

Summary
Severe acute respiratory syndrome coronavirus‐2 is the etiological agent of the ongoing pandemic of coronavirus disease‐2019, a multi‐organ disease that has triggered an unprecedented global health and economic crisis. The virally encoded 3C‐like protease (3CLpro), which is named after picornaviral 3C protease (3Cpro) due to their similarities in substrate recognition and enzymatic activity, is essential for viral replication and has been considered as the primary drug target. However, information regarding the cellular substrates of 3CLpro and its interaction with the host remains scarce, though recent work has begun to shape our understanding more clearly. Here we summarized and compared the mechanisms by which picornaviruses and coronaviruses have evolved to evade innate immune surveillance, with a focus on the established role of 3Cpro in this process. Through this comparison, we hope to highlight the potential action and mechanisms that are conserved and shared between 3Cpro and 3CLpro. In this review, we also briefly discussed current advances in the development of broad‐spectrum antivirals targeting both 3Cpro and 3CLpro.
Keywords: Covid‐19, picornaviruses, SARS‐CoV‐2, 3Cpro , 3CLpro
Abbreviations
- ACLY
ATP‐citrate synthase
- ASCs
apoptosis‐associated speck‐like protein containing a caspase recruitment domain
- avSGs
antiviral stress granules
- CARD
caspase activation and recruitment domain
- cGAS
cyclic GMP‐AMP synthase
- Covid‐19
coronavirus disease‐2019
- CVA6/A16/B3
Coxsackievirus‐A6/A16/B3
- CYLD
cylindromatosis
- CALCOCO2
coiled‐coil domain‐containing protein 2
- DAMPs
danger associated molecular patterns
- DCP1/2
decapping protein ½
- DDX6
DEAD‐box RNA helicase‐6
- DHAV
duck hepatitis A virus
- DHX36
DEAH‐box helicase 36
- dsRNA
double‐stranded RNA
- eIF4E/F/G
eukaryotic initiation factor 4E/F/G
- EDC3/4
enhancer of mRNA decapping protein‐3/4
- EMCV
encephalomyocarditis virus
- ERAV
equine rhinitis A virus
- EV‐A71/D68
enterovirus‐A71/D68
- FMDV
foot‐and‐mouth disease Virus
- GBF1
golgi‐specific brefeldin A‐resistance guanine nucleotide exchange factor
- G3BP1
Ras‐GTPase‐activating SH3 domain binding protein 1
- GSDMD
gasdermin D
- HAV
hepatitis A virus
- HDAC2
histone deacetylase 2
- HRV
human rhinovirus
- hnRNP M/K
heterogeneous nuclear ribonucleoprotein M/K
- IFN‐I
type‐I interferon
- IKKε
IκB kinase‐ε
- IRES
internal ribosomal entry sites
- IRF3/7/9
interferon regulatory factor‐3/7/9
- ISGs
interferon‐stimulating genes
- K63‐linked
lysine‐63‐linked
- M protein
membrane/matrix protein
- MAVS
mitochondrial antiviral signaling protein
- MDA5
melanoma differentiation‐associated protein‐5
- MERS‐CoV
Middle East respiratory syndrome‐CoV
- MEX3C
Mex‐3 RNA binding family member C
- mRNPs
messenger ribonucleoproteins
- NDP52
nuclear dot 10 protein 52
- NLRP3
NLR family PYD containing protein‐3
- N proteins
nucleocapsid proteins
- NEMO
NF‐κB essential modulator
- NFκB
nuclear factor‐κB
- NLRs
(NOD)‐like receptors
- NSP5
nonstructural protein 5
- ORF1a/3a/3b/6/8b
open reading frame 1a/3b/6/8b
- PAMPs
pathogen‐associated molecular patterns
- PDCoV
porcine deltacoronavirus
- PDCD6IP
programmed cell death 6‐interacting protein
- PEDV
porcine epidemic diarrhea virus
- PFAS
phosphoribosylformylglycinamidine synthetase
- PKR
protein kinase R
- PLEKHM1
pleckstrin homology domain and RUN domain containing M1
- PRRs
pathogen recognition receptors
- PRRSV
porcine reproductive and respiratory syndrome virus
- PLpro
papain‐like protease
- PV
poliovirus
- RIP1
receptor‐interacting serine/threonine‐protein kinase 1
- RLRs
RIG‐I‐like receptors
- RdRp
RNA‐dependent RNA polymerase
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus‐2
- SNAP29
synaptosomal‐associated protein 29
- ssRNA
single‐stranded RNA
- STAT2
signal transducer and activator of transcription 2
- STX17
Syntaxin 17
- SQSTM1
sequestosome 1
- SVV
Seneca Valley virus
- TAB1
TAK1 binding protein‐1
- TAILS
terminal amine isotopic labeling of substrates
- TBK1
TANK binding kinase‐1
- TLRs
toll‐like receptors
- TRAF3
TNF receptor associated factor‐3
- TRIM25
tripartite motif containing 25
- TRIF
(TIR)‐domain‐containing adapter‐inducing interferon‐β
- TRMT1
tRNA methyltransferase 1
- UMP
uridine monophosphate
- UTR
untranslated region
- VAMP8
vesicle‐associated membrane protein 8
- VPg
viral protein genome‐linked
- XRN1
5′‐3′ exoribonuclease 1
- 2Apro
2A‐protease
- 3CLpro/3Cpro
3C‐like protease/3C protease
- 3Dpol
3D polymerase
1. INTRODUCTION
Coronavirus disease‐2019 (Covid‐19) is a multiple organ disease that has posed an unprecedented health and economic threat worldwide since its emergence in late 2019. Covid‐19 is caused by a novel virus strain, 1 , 2 namely severe acute respiratory syndrome‐coronavirus‐2 (SARS‐CoV‐2) 3 , categorized within the family Coronaviridae. It can infect various hosts and target multiple organs through the body. 4 Coronaviruses are a family of enveloped, single‐stranded RNA (ssRNA) viruses with positive polarity that can cause respiratory, enteric, cardiovascular, and central nervous system diseases. 5 , 6 This family of RNA viruses features the second largest genome size (27−31 kb) found to date right after planarian nidovirus (∼41.1 kb). 7 Together with SARS‐CoV and Middle East Respiratory Syndrome‐CoV (MERS‐CoV) that caused SARS and MERS outbreaks in 2003 and 2012, respectively, SARS‐CoV‐2 belongs to the genus betacoronavirus. The genome of betacoronavirus encodes more than 20 proteins, including four major structural proteins (i.e., a spike (S) protein that binds to the cell receptor and mediates fusion between virus and cell membrane, a small envelope (E) protein, a highly hydrophobic membrane (M) protein, and a nucleocapsid (N) protein that interacts with viral RNA to form a helical nucleocapsid structure), two cysteine proteases (i.e., a papain‐like cysteine protease (PLpro) and a 3‐chymotrypsin‐like cysteine protease (3CLpro, also known as the main protease, Mpro) that processes viral polyproteins into individual functional proteins, a helicase required for unwinding double‐stranded RNA (dsRNA), a RNA‐dependent RNA polymerase (RdRp) that catalyzes the replication of RNA from RNA template, and other enzymes such as endo‐ and exonucleases essential for viral nucleic acid metabolism. 8
Among these proteins, SARS‐CoV‐2 proteases play a vital role in viral replication and transcription, thereby being recognized as attractive antiviral targets for Covid‐19 treatment. 9 , 10 Of the two known CoV proteases that are encoded by open reading frame 1a (ORF1a), 3CLpro [corresponding to nonstructural protein 5 (NSP5)], which is highly conserved among all CoV 3CLpro, has been identified to be structurally analogous to the 3Cpro of picornaviruses (3CLpro is named after the picornaviral 3Cpro). 11 , 12 Despite subtle structural differences in the active sites, 3CLpro and 3Cpro share a similar chymotrypsin‐like tertiary structure with a catalytic triad (or dyad) site containing a cysteine nucleophile (Figure 1). Moreover, both of the enzymes have a strong preference for glutamine (Gln) at the P1 position of their targets, the most key determining factor for their substrate recognition. The conserved active sites of 3Cpro and 3CLpro have been confirmed by high‐resolution three‐dimensional structural analysis. Therefore, it is proposed to serve as an attractive target for the design of broad‐spectrum antiviral drugs. 13 , 14 , 15 Picornaviruses are small, non‐enveloped viruses containing a positive‐sense, ssRNA genome with a length of 7.0–8.5 kb. This family comprises 29 genera, including Apthovirus (e.g., foot‐and‐mouth disease virus, FMDV), Cardiovirus (e.g., encephalomyocarditis virus, EMCV), Enterovirus (e.g., poliovirus, PV; coxsackievirus A16/B3, CVA16; CVB3; enterovirus‐A71/D68, EV‐A71; EV‐D68), Rhinovirus (e.g., human rhinovirus, HRV), and Hepatovirus (e.g., hepatitis A virus, HAV) genera. 16 Picornavirus genomic RNA at its 5′ end is covalently linked to a small viral protein (VPg, also known as 3B) that serves as a primer for the initiation of viral RNA replication. Further, instead of a cap structure, the genome of picornaviruses possesses an element termed internal ribosome entry site (IRES) in their 5′‐untranslated region (UTR), which is necessary for initiating a cap‐independent translation of viral RNA. The viral genome of picornaviruses contains one open reading frame encoding a single viral polyprotein that undergoes proteolysis by two viral proteases, 2Apro and 3Cpro, with the latter being responsible for the majority of the maturation cleavage events of viral polyprotein similar to coronaviral 3CLpro 17 . In addition to processing viral polyprotein, picornaviral proteases also target cellular proteins to evade the human immune surveillance and facilitate viral infection. 18
FIGURE 1.
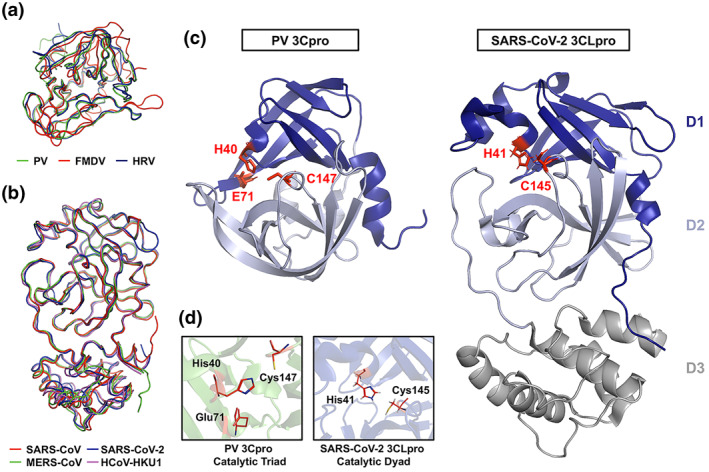
Crystal structures and superposition of picornaviral 3C protease (3Cpro) and coronaviral 3C‐like protease (3CLpro). (a) Ribbon overlay of the picornaviral 3Cpro structures of poliovirus (PV; PDB 1L1N), foot‐and‐mouth disease virus (FMDV; PDB 2BHG), and human rhinovirus (HRV; PDB 1CQQ). (b) Ribbon overlay of human coronaviral 3CLpro structures of severe acute respiratory syndrome‐coronavirus (SARS‐CoV; PDB 2Q6G), Middle East Respiratory syndrome‐CoV (MERS‐CoV; PDB 4YLU), SARS‐CoV‐2 (PDB 6M2N), and HCoV‐HKU1 (PDB 3D23). (c) A side‐by‐side comparison of PV 3Cpro and SARS‐CoV‐2 3CLpro with the two domains of the chymotrypsin‐like fold highlighted and the active site catalytic residues labeled and highlighted (red). (d) Close‐up images of the active site catalytic residues of PV 3Cpro and SARS‐CoV‐3CLpro are shown
Given the common characteristics of 3Cpro and 3CLpro, we postulate that SARS‐CoV‐2, like picornaviruses, is capable of regulating host innate antiviral processes through the catalytic activity of its 3CLpro. The delay or inhibition of multiple host antiviral machineries would allow effective viral growth and subsequently optimal release and infection. Here we will recapitulate some of the scenarios on how picornaviruses utilize its 3Cpro to target major host antiviral mechanisms.
1.1. Structural and functional similarities between picornaviral 3Cpro and coronaviral 3CLpro
Early during the viral replication cycle, the positive‐sense ssRNA (+ssRNA) genomes of picornaviruses and coronaviruses are translated into one or more polyproteins, which include integrated viral protease domains. Maturation cleavage events mediated by virally encoded proteases in both backgrounds are indispensable for virus replication. The picornaviral 3Cpro (working in concert with the 2Apro) mediates the majority of viral cleavage events including autocleavage from the 3D polymerase (3Dpol) domain of the virus. Similarly, coronaviral 3CLpro is responsible for at least 11 maturation cleavages of the viral replicase polyproteins, including its own autoproteolytic cleavage. Beyond these requisite viral polyprotein cleavages, both 3Cpro and 3CLpro target and cleave host cellular proteins. Due to the importance in their respective viral backgrounds, these proteases have been extensively studied as primary targets for viral inhibition for over 30 years.
The picornaviral 3Cpro is a chymotrypsin‐like cysteine protease comprised of two β‐barrel domains of six antiparallel strands which surrounds a conserved Cys‐His‐Asp/Glu catalytic triad (Figure 1). 19 , 20 The protease preferentially cleaves after a P1‐Gln with greater cleavage site variability in the other cleavage site residue positions. 21 During picornaviral replication, the 3CDpro precursor, which is able to process the P1 structural precursor but lacks polymerase activity, is cleaved to release 3Cpro and viral RdRP 3Dpol 22 . The conversion of 3CDpro to 3Cpro plays a critical role in facilitating the transition and regulation from viral translation to replication. 23 , 24 Structurally and functionally analogous to the picornaviral 3Cpro, the coronaviral 3CLpro is an approximately 300 residue, 3‐domain protease. Domains 1 and 2 comprise the substrate‐binding and enzymatic active sites of the protease with dimerization driven by interactions between the structurally unique and largely helical domain 3. 25 , 26 Domains 1 and 2 of 3CLpro form a chymotrypsin‐like fold comprised of antiparallel β‐barrels housing the His‐Cys catalytic dyad residues. 26 , 27 The antiparallel β‐barrel conformation within domains 1 and 2 surrounding and forming the active site of 3CLpro shares structural similarity to the core structure of the picornaviral 3Cpro, albeit with subtle differences in strand positioning (Figure 1). Unlike the picornaviral 3Cpro (monomer with only two catalytic domains), an attached helical third‐domain in 3CLpro facilitates dimerization of the protease, an essential event for its enzymatic activity and viral replication (Figure 1C, Domain 3). 28 While targeting the third domain of 3CLpro serves as a valid strategy to disrupt its dimerization and catalytic activity, any inhibitors identified or designed to do so likely have no impact on picornaviral 3Cpro activity since dimerization is not necessary for its function. In addition, among coronaviruses, there is considerably more structural conservation for the chymotrypsin‐like fold of 3CLpro compared to that of the picornaviral 3Cpro. Based on large part to their structural and functional similarities between the 3Cpro and 3CLpro, it remains unclear to what extent these proteases share common host cellular targets during viral replication and what role these potentially shared cleavages play in viral replication.
2. THE SUBVERSION OF HOST DEFENSE MECHANISMS BY picornaviral 3Cpro
2.1. Targeting type I interferon signaling pathway
The effectiveness of an antiviral innate immunity depends on the accurate recognition of viral moieties, known as the pathogen‐associated molecular patterns (PAMPs), by pattern‐recognition receptors (PRRs) composed of at least three classes: retinoic acid‐inducible gene‐I (RIG‐I)‐like receptors (RLRs), Toll‐like receptors (TLRs), and nucleotide‐binding oligomerization domain (NOD)‐like receptors (NLRs). 29 Upon RNA viral infection, dsRNAs often accumulate in cells in the form of the viral genome or its replication intermediates. The dsRNAs can be recognized by cytosolic viral RNA sensor, RLRs (e.g., RIG‐I (encoded by Ddx58 gene) and melanoma differentiation‐associated proteins (MDA5, encoded by Ifih1 gene)), and/or endosomal viral RNA sensor (e.g., TLR3) to initiate type I interferon (IFN‐I) immune response. While RIG‐I preferentially binds to shorter dsRNA (<1‐2 kb) bearing 5′‐triphosphate group, MDA5 primarily recognizes long dsRNA. 30 Similar to picornavirus, SARS‐CoV‐2 has a long RNA genome. It is therefore expected that SARS‐CoV‐2 RNA favorably binds to MDA5 rather than its paralog RIG‐I. Indeed, MDA5 has been previously shown to be the specific PRR that recognizes murine coronavirus RNA. 31 Interaction between viral RNA and MDA5 forms MDA5 filaments along dsRNAs, which brings together neighboring caspase activation and recruitment domain (CARD) in close proximity to induce oligomerization and activation of the adapter mitochondrial‐antiviral signaling protein (MAVS). 32 Activated MAVS then transmits the signals to its downstream transcription factors, interferon regulatory factor‐3/7 (IRF3/7) and nuclear‐factor‐κB (NF‐κB), through TANK‐binding kinase‐1 (TBK1) and IκB kinase‐ε (IKKε). The homo‐ or hetero‐dimerized IRF3/7 subsequently translocate to the nucleus and induce the expression of IFN‐I‐associated genes (Ifna and Ifnb1), which could further activate the Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling cascade to trigger the expression of antiviral genes, called interferon‐stimulated genes (ISGs). 30 Please see Figure 2 for the details.
FIGURE 2.
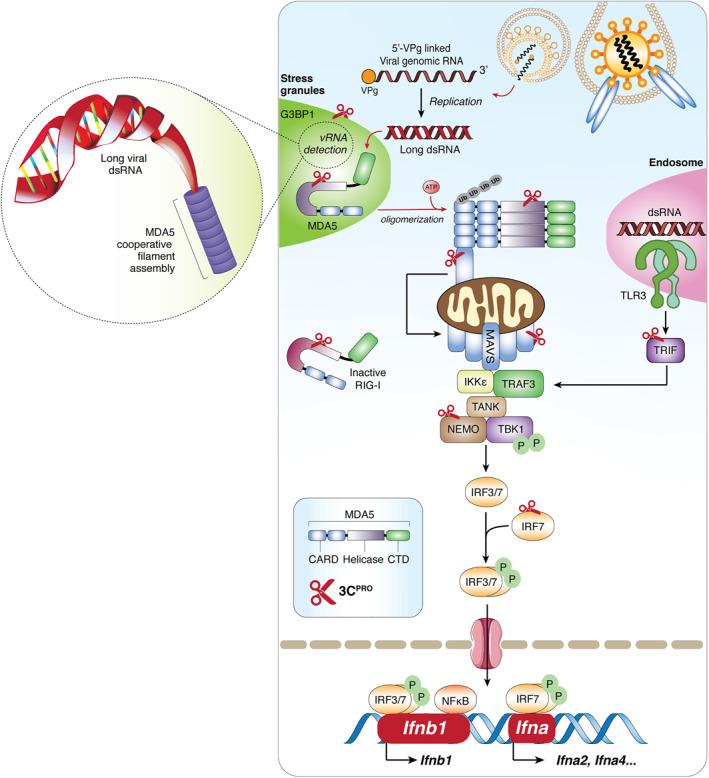
Picornaviruses evade type I interferon immune response via the function of 3C protease (3Cpro). Binding of picornaviruses to their respective receptors facilitates their entry into the cells and release of the 5′‐viral protein genome‐linked‐containing genomic RNA into cytoplasm. Long double‐stranded RNA generated during the replication process binds to MDA5, exposing its CARD and allowing homotypic CARD‐CARD interactions with its downstream adapter, MAVS. Subsequently, MAVS triggers the expression of IFN‐I genes (Ifnb1 and Ifna in dendritic cells) and ISGs for antiviral purposes through the activation of transcription factor IRF3/7 and NF‐κB. To facilitate a robust signaling, more efficient detection of dsRNA occurs in antiviral stress granules. Targets of viral encoded 3Cpro are indicated. CARD, caspase activation and recruitment domain; CTD, C‐terminal binding domain; G3BP1, Ras GTPase‐activating protein‐binding protein 1; IFN‐I, type‐I interferon; IKKε, inhibitor of nuclear factor‐κB (IκB)‐kinase‐ε; IRF3/7, interferon regulatory factors‐3/7; ISGs, interferon‐stimulating genes; MAVS, mitochondrial antiviral signaling protein; MDA5, melanoma‐differentiation associated protein‐5; NF‐κB, nuclear factor‐κB; NEMO, NF‐κB essential modulator; P, phosphate‐group; RIG‐I, retinoic acid‐inducible gene‐I; TBK1, TANK binding kinase‐1; TLR3, Toll‐like receptor 3; TRAF3, TNF‐receptor associated factor‐3; TRIF, Toll/IL‐1 receptor domain‐containing adapter‐inducing interferon‐β
To antagonize human antiviral innate immune response, picornaviruses target critical components of the RLR signaling pathway for degradation. It was reported that infections with Seneca Valley virus (SVV), PV, EV‐A71, EV‐D68, CVB3, CVA16, CVA6, HRV‐1A, EMCV, and HAV cleave MDA5, MAVS, RIG‐I, IRF7, and/or IRF9 through the actions of 3Cpro, leading to a disruption of RLR‐mediated IFN‐I immune responses. 33 , 34 , 35 , 36 , 37 , 38 , 39 In addition to 3Cpro, studies have found that MDA5 and MAVS can also be targeted by 2Apro upon PV, EV‐A71, CVB3, and HRV‐1A infections. 34 , 40 , 41 Moreover, both TAK1 binding protein‐1 (TAB1) and NF‐κB essential modulator (NEMO, an adapter protein bridging the canonical IKKα/β kinases and the noncanonical kinases TBK1/IKKε via the TANK adapter 42 ) are cleaved by 3Cpro following EV‐A71, FMDV and HAV infections, resulting in reduced production of IFN‐I. 43 , 44 , 45 In addition, FMDV utilizes its 3Cpro to inhibit STAT2 function, a component of the IFN‐stimulated gene factor 3 complex, which is also mirrored by the 3CLpro of PDCoV (Table 1, yellow highlighted row). 57 , 58 Another interesting finding associated with this topic is 3Cpro‐induced cleavage of Toll/IL‐1 receptor (TIR)‐domain‐containing adapter‐inducing interferon‐β (TRIF) during CVB3, EV‐A71, and HAV infections. 36 , 47 , 48 TRIF is an adapter protein mediating type I IFN antiviral response downstream of the endosomal TLR3 (a viral RNA sensor) 114 and cleavage causes the loss of its function in host defense.
TABLE 1.
Picornaviruses and coronaviruses proteinase targets [Correction added on 17 February 2021, after first online publication. Table 1 was updated in this version.]
| Proteinase | Virus | Gene symbol | Full gene name | Cleavage site | Implication to host | Ref | |
|---|---|---|---|---|---|---|---|
| Host defense and inflammation | 3C | CVB3, RV, PV, EV‐A71, SVV | RIG‐I | Retinoic acid‐inducible gene‐I | N/A | Dampened cytokines production; enhanced viral propagation | 33, 39 |
| 2A | PV, EV‐A71, CVB3 | MDA5 | Melanoma differentiation‐associated protein 5 | N/A | 37, 40 | ||
| 2A, 3C, 3CLSP | CVB3, HRV, EV‐A71, PV, SVV, PRRSV | MAVS | Mitochondrial antiviral signaling | Q148↓, Q209↓, Q251↓, Q265↓, Q268↓ | 34, 36, 38, 40, 41, 46 | ||
| 3C | EV‐D68, EV‐A71, CVB3, HAV, SVV | TRIF | TIR domain‐containing adapter inducing interferon beta | Q159↓, Q190↓, Q312↓, Q554↓, Q653↓ | 36, 47, 48 | ||
| PL | SARS‐CoV‐2 | IRF3 | Interferon regulatory factor 3 | G270↓ | 49 | ||
| 3C | EV‐D68, EV‐A71 | IRF7 | Interferon regulatory factor 7 | Q167↓, Q189↓ | 35, 50 | ||
| 3C | EV‐A71 | IRF9 | Interferon regulatory factor 9 | N/A | 51 | ||
| 2A, 3C | EV‐A71, CVB3 | NLRP3 | NACHT, LRR, and PYD domain‐containing protein 3 | Q225↓226G, G493↓494L | 52, 53 | ||
| 3CL | SARS‐Cov‐2 | NLRP12 | NACHT, LRR, and PYD domain‐containing protein 12 | Q238↓, Q938↓ | 49 | ||
| 3C | EMCV, SVV | TANK1 | TRAF family member‐associated NF‐κB activator‐1 | E272 and Q291 | 54 | ||
| 2A | EV71 | IFNAR | Interferon alpha and beta receptor subunit 1 | N/A | 55 | ||
| 3C | HRV, PV | C3 | Complement C3 | N/A | 56 | ||
| 3C, 3CL | FMDV, PDCoV | STAT2 | Signal transducer and activator of transcription 2 | Q685↓, Q758↓ | 43, 44, 57, 58, 59, 60, 61 | ||
| 3C, 3CL, 3CLS | FMDV, HAV, PEDV, PRRSV | NEMO | NF‐κB essential modulator | Q304↓, Q383↓, Q231↓, E349↓350S | |||
| 3C | EV‐D68, EV‐A71, CVA6, CVA16 | TAK1 | Transforming growth factor‐β‐activated kinase 1 | Q360↓361S | 37, 45, 49 | ||
| 3C, 3CL | EV‐A71, SARS‐CoV‐2 | TAB1 | TAK1 binding protein 1 | Q414↓415G, Q451↓452S, Q132↓133S, Q444↓445S | |||
| 3C | EV‐A71 | TAB2 | TAK1 binding protein 2 | Q113↓114S | |||
| 3C | EV‐A71 | TAB3 | TAK1 binding protein 3 | Q173↓174G, Q343↓344G | |||
| 3C | CVB3, PV | RIP1, RIP3 | Receptor interacting protein‐1, ‐3 | Q134↓, Q430↓, R118↓119I | 62, 63 | ||
| 3C | EV‐A71 | ZAP | Zinc‐finger antiviral protein | Q369↓370G | 64 | ||
| 3C | EV‐A71 | GSDMD | Gasdermin‐D | Q193↓194G | Inhibition of pyroptosis & promotion of viral growth | 65 | |
| Autophagy | 2A | CVB3 | p62/SQSTM1 | Sequestosome‐1 | G241↓ | Disruption of cellular autophagy pathway | 66, 67, 68, 69, 70 |
| 2A | CVB3 | NBR1 | Neighbor of BRCA1 gene | G402↓, E682↓ | |||
| 3C | CVB3 | CALCOCO2/NDP52 | Calcium binding and coiled‐coil domain containing protein‐2; Nuclear dot 10 protein 52 | Q139 | |||
| 3C | CVB3 | SNAP29 | Synaptosomal‐associated protein 29 | Q161↓ | |||
| 3C | CVB3 | PLEKHM1 | Pleckstrin homology domain and RUN domain Containing M1 | Q668↓ | |||
| Cellular Integrity | 2A | CVB3 | DYSF | Dysferlin | N/A | Disruption of muscle membrane repair | 71 |
| 2A | CVB3, CVB4 | DMD | Dystrophin | Human: (G589↓; G2,435↓)Mouse: (G591↓; G2,428↓) | Muscular dystrophy, especially heart muscle | 72 | |
| 2A | HRV2 | CK‐II | Cytokeratin K8 | G15↓ | Disruption of cytoplasmic cytoskeleton | 73 | |
| 3C | PV | MAP‐4 | Microtubule‐associated protein 4 | Q188↓189G | Collapse of microtubules, disruption of host protein dynamics and interactions | 74 | |
| Transcription | 3C | PV | Oct‐1 | Octomer binding transcription factor‐1 | Q330↓331G | Disruption of polymerase activity, affecting gene transcription | 75 |
| 3C | PV | La/SSB | Lupas autoantigen/Sjogren syndrome antigen B | Q358↓359G | 76 | ||
| 2A | PV | TBP | TATA‐binding protein | Q18↓19G, Q104↓105S | 77, 78, 79, 80 | ||
| 3C | PV | TFIIIA/C | Transcription factor for polymerase III A | N/A | 81, 82 | ||
| 3C | PV | CREB | cAMP response element‐binding protein | Q172↓173G | |||
| 2A | CVB3 | SRF | Serum response factor | G327 | Disruption of SRF‐dependent gene expression (e.g. cardiac contractile and regulatory factors) | 83 | |
| 3C | PV | PTB/p52/hnRNAP‐I | Polypyrimidine tract‐binding protein | Q148/152/321A | Disruption of normal RNA metabolism (e.g. splicing) | 84 | |
| 3C | PV | hnRNP M, K | Heterogenous nuclear ribonucleoprotein M, K | G389↓390E, Q364↓365G | 85 | ||
| 2A | PV | Gemin3 | Gemin3 | G463↓ | 86 | ||
| 3C | CVB3 | TDP‐43 | Transactive response DNA‐binding protein‐43 | Q327↓328A | 87 | ||
| 3C | PV | p65‐RelA | Nuclear factor (NF)‐κB p65 subunit | Q480↓481G | Disruption of p65‐dependent gene expression | 88 | |
| Translation and RNA turn over | 3C | CVB3 | AUF1 | (AU)‐rich element RNA‐binding factor 1 | N/A | Disruption of host mRNA turn over | 89 |
| 2A, 3C | PV, CVB3, HAV, EMCV, FMDV, DHAV | PABP | Poly(A)‐Binding protein | Q537↓, Q367↓368G, M487↓, G488↓, Q437↓438G | Host protein translation shut‐off, disruption of SG formation | 90, 91, 92, 93 | |
| 3C | FMDV | eIF4A | Eukaryotic elongation factor 4A | G674↓675R, R481↓482G | 94, 95, 96, 97, 98, 99 | ||
| L, 2A, 3C | FMDV, PV, CVB3, EV‐A71, RV | eIF4G‐I, ‐II | Eukaryotic elongation factor 4G‐I/4G‐II | ||||
| 3C | PV, CVB3, HRV | eIF5B | Eukaryotic elongation factor 5B | Q478↓479G | 100 | ||
| 2A | CVB3 | DAP5 | Death‐associated protein 5 | G434↓ | 101 | ||
| 3C | PV | PCBP2 | Poly(rC)‐binding protein‐2 | Q253↓, Q306↓, S254↓ | 102 | ||
| 3C | PV | DCP1a | mRNA‐decapping enzyme 1A | N/A | Disruption of P‐body foci; disruption of 5’→3′ guanosine cap removal and Xrn1‐Dcp1‐dependent antiviral pathway | 103 | |
| Others | 3C | CVB3 | IKBα | Inhibitor of κBα | Q249↓250G | Constitutive activation of NFκB and apoptosis | 104 |
| 2A, 3C | CVB3 | GAB1 | Growth factor receptor bound 2‐associated binding protein‐1 | G175↓, G436↓, G238↓ | Disruption the assembly of protein complexes for intracellular signaling | 105 | |
| 2A | HRV, PV | Nup62, Nup98, Nup153, | Nucleoporins | A103↓, G177↓, G201↓, G218↓, G247↓, A298↓, | Temporal blocking of nucleocytoplasmic trafficking | 106, 107 | |
| L, 3C | CVB3, PV, EMCV, FMDV, ERAV | G3BP1, G3BP2 | Ras‐GTPase activating‐SH3‐binding protein‐1 | Q325↓, E284↓ | Disruption of SG and innate immune response | 108, 109, 110, 111, 112, 113 | |
| 3C | CVB3, PV | GBF1 | Golgi‐specific brefeldin A‐resistance guanine exchange factor‐1 | Q1,297↓1298S | Disruption of host secretory pathway | 63 | |
| ACLY | ATP citrate lyase/synthase | Q777↓778A | Disruption of fatty acid biosynthesis | ||||
| p115/USO1 | General vesicular transport factor p115 | Q832↓833G | Disruption of the host peripheral membrane recycling process between cytosol and Golgi | ||||
| ALIX/PDCD6IP | Programmed cell death 6‐interacting protein | Q728↓729S | Disruption of cellular apoptosis | ||||
| PFAS | Phosphoribosylformylglycinamidine synthetase | Q472↓473G | Disruption of host ATP, and L‐glutamine biosynthesis |
Abbreviations: CVA6/A16/B3, Coxsackievirus‐A6/A16/B3; CALCOCO2, coiled‐coil domain‐containing protein 2; DHAV, duck hepatitis A virus; EMCV, encephalomyocarditis virus; ERAV, equine rhinitis A virus; FMDV, foot‐and‐mouth disease virus; GBF1, golgi‐specific brefeldin A‐resistance guanine nucleotide exchange factor; GBF1, Ras‐GTPase‐activating SH3 domain binding protein 1; GSDMDM, gasdermin D; HAV, hepatitis A virus; HRV, human rhinovirus; hnRNP M/K, heterogeneous nuclear ribonucleoprotein M/K; MAVS, mitochondrial antiviral signaling protein; MDA5, melanoma differentiation‐associated protein‐5; NDP52, nuclear dot 10 protein 52; NBR1, neighbor of BRCA1; NLRP3, NLR family PYD containing protein‐3; NEMO, NF‐κB essential modulator; NFκB, nuclear factor‐κB; PDCoV, porcine deltacoronavirus; PEDV, porcine epidemic diarrhea virus; PFAS, phosphoribosylformylglycinamidine synthetase; PLEKHM1, pleckstrin homology domain and RUN domain containing M1; PRRs, pathogen recognition receptors; PRRSV, porcine reproductive and respiratory syndrome virus; RIP1, receptor‐interacting serine/threonine‐protein kinase 1; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2; SSV, Seneca Valley virus; STAT2, signal transducer and activator of transcription 2; TAB1, TAK1 binding protein‐1; TRIF, (TIR)‐domain‐containing adapter‐inducing interferon‐β
Note: Color coding correspond between virus and cleavage site)
In addition to directly targeting components of the IFN‐I pathway, 3Cpro can also modulate the function of proteins that participate in the regulation of this pathway. For instance, the 3Cpro of EV‐A71 downregulates miRNA‐526a, consequently leading to increased expression of cylindromatosis (CYLD, a target of miRNA‐526a). 115 CYLD is a deubiquitinating enzyme negatively regulating the function of RIG‐I by removing K63‐linked polyubiquitin chains from RIG‐I. 116 Ubiquitination is a post‐translational modification required for RIG‐I activation, 117 and deubiquitination results in its inactivation. Together, available evidence reveals that 3Cpro plays a key role in the efforts of picornaviruses in counteracting the host antiviral immune response by cleaving or inactivating essential adapter proteins in the RIG‐I/MDA5‐MAVS and/or the TLR3‐TRIF signaling pathways (Figure 2).
Like picornaviruses, both SARS‐CoV and MERS‐CoV trigger a limited IFN‐I response. 118 , 119 , 120 A mouse model infected with SARS‐CoV demonstrated that a significant delay in IFN production contributes to disease progression and severity. 121 Using different model systems including cells and ferrets infected with SARS‐CoV‐2 and post‐mortem lung tissues from COVID patients, a recent study showed that SARS‐CoV‐2 infections elicit low levels of IFN‐I and no activation of TBK1 and ISGs. 122 Of note, it was found that SARS‐CoV‐2 is highly sensitive to IFN‐I, suggesting an important role for IFN‐I in antiviral defense. 122 , 123 Although experimental data on SARS‐CoV‐2 is still limited, current evidence from SARS‐CoV and MERS‐CoV research revealed that CoVs develop different strategies to overcome the host innate immunity. For example, the PLpro of SARS‐CoV, which has deubiquitinating activities, acts as an IFN‐I antagonist by removing ubiquitin chains from IRF3 and through preventing the phosphorylation of IRF3 124 , 125 . It was also discovered that the ORF3b, ORF6, and N proteins of SARS‐CoV inhibit production and action of IFN‐I. 126 Moreover, M protein of SARS‐CoV was shown to physically associate with RIG‐I, TBK1, IKKε, and TRAF3 and inhibit gene transcription of IFN‐I. 127 Most interestingly, several coronaviruses, including SARS‐CoV‐2, porcine deltacoronavirus (PDCoV), and porcine epidemic diarrhea virus (PEDV) have been reported to cause the cleavage of TAB1 or NEMO through their individual 3CLpro (Table 1, yellow highlighted row), 49 , 59 , 60 suggesting an important role for 3CLpro in antagonizing the host antiviral innate immune response.
The importance of virus‐induced cytoplasmic aggregates, termed antiviral stress granules (avSGs), 128 in RLRs‐mediated innate immunity has been increasingly recognized. Upon viral infection, dsRNAs are generated and accumulate in the cytoplasm, which activate the viral RNA sensor protein kinase R (PKR) and cause phosphorylation of eukaryotic initiation factor 2α and consequent formation of avSGs. Together with key molecules (i.e., Ras‐GTPase‐activating protein SH3 domain‐binding protein 1 (G3BP1) and T cell intracellular antigen 1), avSGs are formed by recruiting multiple antiviral effectors, such as RLRs, Pumilio, DEAH‐box helicase 36, Mex‐3 RNA binding family member C, tripartite motif containing 25, OAS and RNaseL. 128 , 129 , 130 , 131 , 132 The close‐proximity of 5′‐triphosphate containing ssRNA and dsRNA with RLRs within the compact avSG compartment facilitates a more robust RLR‐mediated IFN‐I responses to suppress viral replication. 128 , 129 , 130 , 131 , 132 To bypass this, picornaviruses, including PV, EMCV, CVB3, FMDV, and EV‐D68, utilize 3Cpro to cleave G3BP1 and block avSG formation to prolong viral survival. 108 , 109 , 110 , 111 , 133
CoVs were also found to be able to modulate the formation of avSGs. It was reported that MERS‐CoV accessory protein 4a prevents PKR activation by directly binding viral dsRNA, thereby inhibiting avSG formation allowing for effective viral replication. 134 , 135 However, the role of protein 4a in avSG formation appears to be cell type‐specific. MERS‐CoV mutant with deletion of 4a gene still impedes avSG formation in certain cell types, 134 , 135 suggesting that additional proteins (possibly viral proteases) encoded by MERS‐CoV are required to antagonize avSGs. In a recent report, Grogan and colleagues 8 utilized an affinity purification mass spectrometry proteomics approach to screen for cellular proteins that interact with individual SARS‐CoV‐2 proteins, including 3CLpro. Using both wild‐type and catalytically inactive (C145A) 3CLpro, they identified two high‐confidence interactions with the histone deacetylase 2 (HDAC2) and tRNA methyltransferase 1 (TRMT1), respectively. Of interest, HDAC2 has been previously shown to be required for IFN‐I signaling through histone modification. 136 , 137 Remarkably, in addition to 3CLpro, other SARS‐CoV‐2‐encoded proteins were also found to interact with cellular proteins involved in regulating host innate immunity, including the core avSG protein G3BP1 8 , a known antiviral protein that induces the innate immune response. 129 , 138
Clinical evidence revealed that SARS‐CoV‐2 infections predispose patients to subsequent bacterial or other viral infections, which are associated with poor outcomes including increased severity and fatality. 139 , 140 Based on what is known about the role of 3Cpro during picornaviral infections, at least two mechanisms may contribute to the secondary infections. First, TRIF is not only involved in antiviral but also participates in antibacterial host defense mechanisms. 141 Disruption of TRIF during the initial phase of viral infection could render the host more susceptible to bacterial invasions. Second, recent studies identified G3BP1 as a critical component in cyclic GMP‐AMP synthase (cGAS)‐mediated innate immune responses against DNA viruses. 142 , 143 cGAS is a cytosolic DNA sensor that detects viral DNA to activate the IFN‐I pathway. 144 Cleavage of G3BP1 is thus expected to weaken this mechanism and leave the patients at higher risk from a secondary DNA viral infection. A proposed model that picornaviruses (probably CoVs as well) induce two phases of infection (or co‐infection) and disease progression is depicted in Figure 3.
FIGURE 3.
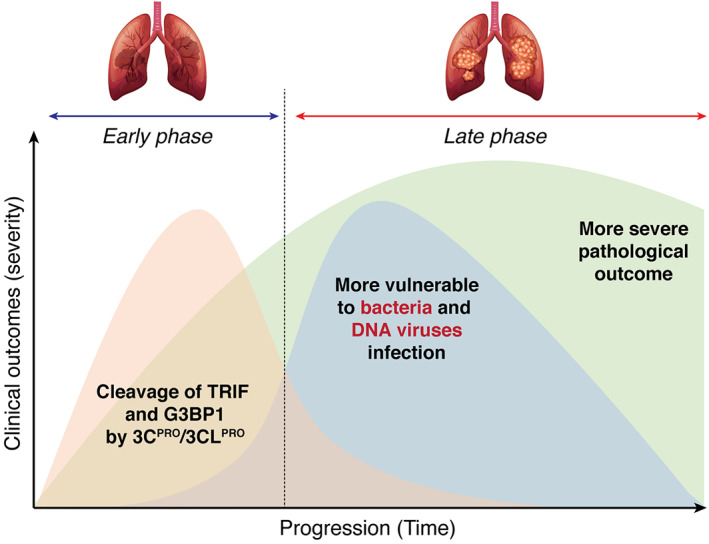
A proposed mechanistic model for the secondary infection after the initial picornavirus or coronavirus infection. During the first phase of infection, proteins (e.g., TRIP and G3BP1) that are involved in a broad range of host defenses (i.e., against not only RNA viral infection, but also bacterial and DNA viral invasion) are targeted by 3C protease or possibly 3C‐like protease for degradation, rendering the patients more vulnerable to secondary infection. Combinatorial infection during the late phase could result in increased disease severity and mortality
2.2. Targeting NLR family PYD containing protein‐3 inflammasome pathway
Similar to RLRs, NLRs are cytosolic PRRs that sense intracellular PAMPs and/or danger‐associated molecular patterns (DAMPs) to induce pro‐inflammatory cytokine production and inflammatory cell death (termed pyroptosis). 145 Under stress, NLRs initiate the formation of a large multiprotein complex, called inflammasome. The best‐studied inflammasome is the NLR family PYD containing protein‐3 (NLRP3) inflammasome, which consists of the sensor NLRP3, the adapter apoptosis‐associated speck‐like protein containing a caspase recruitment domain (ASC), and the effector caspase‐1. Upon sensing viral PAMPs and DAMPs generated during infection, NLRP3 oligomerizes and recruits pro‐caspase‐1 through the adopter protein ASC to form NLRP3 inflammasome. Subsequently, pro‐caspase‐1 undergoes self‐cleavage and activation, promoting maturation and secretion of pro‐inflammatory cytokine interleukin‐1β (IL‐1β) and IL‐18, and inducing cleavage of the pro‐pyroptotic factor gasdermin D (GSDMD). 146 The resulting N‐terminal cleavage product of GSDMD then creates pores on cell membrane, triggering pyroptotic cell death and facilitating release of inflammatory cytokines (IL‐1β and IL‐18). 147 Similar to other types of cell death, pyroptosis limits viral replication by eliminating infected cells.
As NLRP3 plays a vital role in antiviral response, numerous viruses, including picornaviruses, have adopted strategies to counteract its functions. 145 It was reported that NLRP3‐, ASC‐, and caspase‐1‐deficient mice infected with EV‐A71 display more severe disease phenotype and higher virus titers as compared to wild‐type control mice, suggesting a defensive role for the NLRP3 inflammasome against EV‐A71 infection. 52 Further investigation identified that, to overcome the antiviral immune response, EV‐A71 has evolved to inactivate the NLRP3 inflammasome by directly cleaving NLRP3 through the proteolytic activities of 2Apro and 3Cpro 52 In addition, EV‐A71 3Cpro also targets its effector GSDMD for cleavage at a site (Gln193‐Gly194) different from that (Asp275‐Asp276) mediated by caspase‐1. The cleavage products of GSDMD by 3Cpro fail to stimulate pyroptosis; hence, further enhancing EV‐A71 replication. Similarly, following CVB3 infection, NLRP3‐/‐ mice present more rapid disease progression and increased viral loads when compared with wild‐type mice. To evade the host defense, CVB3 3Cpro directly targets NLRP3 and its upstream signaling molecules (RIP1/RIP3, receptor‐interacting protein 1/3) for degradation. 53 , 62 , 148
It is increasingly recognized that early, temporary activation of NLRP3 inflammation has an antiviral role by clearing virus and infected cells, whereas prolonged and extreme activation is harmful, causing disease‐related immunopathology. For example, during hepatitis C viral infection, activation of NLRP3 inflammasome is associated with enhanced inflammation and tissue damage. 149 Persistent activation of NLRP3 during CVB3 infection has also been linked to pathological outcome. 150 , 151 It is well documented that SARS‐CoV‐2 infection leads to cytokine storm, featured by excessive production and secretion of pro‐inflammatory cytokines and chemokines, contributing significantly to disease severity of Covid‐19. 5 , 122 , 152 Several proteins encoded by SARS‐CoV, including E, ORF3a, and ORF8b, have been shown to activate the NLRP3 inflammasome. 153 , 154 , 155 Recent evidence has also revealed that NLRP3 inflammasome is activated in response to SARS‐CoV‐2 infection. 156 It is speculated that overactivation of the inflammasome may be responsible, at least in part, for the observed cytokine storm in Covid‐19 patients.
The NLRP inflammasome pathway and viral manipulation are summarized in Figure 4.
FIGURE 4.
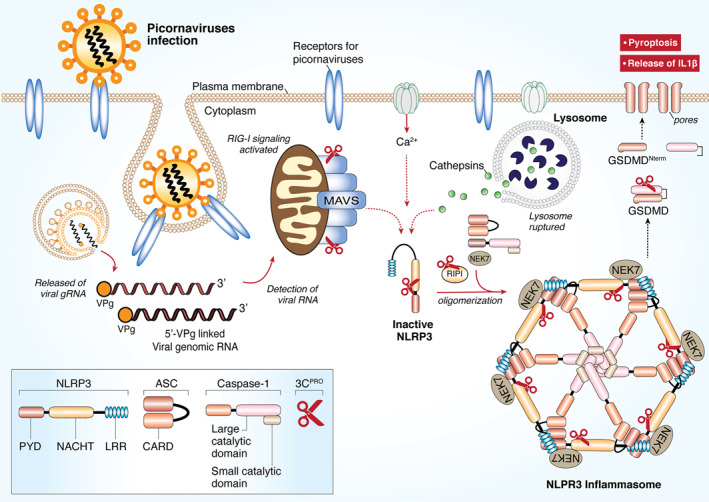
Picornaviral 3Cpro targets the NLRP3 inflammasome for immune evasion. RNA viruses and other DAMPs could activate NLRP3 inflammasome. Formation of the NLRP3‐dependent inflammasome activates caspase 1, which in turn cleaves pro‐IL‐1β and pro‐IL‐18. GSDMD is also cleaved by caspase 1 and the resulting N‐terminal cleavage products are inserted into the plasma membrane, forming multiple pores and inducing pyroptosis and release of pro‐inflammatory cytokine. Upon picornaviral infection, NLRP3, its upstream signaling proteins (RIP1/RIP3), and its downstream effector GSDMD are all targeted by 3Cpro for degradation. As a result, pyroptosis is inhibited for efficient viral replication. CARD, caspase activation and recruitment domain; DAMPs, danger‐associated molecular patterns; GSDMD, Gasdermin D; LRR, Leucine rich repeat; NLRP3, NLR family and pyrin domain‐containing protein 3; PYD, Pyrin domain; RIP, receptor‐interacting protein; NACHT, NAIP, CIITA, HET‐E and TEP1‐associated families; VPg, viral protein genome‐linked; 3Cpro, 3C protease
2.3. Targeting host RNA degradation components
In eukaryotic cells, posttranscriptional processes (e.g., mRNA surveillance, silencing, translational repression, and degradation) play a central role in the regulation of gene expression and ultimately determine the expression levels of a significant fraction of the transcriptome. Recently, it has become apparent that posttranscriptional processes acting on cytoplasmic messenger ribonucleoprotein complexes (mRNPs) are physically tied and can occur in discrete cytoplasmic entity known as processing (P)‐bodies. These compartments are highly conserved across cells derived from vertebrates, invertebrates, yeasts and plants, containing many enzymes involved in mRNA turnover. To date, P‐bodies have been demonstrated to play critical roles in general mRNA degradation, nonsense‐mediated mRNA decay, adenylate‐uridylate‐rich (AU‐rich) element‐mediated mRNA degradation at the 3′ UTRs, and miRNA‐induced RNA silencing pathway. 157
In eukaryotes, the degradation process is initiated firstly by removal of the poly(A) tail by deadenylases. Following deadenylation, mRNAs are subjected to exonucleolytically degradation starting from their 3′‐end by the exosome, a multimeric complex with 3’→5′ exonucleases. In the meantime, the guanosine cap structure at the 5′‐end is removed by several decapping enzymes and coactivators, including decapping protein 1/2 (DCP1/2), DEAD‐box RNA‐helicase‐6 (DDX6), enhancer of mRNA decapping protein‐3 (EDC3) and EDC4, rendering the mRNA susceptible to 5’→3′ degradation by the major cytoplasmic exoribonuclease 1 (XRN1). 158
RNA degradation has emerged as an important antiviral host defense mechanism. 158 The most widely studied viruses in the context of XRN1 are positive‐stranded RNA viruses from the Flaviviridae family, including Dengue virus, West Nile viruses, hepatitis C virus, and Yellow Fever virus. XRN1 acts as an antiviral factor by degrading their genomic RNA. 157 In addition, DCP1/2‐XRN1 were shown to have critical antiviral functions against both picornaviruses and negative‐stranded cytoplasmic RNA viruses. During infection, these viruses generate various RNA intermediate species with defined structures (long/short dsRNA and ssRNA) inside their replication complexes. Both long ssRNA and dsRNA could induce the re‐localization of both DCP1/2 and XRN1 nucleases into the viral replication complexes for viral RNA degradation. 159 This further supports the prior observation that the presence of XRN1 and DCPs potentially poses a threat to enterovirus RNA. To conquer this antiviral activity, PV utilizes its 3Cpro to disperse and destabilize P‐bodies by directly targeting DCP1a for degradation. 103 The interplay between the cellular RNA degradation pathway and the viruses is illustrated in Figure 5.
FIGURE 5.
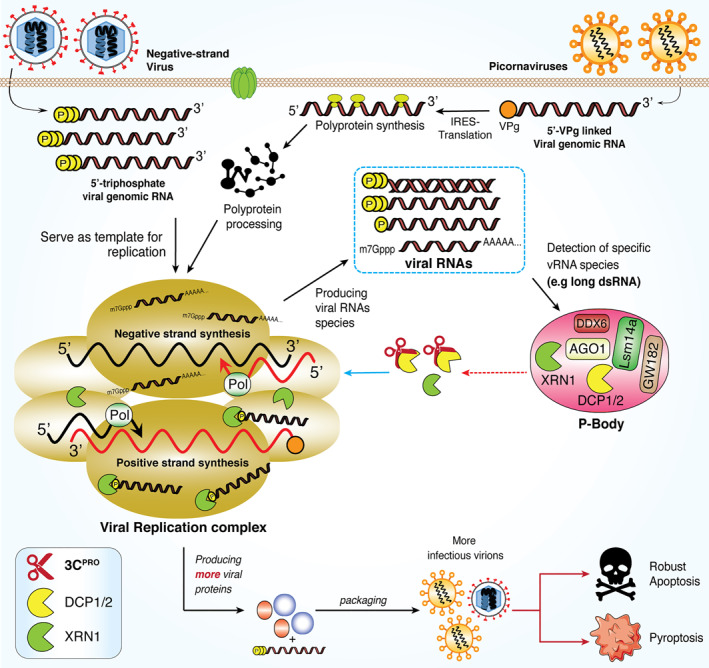
Dispersion of 5′→3′ RNA degradation components within P‐bodies during picornaviral infection. RNA viruses, including picornaviruses, initiate viral replication in a discrete compartment within cytoplasm, generating various RNA species with defined signatures. These includes 5′ppp, 5′p‐ssRNA, dsRNA and viral mRNA. Both picornavirus and coronavirus are able to generate long ssRNA and dsRNA and trigger the translocation of 5’→3′ RNA degradation components, including DCP1, DCP2, and XRN1, into the viral replication complex for degradation of associated viral RNA species. Picornaviral 3Cpro cleaves DCP1, resulting in increased viral particles and infectivity. DCP1/2, decapping protein‐1/2; XRN1, 5’→3′ exoribonuclease‐1; dsRNA, double‐stranded RNA; 3Cpro, 3C protease
While both the genomic and subgenomic SARS‐CoV‐2 mRNAs contain 5′‐cap structure, previous studies have shown that inhibiting several eukaryotic initiation factors family proteins (eIF4E, eIF4F, and eIF4G) could impair coronavirus replication, 160 , 161 highlighting the importance of cap‐dependent translation in SARS‐CoV‐2 mRNA synthesis. Thus, it is not surprising that SARS‐CoV‐2 would intervene the function of host decapping enzyme DCP1/2 and XRN1, possibly through its 3CLpro. However, to date, antiviral roles XRN1 in coronaviral mRNA translation have not been reported.
2.4. Targeting host autophagy machinery
Macroautophagy (or autophagy in short) is a conserved intracellular degradation pathway that is essential in maintaining cellular homeostasis by removing unwanted or dysfunctional cellular components. 162 The process of autophagy is highly regulated by more than 30 “autophagy‐related” proteins and includes three major steps. First, the substrates are sequestered by a crescent‐shaped double‐membrane vesicle called a phagophore. Then, the two ends of the phagophore fuse to form an autophagosome. Finally, autophagosome fuses with a lysosome while the enwrapped cargo is degraded by hydrolysis.
Autophagy plays a significant role in antiviral host defense by directly targeting invading viruses through a process, call virophagy, for clearance. 163 , 164 Virophagy is mediated through the function of autophagy cargo receptors, including sequestosome 1 (SQSTM1)/p62, neighbor of BRCA1 (NBR1), calcium binding and coiled‐coil domain‐containing protein 2 (CALCOCO2)/nuclear dot 10 protein 52 (NDP52), which recruit viral components/particles to autophagosome for degradation. 165 To evade the antiviral efforts of virophagy, many viruses, including picornaviruses, have evolved to disrupt the function of autophagy receptors. 166 For instance, SQSTM1/p62 and CALCOCO2/NDP52 are targeted for degradation by CVB3 2Apro and 3Cpro, respectively. 66 , 67 Cleavage of SQSTM1/p62 was later confirmed upon PV, HRV‐1A, and EV‐D68 infection. 68 Furthermore, NBR1, a homolog of SQSTM1/p62, can also be cleaved by 3Cpro 69 . Remarkably, it was found that cleavage of SQSTM1/p62 and NBR1 not only causes a loss‐of‐function, but also generates dominant‐negative mutants against the function of native proteins. 69 Loss of both SQSTM1/p62 and NBR1 would also impair mitophagy and results in mass production of reactive oxygen species, and IL‐1β through constitutive NLRP3‐independent inflammasome activation (potentially other NLRP family members). 167 The resulting cleavage fragments of SQSTM1/p62 and NBR1 will accumulate to serve as DAMPs signals and further amplify the inflammatory cascade. Acute respiratory pneumonia due to cytokine storm is a hall mark of SARS‐CoV‐2 infection, whether its 3CLpro would manipulate host autophagic system in particular to mass produced IL‐1β in Covid‐19 pathogenesis is certainly worth further investigation.
Decreased autophagic flux as a result of blockage of autophagosome‐lysosome fusion is another mechanism adopted by picornaviruses to escape viral RNA/protein degradation. The fusion process is regulated by multiple proteins involved in membrane trafficking, particularly a group of SNAP29 proteins, including syntaxin 17 (STX17), synaptosomal‐associated protein 29 (SNAP29), and vesicle‐associated membrane protein 8 (VAMP8). 168 It was discovered that upon EV‐D68 and CVB3 infection, the SNAP29 linker protein, SNAP29, is cleaved by 3Cpro, which dissociates the STX17‐interacting domain of SNAP29 from the VAMP8‐binding motif, thereby disrupting the formation of STX17‐SNAP29‐VAMP8 complexes and inhibiting autophagosome‐lysosome fusion. 68 , 70 Figure 6 summarizes the known mechanism by which picornaviral protease subverts the autophagy for immune evasion.
FIGURE 6.
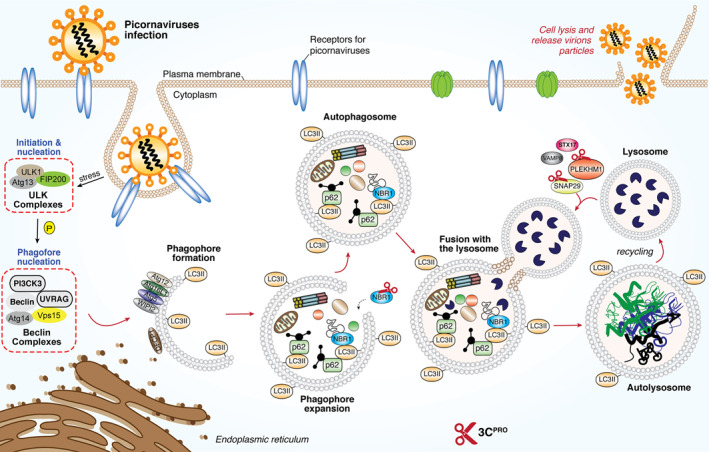
Subversion of host autophagy through picornaviral 3Cpro. Schematic diagram depicted the molecular mechanism for the initiation of host autophagy pathway upon the presence of RNA virus for the clearance of viral‐associated molecules. Picornaviruses utilize its own encoded 2Apro (not shown here) and 3Cpro to cleave key components such as p62, NBR1, SNAP29 and PLEKHM1 to facilitate a more robust replication. ATGs, autophagy‐related genes; DFCP1, double FYVE‐containing protein‐1, FIP200, focal adhesion kinase family interacting protein of 200kD; NBR1, neighbor of BRCA1; SNAP29, synaptosomal‐associated protein‐29; PLEKHM1, Pleckstrin homology and RUN domain containing M1; p62, also known as sequestosome 1 (SQSTM1); STX17, Syntaxin 17; ULK, Unc‐51‐like kinase‐1; UVRAG, UV radiation resistance‐associated gene protein; VAMP8, vesicle‐associated membrane protein‐8; WIPI2, WD‐repeat domain phosphoinositide‐interacting protein‐2; 2Apro, 2A‐protease; 3Cpro, 3C protease
Similar to the observations made with several picornaviruses, it was recently demonstrated that SARS‐CoV‐2 infection impairs autophagic flux by blocking autophagosome fusion with a lysosome and induction of autophagy reduces viral replication. 169 This finding is consistent with an earlier report with MERS‐CoV infection by the same research group. 170 Although the mechanism responsible for decreased autophagosome‐lysosome fusion remains largely unclear, it was previously shown that over‐expression of the membrane‐associated PLpro of SARS‐CoV and MERS‐CoV is sufficient to suppress the fusion between autophagosome and lysosome, 171 whether CoV 3CLpro has a role in regulation of autophagic flux and cargo recognition in general as some picornaviral 3Cpro does has not been explored and requires further investigations. The list of substrates of 3Cpro and 3CLpro is summarized in Table 1.
2.5. High‐throughput identification of cellular substrates of 3Cpro
A proteomics‐based quantitative method, termed N‐terminomic terminal amine isotopic labeling of substrates (TAILS) (Figure 7), 172 has been exploited to globally search for novel cellular substrates of picornaviral proteases. 63 , 85 TAILS, developed by the Overall laboratory at the University of British Columbia, uses an unbiased negative selection approach to identify neo‐N and ‐C termini (named N‐terminomic and C‐terminomic TAILS, respectively). 172 , 173 , 174 , 175 This state‐of‐the‐art approach has several advantages, including quantitative, highly sensitive, and concurrent identification of both the substrates and the cleavage sites, 176 and has been used to analyze substrates of many types of proteases. 177 , 178
FIGURE 7.
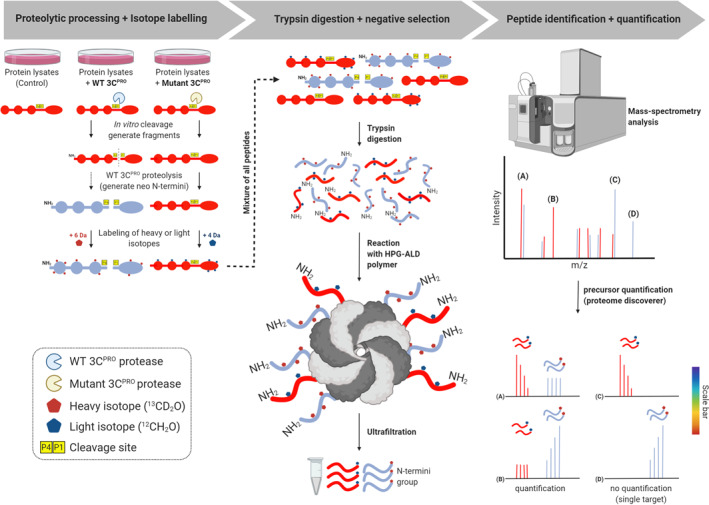
Schematic workflow of TAILS N‐terminomics screening of 3Cpro and 3CLprosubstrates. Schematic diagram depicts the TAILS workflow and scheme for identification of 3Cpro or 3CLpro substrates. In brief, protein samples from whole cell lysates were subjected to in vitro cleavage by either recombinant purified WT 3Cpro/3CLpro or mutant (C147A) 3Cpro/(C145A) 3CLpro, followed by N‐terminal enrichment using TAILS (left panel). Samples were then combined and subjected to pre‐TAILS shotgun‐like mass‐spectrometry analysis after complete digestion with trypsin. The exposed amine groups of N‐termini generated by the trypsin digestion were then removed by covalently coupling to a high‐molecular weight polyaldehyde polyglycerol polymer. This process allowed for selection via negative enrichment of blocked N termini (middle panel). Peptides were subsequently identified and quantified using high‐resolution mass spectrometry (indicated in the right panel). The resultant high‐confidence candidate substrates were determined through the analysis of the quantified heavy/light (H/L) ratio of dimethylation‐labeled semitryptic neo‐N terminus peptides. They will be subjected to further validation through similar in vitro cleavage assay by 3Cpro/3CLpro, followed by immunoblotting using specific antibodies; TAILS, terminal amine isotopic labeling of substrates; 3Cpro, 3C protease; 3CLpro, 3C‐like protease
Jan and colleagues conducted N‐terminomic TAILS on HeLa cell or mouse cardiomyocyte extracts subjected to incubation with purified recombinant PV or CVB3 3Cpro, respectively, to identify cleaved neo‐N‐terminal peptides (see Figure 7 for the detailed procedure). 63 , 85 A list of high confidence candidate proteins was generated for 3Cpro, including peptides corresponding to previously reported substrates at the known cleavage sites (i.e., poly(A) binding protein (PABP), 90 G3BP1 110 , and TAR DNA‐binding protein 43 (TDP‐43) 87 ). Among them, a subset of candidate targets of PV 3Cpro has been validated in vitro and under viral infection, which include four common protein substrates of both PV and CVB3 3Cpro (i.e., heterogeneous nuclear ribonucleoprotein M (hnRNP M), hnRNP K, RIP1, and phosphoribosylformylglycinamidine synthetase [PFAS]), programmed cell death 6‐interacting protein (PDCD6IP, also known as ALIX), general vesicular transport factor p115, ATP‐citrate synthase (ACLY), Golgi‐specific brefeldin A‐resistance guanine nucleotide exchange factor 1 (GBF1). Studies through gene‐silencing or expression of a non‐cleavable mutant form of these substrates have demonstrated a pivotal role for these proteins in regulating viral replication/propagation. 63 , 85
Given the effectiveness of the N‐terminomic TAILS in identifying the cellular targets of 3Cpro, this unbiased proteomics approach is currently being utilized to analyze the cellular targets of MHV and SARS‐CoV‐2 proteases, including 3CLpro. It is anticipated that identification of the full repertoire of host substrates of CoV proteases will provide a more comprehensive understanding of viral tropism, interaction with host cells, and pathogenesis, as well as assist in the development of novel antiviral drugs.
2.6. Current 3Cpro and 3CLpro inhibitors and future challenges
As discussed earlier, both picornaviral 3Cpro and coronaviral 3CLpro are chymotrypsin‐like cysteine proteases with conserved substrate specificity (P1‐P1′ and P4 cleavage sites of the substrates are highly conserved between two enzymes) and active sites. 179 Owing to these similarities, efforts have been made to explore the potential of developing broad‐spectrum antiviral compounds. 13 , 14 , 15 The fact that no known human homologs further increases the feasibility of this strategy.
Rupintrivir (AG‐7088, a protease inhibitor originally developed for HRV to treat common cold) and/or its derivatives/analogs have been found to possess antiviral activities against a wide range of picornaviruses (i.e., HRV, PV, EV‐A71, EV‐D68, CVB, CVA, HAV, and FMDV) 180 , 181 , 182 , 183 and coronaviruses, including SARS‐CoV. 14 , 184 Furthermore, it was reported that dipeptidyl aldehyde (GC373), α‐ketoamide (GC375), and dipeptidyl bisulfite adduct (GC376), are able to inhibit both picornaviral 3Cpro and coronaviral 3CLpro activities and block viral replication. 13 These studies point to the possibility of developing broad‐spectrum antiviral therapeutics against SARS‐CoV‐2. However, rupintrivir failed to show efficacy in natural HRV infection conditions during a clinical phase II trial. 185
Although Remdesivir (a nucleotide analog inhibitor of RdRp, originally developed for the treatment of Ebola virus disease by Gilead Sciences) has been approved for treating severely ill Covid‐19 patients in the United States and the Europe, 186 its safety and antiviral activity are still under extensive investigations and clinical trials. Similarly, despite some early promising findings showing the effectiveness of chloroquine (a classical anti‐malarial and autoimmune disease drugs) against SARS‐CoV‐2 infection in non‐respiratory cells, 187 , 188 recent research has demonstrated that chloroquine fails to block infection of human lung epithelial cells with SARS‐CoV‐2. 189 In addition, it is known that chloroquine may cause severe cardiotoxicity. 190 A number of attempts have been made to design drugs targeting 3CLpro of SARS‐CoV‐2. Different approaches, including laboratory synthesis, virtual screening, drug repositioning, and structure‐based molecular docking have been taken for this purpose. Hilgenfeld laboratory recently elucidated the X‐ray crystallographic structure of SARS‐CoV‐2 3CLpro and synthesized improved α‐ketoamide inhibitors to target 3CL. 12 Using computer‐aided drug design, Jin et al. identified a 3CLpro inhibitor (N3) and determined the crystal structure of its complex with 3CLpro of SARS‐CoV‐2 10 . Through a combined structure‐based virtual and high‐throughput screening, they further showed that six compounds exhibit potent anti‐3CLpro activities. 10 Most recently, Dai et al. reported the design and synthesis of two lead compounds (11a and 11b) targeting 3CLpro and solved the X‐ray crystal structure of these inhibitors bound to 3CL. 9 Although these compounds serve as promising drug candidates, their effectiveness in nature infection remain to be investigated. The list of inhibitors targeting 3Cpro and 3CLpro is summarized in Table 2.
TABLE 2.
Studies of 3Cpro or 3CLpro inhibitors antiviral compounds [Correction added on 17 February 2021, after first online publication. The references were updated throughout Table 2 and in‐text citations.]
| Research phase | Compound | Target | Results | Ref |
|---|---|---|---|---|
| In vitro validation | TG‐0205221 | 3CLpro of SARS‐CoV and HCoV‐229E | Reduces SARS‐CoV and HCoV‐229E replication by titer of 4.7 Log10 | 191 |
| Flavonoids: Apigenin, luteolin, quercetin, amentoflavone, aueretin, daidzein, puerarin, epigallocatechin gallate, gallocatechin gallate, kaempferol,rhoifolin, pectolinarin, herbacetin, flavonol | SARS‐CoV 3CLpro | Inhibit SARS‐CoV 3CLpro FRET protease assay catalytic activity | 192 | |
| Pyrazolone and pyrimidines inhibitors | SARS‐CoV 3CLpro | Show potent inhibitory activities against SARS‐CoV 3CLpro at micromolar range. | 193 | |
| Aryl methylene ketones, Mono‐, and difluorinated methylene ketones groups | SARS‐CoV 3CLpro | Improved version is stable and less toxic to cells. Potently inhibits SARS‐CoV 3CLpro at nanomolar range | 194 | |
| Heteroaromatic esters and benzotriazole esters derivatives | SARS‐CoV 3CLpro | Show potent inhibitory activities against SARS‐CoV 3CLpro at nanomolar range | 195 , 196 , 197 | |
| Boronic | SARS‐CoV 3CLpro | Significantly inhibits SARS‐CoV 3CLpro enzymatic activity in micromolar range | 198 | |
| Aza‐peptide epoxides derivatives | SARS‐CoV 3CLpro | Show irreversible inhibition against SARS‐CoV 3CLpro | 199 , 200 , 201 | |
| Etacrynic acid derivatives | SARS‐CoV PLpro and 3CLpro | Show more than 70% inhibition on SARS‐CoV at concentration of 100μM | 202 , 203 | |
| Peptides aldehydes derivatives | SARS‐CoV and HCoV‐229E 3CLpro | Suppress SARS‐CoV by 4.7 Log10 and HCoV‐229E by 5.2 Log10 | 191 | |
| Modified version of HIV protease inhibitors | SARS‐CoV 3CLpro | Potent inhibitors against SARS‐CoV 3CLpro but not against HIV protease | 204 | |
| Sulfone and dihydroimidazole derivatives | SARS‐CoV 3CLpro | 21 derivatives from these two analogs show EC50 less than 50 μM against SARS‐CoV 3CLpro | 205 , 206 | |
| Michael acceptor peptidomimetics | SARS‐CoV 3CLpro | Show potent inhibitory against SARS‐CoV 3CLpro | 207 , 208 | |
|
Lignoids, di‐ and triterpenoid derivatives: Betulinic acid, savinin, ferruginol, pritimererin, tingenone, iguestrin and triterpenoids celastrol |
SARS‐CoV 3CLpro | Abietane type diterpenoids are the most robust terpenoids on SARS‐CoV (EC50 = 9.1 μM) | 207 , 209 , 211 | |
|
Metal conjugated: Zinc‐ or mercuric based |
SARS‐CoV PLpro and 3CLpro, | Inhibition is pronounced in Zinc‐conjugated compounds | 212 , 213 | |
| α‐Ketoamides: | 3Cpro of CVB3, &; HRV, EV‐D68, EV‐A713CLpro of SARS‐CoV, MERS, 229E | Display low toxicity &; low micromolar of EC50 against tested viruses | 214 | |
| Pyridyl, pyrazyl and Benzotriazole‐derivatives inhibitors | SARS‐CoV PLpro or 3CLpro | Robust inhibition on SARS‐CoV in vitro within micromolar range | 215 , 216 , 217 | |
| Rupintrivir (AG‐7088) | Targeting 3Cpro and 3CLpro encoding viruses | Shows robust activity against SARS‐CoV‐2 in vitro | 218 , 219 | |
| Boceprevir, Calpain inhibitors II, and XII | SARS‐CoV‐2 3CLpro | Inhibit SARS‐CoV‐2 in vitro with EC50 less than 5μM | 220 | |
| 3CLpro‐1 | Originally designed for 3Cpro of EV‐A71. | Shows robust efficacy of EC50 200 nM, effective against SARS‐CoV‐2 and MERS‐CoV. | 221 , 222 | |
| Isatin derivatives | Targeting 3Cpro and SARS‐CoV 3CLpro | Effectively inhibit SARS‐CoV 3CLpro through noncovalent bonding in low micromolar range | 219 , 220 | |
| Anilide derivaties: 2‐chloro‐4‐nitro anilineL‐phenylalanine, 4‐(dimethylamino)benzoic acid | SARS‐CoV 3CLpro | Potent and highly specific inhibitors against SARS‐CoV 3CLpro | 225 | |
| Computational prediction (docking analysis) | Anti‐HIV‐1 drugs: Indinavir, Darunavir | SARS‐CoV‐2 3CLpro | Docking & binding free energy prediction shows high scores & high binding affinities against SARS‐CoV‐2 3CLpro | 226 |
| Decahydroisoquinoline inhibitors | SARS‐CoV‐2 3CLpro | X‐ray crystallization studies confirmed that these inhibitors fit well into the cleft of 3CLpro | 227 | |
| In vitro and in vivo validation | Peptides with halomethyl ketone derivatives | SARS‐CoV 3CLpro | Effectively inhibit SARS‐CoV infection, with low cytotoxicity in cells and in mice. | 228 |
| Widely tested in animals and now under trial on human for coronavirus disease‐2019 | GC376 | Targeting 3Cpro and 3CLpro encoding viruses | Shows robust activity against SARS‐CoV, SARS‐CoV‐2, and Norovirus | 220 |
Abbreviations: HRV, human rhinovirus; MERS‐CoV, Middle East respiratory syndrome‐CoV; PLpro, papain‐like protease; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2 3CLpro/3Cpro, 3C‐like protease/3C protease.
3. CONCLUDING REMARKS
To date, we know very little about the 3CLpro of SARS‐CoV‐2, especially the molecular mechanism of the pathways by which SARS‐CoV‐2 3CLpro blocks. Certainly, we cannot rule out that there are some differences but also a lot of similarities among both families. While we are in the process of understanding the structure and functions of SARS‐CoV‐2 3CLpro, a comparison with the 3Cpro from picornaviruses can provide more insights into the pathogenesis and regulatory mechanisms of Covid‐19. These will serve as a critical foundation for the design of broad‐spectrum anti‐coronaviruses inhibitors, or perhaps anti‐3C/3CLpro expressing viruses.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHORS CONTRIBUTIONS
Chen Seng Ng and Honglin Luo designed and outlined the review. Chen Seng Ng prepared the initial manuscript and figures. Christopher C. Stobart prepared the structural figure and relevant information. All authors contributed to revising and writing the final version.
ACKNOWLEDGEMENT
We thank all the members in Luo lab for insights and discussion. We apologize for not including all related references due to space constraints. C.S.N. is funded by VIROGIN Biotech Ltd. through the MITACS‐Accelerate Program. This work was supported by grants from the Natural Sciences and Engineering Research Council (RGPIN2016‐03811), the Canadian Institutes of Health Research (PJT‐159546 and PJT‐173318), and the Heart & Stroke Foundation of Canada (G‐16‐00013800 and G‐18‐0022051).
Ng CS, Stobart CC, Luo H. Innate immune evasion mediated by picornaviral 3C protease: Possible lessons for coronaviral 3C‐like protease? Rev Med Virol. 2021;31(5):e2206. doi: 10.1002/rmv.2206
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1. Zhou P, Yang X‐L, Wang X‐G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses . The species Severe acute respiratory syndrome‐related coronavirus: classifying 2019‐nCoV and naming it SARS‐CoV‐2. Nat Microbiol. 2020;5:536‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wadman M, Couzin‐Frankel J, Kaiser J, Matacic C. A rampage through the body. Science. 2020;368:356‐360. [DOI] [PubMed] [Google Scholar]
- 5. Huang C, Wang Y, Li X, et al. clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID‐19) outbreak in China: summary of a report of 72 314 cases from the Chinese center for disease control and prevention. J Am Med Assoc. 2020; 323(13):1239–1242. 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 7. Saberi A, Gulyaeva AA, Brubacher JL, Newmark PA, Gorbalenya AE. A planarian nidovirus expands the limits of RNA genome size. PLoS Pathog. 2018;14:e1007314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dai W, Zhang B, Jiang X‐M, et al. Structure‐based design of antiviral drug candidates targeting the SARS‐CoV‐2 main protease. Science. 2020;368:1331‐1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS‐CoV‐2 and discovery of its inhibitors. Nature. 2020;582:289‐293. [DOI] [PubMed] [Google Scholar]
- 11. Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti‐SARS drugs. Science. 2003;300:1763‐1767. [DOI] [PubMed] [Google Scholar]
- 12. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved α‐ketoamide inhibitors. Science. 2020;368:409‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim Y, Lovell S, Tiew K‐C, et al. Broad‐spectrum antivirals against 3C or 3C‐like proteases of picornaviruses, noroviruses, and coronaviruses. J Virol. 2012;86:11754‐11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuo C‐J, Liu H‐G, Lo Y‐K, et al. Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Lett. 2009;583:549‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ramajayam R, Tan K‐P, Liang P‐H. Recent development of 3C and 3CL protease inhibitors for anti‐coronavirus and anti‐picornavirus drug discovery. Biochem Soc Trans. 2011;39:1371‐1375. [DOI] [PubMed] [Google Scholar]
- 16. Lefkowitz EJ, Dempsey DM, Hendrickson RC, et al. Virus taxonomy: the database of the international committee on taxonomy of viruses (ICTV). Nucleic Acids Res. 2018;46:D708‐D717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun D, Chen S, Cheng A, Wang M. Roles of the picornaviral 3C proteinase in the viral life cycle and host cells. Viruses. 2016;8:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Y, Ma L, Stipkovits L, Szathmary S, Li X, Liu Y. The strategy of picornavirus evading host antiviral responses: non‐structural proteins suppress the production of IFNs. Front Microbiol. 2018;9:2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J, Fan T, Yao X, et al. Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. J Virol. 2011;85:10021‐10030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hämmerle T, Hellen CU, Wimmer E. Site‐directed mutagenesis of the putative catalytic triad of poliovirus 3C proteinase. J Biol Chem. 1991;266:5412‐5416. [PubMed] [Google Scholar]
- 21. Zunszain PA, Knox SR, Sweeney TR, et al. Insights into cleavage specificity from the crystal structure of foot‐and‐mouth disease virus 3C protease complexed with a peptide substrate. J Mol Biol. 2010;395:375‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ypma‐Wong MF, Dewalt PG, Johnson VH, Lamb JG, Semler BL. Protein 3CD is the major poliovirus proteinase responsible for cleavage of the P1 capsid precursor. Virology. 1988;166:265‐270. [DOI] [PubMed] [Google Scholar]
- 23. Chan YM, Boehr DD. Allosteric functional switch in poliovirus 3C protease. Biophys J. 2015;108:528a. [Google Scholar]
- 24. Roehl HH, Parsley TB, Ho TV, Semler BL. Processing of a cellular polypeptide by 3CD proteinase is required for poliovirus ribonucleoprotein complex formation. J Virol. 1997;71:578‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R, et al. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha‐helical domain. Embo J. 2002;21:3213‐3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gorbalenya AE, Donchenko AP, Blinov VM, Koonin EV. Cysteine proteases of positive strand RNA viruses and chymotrypsin‐like serine proteases. A distinct protein superfamily with a common structural fold. FEBS Lett. 1989;243:103‐114. [DOI] [PubMed] [Google Scholar]
- 27. Lu Y, Denison MR. Determinants of mouse hepatitis virus 3C‐like proteinase activity. Virology. 1997;230:335‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tomar S, Johnston ML, St John SE, et al. Ligand‐induced dimerization of Middle East respiratory syndrome (MERS) coronavirus nsp5 protease (3CLpro): IMPLICATIONS for nsp5 regulation and the development OF antivirals. J Biol Chem. 2015;290:19403‐19422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Akira, S , Uematsu, S & Takeuchi, O Pathogen recognition and innate immunity. Cell. 2006;124:783–801. [DOI] [PubMed] [Google Scholar]
- 30. Ng CS, Kato H, Fujita T. Fueling type I interferonopathies: regulation and function of type I interferon antiviral responses. J Interferon Cytokine Res. 2019;39:383‐392. [DOI] [PubMed] [Google Scholar]
- 31. Roth‐Cross JK, Bender SJ, Weiss SR. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol. 2008;82:9829‐9838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peisley A, Lin C, Wu B, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci USA. 2011;108:21010‐21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barral PM, Sarkar D, Fisher PB, Racaniello VR. RIG‐I is cleaved during picornavirus infection. Virology. 2009;391:171‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Drahos J, Racaniello VR. Cleavage of IPS‐1 in cells infected with human rhinovirus. J Virol. 2009;83:11581‐11587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lei X, Xiao X, Xue Q, Jin Q, He B, Wang J. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J Virol. 2013;87:1690‐1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mukherjee A, Morosky SA, Delorme‐Axford E, et al. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011;7:e1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rui Y, Su J, Wang H, et al. Disruption of MDA5‐mediated innate immune responses by the 3C proteins of coxsackievirus A16, coxsackievirus A6, and enterovirus D68. J Virol. 2017;91(13):e00546–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang Y, Liang Y, Qu L, et al. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci USA. 2007;104:7253‐7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wen W, Yin M, Zhang H, et al. Seneca Valley virus 2C and 3C inhibit type I interferon production by inducing the degradation of RIG‐I. Virology. 2019;537:122‐129. [DOI] [PubMed] [Google Scholar]
- 40. Feng Q, Langereis MA, Lork M, et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J Virol. 2014;88:3369‐3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang B, Xi X, Lei X, et al. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti‐viral type I interferon responses. PLoS Pathog. 2013;9:e1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao T, Yang L, Sun Q, et al. The NEMO adaptor bridges the nuclear factor‐kappaB and interferon regulatory factor signaling pathways. Nat Immunol. 2007;8:592‐600. [DOI] [PubMed] [Google Scholar]
- 43. Wang D, Fang L, Li K, et al. Foot‐and‐mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J Virol. 2012;86:9311‐9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang D, Fang L, Wei D, et al. Hepatitis A virus 3C protease cleaves NEMO to impair induction of beta interferon. J Virol. 2014;88:10252‐10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lei X, Han N, Xiao X, Jin Q, He B, Wang J. Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB1/TAB2/TAB3 complex. J Virol. 2014;88:9830‐9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dong J, Xu S, Wang J, et al. Porcine reproductive and respiratory syndrome virus 3C protease cleaves the mitochondrial antiviral signalling complex to antagonize IFN‐β expression. J Gen Virol. 2015;96:3049‐3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll‐like receptor 3. J Virol. 2011;85:8811‐8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Qu L, Feng Z, Yamane D, et al. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease‐polymerase processing intermediate, 3CD. PLoS Pathog. 2011;7:e1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moustaqil M, Ollivier E, Chiu H‐P, et al. SARS‐CoV‐2 proteases cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species and the search for reservoir hosts. BioRxiv. 2020. 10.1101/2020.06.05.135699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xiang Z, Liu L, Lei X, Zhou Z, He B, Wang J. 3C protease of enterovirus D68 inhibits cellular defense mediated by interferon regulatory factor 7. J Virol. 2016;90:1613‐1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hung H‐C, et al. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J Infect Dis. 2011;203:1784‐1790. [DOI] [PubMed] [Google Scholar]
- 52. Wang H, et al. Reciprocal regulation between enterovirus 71 and the NLRP3 inflammasome. Cell Rep. 2015;12:42‐48. [DOI] [PubMed] [Google Scholar]
- 53. Wang C, et al. NLRP3 deficiency exacerbates enterovirus infection in mice. FASEB J. 2019;33:942‐952. [DOI] [PubMed] [Google Scholar]
- 54. Huang L, et al. Encephalomyocarditis virus 3C protease relieves TRAF family member‐associated NF‐κB activator (TANK) inhibitory effect on TRAF6‐mediated NF‐κB signaling through cleavage of TANK. J Biol Chem. 2015;290:27618‐27632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lu J, et al. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J Virol. 2012;86:3767‐3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tam JCH, Bidgood SR, McEwan WA, James LC. Intracellular sensing of complement C3 activates cell autonomous immunity. Science. 2014;345:1256070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhu X, Wang D, Zhou J, et al. Porcine deltacoronavirus nsp5 antagonizes type I interferon signaling by cleaving STAT2. J Virol. 2017;91(10):e00003–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Du Y, Bi J, Liu J, et al. 3Cpro of foot‐and‐mouth disease virus antagonizes the interferon signaling pathway by blocking STAT1/STAT2 nuclear translocation. J Virol. 2014;88:4908‐4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang D, Dong J, Xu S, et al. Porcine epidemic diarrhea virus 3C‐like protease regulates its interferon antagonism by cleaving NEMO. J Virol. 2016;90:2090‐2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhu X, Wang D, Zhou J, et al. Porcine deltacoronavirus nsp5 inhibits interferon‐β production through the cleavage of NEMO. Virology. 2017;502:33‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huang C, Zhang Q, Guo X, et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 4 antagonizes beta interferon expression by targeting the NF‐κB essential modulator. J Virol. 2014;88:10934‐10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Harris KG, Morosky SA, Drummond CG, et al. RIP3 regulates autophagy and promotes coxsackievirus B3 infection of intestinal epithelial cells. Cell Host Microbe. 2015;18:221‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jagdeo JM, et al. N‐terminomics TAILS identifies host cell substrates of poliovirus and coxsackievirus B3 3C proteinases that modulate virus infection. J Virol. 2018;92(8):e02211–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xie L, et al. The 3C protease of enterovirus A71 counteracts the activity of host zinc‐finger antiviral protein (ZAP). J Gen Virol. 2018;99:73‐85. [DOI] [PubMed] [Google Scholar]
- 65. Lei X, Zhang Z, Xiao X , et al. Enterovirus 71 inhibits pyroptosis through cleavage of gasdermin D. J Virol. 2017;91(18):e01069–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mohamud Y, Qu J, Xue YC, Liu H, Deng H, Luo H. CALCOCO2/NDP52 and SQSTM1/p62 differentially regulate coxsackievirus B3 propagation. Cell Death Differ. 2019;26:1062‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shi J, Wong J, Piesik P, et al. Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy. 2013;9:1591‐1603. [DOI] [PubMed] [Google Scholar]
- 68. Corona AK, Saulsbery HM, Corona Velazquez AF, Jackson WT. Enteroviruses remodel autophagic trafficking through regulation of host SNARE proteins to promote virus replication and cell exit. Cell Rep. 2018;22:3304‐3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shi J, Fung G, Piesik P, Zhang J, Luo H. Dominant‐negative function of the C‐terminal fragments of NBR1 and SQSTM1 generated during enteroviral infection. Cell Death Differ. 2014;21:1432‐1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mohamud Y, Shi J, Qu J, et al. Enteroviral infection inhibits autophagic flux via disruption of the SNARE complex to enhance viral replication. Cell Rep. 2018;22:3292‐3303. [DOI] [PubMed] [Google Scholar]
- 71. Wang C, Wong J, Fung G, et al. Dysferlin deficiency confers increased susceptibility to coxsackievirus‐induced cardiomyopathy. Cell Microbiol. 2015;17:1423‐1430. [DOI] [PubMed] [Google Scholar]
- 72. Badorff C, Lee GH, Lamphear BJ, et al. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med. 1999;5:320‐326. [DOI] [PubMed] [Google Scholar]
- 73. Seipelt J, Liebig HD, Sommergruber W, Gerner C, Kuechler E. 2A proteinase of human rhinovirus cleaves cytokeratin 8 in infected HeLa cells. J Biol Chem. 2000;275:20084‐20089. [DOI] [PubMed] [Google Scholar]
- 74. Joachims M, Harris KS, Etchison D. Poliovirus protease 3C mediates cleavage of microtubule‐associated protein 4. Virology. 1995;211:451‐461. [DOI] [PubMed] [Google Scholar]
- 75. Yalamanchili P, Weidman K, Dasgupta A. Cleavage of transcriptional activator Oct‐1 by poliovirus encoded protease 3Cpro. Virology. 1997;239:176‐185. [DOI] [PubMed] [Google Scholar]
- 76. Shiroki K, Isoyama T, Kuge S, et al. Intracellular redistribution of truncated La protein produced by poliovirus 3Cpro‐mediated cleavage. J Virol. 1999;73:2193‐2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Clark ME, Lieberman PM, Berk AJ, Dasgupta A. Direct cleavage of human TATA‐binding protein by poliovirus protease 3C in vivo and in vitro. Mol Cell Biol. 1993;13:1232‐1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kundu P, Raychaudhuri S, Tsai W, Dasgupta A. Shutoff of RNA polymerase II transcription by poliovirus involves 3C protease‐mediated cleavage of the TATA‐binding protein at an alternative site: incomplete shutoff of transcription interferes with efficient viral replication. J Virol. 2005;79:9702‐9713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Das S, Dasgupta A. Identification of the cleavage site and determinants required for poliovirus 3CPro‐catalyzed cleavage of human TATA‐binding transcription factor TBP. J Virol. 1993;67:3326‐3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Clark ME, Hämmerle T, Wimmer E, Dasgupta A. Poliovirus proteinase 3C converts an active form of transcription factor IIIC to an inactive form: a mechanism for inhibition of host cell polymerase III transcription by poliovirus. EMBO J. 1991;10:2941‐2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yalamanchili P, Datta U, Dasgupta A. Inhibition of host cell transcription by poliovirus: cleavage of transcription factor CREB by poliovirus‐encoded protease 3Cpro. J Virol. 1997;71:1220‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yalamanchili P, Harris K, Wimmer E, Dasgupta A. Inhibition of basal transcription by poliovirus: a virus‐ encoded protease (3Cpro) inhibits formation of TBP‐TATA box complex in vitro. J Virol. 1996;70:2922‐2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wong J, Zhang J, Yanagawa B, et al. Cleavage of serum response factor mediated by enteroviral protease 2A contributes to impaired cardiac function. Cell Res. 2012;22:360‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Back SH, Kim YK, Kim WJ, et al. Translation of polioviral mRNA is inhibited by cleavage of polypyrimidine tract‐binding proteins executed by polioviral 3C(pro). J Virol. 2002;76:2529‐2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jagdeo JM, Dufour A, Fung G, et al. Heterogeneous nuclear ribonucleoprotein M facilitates enterovirus infection. J Virol. 2015;89:7064‐7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Almstead LL, Sarnow P. Inhibition of U snRNP assembly by a virus‐encoded proteinase. Genes Dev. 2007;21:1086‐1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fung G, Shi J, Deng H, et al. Cytoplasmic translocation, aggregation, and cleavage of TDP‐43 by enteroviral proteases modulate viral pathogenesis. Cell Death Differ. 2015;22:2087‐2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Neznanov N, Chumakov KM, Neznanova L, Almasan A, Banerjee AK, Gudkov AV. Proteolytic cleavage of the p65‐RelA subunit of NF‐kappaB during poliovirus infection. J Biol Chem. 2005;280:24153‐24158. [DOI] [PubMed] [Google Scholar]
- 89. Wong J, Si X, Angeles A, et al. Cytoplasmic redistribution and cleavage of AUF1 during coxsackievirus infection enhance the stability of its viral genome. FASEB J. 2013;27:2777‐2787. [DOI] [PubMed] [Google Scholar]
- 90. Joachims M, Van Breugel PC, Lloyd RE. Cleavage of poly(A)‐binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J Virol. 1999;73:718‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sun D, Wang M, Wen X, et al. Cleavage of poly(A)‐binding protein by duck hepatitis A virus 3C protease. Sci Rep. 2017;7:16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhang B, Morace G, Gauss‐Müller V, Kusov Y. Poly(A) binding protein, C‐terminally truncated by the hepatitis A virus proteinase 3C, inhibits viral translation. Nucleic Acids Res. 2007;35:5975‐5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kobayashi M, Arias C, Garabedian A, Palmenberg AC, Mohr I. Site‐specific cleavage of the host poly(A) binding protein by the encephalomyocarditis virus 3C proteinase stimulates viral replication. J Virol. 2012;86:10686‐10694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Belsham GJ, McInerney GM, Ross‐Smith N. Foot‐and‐mouth disease virus 3C protease induces cleavage of translation initiation factors eIF4A and eIF4G within infected cells. J Virol. 2000;74:272‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Devaney MA, Vakharia VN, Lloyd RE, Ehrenfeld E, Grubman MJ. Leader protein of foot‐and‐mouth disease virus is required for cleavage of the p220 component of the cap‐binding protein complex. J Virol. 1988;62:4407‐4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Medina M, Domingo E, Brangwyn JK, Belsham GJ. The two species of the foot‐and‐mouth disease virus leader protein, expressed individually, exhibit the same activities. Virology. 1993;194:355‐359. [DOI] [PubMed] [Google Scholar]
- 97. Chau DHW, Zhang H, Yuan J, et al. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis. 2007;12:513‐524. [DOI] [PubMed] [Google Scholar]
- 98. Lamphear BJ, Rhoads RE. A single amino acid change in protein synthesis initiation factor 4G renders cap‐dependent translation resistant to picornaviral 2A proteases. Biochemistry. 1996;35:15726‐15733. [DOI] [PubMed] [Google Scholar]
- 99. Lamphear BJ, Kirchweger R, Skern T, Rhoads RE. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. Implications for cap‐dependent and cap‐independent translational initiation. J Biol Chem. 1995;270:21975‐21983. [DOI] [PubMed] [Google Scholar]
- 100. de Breyne S, Bonderoff JM, Chumakov KM, Lloyd RE, Hellen CUT. Cleavage of eukaryotic initiation factor eIF5B by enterovirus 3C proteases. Virology. 2008;378:118‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hanson PJ, Ye X, Qiu Y, et al. Cleavage of DAP5 by coxsackievirus B3 2A protease facilitates viral replication and enhances apoptosis by altering translation of IRES‐containing genes. Cell Death Differ. 2016;23:828‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Perera R, Daijogo S, Walter BL, Nguyen JHC, Semler BL. Cellular protein modification by poliovirus: the two faces of poly(rC)‐binding protein. J Virol. 2007;81:8919‐8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Dougherty JD, White JP, Lloyd RE. Poliovirus‐mediated disruption of cytoplasmic processing bodies. J Virol. 2011;85:64‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zaragoza C, Saura M, Padalko EY, et al. Viral protease cleavage of inhibitor of kappaBalpha triggers host cell apoptosis. Proc Natl Acad Sci USA. 2006;103:19051‐19056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Deng H, Fung G, Shi J, et al. Enhanced enteroviral infectivity via viral protease‐mediated cleavage of Grb2‐associated binder 1. FASEB J. 2015;29:4523‐4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Park N, Skern T, Gustin KE. Specific cleavage of the nuclear pore complex protein Nup62 by a viral protease. J Biol Chem. 2010;285:28796‐28805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Watters K, Palmenberg AC. Differential processing of nuclear pore complex proteins by rhinovirus 2A proteases from different species and serotypes. J Virol. 2011;85:10874‐10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Fung G, Ng CS, Zhang J, et al. Production of a dominant‐negative fragment due to G3BP1 cleavage contributes to the disruption of mitochondria‐associated protective stress granules during CVB3 infection. PLoS One. 2013;8:e79546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ng CS, Jogi M, Yoo J‐S, et al. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J Virol. 2013;87:9511‐9522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. White JP, Cardenas AM, Marissen WE, Lloyd RE. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe. 2007;2:295‐305. [DOI] [PubMed] [Google Scholar]
- 111. Ye X, Pan T, Wang D, et al. Foot‐and‐Mouth disease virus counteracts on internal ribosome entry site suppression by G3BP1 and inhibits G3BP1‐mediated stress granule assembly via post‐translational mechanisms. Front Immunol. 2018;9:1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Visser LJ, Langereis MA, Rabouw HH, et al. Essential role of enterovirus 2A protease in counteracting stress granule formation and the induction of type I interferon. J Virol. 2019;93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Visser LJ, Medina GN, Rabouw HH, et al. Foot‐and‐Mouth disease virus leader protease cleaves G3BP1 and G3BP2 and inhibits stress granule formation. J Virol. 2019;93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ullah MO, Sweet MJ, Mansell A, Kellie S, Kobe B. TRIF‐dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J Leukoc Biol. 2016;100:27‐45. [DOI] [PubMed] [Google Scholar]
- 115. Xu C, He X, Zheng Z, et al. Downregulation of microRNA miR‐526a by enterovirus inhibits RIG‐I‐dependent innate immune response. J Virol. 2014;88:11356‐11368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zhang M, et al. Regulation of IkappaB kinase‐related kinases and antiviral responses by tumor suppressor CYLD. J Biol Chem. 2008;283:18621‐18626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zeng, W et al. Reconstitution of the RIG‐I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Chan RWY, et al. Tropism of and innate immune responses to the novel human betacoronavirus lineage C virus in human ex vivo respiratory organ cultures. J Virol. 2013;87:6604‐6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Menachery VD, et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon‐stimulated gene responses. MBio. 2014;5:e01174‐e011714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zhou J, et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis. 2014;209:1331‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Channappanavar R, et al. Dysregulated type I interferon and inflammatory monocyte‐macrophage responses cause lethal pneumonia in SARS‐CoV‐infected mice. Cell Host Microbe. 2016;19:181‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Blanco‐Melo D, et al. Imbalanced Host Response to SARS‐CoV‐2 Drives Development of COVID‐19. Cell. 2020;181:1036‐1045 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lokugamage KG, Hage A, Vries MD, et al. Type I interferon susceptibility distinguishes SARS‐CoV‐2 from SARS‐CoV. J Virol. 2020;94(23):e01410–e01420. 10.1128/JVI.01410-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Clementz MA, et al. Deubiquitinating and interferon antagonism activities of coronavirus papain‐like proteases. J Virol. 2010;84:4619‐4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Matthews K, Schäfer A, Pham A, Frieman M. The SARS coronavirus papain like protease can inhibit IRF3 at a post activation step that requires deubiquitination activity. Virol J. 2014;11:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kopecky‐Bromberg SA, Martínez‐Sobrido L, Frieman M, Baric RA, Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007;81:548‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Siu K‐L, et al. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem. 2009;284:16202‐16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Onomoto K, et al. Critical role of an antiviral stress granule containing RIG‐I and PKR in viral detection and innate immunity. PLoS One. 2012;7:e43031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kim SS‐Y, Sze L, Liu C, Lam K‐P. The stress granule protein G3BP1 binds viral dsRNA and RIG‐I to enhance interferon‐β response. J Biol Chem. 2019;294:6430‐6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kuniyoshi K, et al. Pivotal role of RNA‐binding E3 ubiquitin ligase MEX3C in RIG‐I‐mediated antiviral innate immunity. Proc Natl Acad Sci USA. 2014;111:5646‐5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Narita R, et al. A novel function of human Pumilio proteins in cytoplasmic sensing of viral infection. PLoS Pathog. 2014;10:e1004417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Yoo J‐S, et al. DHX36 enhances RIG‐I signaling by facilitating PKR‐mediated antiviral stress granule formation. PLoS Pathog. 2014;10:e1004012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Cheng J, Gao S, Zhu C , et al. Typical stress granule proteins interact with the 3’ untranslated region of enterovirus D68 to inhibit viral replication. J Virol. 2020;94(7):e02041–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Rabouw HH, et al. Middle East respiratory coronavirus accessory protein 4a inhibits PKR‐mediated antiviral stress responses. PLoS Pathog. 2016;12:e1005982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Nakagawa K, Narayanan K, Wada M, Makino S. Inhibition of stress granule formation by middle east respiratory syndrome coronavirus 4a accessory protein facilitates viral translation, leading to efficient virus replication. J Virol. 2018;92(20):e00902–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Icardi L, et al. Opposed regulation of type I IFN‐induced STAT3 and ISGF3 transcriptional activities by histone deacetylases (HDACS) 1 and 2. FASEB J. 2012;26:240‐249. [DOI] [PubMed] [Google Scholar]
- 137. Klampfer L, Huang J, Swaby L‐A, Augenlicht L. Requirement of histone deacetylase activity for signaling by STAT1. J Biol Chem. 2004;279:30358‐30368. [DOI] [PubMed] [Google Scholar]
- 138. Reineke LC, Lloyd RE. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J Virol. 2015;89:2575‐2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Chen N, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zhou F, et al. Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054‐1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hyun J, Kanagavelu S, Fukata M. A unique host defense pathway: TRIF mediates both antiviral and antibacterial immune responses. Microbes Infect. 2013;15:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Hu S, et al. PKR‐dependent cytosolic cGAS foci are necessary for intracellular DNA sensing. Sci Signal. 2019;12. [DOI] [PubMed] [Google Scholar]
- 143. Liu Z‐S, et al. G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol. 2019;20:18‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Li X‐D, et al. Pivotal roles of cGAS‐cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341:1390‐1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Zhao C, Zhao W. NLRP3 inflammasome‐A key player in antiviral responses. Front Immunol. 2020;11:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Shi J, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660‐665. [DOI] [PubMed] [Google Scholar]
- 147. Liu X, et al. Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Wang X, et al. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1‐RIP3‐DRP1 signaling pathway. Nat Immunol. 2014;15:1126‐1133. [DOI] [PubMed] [Google Scholar]
- 149. Negash AA, et al. IL‐1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013;9:e1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Tschöpe C, Muller I, Xia Y, et al. NOD2 (nucleotide‐binding oligomerization domain 2) is a major pathogenic mediator of coxsackievirus B3‐induced myocarditis. Circ Heart Fail; 2017;10(9):e003870. [DOI] [PubMed] [Google Scholar]
- 151. Wang Y, Gao B, Xiong S. Involvement of NLRP3 inflammasome in CVB3‐induced viral myocarditis. Am J Physiol Heart Circ Physiol. 2014;307:H1438‐H1447. [DOI] [PubMed] [Google Scholar]
- 152. Chen G, Wu D, Guo W, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130:2620‐2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Siu K‐L, Yuen K‐S, Castaño‐Rodriguez C, et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3‐dependent ubiquitination of ASC. FASEB J. 2019;33:8865‐8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Shi C‐S, Nabar NR, Huang N‐N, Kehrl JH. SARS‐Coronavirus Open Reading Frame‐8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019;5:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Nieto‐Torres JL, Verdiá‐Báguena C, Jimenez‐Guardeño JM, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485;330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Rodrigues TS, Sa KS, Ishimoto AY, et al. Inflammasome activation in COVID‐19 patients. medRxiv. 2020. 10.1101/2020.08.05.20168872. [DOI] [Google Scholar]
- 157. Tsai W‐C, Lloyd RE. Cytoplasmic RNA granules and viral infection. Annu Rev Virol. 2014;1:147‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Abernathy E, Glaunsinger B. Emerging roles for RNA degradation in viral replication and antiviral defense. Virology. 2015:479‐480;600‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ng CS, Kasumba DM, Fujita T, Luo H. Spatio‐temporal characterization of the antiviral activity of the XRN1‐DCP1/2 aggregation against cytoplasmic RNA viruses to prevent cell death. Cell Death Differ. 2020;27:2363‐2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Cencic R, Desforges M, Hall DR, et al. Blocking eIF4E‐eIF4G interaction as a strategy to impair coronavirus replication. J Virol. 2011;85:6381‐6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Cencic R, Hall DR, Robert F, et al. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc Natl Acad Sci USA. 2011;108:1046‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. Embo J. 2017;36:1811‐1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14:243‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kim B‐W, Kwon DH, Song HK. Structure biology of selective autophagy receptors. BMB Rep. 2016;49:73‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Mohamud Y, Luo H. The intertwined life cycles of enterovirus and autophagy. Virulence. 2019;10:470‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Nakamura S, Yoshimori T. New insights into autophagosome‐lysosome fusion. J Cell Sci. 2017;130:1209‐1216. [DOI] [PubMed] [Google Scholar]
- 169. Gassen NC, Papies J, Bajaj T, et al. Analysis of SARS‐CoV‐2‐controlled autophagy reveals spermidine, MK‐2206, and niclosamide as putative antiviral therapeutics. BioRxiv. 2020. 10.1101/2020.04.15.997254. [DOI] [Google Scholar]
- 170. Gassen NC, Niemeyer D, Muth D, et al. SKP2 attenuates autophagy through Beclin1‐ubiquitination and its inhibition reduces MERS‐Coronavirus infection. Nat Commun. 2019;10:5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Chen X, Wang K, Xing Y, et al. Coronavirus membrane‐associated papain‐like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell. 2014;5:912‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Kleifeld O, Doucet A, Prudova A, et al. Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat Protoc. 2011;6:1578‐1611. [DOI] [PubMed] [Google Scholar]
- 173. Schilling O, Overall CM. Proteome‐derived, database‐searchable peptide libraries for identifying protease cleavage sites. Nat Biotechnol. 2008;26:685‐694. [DOI] [PubMed] [Google Scholar]
- 174. Schilling O, Huesgen PF, Barré O. Auf dem Keller U, Overall C M. Characterization of the prime and non‐prime active site specificities of proteases by proteome‐derived peptide libraries and tandem mass spectrometry. Nat Protoc. 2011;6:111‐120. [DOI] [PubMed] [Google Scholar]
- 175. Kleifeld O, Doucet A, auf dem Keller U, et al. Isotopic labeling of terminal amines in complex samples identifies protein N‐termini and protease cleavage products. Nat Biotechnol. 2010;28:281‐288. [DOI] [PubMed] [Google Scholar]
- 176. Huesgen PF, Overall CM. N‐ and C‐terminal degradomics: new approaches to reveal biological roles for plant proteases from substrate identification. Physiol Plant. 2012;145:5‐17. [DOI] [PubMed] [Google Scholar]
- 177. Klein T, Fung S‐Y, Renner F, et al. The paracaspase MALT1 cleaves HOIL1 reducing linear ubiquitination by LUBAC to dampen lymphocyte NF‐κB signalling. Nat Commun. 2015;6:8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Eckhard U, Bandukwala H, Mansfield MJ, et al. Discovery of a proteolytic flagellin family in diverse bacterial phyla that assembles enzymatically active flagella. Nat Commun. 2017;8:521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung S‐H. An overview of severe acute respiratory syndrome‐coronavirus (SARS‐CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem. 2016;59:6595‐6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Lu G, Qi J, Chen Z, et al. Enterovirus 71 and coxsackievirus A16 3C proteases: binding to rupintrivir and their substrates and anti‐hand, foot, and mouth disease virus drug design. J Virol. 2011;85:10319‐10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Binford SL, Maldonado F, Brothers MA, et al. Conservation of amino acids in human rhinovirus 3C protease correlates with broad‐spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob Agents Chemother. 2005;49:619‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Tan J, George S, Kusov Y, et al. 3C protease of enterovirus 68: structure‐based design of Michael acceptor inhibitors and their broad‐spectrum antiviral effects against picornaviruses. J Virol. 2013;87:4339‐4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. van der Linden L, Ulferts R, Nabuurs SB, et al. Application of a cell‐based protease assay for testing inhibitors of picornavirus 3C proteases. Antiviral Res. 2014;103:17‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Yang H, Xie W, Xue X, et al. Design of wide‐spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Hayden FG, Turner RB, Gwaltney JM, et al. Phase II, randomized, double‐blind, placebo‐controlled studies of ruprintrivir nasal spray 2‐percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob Agents Chemother. 2003;47:3907‐3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Grein J, Ohmagari N, Shin D, et al. Compassionate Use of Remdesivir for Patients with Severe Covid‐19. N Engl J Med. 2020;382(24):2327–2336. 10.1056/nejmoa2007016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Touret F, de Lamballerie X. Of chloroquine and COVID‐19. Antiviral Res. 2020;177:104762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Research. 2020;30(3):269–271. 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Hoffmann M, Mösbauer K, Hofmann‐Winkler H, et al. Chloroquine does not inhibit infection of human lung cells with SARS‐CoV‐2. Nature. 2020;585(7826):588–590. 10.1038/s41586-020-2575-3. [DOI] [PubMed] [Google Scholar]
- 190. Chorin E, Rozenbaum Z, Topilsky Y, et al. Tricuspid regurgitation and long‐term clinical outcomes. European Heart Journal ‐ Cardiovascular Imaging. 2019;21:157–165. 10.1093/ehjci/jez216. [DOI] [PubMed] [Google Scholar]
- 191. Yang S, Chen S‐J, Hsu M‐F, et al. Synthesis, crystal structure, structure‐activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J Med Chem. 2006;49:4971‐4980. [DOI] [PubMed] [Google Scholar]
- 192. Jo S, Kim S, Shin DH, Kim M‐S. Inhibition of SARS‐CoV 3CL protease by flavonoids. J Enzyme Inhib Med Chem. 2020;35:145‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Ramajayam R, Tan K‐P, Liu H‐G, Liang P‐H. Synthesis and evaluation of pyrazolone compounds as SARS‐coronavirus 3C‐like protease inhibitors. Bioorg Med Chem. 2010;18:7849‐7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Zhang J, Huitema C, Niu C, et al. Aryl methylene ketones and fluorinated methylene ketones as reversible inhibitors for severe acute respiratory syndrome (SARS) 3C‐like proteinase. Bioorg Chem. 2008;36:229‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Blanchard JE, Elowe NH, Huitema C, et al. High‐throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem. Biol. 2004;11:1445‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Wu C‐Y, King K‐Y, Kuo C‐J, et al. Stable benzotriazole esters as mechanism‐based inactivators of the severe acute respiratory syndrome 3CL protease. Chem. Biol. 2006;13:261‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Verschueren KHG, Pumpor K, Anemüller S, Chen S, Mesters JR, Hilgenfeld R. A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem. Biol. 2008;15:597‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Bacha U, Barrila J, Velazquez‐Campoy A, Leavitt SA, Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry. 2004;43:4906‐4912. [DOI] [PubMed] [Google Scholar]
- 199. Lee T‐W, Cherney MM, Liu J, et al. Crystal structures reveal an induced‐fit binding of a substrate‐like Aza‐peptide epoxide to SARS coronavirus main peptidase. J Mol Biol. 2007;366:916‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Lee T‐W, Cherney MM, Huitema C, et al. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate‐like aza‐peptide epoxide. J Mol Biol. 2005;353:1137‐1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Asgian JL, James KE, Li ZZ, et al. Aza‐peptide epoxides: a new class of inhibitors selective for clan CD cysteine proteases. J Med Chem. 2002;45:4958‐4960. [DOI] [PubMed] [Google Scholar]
- 202. Kaeppler U, Stiefl N, Schiller M, et al. A new lead for nonpeptidic active‐site‐directed inhibitors of the severe acute respiratory syndrome coronavirus main protease discovered by a combination of screening and docking methods. J Med Chem. 2005;48:6832‐6842. [DOI] [PubMed] [Google Scholar]
- 203. Kaeppler U, Schirmeister T. New non‐peptidic inhibitors of papain derived from etacrynic acid. Med Chem. 2005;1:361‐370. [DOI] [PubMed] [Google Scholar]
- 204. Shao Y‐M, Yang W‐B, Peng H‐P, et al. Structure‐based design and synthesis of highly potent SARS‐CoV 3CL protease inhibitors. Chembiochem. 2007;8:1654‐1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Tsai K‐C, Chen S‐Y, Liang P‐H, et al. Discovery of a novel family of SARS‐CoV protease inhibitors by virtual screening and 3D‐QSAR studies. J Med Chem. 2006;49:3485‐3495. [DOI] [PubMed] [Google Scholar]
- 206. Lu I‐L, Mahindroo N, Liang P‐H, et al. Structure‐based drug design and structural biology study of novel nonpeptide inhibitors of severe acute respiratory syndrome coronavirus main protease. J Med Chem. 2006;49:5154‐5161. [DOI] [PubMed] [Google Scholar]
- 207. Ghosh AK, Xi K, Ratia K, et al. Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin‐like protease inhibitors. J Med Chem. 2005;48:6767‐6771. [DOI] [PubMed] [Google Scholar]
- 208. Shie J‐J, Fang J‐M, Kuo T‐H, et al. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic alpha,beta‐unsaturated esters. Bioorg Med Chem. 2005;13:5240‐5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Ryu YB, Jeong HJ, Kim JH, et al. Biflavonoids from Torreya nucifera displaying SARS‐CoV 3CL(pro) inhibition. Bioorg Med Chem. 2010;18:7940‐7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Wen C‐C, Kuo Y‐H, Jan J‐T, et al. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J Med Chem. 2007;50:4087‐4095. [DOI] [PubMed] [Google Scholar]
- 211. Ryu YB, Park S‐J, Kim YM, et al. SARS‐CoV 3CLpro inhibitory effects of quinone‐methide triterpenes from Tripterygium regelii. Bioorg Med Chem Lett. 2010;20:1873‐1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Lee C‐C, Kuo C‐J, Hsu M‐F, et al. Structural basis of mercury‐ and zinc‐conjugated complexes as SARS‐CoV 3C‐like protease inhibitors. FEBS Lett. 2007;581:5454‐5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Hsu JT‐A, Kuo C‐J, Hsieh H‐P, et al. Evaluation of metal‐conjugated compounds as inhibitors of 3CL protease of SARS‐CoV. FEBS Lett. 2004;574:116‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Zhang L, Lin D, Kusov Y, et al. α‐Ketoamides as broad‐spectrum inhibitors of coronavirus and enterovirus replication: structure‐based design, synthesis, and activity assessment. J Med Chem. 2020;63:4562‐4578. [DOI] [PubMed] [Google Scholar]
- 215. Jacobs J, Grum‐Tokars V, Zhou Y, et al. Discovery, synthesis, and structure‐based optimization of a series of N‐(tert‐butyl)‐2‐(N‐arylamido)‐2‐(pyridin‐3‐yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS‐CoV) 3CL protease. J Med Chem. 2013;56:534‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Turlington M, et al. Discovery of N‐(benzo[1,2,3]triazol‐1‐yl)‐N‐(benzyl)acetamido)phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS‐CoV) 3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced‐fit binding. Bioorg Med Chem Lett. 2013;23:6172‐6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Barretto N, et al. The papain‐like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol. 2005;79:15189‐15198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Dragovich PS, et al. Structure‐based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as L‐glutamine replacements. J Med Chem. 1999;42:1213‐1224. [DOI] [PubMed] [Google Scholar]
- 219. Yuan S, et al. Structure of the HRV‐C 3C‐rupintrivir complex provides new insights for inhibitor design. Virol Sin. 2020;35:445–454. 10.1007/s12250-020-00196-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Ma C, et al. Boceprevir, GC‐376, and calpain inhibitors II, XII inhibit SARS‐CoV‐2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Kuo C‐J, Shie J‐J, Fang J‐J, et al. Design, synthesis, and evaluation of 3C protease inhibitors as anti‐enterovirus 71 agents. Bioorg Med Chem. 2008;16:7388‐7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Morse JS, Lalonde T, Xu S, Liu WR. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019‐nCoV. Chembiochem. 2020;21:730‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Liu W, Zhu H‐M, Niu G‐J, et al. Synthesis, modification and docking studies of 5‐sulfonyl isatin derivatives as SARS‐CoV 3C‐like protease inhibitors. Bioorg Med Chem. 2014;22:292‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Chen L‐R, Wang Y‐C, Lin YW, et al. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg Med Chem Lett. 2005;15:3058‐3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Shie J‐J, Fang J‐M, Kuo C‐J, et al. Discovery of potent anilide inhibitors against the severe acute respiratory syndrome 3CL protease. J Med Chem. 2005;48:4469‐4473. [DOI] [PubMed] [Google Scholar]
- 226. Sang P, Tian S, Meng Z, Yang L. Insight derived from molecular docking and molecular dynamics simulations into the binding interactions between HIV‐1 protease inhibitors and SARS‐CoV‐2 3CLpro. 2020. 10.26434/chemrxiv.11932995.v1. [DOI]
- 227. Shimamoto Y, Hattori Y, Kobayashi K, et al. Fused‐ring structure of decahydroisoquinolin as a novel scaffold for SARS 3CL protease inhibitors. Bioorg Med Chem. 2015;23:876‐890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Zhang H‐Z, Zhang H, Kemnitzer W, et al. Design and synthesis of dipeptidyl glutaminyl fluoromethyl ketones as potent severe acute respiratory syndrome coronovirus (SARS‐CoV) inhibitors. J Med Chem. 2006;49:1198‐1201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.