

Abstract
Centriole copy number is tightly maintained by the once‐per‐cycle duplication of these organelles. Centrioles constitute the core of centrosomes, which organize the microtubule cytoskeleton and form the poles of the mitotic spindle. Centrosome amplification is frequently observed in tumors, where it promotes aneuploidy and contributes to invasive phenotypes. In non‐transformed cells, centrosome amplification triggers PIDDosome activation as a protective response to inhibit cell proliferation, but how extra centrosomes activate the PIDDosome remains unclear. Using a genome‐wide screen, we identify centriole distal appendages as critical for PIDDosome activation in cells with extra centrosomes. The distal appendage protein ANKRD26 is found to interact with and recruit the PIDDosome component PIDD1 to centriole distal appendages, and this interaction is required for PIDDosome activation following centrosome amplification. Furthermore, a recurrent ANKRD26 mutation found in human tumors disrupts PIDD1 localization and PIDDosome activation in cells with extra centrosomes. Our data support a model in which ANKRD26 initiates a centriole‐derived signal to limit cell proliferation in response to centrosome amplification.
Keywords: ANKRD26, centriole, centriole amplification, PIDDosome
Subject Categories: Autophagy & Cell Death; Cell Adhesion, Polarity & Cytoskeleton; Cell Cycle
A genome‐wide screen identifies a key role for distal appendage protein ANKRD26 in cell proliferation inhibition in the presence of extra‐centrosomes, which is affected by cancer‐linked mutations.
Introduction
Centrioles are evolutionarily conserved microtubule‐based structures that recruit a surrounding pericentriolar material (PCM) to form the centrosome, an organelle that organizes the interphase microtubule cytoskeleton in many animal cells and forms the poles of the mitotic spindle (Gonczy, 2012; Nigg & Holland, 2018). Centrioles can also dock at the plasma membrane, where they template the formation of cilia that function in cell signaling, fluid movement, and locomotion. Centriole number is tightly controlled in proliferating cells (Firat‐Karalar & Stearns, 2014). G1 cells contain two parent centrioles that differ in age and structure; the younger of the two centrioles was assembled in the previous cell cycle and lacks appendages, while the mature centriole was formed earlier and is decorated with distal appendage structures that are required for membrane docking and ciliogenesis (Nigg & Stearns, 2011; Tanos et al, 2013). At the G1‐S transition, one new procentriole grows perpendicularly from a single site at the proximal end of each existing centriole (Tsou & Stearns, 2006). The procentrioles elongate and, in G2, the two centriole pairs separate and increase PCM recruitment to catalyze the formation of the mitotic spindle. Following chromosome segregation, the two centrosomes are divided such that each daughter cell inherits a pair of centrioles.
The faithful control of centriole number is deregulated in a wide range of tumors, leading to either the acquisition of extra copies of centrosomes or centrosome loss (Chan, 2011; Nigg & Holland, 2018; Wang et al, 2019). The presence of supernumerary centrosomes correlates with advanced disease and poor clinical outcome. Centrosome amplification in cultured cells promotes chromosome segregation errors (Ganem et al, 2009; Silkworth et al, 2009) and leads to the accumulation of DNA damage that can drive structural chromosomal alterations (Janssen et al, 2011; Crasta et al, 2012; Ganem & Pellman, 2012). Moreover, extra centrosomes have also been proposed to promote metastasis through multiple distinct mechanisms (Godinho et al, 2014; Arnandis et al, 2018; Ganier et al, 2018a; Ganier et al, 2018b; LoMastro & Holland, 2019). Centrosome amplification has been modeled in vivo by increasing the levels of Polo‐like kinase 4 (PLK4), the master regulator of centriole duplication. PLK4 overexpression can promote tumorigenesis in both flies and mice (Basto et al, 2008; Coelho et al, 2015; Sercin et al, 2016; Levine et al, 2017). Moreover, the tumors that form in mice with extra centrosomes exhibit complex karyotype changes similar to those observed in human tumors with centrosome amplification (Levine et al, 2017).
Although centrosome amplification is observed across a range of human tumors, extra centrosomes trigger a protective TP53‐dependent proliferative block in non‐transformed cells in culture (Holland et al, 2012; Fava et al, 2017). Consistently, dysregulation of the TP53 pathway facilitates the ability of extra centrosomes to promote tumorigenesis in mice (Coelho et al, 2015; Sercin et al, 2016; Levine et al, 2017). Insight into how cells respond to extra centrosomes came from the observation that centrosome amplification triggers activation of a multi‐protein complex known as the PIDDosome (Fava et al, 2017). The PIDDosome is comprised of caspase‐2 (CASP2), PIDD1, and CRADD. PIDD1 and CRADD assemble into a molecular platform that promotes the autocatalytic, proximity‐induced activation of CASP2 (Tinel & Tschopp, 2004; Sladky et al, 2017). Activated CASP2 then cleaves MDM2, a negative regulator of TP53 stability, resulting in TP53 activation and subsequent upregulation of the cell cycle inhibitor P21 (Oliver et al, 2011; Fava et al, 2017). This signaling network was shown to limit hepatocyte ploidy in mice (Sladky et al, 2020). While activation of the PIDDosome explains how TP53 is stabilized in cells with extra centrosomes, it remains unclear exactly how extra centrosomes activate the PIDDosome and whether inactivation of the PIDDosome increases the long‐term fitness of cells with centrosome amplification.
Here we performed a genome‐wide CRISPR knockout screen to identify genes required to arrest the proliferation of cells with extra centrosomes. Our results confirm a central role of the PIDDosome in arresting the growth of cells with extra centrosomes. Furthermore, we show that PIDDosome activation requires ANKRD26‐dependent recruitment of PIDD1 to the distal appendages of mature parent centrioles. Since cycling cells usually contain only one mature centriole with distal appendages, our data support a model in which extra mature parent centrioles initiate a local signal that activates the PIDDosome in response to centrosome amplification.
Results
A genome‐wide CRISPR screen identifies genes required to arrest proliferation following centrosome amplification
To study the impact of centrosome amplification on cell proliferation, we used hTERT immortalized RPE1 cells that can be induced with doxycycline (dox) to overexpress PLK4 (hereafter referred to as PLK4Dox). PLK4Dox cells efficiently cluster extra centrosomes during mitosis and undergo a robust TP53‐dependent cell cycle arrest in response to PLK4‐induced centrosome amplification (Holland et al, 2012). To determine if the cell cycle arrest that occurs following PLK4 overexpression is due to centrosome amplification or an alternative function of PLK4, we created dox‐inducible PLK4 cells with or without the centriole protein SAS6. To allow for the continued proliferation of SAS6 −/− cells lacking centrioles, we performed experiments in a TRIM37 knockout background (Fong et al, 2016; Meitinger et al, 2016). While TRIM37‐knockout RPE1 cells arrested proliferation after 4 days of PLK4 overexpression, knockout of SAS6 overcame this proliferative arrest (Fig 1A). This suggests the growth arrest induced by PLK4 overexpression is due to centrosome amplification and not PLK4 overexpression per se.
Figure 1. A CRISPR/Cas9 knockout screen identifies novel genes that arrest the proliferation of cells with centrosome amplification.
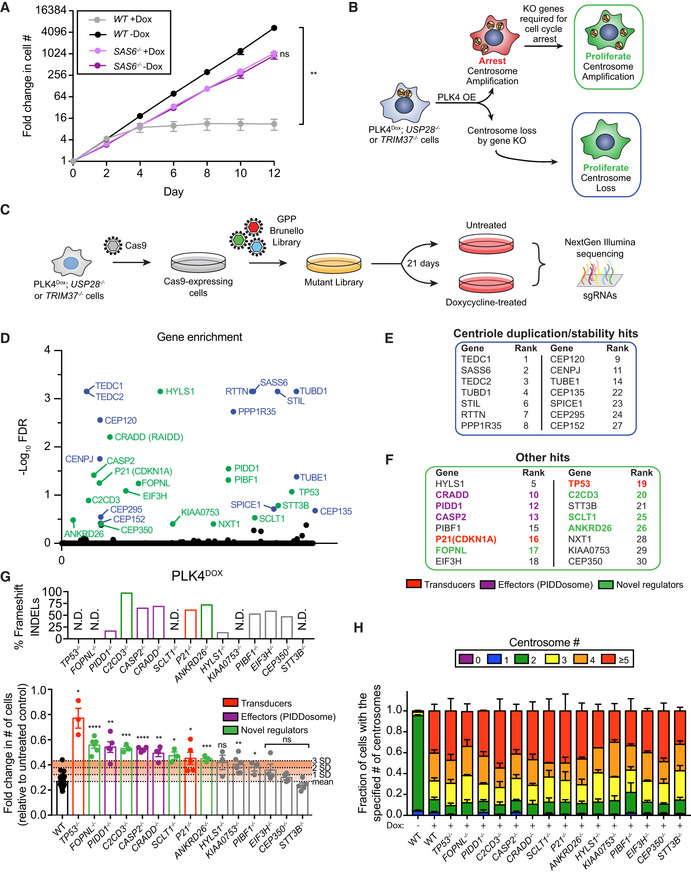
- Growth assay of the indicated cells with and with out doxycycline‐inducible overexpression of PLK4. Experiments were performed in wild‐type or SAS6 monoclonal knockout cells. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
- Schematic overview of the screen design.
- Schematic showing the procedure for a CRISPR/Cas9‐positive selection screen to identify gene knockouts that increase the proliferation of cells with extra centrosomes.
- Top hits that emerged from the screens ranked by MaGeCK FDR value. Blue hits are genes required for centriole duplication or stability. Green hits are genes predicted to be required to arrest the growth of cells with extra centrosomes.
- Hits with a known role in centriole duplication or stability.
- Candidate hits responsible for arresting the proliferation of cells with centrosome amplification. Purple hits correspond to PIDDosome genes. Red hits correspond to downstream effectors. Green hits are novel regulators.
- Top: Graph showing the efficiency of frameshifting INDELs measured using TIDE. N.D. = Not determined. Data shown are from n = 1 biological replicate. Bottom: Graph showing the relative growth of doxycycline‐treated PLK4Dox cells expressing an sgRNA targeting the indicated genes. Each dot displays measurements from a single experiment. Experiments were performed in polyclonal knockout cells. Data acquired across n ≥ 3 biological replicates. Mean ± s.e.m.
- Quantification of centrosome number in PLK4Dox cells expressing an sgRNA targeting the indicated genes. Experiments were performed in polyclonal knockout cells. Data acquired across n ≥ 3 biological replicates. Mean ± s.e.m.
Data information: Asterisks indicate statistically significant differences between measurements (*P < 0.05; **P < 0.01; ***P < 0.001, ****P < 0.0001). Statistics for all Figures were calculated using a two‐tailed Student’s t‐test.
Source data are available online for this figure.
To gain insight into how cells respond to extra centrosomes, we designed a genome‐wide CRISPR‐Cas9 knockout screen to identify genes required to arrest proliferation following centrosome amplification. We utilized PLK4Dox cells carrying a mouse PLK4 transgene that would not be targeted by the human PLK4 sgRNAs encoded in the sgRNA library. We also knocked out USP28 or TRIM37, which allows for cell proliferation following centrosome loss but not following centrosome amplification (Fong et al, 2016; Lambrus et al, 2016; Meitinger et al, 2016). We anticipated two mechanisms by which PLK4‐overexpressing, USP28 or TRIM37 knockout cells could be permitted to proliferate: (i) loss of genes required for centriole duplication or stability and (ii) loss of genes required to arrest the growth of cells with extra centrosomes (Fig 1B).
Cas9‐expressing PLK4Dox; USP28 −/− and PLK4Dox; TRIM37 −/− RPE1 cells were infected with a genome‐wide sgRNA library containing four independent sgRNAs for every human gene (Fig 1C). Transduced cells were selected for 7 days with puromycin, and knockout libraries of cells were treated with dox to induce centrosome amplification and a subsequent cell cycle arrest. After three weeks of dox treatment, cells were harvested, and sgRNA abundance was analyzed. sgRNAs that provide a growth advantage were expected to enrich in dox treated cells compared with untreated controls. The screen was repeated twice for both PLK4Dox; USP28 −/− and PLK4Dox; TRIM37 −/− cells, and the data from the four screens were analyzed together using the MAGeCK method for prioritizing genes and pathways (Li et al, 2014) (Table EV1; Fig 1D).
Twenty‐three of the top 30 hits (FDR ≤ 0.4) identified in our screen were genes that encode proteins reported to localize to the centrosome (Table EV2) (Alves‐Cruzeiro et al, 2014). From the top 30 hits of our screen, 14 genes had firmly established roles in centriole assembly/stability and were not analyzed further (Fig 1E, Table EV2). We performed competition based‐growth assays to validate that knockout of the remaining 16 genes enhanced the proliferation of the PLK4Dox; TRIM37 −/− cells used in our screen (Fig EV1A). The only gene that failed to improve the growth of PLK4Dox; TRIM37 −/− cells in this assay was NXT1, which was excluded from further analysis (Fig EV1B). The remaining 15 genes encoded all of the proteins previously known to be required to arrest the cell cycle in response to centrosome amplification, including all three components of the PIDDosome (CRADD, PIDD1, and CASP2), as well as TP53 and P21 (CDKN1A) (Fig 1F). We therefore assayed these remaining 15 genes for their role in arresting the growth of cells with extra centrosomes.
Figure EV1. PIDD1 is recruited to the centriole distal appendage.
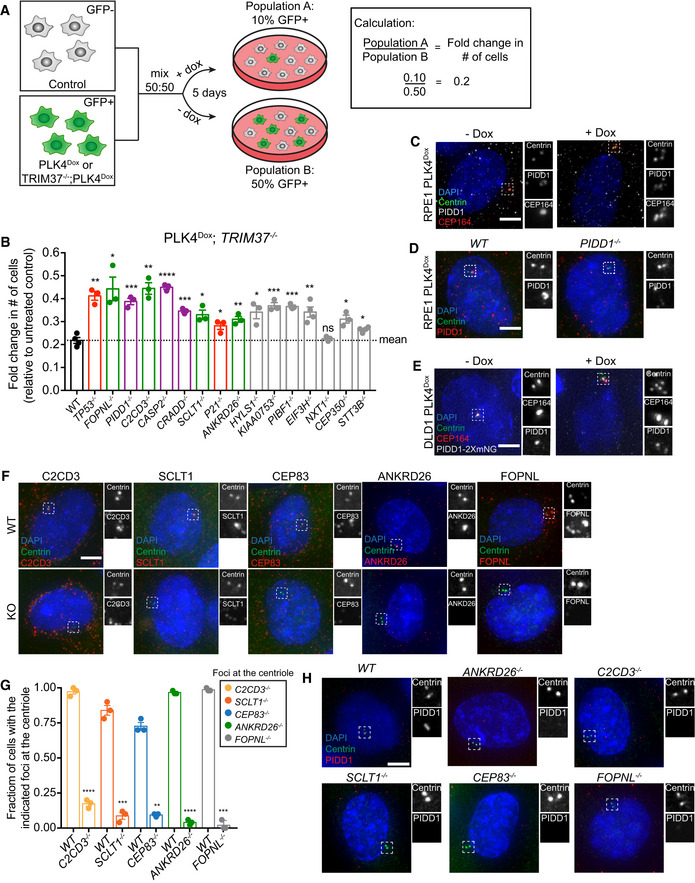
- Schematic outlining the competition growth assay used to measure fold change in the number of GFP+ cells treated with doxycycline compared to control, untreated GFP− cells.
- Graph showing the relative growth of doxycycline‐treated PLK4Dox; TRIM37 −/− cells expressing an sgRNA targeting the indicated genes. Each dot displays measurements from a single experiment. Experiments were performed in polyclonal knockout cells. Data acquired across n ≥ 3 biological replicates. Mean ± s.e.m.
- Representative images of PLK4Dox RPE1 cells treated with and without doxycycline for two days and immunostained with the indicated antibodies. Scale bar = 5 µm.
- Representative images of wild‐type and PIDD1 −/− PLK4Dox RPE1 cells treated with and without doxycycline for two days and immunostained with the indicated antibodies. Scale bar = 5 µm.
- Representative images of PLK4Dox PIDD1‐mNeonGreen DLD1 cells treated with and without doxycycline for 2 days and immunostained with the indicated antibodies. Scale bar = 5 µm.
- Representative images of WT or knockout PLK4Dox cells immunostained with the indicated antibodies. Experiments were performed in PLK4Dox monoclonal knockout cells. Scale bar = 5 µm.
- Quantification of the fraction of cells with the indicated protein localized at the centriole. Experiments were performed in PLK4Dox monoclonal knockout cells. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
- Representative images of WT or knockout PLK4Dox cells immunostained with the indicated antibodies. Experiments were performed in PLK4Dox monoclonal knockout cells. Scale bar = 5 µm.
Data information: Asterisks indicate statistically significant differences between measurements (*P < 0.05; **P < 0.01; ***P < 0.001, ****P < 0.0001). Statistics for all Figures were calculated using a two‐tailed Student’s t‐test.
Source data are available online for this figure.
We tested the ability of each knockout to enhance the proliferation of PLK4Dox cells using competition growth assays. As expected, sgRNAs targeting CRADD, PIDD1, CASP2, TP53, and P21 increased cell proliferation in response to PLK4 overexpression (Fig 1G). Furthermore, sgRNAs targeting four of the remaining 11 genes tested (FOPNL, C2CD3, SCLT1, and ANKRD26) increased the proliferation of cells with centrosome amplification more than three standard deviations above the mean of control cells. Cells expressing sgRNAs targeting each of these genes exhibited high levels of centrosome amplification following PLK4 overexpression, showing that knockout of these genes did not prevent centrosome amplification (Fig 1H). In summary, we identified all the known components of the PIDDosome pathway along with FOPNL, C2CD3, SCLT1, and ANKRD26 as playing a role in suppressing the proliferation of cells with extra centrosomes.
Centriole distal appendages are required to arrest the cell cycle in response to centrosome amplification
PIDD1 has been reported to decorate mature centrioles (Fava et al, 2017). An antibody raised against the PIDD1 death domain showed numerous cytoplasmic foci in addition to a signal that co‐localized with the distal appendage of mature parent centrioles (Fig EV1C). The centriole staining of PIDD1 was lost in PIDD1 −/− RPE1 cells while the cytosolic foci persisted, suggesting that the cytoplasmic staining is likely to be non‐specific (Fig EV1D). Consistent with this interpretation, endogenously tagged PIDD1‐mNeonGreen localized to the centriole distal appendage in DLD1 cells, but did not label cytosolic stuctures. (Fig EV1E). Importantly, PIDD1 was observed to localize to the distal appendage in both cells with normal and amplified centrosome numbers (Fig EV1C and E). 3D STORM revealed that PIDD1 exhibited a nine‐fold localization pattern similar to that of other distal appendage proteins, with an inner and outer diameter of ~ 349 and ~ 595 nm, respectively (Fig 2A–C). PIDD1 localization was most similar to that of ANKRD26 (Bowler et al, 2019), which has previously been shown to localize at the outer portion of the distal appendage and was a hit in our screen.
Figure 2. Distal appendage proteins are required to recruit PIDD1 to the mature parent centriole.
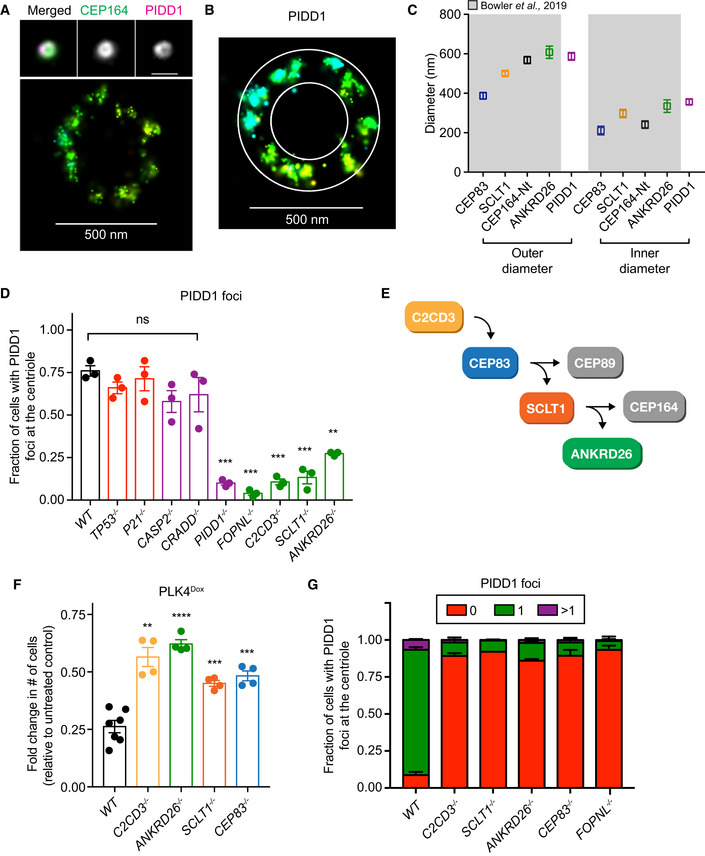
- Top, wide‐field image showing the localization of CEP164 and PIDD1 at the mature mother centriole. Bottom: Representative 3D STORM image of the same centriole showing PIDD1 localization. STORM image colors correspond to the Z‐depth with red being the closest to the coverslip and blue being the most distant.
- 3D STORM image of PIDD1 at the mature mother centriole. The overlaid mask represents the inner and outer diameters of the PIDD1 signal.
- Inner and outer diameter measurements for distal appendage proteins and PIDD1. Measurements in the shaded region were previously reported in Bowler et al (2019). Data displayed as box and whisker plots, where the box represents the upper and lower quartile and the whiskers represent the s.d. Scale bars: 1 μm for all wide‐field images of centrioles and 500 nm for STORM images. Data acquired across n ≥ 6 cells.
- Quantification of the fraction of cells with PIDD1 localized to the mature mother centriole in PLK4Dox cells expressing an sgRNA targeting the indicated genes. A dot displays measurements from each experiment. Experiments were performed in PLK4Dox polyclonal knockout cells. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
- Schematic depicting the hierarchy of recruitment of distal appendage proteins.
- Graph showing the relative growth of doxycycline‐treated PLK4Dox cells that were knocked out for the indicated genes. Each dot displays measurements from a single experiment. Experiments were performed in PLK4Dox monoclonal knockout cells. Data acquired across n ≥ 3 biological replicates. Mean ± s.e.m.
- Quantification of the fraction of cells with PIDD1 localized at the mature mother centriole. Experiments were performed in PLK4Dox monoclonal knockout cells. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
Data information: Asterisks indicate statistically significant differences between measurements (**P < 0.01; ***P < 0.001, ****P < 0.0001). Statistics for all Figures were calculated using a two‐tailed Student’s t‐test.
Source data are available online for this figure.
We tested whether sgRNAs targeting each of the nine genes that function to suppress the growth of cells with extra centrosomes disrupted the centriole recruitment of PIDD1 (Fig 1G). sgRNA‐mediated disruption of CRADD, CASP2, TP53, and P21 did not affect the centriole localization of PIDD1 (Fig 2D). By contrast, sgRNAs targeting the gene FOPNL, which encodes a protein required for cilia formation; C2CD3, encoding a protein required for distal appendage assembly (Ye et al, 2014); or SCLT1 and ANKRD26, which encode distal appendage proteins (Tanos et al, 2013; Yang et al, 2018; Bowler et al, 2019), dramatically reduced the fraction of cells that recruited PIDD1 to the centriole (Fig 2D).
Previous work has established a hierarchy for centriole distal appendage assembly in which C2CD3 is required to recruit CEP83, which recruits SCLT1 and finally ANKRD26 (Fig 2E) (Tanos et al, 2013; Bowler et al, 2019). C2CD3, SCLT1, and ANKRD26 were identified in the top 30 hits of our screens, while CEP83 did not score highly. To examine the role of all of these genes, we established monoclonal PLK4Dox knockout cell lines for C2CD3, ANKRD26, SCLT1, and CEP83 (Fig EV1F and G). Knockout of all four genes improved cell proliferation following centrosome amplification (Fig 2F) and almost completely abolished the recruitment of PIDD1 to the mature parent centriole (Figs 2G and EV1H). These data show that distal appendages are required to recruit PIDD1 to mature centrioles and to inhibit cell growth in response to centrosome amplification.
Cells lacking ANKRD26 show improved long‐term growth following centrosome amplification
In accord with its localization to the peripheral region of the distal appendage, ANKRD26 localization at mature centrioles was abolished in C2CD3 −/−, SCLT1 −/−, and CEP83 −/− cells (Figs 3A and EV2A). Moreover, sgRNA‐mediated disruption of FOPNL also prevented ANKRD26 and PIDD1 recruitment to the centriole, suggesting that FOPNL plays a role in the assembly of the centriole distal appendage (Figs 2G and 3A, and EV1H and EV2A). ANKRD26 −/− cells had a normal distal appendage localization of CEP83, SCLT1, CEP89, and CEP164 (Fig 3B and C). Moreover, ANKRD26 −/− cells formed cilia upon serum starvation at a frequency comparable to wild‐type RPE1 cells (Fig 3D and E). These data suggest that the distal appendages remain mostly intact in cells lacking ANKRD26.
Figure 3. ANKRD26 is required to arrest cell proliferation in response to centrosome amplification.
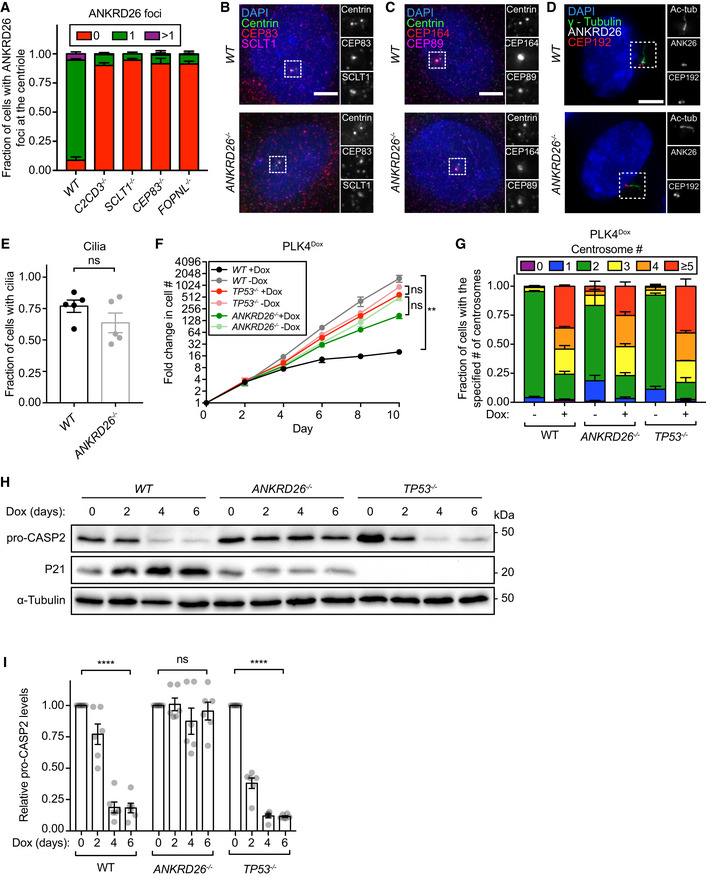
-
AQuantification of the fraction of cells with ANKRD26 localized at the mature mother centriole. Experiments were performed in PLK4Dox monoclonal knockout cells. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
-
B–DRepresentative images of WT or ANKRD26 −/− cells immunostained with the indicated antibodies. Scale bar = 5 µm.
-
EQuantification of the fraction of WT or ANKRD26 −/− hTERT RPE1 cells with cilia. Each dot displays measurements from a single experiment. Data acquired across n = 5 biological replicates. Mean ± s.e.m.
-
FGrowth assay of the indicated cells with and without doxycycline‐inducible overexpression of PLK4. Experiments were performed in PLK4Dox monoclonal knockout cells. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
-
GQuantification of centrosome number in PLK4Dox cells expressing an sgRNA targeting the indicated genes. Experiments were performed in PLK4Dox monoclonal knockout cells. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
-
HWestern blot showing expression of pro‐CASP2 and P21 following treatment with dox for the specified number of days. Experiments were performed in PLK4Dox monoclonal knockout cells.
-
IQuantification of pro‐CASP2 levels following treatment with dox for the specified number of days. Experiments were performed in PLK4Dox monoclonal knockout cells. Each dot displays measurements from a single experiment. Data acquired across n = 6 biological replicates. Mean ± s.e.m.
Data information: Asterisks indicate statistically significant differences between measurements (**P < 0.01; ****P < 0.0001). Statistics for all Figures were calculated using a two‐tailed Student’s t‐test.
Source data are available online for this figure.
Figure EV2. ANKRD26 is not required for the assembly of functional centrosomes.
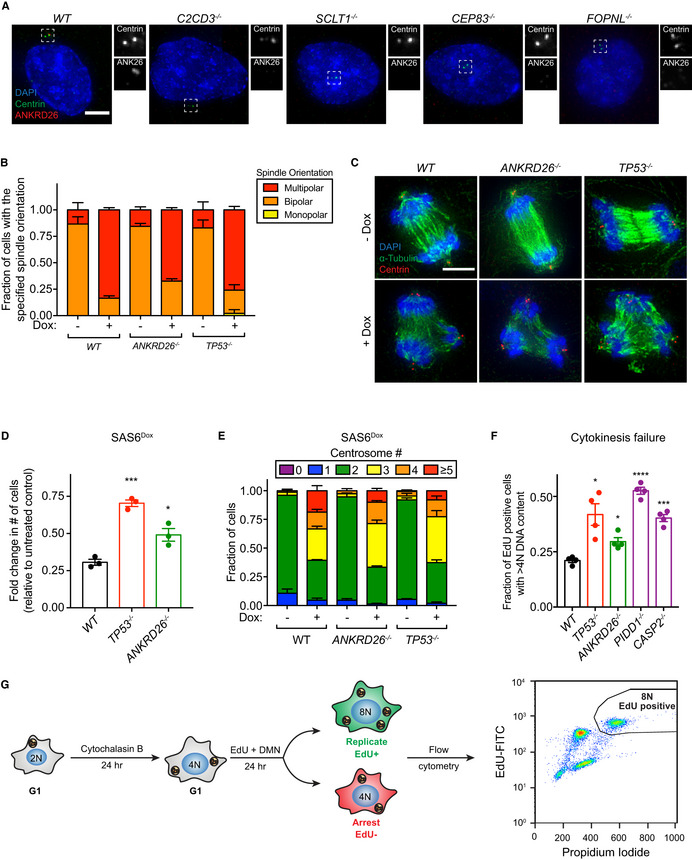
- Representative images of monoclonal PLK4Dox knockout cells. Cells were stained with the indicated antibodies. Scale bar = 5 µm.
- Quantification of the fraction of cells with the indicated mitotic spindle orientation following treatment with the centrosome declustering agent griseofulvin. Experiments were performed in monoclonal PLK4Dox knockout cells. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
- Representative images of PLK4Dox cells treated with and without doxycycline for two days then treated with griseofulvin for 24 h. Experiments were performed in monoclonal PLK4Dox knockout cells. Cells were immunostained with indicated antibodies. Scale bar = 5 µm.
- Graph showing the relative growth of doxycycline‐treated SAS6Dox cells that were knocked out for the indicated genes. Experiments were performed in SAS6Dox monoclonal knockout cells. Each dot displays measurements from a single experiment. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
- Quantification of centrosome number in SAS6Dox cells expressing an sgRNA targeting the indicated genes. Experiments were performed in SAS6Dox monoclonal knockout cells. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
- Quantification of the fold change in cycling cells with a DNA content > 4N following cytokinesis failure. Experiments were performed in PLK4Dox monoclonal knockout cells. Each dot displays measurements from a single experiment. Data acquired across n = 4 biological replicates. Mean ± s.e.m.
- Schematic showing the treatment regime for the cytokinesis failure assay shown in (F). Cells were treated with cytochalasin B for 24 h to induce cytokinesis failure followed by treatment with EdU and DMN to mark S‐phase cells and block progression through mitosis. The fraction of EdU+ cells with a DNA content > 4N was measured using flow cytometry.
Data information: Asterisks indicate statistically significant differences between measurements (*P < 0.05; ***P < 0.001, ****P < 0.0001). Statistics for all Figures were calculated using a two‐tailed Student’s t‐test.
Source data are available online for this figure.
Knockout of ANKRD26 allowed the long‐term growth of cells with extra centrosomes generated by overexpression of PLK4 (Fig 3F). Importantly, ANKRD26 was not required for normal centriole duplication, and knockout of ANKRD26 did not suppress centrosome amplification in cells overexpressing PLK4 (Fig 3G). To test if the extra centrosomes in ANKRD26 −/− cells could act as microtubule organizing centers (MTOCs), we treated PLK4Dox cells with dox for two days and then added the centrosome declustering agent griseofulvin (Rebacz et al, 2007). As expected, extra centrosomes in both wild‐type and ANKRD26 −/− cells nucleated microtubules that led to multi‐polar divisions in the presence of griseofulvin (Fig EV2B and C). We conclude that ANKRD26 is required to restrict the proliferation of cells following centrosome amplification but is not required for cilia assembly, centriole duplication, or PCM recruitment.
To further test if ANKRD26 suppresses the proliferation of cells with extra centrosomes, we used two additional methods to drive the formation of excessive centrosomes that did not require overexpression of PLK4. First, overexpression of the centriole structural component SAS6 led to centrosome amplification that reduced cell proliferation (Holland et al, 2012). This decrease in proliferation was alleviated by knockout of ANKRD26 (Fig EV2D and E). Second, we treated cells with cytochalasin B to induce cytokinesis failure and tested the ability of ANKRD26, PIDD1, and CASP2 to arrest the proliferation of tetraploid cells that have twice the normal centrosome content. Knockout of ANKRD26, PIDD1, or CASP2 increased the fraction of cycling tetraploid cells, indicating that all three of these genes act to restrict the proliferation of newly created tetraploid cells (Fig EV2F and G). We note that the relative increase in growth following loss of ANKRD26 in tetraploid cells was more modest than what was observed in cells with extra centrosomes generated by PLK4 overexpression (Figs 2F and EV2F). This may reflect the shorter duration of the growth assays performed in tetraploid cells (2 days) compared to those carried out in PLK4Dox cells (5 days). Alternatively, tetraploid cells may activate ANKRD26‐independent pathways that restrict cell proliferation (Ganem et al, 2014).
ANKRD26 links extra centrosomes to PIDDosome activation
To test the effect of ANKRD26 knockout on PIDDosome activation, we monitored the loss of pro‐CASP2 and the upregulation of P21 upon centrosome amplification. As expected (Fava et al, 2017), the levels of pro‐CASP2 decreased, and P21 increased in PLK4Dox cells in a time‐dependent manner following dox addition (Fig 3H and I). Knockout of TP53 prevented P21 upregulation but did not affect pro‐CASP2 processing following centrosome amplification. By contrast, knockout of ANKRD26 suppressed both CASP2 activation and P21 upregulation in cells with extra centrosomes (Fig 3H and I). These data show ANKRD26 acts as an upstream activator of the PIDDosome and CASP2 in cells with supernumerary centrosomes.
CASP2 is activated in response to several different upstream signals (Sladky & Villunger, 2020). To investigate if the loss of ANKRD26 globally prevents CASP2 activation, we tested the ability of ANKRD26 −/− and TP53 −/− cells to activate the CASP2 following DNA damage induced by etoposide. While the loss TP53 prevented P21 expression downstream of CASP2 activation, knockout of ANKRD26 did not alter CASP2 activation or P21 induction in cells that experienced DNA damage (Fig EV3A and B). This suggests that ANKRD26 is required to activate CASP2 in cells with extra centrosomes but is not required for CASP2 function per se. Notably, CASP2 processing still occurred in PIDD1 −/− cells treated with etoposide, suggesting that etoposide‐induced CASP2 activation is independent of the PIDDosome (Fig EV3C).
Figure EV3. ANKRD26 is not required to activate the PIDDosome in response to DNA damage.
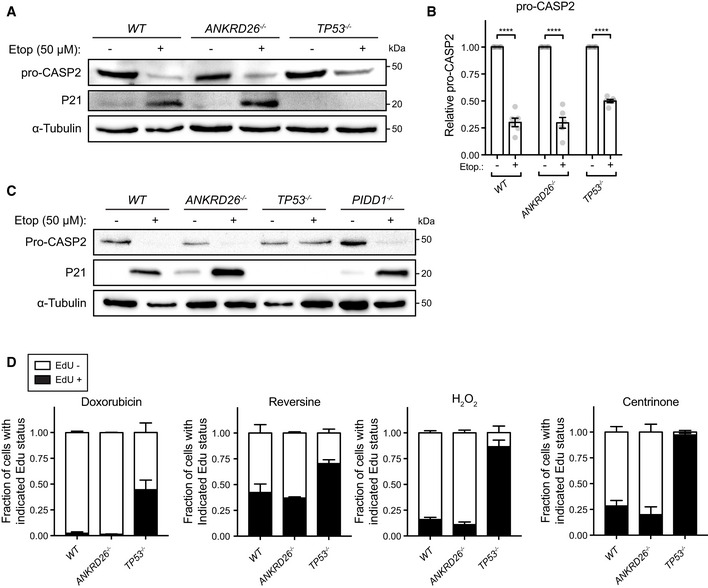
- Western blot showing expression of pro‐CASP2 and P21 following treatment with etoposide. Experiments were performed in monoclonal PLK4Dox knockout cells.
- Quantification of pro‐CASP2 levels following treatment with etoposide. Experiments were performed in monoclonal PLK4Dox knockout cells. Each dot displays measurements from a single experiment. Data acquired across n = 6 biological replicates. Mean ± s.e.m.
- Western blot showing expression of pro‐CASP2 and P21 following treatment with etoposide. Experiments were performed in PLK4Dox cells knocked out for the indicated genes.
- Quantification of the fraction of proliferating cells following treatment with the indicated drugs/reagents. Experiments were performed in monoclonal PLK4Dox knockout cells. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
Data information: Asterisks indicate statistically significant differences between measurements (****P < 0.0001). Statistics for all Figures were calculated using a two‐tailed Student’s t‐test.
Source data are available online for this figure.
We next investigated if ANKRD26 −/− cells were defective in other TP53‐dependent response pathways. Knockout of ANKRD26 −/− did not affect the ability of cells to arrest following DNA damage, centrosome loss, and chromosome segregation errors (Fig EV3D). Moreover, ANKRD26 −/− cells also underwent H2O2‐induced arrest at similar levels to control cells (Fig EV3D). By contrast, TP53 −/− cells exhibited enhanced growth in all of these conditions. Since upstream components of the DNA damage and senescence pathways did not emerge as hits in our screen, we conclude that ANKRD26‐mediated activation of TP53 is functionally distinct from the DNA damage and senescence response pathways.
The ANKRD26 coiled‐coil region binds to the UPA domain of PIDD1
To determine which region of ANKRD26 is required to recruit PIDD1 to the distal appendage, we expressed deletion mutants of mCherry‐ANKRD26 in ANKRD26 −/− cells and monitored PIDD1 recruitment to the centriole. The deletion of the ANKRD26 N‐terminus (amino acids 1–344) led to a partial reduction in the centriole recruitment of ANKRD26 and PIDD1 (Fig EV4A–D). In contrast, deletion of the ANKRD26 C‐terminus (amino acids 1,231–1,710) prevented the centriole localization of both proteins (Fig EV4A–D). This suggests that the N and C‐terminal region of ANKRD26 both play a role in recruitment to the centriole.
Figure EV4. An ANKRD26ΔM2 mutant fails to recruit PIDD1 to the distal appendage.
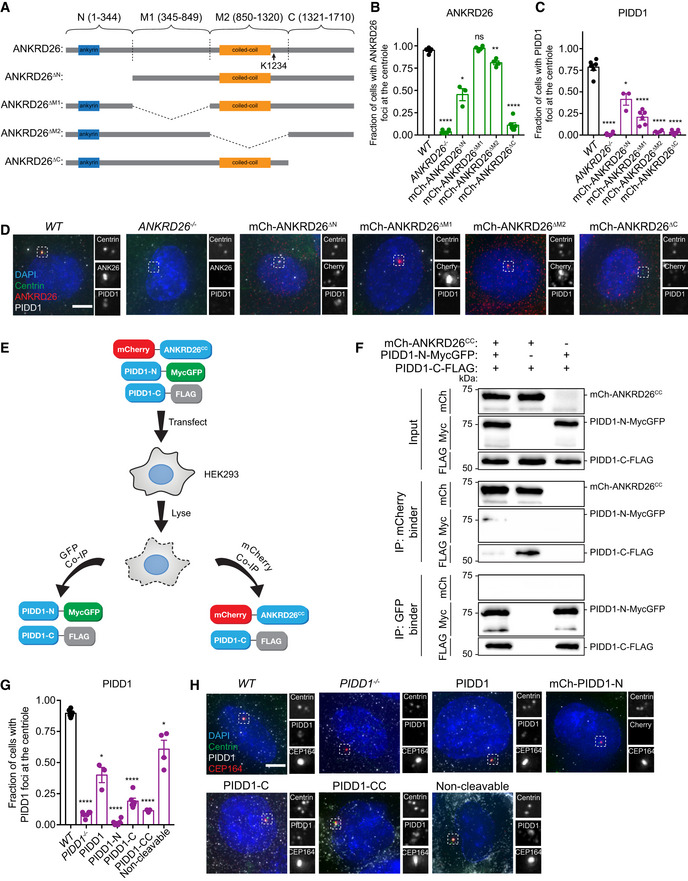
- Schematic representation of wild‐type ANKRD26 and various mutants.
- Quantification of the fraction of cells with ANKRD26 localized to the mature mother centriole in monoclonal ANKRD26 −/− PLK4Dox cells expressing the indicated mCherry‐ANKRD26 transgene. Each dot displays measurements from a single experiment. Data acquired across n ≥ 3 biological replicates. Mean ± s.e.m.
- Quantification of the fraction of cells with PIDD1 localized to the mature mother centriole in monoclonal ANKRD26 −/− PLK4Dox cells expressing the indicated mCherry‐ANKRD26 transgene. Each dot displays measurements from a single experiment. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
- Representative images of monoclonal ANKRD26 −/− PLK4Dox cells expressing the indicated mCherry‐ANKRD26 transgene. Cells were immunostained with indicated antibodies. Scale bar = 5 µm.
- Schematic of the co‐immunoprecipitation procedure performed in (F).
- HEK293FT cells were transfected with the indicated constructs. Cell lysates were split and subjected to co‐immunoprecipitation with mCherry or GFP binder beads.
- Quantification of the fraction of cells with PIDD1 localized to the mature mother centriole in monoclonal PIDD1 −/− PLK4Dox cells expressing the indicated PIDD1 transgene. Each dot displays measurements from a single experiment. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
- Representative images of monoclonal PIDD1 −/− PLK4Dox cells expressing the indicated PIDD1 transgene. Cells were immunostained with indicated antibodies. Scale bar = 5 µm.
Data information: Asterisks indicate statistically significant differences between measurements (ns P > 0.05; *P < 0.05; **P < 0.01; ****P < 0.0001). Statistics for all Figures were calculated using a two‐tailed Student’s t‐test.
Source data are available online for this figure.
The deletion of the ANKRD26 M1 region (amino acids 345–849) had no impact on the centriole recruitment of ANKRD26, but partly compromised the centriole localization of PIDD1. Notably, an ANKRD26ΔM2 mutant lacking amino acids 850–1,320 localized to the centriole, but completely failed in the recruitment of PIDD1 (Fig EV4A–D). These data indicate that the 850–1,320 amino acid region of ANKRD26 is required for the recruitment of PIDD1 to the centriole distal appendage.
The 850–1,320 amino acid region of ANKRD26 contains a conserved coiled‐coil region (amino acids 913‐1216) (Fig EV4A). To establish if this coiled‐coil region is responsible for binding to PIDD1, we overexpressed in cells with a normal centrosome content mCherry‐ANKRD26 coiled‐coil (mCherry‐ANKRD26CC) and wild‐type PIDD1‐FLAG. Purified mCherry‐ANKRD26CC associated with PIDD1‐FLAG, showing that the coiled‐coil region of ANKRD26 is sufficient for PIDD1 binding (Fig 4A).
Figure 4. The coiled‐coil domain of ANKRD26 is required for PIDD1 recruitment to the centriole and PIDDosome activation following centrosome amplification.
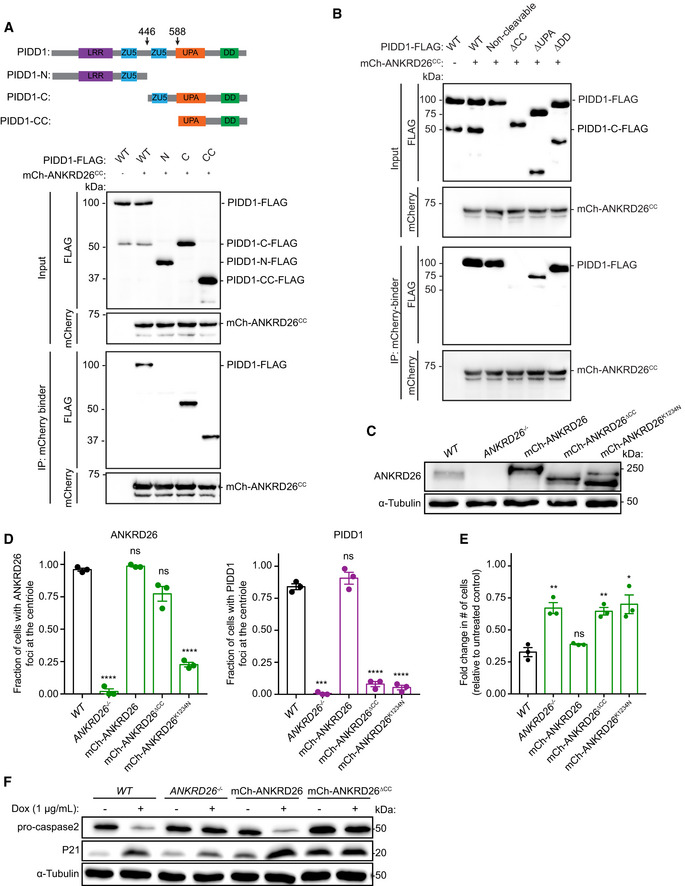
- Top: schematic representation of PIDD1 and its cleavage products. Bottom: HEK293FT cells were transfected with the indicated constructs, subjected to co‐immunoprecipitation and immunoblotted with the indicated antibodies.
- HEK293FT cells were transfected with the indicated constructs, subjected to co‐immunoprecipitation and immunoblotted with the indicated antibodies.
- Western blot showing the expression of ANKRD26. Experiments were performed in monoclonal ANKRD26 −/− PLK4Dox cells expressing the indicated ANKRD26 transgenes.
- Quantification of the fraction of cells with ANKRD26 (left) and PIDD1 (right) localized to the mature mother centriole in monoclonal ANKRD26 −/− PLK4Dox cells expressing the indicated transgenes. Each dot displays measurements from a single experiment. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
- Graph showing the relative growth of doxycycline‐treated, monoclonal ANKRD26 −/− PLK4Dox cells expressing the indicated ANKRD26 transgenes. Each dot displays measurements from a single experiment. Data acquired across n = 3 biological replicates. Mean ± s.e.m.
- Western blot showing expression of pro‐CASP2 and P21 following treatment with dox for 4 days. Experiments were performed in monoclonal ANKRD26 −/− PLK4Dox cells expressing the indicated transgenes.
Data information: Asterisks indicate statistically significant differences between measurements (*P < 0.05; **P < 0.01; ***P < 0.001, ****P < 0.0001). Statistics for all Figures were calculated using a two‐tailed Student’s t‐test.
Source data are available online for this figure.
Full‐length PIDD1 is processed into three fragments: an N‐terminal piece called PIDD1‐N and two C‐terminal fragments, PIDD1‐C and PIDD1‐CC, respectively (Fig 4A). Cleavage occurs at S446 and S588 through an autoproteolytic process similar to the mechanism described by self‐cleaving protein segments such as inteins (Tinel et al, 2007). Full‐length PIDD1 is rapidly processed in cells to form PIDD1‐C, while the second cleavage event that forms the PIDD1‐CC fragment is stimulated by specific inputs (Sladky et al, 2017, 2020). To define the region of PIDD1 that interacts with ANKRD26, we expressed in cells mCherry‐ANKRD26CC along with FLAG‐tagged PIDD1‐N, PIDD1‐C, and PIDD1‐CC. mCherry‐ANKRD26CC interacted with the PIDD1‐C and PIDD1‐CC fragments but failed to bind PIDD1‐N (Fig 4A). Importantly, ANKRD26CC did not bind to PIDD1 lacking the CC fragment (PIDD1ΔCC), demonstrating that the CC fragment of PIDD1 is both necessary and sufficient for ANKRD26 binding (Fig 4B).
The PIDD1‐CC region contains a UPA domain of unknown function (Wang et al, 2009) and a death domain (DD) that binds to CRADD to form the PIDDosome (Park et al, 2007). To establish which of these domains are responsible for associating with ANKRD26, we tested the ability of PIDD1 mutants lacking the UPA domain or DD to bind to ANKRD26CC. ANKRD26CC robustly interacted with PIDD1 lacking the DD (PIDD1ΔDD), but only weakly associated with PIDD1 deleted of the UPA domain (PIDD1ΔUPA) (Fig 4B). This suggests that the coiled‐coil region of ANKRD26 predominantly associates with the PIDD1 UPA domain.
A non‐cleavable mutant of PIDD1 retained its ability to interact with ANKRD26CC, arguing that PIDD1 cleavage is not required to bind to ANKRD26 (Fig 4B). In fact, ANKRD26CC preferentially binds to unprocessed, full‐length PIDD1 (Fig 4B). Since the PIDD1‐N and PIDD1‐C cleavage fragments remain associated in a complex after processing (Tinel et al, 2007), this raised the possibility that ANKRD26 associates with the UPA domain of unprocessed PIDD1 and that following cleavage, PIDD1‐N inhibits the binding of ANKRD26CC to PIDD1‐C. To test this hypothesis, we co‐expressed PIDD1‐N‐MycGFP, mCherry‐ANKRD26CC, and PIDD1‐C‐FLAG, and immunoprecipitated mCherry‐ANKRD26CC or PIDD1‐N‐MycGFP (Fig EV4E and F). As expected, PIDD1‐N‐MycGFP interacted with PIDD1‐C‐FLAG (Tinel et al, 2007). Notably, however, the interaction of PIDD1‐C with ANKRD26CC decreased when PIDD1‐N was expressed. Therefore, PIDD1‐C preferentially associates with PIDD1‐N and this binding inhibits the association of PIDD1‐C with ANKRD26.
The C‐terminal fragment of PIDD1 is required for its centriole localization
To determine which region of PIDD1 is responsible for its recruitment to the centriole, we generated PIDD1 −/− RPE1 cells stably expressing mCherry‐PIDD1‐N or untagged full‐length PIDD1, non‐cleavable PIDD1, PIDD1‐C, or PIDD1‐CC. We evaluated the localization of full‐length PIDD1, non‐cleavable PIDD1, PIDD1‐C, and PIDD1‐CC using an antibody raised against the PIDD1 death domain. The PIDD1‐N fragment lacked the death domain and was visualized using the mCherry tag. As expected, both full‐length and non‐cleavable PIDD1 localized to the mature parent centriole (Fig EV4G and H). The PIDD1‐C and PIDD1‐CC fragments that are capable of binding to ANKRD26 were also recruited to the centriole, but at a significantly diminished level compared with full‐length PIDD1. By contrast, the PIDD1‐N fragment that failed to interact with ANKRD26 showed no centriole localization (Fig EV4G and H). These data suggest that the PIDD1‐CC fragment is responsible for binding to ANKRD26 and recruiting PIDD1 to the centriole.
ANKRD26‐mediated recruitment of PIDD1 to the distal appendage is required for PIDDosome activation
To test the role of ANKRD26‐mediated recruitment of PIDD1 in activating the PIDDosome, we expressed a mCherry‐ANKRD26 or mCherry‐ANKRD26ΔCC transgene in ANKRD26 −/− PLK4Dox cells (Fig 4C). Expression of a wild‐type mCherry‐ANKRD26 transgene rescued PIDD1 recruitment to the mature parent centrioles (Fig 4D). Furthermore, wild‐type mCherry‐ANKRD26 promoted PIDDosome activation and suppressed the proliferation of cells with extra centrosomes (Fig 4E and F). By contrast, although the mCherry‐ANKRD26ΔCC mutant localized to the centriole, it could not recruit PIDD1 to the distal appendage (Fig 4D). Importantly, the mCherry‐ANKRD26ΔCC mutant did not restrict proliferation in cells with extra centrosomes nor did it induce PIDDosome activation (Fig 4E and F). This suggests that ANKRD26‐mediated recruitment of PIDD1 to the distal appendage is required for PIDDosome activation and growth arrest following centrosome amplification.
A recurrent cancer mutation disrupts the ability of ANKRD26 to arrest the proliferation of cells with extra centrosomes
We investigated whether defects in ANKRD26‐mediated PIDDosome activation occur in human cancers. Interestingly, a single nucleotide deletion in ANKRD26 was found in 20 independent tumors from the colon, stomach, uterus, and brain (Fig EV5A) (Cerami et al, 2012). This one base pair deletion resulted in a K1234N mutation and a premature translational stop that truncated ANKRD26 just after the CC domain that interacts with PIDD1 (Fig EV4A). The striking recurrence of the same mutation suggests that the K1234Nfs*19 alteration is selected for in these cancers. To test the impact of this mutation on ANKRD26‐mediated PIDDosome activation, we expressed a mCherry‐ANKRD26 K1234Nfs*19 transgene in ANKRD26 −/− PLK4Dox cells (Fig 4C).
Figure EV5. A recurrent ANKRD26 mutation is observed in human tumors.
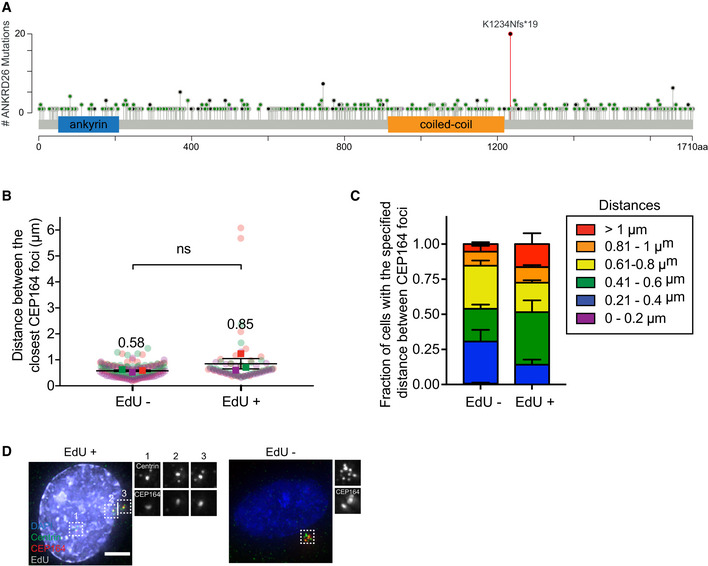
- Schematic showing the location of the 533 mutations in ANKRD26 in human tumors. Black represents truncating mutations; green represents missense mutations; purple represents inframe mutations. The K1234Nfs*19 mutation is shown in red. Data from curated set of non‐redundant studies in cBioPortal (Cerami et al, 2012).
- Plot showing the distance between the closest CEP164 rings in cycling (EdU+) and non‐cycling (EdU− cells). Squares show the mean for each biological replicate; colored circles show individual data points from each of the replicates. Data acquired across n = 3, biological replicates, each with > 20 cells. P values, unpaired two‐tailed t‐test. Mean ± s.e.m.
- Stacked bar graphs of the same data shown in (B). Mean ± s.e.m.
- Representative images of PLK4Dox RPE1 cells treated with and without doxycycline for two days and immunostained with the indicated antibodies. Scale bar = 5 µm.
Source data are available online for this figure.
Similar to what was observed following the deletion of the ANKRD26 C‐terminus (amino acids 1,231–1,710) (Fig EV4A–D), the mCherry‐ANKRD26 K1234Nfs*19 mutant localized inefficiently to the centriole and was defective in recruiting PIDD1 to the distal appendage (Fig 4D). This mutant also failed to promote PIDDosome activation or restrict the proliferation of cells with extra centrosomes. This suggests that the K1234Nfs*19 mutation disrupts the ability of ANKRD26 to localize to the centriole and trigger PIDD1 activation following centrosome amplification (Fig 4E and F).
TP53 functions downstream of ANKRD26 to arrest the growth of cells with extra centrosomes. Given that TP53 loss of function frequently occurs in human tumors and would be expected to overcome the requirement for ANKRD26‐disrupting mutations, we analyzed the fraction of ANKRD26 K1234Nfs*19 tumors that also showed oncogenic TP53 alterations. Of the 20 tumors containing K1234N mutations, 15% also contained an oncogenic TP53 alteration, and an additional 15% have a TP53 variant of unknown significance (Table EV3). This fraction is lower than the overall frequency of TP53 alterations observed for each tumor type subtype. However, since the number of tumors analyzed is small, no definitive conclusions can be drawn at this stage.
Discussion
Centrosome amplification has been shown to activate the PIDDosome and stabilize TP53 to limit cell proliferation (Holland et al, 2012; Fava et al, 2017). However, the mechanism by which excessive numbers of centrosomes activate the PIDDosome remained unclear. Using an unbiased, genome‐wide screen, we discovered five new proteins that restrict the proliferation of cells with extra centrosomes. All of these proteins localize to or are required for the assembly of centriole distal appendages. Moreover, we show that one of these proteins, the distal appendage protein ANKRD26, binds to and recruits PIDD1 to the centriole. This binding interaction is required for PIDDosome activation in response to centrosome amplification. We found that ANKRD26 preferentially interacts with the UPA domain of unprocessed PIDD1. Our data suggest that following auto‐cleavage, the PIDD1‐N fragment outcompetes the binding of ANKRD26 to the PIDD1 UPA domain. This would result in processed PIDD1 being released from binding to ANKRD26 at the distal appendage. Notably, the other PIDDosome components (CRADD and CASP2) do not appear to localize to the centriole but are needed to arrest the cell cycle in response to centrosome amplification. This suggests a model in which ANKRD26 “primes” PIDD1 at the distal appendage and releases processed PIDD1 into the cytoplasm, where it interacts with CRADD to promote CASP2 processing (Fig 5).
Figure 5. A model for PIDDosome activation in cells with extra centrosomes.
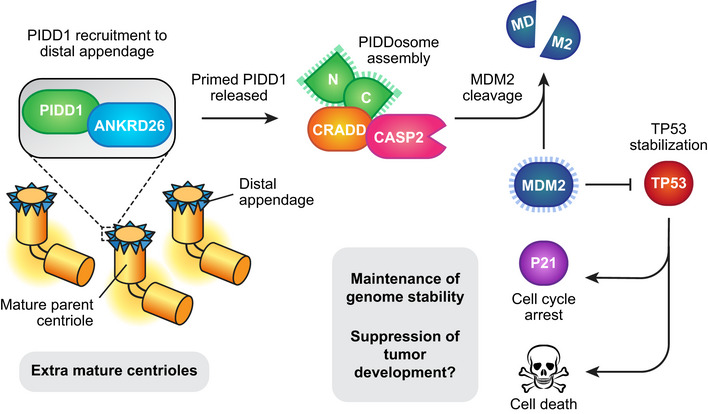
Cycling cells normally contain a single mature parent centriole that is decorated with distal appendages. PIDD1 is recruited by ANKRD26 to the centriole distal appendage. In the presence of extra mature parent centrioles, PIDD1 is activated at the distal appendage and released into the cytoplasm to promote PIDDosome assembly and caspase‐2 activation. Active caspase‐2 cleaves MDM2 leading to stabilization of P53 and subsequent cell cycle arrest or cell death.
The requirement of centriole distal appendages for PIDDosome activation following centrosome amplification suggests that these structures are being actively monitored in cells. This is consistent with a previous model where an increase in the number of mature parent centrioles was shown to be the cue that triggers PIDDosome activation (Fava et al, 2017). PIDD1 decorates the appendages of all mature parent centrioles irrespective of their number, raising the question of how PIDD1 monitors the presence of excessive numbers of mature centrioles. One possibility is that extra mature centrioles dilute out one or more components of the distal appendage, leading to structural aberrations that induce a conformational change in PIDD1 to allow for PIDDosome assembly. In this regard, it will be interesting to examine the distal appendage structure by super‐resolution and electron microscopy in cells with normal and extra mature parent centrioles. An alternative possibility is that the clustering of multiple mature parent centrioles after mitosis brings about a conformational change in PIDD1 at the distal appendage that leads to PIDDosome assembly (Fava et al, 2017). Indeed, in interphase cells, supernumerary mature centrioles are often orientated with their distal appendages ~ 600 nM apart (Fig EV5B). Moreover, we observed a ~ 30% decrease in the average distance between the distal appendages of the closest parent centrioles in arrested cells compared with cycling cells, but this difference did not reach statistical significance (Fig EV5B–D). In the future, it will be important to determine if centriole clustering in interphase is required to trigger PIDDosome activation in cells with extra centrosomes. Finally, it is plausible that a mature parent centriole produces a low‐level of active PIDD1 that is below the threshold level required for PIDDosome assembly. The presence of extra mature centrioles may push the amount of active PIDD1 above the threshold needed to trigger a cell cycle arrest. Identifying the turnover kinetics of endogenous PIDD1 at the centriole distal appendage will be critical to test this model.
Loss of ANKRD26 enables the continued proliferation of cells with extra centrosomes, raising the question of whether inactivation of this pathway is selected for during tumorigenesis. Loss of ANKRD26 would be predicted to offer no fitness advantage in tumor cells that lack TP53 functionality, perhaps explaining why ANKRD26 mutations are not commonly observed in human cancers. Nevertheless, we identified a recurrent frameshift mutation in ANKRD26 (K1234Nfs*19) observed in multiple stomach, brain, uterine, and colon tumors. This mutation leads to the expression of a truncated form of ANKRD26 that fails to recruit PIDD1 to the centriole and activate the PIDDosome in response to centrosome amplification. This suggests that loss of ANKRD26‐mediated PIDD1 signaling is selected for in some human tumors. It is plausible that the K1234Nfs*19 ANKRD26 mutation enables the continued propagation of tumor cells with extra centrosomes, and it will be interesting to examine if selection for this mutation co‐occurs with centrosome amplification in cancer cells.
In addition to a possible role of ANKRD26 mutations in tumorigenesis, mutations in the 5'UTR of the ANKRD26 gene cause an autosomal‐dominant form of thrombocytopenia due to reduced blood platelet production by megakaryocytes (Pippucci et al, 2011; Noris et al, 2013). During their maturation, megakaryocytes undergo several rounds of endomitosis to produce large polyploid cells with extra centrosomes (Moskvin‐Tarkhanov & Onishchenko, 1978; Vitrat et al, 1998; Falcieri et al, 2000). Thrombocytopenia‐associated mutations in ANKRD26 lead to the persistent expression of ANKRD26 during the late stages of megakaryocyte development, and, consequently, megakaryocytes from these patients exhibit reductions in cell ploidy (Bluteau et al, 2014). Based on our observations, it is tempting to speculate that the reduction in megakaryocyte ploidy in these patients arises due to ANKRD26‐mediated activation of the PIDDosome in megakaryocytes with extra centrosomes (Bluteau et al, 2014). Patients with mutations in the 5′UTR of ANKRD26 also exhibit an increased incidence of myeloid malignancies (Pippucci et al, 2011; Noris et al, 2013; Marquez et al, 2014), and N‐terminal truncating mutations in ANKRD26 have been identified in sporadic, adult‐onset AML cases (Marconi et al, 2017). How these ANKRD26 mutations lead to an increased incidence of hematological malignancies remains unclear.
Many human tumors arise from a tetraploid intermediate containing twice the normal DNA and centrosome content (Zack et al, 2013; Bielski et al, 2018). At least two pathways appear to have evolved to restrict the proliferation of genomically unstable tetraploid cells. First, extra centrosomes in tetraploid cells activate the PIDDosome and CASP2 to inactivate MDM2 (Oliver et al, 2011; Fava et al, 2017; Sladky et al, 2020). Second, tetraploid cells hyper‐activate Rac1, which leads to a decline in RhoA‐GTP (Godinho et al, 2014). Decreased RhoA‐GTP leads to activation of the hippo pathway kinase LATS2, which stabilizes p53 through inhibition of MDM2 (Ganem et al, 2014). Correspondingly, RNAi knockdown of LATS2 enables tetraploid cells to overcome a G1 cell cycle arrest (Ganem et al, 2014). However, it is notable that we did not identify LATS2 in our screen for genes that limit the proliferation of cells with extra centrosomes. We envisage several possible explanations for this discrepancy. First, there may be differences in the fitness of LATS2 knockout cells generated using CRISPR/Cas9 in our study and cells depleted of LATS2 by RNAi in the Ganem et al, study. Second, knockdown of LATS2 was shown to enable tetraploid cells to progress into S phase, but may not offer the long‐term growth advantage that would be required to score as a hit in our screen. Third, activation of LATS2 in tetraploid cells may not only be driven by an increased number of centrosomes and could rely on other features of tetraploid cells, such as the increased DNA content or reduced surface area to volume ratio (Ganem et al, 2014). Finally, tetraploid cells are expected to contain two mature parent centrioles. A more dramatic increase in parent centriole number following PLK4 overexpression could lead to more robust PIDDosome activation and a greater reliance on this pathway to suppress proliferation.
In conclusion, we have identified a new role for the centriole distal appendage protein ANKRD26 in signaling to the PIDDosome. Mammalian centrosomes have long been proposed to function as signaling hubs in a manner analogous to the related spindle pole bodies (SPB) of yeast (Arquint et al, 2014). However, while yeast SPBs have established roles in controlling entry into and exit from mitosis (Hagan & Grallert, 2013; Fu et al, 2015), there is a lack of definitive evidence for a role of vertebrate centrosomes in cell signaling. Our finding that distal appendages of mature parent centrioles deliver a signal required to activate the PIDDosome offers new support for a role of centrosomes in controlling cell proliferation in human cells.
Materials and Methods
Cell culture and drug treatments
RPE‐1 cells were grown in DMEM:F12 50:50 medium (Cellgro; Corning) containing 10% fetal bovine essence (Seradigm), 0.348% sodium bicarbonate, 100 U/ml penicillin, 100 U/ml streptomycin, and 2 mM l‐glutamine. HEK293FT and DLD1 cells were grown in DMEM medium (Cellgro; Corning) containing 10% fetal bovine essence (Seradigm), 100 U/ml penicillin, 100 U/ml streptomycin, and 2 mM l‐glutamine. RPE‐1 and DLD1 cells were validated by STR genotyping. Cells were maintained at 37°C in a 5% CO2 atmosphere with 21% oxygen. Doxycycline (Millipore Sigma) was diluted in water and used at a final concentration of 1 µg/ml. Etoposide (Millipore Sigma) was diluted in DMSO and used at a final concentration of 50 µM. Doxorubicin (Sigma‐Aldrich) was diluted in DMSO and used at a final concentration of 50 ng/µl. Reversine (Sigma‐Aldrich) was diluted in DMSO and used at a final concentration of 500 nM. Centrinone (Tocris) was dissolved in DMSO and used at a final concentration of 125 nM. Griseofulvin (Sigma‐Aldrich) was diluted in DMSO and used at a final concentration of 25 µM. Hydrogen peroxide 30% w/w (Sigma‐Aldrich) was diluted in water and used at a final concentration of 150 nM. All cell lines were determined to be free from mycoplasma contamination using DAPI staining.
Lentiviral production and transduction
The pLentiGuide‐puromycin (117986; Addgene), pLentiCRISPR‐v2‐Neomycin (127644; Addgene), Lenti‐CMV‐Zeocin (this study), Tet‐ON mPLK4 or Tet‐ON hSAS6 lentiviral plasmid was co‐transfected into HEK293FT cells with the lentiviral packaging plasmids psPAX2 and pMD2.G (12260 and 12259; Addgene). In brief, 3 × 106 HEK293FT cells were seeded into a poly‐l‐Lysine‐coated 10 cm culture dish the day before transfection. For each 10 cm dish, the following DNA was diluted in 0.6 ml OptiMEM (Thermo Fisher Scientific): 4.5 µg lentiviral vector, 6 µg psPAX2, and 1.5 µg pMD2.G. Separately, 35 µl of 1 µg/µl 25‐kD polyethylenimine (Sigma‐Aldrich) was diluted into 0.6 ml OptiMEM and incubated at room temperature for 5 min. After incubation, the DNA and polyethylenimine mixtures were combined, and incubated at room temperature for 20 min. During this incubation, the culture media was replaced with 8 ml pre‐warmed DMEM + 1% FBS. The transfection mixture was then added dropwise to the 10 cm dish. Viral particles were harvested 48 h after the media change and filtered through a 0.45 µm PVDF syringe filter. The filtered supernatant was used directly to infect cells. Aliquots were snap‐frozen and stored at −80°C. For transduction, lentiviral particles were diluted in complete growth media supplemented with 10 µg/ml polybrene (Sigma‐Aldrich) and added to cells.
Gene targeting and generation of stable cell lines
To create RPE1 PLK4Dox and SAS6Dox cells, puromycin‐sensitive RPE‐1 cells stably expressing Cas9 (Lambrus et al, 2016) were transduced with a Tet‐ON mPLK4 rtTA‐IRES‐GFP‐T2A‐Neo or Tet‐ON hSAS6 rtTA‐IRES‐GFP‐T2A‐Neo lentivirus and selected with 400 µg/ml G418 (P212121 cat # LGB‐418‐10) for 2 weeks. A monoclonal cell line stably expressing Cas9 and the Tet‐ON mPLK4 or Tet‐ON hSAS6 transgene was isolated and used in all further experiments.
ANKRD26 rescue cell lines were generated by transducing monoclonal RPE‐1 PLK4Dox ANKRD26 −/− cells with a CMV‐mCherry‐ANKRD26‐SV40‐Zeo lentivirus. Cells were selected in 400 µg/ml zeocin (Invitrogen) for 10 days. PIDD1 rescue cell lines were generated by transducing monoclonal RPE‐1 PLK4Dox PIDD1 −/− cells with a CMV‐PIDD1‐SV40‐Zeo lentivirus. Cells were selected in 400 µg/ml zeocin (Invitrogen) for 10 days.
To create the DLD1; PIDD1‐2xmNeonGreen cell line, an sgRNA targeting the PIDD1 gene (5′‐cccagagcctgcccaggcct‐3′) was cloned into a pX459 vector (#62988; Addgene). To generate the PIDD1 repair vector, we cloned a 2× mNeonGreen tag followed by a T2A‐neomycin and a translational stop codon into a modified pUC vector. 500 bp 5′ and 500 bp 3′ homology arms were PCR amplified from genomic DLD1 DNA and cloned on either side of the central 2′ mNeonGreen‐T2A‐Neomycin cassette. DLD1 cells were transiently transfected (X‐tremeGENE HP, Roche) with the pX459 plasmid and repair vector. Selection of transfected cells was performed 5 days after transfection with 400 µg/ml G418.
Generation of knockout cell lines
RPE‐1 PLK4Dox or SAS6Dox cells were transduced with a pLentiGuide‐Puromycin lentivirus containing an sgRNA targeting the gene of interest. Cells were then selected in 1 µg/ml puromycin for 2 days. Knockout cell lines were validated by sequencing genomic DNA to characterize frameshift mutations and loss of signal by either immunoblotting or immunofluorescence. To examine cilia formation, hTERT RPE‐1 cells were transduced with a pLentiCRISPR‐v2‐Neomycin lentivirus containing an sgRNA targeting ANKRD26. Cells were then selected with 400 µg/ml Neo for 7 days.
TIDE
TIDE (Tracking of Indels by Decomposition) was used to estimate the INDEL efficiency in the polyclonal Plk4Dox knockout cells shown in Fig 1G and H (Brinkman et al, 2014). To calculate the frequency of frameshift mutations, INDELs in the +3, +6, +9, −3, −6, −9 reading frames were excluded from the final calculation.
Growth assays
For competition growth assays, RPE‐1 PLK4Dox cells constitutively expressing EGFP and non‐fluorescent RPE1 cells were mixed at a 1:1 ratio and seeded into duplicate wells. One well from each pair was treated with doxycycline. After 5 days, each well was trypsinized and analyzed on a Guava easyCyte flow cytometer to determine the fraction of GFP‐positive cells. For each well, the fraction of GFP‐positive cells was divided by the GFP‐negative cells. The value obtained from the doxycycline treated well was then divided by that obtained in the untreated well to determine the fold change in GFP‐positive cells.
For long‐term growth assays, RPE‐1 PLK4Dox cells were seeded into duplicate wells and one well form each pair was treated with 1 µg/ml doxycycline. Cell number was counted every 2 days in triplicate using a LUNA‐II automated cell counter.
Co‐immunoprecipitation
Co‐immunoprecipitation was performed as previously described (Moyer et al, 2015). 2 × 106 HEK293FT cells were seeded into 10 cm2 dishes and 24 h later were transfected with plasmid DNA. 48 h later, transfected cells were lysed in lysis buffer [10 mM Tris pH 7.5, 0.1% Triton X‐100, 250 mM NaCl, 1 mM EDTA, 50 mM NaF, 50 mM β‐glycerophosphate, 1 mM DTT, 500 nM microcystin, 1 mM PMSF and EDTA‐free protease inhibitor tablet (Roche)], sonicated and soluble extracts prepared. The supernatant was incubated with beads coupled to either GFP‐binding or mCherry‐binding protein (Rothbauer et al, 2008). Beads were washed three times in lysis buffer, and immunopurified protein was analyzed by immunoblot.
Immunoblotting and immunofluorescence
Immunoblotting and immunofluorescence were performed as previously described (Moyer et al, 2015). For immunoblot analysis, protein samples were separated by SDS–PAGE, transferred onto nitrocellulose membranes with a Trans‐Blot Turbo Transfer System (BioRad) or by overnight wet‐transfer and probed with the following antibodies: alpha‐tubulin (rat, Y1/2, Invitrogen, MA180017, 1:1,000), ANKRD26 (rabbit, GeneTex, GTX128255, 1:1,000), PIDD1 (mouse, Enzo, ALX‐804837C100, 1:500), caspase 2 (rat, 11B4, Millipore Sigma, MAB3507, 1:1,000), TRIM37 (rabbit, Bethyl Laboratories, A301‐174A‐M, 1:1,000), and p21WAF1 (mouse, Ab‐1, CalBiochem, OP64, 1:100). Blots were blocked with 5% milk in TBST and washed with TBST.
For immunofluorescence, cells were grown on 18‐mm glass coverslips and fixed in 100% −20°C methanol for 10 min. Cells were blocked in 2.5% FBS, 200 mM glycine, and 0.1% Triton X‐100 in PBS for 1 h. Antibody incubations were conducted in the blocking solution for 1 h. DNA was detected using DAPI, and cells were mounted in Prolong Antifade (Invitrogen). Staining was performed with the following primary antibodies: USP28 (rabbit, Proteintech, 17707‐1‐AP, 1:1,000), Centrin (rabbit, directly conjugated, (Moyer & Holland, 2019), 1:1,000), CEP192 (goat, directly labeled, (Moyer & Holland, 2019), 1:1,000), SCLT1 (rat, (Tanos et al, 2013), 1:250), CEP83 (rabbit, Sigma‐Aldrich, HPA038161, 1:200), CEP89 (rat, (Tanos et al, 2013), 1:500), 1:1,000), Polyglutamylation (mouse, GT335, AdipoGen, AG‐20B‐0020‐C100), FOPNL/FOR20 (rat, (Sedjaï et al, 2010), 1:500), C2CD3 (rabbit, Atlas Antibodies, HPA038552, 1:500), and CEP164 (rabbit, Millipore Sigma, AEB2621, 1:1,000). Secondary donkey antibodies were conjugated to Alexa Fluor® 488, 555, or 650 (Life Technologies).
Immunofluorescence images were collected using a Deltavision Elite system (GE Healthcare) controlling a Scientific CMOS camera (pco.edge 5.5). Acquisition parameters were controlled by SoftWoRx suite (GE Healthcare). Images were collected at room temperature using an Olympus 60× 1.42 NA or Olympus 100× 1.4 NA oil objective at 0.2 μm z‐sections and subsequently deconvolved in SoftWoRx suite. Images were acquired using Applied Precision immersion oil (N = 1.516).
Cilia assembly
hTERT RPE‐1 cells were grown to confluency on 18‐mm glass coverslips and then placed in serum‐free media (DMEM:F12 50:50 medium supplemented with 0.348% sodium bicarbonate, 100 U/ml penicillin, 100 U/ml streptomycin, and 2 mM l‐glutamine) for 2 days. Prior to fixation, cells were washed with 1× PBS and incubated in microtubule stabilization buffer (30% glycerol, 100 mM PIPES, 1 mM EGTA, 1 mM MgSO4) for 30 s. Cells were then washed with PBS and processed for immunofluorescence staining.
Cytokinesis failure assay
To look at the effects of cytokinesis failure, PLK4Dox cells were seeded in duplicate wells. One well was treated with 10 µg/ml cytochalasin B (Sigma‐Aldrich), and the other well was treated with DMSO for 24 h. Cells were then washed with media 3 times, and then, fresh media were added to cells supplemented with 10 µM EdU (Invitrogen) and 10 µM Dimethylenastron (DMN) (Sigma‐Aldrich). Cells were incubated for 24 h. Cells were then collected and fixed with 100% ethanol at −20°C for at least 1 h. Each sample was washed with PBS then stained with a Click‐iT reaction for 30 min in the dark. The Click‐iT reaction was prepared following manufacturer’s protocol (Invitrogen). Samples were washed and stained with a 25 µg/ml propidium iodide solution supplemented with RNase A (Sigma‐Aldrich) for 30 min in the dark. Samples were analyzed on a BD FACSCalibur flow cytometer to determine DNA content and EdU incorporation. The percent of EdU‐positive cells with a DNA content of greater than 4N was determined for each sample. The DMSO‐treated sample was subtracted from the cytochalasin B‐treated sample for each cell line.
Griseofulvin assay
Cells were grown on 18‐mm glass coverslips and treated with griseofulvin for 24 h. Cells were then fixed in 100% −20°C methanol for 10 min and processed for immunofluorescence staining. Anaphase cells were imaged, and spindle morphology scored.
EdU incorporation
PLK4Dox cells were seeded in duplicate wells and one was treated with one of the following drugs: doxorubicin for 2 days, reversine for 2 days, centrinone for 5 days or H2O2 for 2 h followed by an overnight incubation in fresh media. Both the treated and untreated wells were seeded onto 18‐mm glass coverslips and incubated in media supplemented with 10 µM EdU for 24 h. Cells were then fixed in 100% −20°C methanol for 10 min, washed with PBST and stained with a Click‐iT reaction for 30 min in the dark to label EdU. Each coverslip was incubated with 197.2 µl 1× PBS, 2.5 µl 100 mM CuSO4, 50 µl 500 mM Ascorbic Acid (prepared fresh), and 0.3 µl 125 µM Azide‐fluor 488 (Millipore Sigma). After 30 min, cells were rinsed with PBS, DAPI stained, and then mounted with Prolong Antifade.
Immunofluorescence for STORM
Cells were grown on 25 mm, 1.5 high tolerance coverslips (Warner Instruments). Cells were fixed in 1.5% formaldehyde for 4 min then permeabilized in 0.05% Triton for 30 s. Samples were washed in 1× PBS and blocked in IF buffer (1% BSA, 0.05% Tween‐20, in 1× PBS) for 15 min. Cells were incubated with primary antibody diluted in IF buffer at 37°C for 3 h. After washing in 1× PBS, cells were incubated with secondary antibody diluted IF buffer at 37°C for 3 h. The following primary antibodies were used: CEP164 (rabbit, recognizing aa: 1–112, Proteintech, 22227‐1‐AP, 1:3,500), PIDD1 (mouse, Enzo, ALX‐804837C100, 1:500). Conjugated secondary antibodies CF647 anti‐mouse (Biotium, 20042) and CF568 anti‐rabbit (Biotium, 20099) were used at 1:800 dilution.
Stochastic optical reconstruction microscopy (STORM)
Samples were layered with 100 nm tetra‐spectral fluorescent spheres (Invitrogen), which served as fiducial markers. Coverslips were mounted in Attofluor Cell chambers (Thermo Fisher) in imaging buffer (25 mM β‐mercaptoethylamine, 0.5 mg/ml glucose oxidase, 67 μg/ml catalase, 10% dextrose, in 100 mM Tris at pH 8.0). 3D STORM imaging was performed on a Nikon N‐STORM4.0 system using an Eclipse Ti inverted microscope, Apo TIRF 100× SA NA 1.49 Plan Apo oil objective, 405, 561, 488, and 647 nm excitation laser launch and a back‐illuminated EMCCD camera (Andor, DU897). The 647 nm laser line (~ 150 mW out of the fiber and ~ 90 mW before the objective lens) was used to promote fluorophore blinking. The 405 nm laser was used to reactivate fluorophores. The 561 nm laser was used to record the signals of fiducial markers. 20,000 to 30,000‐time points were acquired at a 50 Hz frame rate each 16–20 ms. NIS Elements (Nikon) were used to analyze the data.
Prior to STORM imaging, the position of CEP164 and PIDD1 was recorded in wide‐field mode. The original storm Z color coding scheme illustrating the calibrated Z range (from red for the signals closer to the coverslip to blue for the signals further from the coverslip) is preserved on STORM images, which are presented as a projection of the entire 3D volume. The outer and inner diameters of distal appendage proteins were determined by measuring the diameters of the circles outlining the outer and inner edges of the STORM signal (Bowler et al, 2019).
CRISPR/Cas9 genome‐wide screen
CRISPR/Cas9 pooled, knockout screens were performed essentially as described (Shalem et al, 2014; Chen et al, 2015; Lambrus et al, 2016). RPE‐1 PLK4Dox cells were infected with a lentivirus containing an sgRNA targeting TRIM37 or USP28. The sgRNA sequence targeting TRIM37 was CTCTAATTTAAATAGCATGG. The sgRNA sequence targeting USP28 was ATCAACTCTCCTCCAGTCAT. Infected cells were then selected with 400 µg/ml zeocin for 3 weeks and monoclonal knockout lines isolated and validated by immunoblotting.
The human Brunello CRISPR knockout sgRNA library was purchased from Addgene (a gift of David Root and John Doench; #73178) and plasmid DNA amplified according to the manufacturer’s instructions. To produce virus, the Brunello pooled plasmid library and the lentiviral packaging plasmids psPAX2 and pMD2.G were co‐transfected into 40 × 15 cm culture dishes of HEK293FT cells. 6 × 106 HEK293FT cells were seeded into a poly‐l‐Lysine‐coated 15 cm culture dish the day before transfection. For each 15 cm dish, the following DNA was diluted in 1.2 ml OptiMEM (Thermo Fisher Scientific): 9 µg lentiviral vector, 12 µg psPAX2, and 3 µg pMD2.G. Separately, 70 µl of 1 µg/µl 25‐kD polyethylenimine (Sigma‐Aldrich) was diluted into 1.2 ml OptiMEM and incubated at room temperature for 5 min. After incubation, the DNA and polyethylenimine mixtures were combined and incubated at room temperature for 20 min. During this incubation, the culture media were replaced with 16 ml prewarmed DMEM + 1% FBS. The transfection mixture was then added dropwise to the 15 cm dish. Viral particles were harvested at 24, 48, and 72 h after the media change. Media collected from 24, 48, and 72 h was pooled and filtered through a 0.45 µm PVDF syringe filter. The media were then concentrated using Amicon Ultra‐15 Centrifugal Filter Unit with Ultracel‐50 membrane (EMD Millipore Corporation cat# UFC905024). The virus was then frozen and stored at −80°C.
Cells were transduced with the Brunello library via spinfection as previously described (Lambrus et al, 2016). To find the optimal virus volumes for achieving an MOI ~ 0.1, each new batch of virus was titered by spinfecting 3 × 106 cells with several different volumes of virus. Briefly, 3 × 106 cells per well were seeded into a 12 well plate in growth media supplemented with 10 µg/ml polybrene. Each well received a different titrated virus amount (between 5 and 50 µl) along with a no‐transduction control. The plate was centrifuged at 872 g for 2 h at room temperature. After the spin, media were aspirated, and fresh growth media were added. The following day, cells were counted, and each well was split into duplicate wells. One well received 3 µg/ml puromycin (Sigma) for 3 days. Cells were counted and the percent transduction calculated as the cell count from the replicate with puromycin divided by the cell count from the replicate without puromycin multiplied by 100. The virus volume yielding a MOI closest to 0.1 was chosen for large‐scale transductions.
For the pooled screen, a total of 1 × 108 PLK4Dox; TRIM37 −/−; or PLK4Dox; USP28 −/− cells were infected at MOI ∼ 0.1 and selected with puromycin at 3 μg/ml for 3 days. MOI was calculated using a control well infected in parallel following the same procedure outlined above. Infected cells were expanded under puromycin selection for 5 days and then seeded into 80 × 15 cm dishes with 250,000 cells per dish. 40 of the dishes received media supplemented with 400 µg/ml of G418 to maintain selection for the Tet‐ON mPLK4 transgene and the other 40 dishes received 1 µg/ml of doxycycline and 400 µg/ml of G418. Cells were allowed to grow for 21 days without further passaging before being harvested for DNA extraction.
Cell pellets were resuspended in lysis buffer containing 50 mM Tris, 50 mM EDTA, 1% SDS, pH 8, and 30 µl of 20 mg/ml proteinase K and incubated at 55°C overnight. The next day, 30 µl of 10 mg/ml RNase A was added, and the sample was inverted 25 times and incubated at 37°C for 30 min. Samples were cooled on ice before adding 2 ml of chilled 7.5 M ammonium acetate. Samples were then vortexed at high speed for 20 s and centrifuged at > 4,000 g for 10 min. The supernatants were decanted into new 15 ml conical tubes, and 6 ml of 100% isopropanol was added. The tubes were inverted 50 times and centrifuged at > 4,000 g for 10 min. The supernatant was discarded, and 6 ml of freshly prepared 70% ethanol was added to each tube. The tubes were inverted 10 times and centrifuged at > 4,000 g for 1 min. The supernatant was discarded, and the pellet was air dried for 30 min. Finally, 200 µl of 1 × TE buffer was added, and the tube was incubated at 65°C for 1 h. DNA concentration was measured using a Nanodrop.
The sgRNA library for each sample was amplified and prepared for Illumina sequencing using a two‐step PCR procedure as previously described (Lambrus et al, 2016). For the first PCR, a region containing the sgRNA cassette was amplified using primers specific to the sgRNA‐expression vector:
lentiGuide‐PCR‐F: AATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCG
lentiGuide‐PCR1‐R: CTTTAGTTTGTATGTCTGTTGCTATTATGTCTACTATTCTTTCC
The thermocycling parameters for the first PCR were as follows: 98°C for 30 s, 18–24 cycles of (98°C for 1 s, 62°C for 5 s, 72°C for 35 s), and 72°C for 1 min. 1.5 μg of DNA was used in each PCR reaction. Assuming 6.6 pg of DNA per cell, ~ 100× representation of the Brunello library required ~ 53 µg of DNA per sample (36 PCR reactions). The resulting amplicons for each sample were pooled, gel purified, and used for amplification with barcoded second PCR primers. For each sample, we performed 12 reactions.
Primers for the second PCR include both a variable length sequence to increase library complexity and an 8 bp barcode for multiplexing of different biological samples:
F2: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT(4–7 bp random nucleotides) (8 bp barcode)TCTTGTGGAAAGGACGAAACACCG
R2: CAAGCAGAAGACGGCATACGAGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTCTACTATTCTTTCCCCTGCACTGT
5 µl of the product from the first PCR reaction was used, and the thermocycling parameters for the second PCR were as follows: 98°C for 30 s, 18–24 cycles of (98°C for 1 s, 70°C for 5 s, 72°C for 35 s). Second PCR products were pooled, gel purified, and quantified using the Next Library Quantification Kit (NEB). Diluted libraries with 5% PhiX were sequenced with MiSeq (Illumina).
Sequencing data were processed for sgRNA representation using custom scripts. Briefly, sequencing reads were first demultiplexed using the barcodes in the forward primer and then trimmed to leave only the 20 bp sgRNA sequences. The spacer sequences were then mapped to the spacers of the designed sgRNA library using Bowtie (Langmead et al, 2009). For mapping, a maximum of one mismatch was allowed in the 20 bp sgRNA sequence. Mapped sgRNA sequences were then quantified by counting the total number of reads. The total numbers of reads for all sgRNAs in each sample were normalized.
The screen was performed two independent times for both PLK4Dox, TRIM37 −/−, and PLK4Dox, USP28 −/− cells. We used the MaGeCK scoring algorithm (model‐based analysis of genome‐wide CRISPR‐Cas9 knockout) to analyze and the rank the genes from the screens (Li et al, 2014). We noted that some of the hits from the MAGeCK analysis contained sgRNAs with very low representation (Cerami et al, 2012). Among these low count, hits were many mitochondria and ATP production related genes that are likely selected for by the doxycycline treatment. We therefore excluded all genes that did not show a ≥ 0.005% representation for at least two sgRNAs from the doxycycline‐treated population from any transduction of either the PLK4Dox; TRIM37 −/− or PLK4Dox; USP28 −/− cell lines. Genes with an FDR cutoff of ≤ 0.4 were taken forward for further validation.
ANKRD26 cancer mutation
The AKNRD26 K1234Nfs*19 cancer mutation was identified from the curated set of non‐redundant studies in cBioportal (https://www.cbioportal.org) (Cerami et al, 2012).
Quantification and statistical analysis
Statistical analysis was performed using GraphPad Prism software. Differences between samples were tested using a two‐tailed Student’s t‐test and annotated following the nomenclature: ns (P > 0.05), *(P ≤ 0.05), **(P ≤ 0.01), ***(P ≤ 0.001), and ****(P ≤ 0.0001).
Author contributions
AJH and LTE conceived the project. LTE performed and analyzed the majority of the experiments and prepared the figures. TA performed the genome‐wide CRISPR screen. PS performed the co‐immunoprecipitation experiments. KL and JL performed the STORM imaging of PIDD1. AJH and LTE wrote the manuscript. All authors edited the manuscript. AJH conceived and supervised the study.
Conflict of interest
The authors that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
This work was supported by the National Institutes of Health grants R01GM114119 and R01GM133897, an American Cancer Society Scholar grant RSG‐16‐156‐01‐CCG, and an American Cancer Society Mission Boost Grant MBG‐19‐173‐01‐MBG (to A.J.H). L.T. Evans was funded by National Institutes of Health training grant (T32GM007814) and a National Science Foundation Graduate Research Fellowship. J.L. was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute, Center for Cancer Research.
The EMBO Journal (2021) 40: e105106.
See also: L Krenning et al (February 2021)
Data availability
This study includes no data deposited in external repositories.
References
- Alves‐Cruzeiro JM, Nogales‐Cadenas R, Pascual‐Montano AD (2014) CentrosomeDB: a new generation of the centrosomal proteins database for human and Drosophila melanogaster . Nucleic Acids Res 42: D430–D436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnandis T, Monteiro P, Adams SD, Bridgeman VL, Rajeeve V, Gadaleta E, Marzec J, Chelala C, Malanchi I, Cutillas PR et al (2018) Oxidative stress in cells with extra centrosomes drives non‐cell‐autonomous invasion. Dev Cell 47: 409–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arquint C, Gabryjonczyk AM, Nigg EA (2014) Centrosomes as signalling centres. Philos Trans R Soc Lond B Biol Sci 369: 20130464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW (2008) Centrosome amplification can initiate tumorigenesis in flies. Cell 133: 1032–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielski CM, Zehir A, Penson AV, Donoghue MTA, Chatila W, Armenia J, Chang MT, Schram AM, Jonsson P, Bandlamudi C et al (2018) Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet 50: 1189–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluteau D, Balduini A, Balayn N, Currao M, Nurden P, Deswarte C, Leverger G, Noris P, Perrotta S, Solary E et al (2014) Thrombocytopenia‐associated mutations in the ANKRD26 regulatory region induce MAPK hyperactivation. J Clin Invest 124: 580–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler M, Kong D, Sun S, Nanjundappa R, Evans L, Farmer V, Holland A, Mahjoub MR, Sui H, Loncarek J (2019) High‐resolution characterization of centriole distal appendage morphology and dynamics by correlative STORM and electron microscopy. Nat Commun 10: 993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, van Steensel B (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42: e168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JY (2011) A clinical overview of centrosome amplification in human cancers. Int J Biol Sci 7: 1122–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, Scott DA, Song J, Pan JQ, Weissleder R et al (2015) Genome‐wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160: 1246–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho PA, Bury L, Shahbazi MN, Liakath‐Ali K, Tate PH, Wormald S, Hindley CJ, Huch M, Archer J, Skarnes WC et al (2015) Over‐expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol 5: 150209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D (2012) DNA breaks and chromosome pulverization from errors in mitosis. Nature 482: 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcieri E, Bassini A, Pierpaoli S, Luchetti F, Zamai L, Vitale M, Guidotti L, Zauli G (2000) Ultrastructural characterization of maturation, platelet release, and senescence of human cultured megakaryocytes. Anat Rec 258: 90–99 [DOI] [PubMed] [Google Scholar]
- Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA et al (2017) The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev 31: 34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firat‐Karalar EN, Stearns T (2014) The centriole duplication cycle. Philos Trans R Soc Lond B Biol Sci 369: 20130460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong CS, Mazo G, Das T, Goodman J, Kim M, O'Rourke BP, Izquierdo D, Tsou MF (2016) 53BP1 and USP28 mediate p53‐dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife 5: e16270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Hagan IM, Glover DM (2015) The centrosome and its duplication cycle. Cold Spring Harb Perspect Biol 7: a015800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature 460: 278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Pellman D (2012) Linking abnormal mitosis to the acquisition of DNA damage. J Cell Biol 199: 871–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Cornils H, Chiu SY, O'Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D (2014) Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 158: 833–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganier O, Schnerch D, Nigg EA (2018a) Structural centrosome aberrations sensitize polarized epithelia to basal cell extrusion. Open Biol 8: 180044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganier O, Schnerch D, Oertle P, Lim RY, Plodinec M, Nigg EA (2018b) Structural centrosome aberrations promote non‐cell‐autonomous invasiveness. EMBO J 37: e98576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho SA, Picone R, Burute M, Dagher R, Su Y, Leung CT, Polyak K, Brugge JS, Thery M, Pellman D (2014) Oncogene‐like induction of cellular invasion from centrosome amplification. Nature 510: 167–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonczy P (2012) Towards a molecular architecture of centriole assembly. Nat Rev Mol Cell Biol 13: 425–435 [DOI] [PubMed] [Google Scholar]
- Hagan IM, Grallert A (2013) Spatial control of mitotic commitment in fission yeast. Biochem Soc Trans 41: 1766–1771 [DOI] [PubMed] [Google Scholar]
- Holland AJ, Fachinetti D, Zhu Q, Bauer M, Verma IM, Nigg EA, Cleveland DW (2012) The autoregulated instability of Polo‐like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev 26: 2684–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH (2011) Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 333: 1895–1898 [DOI] [PubMed] [Google Scholar]
- Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, Holland AJ (2016) A USP28‐53BP1‐p53‐p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol 214: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MS, Bakker B, Boeckx B, Moyett J, Lu J, Vitre B, Spierings DC, Lansdorp PM, Cleveland DW, Lambrechts D et al (2017) Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev Cell 40: 313–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS (2014) MAGeCK enables robust identification of essential genes from genome‐scale CRISPR/Cas9 knockout screens. Genome Biol 15: 554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoMastro GM, Holland AJ (2019) The emerging link between centrosome aberrations and metastasis. Dev Cell 49: 325–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marconi C, Canobbio I, Bozzi V, Pippucci T, Simonetti G, Melazzini F, Angori S, Martinelli G, Saglio G, Torti M et al (2017) 5'UTR point substitutions and N‐terminal truncating mutations of ANKRD26 in acute myeloid leukemia. J Hematol Oncol 10: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez R, Hantel A, Lorenz R, Neistadt B, Wong J, Churpek JE, Mardini NA, Shaukat I, Gurbuxani S, Miller JL et al (2014) A new family with a germline ANKRD26 mutation and predisposition to myeloid malignancies. Leuk Lymphoma 55: 2945–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitinger F, Anzola JV, Kaulich M, Richardson A, Stender JD, Benner C, Glass CK, Dowdy SF, Desai A, Shiau AK et al (2016) 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol 214: 155–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskvin‐Tarkhanov MI, Onishchenko GE (1978) Centrioles in mouse bone marrow megakaryocytes. Tsitologiia 20: 1436–1438 [PubMed] [Google Scholar]
- Moyer TC, Clutario KM, Lambrus BG, Daggubati V, Holland AJ (2015) Binding of STIL to Plk4 activates kinase activity to promote centriole assembly. J Cell Biol 209: 863–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer TC, Holland AJ (2019) PLK4 promotes centriole duplication by phosphorylating STIL to link the procentriole cartwheel to the microtubule wall. Elife 8: e46054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Stearns T (2011) The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol 13: 1154–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Holland AJ (2018) Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol 19: 297–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noris P, Favier R, Alessi MC, Geddis AE, Kunishima S, Heller PG, Giordano P, Niederhoffer KY, Bussel JB, Podda GM et al (2013) ANKRD26‐related thrombocytopenia and myeloid malignancies. Blood 122: 1987–1989 [DOI] [PubMed] [Google Scholar]
- Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ, Hubbard D, Bhutkar A, Jacks T (2011) Caspase‐2‐mediated cleavage of Mdm2 creates a p53‐induced positive feedback loop. Mol Cell 43: 57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, Wu H (2007) Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128: 533–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippucci T, Savoia A, Perrotta S, Pujol‐Moix N, Noris P, Castegnaro G, Pecci A, Gnan C, Punzo F, Marconi C et al (2011) Mutations in the 5' UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal‐dominant form of inherited thrombocytopenia, THC2. Am J Hum Genet 88: 115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebacz B, Larsen TO, Clausen MH, Ronnest MH, Loffler H, Ho AD, Kramer A (2007) Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype‐based screen. Cancer Res 67: 6342–6350 [DOI] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, Leonhardt H (2008) A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics 7: 282–289 [DOI] [PubMed] [Google Scholar]
- Sedjaï F, Acquaviva C, Chevrier V, Chauvin JP, Coppin E, Aouane A, Coulier F, Tolun A, Pierres M, Birnbaum D et al (2010) Control of ciliogenesis by FOR20, a novel centrosome and pericentriolar satellite protein. J Cell Sci 123(Pt 14): 2391–2401 [DOI] [PubMed] [Google Scholar]
- Sercin O, Larsimont JC, Karambelas AE, Marthiens V, Moers V, Boeckx B, Le Mercier M, Lambrechts D, Basto R, Blanpain C (2016) Transient PLK4 overexpression accelerates tumorigenesis in p53‐deficient epidermis. Nat Cell Biol 18: 100–110 [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG et al (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth WT, Nardi IK, Scholl LM, Cimini D (2009) Multipolar spindle pole coalescence is a major source of kinetochore mis‐attachment and chromosome mis‐segregation in cancer cells. PLoS One 4: e6564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladky V, Schuler F, Fava LL, Villunger A (2017) The resurrection of the PIDDosome – emerging roles in the DNA‐damage response and centrosome surveillance. J Cell Sci 130: 3779–3787 [DOI] [PubMed] [Google Scholar]
- Sladky VC, Knapp K, Soratroi C, Heppke J, Eichin F, Rocamora‐Reverte L, Szabo TG, Bongiovanni L, Westendorp B, Moreno E et al (2020) E2F‐family members engage the PIDDosome to limit hepatocyte ploidy in liver development and regeneration. Dev Cell 52: 335–349 [DOI] [PubMed] [Google Scholar]
- Sladky VC, Villunger A (2020) Uncovering the PIDDosome and caspase‐2 as regulators of organogenesis and cellular differentiation. Cell Death Differ 27: 2037–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanos BE, Yang HJ, Soni R, Wang WJ, Macaluso FP, Asara JM, Tsou MF (2013) Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev 27: 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinel A, Tschopp J (2004) The PIDDosome, a protein complex implicated in activation of caspase‐2 in response to genotoxic stress. Science 304: 843–846 [DOI] [PubMed] [Google Scholar]
- Tinel A, Janssens S, Lippens S, Cuenin S, Logette E, Jaccard B, Quadroni M, Tschopp J (2007) Autoproteolysis of PIDD marks the bifurcation between pro‐death caspase‐2 and pro‐survival NF‐kappaB pathway. EMBO J 26: 197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou MF, Stearns T (2006) Controlling centrosome number: licenses and blocks. Curr Opin Cell Biol 18: 74–78 [DOI] [PubMed] [Google Scholar]
- Vitrat N, Cohen‐Solal K, Pique C, Le Couedic JP, Norol F, Larsen AK, Katz A, Vainchenker W, Debili N (1998) Endomitosis of human megakaryocytes are due to abortive mitosis. Blood 91: 3711–3723 [PubMed] [Google Scholar]
- Wang R, Wei Z, Jin H, Wu H, Yu C, Wen W, Chan LN, Wen Z, Zhang M (2009) Autoinhibition of UNC5b revealed by the cytoplasmic domain structure of the receptor. Mol Cell 33: 692–703 [DOI] [PubMed] [Google Scholar]
- Wang M, Nagle RB, Knudsen BS, Cress AE, Rogers GC (2019) Centrosome loss results in an unstable genome and malignant prostate tumors. Oncogene 39: 399–413 [DOI] [PubMed] [Google Scholar]
- Yang TT, Chong WM, Wang WJ, Mazo G, Tanos B, Chen Z, Tran TMN, Chen YD, Weng RR, Huang CE et al (2018) Super‐resolution architecture of mammalian centriole distal appendages reveals distinct blade and matrix functional components. Nat Commun 9: 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Zeng H, Ning G, Reiter JF, Liu A (2014) C2cd3 is critical for centriolar distal appendage assembly and ciliary vesicle docking in mammals. Proc Natl Acad Sci USA 111: 2164–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH et al (2013) Pan‐cancer patterns of somatic copy number alteration. Nat Genet 45: 1134–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Data Availability Statement
This study includes no data deposited in external repositories.