

Abstract
The application of sequencing technology is shifting from research to clinical laboratories owing to rapid technological developments and substantially reduced costs. However, although thousands of microorganisms are known to infect humans, identification of the etiological agents for many diseases remains challenging as only a small proportion of pathogens are identifiable by the current diagnostic methods. These challenges are compounded by the emergence of new pathogens. Hence, metagenomic next‐generation sequencing (mNGS), an agnostic, unbiased, and comprehensive method for detection, and taxonomic characterization of microorganisms, has become an attractive strategy. Although many studies, and cases reports, have confirmed the success of mNGS in improving the diagnosis, treatment, and tracking of infectious diseases, several hurdles must still be overcome. It is, therefore, imperative that practitioners and clinicians understand both the benefits and limitations of mNGS when applying it to clinical practice. Interestingly, the emerging third‐generation sequencing technologies may partially offset the disadvantages of mNGS. In this review, mainly: a) the history of sequencing technology; b) various NGS technologies, common platforms, and workflows for clinical applications; c) the application of NGS in pathogen identification; d) the global expert consensus on NGS‐related methods in clinical applications; and e) challenges associated with diagnostic metagenomics are described.
Keywords: clinical application, infectious disease, metagenomics, next‐generation sequencing
Sequencing technology is becoming increasingly available in clinic. This review sheds lights on the most commonly used metagenomic next‐generation sequencing (mNGS). History and different platforms, current workflows, and applications of mNGS in pathogens identification, as well as challenges in the diagnostic metagenomics, are discussed. mNGS cannot substitute for traditional methods in the short term, but plays an irreplaceable role in microbiological detection.
1. History of Sequencing Technology
Since the development of first‐generation Sanger sequencing in 1977, DNA sequencing technology has made considerable strides.[ 1 ] From the first‐generation to the fourth‐generation, the length of sequenced reads has changed from long to short, while from the second to the fourth‐generation, the read length has change from short to long. Although the third‐ and fourth‐generation sequencing technologies have developed rapidly,[ 2 ] the current next‐generation short‐read and long‐read sequencing technologies maintain a dominant position in the global sequencing market.[ 3 ] Nevertheless, the development of each generation of sequencing technology has served to significantly improve a myriad of fields in genome and medical research, drug development, infectious diseases, etc. (Table 1 ).
Table 1.
Overview of high throughput sequencing platform parameters in pathogens detection
| Platform | Method | Read length [bp] | Read type | Advantages | Disadvantages | Refs. |
|---|---|---|---|---|---|---|
| Second‐generation sequencing | ||||||
| Illumina MiSeq | Sequencing by synthesis and reversible termination | 150, 250 | PE | The read length is relatively long | The sequencing results were obtained in 24–36 h, low throughput | [ 196 , 215 ] |
| Illumina NextSeq500/550 | 75, 150 | SE, PE | The sequencing results were obtained in 11–29 h | Short read length | [ 25 , 26 , 236 ] | |
| Illumina HiSeq 4000 | 125, 150 | PE | High throughput and long read length | The sequencing results were obtained in 84 h | [ 21 , 25 ] | |
| Illumina NovaSeq 600 | 150 | PE | High throughput and long read length | The sequencing results were obtained in 40 h | [ 237 , 238 ] | |
| BGISEQ‐50 | Combinatorial probe anchor ligation and DNA nanoball | 50 | SE | Low cost, received medical device certification clearance | Short read length | [ 24 , 132 , 239 ] |
| BGISEQ‐100 | 50 | SE | Low cost | [ 29 , 30 ] | ||
| MGISEQ‐2000 | 100 | PE | Low cost, received medical device certification clearance | [ 240 , 241 ] | ||
| Third‐generation sequencing | ||||||
| PacBio Sequel | Single molecule real time (SMRT) sequencing | 1–1.8 kb | SE | Long read length | Low accuracy and high cost | [ 242 ] |
| Oxford Nanopore MinION | Nanopore sequencing | / | 1D | Long read length and rapid sequence time | Low accuracy and high cost | [ 174 ] |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
1.1. First‐Generation Sequencing Technologies
The development of the first‐generation sequencing technologies began in the 1970s. In 1975, Sanger and Coulson established a simple and rapid method for determining DNA sequences using primed synthesis with DNA polymerase.[ 4 ] In 1977, Maxam and Gilbert further developed a DNA sequencing technique via chemical degradation.[ 5 ] Both of these initial platforms relied on manual sequencing. Whereas the development of the fluorescence detection technology and capillary electrophoresis technologies catapulted DNA sequencing into the era of automatic sequencing. At the end of the 20th century, automatic plate electrophoresis sequencing and capillary sequencing were introduced, with the latter playing a particularly important role in the sequencing work performed for the Human Genome Project (HGP).[ 6 ]
The primary feature of first‐generation sequencing technology, known as the gold standard method, is the ability to read DNA with a length of ≈1000 bp with 99.999% accuracy. However, its high cost (half a dollar per 1000 bases), low throughput (96–384 samples per round of sequencing), among other disadvantages, significantly impeded its large‐scale application. Nevertheless, Sanger sequencing continues to be used for whole genome sequencing of pathogens with small genomes, such as humanin fluenza A viruses, and Zika virus, as well as for bacteria 16S rRNA gene sequencing and fungi 18S rRNA gene sequencing.[ 7 , 8 , 9 , 10 ] It is also used to verify the authenticity of microorganisms detected by next‐generation sequencing (NGS). For example, in 2017, Chinese scientists detected Pseudorabies Virus in vitreous humor for the first time using NGS technology, while Sanger sequencing was used to further verify their findings.[ 11 ]
1.2. Second‐Generation Sequencing Technologies
Second‐generation sequencing technologies, also commonly referred to as NGS or high‐throughput sequencing technologies, improved upon the low throughput issue associated with first‐generation sequencing methods. Since the year 2000, massively parallel sequencing technologies have made high‐throughput sequencing possible. Several developments such as pyrosequencing, reversible terminator chemistry sequencing, as well as supported oligonucleotide ligation and sequencing have greatly increased throughput. Simultaneously, the cost of sequencing has been greatly reduced with time.[ 12 ] The most common current applications of NGS in diagnostic microbiology laboratories include targeted NGS with different methods for enrichment including amplification or probe hybridization, and metagenomics next‐generation sequencing (mNGS). Currently, the most widely employed second‐generation sequencer in clinical settings is manufactured by Illumina, followed by Beijing Genomics Institute (BGI).
1.2.1. Illumina Sequencing by Synthesis
Illumina‐based platforms perform sequencing using a synthesis and reversible terminator chemistry strategy,[ 13 , 14 ] in addition to the bridge amplification method, whereby single molecules of DNA are first attached to a flow cell and subsequently amplified locally into a clonal cluster.[ 13 ] Next, a sequencing by synthesis reaction occurs, in which synthesis of complementary DNA occurs via addition of one nucleotide per cycle, and the optical readout of the fluorescently labeled nucleotides then determines its identity (A, G, T, or C).[ 13 ] Illumina offers a popular series of platforms, including HiSeq, MiniSeq, MiSeq, NextSeq, and NovaSeq, which are all suitable for ultralarge‐scale sequencing and to meet the needs of different sequencing scales and applications.[ 15 , 16 , 17 , 18 ] The Illumina MiSeq instrument was the first platform to be applied for clinical pathogen detection after it was successfully used to diagnosis Leptospira infection within 48 h in a boy with severe comprehensive immunodeficiency. Since this time, a new era has begun for the clinical application of mNGS.[ 19 ] Specifically, the HiSeq series platform is generally used to carry out clinical pathogenic detection on large sample sizes due to its high‐throughput and relatively long sequencing read length, however, each run can take up to 3.5 days, which is not suitable for the rapid detection of single samples.[ 20 , 21 , 22 ] Alternatively, NextSeq500 and NextSeq550 series sequencing systems offer the advantages of moderate throughput and short running time (12–30 h per run), making them particularly suitable for the detection of clinical pathogens, for which they are widely used.[ 23 , 24 , 25 , 26 ]
1.2.2. BGI Sequencing
Sequencing using the combinatorial probe anchor ligation (cPAL) and DNA nanoballs (DNB) methods is the core of BGI.[ 27 ] Single‐stranded circular DNA is amplified using rolling circle amplification (RCA) by 2–3 orders of magnitude; the amplification products are designated, DNB. The nanoballs are then fixed on an arrayed silicon chip using DNB loading technology, which relies on joint probe anchoring polymerization. DNA molecular anchors and fluorescent probes are polymerized on the DNB, and the resulting optical signals are collected by a high‐resolution imaging system and digitally processed to obtain the sequence of interest.[ 28 ] The BGI sequencing platform is being increasingly used in clinical pathogen mNGS detection due to its low cost, short sequencing time and other advantages. BGISEQ sequencers are most commonly employed to detect various pathogens, including those causing focal infections, Mycobacterium tuberculosis (MTB), etc.[ 24 , 29 , 30 ]
The development of second‐generation sequencing technologies has not only greatly promoted the development of biological and medical research, but has also ushered in a new era for the clinical detection of pathogens. However, regardless of the platform or technology used, the resulting read lengths using mainstream methods are short at ≈50–300 bp, and subsequent analysis depends on fragment splicing, which may introduce errors. Moreover, PCR amplification is performed during library preparation to amplify low amounts of starting DNA and to aid sequence enrichment. However, sequences with low content may not be amplified, resulting in the loss of certain information. Further, the run time required for second‐generation sequencing is long with most sequencers requiring more than 12 h, hence, the sequencing data cannot be obtained in real‐time. Hence, there remains room for improvement in these sequencing technologies.
1.3. Third‐Generation Sequencing Technology
The third‐generation sequencing technology is also known as single‐molecule sequencing technology.[ 31 ] Compared with NGS, third‐generation sequencing technologies can detect ten nucleotides per second, which greatly reduces the time required for sequencing. Moreover, the full‐length mRNA sequence can be derived from the ultralong read lengths obtained. Third‐generation sequencers can also directly sequence the original DNA/RNA samples without requiring PCR amplification, with no preference for CG nucleotides, and can directly detect and obtain methylation information. However, despite the many advantages of third‐generation sequencing, it has not been widely adopted in clinical applications due to its high error rate and high associated cost.[ 2 ]
1.3.1. Single‐Molecule Real‐Time (SMRT) Sequencing
SMRT sequencing is a representative of third‐generation sequencing technology that applies the principle of sequencing by synthesis. SMRT cell contains nanoscale zero‐mode waveguide (ZMW) wells. Each ZMW contains a DNA polymerase molecule and DNA sample chain, which are template fragments that are processed and ligated to hairpin adapters at each end, resulting in a circular DNA molecule with constant single‐stranded DNA (ssDNA) regions at each end, and the double‐stranded DNA (dsDNA) template in the middle. This platform allows for single‐molecule sequencing and real‐time detection of the fluorescence signal from the nucleotides. Incorporation of nucleotides into the growing chain results in the dissociation of the labeling groups, which reduces steric hindrance and helps maintain the continuous synthesis of DNA chains, prolonging the sequence read length.[ 32 , 33 ]
Additionally, the newly launched SMRT Sequel series by PacBio provides an average read length of 10–20 kb, and can achieve sequencing throughputs of 160 GB. However, in spite of these features, the bulky equipment and expensive hardware make this system unpopular for clinical diagnosis of pathogens. Nevertheless, during the novel coronavirus pandemic, the PacBio system has been employed numerous times for whole genome sequencing and reassembly of SARS‐CoV‐2.[ 34 , 35 ]
1.3.2. Nanopore Sequencing
Nanopore sequencing technology is another representative third‐generation sequencing technology, however, it is also considered to be a fourth‐generation platform as it can perform real‐time data acquisition and analysis.[ 36 ] A core component of nanopore sequencing is the use of nanoscale pores composed of transmembrane proteins, which is a significant departure from the previous sequencing technologies.[ 37 , 38 ] In 2012, Oxford Nanopore Technologies released the GridION and MinION sequencing platforms.[ 39 ] MinION is a mobile single‐molecule nanopore sequencing device that is 4 in. long and can be connected through a USB3.0 port to a laptop computer. It is, therefore, conveniently portable and meets DNA sequencing needs under several conditions. Notably, nanopore DNA sequencing is orders of magnitude faster than other strategies for the diagnosis of infectious diseases. The data output of a single flow cell in the platform is 10–25 Gb, with the sequencing time ranges from 0 to 48 h, and read lengths of 800 kb can be achieved, through the error rate can be as high as 5–40%.[ 40 , 41 , 42 ] Due to the advantages of nanopore sequencing technology, it has been widely adopted in the field of epidemic outbreak investigation to detect infectious pathogens, antimicrobial resistance, as well as other infectious areas of concern.[ 43 , 44 , 45 , 46 ] However, the high number of sequencing errors, and higher per‐read costs compared to other NGS platforms may limit its utility for certain applications.
2. Sequencing Methods and Bioinformatic Analysis
The process of high‐throughput sequencing of pathogens primarily includes two components: experimental manipulations (wet lab) and bioinformatic analysis (dry lab). The wet lab manipulations include sample pretreatment, nucleic acid extraction, library construction, and sequencing. The dry lab bioinformatic analysis includes quality control of data, removal of human sequences, sequence alignment of sequences from microbial species, and analysis of genes for drug resistance or virulence.
However, the diagnosis of infectious diseases requires that prior to pretreatment of samples, specific samples must first be collected from the primary site of infection. For example, bronchoalveolar lavage fluid (BALF) and sputum are generally recommended for pulmonary infection, while cerebrospinal fluid (CSF) is recommended for central nervous system (CNS) infection. Although the library construction, sequencing and bioinformatics analysis of different samples are the same, pretreatment and nucleic acid extraction differs depending on the sample source. The following section will describe the mNGS detection process for samples suitable for different infection systems (Figure 1 ).
Figure 1.
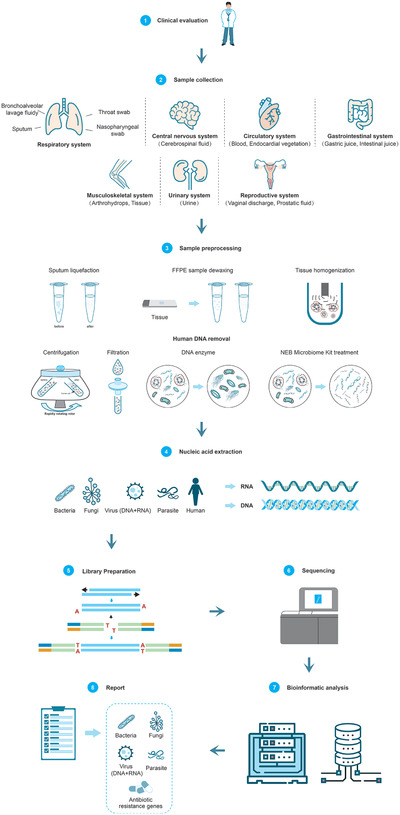
mNGS workflow in clinical application. This workflow consists of eight components. 1) Clinical evaluation: the test is suitable for patients with infectious diseases. 2) Sample collection: collecting samples from the primary site of infection can greatly increase the detection rate. 3) Sample preprocessing: the pretreatment methods for different types of samples are different, sputum needs liquefaction treatment, FFPE samples are dewaxed, and tissue needs homogenate. The percentage of human DNA in samples can be reduced using methods such as filtration, differential centrifugation, DNA enzymatic hydrolysis, and methylation reagent treatment. 4) Nucleic acid extraction: there are differences between DNA and RNA extraction. 5) Library preparation: library construction method is selected according to the sequencing platform and purpose. 6) Sequencing: at present, the mainstream second‐generation sequencing platforms are produced by Illumina and BGI. 7) Bioinformatic analysis: based on the analysis of the raw data, the information of species and antibiotic resistance genes in the samples were obtained. 8) Report: the possible pathogens were screened out according to the analysis results.
2.1. Sample Preprocessing
2.1.1. Sputum
For respiratory tract infections, the most common samples are BALF and sputum. However, the fiberoptic bronchoscopy procedure, required for collection of BALF, is invasive and often deemed intolerable by patients, thus sputum becomes an alternative sample.[ 21 ] Sputum samples are generally highly viscous and heterogeneous; additionally, the distribution of microorganisms in sputum is uneven. Therefore, sputum liquefaction treatment is performed prior to extracting nucleic acids from the samples. The existing sputum liquefaction schemes include the following methods: i) standard N‐acetyl‐l‐cysteine treatment and digestion with 2% NaOH and decontamination; ii) NaOCl liquefaction and sedimentation; iii) chitin solution liquefaction; iv) the use of sputum and an equal volume of phosphate buffer containing 1 g L−1 protease K to liquefy sputum; and v) classical dithiothreitol (DTT) liquefaction, in which a chitin solution homogenizes the mucus sputum more quickly than the N‐acetyl‐l‐cysteine and NaOCl methods.[ 47 , 48 , 49 ] With the advancement of automated processing, a portable, low‐power preprocessing device can perform all of the required steps for sputum preprocessing, including liquefaction, homogenization, dissolution, and inactivation.[ 50 ]
2.1.2. Tissue Sample
Tissue samples account for one of the most common types of clinical samples as they allow for the detection of various infections, including lung, brain, skin, and soft tissue. For patients who require surgery, the relevant tissues can be obtained during the operation without requiring additional sampling steps.[ 51 , 52 , 53 ] Meanwhile, formalin fixed paraffin embedded (FFPE) samples can introduce contaminating microorganisms and cause damage to DNA and RNA due to the complex processing steps required. Therefore, although these samples can be preserved for an extended period of time and are important for the detection of infectious diseases, they can be difficult to process and analyze effectively. FFPE samples are first dewaxed by treating 25 µm thick sections in 1.5 mL tubes with 120 µL of xylene for 10 min twice, followed by 1200 µL of 100% ethanol for 10 min three times with constant gentle agitation for deparaffination.[ 54 ] Alternatively, fresh tissue is commonly used for analysis, which requires tissue homogenization. The FastPrep‐24 Instrument can be used for tissue fragmentation, after which the centrifuged supernatant is then used for further experiments.[ 55 ] Tissue digestion is performed using a lysis buffer composed of 0.5% Tween 20, 2 mg proteinase K, 3.5 × 10−3 m MgCl2, 15 × 10−3 m ammonium sulfate, and 60 × 10−3 m Tris‐HCl at 56 °C for 1 h to obtain transparent cleavage products.[ 54 ] Although tissue samples often have high human cell background content, their sequences can be eliminated from analysis by using an appropriate dehosting process to improve the sensitivity of overall detection.
2.2. Nucleic Acid Extraction
Nucleic acid extraction is an important step in the mNGS detection process. Sufficient DNA or RNA of appropriate quality is extracted from cells for downstream processing. To achieve this, various sample extraction strategies are employed. The raw input varies based on the source and type of sample, as well as the method of preprocessing used.[ 56 ] The extraction strategy for cell‐free DNA (cfDNA) or cell‐free RNA (cfRNA) is generally used in samples from peripheral blood.[ 57 ] Accordingly, each commercial vendors often offers several different kits for manual or automated extraction using liquid‐handling robots. After dewaxing, DNA from FFPE samples can be extracted using nucleic acid extraction kits suitable for different target microorganisms.[ 54 ] With the advancement of nucleic acid extraction kits, mature commercial kits (such as the Molzym Ultra‐Deep Microbiome Prep kit) are now suitable for DNA extraction from various types of samples, including biopsies, which are capable of removing host DNA.[ 54 , 58 ]
2.3. Library Preparation and Sequencing
Currently, several platforms and instruments are available for mNGS, each with different schemes for library construction, the complexity of which is greater for second‐generation sequencing libraries compared to third‐generation. In this section, we introduce the commonly used database construction schemes for Illumina, BGI, Oxford Nanopore technologies, and the PacBio sequencing platforms.
The Illumina sequencing platform is currently the most widely used in the field of mNGS. Its library construction process involves the random breakage of genomic DNA or double‐stranded cDNA (obtained by RNA reverse transcription) into small fragments by enzymatic cleavage or using ultrasound, enzyme‐based flattening of both ends of the DNA molecule, addition of an A base to the 3′‐end using the Klenow enzyme, and adapter ligation.[ 59 ] However, the reliance of all existing RNA sequencing on RNA reverse transcription to produce cDNA, followed by second‐strand synthesis via additional enzymes and purification steps, can introduce sequence‐dependent bias. Recently, Chinese scientists combined the Tn5 transposase with transposon sequencing to develop a new rapid library construction method, called the SHERRY method. Compared to existing methods, it significantly simplifies the process of database construction and reduces the initial template input, which is relevant for samples with low starting concentrations, including that of coronaviruses.[ 60 , 61 ]
Certain similarities and differences exist between the BGISEQ technologies and the Illumina sequencing platforms. In both cases, five sample preparation steps are required: sample fragmentation, end repair, addition of an A nucleotide, adaptors addition, and PCR; however, the specific experimental steps performed and the enzymes used are quite different. In the BGI prescribed method, the DNA overhangs are filled in to form blunt‐ended molecules, and a sequence of connectors is added to the two DNA fragments ends to connect them, forming a ring structure. A special molecule of cyclic DNA is then thermally denatured together, which is added in reverse to a special chain of the PCR product, and the single‐stranded molecule is connected by DNA ligase. The remaining single‐stranded linear molecules are digested by exonuclease to obtain a single‐stranded circular DNA library. PMseq was sequenced by single‐stranded ring DNA amplification to form DNA nanospheres (DNB), which are fixed on arrayed silicon chips by rolling ring amplification (RCA).[ 62 , 63 ]
Oxford Nanopore DNA library construction methods can be divided into three categories: 1D, 1D2, and 2D methods.[ 64 ] The 1D2 library construction method is an improvement over the 1D method, and though a higher throughput can be obtained using the 1D library, a higher sequence accuracy is achieved with the 1D2 library.[ 37 , 65 ] Further, libraries constructed using the Nanopore kits do not require the reverse transcription of RNA into cDNA, as it is possible to directly sequence the single RNA strand. Therefore, construction of the RNA libraries can be divided into two categories based on end‐use: 1) direct sequencing; 2) sequencing of the cDNA library by reverse transcription. The time required to construct the two libraries is similar (115 min vs 125 min, respectively); however, direct sequencing requires ≈500 ng of input, RNA template, whereas the cDNA library only needs ≈50 ng.[ 66 , 67 ]
The SMRT bell libraries used in PacBio sequencers are dumbbell‐shaped and offer the advantage of circularizing whole molecules. Hence, the molecules can be sequenced repeatedly, allowing for the generation of long‐read lengths. The dumbbell‐shaped molecules are composed of two parts: the hairpin adapter and the dsDNA template. Construction of the sequencing library involves DNA fragmentation to obtain dsDNA fragments with sticky ends, and subsequently, AMPure PB magnetic beads used for size selection and purification of the target fragments. Next, damage repair and end repair of the DNA template is performed to enable its use in direct sequencing and to avoid negative effects on read length. Subsequently, the flat end hairpin joints are connected to the DNA template, which is then purified and quantified.[ 68 ] Additionally, Coupland et al.[ 69 ] reported that small DNA molecules can be sequenced directly from 1 ng of DNA without requiring standard library preparation.
2.4. Bioinformatic Analysis
Bioinformatic analysis is the final step required for mNGS‐based detection of pathogenic microorganisms, which enables their rapid and accurate identification. Pathogenic infections require varying treatments, making it imperative that their detection is accurate, direct, and verifiable. Errors and contamination can be introduced throughout the process during sampling, assaying, or sequencing of samples, as well as by the instrument consumables or production environment. Errors may also be introduced during bioinformatic analysis, resulting in false‐negative or false‐positive results. From the sequencing read‐based identification of a species, it is necessary to optimize and verify every step of the process, including quality control, human host subtraction, microbial identification, verification, etc. (Figure 2 ).
Figure 2.
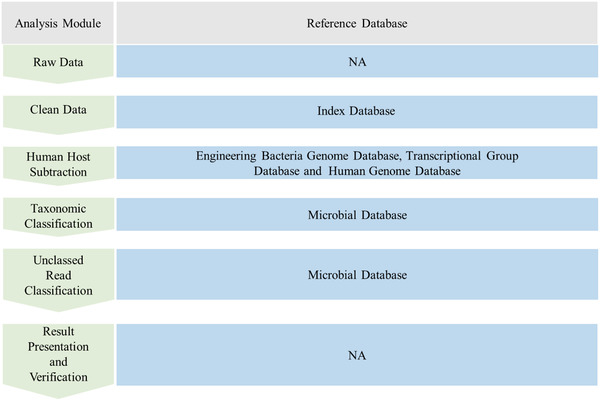
The bioinformatics analysis process starts with the fastq date, including the removal of low‐quality and low‐complex sequences, host and engineering bacteria sequences, and the identification of pathogens. Samples qualified for sequencing (Q30 qualified), the proportion of removing low‐quality and low‐complex sequences is about 5%, which accounts for about 20% of the whole process time. Removal of hosts, engineering bacteria, plasmids, different types of samples and different treatment methods, and the host proportion varies greatly, between 40% and 99%; this step accounts for about half of the whole process time. For taxonomic classification, different alignment software is quite different, such as k‐mer algorithm, there are 30% to 80% of the sequences, which can be assigned to microbes. The proportion of unclassified sequences is high, so can choose to compare with NR database to identify distant sequences. This step accounts for about 30%.
2.4.1. Raw Data
Raw sequencing data can be generated using different platforms, for which the first in silico analysis step involves the separation of data for each sample into independent files per index of the individual sample library. The sequenced reads are usually retrieved as FASTQ files. Particular attention must be paid to the amount of sequencing data for each sample (at least 10 Mb is currently required) and the quality of the sequenced reads (Q20/Q30). The sequencing quantity and quality should be of an acceptable standard before further processing, otherwise additional data may be required.
2.4.2. Clean Data
The next step in the analysis of sequencing data requires removal of low‐quality reads, which may interfere with the downstream analysis or cause false positives. The quality control process includes trimming of reads to remove adaptors/adaptor trimming, quality filtering of reads, removal of low‐quality reads, removal of short reads, discarding reads shorter than 36 nucleotides, low‐complexity read filtering, and removal of duplicate reads. The Trimmomatica or Fastp tools are commonly used for the removal of low‐quality reads, adaptors, and short reads.[ 70 , 71 ] Additionally, tools such as DustMasker are used to mask low‐complexity regions, while duplicate reads can be removed using PRINSEQ, and FastQC can be used to graphically analyze the data for each sample.[ 72 ]
2.4.3. Human Host Subtraction
The presence of contaminating human DNA is common in the sequencing results for various microbes, making it necessary to remove interference from the host sequence to shorten the analysis time. To this end, sequence comparison with the human reference genome can be made, and the host sequence removed, though software and reference genome selection does affect the degree to which host DNA removal is effective.
Next, sequence alignments are performed relative to the reference genome using mapping software, such as Bowtie 2, BWA, HISAT2, etc.[ 73 , 74 , 75 ] HISAT2 is based on the HISAT and Bowtie2 implementations, small indexes, and alignment speed. These tools are widely used in genome resequencing and RNASeq data analysis, providing alignment accuracy, optimization of called mutations, and discarding unmapped reads from the final output. Software such as BMTagger and CS‐SCORE are specifically used to remove host DNA,[ 76 , 77 ] compared to Bowtie2 and BWA, these programs offer better host DNA removal rates.[ 78 ] Further, BMTagger is the standard operating procedure tool used for the Human Microbiome Project.
The human reference genome (current version: GRCh38.p13, release date 2020.04) consists of chromosomal scaffolds, unplaced scaffolds, and “alternate” scaffolds, with the latter representing sequences that are divergent from the primary chromosome sequence. The earliest release was in October 2014; however, the previously released version GRCh37 from April 2011 (updated in December 2013; UCSC version hg19) is also widely used as annotation libraries are available. When removing host DNA, priority should be given to the latest reference genome version. It is advisable to not use versions optimized for mapping, which include “traps,” Hepatitis B virus sequences, and chromosome optimizations. For more efficient removal of the host sequences, the human mRNA reference sequence of human can be added to the reference genome simultaneously. Further, given the differences between populations, different versions of the human genome can be added to the host database for detection of DNA sequences from different populations, as well as sequences from bacteriophages, plasmids, engineered bacteria, and potentially contaminating bacteria, to remove the corresponding interfering sequences.
2.4.4. Reference Databases
For taxonomic classification, alignments are performed relative to reference databases regardless of the algorithm used. Various reference genomes can be used for library comparisons, including the National Center for Biotechnology Information (NCBI) nonredundant nucleotide sequence database (NT/NR), NCBI Reference Sequence (RefSeq) and genome database, NCBI GenBank database, and microbial reference sequences or genome collections.[ 79 ] Based on the sequence similarity and algorithm, the NT/NR library and reference genome library (with higher assembly level) can be selected based on marker specific region recognition. Further, a k‐mer counting algorithm is recommended, as it is fault‐tolerant and enables the analysis of microbial genome sequences, which is helpful to improve the accuracy and positive rate of identification.[ 80 ]
Microbial species are extremely diverse and include viruses, bacteria, fungi, and parasites. To enable their identification, it is necessary to analyze their genomes and construct a microbial reference genome database. However, the construction of reference databases should consider potential discrepancies in the study, as in some cases, the similarity between species is greater than that between strains of the same species when classification is based on morphology, as shown by a comparison of genomic differences.[ 81 ] Further, differences may exist in the reference genome sequence due to variations in genome quality, including at the contig, scaffold, and whole genome levels. Hence, the various methods available for database construction must be carefully analyzed for choice of sequencing platform, sequencing depth, splicing software, and establishment of a standard genome grouping mechanism. These methods must be combined with comparative genome analysis, phylogeny, genome quality assessment using tools such as CheckM, establishment of a filtering mechanism, quality assessment of contig and scaffold sequences in the genome for assessment of contaminating sequences, and removal of low‐quality sequences.[ 82 ] Additionally, publicly available curated microbial genome databases, such as FDA‐Argos and the FDA Reference Viral Database (RVDB), have been made available.[ 83 ] A combined approach that incorporates annotated sequences from multiple databases may encourage greater confidence in the sensitivity and specificity of microorganism identification.
2.4.5. Taxonomic Classification
Metagenomics classification tools, including the Kraken series represented by the k‐mer algorithm include Kraken, Bracken, KrakenUniq, Kraken 2, Centrifuge, and CLARK.[ 80 , 84 , 85 , 86 ] For a certain length (k‐mer length) reference genome, the sliding window technique using 1 bp window slides is used. If the k‐mer appears in two species, it is assigned to the smallest taxon lowest common ancestor (LCA), and the genome sequences of all microorganisms are analyzed in turn, establishing the k‐mer database. During the identification of classifier sequences, the trimming of reads is similar to the defining of k‐mers for database query, and the classification of reads is determined based on the information of each k‐mer. Results containing false positives are a significant challenge in metagenomics classification, which has been addressed by the KrakenUniq algorithm, which optimizes the results in the Kraken classification results. Bracken calculates the abundance of detected species based on Kraken. The Kraken 2 algorithm optimizes the storage index of k‐mers, which reduces the memory required and improves the speed by reducing memory usage by 85%.[ 85 ] Alternatively, the Centrifuge algorithm uses an indexing scheme based on the Burrows–Wheeler transform (BWT) and the Ferragina–Manzini (FM) index, and is optimized specifically to resolve metagenomic classification related issues. Methods, such as MetaPhlAn2, that analyze the composition of microbial communities, use unique marker genes for classification, which can be optimized for specific groups, such as mycobacteria. Although this platform offers superior identification accuracy and more professional library construction, it is possible that certain identified reads are lost.[ 87 ]
In studies investigating macrogenomic communities, most species are unknown, and their identification is conducted by comparing with protein sequences, using tools such as DIAMOND and Kaiju. Samples with a large number of unclassified reads indicate that there may be unknown microbe species present in the community. This can be confirmed using the nr protein library.[ 88 , 89 ] The alignment‐based methods, such as BLAST and Mega BLAST,[ 90 ] are widely used for the identification of taxonomic differences using sequencing data and can be regarded as the gold standard. However, due to the amount of data generated by NGS, the operation speed of such aligners for these datasets is slow, making them not ideal, and not commonly used, for this purpose. Nevertheless, the results obtained from identification analyses can be verified using such aligners.
2.4.6. Report
The mNGS technique is highly sensitive, capable of readily detecting pathogens in samples, and identifying microorganisms in reagents, consumables, and the environment. However, to achieve accurate results, data must be filtered according to a certain threshold. Commonly used filtering indicators include sequencing reads and relative abundance.
The type of pathogen influences the amount of sequencing data obtained. Considering the differences in microbial genome size, the detected sequences are first standardized based on reads per million (RPM). The threshold for viruses is generally at, or above, 3 RPM and exceeds 1000 RPM for retroviruses. For the identification of fungi, the threshold is above 5 RPM, and the credibility of the applied threshold is greater when there is an increase in the number of sequenced reads that can reach 100 RPM. For intracellular bacteria, such as MTB and Legionella, the threshold is relatively low, and credible data can be obtained with a threshold of 1 RPM. With an increase in the number of sequences, credibility gradually increases, however, upon reaching 30 RPM, further increments no longer significantly increase the credibility of the threshold. For parasites, the applied threshold should be above 10 RPM, and the specificity of the sequence should be strictly confirmed.[ 3 , 91 , 92 , 93 ]
Different thresholds may need to be set based on data from the negative control and sequence detection data. For example, in a virus, if the number of detected sequences covers more than three nonoverlapping regions of the virus reference genome, the detected sequences are first standardized as RPM. Next, the ratio (RPM‐r) of the clinical sample RPM to that of the negative quality no template control (NTC) is calculated. An RPM value RPM‐r = RPM sample/NTC ≥ 10 indicates that the species should be included in the clinical report.[ 22 , 94 ]
The pipeline for the evaluation and analysis of microbe testing can include DisCVR for virus identification and real‐time fluorescence quantitative PCR (RT‐PCR) analysis as a reference. The optimal threshold of 850 k‐mers for DisCVR and 150 reads for CLARK and Kraken is appropriate.[ 95 ] Based on relative abundance, viruses in the blood samples can be filtered for detection, and in clinical samples, thresholds of 1% for relative abundance and 0.01% for total reads classified have been applied.[ 96 ]
For some bacteria, it is necessary to focus on distinguishing between colonization, contamination of samples, and the presence of real pathogens and filter them according to the background bacterial database and baseline detection. A more accurate report for pathogens in samples can be derived after excluding human microflora, contamination from the laboratory environment, and reagent consumables using the background microbial databases.[ 97 ]
To evaluate the credibility of the identification results, the reads obtained can be compared with the reference genome of the species to calculate the coverage and sequencing depth by mapping. The higher the coverage and depth, the more reliable the results. Hence, if the coverage is low, but the depth is high, the credibility for identification is relatively low. Meanwhile if both coverage and depth are very low, minimal usable data is available for identification of the species, and other detection methods, such as RT‐PCR, can be used. The data can be visualized using the genome coverage curve, hot spot charts, pie charts, the Sanger chart, etc.[ 98 ]
3. Clinical Application of NGS
Infectious diseases remain a leading cause of human morbidity and mortality worldwide. Fast and accurate diagnosis of the etiologic pathogen can be challenging since a wide variety of microorganisms may cause clinically indistinguishable diseases, whereas the spectrum of detectable pathogens is relative narrow by current methods. Generally, a battery of combined tests, including culture, type‐specific serologic assays, and nucleic acid amplification tests, are required for establishing a diagnosis. Further, these methods may be time‐consuming, for example, delivering results for commonly encountered pathogens by culture can take at least 2–5 days, or even longer (several weeks to months), for more fastidious or insidious organisms, such as mycobacteria, Nocardia, and fungi.[ 24 , 99 ] Yet many common pathogens are impossible to culture in vitro, such as viruses,[ 99 , 100 ] and samples may be difficult to obtain, requiring invasive biopsy.[ 101 , 102 ] Additionally, the administration of antibiotics may affect the sensitivity of pathogens identification by traditional methods, such as culture.[ 103 , 104 ] Meanwhile, the introduction of syndromic multiplex PCR panels, 16S ribosomal DNA detection, and matrix‐assisted laser desorption/ionization‐time‐of‐flight mass spectrometry (MALDI‐TOF MS) have served to dramatically reduce the turnaround time for testing, however, the etiology remains unknown in up to 60% of infectious diseases.[ 105 , 106 ] Moreover, missed or delayed diagnosis, caused by the disadvantages of traditional microbe identification methodologies, drives the abuse of empirical broad spectrum antibiotics or antifungal drugs, thus preventing the use of targeted and curative treatments.[ 3 ] Alternatively, mNGS enables quick detection and comprehensive identification of bacteria, fungi, viruses, and parasites without the need for prior presumption of the causative culprits. Pathogens can be identified directly from various clinical specimens with a higher sensitivity and accuracy, outperforming culture‐based methods, especially for mycobacteria, anaerobes, atypical pathogens, and viruses.[ 29 , 99 ] Furthermore, mNGS is less affected by prior antibiotic exposure.[ 99 ] Therefore, mNGS shows great potential for infectious diseases diagnosis and differential diagnoses, which has been supported by a series of studies (Table 2 ).
Table 2.
Studies of NGS for infectious diseases diagnosis
| Year | Samples and populations | Methods | Results | Conclusions | Refs. |
|---|---|---|---|---|---|
| Big data studies | |||||
| 2018 | 511 specimens (blood, respiratory tract samples, body fluids and pus, CSF a) , urine, and swabs) from 561 cases suspected infections from April 2017 to December 2017. | A retrospective study |
|
|
[ 99 ] |
| 2019 | 132 clinical samples (sputum, CSF, pus, etc.) from 105 patients presenting with suspected active MTB infection between June 1, 2017 and May 21, 2018. | A prospective study |
|
|
[ 24 ] |
| 2019 | 163 specimens (CSF, blood, and throat swabs) from 105 patients suspected with viral encephalitis/meningitis or respiratory infection from May 2017 to June 2019. | A prospective study |
|
|
[ 127 ] |
| Focal infection | |||||
| 2019 | Tissues from 98 suspected focal infection cases. | A single‐center retrospective study |
|
|
[ 29 ] |
| Blood stream infection and febrile illness | |||||
| 2016 | 78 plasma samples from ICU a) patients, and 10 plasma samples from healthy volunteers between July 2014 and August 2014. | A prospective study |
|
|
[ 57 ] |
| 2019 | Plasma samples from 40 returning travelers presenting with a fever of ≥38 °C | A single center, proof‐of‐principle study |
|
|
[ 196 ] |
| 2019 | 60 serum, 90 nasopharyngeal, and 10 stool specimens were collected from 94 children with febrile illness. | A retrospective exploratory study |
|
|
[ 171 ] |
| Central nervous system (CNS) infections | |||||
| 2018 | CSF samples from 99 pediatric bacterial meningitis patients. | A retrospective observational study |
|
|
[ 122 ] |
| 2019 | CSF samples from 135 pediatric bacterial meningitis cases. | A retrospective study |
|
|
[ 128 ] |
| 2019 | CSF samples from 204 pediatric and adult patients at eight hospitals. | A 1 year, multicenter, prospective study |
|
|
[ 22 ] |
| 2020 | CSF samples from 248 adult patients suspected with CNS infections. | A single‐center prospective cohort study |
|
|
[ 117 ] |
| 2020 | CSF samples from 51 patients with suspected tuberculous meningitis from January 2017 to December 2018 | A retrospective analysis |
|
|
[ 132 ] |
| 2020 | CSF samples from 213 patients with infectious and noninfectious CNS diseases from November 2016 to May 2019. | A prospective multicenter study |
|
|
[ 130 ] |
| Respiratory infections | |||||
| 2018 | Samples of sputum, blood, or BALF from 178 severe pneumonia patients in the ICU. | A retrospective study |
|
|
[ 150 ] |
| 2019 | Specimens (pulmonary biopsy and BALF) from 55 cases (36 with mixed and 19 with nonmixed pulmonary infection) collected between July 2018 and March 2019. | A prospective study |
|
|
[ 151 ] |
| 2019 | 88 nasopharyngeal swabs from 63 patients with chronic obstructive pulmonary disease exacerbations. | A prospective study |
|
|
[ 149 ] |
| 2020 | Lung biopsy tissues from 121 patients diagnosed with peripheral pulmonary lesions (PPLs) and lung infection. | A prospective randomized study |
|
[ 157 ] | |
| 2020 | Samples (including lung tissue, BALF, and brush) collected from patients suspected with pulmonary infection from June 2018 to August 2019 | A retrospective study |
|
|
[ 148 ] |
| Bone and joint infections | |||||
| 2020 | 37 patients suspected with periprosthetic joint infection (PJI) who underwent prosthetic joint revision surgery from July 2016 to December 2018 | A retrospective cohort study |
|
|
[ 160 ] |
| 2020 | 44 periprosthetic tissues collected intraoperatively from patients who were suspected of PJI and underwent surgery. | A prospective study |
|
|
[ 51 ] |
| 2017 | 131 sonication fluid samples from patients undergoing revision arthroplasty or removal of other orthopedic devices. | A laboratory method development study |
|
|
[ 162 ] |
| Digestive system infection and urinary tract infection (UTI) | |||||
| 2017 | Serum samples from 204 adult acute liver failure (ALF) patients collected from 1998 to 2010. | A retrospective cohort study |
|
|
[ 173 ] |
| 2018 | Urine/semen/rectal swab samples from 112 patients in different areas of urology for prevention and treatment purpose. | Clinical application of NGS in different clinical phase I–II trials |
|
|
[ 167 ] |
| Infections in immunocompromised patients | |||||
| 2017 | Blood samples, and in some cases, nasopharyngeal swabs and/or biological fluids from 101 immunocompromised adults. | A multicenter, blinded, prospective, proof‐of‐concept study |
|
|
[ 188 ] |
Abbreviations: mNGS, metagenomic next‐generation sequencing; CSF, cerebrospinal fluid; MTB, Mycobacterium tuberculosis; WBC, white blood cell; PPV, positive predictive value; NPV, negative predictive value; AFB, acid‐fast bacilli; TBM, tuberculous meningitis; SSRN, species‐specific read number; GSRN, genus‐specific read number; R‐EBUS, radial endobronchial ultrasound; TBLB, transbronchial lung biopsy.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.1. Infections in Different Organ Systems
3.1.1. Bloodstream Infections
The constitution of causative pathogens varies with septic patients in different clinical backgrounds, with cases of Gram‐negative bacteria, anaerobes, and fungal sepsis increasing over the past decade, although Gram‐positive bacteria remain the most frequently identified.[ 107 ] The causative organisms remain unidentified in approximately half of sepsis patients, namely, culture‐negative sepsis. Alternatively, acute onset and severe sepsis can limit the available time available for diagnosis of the causative pathogen, contributing to high mortality rates,[ 108 ] which is further amplified in patients that receive inappropriate or mismatched antimicrobial therapy.[ 109 ] Hence, there is an urgent need for more rapid and sensitive pathogen identification technologies.
Multiple preliminary studies and case reports have shown that the genomic DNA or RNA fragments from circulating or noncirculating pathogens associated with the infections can be detected as cfDNA or cfRNA in purified plasma,[ 57 , 110 , 111 , 112 ] which allows for the use of mNGS for rapid and precise identity of pathogens causing sepsis, along with information regarding the abundance and genetic relatedness.[ 113 , 114 ] The diagnosis agreement of mNGS with tests based on blood culture in patients with a sepsis can be as high of 93.7% with the results are equivalent to, or superior to, those obtained by other direct molecular diagnostic methods, indicating the potential for mNGS to identify a wide range of infections, including bloodstream infection caused by various etiologies.[ 115 ] Further, multiple studies have reported that the overall diagnostic sensitivity of mNGS is significantly greater than bacterial culture used alone, while offering superior results for culture‐negative sepsis cases.[ 57 , 116 ]
Note, bacterial DNA may periodically exist in the blood of healthy people, which was previously considered a sterile microenvironment, however, the bacterial diversity differs significantly from that of septic patients. A significant predominance of anaerobic bacteria, primarily belonging to the order of Bifidobacteriales, was observed in healthy volunteers, whereas aerobic or microaerophilic microorganisms dominated in septic patients. For example, the abundance of Actinobacteria phyla decreased in sepsis, while Proteobacteria phyla decreased in healthy volunteers.[ 112 ] These results may help interpret mNGS reports for patients with suspected sepsis, while reminding us that bacteria continuously translocate into the blood, but do not always cause sepsis.
To summarize, mNGS has undeniable advantages over culture‐based methods in the diagnosis and treatment guidance of bloodstream infection, as it offers significantly reduced time required for pathogens identification to less than 24 h, regardless of the type of microorganism, and is less affected by antibiotics administration.[ 113 ] Moreover, this approach may prove invaluable in patients infected with fastidious pathogens, or less commonly fungi, Mycobacterium, and parasites, allowing physicians to accurately diagnose and regulate treatment.[ 99 ] In addition, mNGS detects the presence of viral infections[ 117 ] or mixed‐infections,[ 118 , 119 , 120 ] avoiding the abuse of antibiotics in sepsis‐like patients, while informing proper and targeted antibiotics usage in septic patients.
3.1.2. Central Nervous System Infections
Various pathogenic microbes can infect the CNS, presenting as meningitis, encephalitis, and abscess, which are often life‐threatening; however, routine microbiological tests are often insufficient to detect all neuroinvasive pathogens, especially for rare pathogens. Moreover, obtaining the relevant samples to detect the etiologic pathogens require invasive procedures, such as lumbar puncture or brain biopsy, which are limited by the availability and volume of CSF or brain tissues. Therefore, the etiology of CNS infections often remains undiagnosed, which occurs in up to 50% of acute meningoencephalitis cases.[ 121 ] Thus, methods for timely and accurate diagnosis are urgently required, where mNGS shows its superiority in recent years.[ 19 , 122 ] In fact, multiple case studies have reported the detection of viruses,[ 120 , 123 , 124 , 125 , 126 , 127 ] bacteria,[ 19 , 128 ] fungi, and parasites[ 120 , 129 ] using mNGS in CSF and brain tissues.[ 130 ] Additionally, mNGS also proven valuable in diagnosing subacute or chronic meningitis.[ 120 ] Specially, elevated levels of white blood cells (WBCs) and protein, as well as decreased glucose ratio in the CSF may correlate with increased detection of CNS infections via mNGS.[ 117 ] A system review had recommend that NGS be considered as a front‐line diagnostic test for chronic and recurring infections, and as a second‐line technology for acute cases of encephalitis.[ 131 ]
In addition to its reasonably high diagnostic performances, mNGS may also help guide more targeted therapy for CNS infections and accelerate the identification and subsequent treatments for noninfectious causes.[ 22 ] However, although mNGS has an overall superior detection rate compared to conventional method, it cannot replace the necessity for traditional methodologies, such as culture.[ 117 , 130 ] Rather, mNGS is recommended to be applied in combination with conventional microbiological testing to improve the pathogenic diagnosis of CNS infections. Moreover, it may prove useful for ruling out an active infection in patients with suspected autoimmune encephalitis, thereby providing the necessary information for clinicians to confidently initiate immunosuppressive therapies for autoimmune disease with reduced concerns regarding missed detection of microbial infections.[ 22 , 130 ] Furthermore, mNGS results are less affected by prior use of antibiotics before collection of CNS samples,[ 128 ] and therefore the technique has an advantage over other methods in cases whereby disease‐causing microbial detection in patients is greatly affected by antibiotic use. However, it should be noted that with prolonged treatment times, the detection rate of pathogenic microorganisms via mNGS is decreasing.[ 117 ]
Studies have shown that mNGS offers high sensitivity, specificity, and positive predictive value (PPV) in the diagnosis of tuberculous meningitis in the CSF.[ 30 ] Indeed, the sensitivity of mNGS was significantly higher than culture alone, and a combination of mNGS and conventional methods significantly increased the detection rate significantly. Moreover, patients with a significant increase in cell number and protein in their CSF have a higher chance of being MTB positive when detected by NGS.[ 132 ] These results indicate that mNGS has the potential to be used as a first‐line CSF test to detect the presence of mycobacterial DNA. Since MTB complex members exhibit >99.99% genomic sequence similarity, genus stringent mapped reads number (GSMRN) was considered reliable when interpreting reports for the MTB complex.[ 130 ]
mNGS of CSF can also effectively identify fungi causing CNS infections. A large, prospective, multicenter series case study showed that the sensitivity of mNGS using species‐specific read number (SSRN, equivalent of stringent mapped reads number, SMRN) ≥2 in the diagnosis of cryptococcal meningitis and cerebral aspergillosis was 76.92% and 80%, respectively,[ 130 ] which can be significantly improved with the combination of traditional methods, thereby informing the choice of appropriate antifungal agents and therapeutic courses.[ 118 ] However, wide adoption of mNGS for clinical purposes to diagnose cryptococcal meningitis is unlikely as the current diagnostic tests available (CSF CrAg) are highly sensitive and specific, with rapid turnaround times.[ 133 ]
Definitive diagnosis of viral infection in the CNS is dependent upon virus isolation from CSF or brain tissues, which is challenging and generally only performed in the laboratory. Therefore, treatment primarily depends on empirically provided therapy. The identification of certain CNS viral pathogens remains difficult, as many viral families that infect the CNS failed to be identified by the gold standard pathogen‐specific PCR assays. Although DNA viruses (mostly herpes virus) are often identified by mNGS, its prediction of viral encephalitis and meningitis has not significantly improved,[ 134 , 135 , 136 , 137 ] which may be partially due to the absence of RNA detection methods, since RNA‐based mNGS has not been launched extensively.[ 130 ] RNA viruses, such as enteroviruses and the Japanese encephalitis virus, are important causative agents of viral encephalitis and meningitis.[ 138 ] Hence, simultaneous extraction of DNA and RNA followed by cosequencing may improve the detection rates of viruses in CSF samples.[ 139 ]
Further, mNGS also has values for the characterization of complex and rare pathogens present in culture‐negative and undiagnosed cases. mNGS has been shown to help detect the presence of microbes, such as Listeria monocytogenes,[ 140 ] Brucellosis‐causing species,[ 93 , 141 ] Naegleria fowleri,[ 142 ] neurocysticercosis‐causing parasites,[ 143 , 144 ] and Vibrio vulnificus.[ 145 ] In CNS toxoplasmosis, mNGS may be useful in cases when the toxoplasmosis IgG is negative, CSF PCR is negative, and imaging is not classic, or when there is a lack of response to antitoxoplasmosis therapy.[ 119 , 133 ]
Additionally, mNGS can be used to dynamically monitor disease progression using semiquantitative value analysis.[ 117 ] However, negative tests must be interpreted with caution owing to the higher risk of false‐negative results.[ 22 ]
3.1.3. Respiratory Infections
Upper and lower respiratory tract infections are among the most common illnesses leading to medical consultation, and are associated with significant mortality.[ 146 ] Hundreds of pathogens, including bacteria, fungus, viruses, and parasites, can cause pulmonary infection, and the situations in immunocompromised patients may be even more complex. Undoubtedly the identification and characterization of pathogens is crucial for precision treatment and improved prognosis of patients. However, frequent and inappropriate use of antibiotics in respiratory tract infections limits the sensitivity and reliability of culture‐based testing, which often occurs in clinical settings.
mNGS provides relatively fast and precise detection and identification of a large variety of pathogens, contributing to the prompt and accurate treatment of pulmonary infection,[ 147 , 148 , 149 ] particularly for critically ill patients and those with mixed‐infection.[ 150 , 151 ] The most common pathogens causing lower respiratory pulmonary infectious have been identified as bacteria, such as Pseudomonas aeruginosa, Klebsiella pneumoniae, and Acinetobacter baumannii.[ 99 , 152 ] In addition, a greater number of fastidious bacteria, including MTB, nontuberculosis mycobacteria (NTM), Nocardia, and various Actinomycetes, were identified by mNGS compared to conventional culture methods.[ 99 ] Moreover, the use of mNGS has also been shown to improve the diagnosis of pulmonary invasive fungal infections.[ 147 , 151 ] The sensitivity of mNGS is further highlighted in mixed pulmonary infection diagnosis and severe nonresponding pneumonia.[ 153 , 154 , 155 ] In fact, the detection accuracy rate of mNGS may even reach 100% in the analysis of immunocompromised patients.[ 156 ]
Additionally, mNGS provides more strain‐specific information and helps to identify new pathogens.[ 153 ] However, the interpretation of the specificity differs,[ 151 ] depending on whether the pathogens are the causative agents of infection due to the presence of normal flora, commensal oral flora, or colonizers, therefore a tradeoff is generally needed. NGS is currently the preferred methodology for virus discovery, particularly for emerging pneumonia‐causing viruses (i.e., SARS‐CoV, MERS‐CoV, and H7N9), and could potentially help to trace and control outbreaks.[ 154 ]
Interestingly, the specificity of mNGS is reportedly higher for transbronchial lung biopsy (TBLB) tissue than for BALF; however, detection in BALF has been shown to have a higher sensitivity in the diagnosis of peripheral pulmonary infectious lesions.[ 152 ] However, in our experience, the overall sensitivity of mNGS does not differ among respiratory tract sample types and is not superior to that of culture for recognizing common bacteria. Nevertheless, mNGS has been reported to have a significantly higher sensitivity than culture in sputum and lung tissue samples.[ 99 ] Moreover, NTM, rather than MTB, Aspergillus, or Cryptococcus, was more often detected in BALF than sputum by mNGS.[ 99 ] Meanwhile, R‐EBUS‐guided‐TBLB significantly facilitates the accurate insertion of the bronchoscope into the lesions in the pulmonary, thereby improving the positivity rate of mNGS analysis in pathogen detection.[ 157 ]
3.1.4. Bone and Joint Infection (BJI)
BJIs are serious and potentially life‐threatening, particularly for periprosthetic joint infection (PJI), which is a devastating complication that can occur after arthroplasty and multiple revision surgeries.[ 158 ] Conventional diagnostic challenges in BJI due to the fastidious nature of the organisms, biofilm formation on implant surfaces, prior antibiotic administration, or limited sample availability, all contribute to the high rate of culture‐negative PJIs, the microbial etiology for which is challenging to diagnose.[ 159 , 160 ]
In most cases, mNGS, with high sensitivity, specificity, and positive and negative predictive values, can be used as an effective supplemental method to improve the diagnostic efficiency of BJI. Synovial fluid, sonication fluid from explants (i.e., prosthetic joint and other orthopedic devices), and periprosthetic tissue, all can be used to identify the causative pathogens via mNGS.[ 51 , 161 ] Remarkably, sonication fluid is potentially more advantageous than synovial fluid for distinguishing the primary causative pathogens from contaminants, possibly because sonication may enrich the microorganism abundance in samples by dislodging biofilms from the surface of implants.[ 162 , 163 , 164 ] Fastidious bacteria NTM can be detected by mNGS from all sample types, enabling surgeons to collect specimen from various samples. Moreover, assigned reads in the sonicated fluid is significantly higher than in synovial fluid (more than 200‐fold times) and tissues,[ 165 ] which need further research to verify.[ 162 , 163 , 164 ] mNGS should be highly recommended for the cases of culture‐negative BJI, limited sample volume obtained by joint cavity puncture, or when the patient has poor responses to empirical antibiotics therapy or debridement.[ 160 , 165 ]
3.1.5. Urinary Tract Infection (UTI)
UTI is a common community‐acquired infection commonly caused by bacteria, as well as certain fungi or virus (i.e., polyomavirus and the human herpesvirus).[ 166 , 167 , 168 ] Escherichia coli (≈75%) is the most common causative agent for both uncomplicated and complicated UTI, with the latter comprising a complex constitution of causative microorganisms, making it rather challenging to diagnose the pathogens.[ 166 , 169 ] However, mNGS is capable of identifying pathogens more quickly and with more sensitivity than culture methods, providing clinicians with the information necessary to make accurate diagnoses, as well as to determine the preventive, prophylactic, and appropriate treatment options.[ 166 , 167 ]
However, interpretation of urine mNGS reports may prove difficult for various reasons. First, urine samples are readily contaminated by improper procedures before and during urine collection. Second, colonizers inhabit the distal urethra, the skin around the urethral meatus, as well as the vagina, which must be taken into consideration, as specific organisms may become pathogenic in immunocompromised patients. Third, asymptomatically persistence or latent infections often occur. Therefore, even with precise quantitative distribution of all microbial associations, it may be difficult to define which bug or superbug is causative of UTI.[ 166 , 168 ]
3.1.6. Digestive System Infection
The major enteropathogenic bacterial species include invasive enteropathogens originating from outside environments, and pathobionts originating from commensal gut species. Although the study of the intestinal microbiome is a very relevant topic popular among researchers, only a few have attempted to diagnose the associated infectious diseases, such as diarrhea, using mNGS techniques.[ 170 ] As the intestines are normally colonized by commensal bacteria, mNGS of stool samples primarily focus on nonbacterial species, such as Rotavirus A, Cryptosporidium, and the human parechovirus, which are often detected in the stool samples of febrile children.[ 171 ] mNGS has also been used to detect potential causative agents in patients with acute cholecystitis,[ 172 ] screen for the presence of uncommon viruses, or identify co‐infections in patients with acute liver failure.[ 173 ] In addition to pathogen identification, mNGS is also a rapid and agnostic diagnostic approach for investigating resistome, and can be used for identifying antibiotic resistance genes (ARGs) in the gut, helping detect multidrug resistant organisms (MDROs), thus leading to early implementation of infection prevention practices, antimicrobial optimization, and prevention of invasive infections.[ 174 , 175 ]
3.1.7. Complex and Atypical Pathogen Infections
Many cases have demonstrated the superiority of mNGS in the identification of i) complex and atypical infections, such as Chlamydia psittaci,[ 176 , 177 ] Legionella spp.,[ 178 ] anaerobes that are difficult to culture,[ 179 ] and mycobacteria;[ 165 , 180 ] ii) zoonotic pathogens, such as Streptococcus suis,[ 181 ] Orientia tsutsugamushi,[ 182 ] Leptospira santarosai,[ 19 ] and Toxoplasma gondii;[ 119 ] iii) emerging RNA and DNA viruses, such as Bocaparvovirus,[ 183 ] Ebola virus,[ 184 ] Zika and chikungunya viruses,[ 185 ] bornavirus,[ 186 ] and SARS‐CoV‐2.[ 187 ]
3.2. Infections in Special Populations
3.2.1. Infections in Immunocompromised Patients
Infections in immunocompromised hosts have drawn a great deal of attention by clinicians, particularly in recent years, since they are generally more complex and serious with the potential to result in disastrous consequences, such as failure of a transplanted solid organ. mNGS has demonstrated distinct advantages in the detection of pathogens in this group of people,[ 188 ] and was recommended as a first‐line diagnostic tool for patients undergoing solid organ transplantation and hematopoietic stem cell transplantation,[ 23 , 110 , 188 , 189 , 190 ] or as an addition to routine test panels.[ 191 ]
Allograft health monitoring, including infections and transplant rejection, which occur frequently, and sometimes simultaneously, is an important component of post‐transplant therapy. mNGS detection of cfDNA can be applied in monitoring organ transplant rejection via genome transplant dynamics, and may have the potential to replace invasive techniques, such as lung and endomyocardial biopsies as the symptoms of infection and rejection are often difficult to discriminate.[ 110 , 192 ] Additionally, data from mNGS may provide important information regarding the relationship between the human virome, the state of the immune system, and the effects of pharmacological treatment. For example, the total viral load is reportedly more significantly affected by immunosuppression than the bacterial microbiome.[ 193 ] The infecting viruses include those that are common, such as Cytomegalovirus, and Epstein–Barr virus, as well as undiagnosed DNA viruses, such as the human herpesvirus, polyomavirus, adenovirus, and the torque teno virus.[ 23 , 110 , 190 ]
3.2.2. Febrile Illness
The global burden of febrile illness has been difficult to quantify. Identification of etiologies in patients with fever of unknown origin (FUO) is the chief issue, among which infectious factors remain the primary cause.[ 194 ] This poses a challenge for diagnostic and therapeutic determination, and may result in inappropriate antibiotic use, making it difficult to predict, detect, or evaluate potential outbreaks or emerging infections. Unbiased mNGS represents a powerful tool to fill the gaps in our understanding toward the etiology of febrile illness, thus informing the improvement of diagnostic algorithms, therapeutic guidelines, and public health strategies.[ 171 , 195 ]
Viruses play an important role in FUO, and distinctions have been observed between the viromes of febrile and afebrile groups using short‐read Illumina sequencing. The assessment and identification of known, or potentially novel, viruses have improved the medical management of children with FUO, helping to avoid the administration of unnecessary antibiotic therapy in those with viral infections.[ 195 ] Another worldwide challenge is travel‐associated infections, with a broad spectrum of potential etiologies. mNGS has greatly improved the pathogens diagnostic efficiency of returning travelers with acute febrile illness, particularly for viruses, such as dengue virus, Ebola virus, hepatitis E and hepatitis A, and Chikungunya virus.[ 196 ] mNGS is considered to have the potential of being used as an all‐in‐one diagnostic test for FUO patients, helping differentiate infections and noninfections, identify causative agents, and discover novel or emerging pathogens.[ 196 ] However, a cost–benefit study reported that current conditions do not warrant a widespread rush to deploy metagenomic testing to resolve any and all uncertainty, but rather as a front‐line technology that should be used in specific contexts, such as acute and seriously ill cases, or as a supplement to, rather than a replacement for, careful clinical judgment.[ 197 ]
3.2.3. Unexplained Pneumonia
Metagenomic analysis is of great value for the monitoring and response to rapidly emerging infectious diseases, such as unexplained pneumonia, among which COVID‐19 is currently the most popular.[ 198 ] Metagenomic sequencing, with its unbiased nature, plays a crucial role in the etiological diagnosis of unexplained pneumonia, particularly when encountering an atypical or novel causative agent.[ 187 , 199 , 200 ] During the initial outbreak of SARS‐CoV‐2, a commercial sequencing company provided an incorrect report stating the identification of SARS virus, causing widespread panic. Later, upon careful analysis using additional sequencing data, the causative agent was determined to be a novel coronavirus.[ 200 ] Further, combined with multiplex PCR amplification and nanopore sequencing, the phylogenetic structure of the novel coronavirus were analyzing during the early days of the pandemic.[ 201 ]
However, important lessons should be learned from this outbreak; i.e., although metagenomics sequencing lacks the capability of identifying the novel pathogen, it proved valuable for ruling out common pathogens as first‐line diagnosis of unexplained pneumonia.[ 200 ]
3.3. Infection Control
The hospital infection prevention control management is an important aspect of healthcare, nosocomial infections occur worldwide and affect hundreds of millions of patients, leading to tens of billions of dollars lost each year, and remain a particularly aggressive problem in the pediatric healthcare context.[ 202 ] The mNGS of clinical or environment samples is a promising approach for rapidly identifying pathogen sequences for the detection and epidemiological determination of transmission.[ 203 , 204 , 205 ]
4. The Global Expert Consensus for NGS in the Diagnosis of Pathogens
mNGS, with recent advances, reduced costs, user‐friendly data analysis tool, and accurate and comprehensive databases, has been a revolutionary technology that has disrupted traditional clinical diagnostic microbiology.[ 106 ] mNGS can overcome the limitations of traditional diagnostic tests, enabling a hypothesis‐free, culture‐independent, and universal pathogen detection platform, with which clinical specimens can be directly used for microbe identification of bacteria, viruses, fungi, or parasites, while also assisting in the identification of novel organisms.[ 3 ] Yet, few associated expert consensuses for the use of NGS in infectious diseases diagnosis. However, the general consensus is that its relatively high cost is a barrier to mNGS becoming the first‐line of detection method for clinical in the short term. However, it is recommended as the first‐line detection method in situations involving patients with difficult or complicated infectious diseases, including those who are acutely and critically ill, suffering from immunodeficiency,[ 188 ] or in special populations, as well as in situations of infectious diseases outbreaks of unknown origin.[ 187 , 206 ] In most cases, mNGS is recommended as a supplement to conventional detection methods, particularly in cases where traditional diagnostic assays are negative[ 133 ] (Table 3 ).
Table 3.
Global expert consensus and guidelines of NGS in pathogens diagnosis
| Countries/regions | Year | Viewpoint | Refs. |
|---|---|---|---|
| Winnipeg | 2018 | NGS has obvious advantages in the detection and typing of HIV drug resistance genes. | [ 243 ] |
| International | 2018 | NGS provides new means for the cognition of joint microorganisms and is expected to better understand joint colonization. | [ 244 ] |
| UK | 2018 | NGS should be considered as a front‐line diagnostic test in chronic and recurring presentations and, given current sample‐to‐result turn‐around times, as second‐line in acute cases of encephalitis. | [ 131 ] |
| Singapore | 2018 | Current conditions do not warrant a widespread rush to deploy metagenomic testing to resolve any and all uncertainty (i.e., pyrexia of unknown origin PUO), but rather as a front‐line technology that should be used in specific contexts, as a supplement to rather than a replacement for careful clinical judgment. | [ 197 ] |
| China | 2020 | As a comprehensive direct detection method, the current high cost makes it impossible for mNGS to become a clinical first‐line detection method in the short term, but in difficult and complicated diseases, critically ill, immunodeficiency, and other special population, it still has the potential to become a quasi‐first‐line detection method for pathogen diagnosis. | [ 92 ] |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
5. The Challenges for NGS in Pathogens Diagnosis
The multiple achievements made in high‐throughput sequencing technology for the diagnosis of infectious diseases are encouraging; yet, some hurdles remain to be addressed, including the limitations of the technology, clinical application, high costs, the lack of current standardization of methods and data analysis, interpretation guidelines for clinicians, generation of appropriate work pipelines, the regulation, and supervision. Moreover, it remains to be determined how to: i) minimize the influence of human host background and extraneous sources of nucleic acids; ii) differentiate colonization from infection; iii) improve the detection efficiency of intracellular microorganisms; iv) generate more reasonable standardization of the methods, data storage, protection, analysis, and interpretation. Here, we comprehensively discuss the challenges of the clinical application of high‐throughput sequencing technologies and propose possible solutions.
5.1. The Level of Technology
5.1.1. The Influence of Human Host Background
Samples collected for testing samples can be of different types depending on the site of infection, and may be obtained from the CSF, peripheral blood, sputum, BALF, or other sites. As the human genome is much larger than the microbial genome, the host DNA background is also much higher than that of microbes; therefore, even a small number of human cells can greatly affect the accuracy of microbial detection, decreasing the useful coverage of the sequencing data, and increasing the sequencing cost. Currently available methods for the removal of human DNA and enriching microbial DNA include: i) the removal of host cells based on differences in cell size relative to microbial cells using methods such as differential centrifugation and filtration, and ii) the selective lysis of host cells and subsequent treatment with enzymatic or chemical reagents to remove exposed DNA.[ 207 ]
Centrifugation is a simple, convenient, and low‐cost method for the removal of human DNA and is widely used in mNGS‐based detection processes. For example, CSF samples from patients with meningitis are centrifuged at 5000 × g for 15 min to remove cell precipitates, nucleic acid is then extracted from the supernatant, and the sequencing library is constructed. Processing the samples before library construction can reduce the host genome proportion, significantly increase the sequence coverage of pathogens in the samples, and improve the detection sensitivity.[ 20 ] Further, centrifugation and filtration can be combined to enrich pathogenic microorganisms. For second‐generation sequencing to detect viruses, samples can be centrifuged at a low speed of 6000 × g for 10 min, and the supernatant filtered using a 0.80 µm filter to remove eukaryotic and bacterial cells without affecting the extraction of RNA viruses.[ 208 ] For example, recovery of the human genome from saliva samples by sifting through a 5 µm filter was 89.69 ± 0.84% compared to unfiltered original samples 89.29 ± 0.61% recovery. These results indicate that a considerable amount of extracellular host DNA in the samples may not be removed via filtration method based on cell size alone. To overcome this deficiency, human cells can be selectively lysed, and enzymes or chemical reagents can be used to remove exposed DNA. The percentage of human DNA in samples can be reduced using methods such as propidium monoazide treatment after selective mammalian cell lysis by sonication (human DNA recovery is 25.6%), and via osmotic lysis with H2O (1.7%) followed by mammalian cell removal by differential centrifugation (1.4%). The treatment of raw saliva samples with propidium monoazide has been shown to reduce the percentage of human DNA (16.8%), suggesting that the majority of human DNA in saliva is already exposed.[ 209 ] In addition to ultrasonic fragmentation, nonionic surfactant saponins can also be used to lyse human cells and subsequently, the digested DNA by enzymes has been shown to increase the number of sequences retrieved for Cryptococcus neoformans.[ 20 ]
Human genomic DNA undergoes methylation, and methylated host DNA can be excluded from samples using the methyl‐CpG binding domain to enrich for microbial DNA, resulting in a 50‐fold reduction in the number of reads mapping to the human genome.[ 210 ] A study on the removal of host DNA from CSF showed that upon processing the sample using the NEB Next Microbiome DNA Enrichment Kit, the relative proportion of pathogen DNA relative to human reads was enriched by ≈2.5‐fold.[ 211 ] However, results from another study showed that the proportion of the human genome in saliva treated with the same kit remained high at 90.83 ± 0.77%, which was not significantly different than the untreated original samples (89.29 ± 0.61%).[ 207 ] At present, the evaluation of the effect of dehosting nucleic acids based on the characteristics of host DNA methylation is different, and is related to the sample type, host DNA content, and other factors. The true dehosting efficiency remains to be confirmed by additional experimental data. Currently, all dehosting DNA schemes have certain inherent limitations, and a combination of several schemes may be considered in the future to achieve maximum host DNA removal.
5.1.2. mNGS Efficiency in Samples Containing Intracellular Bacteria and Fungi
mNGS can potentially detect the presence of all microorganisms in a sample, and the content of the particular microbial DNA in the sample partially affects the detection rate of the species. The detection efficiency of fungi and intracellular bacteria, such as tuberculosis, is lower than that of other microorganisms. Therefore, a suitable cell lysis method that can efficiently lyse the cell walls of fungi and intracellular bacteria may improve the detection sensitivity of these microorganisms.
At present, three kinds of methods for microbial cell lysis exist: heating and enzymatic digestion of cell walls, mechanical (sonication and using bead‐beaters), and chemical.[ 212 ] In a comparative study of several cell wall disrupting methods, lysozyme was found to be effective in the enzymatic hydrolysis of cells; however, its use is limited to Gram‐positive bacteria that are sensitive to lysozyme. Although bead‐beading is more generally applicable, the cell wall cracking efficiency can be highly variable depending on equipment, buffer, and the cell type. For spores and other cells, such as mycobacteria, that are difficult to destroy through bead‐beading.[ 213 ] In another study using six different methods for cell wall disruption and extraction of MTB DNA, the results showed that Infection Diagnostic, Inc. (IDI) lysing tubes had a higher cleavage and extraction efficiency than the other five treatment methods, including the QIAGEN QIAamp DNA mini kit.[ 214 ] Many schemes can be used to improve the cell wall disruption efficiency in fungi and intracellular bacteria, though it is important to consider the corresponding effect on RNA extraction. In a study by Huang et al., the sample treatment program used underestimated the possibility of RNA virus infection. As the cell walls of fungi and intracellular bacteria are thick, cell wall disrupting methods are used to extract DNA, which may affect the detection of RNA viruses.[ 148 ] Therefore, it is important to consider the use of a variety of cell wall disrupting techniques to improve the efficiency of cell breakage and the detection rate for RNA viruses.
5.1.3. Stability of RNA in the Detection Process
Infections resulting from RNA viruses are common. For example, detection of the novel SARS‐CoV‐2 coronavirus, mNGS technology was used to extract total RNA from BALF samples, resulting in the successful identification of the virus.[ 215 ] RNA forms unstable molecules that are easily degraded by RNases in the environment. Therefore, RNA extraction immediately after sample collection can help to obtain high‐quality RNA. However, samples often need to be sent to other locations for testing, and thus, timely extraction cannot be guaranteed. To ensure the accuracy of the subsequent test results, it is necessary to ensure that the RNA in the samples is not degraded during collection and transportation. At present, RNA samples are transported using dry ice or after the addition of an RNA stabilizer. Although a low‐temperature environment can slow the rate of nucleic acid degradation, multiple freeze–thaw steps leads to the release of cellular nucleases, resulting in RNA degradation.[ 216 ] Commonly used reagents for sample preservation include RNAlater (Thermo Fisher Scientific and Qiagen) and RNAstable (Sigma‐Aldrich).[ 217 ] As transcriptome technology has matured over time, sample processing is carried out in strict accordance with standard operating procedures to ensure the accuracy of the test results.
5.1.4. Detection of Drug Resistance Genes and Virulence Factors
The development of sequencing technology allows for a more in‐depth understanding of the changes and evolution of microorganisms, as well as a comprehensive and accurate description of strain genotypes, virulence, antimicrobial resistance spectra, and systematic genetic background.[ 218 ] Mason et al. analyzed the data for resistance causing genes using whole genome sequencing (WGS) of 1379 isolates of Staphylococcus aureus. Their results showed that the correlation between drug resistance genotypes and phenotypes can be as high as 98.3%, indicating that before obtaining results related to drug sensitivity, drug resistance in infected pathogens can be detected using WGS technology to avoid the ineffective use of drugs.[ 219 ] Greater attention is being paid to virulence genes, and an in‐depth study on virulence factors is likely to provide a strong basis for the distinction between pathogenic and colonized bacteria. A genome‐wide study on the virulence genes of K. pneumoniae clinical isolates identified 21 new genes in addition to the iroBCD/iucABCD and rmpA/A2 genes already known to be associated with virulence. These 21 genes may be factors contributing to the high virulence phenotype of K. pneumoniae.[ 220 ] Although sequencing technology has made various strides in the detection of virulence factors and drug resistance genes, there are certain differences between mNGS‐ and WGS‐based detection, with challenges persisting in the application of mNGS for practical detection. First, samples generally expend a large amount of sequencing output consuming human DNA sequences, leading to insufficient coverage for the detection of pathogen DNA and limiting the comprehensive analysis of drug resistance genes. Second, second‐generation sequencing generates short fragments and even after the detection of drug resistance genes, it may be difficult to determine the pathogen to which the gene belongs due to sequence splicing. Third, the drug resistance mechanism is complex and diverse; the current databases for drug resistance genes and virulence genes require improvement, and there are inconsistencies in the reports of genotypes and phenotypes of pathogens. These considerations have caused resistance to the application of mNGS for the detection of drug resistance genes and virulence factors. Meanwhile, third‐generation sequencing technology offers the advantages of delivering long reads, high‐throughput, and synchronous data analysis, and is a promising technique for the detection of drug resistance genes and virulence factors.
5.2. Biological Information Analysis
5.2.1. Optimization of Microbial Databases
Public databases containing microbial reference genomes are constantly being updated, requiring laboratories to track the version being used at any given time, and to address potential erroneous annotations and other database errors. Larger and more complete databases that contain publicly stored sequences, such as the NCBI nucleotide database, are more comprehensive and more accurate than the limited databases, such as FDA‐Argos or RVDB, though the larger databases may contain more errors. A combinatorial method using multiple database annotation sequences can improve the sensitivity and specificity of microbial identification.
In the laboratory, the analysis software and reference databases used are best determined before verification and clinical use. Many laboratories maintain the production version and the latest development version of the clinical reference database (for example, the NCBI nucleotide database is updated fortnightly), as databases are updated regularly at prespecified intervals. Standardized datasets can be used to validate the database after updates to ensure the accuracy and repeatability of results, as newly saved sequences and clinical metadata may introduce errors.
In addition to the aspects mentioned above, it is also important to take note of the following: i) incomplete sequences of rare pathogens or emerging pathogenic strains, ii) biased references toward species, iii) genetically similar pathogens (e.g., contamination of mycobacterial strains), and iv) the presence of normal flora in reagents, which is a common phenomenon that limits specificity. To provide accurate results, the reported algorithm may need to account for the test process and sample quality, pathogens that are rare in current and previous tests, the relative and absolute abundance of microorganisms, whether there are genetically similar microbes, and microbial genome coverage. The workflow for reporting results requires that the standards for quality control and the interpretation of results should be pre‐established, which may include expert reviews of all cases or a subset of cases that meet the defined criteria or in diseases with unusual outcomes.
5.2.2. Virus Classification
Viruses are the simplest biological organisms with varying genome types, ranging from single‐stranded RNA or single‐stranded DNA, which encode only a few proteins, to dsDNA containing viruses that are similar in size to bacterial genomes. The average genome size of viruses is smaller than 50 kb. Viruses evolve and mutate faster than other pathogens, hence, the classification of viruses has greater challenges compared to those for bacteria or fungi, and there are often different classification rules for different groups of viruses.[ 221 , 222 , 223 ] The International Committee on Taxonomy of Viruses (ICTV) is responsible for updating information relating to virus classification that depends on genomic information.[ 223 , 224 ] The standardization of genome assemblies has resulted in the creation of standards for the quality control of sequencing data, splicing information, quality evaluation after splicing, and additional problems that require manual identification.[ 225 ] Standardization of the genome annotation process includes the prediction of viral genes, while classification and identification criteria are based on the COG protein homologous family, with the accuracy of classification ensured by using the characteristics of genomes with different classification units.[ 226 , 227 ]
RVDB is a standard genome database used for virus classification that contains nucleic acid sequences related to all identified viral, virus‐related, virus‐like, and endogenous nonretroviral elements, as well as endogenous retroviruses and retrotransposons, however, excludes bacteriophages and other nonviral sequences. The virus genomic sequences are checked and verified manually, similarity comparisons are made, redundancies are removed, and the classification of virus species is confirmed by a comparison with clustered representative sequences.[ 228 ] Virus identification software, such as DisCVR, also uses the k‐mer algorithm and compares the length of the k‐mer (18, 22, 22, 26, and 30). Ultimately, 22 is selected and, compared with CLARK and Kraken, it has higher specificity and sensitivity.[ 95 ] Therefore, along with the use of virus genome databases such as NT or NCBI, there is a need for the optimization of the alignment algorithms of existing databases based on the inclusion standard for accurate virus classification. Viral genomes from public databases in the future can be included in the local identification database through the confirmation of classification, filtering of contaminating sequences, providing genome quality ratings, etc., and simultaneously, it is necessary to carry out parallel or confirmatory identification and confirmation.[ 229 ]
5.3. The Level of Interpretation
A well interpreted mNGS report can guide clinicians in diagnosis, differential diagnosis, treatment, and prognosis evaluation. The human microbiome is complex as portions of the human body are naturally colonized by numerous bacteria, fungi, and viruses, primarily within the respiratory and gastrointestinal tract, skin, and vagina, whereas most areas inside the body of healthy individuals are physiologically sterile (i.e., blood) and microorganisms appear only under certain conditions (i.e., sepsis). Therefore, the interpretation of mNGS reports is rather difficult and requires a battery of clinical, laboratory, and microbiological research experience.
Due to the associated technical and clinical complexity, there is no absolutely standard method for interpreting mNGS results. Hence, clinicians working with the sequencing results for an infectious disease face several constraints. To address these challenges, first resumption of working groups should be implemented. Some institutions have assembled teams working on precision medicine, consisting of representatives from clinician groups (particularly infectious diseases), microbiology laboratories, and professional technical teams, who can assess the results of sequencing and provide an appropriate interpretation before the reports of the mNGS are delivered to clinicians. Another strongly advised approach is to have an experienced laboratory or clinician director manually revise the results prior to release.[ 230 ]
Second, to provide accurate results, several constraints should be considered when formulating the algorithms for interpreting the reports. i) the quality of the samples and test process; ii) types and collection methods of samples, whether from a site with colonized flora or not; iii) detection efficiency for different microorganisms, such as common bacteria, mycobacteria, fungi, virus, or parasites; iv) certain pathogens may be similar genetically (i.e., species of mycobacteria); v) the reference databases bias or incompleteness for rare pathogens or emerging strains of pathogens; vi) contamination from the reagents and environment. Laboratories should establish an appropriate workflows depending on its own unique situation, including metrics for quality control and interpreting the findings, an expert review should then be included when defining criteria or reviewing unusual findings.[ 106 , 231 , 232 ]
A large proportion of the samples are collected from the respiratory tract, where the microbiome is particularly difficult to characterize, with diverse pathogens causing infectious diseases, as well as colonizers. For example, the mNGS of lung biopsies or BALF yields reads of bacteria from the oropharyngea flora, as well as viruses or Candida, which may not be considered causes of pulmonary infections in immunocompetent patients, but may be considered pathogenic in immunocompromised hosts.[ 147 ] Thresholds such as the relative abundance of a potential pathogen at 30% of all genera present and the sampling of at least 100 unique reads could be used to indicate the presence of potential pathogens relative to the background lung microbiome. In general, higher coverage rate, relative abundance, and SMRN are typically reliable with true infections, whereas lower indices are somewhat discredited for infections or are considered to be more likely associated with the presence of commensal/colonizer/contaminant microorganisms of unknown clinical significance. Thus, the entire clinical picture must be taken into consideration in any cases when determining the clinical significance of the detected microbes.[ 115 ]
5.4. Limitations Compared with Traditional Pathogen Identification Methods
Massive case reports, case series reports, and studies on clinical application have sprung up and showed the superiority of mNGS over traditional microbial detection methods, such as microscopy and staining, culture, nucleic acid amplification tests, immunological assays, 16S rRNA gene sequencing, and MALDI‐TOF MS, as presented in detail in Table 2. mNGS works better in comprehensively cataloging the individual members comprising a specific microbiome, optimizing the precise treatment strategy and improving the prognosis of patients, and understanding how microbial communities function and influence host–pathogen interactions.[ 233 ] However, at present, it is generally recognized that mNGS will not likely universally replace traditional microbiology techniques in the short term for the following reasons: i) some traditional detection methods are highly sensitive and specific with rapid turnaround times, such as CSF CrAg, rapid and specific PCR methods; ii) indirect approaches such as serological tests will continue to play a key part in the diagnostic workup for infections, especially for the emerging infectious diseases;[ 234 ] iii) functional assays such as culture, phenotypic susceptibility testing, and drug sensitive test will likely always be useful for research studies.[ 235 ]
In summary, there are considerable barriers, including sensitivity, interpretation, actionability, popularity, turnaround time, antimicrobial susceptibility, clinical utility, laboratory workflow, cost, etc., for the adoption of diagnostic metagenomics. Current limitations suggest that mNGS can be a complementary, and perhaps an essential test in certain clinical settings, rather than being an “one‐in‐all” test.[ 148 ]
5.5. Recommendations for the Use of mNGS in Clinical Applications
The application of mNGS in the detection of clinical pathogens remains in the early stage, with no mNGS workflows or methods approved by the Food and Drug Administration or National Medical Products Administration for the diagnosis of infectious diseases. Therefore, the current mNGS processes are highly personalized, indicating the importance and difficulties associated with supervising the clinical applications of this technology. Clinical regulation exists in all aspects, including for testing population selection, method standardization, quality control, and workflow validation. The expert consensus on the application of mNGS technology in the detection of infections of critical importance was released in China in 2019,[ 92 ] which recommended the following six indicators for mNGS detection: i) the pathogen should be identified as soon as possible in critical cases; ii) the pathogen should be identified as soon as possible for special patients such as immunosuppressed hosts, patients with underlying diseases, and patients with severe infection who were hospitalized repeatedly; iii) patients in which the traditional microbiological detection techniques were repeatedly negative and the treatment effect was not good; iv) cases in which the presence of new pathogens is suspected, indicating that there may be a certain degree of clinical infectivity; v) cases where special pathogen infections are suspected, and vi) long‐term fever and/or presence of infection with additional clinical symptoms of unknown causes. Screening the test subjects and selecting the appropriate population for the test is the first step in clinical supervision, as these measures can help avoid wasted of resources and benefit patients who urgently require testing.
The mNGS experimental process is more complicated than other detection techniques as it involves sample preprocessing, nucleic acid extraction, library construction, sequencing, data analysis, and data interpretation. The more complex the steps, the more difficult it is to validate the whole workflow and establish a quality control system. At present, a few research teams have tried to establish quality control standards, however, no unified index has yet been generated.[ 106 ] Quality control is required for various steps in the workflow, including during the extraction, as well as for the concentration and purity of nucleic acids used, nucleic acid fragment distribution range, library concentration, the establishment of reference database standards, sequencing data volume, and pathogen screening principles. Additionally, different quality control standards may need to be established for different types of samples.
Currently, research validating the performance of mNGS assays in clinical applications remains scarce. In 2019, the Miller team performed a validation for mNGS using CSF samples, and comprehensively analyzed and evaluated the lowest detection limit, precision, accuracy, stability, and other indicators.[ 211 ] Similarly, the performance of mNGS in other types of samples such as BALF and sputum requires evaluation. We recommend that laboratories and third‐party testing companies verify the performance of the workflow using different sample types before testing clinical samples to confirm the accuracy and stability of the workflow. This is particularly important as the formation of standardized detection methods that utilize mNGS technology will enable better clinical supervision. For example, this work depends on the joint efforts of relevant government agencies, scientific research institutions, and commercial companies. In May 2019, more than 20 units participated in a joint study launched by the National Institutes of Food and Drug Control to evaluate the quality of the metagenome second‐generation sequencing reagents used for pathogen detection. The testing of different mNGS processes for unified samples promotes the research process for quality control and standardization. At present, there are great differences in mNGS workflows among different units, and there is still a long way to go to achieve standardized detection procedures.
In summary, although there are many reported successful applications of mNGS in the clinical setting, most of them are case reports or retrospective studies. Thus, large‐scale prospective studies with large sample sizes are needed to further demonstrate the reliability of this technology. These studies should focus on when and how to select mNGS, their diagnostic value compared, or combined with, traditional methods, how can mNGS help direct patients’ management, including diagnosis and differential diagnosis of infectious diseases, guide treatment, assess efficacy and prognosis, in addition with cost–benefit analysis. In technology, the main goal should always be the further optimization of the sequencing process, database construction, and bioinformatic analysis, making mNGS more accurate, cost‐effective, and improving its universality. Currently, there are few expert consensuses on the clinical application of mNGS, however, it is generally accepted that mNGS cannot yet replace the place of traditional methods in the short term, but is recommended as an auxiliary or necessary detection method for detecting microorganisms. We hope that high‐quality clinical studies on mNGS will be carried out in the coming years, giving mNGS a greater role in clinical application. In addition, with the emergence of new pathogens in recent decades, mNGS, along with the continuous improvement of the regulatory network and popularization of third‐generation sequencing technology, will also play an irreplaceable role in monitoring and tracking the outbreak of novel pathogen infections.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
N.L. and Q.C. contributed equally to this work. The authors acknowledge the special fund for clinical research of Zhongshan Hospital, Fudan University (No. 2018ZSLC06).
Biographies
Na Li is a resident in the Department of Infectious Diseases of Zhongshan Hospital, Fudan University. She received her Master's degree from Fudan University. Her research interest is focused on molecular diagnosis of infectious diseases and alternative therapeutic strategies for multiple drug resistant organisms.

Bijie Hu, a professor and present director of the Department of Infectious Diseases of Zhongshan Hospital, Fudan University, graduated from Shanghai Medical College of Fudan University with a Ph.D. degree. He is also an expert on multiple drug resistant organisms and nosocomial infection control at the World Health Organization, and a director of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID) collaboration center. He has expertise in various bacterial and fungal infections, especially in the diagnosis of difficult pulmonary infections, as well as accurate and early determination of the pathogens.

Li N., Cai Q., Miao Q., Song Z., Fang Y., Hu B., High‐Throughput Metagenomics for Identification of Pathogens in the Clinical Settings. Small Methods 2021, 5, 2000792 10.1002/smtd.202000792
References
- 1. Sanger F., Nicklen S., Coulson A. R., Proc. Natl. Acad. Sci. USA 1977, 74, 5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Charalampous T., Kay G. L., Richardson H., Aydin A., Baldan R., Jeanes C., Rae D., Grundy S., Turner D. J., Wain J., Leggett R. M., Livermore D. M., O'Grady J., Nat. Biotechnol. 2019, 37, 783. [DOI] [PubMed] [Google Scholar]
- 3. Simner P. J., Miller S., Carroll K. C., Clin. Infect. Dis. 2018, 66, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanger F., Coulson A. R., J. Mol. Biol. 1975, 94, 441. [DOI] [PubMed] [Google Scholar]
- 5. Maxam A. M., Gilbert W., Proc. Natl. Acad. Sci. USA 1977, 74, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Woolley A. T., Mathies R. A., Anal. Chem. 1995, 67, 3676. [DOI] [PubMed] [Google Scholar]
- 7. Deng Y. M., Spirason N., Iannello P., Jelley L., Lau H., Barr I. G., J. Clin. Virol. 2015, 68, 43. [DOI] [PubMed] [Google Scholar]
- 8. Cabral G. B., Ferreira J. L. P., Souza R. P., Cunha M. S., Luchs A., Figueiredo C. A., Brigido L. F. M., Mem. Inst. Oswaldo Cruz 2018, 113, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sabat A. J., van Zanten E., Akkerboom V., Wisselink G., van Slochteren K., de Boer R. F., Hendrix R., Friedrich A. W., Rossen J. W. A., Kooistra‐Smid A., Sci. Rep. 2017, 7, 3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Banos S., Lentendu G., Kopf A., Wubet T., Glockner F. O., Reich M., BMC Microbiol. 2018, 18, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ai J. W., Weng S. S., Cheng Q., Cui P., Li Y. J., Wu H. L., Zhu Y. M., Xu B., Zhang W. H., Emerging Infect. Dis. 2018, 24, 1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Han D., Li Z., Li R., Tan P., Zhang R., Li J., Crit. Rev. Microbiol. 2019, 45, 668. [DOI] [PubMed] [Google Scholar]
- 13. Bentley D. R., Balasubramanian S., Swerdlow H. P., Smith G. P., Milton J., Brown C. G., Hall K. P., Evers D. J., Barnes C. L., Bignell H. R., Boutell J. M., Bryant J., Carter R. J., Keira Cheetham R., Cox A. J., Ellis D. J., Flatbush M. R., Gormley N. A., Humphray S. J., Irving L. J., Karbelashvili M. S., Kirk S. M., Li H., Liu X., Maisinger K. S., Murray L. J., Obradovic B., Ost T., Parkinson M. L., Pratt M. R., Rasolonjatovo I. M., Reed M. T., Rigatti R., Rodighiero C., Ross M. T., Sabot A., Sankar S. V., Scally A., Schroth G. P., Smith M. E., Smith V. P., Spiridou A., Torrance P. E., Tzonev S. S., Vermaas E. H., Walter K., Wu X., Zhang L., Alam M. D., Anastasi C., Aniebo I. C., Bailey D. M., Bancarz I. R., Banerjee S., Barbour S. G., Baybayan P. A., Benoit V. A., Benson K. F., Bevis C., Black P. J., Boodhun A., Brennan J. S., Bridgham J. A., Brown R. C., Brown A. A., Buermann D. H., Bundu A. A., Burrows J. C., Carter N. P., Castillo N., Chiara E. C. M., Chang S., Neil Cooley R., Crake N. R., Dada O. O., Diakoumakos K. D., Dominguez‐Fernandez B., Earnshaw D. J., Egbujor U. C., Elmore D. W., Etchin S. S., Ewan M. R., Fedurco M., Fraser L. J., Fuentes Fajardo K. V., Scott Furey W., George D., Gietzen K. J., Goddard C. P., Golda G. S., Granieri P. A., Green D. E., Gustafson D. L., Hansen N. F., Harnish K., Haudenschild C. D., Heyer N. I., Hims M. M., Ho J. T., Horgan A. M., Hoschler K., Hurwitz S., Ivanov D. V., Johnson M. Q., James T., Huw Jones T. A., Kang G. D., Kerelska T. H., Kersey A. D., Khrebtukova I., Kindwall A. P., Kingsbury Z., Kokko‐Gonzales P. I., Kumar A., Laurent M. A., Lawley C. T., Lee S. E., Lee X., Liao A. K., Loch J. A., Lok M., Luo S., Mammen R. M., Martin J. W., McCauley P. G., McNitt P., Mehta P., Moon K. W., Mullens J. W., Newington T., Ning Z., Ling Ng B., Novo S. M., O'Neill M. J., Osborne M. A., Osnowski A., Ostadan O., Paraschos L. L., Pickering L., Pike A. C., Pike A. C., Chris Pinkard D., Pliskin D. P., Podhasky J., Quijano V. J., Raczy C., Rae V. H., Rawlings S. R., Chiva Rodriguez A., Roe P. M., Rogers J., Rogert Bacigalupo M. C., Romanov N., Romieu A., Roth R. K., Rourke N. J., Ruediger S. T., Rusman E., Sanches‐Kuiper R. M., Schenker M. R., Seoane J. M., Shaw R. J., Shiver M. K., Short S. W., Sizto N. L., Sluis J. P., Smith M. A., Ernest Sohna Sohna J., Spence E. J., Stevens K., Sutton N., Szajkowski L., Tregidgo C. L., Turcatti G., Vandevondele S., Verhovsky Y., Virk S. M., Wakelin S., Walcott G. C., Wang J., Worsley G. J., Yan J., Yau L., Zuerlein M., Rogers J., Mullikin J. C., Hurles M. E., McCooke N. J., West J. S., Oaks F. L., Lundberg P. L., Klenerman D., Durbin R., Smith A. J., Nature 2008, 456, 53.18987734 [Google Scholar]
- 14. Hu Z., Cheng L., Wang H., Methods Mol. Biol. 2015, 1231, 91. [DOI] [PubMed] [Google Scholar]
- 15. Wright M. N., Gola D., Ziegler A., Methods Mol. Biol. 2017, 1666, 629. [DOI] [PubMed] [Google Scholar]
- 16. Prakash G., Kumar A., Sheoran N., Aggarwal R., Satyavathi C. T., Chikara S. K., Ghosh A., Jain R. K., Microbiol. Resour. Announce. 2019, 8, e01499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Korostin D., Kulemin N., Naumov V., Belova V., Kwon D., Gorbachev A., PLoS One 2020, 15, e0230301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marine R. L., Magana L. C., Castro C. J., Zhao K., Montmayeur A. M., Schmidt A., Diez‐Valcarce M., Ng T. F. F., Vinje J., Burns C. C., Nix W. A., Rota P. A., Oberste M. S., J. Virol. Methods 2020, 280, 113865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilson M. R., Naccache S. N., Samayoa E., Biagtan M., Bashir H., Yu G., Salamat S. M., Somasekar S., Federman S., Miller S., Sokolic R., Garabedian E., Candotti F., Buckley R. H., Reed K. D., Meyer T. L., Seroogy C. M., Galloway R., Henderson S. L., Gern J. E., DeRisi J. L., Chiu C. Y., N. Engl. J. Med. 2014, 370, 2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ji X. C., Zhou L. F., Li C. Y., Shi Y. J., Wu M. L., Zhang Y., Fei X. F., Zhao G., J. Mol. Neurosci. 2020, 70, 659. [DOI] [PubMed] [Google Scholar]
- 21. Langelier C., Kalantar K. L., Moazed F., Wilson M. R., Crawford E. D., Deiss T., Belzer A., Bolourchi S., Caldera S., Fung M., Jauregui A., Malcolm K., Lyden A., Khan L., Vessel K., Quan J., Zinter M., Chiu C. Y., Chow E. D., Wilson J., Miller S., Matthay M. A., Pollard K. S., Christenson S., Calfee C. S., DeRisi J. L., Proc. Natl. Acad. Sci. USA 2018, 115, E12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wilson M. R., Sample H. A., Zorn K. C., Arevalo S., Yu G., Neuhaus J., Federman S., Stryke D., Briggs B., Langelier C., Berger A., Douglas V., Josephson S. A., Chow F. C., Fulton B. D., DeRisi J. L., Gelfand J. M., Naccache S. N., Bender J., Dien Bard J., Murkey J., Carlson M., Vespa P. M., Vijayan T., Allyn P. R., Campeau S., Humphries R. M., Klausner J. D., Ganzon C. D., Memar F., Ocampo N. A., Zimmermann L. L., Cohen S. H., Polage C. R., DeBiasi R. L., Haller B., Dallas R., Maron G., Hayden R., Messacar K., Dominguez S. R., Miller S., Chiu C. Y., N. Engl. J. Med. 2019, 380, 2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carpenter M. L., Tan S. K., Watson T., Bacher R., Nagesh V., Watts A., Bentley G., Weber J., Huang C., Sahoo M. K., Hinterwirth A., Doan T., Carter T., Dong Q., Gourguechon S., Harness E., Kermes S., Radhakrishnan S., Wang G., Quiroz‐Zarate A., Ching J., Pinsky B. A., J. Clin. Microbiol. 2019, 57, e01113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou X., Wu H., Ruan Q., Jiang N., Chen X., Shen Y., Zhu Y. M., Ying Y., Qian Y. Y., Wang X., Ai J. W., Zhang W. H., Front. Cell. Infect. Microbiol. 2019, 9, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Boheemen S., van Rijn A. L., Pappas N., Carbo E. C., Vorderman R. H. P., Sidorov I., van 't Hof P. J., Mei H., Claas E. C. J., Kroes A. C. M., de Vries J. J. C., J. Mol. Diagn. 2020, 22, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen L., Xu Y., Liu C., Huang H., Zhong X., Ma C., Zhao H., Chen Y., BMC Infect. Dis. 2020, 20, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Drmanac R., Sparks A. B., Callow M. J., Halpern A. L., Burns N. L., Kermani B. G., Carnevali P., Nazarenko I., Nilsen G. B., Yeung G., Dahl F., Fernandez A., Staker B., Pant K. P., Baccash J., Borcherding A. P., Brownley A., Cedeno R., Chen L., Chernikoff D., Cheung A., Chirita R., Curson B., Ebert J. C., Hacker C. R., Hartlage R., Hauser B., Huang S., Jiang Y., Karpinchyk V., Koenig M., Kong C., Landers T., Le C., Liu J., McBride C. E., Morenzoni M., Morey R. E., Mutch K., Perazich H., Perry K., Peters B. A., Peterson J., Pethiyagoda C. L., Pothuraju K., Richter C., Rosenbaum A. M., Roy S., Shafto J., Sharanhovich U., Shannon K. W., Sheppy C. G., Sun M., Thakuria J. V., Tran A., Vu D., Zaranek A. W., Wu X., Drmanac S., Oliphant A. R., Banyai W. C., Martin B., Ballinger D. G., Church G. M., Reid C. A., Science 2010, 327, 78.19892942 [Google Scholar]
- 28. Zhu F. Y., Chen M. X., Ye N. H., Qiao W. M., Gao B., Law W. K., Tian Y., Zhang D., Zhang D., Liu T. Y., Hu Q. J., Cao Y. Y., Su Z. Z., Zhang J., Liu Y. G., Plant Methods 2018, 14, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang H. C., Ai J. W., Cui P., Zhu Y. M., Hong‐Long W., Li Y. J., Zhang W. H., J. Infect. 2019, 79, 419. [DOI] [PubMed] [Google Scholar]
- 30. Wang S., Chen Y., Wang D., Wu Y., Zhao D., Zhang J., Xie H., Gong Y., Sun R., Nie X., Jiang H., Zhang J., Li W., Liu G., Li X., Huang K., Huang Y., Li Y., Guan H., Pan S., Hu Y., Front. Microbiol. 2019, 10, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Petersen L. M., Martin I. W., Moschetti W. E., Kershaw C. M., Tsongalis G. J., J. Clin. Microbiol. 2019, 58, e01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goodwin S., McPherson J. D., McCombie W. R., Nat. Rev. Genet. 2016, 17, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B., Bibillo A., Bjornson K., Chaudhuri B., Christians F., Cicero R., Clark S., Dalal R., Dewinter A., Dixon J., Foquet M., Gaertner A., Hardenbol P., Heiner C., Hester K., Holden D., Kearns G., Kong X., Kuse R., Lacroix Y., Lin S., Lundquist P., Ma C., Marks P., Maxham M., Murphy D., Park I., Pham T., Phillips M., Roy J., Sebra R., Shen G., Sorenson J., Tomaney A., Travers K., Trulson M., Vieceli J., Wegener J., Wu D., Yang A., Zaccarin D., Zhao P., Zhong F., Korlach J., Turner S., Science 2009, 323, 133. [DOI] [PubMed] [Google Scholar]
- 34. Yu X., Jiang W., Shi Y., Ye H., Lin J., J. Cell. Mol. Med. 2019, 23, 7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gonzalez‐Reiche A. S., Hernandez M. M., Sullivan M. J., Ciferri B., Alshammary H., Obla A., Fabre S., Kleiner G., Polanco J., Khan Z., Alburquerque B., van de Guchte A., Dutta J., Francoeur N., Melo B. S., Oussenko I., Deikus G., Soto J., Sridhar S. H., Wang Y. C., Twyman K., Kasarskis A., Altman D. R., Smith M., Sebra R., Aberg J., Krammer F., Garcia‐Sastre A., Luksza M., Patel G., Paniz‐Mondolfi A., Gitman M., Sordillo E. M., Simon V., van Bakel H., Science 2020, 369, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Feng Y., Zhang Y., Ying C., Wang D., Du C., Genomics, Proteomics Bioinf. 2015, 13, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leggett R. M., Clark M. D., J. Exp. Bot. 2017, 68, 5419. [DOI] [PubMed] [Google Scholar]
- 38. Bowden R., Davies R. W., Heger A., Pagnamenta A. T., de Cesare M., Oikkonen L. E., Parkes D., Freeman C., Dhalla F., Patel S. Y., Popitsch N., Ip C. L. C., Roberts H. E., Salatino S., Lockstone H., Lunter G., Taylor J. C., Buck D., Simpson M. A., Donnelly P., Nat. Commun. 2019, 10, 1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eisenstein M., Nat. Biotechnol. 2012, 30, 295. [DOI] [PubMed] [Google Scholar]
- 40. Goodwin S., Gurtowski J., Ethe‐Sayers S., Deshpande P., Schatz M. C., McCombie W. R., Genome Res. 2015, 25, 1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ip C. L. C., Loose M., Tyson J. R., de Cesare M., Brown B. L., Jain M., Leggett R. M., Eccles D. A., Zalunin V., Urban J. M., Piazza P., Bowden R. J., Paten B., Mwaigwisya S., Batty E. M., Simpson J. T., Snutch T. P., Birney E., Buck D., Goodwin S., Jansen H. J., O'Grady J., Olsen H. E., MinION Analysis and Reference Consortium , F1000Research 2015, 4, 1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rang F. J., Kloosterman W. P., de Ridder J., Genome Biol. 2018, 19, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Quick J., Loman N. J., Duraffour S., Simpson J. T., Severi E., Cowley L., Bore J. A., Koundouno R., Dudas G., Mikhail A., Ouedraogo N., Afrough B., Bah A., Baum J. H., Becker‐Ziaja B., Boettcher J. P., Cabeza‐Cabrerizo M., Camino‐Sanchez A., Carter L. L., Doerrbecker J., Enkirch T., Dorival I. G. G., Hetzelt N., Hinzmann J., Holm T., Kafetzopoulou L. E., Koropogui M., Kosgey A., Kuisma E., Logue C. H., Mazzarelli A., Meisel S., Mertens M., Michel J., Ngabo D., Nitzsche K., Pallash E., Patrono L. V., Portmann J., Repits J. G., Rickett N. Y., Sachse A., Singethan K., Vitoriano I., Yemanaberhan R. L., Zekeng E. G., Trina R., Bello A., Sall A. A., Faye O., Faye O., Magassouba N., Williams C. V., Amburgey V., Winona L., Davis E., Gerlach J., Washington F., Monteil V., Jourdain M., Bererd M., Camara A., Somlare H., Camara A., Gerard M., Bado G., Baillet B., Delaune D., Nebie K. Y., Diarra A., Savane Y., Pallawo R. B., Gutierrez G. J., Milhano N., Roger I., Williams C. J., Yattara F., Lewandowski K., Taylor J., Rachwal P., Turner D., Pollakis G., Hiscox J. A., Matthews D. A., O'Shea M. K., Johnston A. M., Wilson D., Hutley E., Smit E., Di Caro A., Woelfel R., Stoecker K., Fleischmann E., Gabriel M., Weller S. A., Koivogui L., Diallo B., Keita S., Rambaut A., Formenty P., Gunther S., Carroll M. W., Nature 2016, 530, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prazsak I., Moldovan N., Balazs Z., Tombacz D., Megyeri K., Szucs A., Csabai Z., Boldogkoi Z., BMC Genomics 2018, 19, 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bainomugisa A., Duarte T., Lavu E., Pandey S., Coulter C., Marais B. J., Coin L. M., Microb. Genomics 2018, 4, e000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Runtuwene L. R., Tuda J. S. B., Mongan A. E., Makalowski W., Frith M. C., Imwong M., Srisutham S., Nguyen Thi L. A., Tuan N. N., Eshita Y., Maeda R., Yamagishi J., Suzuki Y., Sci. Rep. 2018, 8, 8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Farnia P., Mohammadi F., Zarifi Z., Tabatabee D. J., Ganavi J., Ghazisaeedi K., Farnia P. K., Gheydi M., Bahadori M., Masjedi M. R., Velayati A. A., J. Clin. Microbiol. 2002, 40, 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Terranova L., Oriano M., Teri A., Ruggiero L., Tafuro C., Marchisio P., Gramegna A., Contarini M., Franceschi E., Sottotetti S., Cariani L., Bevivino A., Chalmers J. D., Aliberti S., Blasi F., Int. J. Mol. Sci. 2018, 19, 3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guiot J., Demarche S., Henket M., Paulus V., Graff S., Schleich F., Corhay J. L., Louis R., Moermans C., J. Visualized Exp. 2017, 130, e56612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Park H. J., Woo A., Cha J. M., Lee K. S., Lee M. Y., Sci. Rep. 2018, 8, 17994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cai Y., Fang X., Chen Y., Huang Z., Zhang C., Li W., Yang B., Zhang W., Int. J. Infect. Dis. 2020, 96, 573. [DOI] [PubMed] [Google Scholar]
- 52. Wang Q., Miao Q., Pan J., Jin W., Ma Y., Zhang Y., Yao Y., Su Y., Huang Y., Li B., Wang M., Li N., Cai S., Luo Y., Zhou C., Wu H., Hu B., Int. J. Infect. Dis. 2020, 100, 414. [DOI] [PubMed] [Google Scholar]
- 53. Ramachandran P. S., Wilson M. R., Nat. Rev. Neurol. 2020, 16, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Frickmann H., Kunne C., Hagen R. M., Podbielski A., Normann J., Poppert S., Looso M., Kreikemeyer B., BMC Microbiol. 2019, 19, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kohl C., Brinkmann A., Dabrowski P. W., Radonic A., Nitsche A., Kurth A., Emerging Infect. Dis. 2015, 21, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dedhia P., Tarale S., Dhongde G., Khadapkar R., Das B., Asian Pac. J. Cancer Prev. 2007, 8, 55. [PubMed] [Google Scholar]
- 57. Long Y., Zhang Y., Gong Y., Sun R., Su L., Lin X., Shen A., Zhou J., Caiji Z., Wang X., Li D., Wu H., Tan H., Arch. Med. Res. 2016, 47, 365. [DOI] [PubMed] [Google Scholar]
- 58. Helmersen K., Aamot H. V., Sci. Rep. 2020, 10, 2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bennett E. A., Massilani D., Lizzo G., Daligault J., Geigl E. M., Grange T., BioTechniques 2014, 56, 289. [DOI] [PubMed] [Google Scholar]
- 60. Di L., Fu Y., Sun Y., Li J., Liu L., Yao J., Wang G., Wu Y., Lao K., Lee R. W., Zheng G., Xu J., Oh J., Wang D., Xie X. S., Huang Y., Wang J., Proc. Natl. Acad. Sci. USA 2020, 117, 2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lu B., Dong L., Yi D., Zhang M., Zhu C., Li X., Yi C., eLife 2020, 9, e54919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huang J., Liang X., Xuan Y., Geng C., Li Y., Lu H., Qu S., Mei X., Chen H., Yu T., Sun N., Rao J., Wang J., Zhang W., Chen Y., Liao S., Jiang H., Liu X., Yang Z., Mu F., Gao S., GigaScience 2017, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wu J., Zhang W., Huang S., He Z., Cheng Y., Wang J., Lam T. W., Peng Z., Yiu S. M., Bioinformatics 2013, 29, 2971. [DOI] [PubMed] [Google Scholar]
- 64. de Lannoy C., de Ridder D., Risse J., F1000Research 2017, 6, 1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lu H., Giordano F., Ning Z., Genomics, Proteomics Bioinf. 2016, 14, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Seki M., Katsumata E., Suzuki A., Sereewattanawoot S., Sakamoto Y., Mizushima‐Sugano J., Sugano S., Kohno T., Frith M. C., Tsuchihara K., Suzuki Y., DNA Res. 2019, 26, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Soneson C., Yao Y., Bratus‐Neuenschwander A., Patrignani A., Robinson M. D., Hussain S., Nat. Commun. 2019, 10, 3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rhoads A., Au K. F., Genomics, Proteomics Bioinf. 2015, 13, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Coupland P., Chandra T., Quail M., Reik W., Swerdlow H., BioTechniques 2012, 53, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bolger A. M., Lohse M., Usadel B., Bioinformatics 2014, 30, 2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen S., Zhou Y., Chen Y., Gu J., Bioinformatics 2018, 34, i884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schmieder R., Edwards R., Bioinformatics 2011, 27, 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Piatigorsky J., Tyler A., J. Cell Sci. 1968, 3, 515. [DOI] [PubMed] [Google Scholar]
- 74. Li H., Durbin R., Bioinformatics 2009, 25, 1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Langmead B., Salzberg S. L., Nat. Methods 2012, 9, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Eisenstein M., Nat Biotechnol. 2012, 30, 295. [DOI] [PubMed] [Google Scholar]
- 77. Haque M. M., Bose T., Dutta A., Reddy C. V., Mande S. S., Genomics 2015, 106, 116. [DOI] [PubMed] [Google Scholar]
- 78. Czajkowski M. D., Vance D. P., Frese S. A., Casaburi G., Bioinformatics 2019, 35, 2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Haft D. H., DiCuccio M., Badretdin A., Brover V., Chetvernin V., O'Neill K., Li W., Chitsaz F., Derbyshire M. K., Gonzales N. R., Gwadz M., Lu F., Marchler G. H., Song J. S., Thanki N., Yamashita R. A., Zheng C., Thibaud‐Nissen F., Geer L. Y., Marchler‐Bauer A., Pruitt K. D., Nucleic Acids Res. 2018, 46, D851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ounit R., Wanamaker S., Close T. J., Lonardi S., BMC Genomics 2015, 16, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wittouck S., Wuyts S., Meehan C. J., van Noort V., Lebeer S., mSystems 2019, 4, e00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W., Genome Res. 2015, 25, 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sichtig H., Minogue T., Yan Y., Stefan C., Hall A., Tallon L., Sadzewicz L., Nadendla S., Klimke W., Hatcher E., Shumway M., Aldea D. L., Allen J., Koehler J., Slezak T., Lovell S., Schoepp R., Scherf U., Nat. Commun. 2019, 10, 3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Breitwieser F. P., Baker D. N., Salzberg S. L., Genome Biol. 2018, 19, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wood D. E., Lu J., Langmead B., Genome Biol. 2019, 20, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kim D., Song L., Breitwieser F. P., Salzberg S. L., Genome Res. 2016, 26, 1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Truong D. T., Franzosa E. A., Tickle T. L., Scholz M., Weingart G., Pasolli E., Tett A., Huttenhower C., Segata N., Nat. Methods 2015, 12, 902. [DOI] [PubMed] [Google Scholar]
- 88. Menzel P., Ng K. L., Krogh A., Nat. Commun. 2016, 7, 11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Buchfink B., Xie C., Huson D. H., Nat. Methods 2015, 12, 59. [DOI] [PubMed] [Google Scholar]
- 90. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., J. Mol. Biol. 1990, 215, 403. [DOI] [PubMed] [Google Scholar]
- 91. Allicock O. M., Guo C., Uhlemann A. C., Whittier S., Chauhan L. V., Garcia J., Price A., Morse S. S., Mishra N., Briese T., Lipkin W. I., mBio 2018, 9, e02007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Consensus Group of Experts on Application of Metagenomic Next Generation Sequencing in the Pathogen Diagnosis in Clinical Moderate and Severe Infections , Professional Committee of Sepsis and Shock, Chinese Research Hospital Association , Professional Committee of Microbial Toxins Chinese Society for Microbiology , Professional Committee of Critical Care Medicine Shenzhen Medical Association , Zhonghua Weizhongbing Jijiu Yixue 2020, 32, 531. [Google Scholar]
- 93. Mongkolrattanothai K., Naccache S. N., Bender J. M., Samayoa E., Pham E., Yu G., Dien Bard J., Miller S., Aldrovandi G., Chiu C. Y., J. Pediatr. Infect. Dis. Soc. 2017, 6, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Breitwieser F. P., Salzberg S. L., Bioinformatics 2020, 36, 1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Maabar M., Davison A. J., Vucak M., Thorburn F., Murcia P. R., Gunson R., Palmarini M., Hughes J., Virus Evol. 2019, 5, vez033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Watts G. S., Thornton J. E. Jr., Youens‐Clark K., Ponsero A. J., Slepian M. J., Menashi E., Hu C., Deng W., Armstrong D. G., Reed S., Cranmer L. D., Hurwitz B. L., PLoS Comput. Biol. 2019, 15, e1006863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Strong M. J., Xu G., Morici L., Splinter Bon‐Durant S., Baddoo M., Lin Z., Fewell C., Taylor C. M., Flemington E. K., PLoS Pathog. 2014, 10, e1004437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ondov B. D., Bergman N. H., Phillippy A. M., BMC Bioinformatics 2011, 12, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Miao Q., Ma Y., Wang Q., Pan J., Zhang Y., Jin W., Yao Y., Su Y., Huang Y., Wang M., Li B., Li H., Zhou C., Li C., Ye M., Xu X., Li Y., Hu B., Clin. Infect. Dis. 2018, 67, S231. [DOI] [PubMed] [Google Scholar]
- 100. Mancini N., Carletti S., Ghidoli N., Cichero P., Burioni R., Clementi M., Clin. Microbiol. Rev. 2010, 23, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Fishman J. A., N. Engl. J. Med. 2007, 357, 2601. [DOI] [PubMed] [Google Scholar]
- 102. Tomblyn M., Chiller T., Einsele H., Gress R., Sepkowitz K., Storek J., Wingard J. R., Young J. A., Boeckh M. J., Center for International Blood and Marrow Research , National Marrow Donor Program , European Blood and Marrow Transplant Group , American Society of Blood and Marrow Transplantation , Canadian Blood and Marrow Transplant Group , Infectious Diseases Society of America , Society for Healthcare Epidemiology of America , Association of Medical Microbiology and Infectious Disease Canada , Centers for Disease Control and Prevention , Biol. Blood Marrow Transplant. 2009, 15, 1143.19747629 [Google Scholar]
- 103. Barlam T. F., Cosgrove S. E., Abbo L. M., MacDougall C., Schuetz A. N., Septimus E. J., Srinivasan A., Dellit T. H., Falck‐Ytter Y. T., Fishman N. O., Hamilton C. W., Jenkins T. C., Lipsett P. A., Malani P. N., May L. S., Moran G. J., Neuhauser M. M., Newland J. G., Ohl C. A., Samore M. H., Seo S. K., Trivedi K. K., Clin. Infect. Dis. 2016, 62, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Paul M., Shani V., Muchtar E., Kariv G., Robenshtok E., Leibovici L., Antimicrob. Agents Chemother. 2010, 54, 4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Glaser C. A., Honarmand S., Anderson L. J., Schnurr D. P., Forghani B., Cossen C. K., Schuster F. L., Christie L. J., Tureen J. H., Clin. Infect. Dis. 2006, 43, 1565. [DOI] [PubMed] [Google Scholar]
- 106. Schlaberg R., Chiu C. Y., Miller S., Procop G. W., Weinstock G., The Professional Practice Committee and Committee on Laboratory Practices of the American Society for Microbiology , The Microbiology Resource Committee of the College of American Pathologists , Arch. Pathol. Lab. Med. 2017, 141, 776. [DOI] [PubMed] [Google Scholar]
- 107. Martin G. S., Mannino D. M., Eaton S., Moss M., N. Engl. J. Med. 2003, 348, 1546. [DOI] [PubMed] [Google Scholar]
- 108. Gupta S., Sakhuja A., Kumar G., McGrath E., Nanchal R. S., Kashani K. B., Chest 2016, 150, 1251. [DOI] [PubMed] [Google Scholar]
- 109. Kumar A., Ellis P., Arabi Y., Roberts D., Light B., Parrillo J. E., Dodek P., Wood G., Kumar A., Simon D., Peters C., Ahsan M., Chateau D., Cooperative Antimicrobial Therapy of Septic Shock Database Research Group , Chest 2009, 136, 1237. [DOI] [PubMed] [Google Scholar]
- 110. De Vlaminck I., Martin L., Kertesz M., Patel K., Kowarsky M., Strehl C., Cohen G., Luikart H., Neff N. F., Okamoto J., Nicolls M. R., Cornfield D., Weill D., Valantine H., Khush K. K., Quake S. R., Proc. Natl. Acad. Sci. USA 2015, 112, 13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pan W., Ngo T. T. M., Camunas‐Soler J., Song C. X., Kowarsky M., Blumenfeld Y. J., Wong R. J., Shaw G. M., Stevenson D. K., Quake S. R., Clin. Chem. 2017, 63, 1695. [DOI] [PubMed] [Google Scholar]
- 112. Gosiewski T., Ludwig‐Galezowska A. H., Huminska K., Sroka‐Oleksiak A., Radkowski P., Salamon D., Wojciechowicz J., Kus‐Slowinska M., Bulanda M., Wolkow P. P., Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Abril M. K., Barnett A. S., Wegermann K., Fountain E., Strand A., Heyman B. M., Blough B. A., Swaminathan A. C., Sharma‐Kuinkel B., Ruffin F., Alexander B. D., McCall C. M., Costa S. F., Arcasoy M. O., Hong D. K., Blauwkamp T. A., Kertesz M., Fowler V. G. Jr., Kraft B. D., Open Forum Infect. Dis. 2016, 3, ofw144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hong D. K., Blauwkamp T. A., Kertesz M., Bercovici S., Truong C., Banaei N., Diagn. Microbiol. Infect. Dis. 2018, 92, 210. [DOI] [PubMed] [Google Scholar]
- 115. Blauwkamp T. A., Thair S., Rosen M. J., Blair L., Lindner M. S., Vilfan I. D., Kawli T., Christians F. C., Venkatasubrahmanyam S., Wall G. D., Cheung A., Rogers Z. N., Meshulam‐Simon G., Huijse L., Balakrishnan S., Quinn J. V., Hollemon D., Hong D. K., Vaughn M. L., Kertesz M., Bercovici S., Wilber J. C., Yang S., Nat. Microbiol. 2019, 4, 663. [DOI] [PubMed] [Google Scholar]
- 116. Crawford E., Kamm J., Miller S., Li L. M., Caldera S., Lyden A., Yokoe D., Nichols A., Tran N. K., Barnard S. E., Conner P. M., Nambiar A., Zinter M. S., Moayeri M., Serpa P. H., Prince B. C., Quan J., Sit R., Tan M., Phelps M., DeRisi J. L., Tato C. M., Langelier C., Clin. Infect. Dis. 2019, 71, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zhang Y., Cui P., Zhang H. C., Wu H. L., Ye M. Z., Zhu Y. M., Ai J. W., Zhang W. H., J. Transl. Med. 2020, 18, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Xing X. W., Zhang J. T., Ma Y. B., Zheng N., Yang F., Yu S. Y., J. Med. Microbiol. 2019, 68, 1204. [DOI] [PubMed] [Google Scholar]
- 119. Hu Z., Weng X., Xu C., Lin Y., Cheng C., Wei H., Chen W., Ann. Clin. Microbiol. Antimicrob. 2018, 17, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wilson M. R., O'Donovan B. D., Gelfand J. M., Sample H. A., Chow F. C., Betjemann J. P., Shah M. P., Richie M. B., Gorman M. P., Hajj‐Ali R. A., Calabrese L. H., Zorn K. C., Chow E. D., Greenlee J. E., Blum J. H., Green G., Khan L. M., Banerji D., Langelier C., Bryson‐Cahn C., Harrington W., Lingappa J. R., Shanbhag N. M., Green A. J., Brew B. J., Soldatos A., Strnad L., Doernberg S. B., Jay C. A., Douglas V., Josephson S. A., DeRisi J. L., JAMA Neurol. 2018, 75, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Granerod J., Ambrose H. E., Davies N. W., Clewley J. P., Walsh A. L., Morgan D., Cunningham R., Zuckerman M., Mutton K. J., Solomon T., Ward K. N., Lunn M. P., Irani S. R., Vincent A., Brown D. W., Crowcroft N. S., UK Health Protection Agency (HPA) Aetiology of Encephalitis Study Group , Lancet Infect. Dis. 2010, 10, 835. [DOI] [PubMed] [Google Scholar]
- 122. Guo L. Y., Li Y. J., Liu L. L., Wu H. L., Zhou J. L., Zhang Y., Feng W. Y., Zhu L., Hu B., Hu H. L., Chen T. M., Guo X., Chen H. Y., Yang Y. H., Liu G., J. Infect. 2019, 78, 323. [Google Scholar]
- 123. Wilson M. R., Suan D., Duggins A., Schubert R. D., Khan L. M., Sample H. A., Zorn K. C., Rodrigues Hoffman A., Blick A., Shingde M., DeRisi J. L., Ann. Neurol. 2017, 82, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wilson M. R., Zimmermann L. L., Crawford E. D., Sample H. A., Soni P. R., Baker A. N., Khan L. M., DeRisi J. L., Am. J. Transplant. 2017, 17, 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Chiu C. Y., Coffey L. L., Murkey J., Symmes K., Sample H. A., Wilson M. R., Naccache S. N., Arevalo S., Somasekar S., Federman S., Stryke D., Vespa P., Schiller G., Messenger S., Humphries R., Miller S., Klausner J. D., Emerging Infect. Dis. 2017, 23, 1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Murkey J. A., Chew K. W., Carlson M., Shannon C. L., Sirohi D., Sample H. A., Wilson M. R., Vespa P., Humphries R. M., Miller S., Klausner J. D., Chiu C. Y., Open Forum Infect. Dis. 2017, 4, ofx121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kufner V., Plate A., Schmutz S., Braun D. L., Gunthard H. F., Capaul R., Zbinden A., Mueller N. J., Trkola A., Huber M., Genes 2019, 10, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zhang X. X., Guo L. Y., Liu L. L., Shen A., Feng W. Y., Huang W. H., Hu H. L., Hu B., Guo X., Chen T. M., Chen H. Y., Jiang Y. Q., Liu G., BMC Infect. Dis. 2019, 19, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wilson M. R., Shanbhag N. M., Reid M. J., Singhal N. S., Gelfand J. M., Sample H. A., Benkli B., O'Donovan B. D., Ali I. K., Keating M. K., Dunnebacke T. H., Wood M. D., Bollen A., DeRisi J. L., Ann. Neurol. 2015, 78, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Xing X. W., Zhang J. T., Ma Y. B., He M. W., Yao G. E., Wang W., Qi X. K., Chen X. Y., Wu L., Wang X. L., Huang Y. H., Du J., Wang H. F., Wang R. F., Yang F., Yu S. Y., Front. Cell. Infect. Microbiol. 2020, 10, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Brown J. R., Bharucha T., Breuer J., J. Infect. 2018, 76, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Yan L., Sun W., Lu Z., Fan L., Int. J. Infect. Dis. 2020, 96, 270. [DOI] [PubMed] [Google Scholar]
- 133. Thakur K. T., Curr. HIV/AIDS Rep. 2020, 17, 507. [DOI] [PubMed] [Google Scholar]
- 134. Tyler K. L., N. Engl. J. Med. 2018, 379, 557. [DOI] [PubMed] [Google Scholar]
- 135. Fang M., Weng X., Chen L., Chen Y., Chi Y., Chen W., Hu Z., BMC Infect. Dis. 2020, 20, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Xia H., Guan Y., Zaongo S. D., Xia H., Wang Z., Yan Z., Ma P., Front. Neurol. 2019, 10, 1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Guan H., Shen A., Lv X., Yang X., Ren H., Zhao Y., Zhang Y., Gong Y., Ni P., Wu H., Zhu Y., Cui L., J. NeuroVirol 2016, 22, 240. [DOI] [PubMed] [Google Scholar]
- 138. Ai J., Xie Z., Liu G., Chen Z., Yang Y., Li Y., Chen J., Zheng G., Shen K., BMC Infect. Dis. 2017, 17, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Salzberg S. L., Breitwieser F. P., Kumar A., Hao H., Burger P., Rodriguez F. J., Lim M., Quinones‐Hinojosa A., Gallia G. L., Tornheim J. A., Melia M. T., Sears C. L., Pardo C. A., Neurol.: Neuroimmunol. Neuroinflammation 2016, 3, e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Yao M., Zhou J., Zhu Y., Zhang Y., Lv X., Sun R., Shen A., Ren H., Cui L., Guan H., Wu H., J. Clin. Neurol. 2016, 12, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Fan S., Ren H., Wei Y., Mao C., Ma Z., Zhang L., Wang L., Ge Y., Li T., Cui L., Wu H., Guan H., Int. J. Infect. Dis. 2018, 67, 20. [DOI] [PubMed] [Google Scholar]
- 142. Wang Q., Li J., Ji J., Yang L., Chen L., Zhou R., Yang Y., Zheng H., Yuan J., Li L., Bi Y., Gao G. F., Ma J., Liu Y., BMC Infect. Dis. 2018, 18, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Liu P., Weng X., Zhou J., Xu X., He F., Du Y., Wu H., Gong Y., Peng G., BMC Infect. Dis. 2018, 18, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Fan S., Qiao X., Liu L., Wu H., Zhou J., Sun R., Chen Q., Huang Y., Mao C., Yuan J., Lu Q., Ge Y., Li Y., Ren H., Wang J., Cui L., Zhao W., Guan H., Front. Neurol. 2018, 9, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. He R., Zheng W., Long J., Huang Y., Liu C., Wang Q., Yan Z., Liu H., Xing L., Hu Y., Xie H., J. NeuroVirol. 2019, 25, 127. [DOI] [PubMed] [Google Scholar]
- 146. Milucky J., Pondo T., Gregory C. J., Iuliano D., Chaves S. S., McCracken J., Mansour A., Zhang Y., Aleem M. A., Wolff B., Whitaker B., Whistler T., Onyango C., Lopez M. R., Liu N., Rahman M. Z., Shang N., Winchell J., Chittaganpitch M., Fields B., Maldonado H., Xie Z., Lindstrom S., Sturm‐Ramirez K., Montgomery J., Wu K. H., Van Beneden C. A., Adult TAC Working Group , PLoS One 2020, 15, e0240309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Li H., Gao H., Meng H., Wang Q., Li S., Chen H., Li Y., Wang H., Front. Cell. Infect. Microbiol. 2018, 8, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Huang J., Jiang E., Yang D., Wei J., Zhao M., Feng J., Cao J., Infect. Drug Resist. 2020, 13, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. van Rijn A. L., van Boheemen S., Sidorov I., Carbo E. C., Pappas N., Mei H., Feltkamp M., Aanerud M., Bakke P., Claas E. C. J., Eagan T. M., Hiemstra P. S., Kroes A. C. M., de Vries J. J. C., PLoS One 2019, 14, e0223952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Xie Y., Du J., Jin W., Teng X., Cheng R., Huang P., Xie H., Zhou Z., Tian R., Wang R., Feng T., J. Infect. 2018, 78, 158. [DOI] [PubMed] [Google Scholar]
- 151. Wang J., Han Y., Feng J., BMC Pulm. Med. 2019, 19, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Liu N., Kan J., Cao W., Cao J., Jiang E., Zhou Y., Zhao M., Feng J., J. Int. Med. Res. 2019, 47, 4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wang H., Lu Z., Bao Y., Yang Y., de Groot R., Dai W., de Jonge M. I., Zheng Y., PLoS One 2020, 15, e0232610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Li Y., Deng X., Hu F., Wang J., Liu Y., Huang H., Ma J., Zhang J., Zhang F., Zhang C., J Infect. 2018, 76, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Xie Y., Du J., Jin W., Teng X., Cheng R., Huang P., Xie H., Zhou Z., Tian R., Wang R., Feng T., J. Infect. 2019, 78, 158. [DOI] [PubMed] [Google Scholar]
- 156. Li Y., Sun B., Tang X., Liu Y. L., He H. Y., Li X. Y., Wang R., Guo F., Tong Z. H., Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Li G., Huang J., Li Y., Feng J., Can. Respir. J. 2020, 2020, 2367505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Malizos K. N., J. Bone Jt. Surg., Am. Vol. 2017, 99, e20. [DOI] [PubMed] [Google Scholar]
- 159. Thoendel M., Jeraldo P., Greenwood‐Quaintance K. E., Chia N., Abdel M. P., Steckelberg J. M., Osmon D. R., Patel R., Clin. Infect. Dis. 2017, 65, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Fang X., Cai Y., Shi T., Huang Z., Zhang C., Li W., Zhang C., Yang B., Zhang W., Guan Z., Int. J. Infect. Dis. 2020, 99, 108. [DOI] [PubMed] [Google Scholar]
- 161. Huang Z., Li W., Lee G. C., Fang X., Xing L., Yang B., Lin J., Zhang W., Bone Jt. Res 2020, 9, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Street T. L., Sanderson N. D., Atkins B. L., Brent A. J., Cole K., Foster D., McNally M. A., Oakley S., Peto L., Taylor A., Peto T. E. A., Crook D. W., Eyre D. W., J. Clin. Microbiol. 2017, 55, 2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Ivy M. I., Thoendel M. J., Jeraldo P. R., Greenwood‐Quaintance K. E., Hanssen A. D., Abdel M. P., Chia N., Yao J. Z., Tande A. J., Mandrekar J. N., Patel R., J. Clin. Microbiol. 2018, 56, e00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Huang Z., Zhang C., Li W., Fang X., Wang Q., Xing L., Li Y., Nie X., Yang B., Zhang W., J. Bone Jt. Infect. 2019, 4, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Huang Z., Zhang C., Fang X., Li W., Zhang C., Zhang W., Yang B., J. Infect. 2019, 78, 158. [DOI] [PubMed] [Google Scholar]
- 166. Li M., Yang F., Lu Y., Huang W., BMC Infect. Dis. 2020, 20, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Mouraviev V., McDonald M., Can. J. Urol. 2018, 25, 9349. [PubMed] [Google Scholar]
- 168. Li Na M. Q., Qingqing W., Sishi C., Yuyan M., Yi S., Jue P., Bijie H., Natl. Med. J. China 2020, 100, 2198. [Google Scholar]
- 169. Flores‐Mireles A. L., Walker J. N., Caparon M., Hultgren S. J., Nat. Rev. Microbiol. 2015, 13, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Zhou Y., Wylie K. M., El Feghaly R. E., Mihindukulasuriya K. A., Elward A., Haslam D. B., Storch G. A., Weinstock G. M., J. Clin. Microbiol. 2016, 54, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ramesh A., Nakielny S., Hsu J., Kyohere M., Byaruhanga O., de Bourcy C., Egger R., Dimitrov B., Juan Y. F., Sheu J., Wang J., Kalantar K., Langelier C., Ruel T., Mpimbaza A., Wilson M. R., Rosenthal P. J., DeRisi J. L., PLoS One 2019, 14, e0218318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Kujiraoka M., Kuroda M., Asai K., Sekizuka T., Kato K., Watanabe M., Matsukiyo H., Saito T., Ishii T., Katada N., Saida Y., Kusachi S., Front. Microbiol. 2017, 8, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Somasekar S., Lee D., Rule J., Naccache S. N., Stone M., Busch M. P., Sanders C., Lee W. M., Chiu C. Y., Clin. Infect. Dis. 2017, 65, 1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Yee R., Breitwieser F. P., Hao S., Opene B. N. A., Workman R. E., Tamma P. D., Dien‐Bard J., Timp W., Simner P. J., Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1011. [DOI] [PubMed] [Google Scholar]
- 175. Sukhum K. V., Diorio‐Toth L., Dantas G., Clin. Pharmacol. Ther. 2019, 106, 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Chen X., Cao K., Wei Y., Qian Y., Liang J., Dong D., Tang J., Zhu Z., Gu Q., Yu W., Infection 2020, 48, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Gu L., Liu W., Ru M., Lin J., Yu G., Ye J., Zhu Z. A., Liu Y., Chen J., Lai G., Wen W., BMC Pulm. Med. 2020, 20, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Huang Y., Ma Y., Miao Q., Pan J., Hu B., Gong Y., Lin Y., Ann. Transl. Med. 2019, 7, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Jin W., Miao Q., Wang M., Zhang Y., Ma Y., Huang Y., Wu H., Lin Y., Hu B., Pan J., Ann. Transl. Med. 2020, 8, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Quan M., Liu L., Zhou T., Jiang Y., Wang X., Zong Z., Wien. Klin. Wochenschr. 2020, 132, 589. [DOI] [PubMed] [Google Scholar]
- 181. Dai Y., Chen L., Chang W., Lu H., Cui P., Ma X., Front. Public Health 2019, 7, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Wu J., Wu Y., Huang M., Int. J. Infect. Dis. 2020, 90, 1. [DOI] [PubMed] [Google Scholar]
- 183. Kumar D., Chaudhary S., Lu N., Duff M., Heffel M., McKinney C. A., Bedenice D., Marthaler D., Viruses 2019, 11, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Li T., Mbala‐Kingebeni P., Naccache S. N., Theze J., Bouquet J., Federman S., Somasekar S., Yu G., Sanchez‐San Martin C., Achari A., Schneider B. S., Rimoin A. W., Rambaut A., Nsio J., Mulembakani P., Ahuka‐Mundeke S., Kapetshi J., Pybus O. G., Muyembe‐Tamfum J. J., Chiu C. Y., J. Clin. Microbiol. 2019, 57, e00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Sardi S. I., Somasekar S., Naccache S. N., Bandeira A. C., Tauro L. B., Campos G. S., Chiu C. Y., J. Clin. Microbiol. 2016, 54, 2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Hoffmann B., Tappe D., Hoper D., Herden C., Boldt A., Mawrin C., Niederstrasser O., Muller T., Jenckel M., van der Grinten E., Lutter C., Abendroth B., Teifke J. P., Cadar D., Schmidt‐Chanasit J., Ulrich R. G., Beer M., N. Engl. J. Med. 2015, 373, 154. [DOI] [PubMed] [Google Scholar]
- 187. Wu F., Zhao S., Yu B., Chen Y. M., Wang W., Song Z. G., Hu Y., Tao Z. W., Tian J. H., Pei Y. Y., Yuan M. L., Zhang Y. L., Dai F. H., Liu Y., Wang Q. M., Zheng J. J., Xu L., Holmes E. C., Zhang Y. Z., Nature 2020, 579, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Parize P., Muth E., Richaud C., Gratigny M., Pilmis B., Lamamy A., Mainardi J.‐L., Cheval J., de Visser L., Jagorel F., Ben Yahia L., Bamba G., Dubois M., Join‐Lambert O., Leruez‐Ville M., Nassif X., Lefort A., Lanternier F., Suarez F., Lortholary O., Lecuit M., Eloit M., Clin Microbiol. Infect. 2017, 23, 574.e1. [DOI] [PubMed] [Google Scholar]
- 189. Ye M., Wei W., Yang Z., Li Y., Cheng S., Wang K., Zhou T., Sun J., Liu S., Ni N., Jiang H., Jiang H., BMC Infect. Dis. 2016, 16, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Bal A., Sarkozy C., Josset L., Cheynet V., Oriol G., Becker J., Vilchez G., Sesques P., Mallet F., Pachot A., Morfin F., Lina B., Salles G., Reynier F., Trouillet‐Assant S., Brengel‐Pesce K., Viruses 2018, 10, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Lewandowska D. W., Schreiber P. W., Schuurmans M. M., Ruehe B., Zagordi O., Bayard C., Greiner M., Geissberger F. D., Capaul R., Zbinden A., Boni J., Benden C., Mueller N. J., Trkola A., Huber M., PLoS One 2017, 12, e0177340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. De Vlaminck I., Valantine H. A., Snyder T. M., Strehl C., Cohen G., Luikart H., Neff N. F., Okamoto J., Bernstein D., Weisshaar D., Quake S. R., Khush K. K., Sci. Transl. Med. 2014, 6, 241ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. De Vlaminck I., Khush K. K., Strehl C., Kohli B., Luikart H., Neff N. F., Okamoto J., Snyder T. M., Cornfield D. N., Nicolls M. R., Weill D., Bernstein D., Valantine H. A., Quake S. R., Cell 2013, 155, 1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Fusco F. M., Pisapia R., Nardiello S., Cicala S. D., Gaeta G. B., Brancaccio G., BMC Infect. Dis. 2019, 19, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Wylie K. M., Mihindukulasuriya K. A., Sodergren E., Weinstock G. M., Storch G. A., PLoS One 2012, 7, e27735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Jerome H., Taylor C., Sreenu V. B., Klymenko T., Filipe A. D. S., Jackson C., Davis C., Ashraf S., Wilson‐Davies E., Jesudason N., Devine K., Harder L., Aitken C., Gunson R., Thomson E. C., J. Infect. 2019, 79, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Chai J. H., Lee C. K., Lee H. K., Wong N., Teo K., Tan C. S., Thokala P., Tang J. W., Tambyah P. A., Oh V. M. S., Loh T. P., Yoong J., PLoS One 2018, 13, e0194648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Peddu V., Shean R. C., Xie H., Shrestha L., Perchetti G. A., Minot S. S., Roychoudhury P., Huang M. L., Nalla A., Reddy S. B., Phung Q., Reinhardt A., Jerome K. R., Greninger A. L., Clin. Chem. 2020, 66, 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Zhou P., Yang X. L., Wang X. G., Hu B., Zhang L., Zhang W., Si H. R., Zhu Y., Li B., Huang C. L., Chen H. D., Chen J., Luo Y., Guo H., Jiang R. D., Liu M. Q., Chen Y., Shen X. R., Wang X., Zheng X. S., Zhao K., Chen Q. J., Deng F., Liu L. L., Yan B., Zhan F. X., Wang Y. Y., Xiao G. F., Shi Z. L., Nature 2020, 579, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Ai J. W., Zhang Y., Zhang H. C., Xu T., Zhang W. H., Emerg Microbes Infect. 2020, 9, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Lu J., du Plessis L., Liu Z., Hill V., Kang M., Lin H., Sun J., Francois S., Kraemer M. U. G., Faria N. R., McCrone J. T., Peng J., Xiong Q., Yuan R., Zeng L., Zhou P., Liang C., Yi L., Liu J., Xiao J., Hu J., Liu T., Ma W., Li W., Su J., Zheng H., Peng B., Fang S., Su W., Li K., Sun R., Bai R., Tang X., Liang M., Quick J., Song T., Rambaut A., Loman N., Raghwani J., Pybus O. G., Ke C., Cell 2020, 181, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Allegranzi B., Bagheri Nejad S., Combescure C., Graafmans W., Attar H., Donaldson L., Pittet D., Lancet 2011, 377, 228. [DOI] [PubMed] [Google Scholar]
- 203. Greninger A. L., Zerr D. M., Qin X., Adler A. L., Sampoleo R., Kuypers J. M., Englund J. A., Jerome K. R., J. Clin. Microbiol. 2017, 55, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Greninger A. L., Naccache S. N., Messacar K., Clayton A., Yu G., Somasekar S., Federman S., Stryke D., Anderson C., Yagi S., Messenger S., Wadford D., Xia D., Watt J. P., Van Haren K., Dominguez S. R., Glaser C., Aldrovandi G., Chiu C. Y., Lancet Infect. Dis. 2015, 15, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Greninger A. L., Waghmare A., Adler A., Qin X., Crowley J. L., Englund J. A., Kuypers J. M., Jerome K. R., Zerr D. M., J. Pediatr. Infect. Dis. Soc. 2017, 6, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Loman N. J., Constantinidou C., Christner M., Rohde H., Chan J. Z., Quick J., Weir J. C., Quince C., Smith G. P., Betley J. R., Aepfelbacher M., Pallen M. J., JAMA, J. Am. Med. Assoc. 2013, 309, 1502. [DOI] [PubMed] [Google Scholar]
- 207. Marotz C. A., Sanders J. G., Zuniga C., Zaramela L. S., Knight R., Zengler K., Microbiome 2018, 6, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Bal A., Pichon M., Picard C., Casalegno J. S., Valette M., Schuffenecker I., Billard L., Vallet S., Vilchez G., Cheynet V., Oriol G., Trouillet‐Assant S., Gillet Y., Lina B., Brengel‐Pesce K., Morfin F., Josset L., BMC Infect. Dis. 2018, 18, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Jacob J. J., Veeraraghavan B., Vasudevan K., Indian J. Med. Microbiol. 2019, 37, 133. [DOI] [PubMed] [Google Scholar]
- 210. Feehery G. R., Yigit E., Oyola S. O., Langhorst B. W., Schmidt V. T., Stewart F. J., Dimalanta E. T., Amaral‐Zettler L. A., Davis T., Quail M. A., Pradhan S., PLoS One 2013, 8, e76096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Miller S., Naccache S. N., Samayoa E., Messacar K., Arevalo S., Federman S., Stryke D., Pham E., Fung B., Bolosky W. J., Ingebrigtsen D., Lorizio W., Paff S. M., Leake J. A., Pesano R., DeBiasi R., Dominguez S., Chiu C. Y., Genome Res. 2019, 29, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Zhang S. S., Chen D., Lu Q., Genet. Mol. Res. 2012, 11, 1532. [DOI] [PubMed] [Google Scholar]
- 213. de Bruin O. M., Birnboim H. C., BMC Microbiol. 2016, 16, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Aldous W. K., Pounder J. I., Cloud J. L., Woods G. L., J. Clin. Microbiol. 2005, 43, 2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Chen L., Liu W., Zhang Q., Xu K., Ye G., Wu W., Sun Z., Liu F., Wu K., Zhong B., Mei Y., Zhang W., Chen Y., Li Y., Shi M., Lan K., Liu Y., Emerging Microbes Infect. 2020, 9, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Gu W., Miller S., Chiu C. Y., Annu. Rev. Pathol. 2019, 14, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Pereira I. E., M. A., Guedes , R. L. M., 2017.
- 218. Pareek C. S., Smoczynski R., Tretyn A., J. Appl. Genet. 2011, 52, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. The Arabidopsis Genome Initiative , Nature 2000, 408, 796. [DOI] [PubMed] [Google Scholar]
- 220. Ye M., Tu J., Jiang J., Bi Y., You W., Zhang Y., Ren J., Zhu T., Cao Z., Yu Z., Shao C., Shen Z., Ding B., Yuan J., Zhao X., Guo Q., Xu X., Huang J., Wang M., Front. Cell. Infect. Microbiol. 2016, 6, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Seto D., Viruses 2010, 2, 2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Stano M., Beke G., Klucar L., Database 2016, 2016, baw162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Lefkowitz E. J., Dempsey D. M., Hendrickson R. C., Orton R. J., Siddell S. G., Smith D. B., Nucleic Acids Res. 2018, 46, D708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Calisher C. H., Briese T., Brister J. R., Charrel R. N., Durrwald R., Ebihara H., Fulhorst C. F., Gao G. F., Groschup M. H., Haddow A. D., Hyndman T. H., Junglen S., Klempa B., Klingstrom J., Kropinski A. M., Krupovic M., LaBeaud A. D., Maes P., Nowotny N., Nunes M. R. T., Payne S. L., Radoshitzky S. R., Rubbenstroth D., Sabanadzovic S., Sasaya T., Stenglein M. D., Varsani A., Wahl V., Weaver S. C., Zerbini F. M., Vasilakis N., Kuhn J. H., Syst. Biol. 2019, 68, 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Roux S., Adriaenssens E. M., Dutilh B. E., Koonin E. V., Kropinski A. M., Krupovic M., Kuhn J. H., Lavigne R., Brister J. R., Varsani A., Amid C., Aziz R. K., Bordenstein S. R., Bork P., Breitbart M., Cochrane G. R., Daly R. A., Desnues C., Duhaime M. B., Emerson J. B., Enault F., Fuhrman J. A., Hingamp P., Hugenholtz P., Hurwitz B. L., Ivanova N. N., Labonte J. M., Lee K. B., Malmstrom R. R., Martinez‐Garcia M., Mizrachi I. K., Ogata H., Paez‐Espino D., Petit M. A., Putonti C., Rattei T., Reyes A., Rodriguez‐Valera F., Rosario K., Schriml L., Schulz F., Steward G. F., Sullivan M. B., Sunagawa S., Suttle C. A., Temperton B., Tringe S. G., Thurber R. V., Webster N. S., Whiteson K. L., Wilhelm S. W., Wommack K. E., Woyke T., Wrighton K. C., Yilmaz P., Yoshida T., Young M. J., Yutin N., Allen L. Z., Kyrpides N. C., Eloe‐Fadrosh E. A., Nat. Biotechnol. 2019, 37, 29. [DOI] [PubMed] [Google Scholar]
- 226. Mahmoudabadi G., Phillips R., eLife 2018, 7, e31955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Zhang K. Y., Gao Y. Z., Du M. Z., Liu S., Dong C., Guo F. B., Front. Microbiol. 2019, 10, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Goodacre N., Aljanahi A., Nandakumar S., Mikailov M., Khan A. S., mSphere 2018, 3, e00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Brister J. R., Ako‐Adjei D., Bao Y., Blinkova O., Nucleic Acids Res. 2015, 43, D571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Zinter M. S., Dvorak C. C., Mayday M. Y., Iwanaga K., Ly N. P., McGarry M. E., Church G. D., Faricy L. E., Rowan C. M., Hume J. R., Steiner M. E., Crawford E. D., Langelier C., Kalantar K., Chow E. D., Miller S., Shimano K., Melton A., Yanik G. A., Sapru A., DeRisi J. L., Clin. Infect. Dis. 2019, 68, 1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Naccache S. N., Greninger A. L., Lee D., Coffey L. L., Phan T., Rein‐Weston A., Aronsohn A., Hackett J. Jr., Delwart E. L., Chiu C. Y., J. Virol. 2013, 87, 11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Salter S. J., Cox M. J., Turek E. M., Calus S. T., Cookson W. O., Moffatt M. F., Turner P., Parkhill J., Loman N. J., Walker A. W., BMC Biol. 2014, 12, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Malla M. A., Dubey A., Kumar A., Yadav S., Hashem A., Abd Allah E. F., Front. Immunol. 2018, 9, 2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Cheng M. P., Papenburg J., Desjardins M., Kanjilal S., Quach C., Libman M., Dittrich S., Yansouni C. P., Ann. Intern. Med. 2020, 172, 726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Greninger A. L., Expert Rev. Mol. Diagn. 2018, 18, 605. [DOI] [PubMed] [Google Scholar]
- 236. Shi C. L., Han P., Tang P. J., Chen M. M., Ye Z. J., Wu M. Y., Shen J., Wu H. Y., Tan Z. Q., Yu X., Rao G. H., Zhang J. P., J. Infect. 2020, 81, 567. [DOI] [PubMed] [Google Scholar]
- 237. Saha S., Ramesh A., Kalantar K., Malaker R., Hasanuzzaman M., Khan L. M., Mayday M. Y., Sajib M. S. I., Li L. M., Langelier C., Rahman H., Crawford E. D., Tato C. M., Islam M., Juan Y. F., de Bourcy C., Dimitrov B., Wang J., Tang J., Sheu J., Egger R., De Carvalho T. R., Wilson M. R., Saha S. K., DeRisi J. L., mBio 2019, 10, e02877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Zhou H., Chen X., Hu T., Li J., Song H., Liu Y., Wang P., Liu D., Yang J., Holmes E. C., Hughes A. C., Bi Y., Shi W., Curr. Biol. 2020, 30, 2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Chen P., Sun W., He Y., J. Thorac. Dis. 2020, 12, 4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Zhao M., Tang K., Liu F., Zhou W., Fan J., Yan G., Qin S., Pang Y., Front. Microbiol. 2020, 11, 2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Chen J., He T., Li X., Wang X., Peng L., Ma L., Infect. Drug Resist. 2020, 13, 2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Ardui S., Ameur A., Vermeesch J. R., Hestand M. S., Nucleic Acids Res. 2018, 46, 2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Ji H., Enns E., Brumme C. J., Parkin N., Howison M., Lee E. R., Capina R., Marinier E., Avila‐Rios S., Sandstrom P., Van Domselaar G., Harrigan R., Paredes R., Kantor R., Noguera‐Julian M., J. Int. AIDS Soc. 2018, 21, e25193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Elkins J. M., Kates S., Lange J., Lange J., Lichstein P., Otero J., Soriano A., Wagner C., Wouthuyzen‐Bakker M., J. Arthroplasty 2019, 34, S181. [DOI] [PubMed] [Google Scholar]