

Abstract
Wnt/β‐catenin signaling is frequently activated in advanced prostate cancer and contributes to therapy resistance and metastasis. However, activating mutations in the Wnt/β‐catenin pathway are not common in prostate cancer, suggesting alternative regulations may exist. Here, we report that the expression of endothelial cell‐specific molecule 1 (ESM1), a secretory proteoglycan, is positively associated with prostate cancer stemness and progression by promoting Wnt/β‐catenin signaling. Elevated ESM1 expression correlates with poor overall survival and metastasis. Accumulation of nuclear ESM1, instead of cytosolic or secretory ESM1, supports prostate cancer stemness by interacting with the ARM domain of β‐catenin to stabilize β‐catenin–TCF4 complex and facilitate the transactivation of Wnt/β‐catenin signaling targets. Accordingly, activated β‐catenin in turn mediates the nuclear entry of ESM1. Our results establish the significance of mislocalized ESM1 in driving metastasis in prostate cancer by coordinating the Wnt/β‐catenin pathway, with implications for its potential use as a diagnostic or prognostic biomarker and as a candidate therapeutic target in prostate cancer.
Keywords: cancer stemness, tumor metastasis, Wnt‐β‐catenin
Subject Categories: Cancer, Signal Transduction, Regenerative Medicine
Mislocalised secretory proteoglycan ESM1 stabilizes β‐catenin/TCF4 complexes to enhance Wnt activation in cancer stem cells.
Introduction
Although different strategies of androgen deprivation therapies (ADT) have been developed for advanced prostate cancer (PCa), the disease eventually progresses to castration‐resistant PCa (CRPC). In this stage, the mean survival period of PCa patients is only 16–18 month and 90% of them develop distant metastases (Cookson, Roth et al, 2013). During long‐term ADT treatment, PCa develops mechanisms to bypass its dependency on androgen/androgen receptor (AR) signaling. The activation of Wnt/β‐catenin signaling has been found to participate in neuroendocrine differentiation (Yu, Wang et al, 2011; Ciarlo, Benelli et al, 2012), induce epithelial to mesenchymal transition (Liu, Yin et al, 2015), and promote cancer stem cell (CSC) renewal and expansion in PCa (Bisson & Prowse, 2009; Zhang, Guo et al, 2017), all of which contribute to overriding androgen/AR signaling (Yeh, Guo et al, 2019). Increasing evidence also indicates that Wnt/β‐catenin signaling is highly activated in late‐stage or metastatic PCa (Chen, Shukeir et al, 2004; Patel, Brzezinska et al, 2020). However, although nuclear β‐catenin can frequently be observed in metastatic and hormone‐refractory PCa tissues (Chen et al, 2004; Wan, Liu et al, 2012), activating mutations of Wnt/β‐catenin signaling can only be found in 12–22% of advanced PCa (Grasso, Wu et al, 2012; Beltran, Yelensky et al, 2013, Robinson et al., 2015). Moreover, no β‐catenin mutations can be detected in established metastatic PCa cells (Voeller, Truica et al, 1998). Even in tumors carrying β‐catenin activating mutations, accumulation and nuclear localization of β‐catenin is distributed in a heterogeneous fashion (Chesire, Ewing et al, 2000). In some PCa specimens, for example, nuclear β‐catenin can only be observed in the invasive front (Chen et al, 2004). These observations indicate that other aberrant factors might perturb this pathway in PCa.
Endothelial cell‐specific molecule‐1 (ESM‐1), also known as endocan, not only is regarded as a marker of angiogenesis, but it also plays an important role in endothelium‐dependent pathological disorders and inflammatory reactions (Rocha, Schiller et al, 2014). As a key factor in vascular development, neogenesis, and angiogenesis, ESM1 is secreted by vascular endothelial cells and its expression is regulated by a series of cytokines or cell factors. A variety of inflammatory mediators, such as interleukin‐1 and tumor necrosis factor‐α, promote endocan overexpression (Lassalle, Molet et al, 1996; Kali & Shetty, 2014). Apart from angiogenesis, ESM1 is reported to regulate tumor progression (Roudnicky, Poyet et al, 2013). Several studies have shown that ESM1 is dramatically overexpressed in many cancer types including non‐small‐cell lung cancer, clear cell renal cell carcinoma, and ovarian cancer (Grigoriu, Depontieu et al, 2006; Leroy, Aubert et al, 2010; Laloglu, Kumtepe et al, 2017; Kim, Lee et al, 2018). Moreover, ESM1 significantly affects cell proliferation and migration in colorectal, gastric, and hepatocellular carcinomas (Liu, Zhang et al, 2010; Kang, Ji et al, 2012). It appears to be the connecting molecule that regulates both angiogenesis and tumor progression (Kali & Shetty, 2014; Yang, Yang et al, 2015). However, the expression levels and functions of ESM1 in tumor cells are controversial and yet not fully investigated.
In this study, we demonstrate the occurrence of aberrant subcellular localization of ESM1 and provide evidence that this phenomenon may contribute to aggressive features in PCa. Unlike its original characterization as a secretory protein, we observed that ESM1 was predominantly expressed in the nucleus. Moreover, this nuclear ESM1 activated β‐catenin by inducing the interaction between β‐catenin and its coregulator TCF4, thereby promoting cancer stemness, drug resistance, and further tumor metastasis. Our study also shows that β‐catenin directly interacts with ESM1 through association with its armadillo repeat domain. Examination of ESM1 and β‐catenin demonstrates that ESM1 is a novel coregulator of β‐catenin. Our study not only explains how ESM1 induces tumor metastasis but also provides a possible therapeutic target for advanced PCa.
Results
ESM1 is required for the process of metastasis in PCa
Our previous PRECOG analysis had found that ESM1 has prognostic potential in different types of cancer, and PCa was identified among the top‐ranked cancer types (Yang, Pan et al, 2020). To evaluate the clinical characteristics of ESM1, we extended our analysis to other online published clinical databases for ESM1 mRNA expression profiles. The prognostic value of ESM1 was also confirmed in three independent clinical databases (Appendix Fig S1A–C). The expression of ESM1 mRNA was significantly higher in metastatic PCa tissues compared with benign prostate and primary PCa tissues (Fig 1A, Appendix Fig S1D and E). To further determine whether the upregulation of ESM1 was correlated with metastasis, we analyzed ESM1 expression levels in two series of PCa cell lines that recapitulate the properties of primary and metastatic PCa cells (Kozlowski, Fidler et al, 1984; Tsai, Tzeng et al, 2016). As can be seen, both mRNA and protein levels of ESM1 were higher in 22Rv1‐M and PC3‐M cells, two highly metastatic cell lines, compared with their parental cell lines, 22Rv1‐P and PC3‐P, respectively (Fig 1B). These results indicate that there might be a link between ESM1 expression and PCa metastasis. Since the invasion ability of these pairs of cell models has been reported to reflect their metastatic potential (Kozlowski et al, 1984; Tsai et al, 2016), Transwell invasion assays were conducted. Stable ESM1 knockdown, either by shRNAs or by locked nucleic acid (LNA), led to a significant decrease in the ability of cells to invade through an extracellular matrix coating (Figs 1C and EV1, EV2, EV3, EV4, EV5). Similar results were also obtained when we performed an in vivo orthotopic metastasis mouse model. The tumor weights were significantly lower in ESM1‐silencing xenografts compared with the control xenografts. Moreover, metastases in the lung, liver, intestine, and kidney were also fewer in ESM1‐silencing groups compared with the shScramble group (Fig 1D, Appendix Fig S1F). In contrast, overexpressing ESM1 promoted PCa cell invasion and tumor metastasis (Fig EV1, EV2, EV3, EV4, EV5, Appendix Fig S1G). Taken together, these data suggest that ESM1 may play an important role in the metastatic process of PCa.
Figure 1. ESM1 is required for the process of metastasis in PCa.
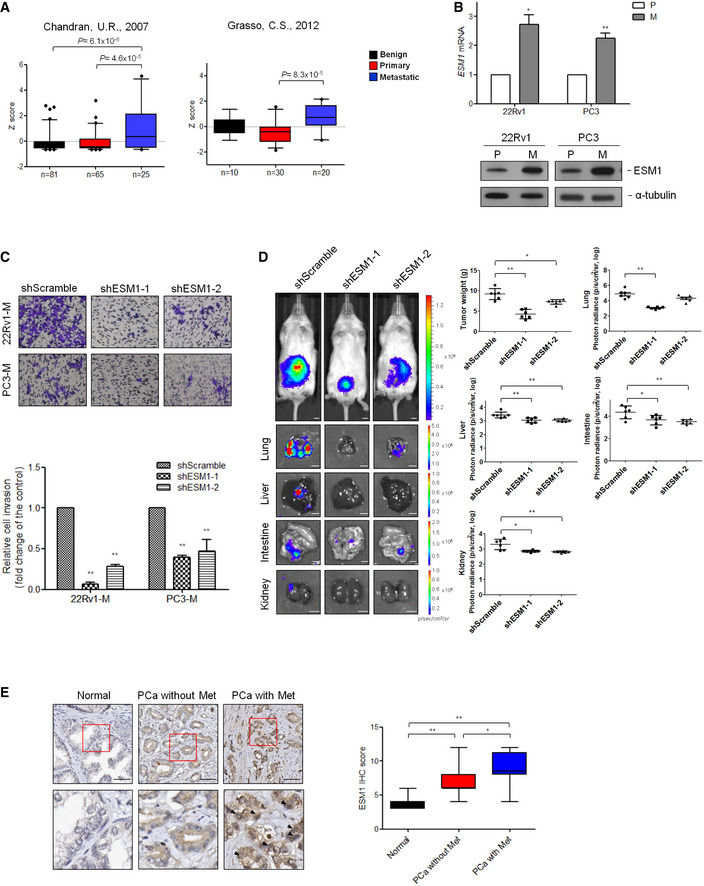
- Graph derived from two published data available in the PubMed GEO database (GSE6919 and GSE35988). The box charts depict the relative expression of ESM1 in benign, primary, and metastatic prostate cancer patients. The center line donates the median value while the box contains the 25th–75th percentiles of dataset. The whiskers mark the 5th and 95th percentiles, and values beyond these upper and lower bounds are considered outliers. P‐value was obtained by two‐tailed Student’s t‐test.
- Relative ESM1 mRNA expression and ESM1 protein expression in two sets of human metastatic PCa cell lines. P: parental cells, M: metastatic cells.
- Cell invasion assays of 22Rv1‐M and PC3‐M cells transfected with either Scramble‐shRNA or ESM1‐shRNA. The data are shown as the relative fold change of invasive cells compared with the shScramble group. Scale bar: 100 μm.
- Representative images of lungs, livers, intestines, and kidneys from mice 8 weeks after orthotopical injection of 22Rv1‐M cells transfected with either Scramble‐shRNA or ESM1‐shRNA. Scale bar: 0.5 cm. Tumors were removed and weighed. Quantitative summary of tumor luminescence in different metastases measured in photons/second is shown.
- Formalin‐fixed, paraffin‐embedded tissue microarray sections of 59 normal prostate tissues, and 65 prostate adenocarcinoma tissues with and without metastasis (n = 16 and n = 49, respectively) were stained with an anti‐ESM1 antibody. Tissue‐bound ESM1 is shown in brown. Scale bar: 50 μm. Arrowheads indicate positively stained nucleus of cancer cells. Plot representation of scores according to immunohistochemical expression of ESM1 in normal prostate tissue related to the prostate adenocarcinoma tissue with and without metastasis. The scores are calculated by intensity (score 1–3) × percentage (score 1–4) of stained cells. The line in the middle of the box is plotted at the median. The box extends from the 25th to 75th percentiles. Whiskers above and below the box indicate the 5th and 95th percentiles. P‐value was obtained by two‐tailed Student’s t‐test.
Data information: Differences in (B) and (C) are shown as fold change compared with control cells presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 when compared to control group by two‐tailed Student’s t‐test. Source data are available online for this figure.
Figure EV1. Manipulating ESM1 expression is correlated with cell invasion and tumor metastasis (related to Fig 2).
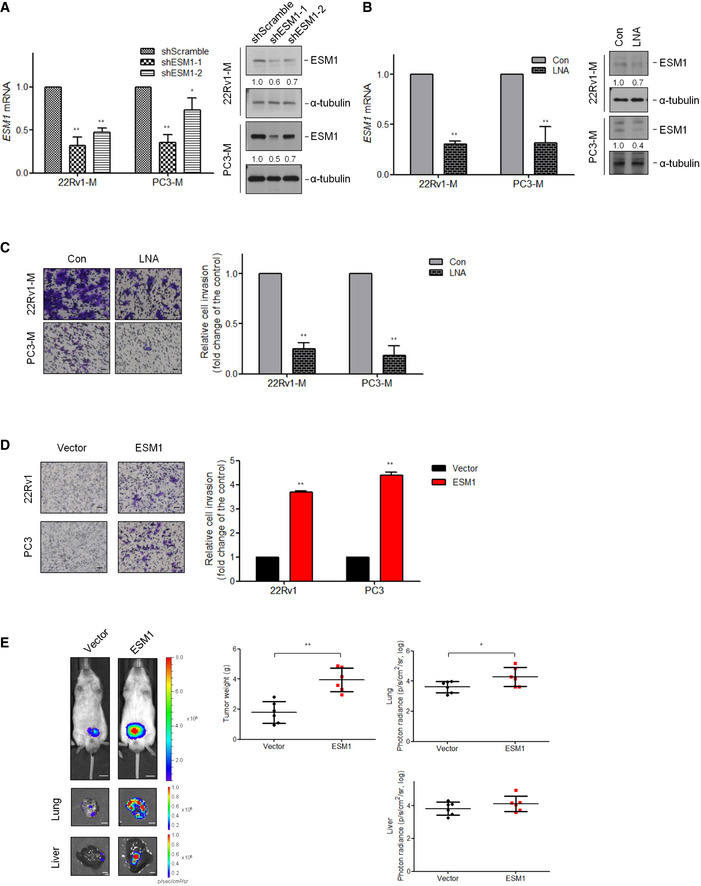
- Relative ESM1 mRNA expression (left) and ESM1 protein expression (right) in human metastatic PCa cell lines‐22Rv1‐M and PC3‐M transfected with either Scramble‐shRNA or ESM1‐shRNA. Differences in mRNA levels compared with shScramble cells are shown as fold changes presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 when compared to shScramble cells by two‐tailed Student’s t‐test. The normalized signal intensity of the target band is shown below the blots as fold changes compared with shScramble group.
- Relative ESM1 mRNA expression (left) and ESM1 protein expression (right) in human metastatic PCa cells treated with either control or ESM1‐locked nucleic acid (LNA). Bars are the mean ± SD of three independent experiments. **P < 0.01 when compared to control group by two‐tailed Student’s t‐test. The normalized signal intensity of the target band is shown below the blots as fold changes compared with control group.
- Left, representative views of cells in the Transwell invasion assay. Scale bar: 100 μm. Right, quantification of cells that invaded through a Matrigel‐coated membrane following treatment with control or ESM1‐LNA. Bars are the mean ± SD of three independent experiments. **P < 0.01 when compared to control group by two‐tailed Student’s t‐test.
- Left, representative views of cells in the Transwell invasion assay. Scale bar: 100 μm. Right, statistical data regarding invasive ability of ESM1‐overexpressing cells. Bars are the mean ± SD of three independent experiments. **P < 0.01 when compared to vector cells by two‐tailed Student’s t‐test.
- Representative images of lungs and livers from mice 8 weeks after orthotopical injection of 22Rv1‐P cells transfected with either vector control or ESM1. Scale bar: 0.5 cm. Tumors were removed and weighed. Quantitative summary of tumor luminescence in photons per second of different metastases. *P < 0.05, **P < 0.01 when compared to vector cells by two‐tailed Student’s t‐test and error bars represent the standard deviation of six independent experiments.
Source data are available online for this figure.
Figure EV2. Characteristics of CSCs are elevated in metastatic PCa cells (related to Fig 3).
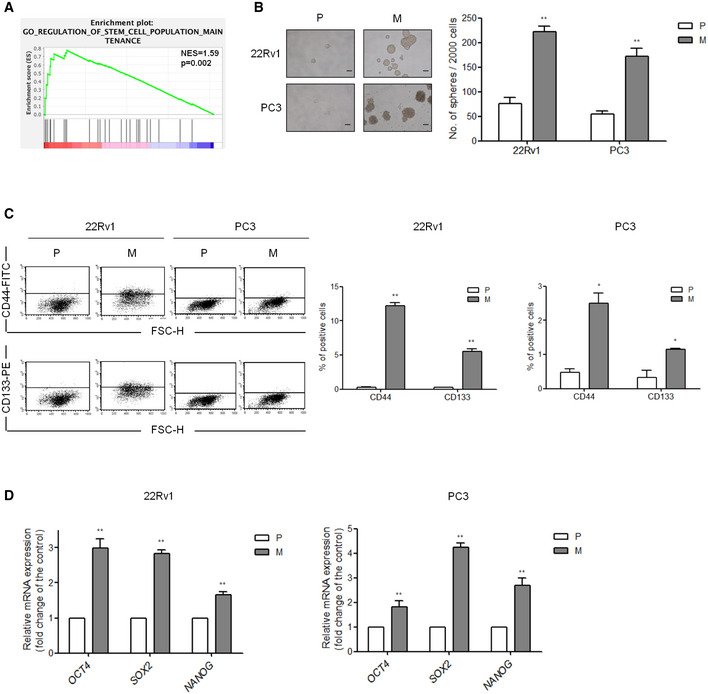
- GSEA demonstrating the enrichment of gene sets related to “regulation of stem cell population maintenance” signatures in the ranked gene list of ESM1‐high vs. ESM1‐low PCa patients from the TCGA database. NES, normalized enrichment score. The P‐values for the GSEA test statistics are calculated by permutation. The original test statistics for the features are permuted, and new test statistics are calculated for each category, based on the permuted feature test statistics.
- Spheroid formation in 22Rv1‐P, 22Rv1‐M, PC3‐P, and PC3‐M cells. P: parental cells, M: metastatic cells. Left, spheroids formed by these four cell lines. Scale bar: 100 μm. Right, total number of spheroid cells was calculated at day 7 in PC3‐P and PC3‐M cells and at day 14 in 22Rv1‐P and 22Rv1‐M cells were calculated. Quantitative data comparing the average number of spheres formed are presented as the mean ± SD of three independent experiments. **P < 0.01 when compared to parental cells by two‐tailed Student’s t‐test.
- Flow cytometry analysis of the ratio of CD44+ and CD133+ cells in 22Rv1‐P/22Rv1‐M and PC3‐P/PC3‐M pairs. Multiples of difference compared with parental cells are presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 when compared to parental cells by two‐tailed Student’s t‐test.
- mRNA expression of PCa stem cell markers, Oct4, Sox2, and Nanog in two sets of human metastatic PCa cell lines. Left, statistical analysis of CSC markers in 22Rv1‐P/22Rv1‐M pairs. Right, statistical analysis of CSC markers in PC3‐P/PC3‐M pairs. Differences in mRNA levels compared with parental cells are shown as fold changes presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 when compared to parental cells by two‐tailed Student’s t‐test.
Figure EV3. Manipulating ESM1 expression is correlated with spheroid formation (related to Fig 3).
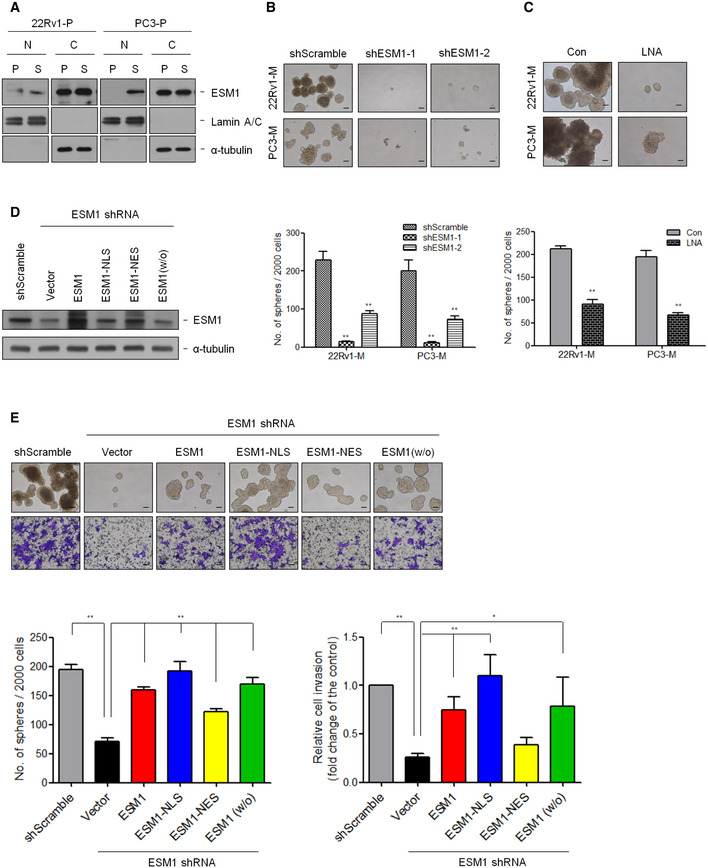
- Nuclear (N) and cytosolic (C) extracts of adherent (P) and spheroid (S) cells in 22Rv1‐P and PC3‐P cells were prepared, and the levels of indicated proteins were detected by immunoblotting.
- Representative images of tumorspheres are shown. Spheres were cultured for 14 days in 22Rv1‐M cells and 7 days in PC3‐M cells before counting. Scale bar: 100 μm. Quantitative data comparing the average number of spheres formed are presented as the mean ± SD of three independent experiments. **P < 0.01 when compared to shScramble cells by two‐tailed Student’s t‐test.
- Representative images of tumorspheres are shown. 22Rv1‐M and PC3‐M cells were treated with or without ESM1‐LNA, and spheroid formation assay was performed. Histogram shows the mean number of spheres cultured as the mean ± SD. Scale bar: 100 μm. **P < 0.01 when compared to control group by two‐tailed Student’s t‐test and n = 3 biologically independent samples per group.
- 22Rv1‐M cells with stable ESM1 knockdown were transfected with different ESM1‐expression pattern constructs, and immunoblotting analysis was performed.
- Cell invasion and spheroid formation assays were performed in the 22Rv1‐M cells with different ESM1 expression patterns. Scale bar: 100 μm. Bars are the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 when compared to shScramble or shESM1‐vector cells by two‐tailed Student’s t‐test.
Source data are available online for this figure.
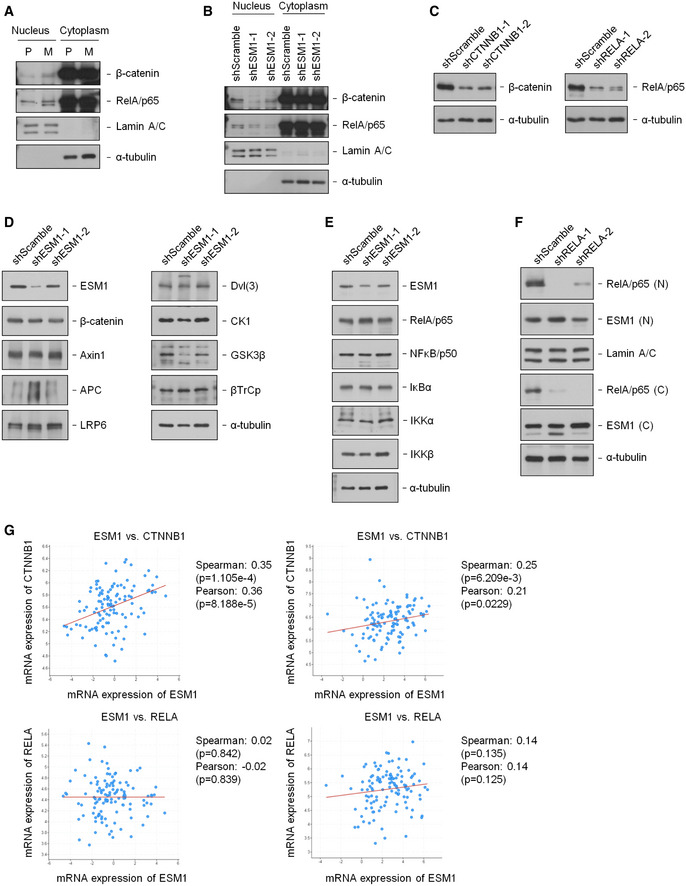
Figure EV4. ESM1 mainly induces nuclear translocation of β‐catenin (related to Fig 5).
-
ANuclear and cytosolic extracts of 22Rv1‐P and 22Rv1‐M cells were prepared, and the levels of indicated proteins were detected by immunoblotting.
-
BNuclear and cytosolic extracts of 22Rv1‐M cells transfected with either Scramble‐shRNA or ESM1‐shRNA were prepared, and the levels of indicated proteins were detected by immunoblotting.
-
CSuppression of β‐catenin or NFκB by shRNA was analyzed.
-
D, EImmunoblotting analysis of 22Rv1‐M cells transfected with either shScramble or shESM1 was performed with the antibodies indicated.
-
FNuclear and cytosolic extracts of 22Rv1‐M cells transfected with either shScramble or shRELA were prepared, and the levels of indicated proteins were detected by immunoblotting.
-
GGraph derived from co‐expression analysis of published data available in the cBioPortal (EGAS00001002923 and phs000915.v1.p1). P‐value was obtained by two‐tailed Student’s t‐test.
Source data are available online for this figure.
Figure EV5. ESM1 stabilizes β‐catenin/TCF4 complex through stereochemically blocking the dissociation of TCF4 from β‐catenin (related to Fig 7).
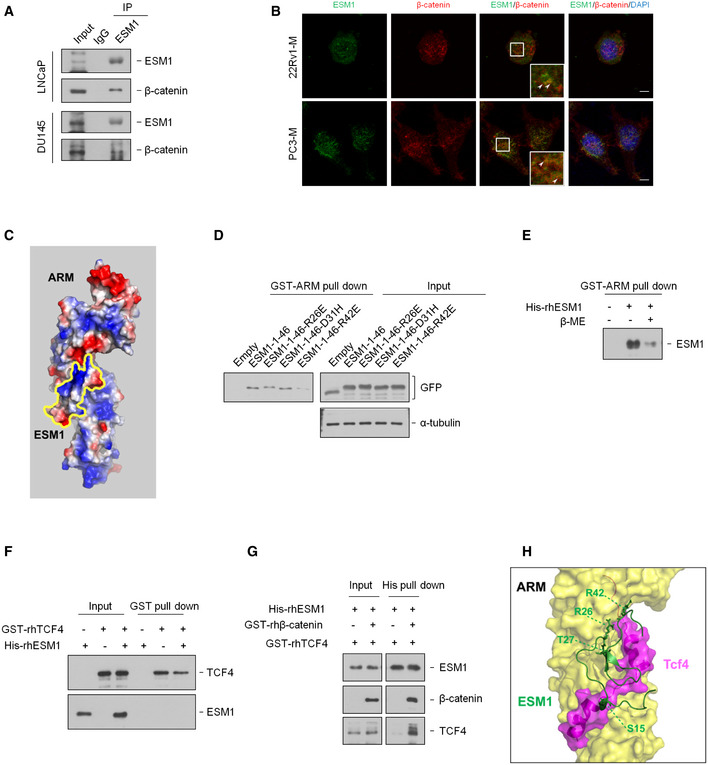
- Cell extracts of LNCaP and DU145 cells were immunoprecipitated with an ESM1 antibody.
- Representative photographs for immunofluorescence staining of endogenous ESM1 and β‐catenin in 22Rv1‐M and PC3‐M cells. Arrowheads indicate the colocalization of ESM1/β‐catenin (yellow). Nuclei were counterstained with DAPI. Scale bar: 10 μm.
- Prediction of the surface charge of ESM1 and β‐catenin interaction using HADDOCK.
- HEK293T cells were transfected with the indicated ESM1 point mutant constructs. Cell extracts were pulled down with purified GST‐ARM.
- Human recombinant His‐ESM1 was treated with β‐mercaptoethanol and pulled down with purified GST‐ARM.
- Human recombinant His‐ESM1 and GST‐TCF4 proteins were pulled down with Glutathione sepharose.
- Human recombinant His‐ESM1, GST‐TCF4, and GST‐β‐catenin proteins were pulled down with Ni sepharose.
- Prediction of the ESM1 and β‐catenin/TCF4 complex interaction using HADDOCK.
Source data are available online for this figure.
ESM1 predominantly expressed in the nucleus in advanced PCa
To further obtain evidence to support our observations, we analyzed ESM1 expression in PCa tissue sections. By using tissue microarrays (TMAs) that contain 124 PCa specimens, including 65 cases of adenocarcinoma and 59 cases of benign prostate tissue, we aimed to detect ESM1 expression in different stages of prostate adenocarcinoma and normal prostate epithelium. Tumors with metastasis had higher expression levels of ESM1 compared to those without metastasis. ESM1 expression was not only correlated with stages and tumor status of the patients, but also correlated with lymph nodal status and metastasis (Fig 1E, Table EV1). Furthermore, unlike its original characterization as a secretory protein, we have observed that ESM1 was expressed in the nucleus of some specimens, especially in tumor tissue with metastasis (Fig 1E). To confirm this unexpected discovery, we examined the secretion and subcellular localization of ESM1 in PCa cell models. Similar to the cellular expression pattern as shown in Fig 1B, secretory levels of ESM1 were also higher in metastatic sublines compared with their parental cell lines. Interestingly, although the cytosolic distribution pattern of ESM1 in the PC3‐P/PC3‐M and 22Rv1‐P/22Rv1‐M pairs was different, the nuclear ESM1 of both was higher in metastatic sublines compared with the parental cell lines (Fig 2A, Appendix Fig S1H). These observations demonstrate that nuclear ESM1 may contribute to aggressive features in PCa.
Figure 2. Nuclear ESM1, instead of secreted ESM1, promotes PCa invasion.
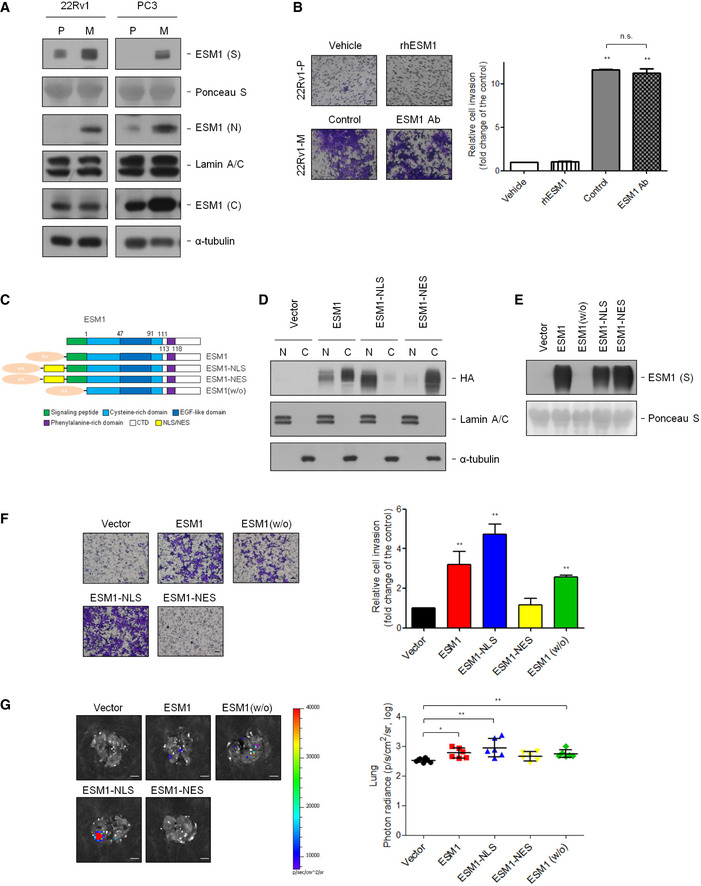
- Immunoblotting analysis of ESM1 in supernatant (S), nucleus (N), and cytoplasm (C) was performed. Ponceau S, Lamin A/C, and α‐tubulin were used as loading control in supernatant, nucleus, and cytoplasm, respectively. P: parental cells, M: metastatic cells.
- Left, representative images of cells in the Transwell invasion assay. Right, quantification of cells that invaded through the Matrigel‐coated membrane following treatment with PBS or human recombinant ESM1 (rhESM1) or normal goat IgG control or anti‐ESM1 neutralizing antibody. Scale bar: 100 μm.
- Schematic diagram of ESM1 expression constructs.
- Elicitation of ESM1 overexpression using HA‐tag antibody results in detectable ESM1 expression in the nucleus and cytoplasm of 22Rv1‐P cells. Lamin A/C and α‐tubulin were used as loading control in the nucleus and cytoplasm, respectively.
- Immunoblotting of ESM1 expression with different expressing patterns in the supernatant (S) of 22Rv1‐P cells.
- Left, representative images of 22Rv1‐P cells in the Transwell invasion assay. Right, statistic data of invasive ability of ESM1‐expressing cells. Scale bar: 100 μm.
- Left, representative bioluminescence images of lung metastasis in mice after tail vein injection of 22Rv1‐Luc cells with different ESM1 expression patterns. Scale bar: 0.5 cm. Right, quantification of the lung bioluminescence by photons/ seconds.
Data information: Differences are shown compared with control cells presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 when compared to control group by two‐tailed Student’s t‐test. Source data are available online for this figure.
Nuclear ESM1, rather than secreted ESM1, promotes PCa invasion
Since ESM1 has been identified as a secretory proteoglycan that executes its functions in the extracellular space (Rocha et al, 2014), the potential influence of secretory ESM1 in PCa cell invasiveness was first examined. As shown in Fig 2B, neither treatment with human recombinant ESM1 nor with polyclonal ESM1 neutralizing antibody affected the ability of cells to invade. Cell invasive abilities of parental cells after induction with condition media from metastatic sublines were also not affected by the pretreatment with the ESM1 neutralizing antibody (Appendix Fig S2A and B). These observations suggested that ESM1 may regulate PCa invasiveness in a non‐secretory manner previously undiscovered. To investigate whether the subcellular localization of ESM1 was important in regulating PCa cell invasiveness, we generated ESM1 expression plasmids with different expression patterns (Fig 2C). Western blot and immunofluorescence results showed that PCa cells overexpressing ESM1‐NLS (ESM1 conjugated with a nuclear localization signal (NLS) peptide) had the majority of their ESM1 located in the nucleus, while PCa cells overexpressing ESM1‐NES (ESM1 conjugated with a nuclear export signal peptide) had only barely detectable ESM1 in the nucleus (Fig 2D, Appendix Fig S2C). ESM1 without any signal peptide (ESM1(w/o)) did not secrete into the medium (Fig 2E). In Fig 2F and Appendix Fig S2D, ESM1‐NLS overexpression increased the invasive ability of 22Rv1‐P and PC3‐P cells to a higher level compared with wild‐type ESM1. On the contrary, overexpressing ESM1‐NES had no significant effect on cell invasiveness. Moreover, after deleting the signal peptide, ESM1 (w/o) could still induce the cell invasive ability. To further substantiate the role of ESM1 in driving PC metastasis, testing of a tail vein metastasis model was conducted in vivo. 22Rv1‐P‐Luc cells with ESM1 or ESM1‐NLS stably overexpressed displayed a superior ability to metastasize to the lung compared with vector controls and those with stably overexpressed ESM1‐NES (Fig 2G, Appendix Fig S2E). Furthermore, similar protein stabilities of ESM1 constructs as observed by half‐life experiments excluded that the disparity of cell invasive abilities between these groups arise from different protein stabilities of ESM1 constructs (Appendix Fig S3A). These data suggest that higher ESM1 expression, especially in the nucleus, may contribute to the metastatic process of PCa.
Nuclear ESM1 regulates PCa metastasis by promoting cancer stem cell (CSC) properties
Evidence supports PCa CSCs as being critical in PCa metastasis and CRPC development (Yun, Zhou et al, 2016; Jung, Cackowski et al, 2018). We ranked 20502 genes from PCa samples in the Cancer Genome Atlas Prostate Adenocarcinoma (TCGA‐PRAD) dataset by their relative ESM1 expression for gene‐set enrichment analysis (GSEA). ESM1Hi tumor samples were enriched in the expression of gene signatures associated with “Regulation of stem cell population maintenance” in comparison with ESM1Low samples (Fig EV2A). Based on our analysis, the metastatic PCa cells had a significant increase in their percentage of CSCs compared with parental cells (Fig EV2, EV3, EV4, EV5). Furthermore, as shown in Fig EV3A, the expression of nuclear ESM1 was higher in PCa cells from the spheroid culture, a classical CSC enrichment condition (Ishiguro, Ohata et al, 2017; Herheliuk, Perepelytsina et al, 2019). We thus hypothesize that the enrichment of CSCs in metastatic PCa may be related to ESM1 expression. To examine whether overexpression of ESM1, especially nuclear ESM1, can promote self‐renewal and confer resistance to chemotherapy, spheroid‐forming assays, and MTT assays were performed. Treatment with human recombinant ESM1 or polyclonal ESM1 neutralizing antibody, which aimed to exclude the functional effect of secretory ESM1, did not affect the self‐renewal ability of PCa cells (Fig 3A). Spheroid formation abilities of 22Rv1‐P cells after induction with condition media from 22Rv1‐M cells were also not affected after ESM1 depletion. Similar effects were also found in the pairs of PC3‐P and PC3‐M cells (Appendix Fig S3B). However, overexpression of different ESM1 constructs in 22Rv1‐P and PC3‐P cells enhanced the self‐renewal ability to different levels. Stably overexpressing ESM1 or ESM1‐NLS led to a significant increase in spheroid formation. On the contrary, ESM1‐NES had no significant effect on the number of spheroids formed compared with the vector group, although ESM1‐NES overexpression did increase the size of spheroids. Interestingly, the spheroid‐forming ability in cells overexpressing ESM1(w/o) was still significantly increased (Fig 3B, Appendix Fig S3C). In contrast, ESM1‐silenced PCa cells formed much fewer spheroids than the control group (Fig EV3B and C). Rescue of ESM1 expression especially in the ESM1‐NLS group significantly increased spheroid formatting and cell invasive ability in stable ESM1 knockdown cells (Fig EV3, EV4, EV5). To examine whether ESM1 confers a chemoresistance advantage, 22Rv1‐P cells with modulated ESM1 expression were treated with varying doses of docetaxel (0, 1, 10, 100, and 1,000 nM). The cell viability index of ESM1‐NLS‐expressing 22Rv1‐P cells showed the highest resistance to docetaxel compared with other cells (Fig 3C). To further investigate the tumorigenicity of ESM1‐overexpressing cells, subcutaneous tumorigenicity was conducted in vivo. Although the angiogenic effects of ESM1 cannot be fully excluded, PC3‐P cells with ESM1‐NLS stably overexpressed formed larger tumors as compared with vector control cells when injected subcutaneously into immunodeficient mice. The tumor incidence was also higher in ESM1‐NLS groups even when only 10 cells were injected (Fig 3D). Furthermore, we also analyzed the percentage of CD44+ and CD133+ cells, as both molecules have been used as stem cell biomarkers for isolation of stem‐like cells from PCa (Liu, Liu et al, 2017). Figure 3E showed that the percentage of CD44+ and CD133+ cells were both significantly higher in ESM1‐NLS‐overexpressing cells compared with vector control or ESM1‐overexpressing cells. Nuclear markers of CSCs, including Oct4, Nanog, and ABCG2, were also expressed at higher levels in ESM1‐NLS‐overexpressing cells (Fig 3F). In view of the above‐mentioned results, we concluded that ESM1, especially that which is distributed into the nucleus, may promote PCa progression by enriching or maintaining the CSC population.
Figure 3. Nuclear ESM1 regulates PCa metastasis by promoting cancer stem cell properties.
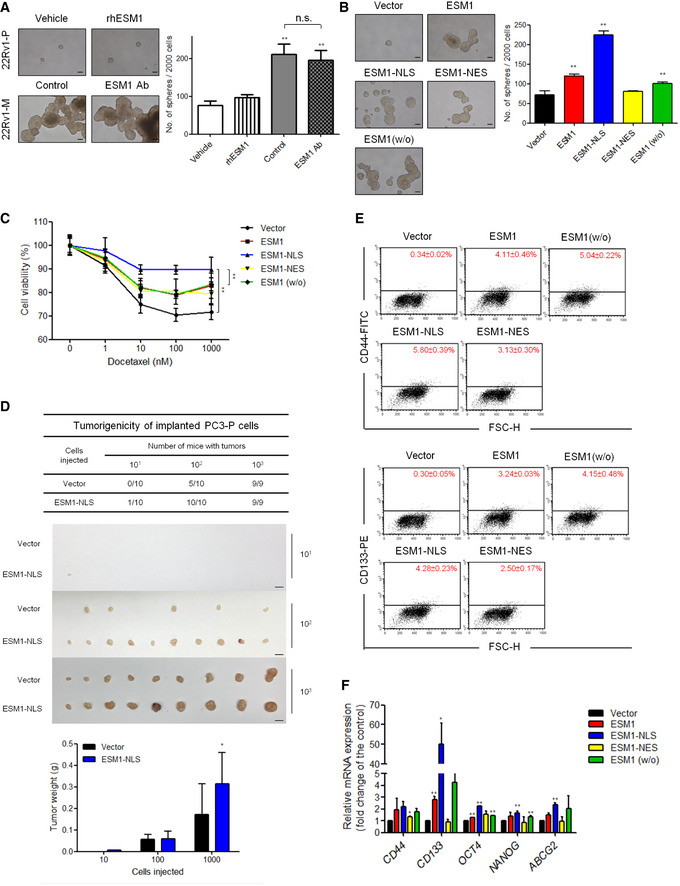
- Tumor spheroid formation. Representative images of tumorspheres formed and quantitative data comparing the average number of spheres formed in the indicated cells treated with PBS or human recombinant ESM1 (rhESM1) or normal goat IgG control or anti‐ESM1 neutralizing antibody. Scale bar: 100 μm. Bars are the mean ± SD of three independent experiments. **P < 0.01 when compared to the vehicle group of 22Rv1‐P cells by two‐tailed Student’s t‐test.
- Representative images of tumorspheres. Spheres were cultured for 14 days before counting. Histogram shows the mean numbers of spheres cultured. Scale bar: 100 μm. Bars are the mean ± SD of three independent experiments. **P < 0.01 when compared to vector cells by two‐tailed Student’s t‐test.
- 22Rv1‐P cells stably expressing ESM1 were treated with docetaxel at the indicated concentrations for 48 h. **P < 0.01 when compared to vector cells by two‐tailed Student’s t‐test and error bars represent the standard deviation of three independent experiments.
- Cells with or without ESM1‐NLS overexpression were subcutaneously injected (10, 100, 1,000 cells per mouse) into NOD/SCID mice. Tumor formation ability and tumor weight were analyzed. Scale bar: 1 cm. Bars are the mean ± SD of indicated (n) independent experiments. *P < 0.05 when compared to vector cells by two‐tailed Student’s t‐test.
- Flow cytometry analysis of the ratio of CD44+ and CD133+ cells in the indicated cells with different subcellular localization types of ESM1 overexpression was performed.
- qRT–PCR analysis of the mRNA levels of the indicated genes in cells with different types of ESM1 overexpression. Differences in mRNA levels compared with vector cells are shown as fold changes presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 when compared to vector cells by two‐tailed Student’s t‐test.
Nuclear ESM1 drives PCa metastasis through Wnt/β‐catenin signaling pathway
To better understand details of the molecular mechanisms underlying ESM1‐driven PCa stemness, we then performed transcriptomic analysis by RNA‐seq on ESM1‐overexpressing and control 22Rv1‐P cells. A fold change cut‐off of ≧ 1.5 and ≦ 0.5 was used for pathway analysis carried out using ingenuity pathway analysis (IPA). For the de‐regulated differentially expressed genes (DEGs) in modulated ESM1‐overexpressing cells, IPA identified activated potential upstream regulators which were further compared by their activated z‐scores. The activation z‐score fluctuation of 14 upstream transcriptional regulators is dramatically highest in the ESM1‐NLS group compared with the ESM1 and ESM1‐NES groups (Fig 4A). Among these upstream regulators, RELA(RelA/p65) and CTNNB1(β‐catenin), which were the key molecules of NFκB and Wnt signaling, respectively, have been found to be activated in PCa CSCs (Rajasekhar, Studer et al, 2011; Zhang et al, 2017; Schneider & Logan, 2018; Kaltschmidt, Banz‐Jansen et al, 2019). Meanwhile, two other Wnt signaling regulators, Wnt3A and α‐catenin, also emerged in our IPA analysis and 65 downstream genes were indicated to be regulated by β‐catenin in our RNA‐seq data (Fig 4B). Moreover, when GSEA was performed on our RNA‐seq data, it also found that ESM1 expression was positively correlated with genes regulated by the Wnt signaling pathway (Fig 4C). GSEA of two PubMed GEO datasets showed similar results that revealed upregulated genes with β‐catenin overexpressing were among the top pathways that were induced in ESM1Hi PRAD patients (Fig 4D). However, the correlation of ESM1 and NFκB signaling was not found in the GSEA results. To further assess the correlation between ESM1 expression and Wnt/β‐catenin or NFκB signaling status, we performed luciferase reporter assays. The β‐catenin transcriptional activity was shown as a ratio of TOP‐FLASH to FOP‐FLASH luciferase‐mediated signals. The results showed that β‐catenin activity in ESM1‐NLS‐overexpressing cells was higher than that in the ESM1 and ESM1‐NES groups (Fig 4E, Appendix Fig S3D). Overexpressing ESM1‐NLS also induced NFκB transcriptional activity in the 22Rv1‐P and PC3‐P cells (Fig 4F, Appendix Fig S3E). Taken together, our analysis suggests that β‐catenin and RelA/p65 may act as critical effectors of ESM1‐mediated PCa stemness.
Figure 4. Nuclear ESM1 drives PCa metastasis through β‐catenin and NFκB signaling pathway.
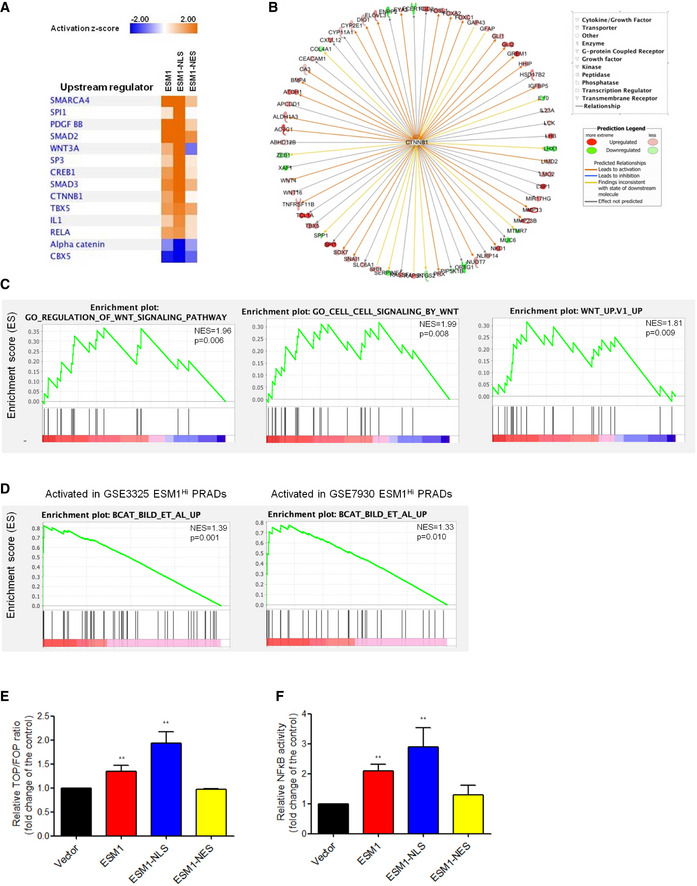
- IPA analysis of RNA‐seq data showing the 14 top‐predicted upstream regulators of differentially expressed genes. Positive Z‐scores (orange) represent activated upstream regulators, and negative Z‐scores (blue) represent inhibited upstream regulators.
- Known regulators of differentially expressed genes in the ESM1‐NLS cells giving rise to the prediction of CTNNB1 as an upstream regulator. Observed upregulation and downregulation of mRNA are shown in red and green, respectively. The predicted relationships were shown as arrow lines with different colors. The detail of predicted relationships of all the genes to the upstream regulator and the color intensity scale was described in the prediction legend.
- GSEA of RNA‐seq data demonstrating the enrichment of gene sets related to Wnt signaling in 22Rv1‐P cells expressing ESM1‐NLS. NES, normalized enrichment score. The p‐values for the GSEA test statistics are calculated by permutation. The original test statistics for the features are permuted, and new test statistics are calculated for each category, based on the permuted feature test statistics.
- GSEA analysis identified β‐catenin oncogenic signatures as the top induced pathways of PRAD in the PubMed GEO datasets (GSE3325 and GSE7930). NES, normalized enrichment score. The p‐values for the GSEA test statistics are calculated by permutation. The original test statistics for the features are permuted, and new test statistics are calculated for each category, based on the permuted feature test statistics.
- The transcriptional activity of β‐catenin in 22Rv1‐P cells with different patterns of ESM1 overexpression. Bars are the mean ± SD of three independent experiments.
- Manipulating ESM1 expression impacts transcriptional activity of NFκB. Bars are the mean ± SD of three independent experiments.
Data information: β‐catenin and NFκB promoter reporter activity were normalized by comparison with Renilla luciferase activity. **P < 0.01 when compared to vector cells by two‐tailed Student’s t‐test.
The regulation of β‐catenin and NFκB activity relies on the accumulation of these two proteins in the nucleus to promote the transcription of many oncogenes. To investigate whether these two signaling pathways were regulated, we performed qPCR to examine the mRNA level of downstream target genes. Expression profiles of these genes in ESM1‐NLS‐expressing 22Rv1‐P cells were significantly higher than that of vector control cells (Fig 5A and B). Moreover, nuclear and cytoplasmic fractions were determined to investigate the translocation of these two molecules. As a result, we found that the expression of β‐catenin and RelA/p65 was significantly increased in metastatic cells while stable ESM1 knockdown led to a significant decrease in the expression of β‐catenin and RelA/p65 protein in the nucleus (Fig EV4A and B). In addition, the expression of β‐catenin in the nucleus was significantly induced in ESM1‐NLS‐expressing 22Rv1‐P cells while RelA/p65 was equally translocated in the nucleus in ESM1‐ and ESM1‐NLS‐expressing cells (Fig 5C). To further validate the role of β‐catenin and NFκB activation in ESM1‐NLS‐induced metastasis and stemness, we analyzed the impact of introducing β‐catenin and RelA/p65 shRNAs into PCa cells with ESM1‐NLS stably overexpressed. Successful suppression of either β‐catenin or RelA/p65 in stably overexpressed ESM1‐NLS cells was confirmed by Western blotting (Fig EV4C). These suppressions inhibited the invasive and stem cell‐like properties conferred by ESM1‐NLS overexpression, as evidenced by diminished abilities of PCa to invade, to form spheroids, and to resist docetaxel (Fig 5D–F). Of note, spheroids formed by PCa cells with RelA/p65 knockdown still grew into comparable sizes as those formed by PCa cells with only ESM1‐NLS overexpression (Fig 5E), suggesting that Wnt/β‐catenin signaling may act as a more important downstream regulator of nuclear ESM1 during PCa metastasis.
Figure 5. Nuclear ESM1 promotes PCa stemness through β‐catenin signaling pathway.
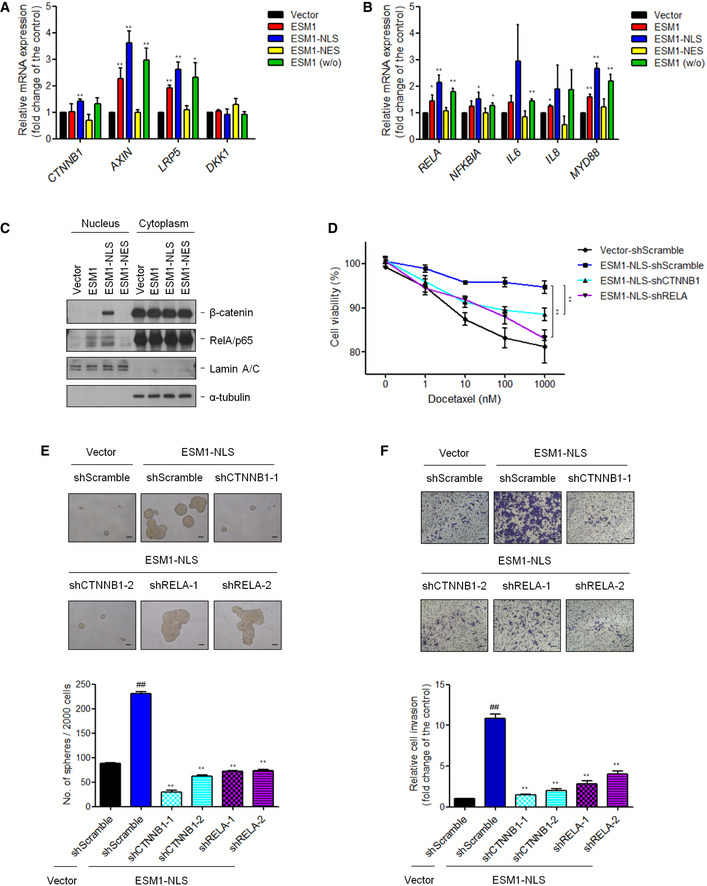
- qRT–PCR analysis of the mRNA levels of the genes related to β‐catenin downstream signaling pathway. Differences in mRNA levels compared with vector cells are shown as fold changes presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 when compared to vector cells by two‐tailed Student’s t‐test.
- qRT–PCR analysis of the mRNA levels of NFκB targeting genes. Differences in mRNA levels compared with vector cells are shown as fold changes presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 when compared to vector cells by two‐tailed Student’s t‐test.
- Immunoblotting analysis of 22Rv1‐P cells with different ESM1 overexpression in the nucleus and cytoplasm was performed with the antibodies indicated.
- Suppression of β‐catenin or NFκB in 22Rv1‐P cells stably expressing ESM1‐NLS was treated with docetaxel at the indicated concentrations for 48 h. **P < 0.01 when compared to vector‐shScramble cells by two‐tailed Student’s t‐test and error bars represent the standard deviation of three independent experiments.
- Tumorspheres formed by 22Rv1‐P cells stably expressing ESM1‐NLS with either β‐catenin or NFκB shRNAs. Scale bar: 100 μm. Bars are the mean ± SD of three independent experiments. ## P < 0.01 when compared to vector‐shScramble cells, **P < 0.01 when compared to ESM1‐NLS‐shScramble cells by two‐tailed Student’s t‐test.
- Representative images and quantitative data comparing cell invasion of the indicated cells. Scale bar: 100 μm. Bars are the mean ± SD of three independent experiments. ##P < 0.01 when compared to vector‐shScramble cells, **P < 0.01 when compared to ESM1‐NLS‐shScramble cells by two‐tailed Student’s t‐test.
Source data are available online for this figure.
ESM1 associates with the ARM domain of β‐catenin
To elucidate the nature and direction of association between ESM1 regulation and Wnt/β‐catenin as well as NFκB signaling, we first examined whether silencing ESM1 affects the expression of Wnt/β‐catenin and NFκB signaling components. The expression of β‐catenin or any other member of the Wnt signaling pathway was not altered (Fig EV4D), nor was there a difference in the expression of NFκB components after silencing ESM1 (Fig EV4E). On the contrary, to our surprise, depletion of β‐catenin, but not RelA/p65, decreased the expression level of ESM1 in the nucleus (Figs 6A and Fig EV4F). We further queried the clinical PCa datasets from the cBioPortal online platform to perform co‐expression analysis. The results shown in Fig EV4G suggested that ESM1 expression was correlated with β‐catenin but not with RelA/p65. β‐catenin harbored a correlation, with both Pearson and Spearman correlation coefficients higher than 0.35, while the Pearson and Spearman correlation coefficients of RelA/p65 were less than 0.05. We thus focused on the mechanism of ESM1 regulation by the Wnt signaling pathway in PCa cells.
Figure 6. ESM1 associates with the ARM domain of β‐catenin.
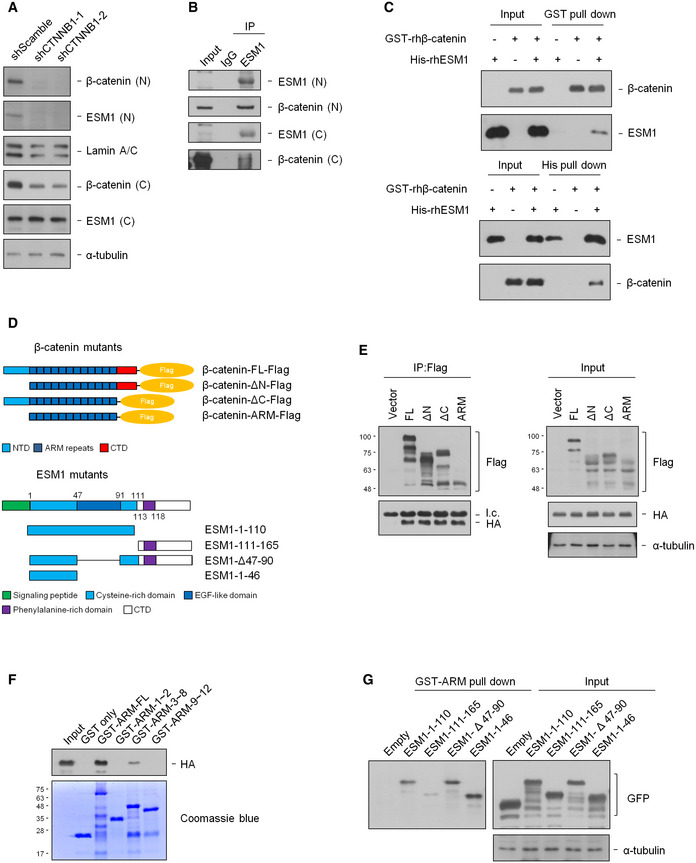
- Nuclear and cytosolic extracts of 22Rv1‐M cells transfected with either shScramble or shCTNNB1 were prepared, and the levels of indicated proteins were detected by immunoblotting.
- Nuclear or cytosolic extracts of 22Rv1‐M cells were immunoprecipitated with an ESM1 antibody.
- Human recombinant His‐ESM1 and GST‐β‐catenin proteins were pull down with either Ni sepharose or glutathione sepharose.
- Upper, mapping β‐catenin regions binding to ESM1. Schematic diagram of full‐length β‐catenin and deletion mutants. Lower, mapping ESM1 regions binding to β‐catenin. Schematic diagram of full‐length ESM1 and deletion mutants.
- HEK293T cells were co‐transfected with the indicated β‐catenin‐Flag and ESM1‐HA constructs. Cell extracts were immunoprecipitated with Flag‐M2 agarose beads. Light chain was labeled as l.c.
- HEK293T cells were transfected with ESM1‐HA constructs, and pull‐down was carried out with different purified GST‐ARM fragments.
- HEK293T cells were transfected with the indicated ESM1‐GFP constructs. Cell extracts were pulled down with purified GST‐ARM.
Source data are available online for this figure.
Since β‐catenin has been found to directly interact with the nuclear pore complexes and mediate its own nuclear entry, and since no classical nuclear localization signal (NLS) can be found in ESM1, we hypothesized that endogenous ESM1 may associate with β‐catenin and thus enter the nucleus. Indeed, the presence of β‐catenin was detected in the immune complex precipitated by the anti‐ESM1 antibody from either the nuclear or cytoplasmic fraction of 22Rv1‐M cells (Fig 6B). The association of ESM1 and β‐catenin was also observed when we performed immunoprecipitation assay in LNCaP and DU145 cells (Fig EV5A). Confocal immunofluorescence analysis was further conducted, and the colocalization of ESM1/β‐catenin was observed (Fig EV5B). The direct binding of recombinant human ESM1 (rhESM1) and GST‐tagged β‐catenin protein, as confirmed in the reciprocal pull‐down assays (Fig 6C), further supports our hypothesis. We next deciphered the domains responsible for binding and their association to better understand how ESM1 and β‐catenin interacted. β‐catenin protein contains a N‐terminal domain (NTD), a C‐terminal domain (CTD), and an armadillo repeat domain (ARM), which mediate its interactions with several transcription factors and other cofactors. We constructed several deletion mutants of β‐catenin and examined their interaction with ESM1 (Fig 6D). The result showed that the deletion of either N‐ or C‐terminal domain of β‐catenin did not affect their interaction (Fig 6E), suggesting that ESM1 may bind to the ARM domain of β‐catenin. Given that different interacting proteins bind with different fragments of the 12 armadillo repeats within the ARM domain of β‐catenin (Xu & Kimelman, 2007), we further performed pull‐down assays with purified GST protein conjugating with different ARM fragments to identify which repeats of the ARM domain interact with ESM1. As shown in Fig 6F, only the fragment containing repeats 3–8 interacted with ESM1. These data demonstrate that β‐catenin interacts with ESM1 through its ARM domain, primarily with the region of repeats 3–8. In addition, we also conducted pull‐down assays with purified GST‐ARM proteins and different ESM1 deletion mutants and determined that ESM1 interacts with β‐catenin by its cysteine‐rich region, especially the region of amino acids 1–46 (Fig 6G). Collectively, our results suggest that ESM1 may regulate Wnt/β‐catenin signaling through direct interaction with β‐catenin protein in the nucleus of PCa cells.
ESM1 promotes Wnt activation by facilitating its association with TCF4/β‐catenin complex
To reveal the detailed mechanism of the ESM1‐β‐catenin interaction in the regulation of Wnt/β‐catenin signaling in PCa CSCs, we docked the ESM1 protein, created by homology modeling, with the β‐catenin protein from Protein Data Bank (PDB, 1jpw) using HADDOCK (High Ambiguity‐Driven biomolecular Docking). The distribution of surface charge on ESM1 (fragments 1–46) and the ARM domain suggested that the surface charge of these proteins is critical for their interaction (Fig EV5C). ESM1 was predicted to bind near the long positively charged groove constituted by the 12 repeats of the ARM domain in the molecular docking model (Fig 7A). By PISA (protein interfaces, structures, and assemblies) analysis, the thermodynamic stability of ESM1‐β‐catenin complex was estimated to predict possible interaction residues and calculate binding surface areas. Three residues including Arg26, Asp31, and Arg42 in ESM1 were predicted to be crucial in the binding region. We thus used site‐directed mutagenesis to generate R26E, D31H, and R42E mutants and examined their affinities with purified GST‐ARM domain. As shown in Fig EV5D, the ESM1 R26E and R42E mutants, but not the D31H mutant, exhibited decreased interaction with the ARM domain compared with wild‐type ESM1. Moreover, the R26/42E double mutant almost completely lost its ability to bind to the ARM domain (Fig 7B). In building the homology model of ESM1, two disulfide bonds were formed in the region of residues 1–46. To confirm the importance of the side‐chain orientation of the region of residues 1‐46 for ESM1‐β‐catenin interaction, the pull‐down ability of GST‐ARM to rhESM1 was examined in the presence of a reducing agent, β‐mercaptoethanol. Compared to the control group, the interaction of rhESM1 with GST‐ARM drastically decreased under the reducing condition (Fig EV5E). Collectively, these data suggest that the interaction between ESM1 and β‐catenin may depend on charge–charge interactions at the protein interface.
Figure 7. ESM1 stabilizes the association of TCF4/β‐catenin complex.
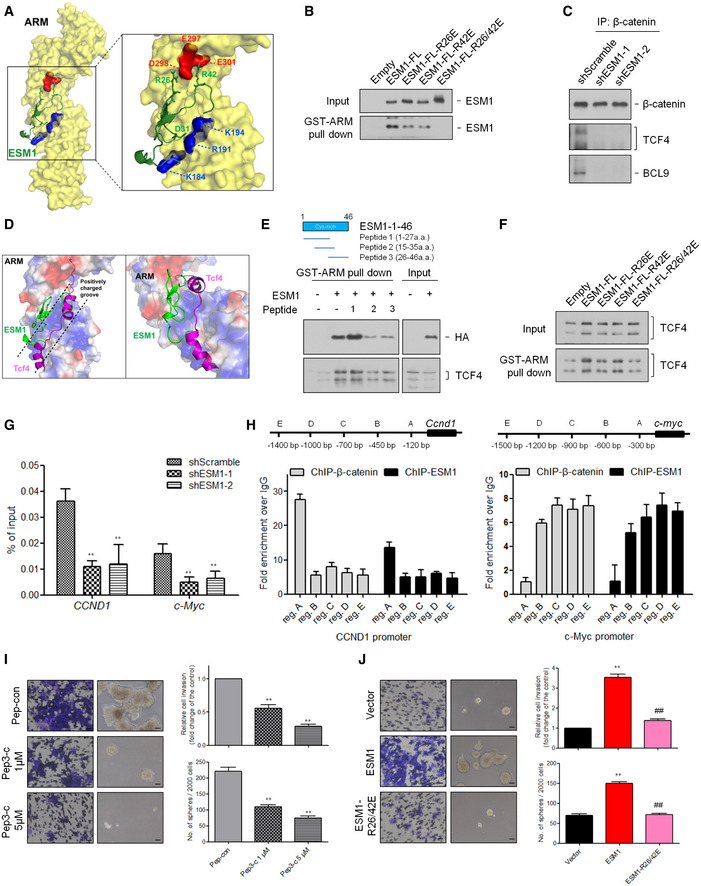
- Prediction of ESM1 and β‐catenin interaction using HADDOCK.
- HEK293T cells were transfected with the indicated ESM1 point mutant constructs. Cell extracts were pulled down with purified GST‐ARM.
- Cell extracts of 22Rv1‐M transfected with either Scramble‐shRNA or ESM1‐shRNA were immunoprecipitated with an β‐catenin antibody and blotted with anti‐TCF4 and anti‐BCL9 antibody.
- Prediction of the ESM1, β‐catenin, and TCF4 interaction using HADDOCK. The surface representation of ARM is colored red and blue for negative and positive charges, respectively, and gray color represents neutral residues.
- Upper, schematic diagram of ESM1‐targeting short peptides. Lower, HEK293T cells were transfected with ESM1‐HA constructs. Cell extracts were treated with the short peptides, pulled down with purified GST‐ARM, and blotted with anti‐HA and anti‐TCF4 antibodies.
- HEK293T cells were transfected with the indicated ESM1 point mutant constructs. Cell extracts were pulled down with purified GST‐ARM and blotted with an anti‐TCF4 antibody.
- ChIP‐qPCR validation of β‐catenin binding to the CCND1 and c‐Myc promoter in 22Rv1‐M cells transfected with either Scramble‐shRNA or ESM1‐shRNA. Bars are the mean ± SD of three independent experiments. **P < 0.01 when compared to shScramble cells by two‐tailed Student’s t‐test.
- Upper, schematic diagram of ChIP‐qPCR primers targeting regions upstream of the CCND1 and c‐Myc transcription start sites (TSSs). Bar graphs show β‐catenin and ESM1 binding at the CCND1 and c‐Myc promoter regions. Differences in DNA levels compared with IgG control are shown as fold changes presented as the mean ± SD of three independent experiments.
- The effects of ESM1 targeting peptides on tumor invasion and spheroid formation. Scale bar: 100 μm. Bars are the mean ± SD of three independent experiments. **P < 0.01 when compared to Pep‐con group by two‐tailed Student’s t‐test.
- The effects of ESM1 point mutants on tumor invasion and spheroid formation. Scale bar: 100 μm. Bars are the mean ± SD of three independent experiments. **P < 0.01 when compared to vector cells, ##P < 0.01 when compared to ESM1 cells by two‐tailed Student’s t‐test.
Source data are available online for this figure.
We next examined the impact of ESM1‐β‐catenin interaction on the transcriptional activity of β‐catenin. Upon activation, β‐catenin recruits its cofactor TCF4 to load onto the DNA as a complex (Ravindranath, Yuen et al, 2011; Schepeler, Holm et al, 2012). Since ESM1 has no identifiable DNA binding motif, we examined whether the existence of ESM1 stabilizes the β‐catenin–TCF4 complex and further contributes to β‐catenin transcriptional activity. As expected, the association of TCF4 and β‐catenin was diminished in the absence of ESM1 in the co‐immunoprecipitation assays (Fig 7C). Interestingly, although TCF4 shared the same binding region with ESM1 on the β‐catenin ARM domain (Xu & Kimelman, 2007), our docking model suggests that they may not compete with each other. As shown in Fig 7D, TCF4 binds to the positively charged groove of the ARM domain, while ESM1 binds to the edge of the groove. Moreover, although ESM1 and TCF4 did not directly bind with each other as suggested by our in vitro binding experiment (Fig EV5F), the presence of β‐catenin did assist the formation of the ESM1–β‐catenin–TCF4 complex (Fig EV5G). We therefore speculated that ESM1 may stabilize β‐catenin–TCF4 complex by stereochemically blocking the dissociation of TCF4 from β‐catenin or by increasing the molecular attraction within the ESM1–β‐catenin–TCF4 complex. Accordingly, as we docked the ESM1 (1–46 fragments) onto the β‐catenin–TCF4 co‐crystallized structure, salt bridges and hydrogen bonds were predicted to form between ESM1‐β‐catenin and ESM1‐TCF4 (Fig EV5H).
To verify the critical role of the ESM1‐β‐catenin interaction in the regulation of Wnt/β‐catenin signaling and PCa stemness, we designed short peptides to interrupt ESM1‐β‐catenin interaction in PCa cells. Peptides 2 and 3, which contain ESM1 residues 15–35 and 26–46, respectively, each diminished the interaction of ARM and ESM1 (Fig 7E). Moreover, the effect of disruption also occurred in the binding of ARM and TCF4. In contrast, the peptide containing ESM1 residues 1–27 did not affect the association of ARM with ESM1 or TCF4. These results support that interruption of charge–charge interaction mediated by the region containing R26/42 residues may result in the collapse of the ESM1–β‐catenin–TCF4 complex. In agreement with this hypothesis, the binding of TCF4 and β‐catenin was reduced in the presence of R26E, R42E, or R26/42E mutants in comparison with wild‐type ESM1 protein (Fig 7F). We next analyzed whether ESM1 affected the transcription activity of β‐catenin–TCF4 complex via ChIP assay. The results demonstrated that silencing ESM1 suppressed the binding of β‐catenin with the promoter of two of its target genes, CCND1 and c‐Myc (Fig 7G). Detailed analysis within the 2‐kb regions upstream of the CCND1 and c‐Myc gene transcription start sites (TSSs) revealed that ESM1 and β‐catenin occupied similar promoter regions (Fig 7H). These data suggest that positive regulation of the ESM1/β‐catenin/TCF4 axis supports PCa stemness.
Based on the above results, the potential of targeting ESM1 as a therapeutic strategy impelled us to further validate whether or not interrupting ESM1‐β‐catenin interaction does affect PCa invasiveness and stemness. A fused peptide was designed by linking a cell‐penetrating and nuclear‐localizing peptide (Chauhan, Tikoo et al, 2007; Heitz, Morris et al, 2009) to peptide 3 (Pep3‐c). As can be seen in Fig 7I, Pep3‐c significantly reduced the ability of PCa cells to invade and to form spheroids. Similar functional observations were also noted when ESM1‐R26/42E was introduced into the cells. Stably overexpressing ESM1‐R26/42E led to a significant decrease in spheroid formation and cell invasion compared with the wild‐type ESM1 group (Fig 7J). These findings verify the therapeutic potential of interrupting the ESM1‐β‐catenin interaction in metastatic PCa. Taken together, our results suggest that ESM1 contributes to PCa metastasis through stimulating β‐catenin activation, strengthening β‐catenin/TCF4 interaction, and modulating the downstream signaling of stemness.
Discussion
Aberrant activation of Wnt/β‐catenin signaling has been found in different kinds of cancer and has been shown to contribute to tumor progression (Zhan, Rindtorff et al, 2017; Madueke, Hu et al, 2019). Despite the infrequent activation of Wnt/β‐catenin signaling in PCa, recent studies have discovered that activation of Wnt/β‐catenin signaling is more frequently observed in advanced tumors and CRPC than in naïve PCa (Kypta & Waxman, 2012; Yokoyama, Shao et al, 2014; Hu, Hu et al, 2017). Furthermore, Wnt/β‐catenin signaling has been shown to promote PCa stem cell renewal and expansion (Bisson & Prowse, 2009). These fundamental results provide a rationale for targeting Wnt/β‐catenin signaling to eliminate PCa CSCs and reduce the development of tumor resistance to therapy. However, only a few inhibitors targeting Wnt/β‐catenin signaling have currently attained early phase clinical trials (Krishnamurthy & Kurzrock, 2018). Therefore, identification of novel drug targets in this pivotal pathway may help to develop novel therapeutic strategies. In this work, we determined that upon Wnt/β‐catenin activation in PCa CSCs, ESM1 was imported into the nucleus via association with β‐catenin. While in the nucleus, ESM1 facilitated the association of β‐catenin and TCF4, which in turn led to retention of β‐catenin in the nucleus, resulting in the maintenance of Wnt/β‐catenin activation to support PCa stemness (Fig 8). Nuclear mislocalization of ESM1 may thus contribute to PCa metastasis and represent a novel target of Wnt/β‐catenin signaling that can be used in the development of a future treatment for advanced PCa.
Figure 8. ESM1 drives PCa metastasis by coordinating with the Wnt/β‐catenin pathway.
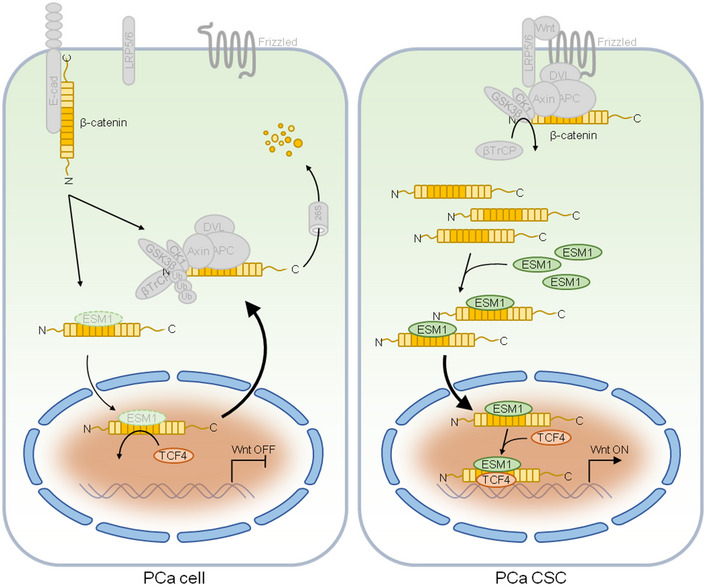
Schematic diagram illustrating how elevated ESM1 expression promotes development of PCa by raising cancer stem cell populations. The interaction between ESM1 and β‐catenin leads to the stabilization of β‐catenin–TCF4 complex and recruitment of this complex to the DNA. Stabilized β‐catenin and ESM1 promote the binding of TCF4 to DNA, Wnt activation of downstream gene expression, cancer cell stemness, and tumor metastasis.
Recently, Chen et al. have reported an opposite effect of ESM1 in regulating tumorigenicity and metastasis of PCa cells (Chen, Lin et al, 2017). However, they used only one shRNA to suppress ESM1 expression, and off‐target effect could not be excluded. Here, we have tried several shRNAs and another gene silencing strategy, the LNAs, to suppress the expression of ESM1 in our cell model. The cell invasive abilities decreased in a similar way after different ESM1 silencing strategies. Consistent with our findings, a recent study also indicated that silencing ESM1 with siRNA may suppress the migration ability of PC3 cells (Rebollo, Geliebter et al, 2017). Based on the multiple lines of evidence, we suggested that ESM1 may act as a positive regulator rather than an inhibitor to the tumorigenicity and metastasis of PCa.
The interaction of ESM1 and β‐catenin was observed in PCa cells with or without AR expression. It suggested that the ESM1‐β‐catenin regulatory axis may operate independently to the AR signaling. However, AR was also reported to associate with β‐catenin (Truica, Byers et al, 2000), despite the consequences of their interaction in PCa remain controversial. Although β‐catenin was found to bind to the ligand‐binding domain (LBD) of AR and enhance its transcriptional activity (Song, Herrell et al, 2003), activated AR was also found to compete with TCF/LEF‐1 to bind to β‐catenin (Chesire & Isaacs, 2002). Moreover, while AR promoted nuclear translocation of β‐catenin in the presence of androgen (Mulholland, Cheng et al, 2002), other reports show that AR activation resulted in the reduction in β‐catenin‐dependent transcription (Kretzschmar, Cottle et al, 2015; Lee, Ha et al, 2015). Surprisingly, we also observed an association of ESM1 and AR in AR+ PCa (unpublished observations), indicating that ESM1 may also participate in the AR/β‐catenin crosstalk. Further investigation will be required to reveal the molecular and functional interplays among these important molecules in PCa.
β‐catenin executes drastically different functions when distributed in different cellular compartments. Upon Wnt activation, accumulated β‐catenin is translocated into the nucleus and transforms into a transcriptional factor that regulates Wnt‐target genes. This conversion exerts further control over a wide range of cellular processes (Murillo‐Garzon & Kypta, 2017). Interestingly, the comprehensive regulation of β‐catenin localization is highly reliant on its ARM domain. E‐cadherin, APC, and TCF4 compete for β‐catenin interaction in the same region of the ARM domain and thus sequester β‐catenin from other interaction partners. Here, we discovered ESM1 to be a new β‐catenin interacting partner that also binds to the ARM domain. However, although ESM1 binds to the same region of the ARM domain as TCF4 does, ESM1 did not interrupt but rather enhanced the β‐catenin–TCF4 binding in the nucleus of PCa cells. According to our ESM1 docking models with ARM or ARM‐TCF4 complex, ESM1 may reinforce β‐catenin–TCF4 binding via establishing an extra salt bridge or hydrogen bond with β‐catenin and TCF4. Unlike the originally identified ligand‐like functions of ESM1 in the extracellular space (Bechard, Gentina et al, 2001; Bechard, Scherpereel et al, 2001; Rocha et al, 2014), nuclear ESM1 acts more like a transcriptional cofactor since it also loaded onto promoter regions of Wnt‐target genes with β‐catenin–TCF4 complex. The nuclear function of mislocalized ESM1 that we have identified here not only demonstrates the complexity of ARM domain‐dependent β‐catenin regulation but also highlights the importance of specifying protein functions according to their subcellular localization.
ESM1 has never been reported to localize in the nucleus. To uncover the mechanism by which ESM1 translocates to the nucleus, the nuclear transport pathways were analyzed. The nuclear transport cycle consists of several important factors including nucleoporins, RanGTPase, karyopherins (importin/exportin/transportin), and nuclear localization signals (NLSs) or nuclear export signals (NESs) in cargo molecules (Kim, Han et al, 2017). After binding to importin, the cargo protein forms cargo‐importin complex and further interacts with nucleoporins to pass through the nuclear pore. However, after surveying the sequence of ESM1, we did not find a region resembling nuclear localization signals. We also immunoprecipitated ESM1 to analyze whether an interaction between ESM1 and importins exists, yet ESM1 did not bind to either importin‐α, importin‐β, or Ran protein (unpublished observations). These results impelled us to exclude the participation of the classic nuclear transport route. A previous study reported that β‐catenin can directly contact the nuclear pore complex (NPC) to self‐regulate its own entry into the nucleus (Jamieson, Sharma et al, 2014). This mechanism is independent of the classical Ran/importin import machinery. Moreover, the β‐catenin–NPC interaction implicates the potential that β‐catenin itself could be regarded as a specialized transport receptor. Indeed, β‐catenin can transport specific cargo proteins and when overexpressed in cancer cells may lead to mislocalization of key tumor suppressors or oncogenes, such as lymphocyte enhancer factor‐1 (lef‐1; Asally & Yoneda, 2005; Sharma, Jamieson et al, 2012). We then surmised that β‐catenin is not only an interaction partner of ESM1, but it may also serve as a transporter that transfers ESM1 to the nucleus.
Nuclear accumulation of β‐catenin is a hallmark of Wnt signaling (Reya & Clevers, 2005; Anastas & Moon, 2013). According to our results, the expression of ESM1 also affected the nuclear levels of β‐catenin in PCa cells. Although the extra NLS peptide we added in the ESM1 construct may have provided an additional nuclear influx of β‐catenin via the classic nuclear transport route in our overexpression experiments, the knockdown of endogenous ESM1 in PCa cells indeed results in the reduction in nuclear β‐catenin. Previous studies indicated that the subcellular localization of β‐catenin was mainly regulated by retaining it in the compartment in which they are localized, rather than by active transport into or out of the nucleus (Krieghoff, Behrens et al, 2006; Lu, Xie et al, 2017; Shang, Hua et al, 2017). Several β‐catenin interaction partners including TCF4 and BCL9 have been found to promote β‐catenin nuclear retention (Henderson & Fagotto, 2002; Krieghoff et al, 2006). Since our results showed that ESM1 may facilitate TCF4 and BCL9 association with β‐catenin in PCa cells, the observed ESM1‐elevated nuclear β‐catenin may arise from the increasing amount of β‐catenin–TCF4–BCL9 complex. Taken together, ESM1 and β‐catenin may mutually regulate each other’s nuclear level in a collaborative manner and thus result in the activation of Wnt signaling in PCa cells.
In summary, the interplay between ESM1 and β‐catenin emerges as a new mechanism that regulates stemness and metastasis in tumors. We propose that the nuclear translocation of ESM1, which may correlate with metastasis, may in turn regulate β‐catenin‐induced cancer stemness. Our finding highlights the importance of ESM1 mislocalization to control not only the localization of β‐catenin but also the promotion of its interaction with co‐activators. Both mechanisms are intrinsically associated and are likely to contribute to metastasis in prostate tumors. Our study demonstrates the importance of the ESM1‐β‐catenin axis in the progression of PCa metastasis and reveals ESM1 to be a novel therapeutic target for advanced PCa patients.
Materials and Methods
DNA constructions, antibodies, cell culture, and transfection
Plasmids bearing ESM1 or β‐catenin constructs were engineered by PCR and subcloned into a pWPI, pCDNA3.1, or pEGFP vector. The point mutations in the cysteine‐rich domain of ESM1 (R26E, D31H, R42E, and R26/42E) were obtained by using QuikChange® mutagenesis kit. The plasmids expressing the GST fusion proteins composed of ARM‐FL, ARM‐1 ~2, ARM‐3 ~8, or ARM‐9 ~12 were engineered by PCR and subcloned into a pGEX‐3X vector.
The following antibodies were used as follows: ESM1 (ab56914, Abcam) for immunohistochemistry staining, ESM1 (LIA‐1001, Lunginnov; Clone 37/customized antibody, Leadgene Biomedical, Inc.) for immunoblotting, ESM1 (HPA036660, Sigma) for immunofluorescence staining, ESM1 (126460, US Biological) for immunoprecipitation, ESM1 (Clone 37/customized antibody, Leadgene Biomedical, Inc.) for ChIP assay, mouse anti‐HA (Sigma), Flag (8146, Cell signaling), GFP (sc8334, Santa Cruz), β‐catenin (sc1496, Santa Cruz), TCF4 (2569, Cell signaling), BCL9 (15096, Cell signaling), RelA/p65 (8242, Cell signaling), NFκB/p50 (GTX100772, GeneTex), Axin1 (2087, Cell signaling), APC (2504, Cell signaling), LRP6 (2560, Cell signaling), Dvl3 (3218, Cell signaling), CK1 (2655, Cell signaling), GSK3β (GTX59576, GeneTex), βTrCP (4394, Cell signaling), IκBα (4814, Cell signaling), IKKα (2682, Cell signaling), and IKKβ (2684, Cell signaling).
Human prostate cancer cell lines PC3‐P and PC3‐M were purchased from Level Biotechnology Inc. and grown in MEM medium supplemented with 1% penicillin–streptomycin and 10% fetal bovine serum at 37°C with 5% CO2. 22Rv1‐P and 22Rv1‐M were gifts from Dr. Ming‐Shyue Lee and grown in RPMI‐1640 medium supplemented with 1% penicillin–streptomycin, 10% fetal bovine serum at 37°C with 5% CO2. All the cell lines have been tested for mycoplasma contamination. Human recombinant ESM1 protein used for functional studies was purchased from R&D Systems (1810‐EC‐050). Transient transfection using jetPRIME (Polyplus transfection) was performed according to the manufacturer’s instructions.
ShRNA sequences
The shRNA constructs against ESM1, CTNNB1, and RELA were purchased from National RNAi Core Facility. The target sequences are as follows:
shESM1‐1: 5’‐TGG CAT CTG GAG ATG GCA ATA‐3’;
shESM1‐2: 5’‐AGA CCG CAG TGA GTC AAA TTA‐3’;
shCTNNB1‐1: 5’‐GCT TGG AAT GAG ACT GCT GAT‐3’;
shCTNNB1‐2: 5’‐TCT AAC CTC ACT TGC AAT AAT‐3’;
shRELA‐1: 5’‐AGA GGA CAT TGA GGT GTA TTT‐3’;
shRELA‐2: 5’‐CGG ATT GAG GAG AAA CGT AAA‐3’;
Lentivirus production and infection
To prepare lentiviral particles, 293T human embryonic kidneys were transfected using calcium phosphate transfection. Briefly, 293T cells were cultured into 10‐cm2 dishes one day before transfection. Then, the cells were transfected with 10 μg DNA together with 10 μg of pCMVΔR8.91 (packaging vector) and 1 μg of pMD.G (envelope vector). After 16 h of incubation, the transfection medium was replaced with fresh culture medium. Forty‐eight hours later, the lentivirus‐containing medium was collected from transfection, spun down at 380 g for 5 min to pellet the cell debris, the supernatant was filtered with a 0.45‐μm filter, and the target cells, 22Rv1, and PC3 cells were infected with the lentivirus‐containing medium (supplemented with 8 μg/ml polybrene) for 48 h.
Gene expression analysis from public databases
Overall survival analysis of PCa patients from the TCGA‐PRAD database, the PubMed GEO database (GSE16560), and the cBioPortal database was performed using the Kaplan–Meier plotter. Patients were subsequently subgrouped into ESM1 high and ESM low based on their relative expression of ESM1. For the gene expression analysis, we used four datasets (GSE6919, GSE35988, GSE21034, and cBioportal). Patients were subsequently subgrouped based on their metastatic sites.
qRT–PCR
Cellular total RNA was isolated with NucleoZOL reagent (Macherey‐Nagel). The cDNA was synthesized with a PrimerScript RT reagent kit (Takara). The real‐time PCR was performed according to the protocol of iTaq Universal SYBR Green Supermix (Bio‐Rad) using the CFX Connect Real‐Time PCR System (Bio‐Rad). The fluorescence data of the detected genes were normalized to the expression of actin using the 2‐ΔΔCT method. The primers utilized in our investigation are listed in Appendix Table S1.
Immunoblotting
Cells were harvested and lysed in RIPA buffer and heated at 95°C for 10 min. Protein samples were separated by SDS–PAGE, transferred to a polyvinylidene difluoride membrane (PVDF), blocked, and incubated with the indicated primary antibody and horseradish peroxidase (HRP)‐conjugated secondary antibody. The immune complexes for Western blot were visualized using an enhanced ECL system.
Two‐chamber invasion assay
Cell invasion ability was determined using a modified two‐chamber invasion assay (8 μm pore size, Merck Millipore) according to the manufacturer’s instructions. About 3 × 105 cells were seeded into the upper chamber which was coated with 40 μl Matrigel, and the cells were allowed to invade into the lower chamber for 24 h. Cells in the upper chamber were carefully removed using cotton buds, and cells at the bottom of the membrane were fixed and stained with crystal violet 0.2%/ methanol 20%. Quantification was performed by counting the stained cells.
In vivo metastasis assay
Five groups of six male NOD‐SCID mice were injected with 22Rv1‐M‐shScramble cells, 22Rv1‐M‐shESM1‐1 cells, 22Rv1‐M‐shESM1‐2 cells, 22Rv1‐P‐vector cells, and 22Rv1‐P‐ESM1 cells, respectively. Cells (2 × 105 for each group) in 10 μl of Matrigel were injected orthotopically into the anterior lobe of the prostate gland. After inoculation for 8 weeks, all mice were then euthanized and their lungs, liver, kidneys, and intestines were removed. Luciferase activity in the excised organs (measured in photons) was determined using an IVIS Spectrum Imaging System (the Xenogen IVIS® Spectrum system in the Animal Center, NTU). Another five groups of six male NOD‐SCID mice were injected with 22Rv1‐P‐vector cells, 22Rv1‐P‐ESM1 cells, 22Rv1‐P‐ESM1‐NLS cells, 22Rv1‐P‐NES cells, and 22Rv1‐P‐ESM1(w/o) cells, respectively. Cells (1 × 106 for each group) in 100 μl of PBS were injected intravenously via the lateral tail vein. At 7 weeks after injection, all mice were euthanized and their lungs were removed. Luciferase activity in the excised organs (measured in photons) was determined using an IVIS Spectrum Imaging System (the Xenogen IVIS® Spectrum system in the Animal Center, NTU). Mouse lungs were fixed, sectioned, and stained with hematoxylin and eosin (H&E). All animal procedures were using protocols approved by the Nation Taiwan University College of Medicine and College of Public Health Institutional Animal Care and Use Committee, with the IACUC Approval number: 20170354.
Immunohistochemistry
Prostate tumor tissue microarrays were purchased from Biomax (PR8011a, PR8011b, and PR483d), including prostate tumor samples and normal prostate tissue samples. Briefly, samples were deparaffinized and rehydrated followed by antigen retrieving used 0.1 M EDTA (pH 8.0) at a sub‐boiling temperature for 10 min. The sections were then incubated with 3% hydrogen peroxide for 10 min to block endogenous peroxidase activity. After 1 h of preincubation in 3% normal horse serum, the samples were incubated with ESM1 antibody at 4°C overnight. The sections were then washed three times and incubated with a secondary antibody. Then, the slides were developed with a DAB HRP substrate kit and the counterstaining color was carried out using hematoxylin. All immunostained slides were scanned for quantification by digital images.
Spheroid formation
The cells were added to a stem cell culture medium, which was composed of RPMI‐1640 or MEM, supplemented with 50 ng/ml recombinant human EGF (rhEGF) and 20 ng/ml of basic fibroblast growth factor (bFGF) together with B27 and N2. Incubation time depended on the size of spheroids. The number of spheroids was counted when the average diameter of spheroids reached 100 μm.
Cell toxicity
The cell toxicity assay of docetaxel was performed in 96‐well format. Cells were treated with docetaxel at the indicated concentrations for 48 h, and the cell viability index was determined by MTT according to the manufacturer’s instructions.
Flow cytometry
Antibodies for the human antigens CD44 (FITC, eBioscience) and CD133 (PE, eBioscience) were used for flow cytometry analysis. Antibodies were validated according to the manufacturer’s website. Briefly, cells were digested with trypsin, washed, and centrifuged for 3 min at 168 g. Fluorescein isothiocyanate‐conjugated anti‐CD44 (1:100) and phycoerythrin‐conjugated anti‐CD133 (1:100) antibodies were added to the samples, which were next shielded from light and left undisturbed for 30 min at 4°C. Flow cytometry data were collected using BD FACSCalibur and analyzed using CellQuest software.
Xenograft tumor models
Tumor cells were suspended in PBS with 50% Matrigel on ice before the xenograft procedure. The indicated numbers of PC3‐P‐vector or PC3‐P‐ESM1‐NLS cells were subcutaneously inoculated into two groups of nine or ten male NOD‐SCID mice. Tumorigenesis was analyzed at 7 or 8 weeks after injection. The tumors were collected, weighed, and photographed at the end of the experiments. All animal procedures were using protocols approved by the Nation Taiwan University College of Medicine and College of Public Health Institutional Animal Care and Use Committee, with the IACUC Approval number: 20170354.
Luciferase reporter assay
Cells were seeded and transfected with the TOP‐FLASH, FOP‐FLASH, and NFκB‐Luc reporter vectors together with a Renilla luciferase plasmid at a ratio of 10:1. The luciferase activity of the cells was analyzed with the dual‐luciferase® reporter assay system according to the manufacturer’s instructions (Promega). The relative levels of luciferase activity were normalized to the Renilla luciferase activity levels.
Immunoprecipitation
Cells were lysed on ice for 30 min in immunoprecipitation buffer (0.5% NP‐40, 20 mM Tri–HCl, 150 mM NaCl, and 1 mM EDTA) containing protease inhibitor cocktail. Protein concentration was determined by the BCA assay. Cell lysates (1 mg) were incubated with 1 μg of indicated antibodies or control IgG at 4°C overnight with rotary agitation. Protein A/G‐sepharose beads were then added to the lysates and incubated for another 2 h. Beads were washed five times with ice‐cold immunoprecipitation buffer and boiled for 10 min in 1× sample loading buffer. Whole‐cell lysates and immunoprecipitates were resolved by SDS–PAGE and analyzed by Western blotting.
GST and His pull‐down assay
GST‐fused ARM‐FL and different fragments of ARM were expressed in BL21 E. coli and purified using glutathione‐sepharose 4B beads (GE Healthcare) according to the standard protocols. Coomassie blue staining was performed to make possible quantification of minute changes in protein expression. Cell lysates were mixed with purified GST‐ARM‐FL, GST‐ARM‐1 ~2, GST‐ARM‐3 ~8, GST‐ARM‐9 ~12, or GST‐only beads at 4°C overnight. Beads were then washed 5 times with IP buffer before immunoblotting analysis. Ni Sepharose High‐Performance histidine‐tagged protein purification resin (GE Healthcare) was incubated with 1 μg of His‐rhESM1 and GST‐rhβ‐catenin or GST‐rhTCF4 at 4°C overnight. Bound protein was washed 5 times with IP buffer and was subjected to SDS–PAGE and detected by using the indicated antibody.
Immunofluorescence microscopy
Cells were grown in 8‐well chamber slides and fixed in 4% paraformaldehyde, permeabilized, and stained with primary antibodies followed by secondary FITC or Alexa Fluor 594‐conjugated mouse or rabbit antibodies. Slides were examined and photographed using Zeiss LSM 510 META microscope (Carl ZEISS, Jena, Germany). Nuclei were counterstained with 4’,6‐diamidino‐2‐phenylindole (DAPI).
ChIP assay
Cells were cultured up to a confluence of 90–95% and were crosslinked with 1% formalin for 10 min followed by the addition of glycine for 5 min at room temperature. The cells were washed twice with ice‐cold phosphate‐buffered saline. Nuclear extracts were sonicated using Bioruptor® Pico sonication device to sheer crosslinked DNA to an average fragment size of 150–300 bp. Sonicated chromatin was incubated with protein A/G magnetic beads and the indicated antibodies for 18 h at 4°C on a rotor. After incubation, beads were washed thoroughly and the chromatin was eluted from the beads. Crosslinks were removed by incubation with Proteinase K at 62°C for 2 h. DNA was then purified using MinElute PCR Purification Kits (Qiagen). ChIP‐qPCR was performed using iTaq Universal SYBR Green Supermix (Bio‐Rad) on a CFX connect Real‐Time PCR system (Bio‐Rad). The primers utilized in our investigation are listed in Appendix Table S2. The analysis of co‐precipitated DNA showed the presence of DNA fragments corresponding to c‐Myc and CCND1 genes for each of the transcription factors, attesting to the functionality of the ChIP assay.
RNA‐seq analysis
ESM1/ESM1‐NLS/ESM1‐NES‐overexpressing or vector 22Rv1‐P cells were collected, and the total RNA was extracted and purified using an RNeasy Mini kit (Qiagen) according to the manufacturer’s instruction. RNA quantitation and quality control were conducted with a Bioanalyzer 2100 (Agilent Technologies). Construction of stranded RNA‐seq libraries for high‐throughput sequencing was performed on an Illumina HiSeq 4000 following the manufacturer’s protocol. RNA‐seq reads were mapped to the reference genome of Illumina Ensembl genome GRCh37. P‐value was calculated by Student’s t‐test.
Ingenuity pathway analysis
The log2‐fold change was calculated for ESM1, ESM1‐NLS, and ESM1‐NES versus vector control samples based on RNA‐seq gene expression values in FPKM (fragments per kilobase of exon per million fragments mapped). These values were imported to IPA for upstream regulator analysis. Activated upstream regulators are colored orange, while inhibited upstream regulators are colored blue by IPA software.
Gene‐set enrichment analysis
Publicly available TCGA gene expression data of PCa samples were downloaded from the UCSC Xena Platform. The gene expression data of PCa samples from two PubMed GEO datasets (GSE3325 and GSE7930) were also downloaded. These cases were subgrouped into ESM1 high and ESM low based on their relative expression of ESM1, and GSEA was performed using Gene Ontology gene sets. GSEA was also performed on our RNA‐seq gene expression data, comparing ESM1‐NLS samples versus vector control.
Molecular docking
The homology models of ESM1 were built with Htra1‐N (PDB entry 3tjq) as a template on the SWISS‐MODEL online server. A complex model of the ESM1‐β‐catenin complex was calculated by an information‐driven method using the High Ambiguity‐Driven biomolecular Docking (HADDOCK) program. On the basis of surface charge analysis, residues Lys184, Arg191, and Lys194 of β‐catenin were selected as active residues that may interact with Asp31 of ESM1 and the ambiguous interaction restraints (AIRs) were generated in a pairwise manner. Similarly, residues Glu297, Asp298, and Glu301 of β‐catenin were selected as active residues that may interact with Arg26 and Arg42 of ESM1, respectively.
Statistical analysis
All data were statistically analyzed with GraphPad Prism 5 and SigmaPlot 10.0 software. A two‐tailed t‐test was utilized to analyze the difference between two groups. Data were presented as mean ± SD. All findings were considered significant at a P‐value threshold of < 0.05. Where results of statistical test are shown, *P < 0.05, **P < 0.01 unless otherwise indicated.
Author contributions
K.‐f.P. participated in the overall design of the study, performed experiments, and wrote the manuscript. W.‐j.L. participated in the xenograft tumor experiments. C.‐c.C. performed the molecular docking experiments. M.‐h.C., Y.‐c.C., and Y.‐c.Y. analyzed the data. M.H. provided additional reagents and analysis tools for the study. K.‐t.H. supervised and coordinated the whole study. All authors read and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
This study was supported by grants from the Ministry of Sciences and Technology, Taiwan (107‐2320‐B‐002‐060‐MY3, 108‐2628‐B‐002‐014, and 109‐2628‐B‐002‐042). We thank the National RNAi Core Facility at Academia Sinica in Taiwan for providing shRNA reagents and related services. We thank the Flow Cytometric Analyzing and Sorting Core of the First Core Laboratory, College of Medicine, National Taiwan University for technical support. We are grateful for the technical support provided by the Microscopy Core Facility, Department of Medical Research, National Taiwan University Hospital.
The EMBO Journal (2021) 40: e105450.
Data availability
RNA sequencing data reported here are available at GEO, under the superseries, GSE157496. The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Anastas JN, Moon RT (2013) WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 13: 11–26 [DOI] [PubMed] [Google Scholar]
- Asally M, Yoneda Y (2005) Beta‐catenin can act as a nuclear import receptor for its partner transcription factor, lymphocyte enhancer factor‐1 (lef‐1). Exp Cell Res 308: 357–363 [DOI] [PubMed] [Google Scholar]
- Bechard D, Gentina T, Delehedde M, Scherpereel A, Lyon M, Aumercier M, Vazeux R, Richet C, Degand P, Jude B et al (2001) Endocan is a novel chondroitin sulfate/dermatan sulfate proteoglycan that promotes hepatocyte growth factor/scatter factor mitogenic activity. J Biol Chem 276: 48341–48349 [DOI] [PubMed] [Google Scholar]
- Bechard D, Scherpereel A, Hammad H, Gentina T, Tsicopoulos A, Aumercier M, Pestel J, Dessaint JP, Tonnel AB, Lassalle P (2001) Human endothelial‐cell specific molecule‐1 binds directly to the integrin CD11a/CD18 (LFA‐1) and blocks binding to intercellular adhesion molecule‐1. J Immunol 167: 3099–3106 [DOI] [PubMed] [Google Scholar]
- Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus DM et al (2013) Targeted next‐generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol 63: 920–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson I, Prowse DM (2009) WNT signaling regulates self‐renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res 19: 683–697 [DOI] [PubMed] [Google Scholar]
- Chauhan A, Tikoo A, Kapur AK, Singh M (2007) The taming of the cell penetrating domain of the HIV Tat: myths and realities. J Control Release 117: 148–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CM, Lin CL, Chiou HL, Hsieh SC, Lin CL, Cheng CW, Hung CH, Tsai JP, Hsieh YH (2017) Loss of endothelial cell‐specific molecule 1 promotes the tumorigenicity and metastasis of prostate cancer cells through regulation of the TIMP‐1/MMP‐9 expression. Oncotarget 8: 13886–13897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Shukeir N, Potti A, Sircar K, Aprikian A, Goltzman D, Rabbani SA (2004) Up‐regulation of Wnt‐1 and beta‐catenin production in patients with advanced metastatic prostate carcinoma: potential pathogenetic and prognostic implications. Cancer 101: 1345–1356 [DOI] [PubMed] [Google Scholar]
- Chesire DR, Ewing CM, Sauvageot J, Bova GS, Isaacs WB (2000) Detection and analysis of beta‐catenin mutations in prostate cancer. Prostate 45: 323–334 [DOI] [PubMed] [Google Scholar]
- Chesire DR, Isaacs WB (2002) Ligand‐dependent inhibition of beta‐catenin/TCF signaling by androgen receptor. Oncogene 21: 8453–8469 [DOI] [PubMed] [Google Scholar]
- Ciarlo M, Benelli R, Barbieri O, Minghelli S, Barboro P, Balbi C, Ferrari N (2012) Regulation of neuroendocrine differentiation by AKT/hnRNPK/AR/beta‐catenin signaling in prostate cancer cells. Int J Cancer 131: 582–590 [DOI] [PubMed] [Google Scholar]
- Cookson MS, Roth BJ, Dahm P, Engstrom C, Freedland SJ, Hussain M, Lin DW, Lowrance WT, Murad MH, Oh WK et al (2013) Castration‐resistant prostate cancer: AUA Guideline. J Urol 190: 429–438 [DOI] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC et al (2012) The mutational landscape of lethal castration‐resistant prostate cancer. Nature 487: 239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriu BD, Depontieu F, Scherpereel A, Gourcerol D, Devos P, Ouatas T, Lafitte JJ, Copin MC, Tonnel AB, Lassalle P (2006) Endocan expression and relationship with survival in human non‐small cell lung cancer. Clin Cancer Res 12: 4575–4582 [DOI] [PubMed] [Google Scholar]
- Heitz F, Morris MC, Divita G (2009) Twenty years of cell‐penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol 157: 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson BR, Fagotto F (2002) The ins and outs of APC and beta‐catenin nuclear transport. EMBO Rep 3: 834–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herheliuk T, Perepelytsina O, Ugnivenko A, Ostapchenko L, Sydorenko M (2019) Investigation of multicellular tumor spheroids enriched for a cancer stem cell phenotype. Stem Cell Investig 6: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WY, Hu DP, Xie L, Li Y, Majumdar S, Nonn L, Hu H, Shioda T, Prins GS (2017) Isolation and functional interrogation of adult human prostate epithelial stem cells at single cell resolution. Stem Cell Res 23: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro T, Ohata H, Sato A, Yamawaki K, Enomoto T, Okamoto K (2017) Tumor‐derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci 108: 283–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson C, Sharma M, Henderson BR (2014) Targeting the beta‐catenin nuclear transport pathway in cancer. Semin Cancer Biol 27: 20–29 [DOI] [PubMed] [Google Scholar]
- Jung Y, Cackowski FC, Yumoto K, Decker AM, Wang J, Kim JK, Lee E, Wang Y, Chung JS, Gursky AM et al (2018) CXCL12gamma promotes metastatic castration‐resistant prostate cancer by inducing cancer stem cell and neuroendocrine phenotypes. Cancer Res 78: 2026–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kali A, Shetty KS (2014) Endocan: a novel circulating proteoglycan. Indian J Pharmacol 46: 579–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt C, Banz‐Jansen C, Benhidjeb T, Beshay M, Forster C, Greiner J, Hamelmann E, Jorch N, Mertzlufft F, Pfitzenmaier J et al (2019) A role for NF‐kappaB in organ specific cancer and cancer stem cells. Cancers 11: 655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YH, Ji NY, Han SR, Lee CI, Kim JW, Yeom YI, Kim YH, Chun HK, Kim JW, Chung JW et al (2012) ESM‐1 regulates cell growth and metastatic process through activation of NF‐kappaB in colorectal cancer. Cell Signal 24: 1940–1949 [DOI] [PubMed] [Google Scholar]
- Kim KH, Lee HH, Yoon YE, Na JC, Kim SY, Cho YI, Hong SJ, Han WK (2018) Clinical validation of serum endocan (ESM‐1) as a potential biomarker in patients with renal cell carcinoma. Oncotarget 9: 662–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Han ME, Oh SO (2017) The molecular mechanism for nuclear transport and its application. Anat Cell Biol 50: 77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski JM, Fidler IJ, Campbell D, Xu ZL, Kaighn ME, Hart IR (1984) Metastatic behavior of human tumor cell lines grown in the nude mouse. Cancer Res 44: 3522–3529 [PubMed] [Google Scholar]
- Kretzschmar K, Cottle DL, Schweiger PJ, Watt FM (2015) The androgen receptor antagonizes Wnt/beta‐catenin signaling in epidermal stem cells. J Invest Dermatol 135: 2753–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieghoff E, Behrens J, Mayr B (2006) Nucleo‐cytoplasmic distribution of beta‐catenin is regulated by retention. J Cell Sci 119: 1453–1463 [DOI] [PubMed] [Google Scholar]
- Krishnamurthy N, Kurzrock R (2018) Targeting the Wnt/beta‐catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev 62: 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kypta RM, Waxman J (2012) Wnt/beta‐catenin signalling in prostate cancer. Nat Rev Urol 9: 418–428 [DOI] [PubMed] [Google Scholar]
- Laloglu E, Kumtepe Y, Aksoy H, Topdagi Yilmaz EP (2017) Serum endocan levels in endometrial and ovarian cancers. J Clin Lab Anal 31: e22079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassalle P, Molet S, Janin A, Heyden JV, Tavernier J, Fiers W, Devos R, Tonnel AB (1996) ESM‐1 is a novel human endothelial cell‐specific molecule expressed in lung and regulated by cytokines. J Biol Chem 271: 20458–20464 [DOI] [PubMed] [Google Scholar]
- Lee E, Ha S, Logan SK (2015) Divergent androgen receptor and beta‐catenin signaling in prostate cancer cells. PLoS One 10: e0141589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy X, Aubert S, Zini L, Franquet H, Kervoaze G, Villers A, Delehedde M, Copin MC, Lassalle P (2010) Vascular endocan (ESM‐1) is markedly overexpressed in clear cell renal cell carcinoma. Histopathology 56: 180–187 [DOI] [PubMed] [Google Scholar]
- Liu C, Liu R, Zhang D, Deng Q, Liu B, Chao HP, Rycaj K, Takata Y, Lin K, Lu Y et al (2017) MicroRNA‐141 suppresses prostate cancer stem cells and metastasis by targeting a cohort of pro‐metastasis genes. Nat Commun 8: 14270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Yin J, Wang H, Jiang G, Deng M, Zhang G, Bu X, Cai S, Du J, He Z (2015) FOXO3a modulates WNT/beta‐catenin signaling and suppresses epithelial‐to‐mesenchymal transition in prostate cancer cells. Cell Signal 27: 510–518 [DOI] [PubMed] [Google Scholar]
- Liu N, Zhang LH, Du H, Hu Y, Zhang GG, Wang XH, Li JY, Ji JF (2010) Overexpression of endothelial cell specific molecule‐1 (ESM‐1) in gastric cancer. Ann Surg Oncol 17: 2628–2639 [DOI] [PubMed] [Google Scholar]
- Lu Y, Xie S, Zhang W, Zhang C, Gao C, Sun Q, Cai Y, Xu Z, Xiao M, Xu Y et al (2017) Twa1/Gid8 is a beta‐catenin nuclear retention factor in Wnt signaling and colorectal tumorigenesis. Cell Res 27: 1422–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madueke I, Hu WY, Hu D, Swanson SM, Vander Griend D, Abern M, Prins GS (2019) The role of WNT10B in normal prostate gland development and prostate cancer. Prostate 79: 1692–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland DJ, Cheng H, Reid K, Rennie PS, Nelson CC (2002) The androgen receptor can promote beta‐catenin nuclear translocation independently of adenomatous polyposis coli. J Biol Chem 277: 17933–17943 [DOI] [PubMed] [Google Scholar]
- Murillo‐Garzon V, Kypta R (2017) WNT signalling in prostate cancer. Nat Rev Urol 14: 683–696 [DOI] [PubMed] [Google Scholar]
- Patel R, Brzezinska EA, Repiscak P, Ahmad I, Mui E, Gao M, Blomme A, Harle V, Tan EH, Malviya G et al (2020) Activation of beta‐catenin cooperates with loss of Pten to drive AR‐independent castration‐resistant prostate cancer. Cancer Res 80: 576–590 [DOI] [PubMed] [Google Scholar]
- Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI (2011) Tumour‐initiating stem‐like cells in human prostate cancer exhibit increased NF‐kappaB signalling. Nat Commun 2: 162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindranath A, Yuen HF, Chan KK, Grills C, Fennell DA, Lappin TR, El‐Tanani M (2011) Wnt‐beta‐catenin‐Tcf‐4 signalling‐modulated invasiveness is dependent on osteopontin expression in breast cancer. Br J Cancer 105: 542–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo J, Geliebter J, Reyes N (2017) ESM‐1 siRNA knockdown decreased migration and expression of CXCL3 in prostate cancer cells. Int J Biomed Sci 13: 35–42 [PMC free article] [PubMed] [Google Scholar]
- Reya T, Clevers H (2005) Wnt signalling in stem cells and cancer. Nature 434: 843–850 [DOI] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G et al (2015) Integrative clinical genomics of advanced prostate cancer. Cell 161: 1215–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha SF, Schiller M, Jing D, Li H, Butz S, Vestweber D, Biljes D, Drexler HC, Nieminen‐Kelha M, Vajkoczy P et al (2014) Esm1 modulates endothelial tip cell behavior and vascular permeability by enhancing VEGF bioavailability. Circ Res 115: 581–590 [DOI] [PubMed] [Google Scholar]
- Roudnicky F, Poyet C, Wild P, Krampitz S, Negrini F, Huggenberger R, Rogler A, Stohr R, Hartmann A, Provenzano M et al (2013) Endocan is upregulated on tumor vessels in invasive bladder cancer where it mediates VEGF‐A‐induced angiogenesis. Cancer Res 73: 1097–1106 [DOI] [PubMed] [Google Scholar]
- Schepeler T, Holm A, Halvey P, Nordentoft I, Lamy P, Riising EM, Christensen LL, Thorsen K, Liebler DC, Helin K et al (2012) Attenuation of the beta‐catenin/TCF4 complex in colorectal cancer cells induces several growth‐suppressive microRNAs that target cancer promoting genes. Oncogene 31: 2750–2760 [DOI] [PubMed] [Google Scholar]
- Schneider JA, Logan SK (2018) Revisiting the role of Wnt/beta‐catenin signaling in prostate cancer. Mol Cell Endocrinol 462: 3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang S, Hua F, Hu ZW (2017) The regulation of beta‐catenin activity and function in cancer: therapeutic opportunities. Oncotarget 8: 33972–33989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Jamieson C, Johnson M, Molloy MP, Henderson BR (2012) Specific armadillo repeat sequences facilitate beta‐catenin nuclear transport in live cells via direct binding to nucleoporins Nup62, Nup153, and RanBP2/Nup358. J Biol Chem 287: 819–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Song LN, Herrell R, Byers S, Shah S, Wilson EM, Gelmann EP (2003) Beta‐catenin binds to the activation function 2 region of the androgen receptor and modulates the effects of the N‐terminal domain and TIF2 on ligand‐dependent transcription. Mol Cell Biol 23: 1674–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truica CI, Byers S, Gelmann EP (2000) Beta‐catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res 60: 4709–4713 [PubMed] [Google Scholar]
- Tsai CH, Tzeng SF, Chao TK, Tsai CY, Yang YC, Lee MT, Hwang JJ, Chou YC, Tsai MH, Cha TL et al (2016) Metastatic progression of prostate cancer is mediated by autonomous binding of galectin‐4‐O‐glycan to cancer cells. Cancer Res 76: 5756–5767 [DOI] [PubMed] [Google Scholar]
- Voeller HJ, Truica CI, Gelmann EP (1998) Beta‐catenin mutations in human prostate cancer. Cancer Res 58: 2520–2523 [PubMed] [Google Scholar]
- Wan X, Liu J, Lu JF, Tzelepi V, Yang J, Starbuck MW, Diao L, Wang J, Efstathiou E, Vazquez ES et al (2012) Activation of beta‐catenin signaling in androgen receptor‐negative prostate cancer cells. Clin Cancer Res 18: 726–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Kimelman D (2007) Mechanistic insights from structural studies of beta‐catenin and its binding partners. J Cell Sci 120: 3337–3344 [DOI] [PubMed] [Google Scholar]
- Yang J, Yang Q, Yu S, Zhang X (2015) Endocan: a new marker for cancer and a target for cancer therapy. Biomed Rep 3: 279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YC, Pan KF, Lee WJ, Chang JH, Tan P, Gu CC, Chang WM, Yang SF, Hsiao M, Hua KT et al (2020) Circulating proteoglycan endocan mediates EGFR‐driven progression of non‐small cell lung cancer. Cancer Res 80: 3292–3304 [DOI] [PubMed] [Google Scholar]
- Yeh Y, Guo Q, Connelly Z, Cheng S, Yang S, Prieto‐Dominguez N, Yu X (2019) Wnt/Beta‐catenin signaling and prostate cancer therapy resistance. Adv Exp Med Biol 1210: 351–378 [DOI] [PubMed] [Google Scholar]
- Yokoyama NN, Shao S, Hoang BH, Mercola D, Zi X (2014) Wnt signaling in castration‐resistant prostate cancer: implications for therapy. Am J Clin Exp Urol 2: 27–44 [PMC free article] [PubMed] [Google Scholar]
- Yu X, Wang Y, DeGraff DJ, Wills ML, Matusik RJ (2011) Wnt/beta‐catenin activation promotes prostate tumor progression in a mouse model. Oncogene 30: 1868–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun EJ, Zhou J, Lin CJ, Hernandez E, Fazli L, Gleave M, Hsieh JT (2016) Targeting cancer stem cells in castration‐resistant prostate cancer. Clin Cancer Res 22: 670–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan T, Rindtorff N, Boutros M (2017) Wnt signaling in cancer. Oncogene 36: 1461–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Guo Y, Wang X, Zhao H, Ji Z, Cheng C, Li L, Fang Y, Xu D, Zhu HH et al (2017) WNT/beta‐catenin directs self‐renewal symmetric cell division of hTERT(high) prostate cancer stem cells. Cancer Res 77: 2534–2547 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
RNA sequencing data reported here are available at GEO, under the superseries, GSE157496. The data that support the findings of this study are available from the corresponding author upon reasonable request.