

Abstract
Autophagy is a lysosome‐dependent degradation pathway essential to maintain cellular homeostasis. Therefore, either defective or excessive autophagy may be detrimental for cells and tissues. The past decade was characterized by significant advances in molecular dissection of stimulatory autophagy inputs; however, our understanding of the mechanisms that restrain autophagy is far from complete. Here, we describe a negative feedback mechanism that limits autophagosome biogenesis based on the selective autophagy‐mediated degradation of ATG13, a component of the ULK1 autophagy initiation complex. We demonstrate that the centrosomal protein OFD1 acts as bona fide autophagy receptor for ATG13 via direct interaction with the Atg8/LC3/GABARAP family of proteins. We also show that patients with Oral‐Facial‐Digital type I syndrome, caused by mutations in the OFD1 gene, display excessive autophagy and that genetic inhibition of autophagy in a mouse model of the disease, significantly ameliorates polycystic kidney, a clinical manifestation of the disorder. Collectively, our data report the discovery of an autophagy self‐regulated mechanism and implicate dysregulated autophagy in the pathogenesis of renal cystic disease in mammals.
Keywords: autophagy receptor, OFD1, polycystic kidney, selective autophagy
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport
Centrosomal protein OFD1 restrains autophagy and prevents pathogenesis of renal cystic disease in mammals through selective autophagy‐mediated degradation of the ULK1‐complex component ATG13.

Introduction
Autophagy is a cellular degradation pathway in which cellular components are engulfed into double‐membrane vesicles, called autophagosomes, which then fuse with lysosomes to degrade their content and recycle nutrients back into the cytoplasm. Autophagy is active at a basal level in most cell types and is strongly induced by starvation (Levine & Kroemer, 2008; Mizushima & Komatsu, 2011). Bulk autophagy is non‐selective, while selective autophagy involves recognition and removal of specific targets through autophagy receptors. These receptors link their cargo to autophagosomes through an LC3B‐interacting region (LIR) that mediates binding to autophagosomal membrane proteins LC3 and/or GABARAP, key proteins involved in phagophore maturation, thus enabling cargo sequestration and degradation (Stolz et al, 2014; Gatica et al, 2018). Autophagy plays a crucial role in human health and disease (Mizushima, 2018) with a protective effect in neurodegeneration and lysosomal storage disorders (Levine & Kroemer, 2019). On the other side, excessive autophagy has been observed in different types of cancers (Amaravadi et al, 2016), or in myocardial and cerebral ischemia where cell death is a consequence of excessive self‐digestion (Marino et al, 2014; Antonioli et al, 2017).
In the past decade, we witnessed a rapid comprehension of the molecular pathways that regulate activation of autophagy, autophagosome formation, and lysosome fusion. However, our understanding of the molecular mechanisms that inhibit autophagy is still limited (Antonioli et al, 2017). Here, we describe a novel negative feedback mechanism that limits autophagosome biogenesis based on selective autophagy‐mediated degradation of ATG13, a component of the unc‐51‐like kinase (ULK1) complex. The ULK1 complex, that comprises the serine/threonine kinase ULK1 and the regulatory components ATG13, FIP200, and ATG101 (Mercer et al, 2018), plays a crucial role in the initiation steps of autophagosome biogenesis, through ULK1‐mediated phosphorylation of downstream targets (Wirth et al, 2013; Wong et al, 2013). We show that a centrosomal/centriolar satellite protein as OFD1 (Romio et al, 2004; Ferrante et al, 2006; Giorgio et al, 2007; Singla et al, 2010) inhibits early phases of the LC3‐mediated autophagic cascade. OFD1 localizes to distal ends of centrioles, constrains centriole elongation, and controls primary cilia formation (Ferrante et al, 2006; Singla et al, 2010; Tang et al, 2013; Morleo & Franco, 2020). Mutations in the OFD1 gene causes the Oral‐Facial‐Digital (OFD) type I syndrome (Ferrante et al, 2001), a X‐linked dominant male‐lethal disorder, characterized by a variable phenotype displaying malformations of the face, oral cavity and digits, and cystic kidneys (Ferrante et al, 2001; Prattichizzo et al, 2008; Saal et al, 2010). We also show that patients with OFD type I syndrome display unrestrained autophagy and demonstrate that autophagy inhibition significantly ameliorates the renal cystic phenotype in Ofd1 mutant mice, thus paving the way to the exploitation of autophagy modulation as possible therapeutic strategy for renal cystic disease in mammals.
Results and Discussion
In a previous tandem mass spectrometry study (Iaconis et al, 2017), we identified FIP200 as an OFD1 interactor, and we thus examined whether OFD1 interacts with other members of the ULK1 complex by co‐immunoprecipitation (co‐IP). Interestingly, we observed that in Human Kidney (HK)2 cells endogenous OFD1 interacts with both FIP200 and ATG13, in nutrient‐rich and nutrient‐depleted conditions (Fig 1A). OFD1 predominantly localizes at centrosomes in HK2 cells (Fig EV1A). By immunofluorescence (IF), we observed a partial colocalization of HA‐OFD1 with endogenous ATG13‐ and FIP200‐positive puncta mainly at centrosomes (labeled with γTUBULIN) in nutrient‐starved HK2 cells (Fig 1B and C). Transfection of cells with siRNAs directed against ATG13 and RB1CC1 (FIP200 codifying gene) demonstrated the specificity of the centrosomal signal observed with anti‐ATG13 and anti‐FIP200 antibodies (Fig EV1B and C). We thus examined the protein levels of components of the ULK1 complex in OFD1 knockout HK2 cells (KO‐OFD1). The levels of FIP200, ATG13, ULK1, and ATG101 were higher in KO‐OFD1 compared with wild‐type (wt) cells in both nutrient‐rich and nutrient‐depleted conditions (Fig 2A). We also analyzed mRNA expression levels of ULK1, ATG13, RB1CC1, and ATG101 and observed no differences between KO‐OFD1 cells and controls (Fig EV2). Conversely, cycloheximide chase analysis revealed a slower decay of protein levels for all components of the ULK1 complex in KO‐OFD1 cells compared with wt (Fig 2B), suggesting that in the absence of OFD1 the complex is stabilized. In line with these findings, we also observed an increased number of ULK1 and ATG13 puncta in KO‐OFD1 cells (Fig 2C). Moreover, when OFD1 was introduced back into KO‐OFD1 cells we observed a reduction of ULK1 complex components levels (Fig 2D). These data indicate that OFD1 interacts with components of the ULK1 complex and controls their degradation in steady state conditions and during starvation.
Figure 1. OFD1 interacts with components of the ULK1 complex.
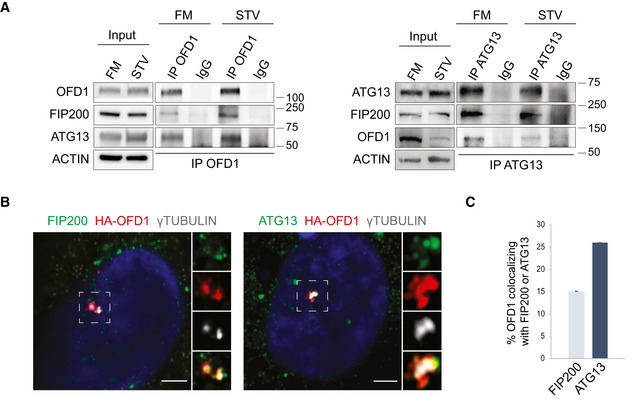
- Lysates of HK2 cells cultured in FM or STV (2 h) were immunoprecipitated with anti‐OFD1 (left) and anti‐ATG13 (right) antibodies or IgG and analyzed by Western blot (WB); ACTIN was used as loading control. n = 3 independent experiments. IP = immunoprecipitation, FM = full medium, STV = HBSS starvation.
- Airyscan confocal analysis of endogenous FIP200/γTUBULIN and ATG13/γTUBULIN colocalization with lentiviral delivered HA‐OFD1 in HK2 cells. Cells were cultured in HBSS (90 min). Green, FIP200 and ATG13; red, HA‐OFD1; gray, γTUBULIN; blue, Hoechst for nuclei. Insets show magnifications and single‐color channels of selected areas. Scale bar = 3.5 μm.
- Quantification of HA‐OFD1/ATG13 and HA‐OFD1/FIP200 colocalization, expressed as % of total OFD1 fluorescence (mean ± SEM). n = 3 independent experiments; n = 53 cells for ATG13, and n = 39 cells for FIP200 were analyzed.
Source data are available online for this figure.
Figure EV1. Centrosomal localization of OFD1, ATG13, and FIP200.

- Representative confocal images of OFD1 (green) and γTUBULIN (red) staining in wt and KO‐OFD1 cells. Hoechst (blue) labels nuclei. Insets show colocalization in selected (dotted) areas at higher magnification. Scale bar = 10 μm.
- Immunofluorescence staining of HK2 cells transfected with siRNAs against ATG13 (siATG13) or scramble (Ctrl) for 72 h, after 90 min of starvation in HBSS. Insets show colocalization signals at higher magnification of selected (dotted) areas. Green, ATG13; red, γTUBULIN; blue, Hoechst to label nuclei. Scale bar = 10 μm. n = 50 cells analyzed.
- Immunofluorescence staining of HK2 cells transfected with siRNAs against RB1CC1 (siRB1CC1), the gene codifying for FIP200, or scramble (Ctrl) for 72 h, after 90 min of starvation with HBSS. Insets show colocalization in selected areas at higher magnification. Green, FIP200; red, γTUBULIN; blue, Hoechst to label nuclei. Scale bar = 10 μm. n = 50 cells analyzed.
Figure 2. OFD1 controls ULK1 complex stability through autophagy.

- Top. Western blot of ULK1‐complex components in wt and KO‐OFD1 cells assessed in FM (−) or STV (2, 4 h). Bands at same exposure, noncontiguous on the same gel. Bottom. ACTIN‐normalized protein levels are expressed as fold increase compared with wt cells, represented by the dashed line (mean ± SEM); n = 5 independent experiments. FM = full medium, STV = HBSS starvation.
- Top. Wild‐type and KO‐OFD1 cells were incubated with 50 µg/ml CHX. Immunoblots were probed with indicated antibodies. Bottom. Quantification of βTUBULIN and ACTIN‐normalized protein levels vs. untreated conditions (−), data are expressed as mean ± SEM; n = 4 independent experiments. CHX = cycloheximide, ns = not significative.
- Immunofluorescence of ULK1 and ATG13 puncta in wt and KO‐OFD1 cells cultured in HBSS (90 min). Green, ATG13 and ULK1; blue, Hoechst for nuclei. Scale bar = 10 μm. Right. Graphs display quantification of puncta/cell, data are expressed as mean ± SEM; n = 5 independent experiments, n ≥ 200 cells/antibody.
- Western blot of ULK1 complex components in KO‐OFD1 cells transfected with empty vector (Empty) or 3xFLAG‐OFD1 (OFD1). Histograms show quantification of band intensities normalized vs. ACTIN expressed as mean ± SEM. n = 5 independent experiments.
- Western blot of ULK1‐complex components in KO‐OFD1 cells transfected with 3xFLAG‐OFD1 (OFD1) or empty vector (Empty) and treated (+) or not (−) with SAR405 (10 μM, 6 h). ACTIN is the loading control. On the right, ATG13 levels are expressed as fold increase compared with empty vector (mean ± SEM); n = 3 independent experiments.
- Representative confocal images of co‐staining of 3xFLAG‐OFD1 with ATG13 in KO‐OFD1 cells incubated in HBSS, treated (+) or not (−) with Baf‐A1 (100 nM, 90 min). Green, ATG13; red, 3xFLAG‐OFD1; blue, Hoechst for nuclei. Scale bar = 10 μm. Right. ATG13 puncta/cell are quantified and expressed as mean ± SEM; n = 3 independent experiments, n ≥ 100 cells.
Data information: Statistical analysis: One‐tailed Student’s t‐test was applied in (A, B, D, and E); the likelihood ratio test for Negative Binomial generalized linear models was applied in (C and F). *P ≤ 0.05, **P ≤ 0.01, ns = not significative.
Source data are available online for this figure.
Figure EV2. Expression levels of genes codifying for components of the ULK1 complex do not increase in KO‐OFD1 cells.

qRT–PCR analysis of mRNA expression levels of ULK1, RB1CC1, ATG13, and ATG101 in wt and KO‐OFD1 cells assessed in fed condition (FM) or following 2 h of HBSS starvation (STV). qRT–PCR expression analysis is expressed as fold change after normalization on different reference genes (ACTIN, B2M, RPL13A mRNA), as mean ± SEM of n ≥ 3 independent biological replicates. One‐tailed Student's t‐test was applied; *P ≤ 0.05; ns = not significative.
We next sought to determine whether OFD1‐mediated ULK1 complex degradation occurred through the proteasome or the autophagic pathway. To inhibit the autophagic pathway, we treated KO‐OFD1 cells transfected with 3xFLAG‐OFD1 with SAR405, an inhibitor of autophagosomes assembly. Autophagy inhibition resulted in a significant rescue of ATG13 levels (Fig 2E). These results were also confirmed by treating 3xFLAG‐OFD1 transfected cells with bafilomycin‐A1 (Baf‐A1), an inhibitor of autophagosome‐lysosome fusion (Fig EV3A). Consistently, the number of ATG13 puncta, which decreased in 3xFLAG‐OFD1 overexpressing cells compared with controls (Fig 2F), showed no significant variations between Baf‐A1–treated 3xFLAG‐OFD1 overexpressing cells and controls (Fig 2F). Although proteasome‐mediated mechanisms for ULK1 kinase degradation have been extensively studied in mammals (Liu et al, 2016; Nazio et al, 2016), relatively little is known regarding ATG13 turnover. As previous reports in yeast and Arabidopsis, showed that ATG13 is targeted to autophagy‐dependent degradation in vacuoles (Suttangkakul et al, 2011; Kraft et al, 2012), we confirmed this observation in mammals through both pharmacological treatment (SAR405, Baf‐A1, and proteasome inhibitors MG132 and bortezomib) and genetic approaches (KO‐ATG9, Saos‐2 cells depleted for ATG9, a key regulator of the autophagic cascade) (Fig EV3, EV4, EV5). Conversely, we showed, consistent with previous reports, that ULK1 kinase is mainly degraded through the proteasome in HK2 and Saos‐2 cells (Fig EV3, EV4, EV5). Taken together, these results demonstrate that OFD1 promotes ATG13 degradation through autophagy.
Figure EV3. ATG13 is degraded through autophagy.
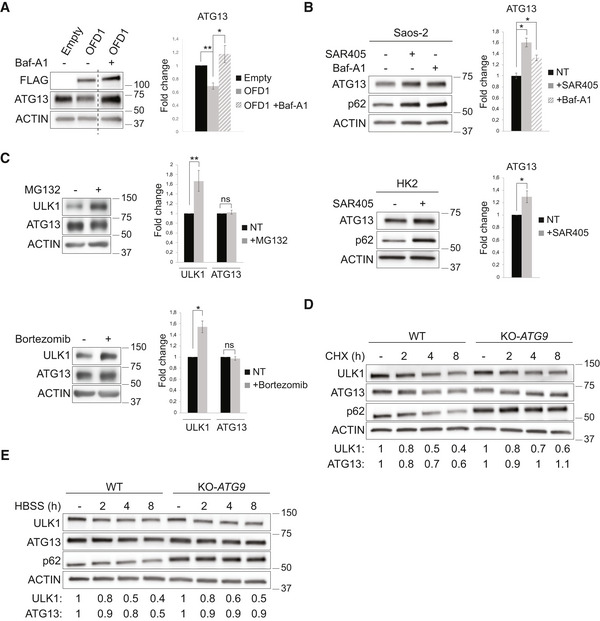
- Western blot analysis of ATG13 in KO‐OFD1 cells transfected with 3xFLAG‐OFD1 (OFD1) or empty vector (Empty) and treated (+) or not (−) with Baf‐A1 (100 nM, 2 h). Bands at same exposure, noncontiguous on the same gel. Right. Protein levels values, normalized vs. ACTIN (loading control), are expressed as fold increase (mean ± SEM) compared with the empty vector; n = 3 independent experiments. One‐tailed Student’s t‐test was applied; *P ≤ 0.05, **P ≤ 0.01. Baf‐A1 = bafilomycin.
- Western blot of ATG13 and p62 in human osteosarcoma (Saos‐2, top) and HK2 (bottom) cells treated (+) or not (−) with SAR405 (10 µM, 6 h) or Baf‐A1 (100 nM, 6 h). Quantification of ATG13 protein levels, normalized vs. ACTIN (loading control), is expressed as fold change (mean ± SEM) compared with untreated conditions (−); n = 3 independent experiments. One‐tailed Student’s t‐test was applied; *P ≤ 0.05. Baf‐A1 = bafilomycin.
- Western blot of ULK1 and ATG13 levels in HK2 cells treated (+) or not (−) with MG132 (10 µM, 2 h, top) or Bortezomib (100 nM, 2 h, bottom). Quantification of protein levels normalized vs. ACTIN (loading control) is expressed as fold change (mean ± SEM) compared with untreated conditions (−); n = 3 independent experiments. One‐tailed Student’s t‐test was applied; *P ≤ 0.05, **P ≤ 0.01.
- Representative blot of the indicated proteins in wt and KO‐ATG9 Saos‐2 cells which were incubated with 50 µg/ml cycloheximide (CHX) and collected at the indicated time points. Quantification of ATG13 and ULK1 protein levels, normalized vs. ACTIN (loading control), is expressed as fold change compared with untreated conditions (−).
- Representative blot of time course evaluating levels of the indicated proteins in wt and KO‐ATG9 Saos‐2 cells during starvation (HBSS treatment) and collected at the indicated time points. Quantification of ATG13 and ULK1 protein levels, normalized vs. ACTIN (loading control), is expressed as fold change compared with full medium condition (−).
Data information: For D and E: quantification of ATG13/ULK1 protein levels is referred to the blots shown in the figure, which are representative of three independent experiments.
Source data are available online for this figure.
Figure EV4. The increased autophagic flux in KO‐OFD1 cells is mTOR independent.
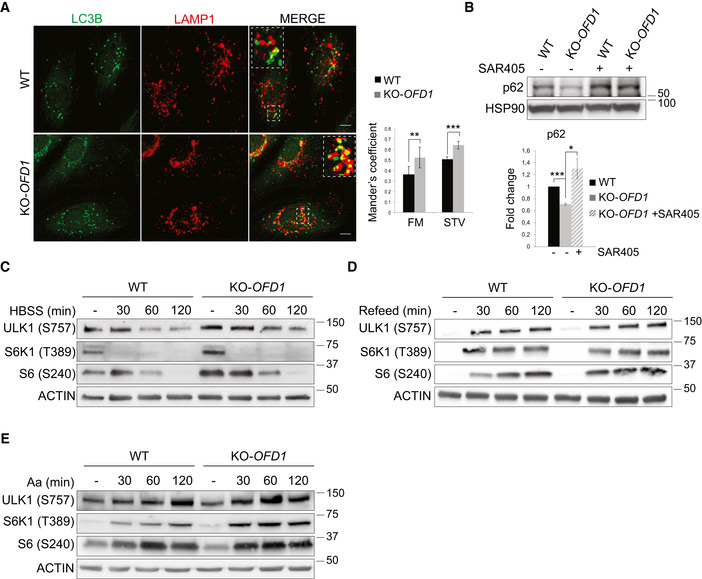
- Immunofluorescence analysis of LAMP1 (red) and LC3B (green) in wt and KO‐OFD1 cells following 90 min of starvation in HBBS. Square dotted areas show colocalization at higher magnification. Scale bars = 10 μm. Histograms on the right show quantification of LAMP1‐LC3B colocalization in fed (FM) or starved (STV) conditions expressed as mean ± SEM. n = 3 independent experiments; n = 200 cells analyzed.
- Western blot analysis of p62 in wt and KO‐OFD1 cells assessed following 2 h of starvation and treated (+) or not treated (−) with SAR405 (10 μM, 4 h). Bottom. Protein level values, normalized vs. HSP90 (loading control), are expressed as fold increase compared with untreated wt cells (mean ± SEM). n = 3 independent experiments.
- Western blot analysis of mTORC1 signaling upon nutrients deprivation in wt and KO‐OFD1 cells fed in full medium (−) and then starved in HBSS and collected at the indicated time points. Upon treatment, cell extracts were analyzed with the indicated antibodies.
- Western blot analysis of mTORC1 signaling upon nutrients stimulation in wt and KO‐OFD1 cells starved for 2 h in HBSS (−) and then refed with complete medium and collected at the indicated time points. Upon treatment, cell extracts were analyzed with the indicated antibodies.
- Wild‐type and KO‐OFD1 cells were deprived of amino acids (Aa) for 60 min (−) and then stimulated with amino acids for the indicated duration (min). Cell extracts were analyzed by WB with the indicated antibodies.
Data information: Data are expressed as mean values derived from the indicated independent experiments. Error bars indicate the SEM. One‐tailed Student’s t‐test was applied (A, B). *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. FM = full medium, STV = starvation, Ctrl = control, Aa = amino acids.
Source data are available online for this figure.
Figure EV5. Autophagosome biogenesis increases in KO‐OFD1 cells in ciliated conditions.
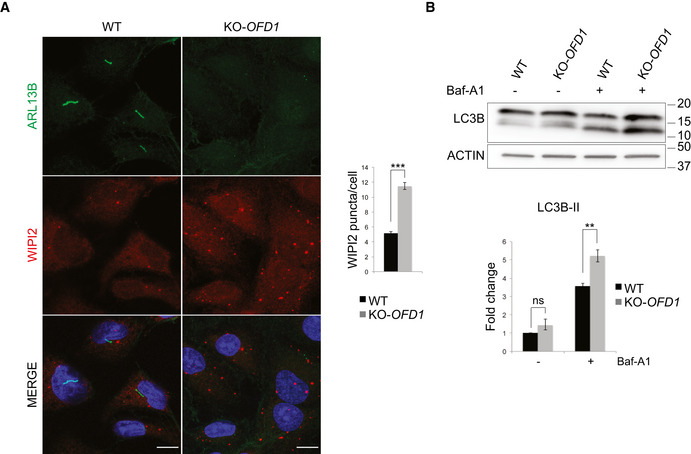
- Representative confocal images of primary cilia, marked by ARL13B, and of WIPI2 puncta in wt and KO‐OFD1 cells after 24 h of serum starvation. Green, ARL13B; red, WIPI2; blue, Hoechst labels nuclei. Scale bar = 10 μm. Histograms on the right show quantification of WIPI2 puncta which is expresses as mean ± SEM. ≥ 300 cells analyzed/sample, n = 5 independent experiments. The likelihood ratio test for Negative Binomial generalized linear models was applied; ***P ≤ 0.001.
- Wild‐type and KO‐OFD1 cells were serum starved for 24 h and treated (+) or not treated (−) with Baf‐A1 (100 nM, 2 h), and total lysates were analyzed by immunoblotting using anti‐LC3B and ‐ACTIN (loading control) antibodies. Bottom. Quantification of the relative amount of ACTIN‐normalized LC3B‐II is expressed as fold change vs. the untreated condition (−) of wt cells which was defined as 1.0. Data are expressed as the mean values; error bars indicate the SEM; n = 3 independent experiments. One‐tailed Student’s t‐test was applied. **P ≤ 0.01. Baf‐A1 = bafilomycin, ns = not significative.
Source data are available online for this figure.
Previous studies demonstrated that OFD1 co‐immunoprecipitates with autophagosomal membrane components LC3/GABARAP in HEK293 cells (Tang et al, 2013; Holdgaard et al, 2019). We thus postulated that OFD1 might function as an autophagy receptor for ATG13. To validate our hypothesis, we first confirmed the physical interaction of OFD1 with LC3B using GST‐pull down assays (Fig 3A). We then showed that OFD1 was not able to bind mutant forms of LC3B (dN LC3 dG and LC3B F52A‐V53A dG) that carry mutations impairing LC3B interactions (Shvets et al, 2008; Grumati et al, 2017), suggesting that OFD1 interacts with LC3B through a classical LIR motif (Fig 3A). We also demonstrated that 3xFLAG‐OFD1 was able to bind to purified GABARAP‐L1 (Fig 3A) and that endogenous OFD1 colocalizes with LC3B and GABARAP by IF (Fig 3B and Appendix Figs S1A and B, and S2). Analysis of the OFD1 protein sequence (Refseq NP_003602) using the iLIR tool (Jacomin et al, 2016) predicted a conserved LIR motif in a putative intrinsically disordered region (Popelka & Klionsky, 2015), as determined by the PONDR‐FIT algorithm (Xue et al, 2010; Fig 3C). Mutation of the LIR domain (EKYMKI to EKAMKA, OFD1ΔLIR) almost completely abolished binding to GST‐LC3B and GST‐GABARAP‐L1 indicating that indeed, the LIR motif is functional and binds both LC3B and GABARAP‐L1 (Fig 3D). Consistently, IF revealed that OFD1ΔLIR no longer colocalizes with LC3B and GABARAP (Fig 3E). Moreover, overexpression of OFD1ΔLIR was unable to induce degradation of ATG13, as indicated by quantification of ATG13 puncta and analysis of ATG13 protein levels (Fig 4A and B), demonstrating that OFD1‐induced ATG13 degradation is dependent on LIR‐mediated OFD1‐LC3B/GABARAP‐L1 binding. To further validate the role of OFD1 as an autophagy receptor for ATG13, we confirmed the OFD1/ATG13 interaction by GST‐pull down assays (Fig 4C). In particular, we showed that the OFD1b construct, containing the central portion of the protein (Giorgio et al, 2007) specifically binds ATG13 (Fig 4C) while OFD1a and OFD1c, containing the N‐ and C‐terminal regions of the protein, respectively, are not involved. We also demonstrated that the interaction between OFD1 and ATG13 is maintained in the absence of the OFD1 LIR domain, which localizes at the C‐terminal (OFD1c) (Appendix Fig S3). In addition, IF showed that ATG13 molecules are more abundant in the lumen of swollen lysosomes in wt compared with KO‐OFD1 cells (Fig 4D). Finally, since selective autophagy receptors have been shown to be degraded with their cargoes (Stolz et al, 2014; Gatica et al, 2018), we verified whether OFD1 is degraded through autophagy in HK2 cells. Indeed, our results indicate that OFD1 colocalizes with LAMP1, moreover, when wt HK2 cells were treated with Baf‐A1, OFD1 molecules were observed in the lumen of swollen lysosomes (Appendix Fig S4) suggesting that OFD1 is degraded through the autophagy‐lysosomal pathway. In addition, time course experiments using Baf‐A1 showed that wt OFD1 was degraded by lysosomes in HK2 cells (Fig 4E), while OFD1ΔLIR was insensible to Baf‐A1 treatment (Fig 4F), indicating that the autophagy‐mediated degradation of OFD1 is LIR dependent. Altogether, our data demonstrate that OFD1 is a novel autophagy receptor which selectively recognizes ATG13 for degradation within lysosomes.
Figure 3. OFD1 interacts with LC3B and GABARAP‐L1 through a classical LIR motif.

- Middle. Lysates from 3xFLAG‐OFD1 overexpressing HEK293 cells were added to beads with immobilized GST‐fused LC3B, LC3B F52A‐V53A dG, dN LC3B dG and GST alone. Right. Lysates from 3xFLAG‐OFD1(OFD1) and empty vector (Empty) overexpressing HEK293 cells were added to GST‐GABARAP‐L1 and GST. Left. Western blot of total lysates (input). All panels display WB for FLAG, ACTIN is the loading control.
- Representative confocal images of endogenous OFD1 (green) co‐staining with LC3B (red), and of 3xFLAG‐OFD1 (red) with GABARAP (green) in HK2 cells (HBSS, 90 min). Hoechst labels nuclei (blue). Insets show magnifications and single‐color channels of selected areas. Scale bar = 10 μm; n ≥ 50 cells/sample.
- OFD1 domains architecture and alignment of LIR motif (boxed) with orthologous sequences. Residues with full (*), strong (:), weak (.) conservation are indicated. LisH = Lis Homology domain, CC = Coiled coil, LIR = LC3 interacting region.
- Lysates from 3xFLAG‐OFD1(OFD1), 3xFLAG‐OFD1∆LIR(OFD1∆LIR) and empty vector (Empty) overexpressing cells were added to GST‐GABARAP‐L1, GST‐LC3B, and GST.
- Representative confocal images of OFD1∆LIR co‐staining with LC3B (left) and GABARAP (right) in HK2 cells (HBSS, 90 min). Green, LC3B and GABARAP; red, 3xFLAG‐OFD1∆LIR; blue, Hoechst for nuclei. Scale bar = 5 μm. n ≥ 50 cells/sample.
Source data are available online for this figure.
Figure 4. OFD1 is an autophagy receptor for ATG13.
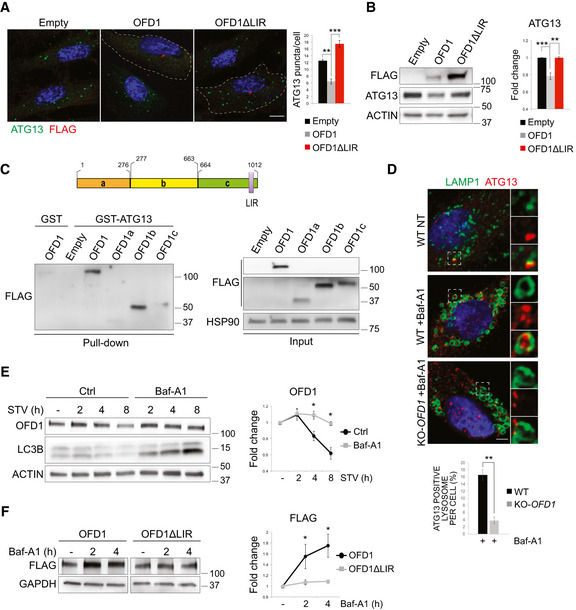
- Representative confocal images of 3xFLAG‐OFD1 (OFD1) and 3xFLAG‐OFD1∆LIR (OFD1∆LIR) co‐staining with ATG13 in KO‐OFD1 cells (HBSS, 90 min). Green, ATG13; red, FLAG; blue, Hoechst for nuclei. Scale bar = 10 μm. Right. ATG13 puncta/cell are quantified and expressed as mean ± SEM; n = 4 independent experiments, n ≥ 100 cells.
- Western blot of ATG13 in KO‐OFD1 cells transfected with 3xFLAG‐OFD1 (OFD1) or 3xFLAG‐OFD1∆LIR (OFD1∆LIR) or empty vector (Empty). Right. ACTIN‐normalized ATG13 protein levels are expressed as fold increase compared with empty vector (mean ± SEM); n = 3 independent experiments.
- Top. OFD1 fragments (a, b and c). Bottom left. Lysates from empty vector (Empty), 3xFLAG‐OFD1(OFD1), 3xFLAG‐OFD1a (OFD1a), 3xFLAG‐OFD1b (OFD1b), and 3xFLAG‐OFD1c (OFD1c) overexpressing HEK293 cells were added to GST‐ATG13; lysates from 3xFLAG‐OFD1(OFD1) overexpressing HEK293 cells were added to GST as control. Bottom right. Western blot of total lysates (input). All panels display WB for FLAG, HSP90 is the loading control. LIR = LC3 interacting region.
- Scanning confocal microscopy analysis of wt and KO‐OFD1 cells incubated in HBSS (8 h), treated (+) or not (−) with Baf‐A1 (50 nM, 8 h). Green, LAMP1; red, ATG13; blue, Hoechst for nuclei. Scale bar = 6 μm. Insets show higher magnification and single color channels of the boxed area. Bottom. Bar graphs show quantification of lysosomes containing ATG13 expressed as % of total amount of LAMP1/cell (mean ± SEM). n = 30 wt cells, n = 28 KO‐OFD1 cells counted; three independent experiments.
- Time course of OFD1 levels in HBSS‐starved wt HK2 cells treated with Baf‐A1 (100 nM) and collected at different time points. LC3B is the control for Baf‐A1 treatment. Right. Graphs show ACTIN‐normalized OFD1 levels vs. FM condition (−) expressed as mean ± SEM; n = 4 independent experiments. STV = starvation, Baf‐A1 = bafilomycin, Ctrl = control.
- Western blot of FLAG in KO‐OFD1 cells transfected with 3xFLAG‐OFD1 (OFD1) or 3xFLAG‐OFD1∆LIR (OFD1∆LIR) and treated or not (−) with Baf‐A1 (100 nM, 2 h). Right. Quantification of GAPDH‐normalized FLAG levels vs. untreated (−) is expressed as mean ± SEM; n = 3 independent experiments. Baf‐A1 = bafilomycin.
Data information: Statistical analysis: One‐tailed Student’s t‐test was applied in (B, D, E, and F); the likelihood ratio test for Negative Binomial generalized linear models was applied in (A). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Source data are available online for this figure.
The ULK1 complex is a key player in autophagy initiation (Wirth et al, 2013; Wong et al, 2013; Mercer et al, 2018), and we thus evaluated whether OFD1 plays a functional role in autophagy regulation. At first, we tested ULK1 kinase activity in KO‐OFD1 cells by measuring VPS34 (p‐VPS34 S249) (Egan et al, 2015) and ATG13 (p‐ATG13 S318) (Joo et al, 2011) phosphorylation and observed increased levels of the phosphorylated forms compared with controls (Fig 5A), indicating that the enhanced protein stability we observed is associated with increased ULK1 kinase activity. We then analyzed autophagy in KO‐OFD1 cells by monitoring the number of autophagosome vesicles (AVs) scoring the number of LC3B‐positive puncta and by measuring the conversion of LC3B‐I to the phosphatidylethanolamine‐conjugated LC3B‐II form, which decorates autophagosomes (Tanida et al, 2005). When we blocked LC3B‐II clearance by Baf‐A1 or by preventing lysosomal degradation using chloroquine (CQ), we observed increased LC3B‐II levels (Fig 5B) in OFD1‐depleted cells, suggesting that autophagosome biogenesis is enhanced compared with controls. Consistently, loss of OFD1 resulted in a higher number of AVs and WIPI2‐positive structures [autophagosome precursors called phagophores (Polson et al, 2010)] in both nutrient‐rich and nutrient‐depleted media (Fig 5C). Furthermore, transmission electron microscopy (EM) analysis demonstrated the presence of an increased number of AVs in KO‐OFD1 cells in response to nutrient deprivation compared with controls (Fig 5D). To verify whether OFD1 depletion enhances the entire autophagic flux, we monitored lysosome‐mediated autophagosome clearance by transfecting siRNA OFD1‐depleted HeLa cells and controls, with the pH‐sensitive reporter mRFP‐eGFP‐LC3B. In this system, autophagosomes appear as yellow puncta (GFP‐ and RFP‐positive) while autophagolysosomes are represented as red puncta (GFP‐negative and RFP‐positive) due to quenching of the GFP signal inside the acidic lysosomal compartment (Kimura et al, 2007). OFD1 depleted cells displayed a significantly higher number of autophagolysosomes (Fig 5E), indicating an enhanced autophagic flux. KO‐OFD1 cells also displayed a higher percentage of autophagosomal and lysosomal markers colocalization (Fig EV4A), and decreased protein levels of the autophagic substrate p62 (Ichimura et al, 2008; Klionsky et al, 2021), which were both rescued by SAR405 treatment (Fig EV4B), confirming the enhanced autophagic flux.
Figure 5. OFD1 regulates autophagy.

- Representative blots of ATG13 (left) and VPS34 (right) phosphorylation levels in wt and KO‐OFD1 cells transiently expressing GFP‐ATG13 and FLAG‐VPS34 in FM.
- Wild‐type and KO‐OFD1 cells were starved in HBSS with/without Baf‐A1 (100 nM, 2 h) and chloroquine (CQ) (50 μM, 2 h), and total lysates were analyzed by immunoblotting using anti‐OFD1, ‐LC3B (LC3B‐I 18 kDa and LC3B‐II 16 kDa) and anti‐ACTIN antibodies. Right. ACTIN‐normalized LC3B‐II levels are expressed as fold change vs. untreated conditions (−) of wt cells. n = 5 independent experiments.
- Representative images of LC3B and WIPI2 puncta in wt and KO‐OFD1 cells (HBSS, 90 min). Green, LC3B; red, WIPI2; blue, Hoechst, nuclei. Scale bar = 10 μm. Right. Quantification of LC3B and WIPI2 puncta is shown. ≥100 cells analyzed/sample, n = 5 independent experiments.
- Transmission electron microscopy (left) and quantification (right) of autophagic vacuoles (arrows) in wt and KO‐OFD1 cells (HBSS, 90 min).
- Representative images of siOFD1‐depleted and control (Ctrl) HeLa cells transiently expressing mRFP–eGFP–LC3B (HBSS, 90 min). Insets show higher magnification of colocalization in selected areas. Scale bar = 10 μm. Bottom, tandem structure of the mRFP‐GFP‐LC3B construct and quantification of RFP+GFP+ and RFP+GFP− puncta in siOFD1‐depleted cells and controls, cultured in full medium (FM) or kept in HBSS for 2 h (STV) are depicted. n ≥ 90 cells/sample analyzed, n = 5 independent experiments.
- Representative images of WIPI2 staining in wt HK2 cells transiently expressing eGFP‐OFD1 (HBSS, 90 min). Green, eGFP‐OFD1, red, WIPI2; blue, Hoechst labels nuclei. On the right, quantification of WIPI2 puncta in eGFP‐OFD1‐transfected cells compared with non‐transfected cells (Ctrl) is shown. Scale bar = 10 μm. n ≥ 90 cells analyzed/sample, n = 3 independent experiments.
- Representative images of WIPI2 and OFD1 co‐staining in KO‐OFD1 cells transfected with empty vector (Empty), 3xFLAG‐OFD1 (OFD1) or 3xFLAG‐OFD1∆LIR (OFD1∆LIR) (HBSS, 90 min). Red, WIPI2; green, OFD1; blue, Hoechst labels nuclei. Scale bars = 10 μm. On the right, quantification of WIPI2 puncta. ≥100 cells analyzed/sample, n = 4 independent experiments.
Data information: Data are expressed as mean values from the indicated independent experiments. Error bars indicate SEM. One‐tailed Student’s t‐test (B and D) and the likelihood ratio test for Negative Binomial generalized linear models (C, E, F, and G) were applied; *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. FM = full medium, STV = starvation, Baf‐A1 = bafilomycin, Ctrl = control.
Source data are available online for this figure.
Consistent with these observations, eGFP‐OFD1 overexpression resulted in decreased autophagosome biogenesis as indicated by the diminished number of WIPI2 puncta in wt HK2 cells compared with controls (Fig 5F). Remarkably, the OFD1ΔLIR mutant had no effect on autophagosome biogenesis, as demonstrated by quantification of WIPI2 puncta (Fig 5G).
Autophagy is negatively regulated by the mammalian target of rapamycin (mTOR) kinase pathway (Saxton & Sabatini, 2017); we thus investigated whether loss of OFD1 enhanced autophagy by decreasing mTOR kinase activity. Interestingly, phosphorylation of direct targets of mTOR, S6K1 (Thr389) (Romanelli et al, 2002) and ULK1 (Ser757) (Kim et al, 2011), was normal as well as those of S6 (Ser240), a S6K1 target (Pende et al, 2004; Fig EV4, EV5). These data demonstrated that the increased autophagy observed in KO‐OFD1 cells is independent of mTOR signaling. Taken together, our findings demonstrate that the inhibition of autophagy exerted by OFD1 is dependent on OFD1‐LC3/GABARAP binding through LIR.
We also observed a dramatic increase of ATG13 protein levels and of autophagy in lymphoblasts isolated from OFD type I patients, both in steady state and under serum starvation (Fig 6A–C). We thus asked whether the role exerted by OFD1 in autophagy could be relevant for polycystic kidney disease, a clinical manifestation of OFD type I syndrome. We crossed Ofd1 fl/fl mice, in which Ofd1 exons were flanked by two loxP sites (Ferrante et al, 2006), with the Ksp‐Cre transgenic line in which the Cre recombinase is expressed under the control of the Ksp‐cadherin gene promoter in renal tubular epithelial cells of the nephrons (Shao et al, 2002). Ofd1 fl/y;creKsp mice show dilated distal tubules which are visible from postnatal day (P)14 (Zullo et al, 2010). We quantified autophagosome‐associated LC3B‐II levels in renal samples and observed that inhibition of lysosomal functions, induced by leupeptin administration, increased LC3B‐II levels in kidneys at pre‐cystic stages in Ofd1 fl/y;creKsp mice compared with wt (Ofd1 fl/y) (Fig 7A). Importantly, Ofd1 fl/y;creKsp;GFP‐LC3 mice generated by crossing Ofd1 fl/fl mice with the transgenic line expressing the fluorescent autophagy reporter GFP‐LC3 to monitor autophagy in vivo (Mizushima, 2009) and the creKsp showed a significant increase of AVs in renal distal tubules at pre‐cystic stages compared with controls (Ofd1fl / y;GFP‐LC3) (Fig 7B). The increased LC3B‐II levels were also confirmed in pre‐cystic kidneys from a tamoxifen‐inducible model (Ofd1‐IND) (Fig 7C) obtained by crossing Ofd1 fl/fl with TgCAGG‐CreERTM mice, in which the Cre recombinase is ubiquitously expressed under the ß‐actin promoter/enhancer (CAGG promoter) (Hayashi & McMahon, 2002). In this model, Ofd1 inactivation is achieved at P0, and cysts are visible from P10 (D'Angelo et al, 2012; Iaconis et al, 2017). Moreover, also Atg13 protein levels were significantly increased in kidneys from Ofd1 fl/y;creKsp, both at pre‐cystic (P8) and cystic (P22) stages (Fig 7D), thus demonstrating that OFD1 is a critical regulator of autophagy in vivo.
Figure 6. Autophagy is increased in OFD type I patients.

- Western blot of LC3B in lymphoblasts of OFD type I patients and controls, either untreated (−) or exposed to Baf‐A1 (10 nM, 2 h) (+). Cells were cultured in complete medium (FM, top) or under serum starvation for 4 h (STV, bottom). ACTIN and GAPDH were used as loading controls.
- Left. Graphs show LC3B‐II levels normalized on ACTIN in patients’ lymphoblasts and controls cultured in complete medium (FM). Right. Graphs show LC3B‐II levels normalized on GAPDH in patients’ lymphoblasts and controls in serum starvation for 4 h (STV). Normalized LC3B‐II levels are expressed as fold change vs. untreated conditions (−) of control cells, n = 4 patients and 3 controls.
- Western blot of ATG13 and ATG101 proteins in serum starved lymphoblasts from controls and OFD type I patients. Right. Protein levels relative to ACTIN (loading control) are expressed as fold change vs. controls, n = 4 patients and 3 controls.
Data information: Data are expressed as mean values, error bars indicate SEM. One‐tailed Student’s t‐test was applied (B and C); *P ≤ 0.05, **P ≤ 0.01. FM = full medium, STV = starvation, Baf‐A1 = bafilomycin, ns = not significative.
Source data are available online for this figure.
Figure 7. Excessive autophagy contributes to renal cystogenesis in OFD type I syndrome.

- Western blot of LC3B in kidney homogenates from leupeptin‐treated (40 mg/kg, 6 h) fasted Ofd1 fl/y;creKsp and control mice. Right. LC3B‐II protein levels normalized to Actin (loading control) are expressed as fold increase vs. control mice, n = 4 mice/group.
- Representative images of GFP–LC3 puncta (autophagosomes, green) in aquaporin2‐positive (AQP2, red) renal distal tubules from P8 Ofd1fl/y;creKsp;GFP‐LC3 and Ofd1fl/ y;GFP‐LC3 mice. Scale bar = 10 μm. Hoechst labels nuclei. Bar graphs on the left show quantitative analysis of GFP‐LC3 puncta; n = 4 mice/group for a total of 300 AQP2‐positive nuclei/group analyzed.
- Western blot of LC3B in kidney homogenates from leupeptin‐treated (40 mg/kg, 6 h) fasted Ofd1‐IND and control mice. Right. LC3B‐II protein levels relative to βTubulin (loading control) are expressed as fold increase compared with control mice, n = 5 mutant/8 control mice.
- Western blot of Atg13 in Ofd1 fl/y;creKsp and control kidneys at pre‐cystic (P8, left) and cystic (P22, right) stages. Gapdh was used as loading control. Atg13 protein levels are expressed as fold change compared with control mice; n = 7 mutant/5 control mice at P8, n = 4 mice/group at P22.
- Hematoxylin and eosin staining of kidney sections from Ofd1 fl/y;Atg7+/+;creKsp and Ofd1 fl/y;Atg7 fl/fl;creKsp mice. Right. Graph shows fold change of the cysts number observed in Ofd1 fl/y;Atg7 fl/fl;creKsp animals compared with Ofd1 fl/y;Atg7+/+;creKsp; n = 12 sections/animal were analyzed; n = 6 mice/group. Scale bars = 1 mm.
- Blood Urea Nitrogen was quantified on blood withdrawn from Ofd1 fl/y;Atg7 fl/fl;creKsp, Ofd1 fl/y;Atg7+/+;creKsp and control mice at P45. n = 8 mice per group.
Data information: Data are expressed as mean values from the indicated independent experiments. Error bars indicate SEM. One‐tailed Student’s t‐test was applied; *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. BUN = blood urea nitrogen.
Source data are available online for this figure.
To determine whether autophagy might have a role in renal cystogenesis, we deleted Atg7, a key player in the autophagic cascade (Komatsu et al, 2005), in renal tubules of Ofd1 fl/y;creKsp mice. Simultaneous inactivation of Ofd1 and Atg7 (Appendix Fig S5A) and decreased autophagy (Appendix Fig S5B) were validated in kidneys of Ofd1 fl/y;Atg7 fl/fl;creKsp mice. Ofd1 fl/y ;Atg7 fl/fl;creKsp and Ofd1 fl/y ;Atg7 +/+;creKsp mice were born at the expected Mendelian ratio. Hematoxylin–eosin analysis of kidneys at P23 from Ofd1 fl/y;Atg7 fl/fl;creKsp animals showed a significative reduction in the number of cysts compared with Ofd1 fl/y;Atg7+/+;creKsp (Fig 7E). The renal function in mutants was assessed by blood urea nitrogen (BUN) quantification, which revealed a partially preserved renal function in Ofd1 fl/y;Atg7 fl/fl;creKsp animals at P45 compared with Ofd1 fl/y;Atg7+/+;creKsp mice (Fig 7F). Taken together, these data show that enhanced autophagy contributes to the renal cystic disease associated with OFD type I syndrome.
Autophagy is a self‐limiting process (Yu et al, 2010). Despite the fact that the mechanisms that regulate autophagy induction have been intensively studied, relatively little is known on the mechanisms involved in turning autophagy off. Our study unveils a novel negative feedback regulation that enables cells to calibrate their self‐degradative process based on selective autophagy‐mediated degradation of autophagic proteins. We demonstrate that the centrosomal OFD1 protein inhibits autophagosome biogenesis by acting as a receptor for ULK1 complex turnover, which is mediated by controlling ATG13 protein stability. However, a role for FIP200 in OFD1‐mediated autophagy restrain cannot be excluded. Indeed, we demonstrated that OFD1 co‐immunoprecipitates and colocalizes with FIP200; thus, we cannot formally exclude that FIP200 might influence, either directly or indirectly, the regulation exerted on ATG13 by OFD1. Further experiments will be needed to address this issue.
A centrosome‐autophagosome crosstalk has been previously described showing that GABARAP stability and recruitment to centrosomes are controlled by the centriolar satellite protein PCM1 (Joachim et al, 2017). In addition, an important function of the centrosome is the nucleation of ciliogenesis in confluent and mitotic quiescent cells (Joukov & De Nicolo, 2019) and the OFD1 protein controls primary cilia formation (Ferrante et al, 2006; Singla et al, 2010; Tang et al, 2013). The ciliary membrane has been proposed as a nucleation site for autophagosomes (Pampliega et al, 2013) and experimental evidence demonstrated that loss of cilia results in decreased autophagy (reviewed in Morleo & Franco, 2019). Analysis of autophagy in conditions promoting ciliogenesis in KO‐OFD1 cells, that are not capable of generating primary cilia, reproduced the same autophagic phenotype observed in conditions that do not allow cilia formation (i.e., cycling and subconfluent cells) (Fig EV5). These findings indicate that OFD1 controls autophagy independently from cilia. This observation was confirmed by analysis performed on OFD type I patients’ lymphoblasts that grow in suspension and are never ciliated (Fig 6). Unrestrained autophagy can contribute to the development of pathological conditions (Antonioli et al, 2017). We demonstrated that enhanced autophagy in vivo contributes to renal cystic disease in Ofd1 mutants. Although OFD type I syndrome is a rare syndromic disorder, over 7 millions of individuals worldwide display or are at risk of developing complications associated with renal cystic disease for which no effective pharmacological therapies are available to date (Kagan et al, 2017). Our work implicates autophagy in the pathogenesis of renal cysts and proposes autophagy modulation as a therapeutic approach for renal cystic disease in mammals.
Materials and Methods
Cell culture and treatments
Human Kidney 2 (HK2), osteosarcoma (Saos‐2), HEK293, and HeLa cell lines were from the American Type Culture Collection (ATCC). HK2 cells were cultured in DMEM/F12 medium (Gibco), HEK293 and HeLa cell lines were cultured in DMEM medium (Gibco), lymphoblastoid cells were maintained in RPMI medium (Gibco), and Saos‐2 cells were cultured in McCoy's 5a Medium Modified (Gibco). Cells medium was supplemented with 10% FBS, 1 mM l‐glutamine, 1% penicillin/streptomycin, and HK2 cells medium was supplemented also with 1% insulin‐transferrin‐sodium selenite (Sigma I1884). To study mTOR activity, cells were cultured in amino acid‐free RPMI 1640 medium (US Biological) supplemented with 10% dialyzed FBS (Invitrogen, Thermo Fisher Scientific) for 1 h, and then, a complete RPMI 1640 medium (with amino acids) was added back to cells. Cells were treated with 50 µg/ml of CHX (SIGMA), 10 µM of SAR405 (ApexBIO), 100 nM of Baf‐A1 (SIGMA), 10 µM of MG132 (SIGMA), and 100 nM of Bortezomib (SIGMA) for the indicated time periods. For each treatment, cells were plated the day before to a final confluence of 70%. HK2 cells were grown to 100% density and brought to quiescence by serum starvation to induce ciliogenesis.
Cloning procedures, DNA mutagenesis and cell transfections
Portions of the full‐length human OFD1 protein, aa 1–276 (OFD1a), aa 277–663 (OFD1b), and aa 664–1,012 (OFD1c) described in Giorgio et al (2007), and the OFD1ΔLIR were tagged at the N terminus with a 3xFLAG tag by cloning it into the CMV10 vector and the plasmids were sequence‐verified. Mutations were generated via site‐directed mutagenesis according to standard protocols. All cDNAs used are reported in Appendix Table S1. For transient expression, DNA plasmids were transfected with TransIT®‐LT1 Transfection Reagent (Mirus) according to manufacturer’s instructions.
RNAi
HeLa cells were transfected with 25 nM of ON‐TARGETplusSMARTpool against OFD1 (L‐009300–00‐0020; Thermo Scientific Dharmacon) using Interferin (Polyplus) for 96 h. Cells were transfected with mRFP‐eGFP‐LC3 for 24 h. HK2 cells were transfected with 25 nM of ON‐TARGETplus siRNA SMARTpool against ATG13 (L‐020765‐01‐0005; Thermo Scientific Dharmacon) and RB1CC1 (L‐021117‐00‐0005; Thermo Scientific Dharmacon) using Lipofectamine RNAiMAX (Invitrogen) for 72 h. Transfectants used following manufacturer’s recommendations. Cells were then collected for Western blot analysis or fixed for IF staining.
Transmission electron microscopy
For routine EM, cells were grown in 12‐well plates in monolayer and then treated as described (Polishchuk & Polishchuk, 2019). For each sample, 60 nm sections were obtained using a Leica EM UC7 (Leica Mycrosystems, Vienna, Austria). Images were acquired using a FEI Tecnai‐12 electron microscope (FEI, Eindhoven, Netherlands) equipped with a VELETTA CCD digital camera (Soft Imaging Systems GmbH, Munster, Germany). Electron microscopy images were acquired at the same magnification. Twenty‐five cells/each sample randomly distributed on single sections were considered. Autophagic‐like structures were manually counted on EM images within 16 μm square field of view.
Antibodies used for WB, immunoprecipitation and IF staining
Primary antibodies are reported in Appendix Table S2. Western blot secondary antibody HRP‐conjugated was from Amersham ECL. For IF staining, the following antibodies were used: Alexa Fluor‐labeled donkey anti‐rabbit 488 (Life Technology; A‐21206; Ober‐Olm, Germany), donkey anti‐mouse 568 (Life Technology; A10037), donkey anti‐mouse 647 (Life Technology; A31571), and goat anti‐rat 568 (Life Technology; A11077).
Immunofluorescence
Cells were grown on glass coverslips, fixed in cold methanol, and permeabilized in 0.05% (w/v) saponin, 0.5% (w/v) BSA, 50 mM NH4Cl, and 0.02% NaN3 in PBS (blocking buffer) for 30 min. Cells were incubated with primary antibodies either at 4°C ON or at room temperature for 2 h and then with appropriate secondary antibodies for 1 h at RT. DNA was stained with Hoechst (33342, Sigma). The experiments were repeated at least three times, and representative images are shown.
Confocal fluorescence microscopy, image processing and colocalization analysis
Samples were examined under LSM700 or LSM800 high‐resolution confocal laser‐scanning microscopes (Zeiss). Optical sections were obtained under a 63× oil‐immersion objective at a definition of 1,024 × 1,024 pixels, adjusting the pinhole diameter to 1 Airy unit for each emission channel. Airyscan microscopy was performed using a LSM 880 (Zeiss) confocal microscope using a 63× Plan‐Apochromat 1.4NA DIC oil‐immersion objective. Images were subjected to Airyscan processing performed with Zen Blue software. To perform quantitative image analysis, 10–15 randomly chosen fields that included 8–10 cells each were scanned, using the same setting parameters (i.e., laser power and detector amplification) below pixel saturation between samples of interest and controls. Dots analysis was performed using ImageJ software on Z‐Stacks images with a slice thickness of 0.5 μm. Single channels from each image were converted into 8‐bit grayscale images and thresholded in order to subtract background. Counting was performed either automatically or manually using the tool available at https://imagej.nih.gov/ij/docs/guide/146‐19.html. The levels of colocalization of OFD1/LC3B, LAMP1/LC3B, HA‐OFD1/ATG13 and HA‐OFD1/FIP200 were calculated by acquiring confocal sections at the same laser power and photomultiplier gain. Images were processed using the ImageJ software. Single channels from each image were converted into 8‐bit grayscale images and thresholded in order to subtract background. The ImageJ “JACoP” plugin was then used to quantify Manders' overlap coefficient. Values range from 0 (corresponding to non‐overlapping images) to 1 (as 100% colocalization between both images) and express the ratio of the green channel intensities overlapping the red one for image.
Western blot analysis
Cells and renal tissues were lysed in RIPA buffer. Western blot analysis was performed as reported (Massa et al, 2019). Immunoblot bands were quantified using ImageJ (Gels and Plot Lanes plugin).
Co‐immunoprecipitation
Cells were lysed in lysis buffer (50 mM Tris–HCl, 1 mM EDTA, 10mM MgCl2, 5 mM EGTA, 0.5% Triton X‐100, pH 7.28). An equal amount of each protein lysate was incubated ON at 4°C with 5 μg of the indicated antibodies and IgG (Cell Signaling), followed by incubation with 50 μl of Protein A/G PLUS‐Agarose beads (Santa Cruz sc‐2003) for 3 h at 4°C. For HA‐OFD1wt and HA‐OFD1ΔLIR clones, protein lysate was incubated ON at 4°C with immobilized anti‐HA affinity resin (Pierce, Thermo Fisher Scientific). Beads were then washed, resuspended in Laemmli buffer, and boiled. Supernatants were loaded on SDS–PAGE.
GST pull down
GST, LC3B‐, dN LC3B dG‐, LC3B F52A‐V53A dG‐, GABARAP‐L1‐, and ATG13‐GST fusion proteins were expressed in Escherichia coli BL21 (DE3) cells in LB medium. Expression was induced by addition of 0.5 mM IPTG, and cells were incubated at 37°C for 5 h. Harvested cells were sonicated in lysis buffer (20 mM Tris–HCl pH 7.5, 10 mM EDTA, 5 mM EGTA, 150 mM NaCl) and GST‐fused proteins were immunoprecipitated using Glutathione Sepharose 4B beads (GE Healthcare) and used in GST‐pull down assays. Transfected HEK293 cells were lysed in lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X‐100, 10% glycerol, 25 mM NaF) and incubated with GST fusion protein‐loaded beads ON at 4°C. Beads were washed, resuspended in Laemmli buffer, and boiled. Supernatants were loaded on SDS–PAGE.
Quantitative RT–PCR
Total RNA extraction, reverse transcription reaction, and quantitative real‐time PCR analysis (qRT–PCR) were performed as reported (Massa et al, 2019). RT‐minus controls confirmed the absence of genomic contamination. ACTIN, B2M and RPL13A (human samples) or Hprt (murine samples) genes were used as endogenous references. All results are shown as means ± SEM of at least three independent biological replicates. Primer sequences are reported in Appendix Table S3.
Generation of stable cell lines
KO‐OFD1 clone
HK2 cells were transfected with an all‐in‐one vector containing the sgRNA of interest (target site sequence: TTAGTCCAACATGTTTACCG with predicted no off‐target sites), whose expression was driven by the U6 promoter, a recombinant form of Cas9 protein under the control of the CMV promoter, and an mCherry reporter gene under the control of the SV40 promoter (Genecopoeia). Cells were transfected with Lipofectamine CRISPRMAX Cas9 Transfection Reagent (Invitrogen, Thermo Fisher Scientific) according to manufacturer’s instructions. Forty‐eight hours after transfection, putative positive clones were FACS sorted for mCherry expression using the BD FACSAria flow cytometer. Sorted cells were kept in culture until confluence, and the GeneArt Genomic Cleavage Detection (GCD) Kit (Invitrogen, Thermo Fisher Scientific) was used to check editing efficiency; different clones were selected and sequence verified.
HA‐OFD1wt and HA‐OFD1ΔLIR clones
Wild‐type and LIR mutant OFD1 sequences were cloned into pDONR223 vector using the BP Clonase Reaction Kit (Invitrogen) and further recombined into the doxycycline inducible lentiviral GATEWAY destination vector HA‐pLTD using In Fusion Cloning kit (Takara). Primers were designed following the indication available at https://www.takarabio.com/products/cloning/in‐fusion‐cloning. Virus was made in HEK293T cells, and cells were infected for 48 h before selection.
GFP‐LC3 clone
HK2 cells transient transfections with eGFP‐LC3B were performed using TransIT®‐LT1 Transfection Reagent. Forty‐eight hours after transfection, cells were selected with medium containing hygromycin until resistant cell clones were observed. After selection, GFP‐positive cells were FACS sorted to obtain single cell clones.
Animals
All mice were maintained in a C57BL/6 strain background. The Ofd1‐floxed, Ofd1‐IND, GFP‐LC3, and Atg7‐floxed mouse lines were previously described (Komatsu et al, 2005; Ferrante et al, 2006; Mizushima 2009; D'Angelo et al, 2012; Iaconis et al, 2017). The Ksp‐Cre mouse transgenic line was provided by The Jackson Laboratory (B6.Cg‐Tg(Cdh16‐cre)91Igr/J, stock No: 012237). Mice were fasted for 7 h and then intraperitoneally injected with 40 mg/kg of leupeptin (SIGMA) 6 h prior to sacrifice. For GFP‐LC3 puncta analysis on AQP2‐positive tubules, mice were fasted for 16 h. The number of mice used in each experiment is specified in the figure legends. All animal treatments were reviewed and approved in advance by the Ethics Committee of the Animal House facility of the Telethon Institute of Genetics and Medicine (Pozzuoli, Naples, Italy) and approved by the Italian Ministry of Health (protocol number: 870/2015‐PR; approval date August 24, 2015).
Histology and BUN test
Kidneys were harvested at P23 and fixed with 4% (w/v) paraformaldehyde (PFA) solution ON at 4°C, and then, they were dehydrated in graded series of ethanol, cleared with xylene, and infiltrated with paraffin. Sections (6 μm) were processed for staining with routine hematoxylin and eosin (H&E) analyses. Light microscope photographs were taken using a Leica DM5000 upright camera under the same magnification. The cysts number was calculated using ImageJ software on at least 12 comparable paraffin sections/each animal. The number of cysts was counted manually as the cumulative number of cystic structures per total area of the kidney.
Blood Urea Nitrogen (BUN) was measured in plasma samples of the indicated number of mice at P45. Adequate blood volume was collected from retro‐orbital plexus. The measurement was performed by Laboratorio Tecniche Separative at San Raffaele hospital (Milano, Italy).
Statistical analysis
In experiments requiring counts of ATG13, ULK1, LC3B, and WIPI2 puncta, the likelihood ratio test for Negative Binomial was applied. In all remaining experiments, statistical significance between two groups was evaluated by one‐tailed Student’s t‐test, and P < 0.05 was considered significant. Quantitative data are presented as the mean ± SEM (Standard Error of the Mean).
Author contributions
Study conception, experiment design, data analysis, and manuscript writing: MM and BF; Experiments and data analysis: SB, UF, DI, and ASM; Technical support: LF, FC, AP, and VB; Reagents, expertise, and feedback: CS and PG; Result discussion and comment on the manuscript: All authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank A Ballabio, G Diez Roux, and G Napolitano for suggestions and critical reading of the manuscript; A Indrieri for helpful discussion; E Damiano for generation of KO‐OFD1 and GFP‐LC3 cells; E Polishchuk for EM analysis; R Tammaro for technical support; I Peluso for molecular cloning of HA‐OFD1wt and HA‐OFD1ΔLIR; C De Leonibus for support with Airyscan confocal analysis; the TIGEM bioinformatic core for statistical analysis; I Dikic for providing GST‐LC3B, GST‐dN LC3B dG, GST‐LC3B F52A‐V53A dG, pGEX‐4T1, and GST‐GABARAP‐L1 constructs; A De Matteis for the mRFP‐eGFP‐LC3B construct, Ruey‐Hwa Chen for the VPS34 construct; A Ernst for the lentiviral vector HA‐pLTD; W Harper for GABARAP family‐deficient HeLa cells; and N Mizushima, for the Atg7‐floxed and the GFP‐LC3 transgenic lines used in this study. This work was supported by the Italian Telethon Foundation, the PKD foundation [Grant number 203g16a to BF] and by the STAR Programme, supported by UniNA and Compagnia di San Paolo L1 2014 [to MM].
The EMBO Journal (2021) 40: e105120.
Contributor Information
Manuela Morleo, Email: morleo@tigem.it.
Brunella Franco, Email: franco@tigem.it.
Data availability
The interactions data from this publication have been deposited to the IMEx database (http://www.imexconsortium.org) through MINT (IMEx ID: IM‐28515).
References
- Amaravadi R, Kimmelman AC, White E (2016) Recent insights into the function of autophagy in cancer. Genes Dev 30: 1913–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonioli M, Di Rienzo M, Piacentini M, Fimia GM (2017) Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci 42: 28–41 [DOI] [PubMed] [Google Scholar]
- D'Angelo A, De Angelis A, Avallone B, Piscopo I, Tammaro R, Studer M, Franco B (2012) Ofd1 controls dorso‐ventral patterning and axoneme elongation during embryonic brain development. PLoS One 7: e52937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra‐Panickar D, Yang CC, Sheffler DJ et al (2015) Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell 59: 285–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf AS et al (2001) Identification of the gene for oral‐facial‐digital type I syndrome. Am J Hum Genet 68: 569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, Dolle P, Franco B (2006) Oral‐facial‐digital type I protein is required for primary cilia formation and left‐right axis specification. Nat Genet 38: 112–117 [DOI] [PubMed] [Google Scholar]
- Gatica D, Lahiri V, Klionsky DJ (2018) Cargo recognition and degradation by selective autophagy. Nat Cell Biol 20: 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio G, Alfieri M, Prattichizzo C, Zullo A, Cairo S, Franco B (2007) Functional characterization of the OFD1 protein reveals a nuclear localization and physical interaction with subunits of a chromatin remodeling complex. Mol Biol Cell 18: 4397–4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati P, Morozzi G, Holper S, Mari M, Harwardt MI, Yan R, Muller S, Reggiori F, Heilemann M, Dikic I (2017) Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 6: e25555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP (2002) Efficient recombination in diverse tissues by a tamoxifen‐inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244: 305–318 [DOI] [PubMed] [Google Scholar]
- Holdgaard SG, Cianfanelli V, Pupo E, Lambrughi M, Lubas M, Nielsen JC, Eibes S, Maiani E, Harder LM, Wesch N et al (2019) Selective autophagy maintains centrosome integrity and accurate mitosis by turnover of centriolar satellites. Nat Commun 10: 4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaconis D, Monti M, Renda M, van Koppen A, Tammaro R, Chiaravalli M, Cozzolino F, Pignata P, Crina C, Pucci P et al (2017) The centrosomal OFD1 protein interacts with the translation machinery and regulates the synthesis of specific targets. Sci Rep 7: 1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Kominami E, Tanaka K, Komatsu M (2008) Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 4: 1063–1066 [DOI] [PubMed] [Google Scholar]
- Jacomin AC, Samavedam S, Promponas V, Nezis IP (2016) iLIR database: a web resource for LIR motif‐containing proteins in eukaryotes. Autophagy 12: 1945–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachim J, Razi M, Judith D, Wirth M, Calamita E, Encheva V, Dynlacht BD, Snijders AP, O'Reilly N, Jefferies HBJ et al (2017) Centriolar satellites control GABARAP ubiquitination and GABARAP‐mediated autophagy. Curr Biol 27: 2123–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo JH, Dorsey FC, Joshi A, Hennessy‐Walters KM, Rose KL, McCastlain K, Zhang J, Iyengar R, Jung CH, Suen DF et al (2011) Hsp90‐Cdc37 chaperone complex regulates Ulk1‐ and Atg13‐mediated mitophagy. Mol Cell 43: 572–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukov V, De Nicolo A (2019) The centrosome and the primary cilium: the yin and yang of a hybrid organelle. Cells 8: 701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan KO, Dufke A, Gembruch U (2017) Renal cystic disease and associated ciliopathies. Curr Opin Obstet Gynecol 29: 85–94 [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent‐tagged LC3. Autophagy 3: 452–460 [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdel‐Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu YP, Acevedo‐Arozena A et al (2021) Guidelines for the use and interpretation of assays for monitoring autophagy (4th edn). Autophagy, in press.
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y et al (2005) Impairment of starvation‐induced and constitutive autophagy in Atg7‐deficient mice. J Cell Biol 169: 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C, Kijanska M, Kalie E, Siergiejuk E, Lee SS, Semplicio G, Stoffel I, Brezovich A, Verma M, Hansmann I et al (2012) Binding of the Atg1/ULK1 kinase to the ubiquitin‐like protein Atg8 regulates autophagy. EMBO J 31: 3691–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132: 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G (2019) Biological functions of autophagy genes: a disease perspective. Cell 176: 11–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Lin YC, Chen YH, Chen CM, Pang LY, Chen HA, Wu PR, Lin MY, Jiang ST, Tsai TF et al (2016) Cul3‐KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol Cell 61: 84–97 [DOI] [PubMed] [Google Scholar]
- Marino G, Niso‐Santano M, Baehrecke EH, Kroemer G (2014) Self‐consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 15: 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa F, Tammaro R, Prado MA, Cesana M, Lee BH, Finley D, Franco B, Morleo M (2019) The deubiquitinating enzyme Usp14 controls ciliogenesis and Hedgehog signaling. Hum Mol Genet 28: 764–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TJ, Gubas A, Tooze SA (2018) A molecular perspective of mammalian autophagosome biogenesis. J Biol Chem 293: 5386–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N (2009) Methods for monitoring autophagy using GFP‐LC3 transgenic mice. Methods Enzymol 452: 13–23 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741 [DOI] [PubMed] [Google Scholar]
- Mizushima N (2018) A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol 20: 521–527 [DOI] [PubMed] [Google Scholar]
- Morleo M, Franco B (2019) The Autophagy‐Cilia axis: an intricate relationship. Cells 8: 905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morleo M, Franco B (2020) OFD Type I syndrome: lessons learned from a rare ciliopathy. Biochem Soc Trans 48: 1929–1939 [DOI] [PubMed] [Google Scholar]
- Nazio F, Carinci M, Valacca C, Bielli P, Strappazzon F, Antonioli M, Ciccosanti F, Rodolfo C, Campello S, Fimia GM et al (2016) Fine‐tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J Cell Biol 215: 841–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampliega O, Orhon I, Patel B, Sridhar S, Diaz‐Carretero A, Beau I, Codogno P, Satir BH, Satir P, Cuervo AM (2013) Functional interaction between autophagy and ciliogenesis. Nature 502: 194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G (2004) S6K1(‐/‐)/S6K2(‐/‐) mice exhibit perinatal lethality and rapamycin‐sensitive 5'‐terminal oligopyrimidine mRNA translation and reveal a mitogen‐activated protein kinase‐dependent S6 kinase pathway. Mol Cell Biol 24: 3112–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polishchuk EV, Polishchuk RS (2019) Pre‐embedding labeling for subcellular detection of molecules with electron microscopy. Tissue Cell 57: 103–110 [DOI] [PubMed] [Google Scholar]
- Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, Tooze SA (2010) Mammalian Atg18 (WIPI2) localizes to omegasome‐anchored phagophores and positively regulates LC3 lipidation. Autophagy 6: 506–522 [DOI] [PubMed] [Google Scholar]
- Popelka H, Klionsky DJ (2015) Analysis of the native conformation of the LIR/AIM motif in the Atg8/LC3/GABARAP‐binding proteins. Autophagy 11: 2153–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prattichizzo C, Macca M, Novelli V, Giorgio G, Barra A, Franco B (2008) Mutational spectrum of the oral‐facial‐digital type I syndrome: a study on a large collection of patients. Hum Mutat 29: 1237–1246 [DOI] [PubMed] [Google Scholar]
- Romanelli A, Dreisbach VC, Blenis J (2002) Characterization of phosphatidylinositol 3‐kinase‐dependent phosphorylation of the hydrophobic motif site Thr(389) in p70 S6 kinase 1. J Biol Chem 277: 40281–40289 [DOI] [PubMed] [Google Scholar]
- Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA (2004) OFD1 is a centrosomal/basal body protein expressed during mesenchymal‐epithelial transition in human nephrogenesis. J Am Soc Nephrol 15: 2556–2568 [DOI] [PubMed] [Google Scholar]
- Saal S, Faivre L, Aral B, Gigot N, Toutain A, Van Maldergem L, Destree A, Maystadt I, Cosyns JP, Jouk PS et al (2010) Renal insufficiency, a frequent complication with age in oral‐facial‐digital syndrome type I. Clin Genet 77: 258–265 [DOI] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM (2017) mTOR Signaling in growth, metabolism, and disease. Cell 168: 960–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X, Somlo S, Igarashi P (2002) Epithelial‐specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol 13: 1837–1846 [DOI] [PubMed] [Google Scholar]
- Shvets E, Fass E, Scherz‐Shouval R, Elazar Z (2008) The N‐terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci 121: 2685–2695 [DOI] [PubMed] [Google Scholar]
- Singla V, Romaguera‐Ros M, Garcia‐Verdugo JM, Reiter JF (2010) Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev Cell 18: 410–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501 [DOI] [PubMed] [Google Scholar]
- Suttangkakul A, Li F, Chung T, Vierstra RD (2011) The ATG1/ATG13 protein kinase complex is both a regulator and a target of autophagic recycling in Arabidopsis . Plant Cell 23: 3761–3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q (2013) Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502: 254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Minematsu‐Ikeguchi N, Ueno T, Kominami E (2005) Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 1: 84–91 [DOI] [PubMed] [Google Scholar]
- Wirth M, Joachim J, Tooze SA (2013) Autophagosome formation–the role of ULK1 and Beclin1‐PI3KC3 complexes in setting the stage. Semin Cancer Biol 23: 301–309 [DOI] [PubMed] [Google Scholar]
- Wong PM, Puente C, Ganley IG, Jiang X (2013) The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9: 124–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN (2010) PONDR‐FIT: a meta‐predictor of intrinsically disordered amino acids. Biochim Biophys Acta 1804: 996–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F et al (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465: 942–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zullo A, Iaconis D, Barra A, Cantone A, Messaddeq N, Capasso G, Dolle P, Igarashi P, Franco B (2010) Kidney‐specific inactivation of Ofd1 leads to renal cystic disease associated with upregulation of the mTOR pathway. Hum Mol Genet 19: 2792–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The interactions data from this publication have been deposited to the IMEx database (http://www.imexconsortium.org) through MINT (IMEx ID: IM‐28515).