 2.18, 2.88, 2.96, 2.87, 2.81 | Arg68, Val133, Tyr123, Ala134, Ile131 , Phe66 , Lys65 | Trp132, Phe66, Asn126, Ile130, Glu136 | 2.73, 2.92, 2.89, 2.58, 2.71 | Tyr123, Arg68, Ala134, Trp132, Ile131 |
| SARS‐CoV‐2 N‐NTD & Camostat | Glu136, Phe66 , Gln69, Ala134, Ala138, Lys65 , Arg68, Ile131 , Ala125 | 2.36, 2.11, 1.85, 2.20, 2.15, 2.86, 2.94, 1.85, 3.66 | Trp132, Arg68, Ala134 | Glu136, Phe66, Gly137, Ala138, Tyr123, Lys65, Ile131 , Ile130 | 2.18, 2.38, 2.28, 1.89, 1.94, 2.44, 2.78, 2.33 | Lys65, Ala134 |
| SARS‐CoV‐2 N‐NTD & Trametinib | Gln69, Ala134, Arg68, Trp132 | 2.16, 1.90, 2.67, 2.81 | Tyr123, Arg68, Val133, Trp132, Ala134 | Lys65, Phe66, Gln69, Ala134, Lys65, Val133 | 2.28, 2.50, 2.13, 2.51, 2.98, 2.71 | Val133 , Ile131, Phe66, Ala134 |
| SARS‐CoV‐2 N‐NTD & Nafamostat | Ala134, Gln70, Pro67, Asn126, Ile130, Gln124, Trp132 | 2.09, 1.93, 1.88, 2.02, 2.61, 2.69, 1.84 | Trp132 | Asp63, Trp132, Thr135, Asp128, Asn126 | 2.13, 2.43, 1.95, 1.85, 2.14 | Arg68, Val133, Ile131 |
FIGURE 3.
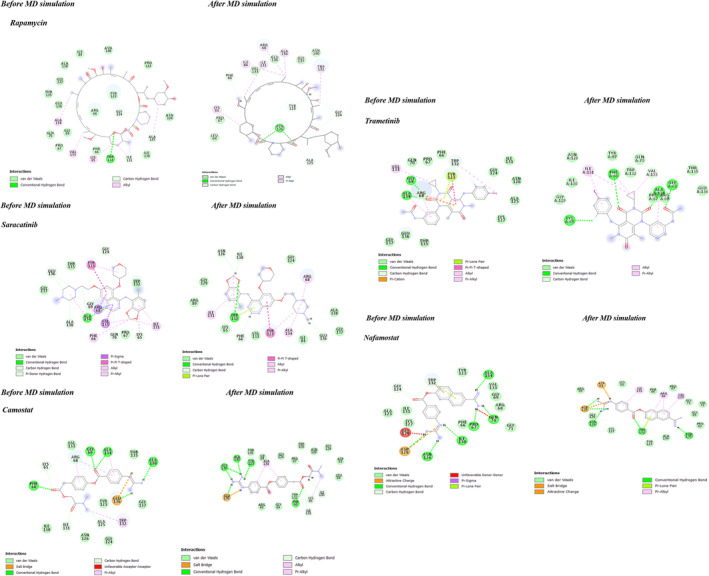
Two‐dimensional (2D) analysis of the lowest energy binding conformations of SARS‐CoV‐2‐N‐NTD and the top five compounds is given
Thus, it can be may suggested that conserved amino acids involved in the non‐covalent interactions may contribute to increase ligand binding affinity for human CoV proteins, facilitating the binding of drug molecules to the NTD‐N‐protein, thereby reducing the efficacy of the N protein to prevent coronavirus infections.
3.2. Molecular dynamics simulation analysis of SARS‐CoV‐2 N with most effective compounds
The molecular dynamic simulations were carried out on these protein‐ligand complex to probe the stabilities of ligand binding modes for most effective compounds. Accordingly, the non‐covalent interaction analysis, RMSF, RMSD, the average of total energy, and potential energy for each structure were calculated during the MD simulation (see Table 2 and Figures 3 and 4, Table S2).
FIGURE 4.
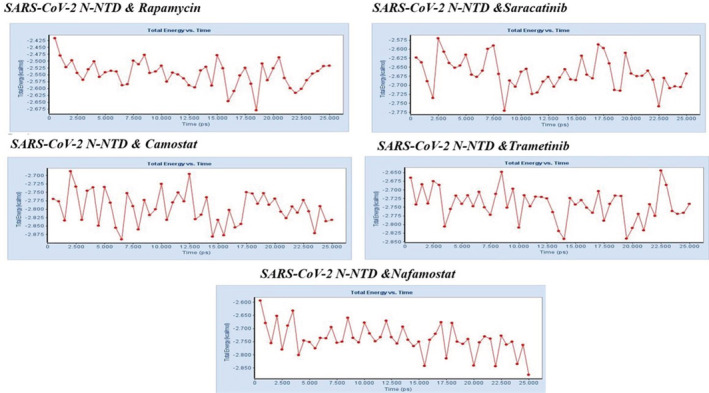
The total energy trajectory of SARS‐CoV‐2 N‐NTD and top 5 compounds at the phase (25 ns) in the simulation
Non‐covalent interactions such as hydrogen bonding and hydrophobic interactions, play a key role in stabilizing energetically favored ligands at the active site of a protein structure, and help improve binding affinity and drug efficacy. Thus, we also analyzed and compared the stabilization of these interactions in the dynamic process at the initial and final stages to understand that the protein‐ligand complex system was stably preserved during the MD simulation analysis. Our analyses indicated that rapamycin, which was most efficient according to molecular docking analysis, continued to maintain interactions with Lys65, Gly124, Ala125, Ile131, and Ala134 of SARS‐CoV‐2 during the 25‐ns MD simulation period. Especially, the hydrogen bond interaction with Gly124 and Ala125 was stable and stronger in the SARS‐CoV‐2 N‐rapamycin complex structure (bond length: 2.68 to 2.36 and, 3.34 to 2.95) after the MD simulation. Likewise, the hydrogen bond interaction with Gly136 in SARS‐CoV‐2 N‐saracatinib and camostat, Gln69 and Ala134 in SARS‐CoV‐2 N‐trametinib, Asn126, and Trp132 in SARS‐CoV‐2 N‐nafamostat continued to interact steadily during the MD simulation. The results of this analysis showed that these important contacts were preserved in the SARS‐CoV‐2 N‐potent compounds (see in Figure 3 and Table 2). In addition, occupancy values were calculated for SARS‐CoV‐2 residues and hydrogen bond interactions that were below 5.0 Å between compounds. Higher occupancy values were obtained for shorter and, therefore, more stable interactions. According to the results of this analysis, these values were found to be high in important amino acids interacting in the binding site (see in Table S3).
Furthermore, the RMSF values of Phe66, Arg68, Gly69, Tyr123, Ile131, Trp132, Ile131, Trp132, Val133, and Ala134, which play an important role in protein‐ligand binding affinity, were between 0.42 and 1.32 nm. In addition, the RMSD values of backbone Cα atoms were calculated to be in the range of 1.87 to 2.70 Å between the initial and the final simulated structures (Table S2 and Figure S1). The results of this analysis show that the stability of the complex structure is reliable and that the interactions that play an important role in the binding affinity of all potent molecules with the protein remain stable throughout the simulation.
4. CONCLUSIONS
Many studies have reported that N proteins will be important candidates for drug‐targeting in other CoVs as they process various critical functions, such as RNA genomic packing, viral replication and transcription, and assembly in infectious cells. 21 , 22 , 25 , 29 , 30 , 44 , 45 , 46 , 47 , 48 Few in silico studies have attempted to design a new drug targeting to inhibit interactions between the SARS‐CoV‐2 N and RNA at the molecular level. For instance, Sarma et al showed that theophylline and pyrimidine drug groups, 21 while Amin et al 49 showed that chloroquine and hydroxychloroquine drug groups are effective against SARS‐CoV‐2 N‐NTD domain. However, the basis of the molecular mechanism of these functions of the newly emerging new SARS‐CoV‐2 N protein is largely unknown. Knowledge of these aspects will contribute significantly to the development of agents that specifically inhibit CoVs replication, transcription, and viral assembly.
In this study, we investigated the interaction mechanism of therapeutic antiviral compounds against SARS‐CoV‐2 N RNA binding domain at the molecular level using structure‐based computational methods. SBDD has been shown as an advanced approach for the discovery and refinement of therapeutic agents 21 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 because it is quite difficult and costly to examine the interaction of many compounds with enzymes at the molecular level under experimental conditions. Thus, these computational methods are providing a significant advantage in the hit identification phase of the costly drug discovery process.
These study results indicated that rapamycin, saracatinib, camostat, trametinib, and nafamostat compounds effectively inhibited the SARS‐CoV‐2 N protein. Rapamycin (Sirolimus) is an agent that can inhibit the mammalian target of rapamycin (mTOR) via the phosphoinositide‐3‐kinase/protein kinase B/mTOR (PI3K/AKT/mTOR) pathway. This pathway plays an important role in CoVs infection and is a new drug target for therapeutic intervention strategies. In experimental studies, it has been reported that rapamycin can inhibit the expansion of Th17 cells in COVID‐19 patients, as in patients with Systemic Lupus Erythematosus. 55 , 56 Likewise, the approved drugs (saracatinib, camostat, trametinib, and nafamostat) are effective in treating HCoV‐229E, MERS‐CoV, SARS‐CoV, SARS‐CoV‐2 infections. 2 , 21 , 28 , 29 , 34
Understanding non‐covalent interactions in protein‐ligand complexes contributes significantly to modern biochemistry and the discovery of new drugs. The affinity of a ligand is related to the binding constant and corresponds to a free energy determined by enthalpic entropic effects. The binding enthalpy is generally treated as the changes in energy resulting from the formations of non‐covalent interactions at the binding interface. 57 Therefore, in this study, we investigated the effects of non‐covalent interactions on binding affinity between protein‐ligand complexes through dynamic processes at the molecular level. In light of this information, our study indicated that energetically favored ligands based on molecular docking analysis, formed hydrogen and hydrophobic interactions with Phe66, Arg68, Gly69, Tyr123, Ile131, Trp132, Val133, and Ala134 in the SARS‐CoV‐2 N‐NTD domain. These interactions continued to persist during the MD simulation period. Amino acid residues that play a role in these interactions may the most important contribution to binding affinity in protein‐ligand complexes.
In conclusion, this study offers a novel testable hypothesis with systematic drug repositioning for rapid and low‐cost identification of approved drugs using computational approaches for the potential treatment of 2019‐nCoV. It also provides better understanding of potential drug targeting sites for the SARS‐CoV‐2 N protein at the molecular level, thus opening new avenues for in vitro validations.
AUTHOR CONTRIBUTIONS
Gizem Tatar: Conceptualization; Methodology; resources. Ezgi Ozyurt: Methodology; resources. Kemal Turhan: Project administration; supervision.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/btpr.3110.
Supporting information
Appendix S1. Supporting Information.
ACKNOWLEDGMENTS
The numerical calculations reported in this abstract were performed at The Scientific and Technological Research Council of Turkey (TUBITAK) ULAKBIM High Performance and Grid Computing Center (TRUBA resources).
Tatar G, Ozyurt E, Turhan K. Computational drug repurposing study of the RNA binding domain of SARS‐CoV‐2 nucleocapsid protein with antiviral agents. Biotechnol Progress. 2021;37:e3110. 10.1002/btpr.3110
[Correction added on 06 January 2021, after first online publication.]
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article and if further data are needed, they are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Perlman S, Netland J. Coronaviruses post‐SARS: update on replication and pathogenesis. Nat Rev Microbiol. 2009;7(6):439‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sawicki SG, Sawicki DL, Siddell SG. A contemporary view of coronavirus transcription. J Virol. 2007;81(1):20‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wijegoonawardane PKM, Cowley JA, Phan T, et al. Genetic diversity in the yellow head nidovirus complex. Virology. 2008;380(2):213‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brian DA, Baric RS. Coronavirus genome structure and replication. In: Enjuanes L, ed. Coronavirus Replication and Reverse Genetics. Vol 287. Current Topics in Microbiology and Immunology. Berlin Heidelberg: Springer; 2005:1‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gorbalenya AE, Snijder EJ, Ziebuhr J. Virus‐encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol. 2000;81(4):853‐879. [DOI] [PubMed] [Google Scholar]
- 6. Lai MMC, Cavanagh D. The molecular biology of coronaviruses. Adv Virus Res. 1997;48:1‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Skariyachan S, Challapilli SB, Packirisamy S, Kumargowda ST, Sridhar VS. Recent aspects on the pathogenesis mechanism, animal models and novel therapeutic interventions for Middle East respiratory syndrome coronavirus infections. Front Microbiol. 2019;10:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dhama K, Patel SK, Sharun K, et al. SARS‐CoV‐2 jumping the species barrier: zoonotic lessons from SARS, MERS and recent advances to combat this pandemic virus. Travel Med Infect Dis. 2020;37:101830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perlman S. Another decade, another coronavirus. N Engl J Med. 2020;382(8):760‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morse JS, Lalonde T, Xu S, Liu WR. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019‐nCoV. Chembiochem. 2020;21(5):730‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jawaid Akhtar M. COVID19 inhibitors: a prospective therapeutics. Bioorg Chem. 2020;101:104027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019‐nCoV). Nat Rev Drug Discov. 2020;19(3):149‐150. [DOI] [PubMed] [Google Scholar]
- 14. Zheng W, Sun W, Simeonov A. Drug repurposing screens and synergistic drug‐combinations for infectious diseases: drug repurposing for infectious diseases. Br J Pharmacol. 2018;175(2):181‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bisson WH. Drug repurposing in chemical genomics: can we learn from the past to improve the future? Curr Top Med Chem. 2012;12(17):1883‐1888. [DOI] [PubMed] [Google Scholar]
- 16. Thiel V, Ivanov KA, Putics Á, et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol. 2003;84(9):2305‐2315. [DOI] [PubMed] [Google Scholar]
- 17. de Haan CAM, Rottier PJM. Molecular interactions in the assembly of coronaviruses. Advances in Virus Research. Vol 64. United States: Elsevier; 2005:165‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zúñiga S, Sola I, Moreno JL, Sabella P, Plana‐Durán J, Enjuanes L. Coronavirus nucleocapsid protein is an RNA chaperone. Virology. 2007;357(2):215‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen C‐Y, Chang C, Chang Y‐W, et al. Structure of the SARS coronavirus nucleocapsid protein RNA‐binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J Mol Biol. 2007;368(4):1075‐1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kang S, Yang M, Hong Z, et al. Crystal structure of SARS‐CoV‐2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm Sin B. 2020;10(7):1228‐1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sarma P, Shekhar N, Prajapat M, et al. In‐silico homology assisted identification of inhibitor of RNA binding against 2019‐nCoV N‐protein (N terminal domain). J Biomol Struct Dynam. 2020;18:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McBride R, van Zyl M, Fielding B. The coronavirus nucleocapsid is a multifunctional protein. Viruses. 2014;6(8):2991‐3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fung TS, Liu DX. Post‐translational modifications of coronavirus proteins: roles and function. Future Virol. 2018;13(6):405‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hsieh P‐K, Chang SC, Huang C‐C, et al. Assembly of severe acute respiratory syndrome coronavirus RNA packaging signal into virus‐like particles is nucleocapsid dependent. J Virol. 2005;79(22):13848‐13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Surjit M, Liu B, Chow VTK, Lal SK. The Nucleocapsid protein of severe acute respiratory syndrome‐coronavirus inhibits the activity of cyclin‐cyclin‐dependent kinase complex and blocks S phase progression in mammalian cells. J Biol Chem. 2006;281(16):10669‐10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liang F‐Y, Lin L‐C, Ying T‐H, et al. Immunoreactivity characterisation of the three structural regions of the human coronavirus OC43 nucleocapsid protein by Western blot: implications for the diagnosis of coronavirus infection. J Virol Methods. 2013;187(2):413‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saikatendu KS, Joseph JS, Subramanian V, et al. Ribonucleocapsid formation of severe acute respiratory syndrome coronavirus through molecular action of the N‐terminal domain of N protein. J Virol. 2007;81(8):3913‐3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grossoehme NE, Li L, Keane SC, et al. Coronavirus N protein N‐terminal domain (NTD) specifically binds the transcriptional regulatory sequence (TRS) and melts TRS‐cTRS RNA duplexes. J Mol Biol. 2009;394(3):544‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keane SC, Liu P, Leibowitz JL, Giedroc DP. Functional transcriptional regulatory sequence (TRS) RNA binding and helix destabilizing determinants of murine hepatitis virus (MHV) nucleocapsid (N) protein. J Biol Chem. 2012;287(10):7063‐7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tatar G, Tok T. Clarification of interaction mechanism of mouse hepatitis virus (MHV) N and nsp3 protein with homology modeling and protein‐protein docking analysis. CAD. 2016;12(2):98‐106. [PubMed] [Google Scholar]
- 31. Tan YW, Fang S, Fan H, Lescar J, Liu DX. Amino acid residues critical for RNA‐binding in the N‐terminal domain of the nucleocapsid protein are essential determinants for the infectivity of coronavirus in cultured cells. Nucleic Acids Res. 2006;34(17):4816‐4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jurrus E, Engel D, Star K, et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018;27(1):112‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dassault Systèmes BIOVIA . Discovery Studio Modeling Environment, Release 2020. San Diego: Dassault Systèmes; 2020. [Google Scholar]
- 34. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785‐2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kindrachuk J, Ork B, Hart BJ, et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for middle east respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob Agents Chemother. 2015;59(2):1088‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shin JS, Jung E, Kim M, Baric RS, Go YY. Saracatinib inhibits middle east respiratory syndrome‐coronavirus replication in vitro. Viruses. 2018;10(6):283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou Y, Vedantham P, Lu K, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015;116:76‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res. 2020;30(3):269‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ito K, Yotsuyanagi H, Sugiyama M, et al. Geographic distribution and characteristics of genotype a hepatitis B virus infection in acute and chronic hepatitis B patients in Japan. J Gastroenterol Hepatol. 2016;31(1):180‐189. [DOI] [PubMed] [Google Scholar]
- 40. Xu J, Zhao S, Teng T, et al. Systematic comparison of two animal‐to‐human transmitted human coronaviruses: SARS‐CoV‐2 and SARS‐CoV. Viruses. 2020;12(2):244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang C‐H, Chung F‐T, Lin S‐M, et al. Adjuvant treatment with a mammalian target of rapamycin inhibitor, sirolimus, and steroids improves outcomes in patients with severe H1N1 pneumonia and acute respiratory failure*. Crit Care Med. 2014;42(2):313‐321. [DOI] [PubMed] [Google Scholar]
- 42. Yamamoto M, Matsuyama S, Li X, et al. Identification of nafamostat as a potent inhibitor of middle east respiratory syndrome coronavirus S protein‐mediated membrane fusion using the split‐protein‐based cell‐cell fusion assay. Antimicrob Agents Chemother. 2016;60(11):6532‐6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. 2012;86(12):6537‐6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chang C, Chen C‐MM, Chiang M, Hsu Y, Huang T. Transient oligomerization of the SARS‐CoV N protein—implication for virus ribonucleoprotein packaging. PLoS One. 2013;8(5):e65045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen I‐J, Yuann J‐MP, Chang Y‐M, et al. Crystal structure‐based exploration of the important role of Arg106 in the RNA‐binding domain of human coronavirus OC43 nucleocapsid protein. Biochim Biophys Acta Proteins Proteom. 2013;1834(6):1054‐1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cong Y, Ulasli M, Schepers H, et al. Nucleocapsid protein recruitment to replication‐transcription complexes plays a crucial role in coronaviral life cycle. J Virol. 2020;94(4):e01925‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kandwal S, Fayne D. Repurposing drugs for treatment of SARS‐CoV‐2 infection: computational design insights into mechanisms of action. J Biomol Struct Dynam. 2020;23:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yadav R, Imran M, Dhamija P, Suchal K, Handu S. Virtual screening and dynamics of potential inhibitors targeting RNA binding domain of nucleocapsid phosphoprotein from SARS‐CoV‐2. J Biomol Struct Dynam. 2020;1‐16. [Online ahead of Print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Amin M, Abbas G. Docking study of chloroquine and hydroxychloroquine interaction with RNA binding domain of nucleocapsid phospho‐protein—an in silico insight into the comparative efficacy of repurposing antiviral drugs. J Biomol Struct Dynam. 2020;1‐13. [Online ahead of Print]. [DOI] [PubMed] [Google Scholar]
- 50. Bayel Secinti B, Tatar G, Taskin TT. Determination of potential selective inhibitors for ROCKI and ROCKII isoforms with molecular modeling techniques: structure based docking, ADMET and molecular dynamics simulation. J Biomol Struct Dynam. 2018;37:1‐7. [DOI] [PubMed] [Google Scholar]
- 51. Bhowmik D, Nandi R, Jagadeesan R, Kumar N, Prakash A, Kumar D. Identification of potential inhibitors against SARS‐CoV‐2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infect Genet Evol. 2020;84:104451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meyer‐Almes F‐J. Repurposing approved drugs as potential inhibitors of 3CL‐protease of SARS‐CoV‐2: virtual screening and structure based drug design. Comput Biol Chem. 2020;88:107351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tatar G, Tok TT. Structure prediction of eukaryotic elongation factor‐2 kinase and identification of the binding mechanisms of its inhibitors: homology modeling, molecular docking and molecular dynamics simulation. J Biomol Struct Dynam. 2019;1‐16. [Online ahead of Print]. [DOI] [PubMed] [Google Scholar]
- 54. Nimgampalle M, Devanathan V, Saxena A. Screening of chloroquine, hydroxychloroquine and its derivatives for their binding affinity to multiple SARS‐CoV‐2 protein drug targets. J Biomol Struct Dynam. 2020;1‐13. [Online ahead of Print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lai Z‐W, Borsuk R, Shadakshari A, et al. Mechanistic target of rapamycin activation triggers IL‐4 production and necrotic death of double‐negative T cells in patients with systemic lupus erythematosus. J Immunol. 2013;191(5):2236‐2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lai Z‐W, Kelly R, Winans T, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single‐arm, open‐label, phase 1/2 trial. Lancet. 2018;391(10126):1186‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Klebe G, Böhm HJ. Energetic and entropic factors determining binding affinity in protein‐ligand complexes. J Recept Signal Transduct Res. 1997;17(1–3):459‐473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article and if further data are needed, they are available from the corresponding author upon reasonable request.